

Significance
Maturation of early to late endosomes is crucial for maintaining endocytosis in eukaryotic cells. One hallmark of endosomal maturation is the transition from the early endosomal Rab5 to the late endosomal Rab7. Rab GTPases serve as identity markers for organelles. One key regulator of the endosomal Rab transition is the conserved Rab7–GEF (guanine nucleotide exchange factor) complex Mon1–Ccz1. It is involved in the inactivation of Rab5 and the activation of Rab7. It has been clarified how Mon1–Ccz1 activates Rab7. However, how the activity of this GEF is regulated is poorly understood. Here, we focused on Mon1 and identified two distinct regulatory sites involved in the correct spatiotemporal activation of this GEF complex, which ensures accurate endosomal Rab transition.
Keywords: endosome, lysosome, Mon1, Rab–cascade, endosomal maturation
Abstract
Maturation from early to late endosomes depends on the exchange of their marker proteins Rab5 to Rab7. This requires Rab7 activation by its specific guanine nucleotide exchange factor (GEF) Mon1–Ccz1. Efficient GEF activity of this complex on membranes depends on Rab5, thus driving Rab–GTPase exchange on endosomes. However, molecular details on the role of Rab5 in Mon1–Ccz1 activation are unclear. Here, we identify key features in Mon1 involved in GEF regulation. We show that the intrinsically disordered N-terminal domain of Mon1 autoinhibits Rab5-dependent GEF activity on membranes. Consequently, Mon1 truncations result in higher GEF activity in vitro and alterations in early endosomal structures in Drosophila nephrocytes. A shift from Rab5 to more Rab7-positive structures in yeast suggests faster endosomal maturation. Using modeling, we further identify a conserved Rab5-binding site in Mon1. Mutations impairing Rab5 interaction result in poor GEF activity on membranes and growth defects in vivo. Our analysis provides a framework to understand the mechanism of Ras-related in brain (Rab) conversion and organelle maturation along the endomembrane system.
All eukaryotic cells utilize endocytosis for homeostasis of their plasma membrane and exchange of materials with the cell’s surroundings. Endocytic cargo is packed into endocytic vesicles, which pinch off at the plasma membrane and fuse with early endosomes. Early endosomes fuse and mature into late endosomes, finally delivering endocytic cargo to the degradative lysosome. Two members of the Rab subfamily of small GTPases, Rab5 and Rab7, are critical identity markers within the endolysosomal pathway (1). Rab5 decorates early endosomal compartments. During maturation of early to late endosomes, Rab5 is replaced by Rab7. This transition is critical for the functionality of each organelle, and a shift in this fine balance can lead to diseases like Charcot–Marie–Tooth syndrome type 2 (2).
Rab GTPases (Rabs) are guanine-nucleotide-binding proteins with a weak intrinsic GTPase activity. Rabs are prenylated at their C terminus to allow for membrane association and cycle between a soluble GDP- and a membrane-anchored GTP-bound state. Within the cytosol, the GDP dissociation inhibitor (GDI) binds to the GDP-bound form and covers its prenyl anchor. The region between the GTPase domain and prenyl anchor, called the hypervariable domain (HVD), is rather flexible. Rabs can dissociate from GDI and thereby sample membranes in the cell. If they encounter their cognate guanine nucleotide exchange factor (GEF) at their target organelle, GDP is exchanged for the more abundant guanosine triphosphate (GTP). In this active GTP-bound state, Rab–GTP stably associates with the membrane and can recruit and bind effector proteins, for instance, tethers or motor proteins. Specific GTPase activating proteins (GAP) can bind to their respective Rab and catalyze the GTP-hydrolysis, thereby transferring the Rab back to its inactive state which can be extracted by GDI (1, 3, 4).
On early endosomes, and possibly already on endocytic vesicles, Rab5 is activated by its GEF Rabex-5. Active Rab5 recruits, among others, its effector Rabaptin-5. Rabaptin-5 in turn interacts with Rabex-5 resulting in a positive feedback loop establishing Rab5 domains on endosomal membranes (5, 6). Other effectors of Rab5 are, for instance, the tethering complex CORVET and the phosphatidylinositol-3-phosphate (PI3P) kinase complex II (7, 8).
An additional Rab5 effector is the GEF for the late endosomal Rab7, the Mon1–Ccz1 complex (9–12), a heterodimer in yeast, and a heterotrimer in metazoan cells (13–16). The third subunit, called RMC1/C18orf8 in humans or Bulli in Drosophila, is required for a functional endosomal system and autophagy in metazoan cells (14–16). However, GEF activity of the Drosophila Mon1–Ccz1 dimer and trimer is comparable, suggesting that the third subunit functions in other processes rather than regulating GEF activity (10, 16). Mon1–Ccz1 belongs to the family of Tri-Longin-GEFs, in which each subunit consists of three Longin domains (LDs). This family includes the BLOC3 complex of Hps1 and Hps4, a GEF of Rab32 and 38 involved in the biogenesis of lysosome-related organelles (17), and the CPLANE complex with its three subunits Fuzzy, Inturned, and Wdpcp, which also binds Rsg1 as a noncanonical Rab–like protein (18). Within Mon1–Ccz1, LD1 of both subunits forms the active site (19, 20), whereas LD2 and LD3 of both proteins form a layer responsible for membrane association (21). Rab7 activation by Mon1–Ccz1 is necessary for fusion events of late endosomes and autophagosomes with the lysosome.
Transition from a Rab5-positive to a Rab7-positive domain on the verge from early to late endosomes has been termed Rab cascade (1, 3, 4, 22, 23). Mon1–Ccz1 is an effector of Rab5 and is recruited to Rab5-positive membranes and subsequently activates Rab7 (Fig. 1A) (9–11). Using reconstitution, we showed before that the presence of active Rab5 on membranes increases GEF activity of Mon1–Ccz1 suggesting an activation of the GEF complex on endosomal membranes on the verge to late endosomes (10, 21). However, it remains unclear how binding to Rab5 activates Mon1–Ccz1. It is also unresolved how a sharp spatiotemporal transition between Rab5 and Rab7 domains in the endolysosomal pathway is achieved and why Mon1–Ccz1 is not activated by Rab5 on early endosomal structures.
Fig. 1.
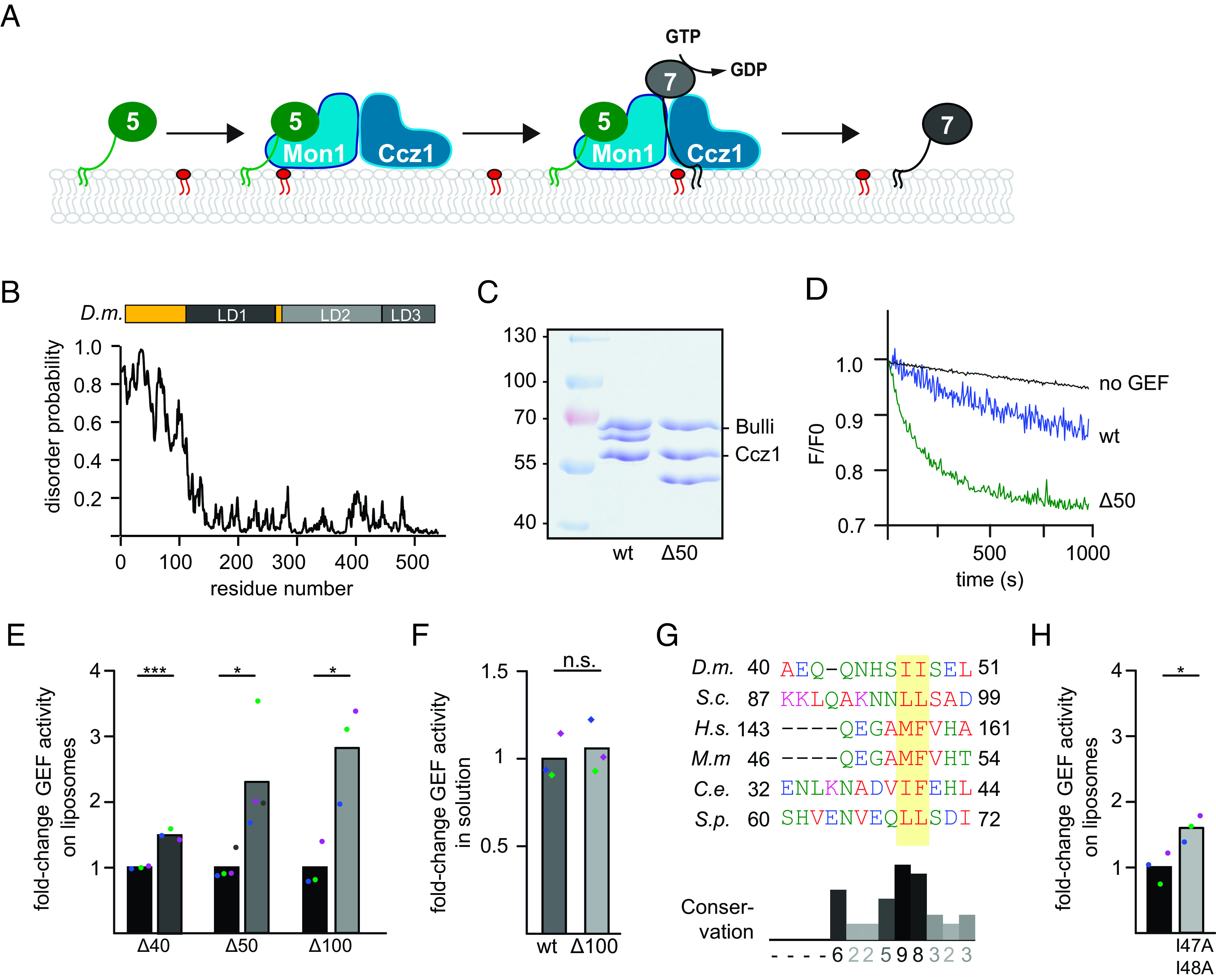
The disordered N-terminal region of Drosophila Mon1 regulates Mon1–Ccz1 GEF activity. (A) Overview of the Rab5-Rab7 cascade on endosomes. Active Rab5 (green) recruits the Mon1–Ccz1 GEF complex (blue) to PI3P-positive (red) endosomal membranes and promotes Rab7 (gray) recruitment and activation. For details, see text. (B) The N-terminal region of D.m. Mon1 is disordered. The disorder probability of each residue of D.m. Mon1 was determined using the IUPred2A web interface (24, 25). Longin domains (LDs) 1 to 3 are depicted in gray shades. Values >0.5 are considered disordered. (C) Truncation of Mon1 does not affect the complex stability of the trimeric Mon1Δ1-50–Ccz1–Bulli complex (∆50) compared to wild-type complex (wt). GEF complexes were purified as described in the Materials and Methods section and analyzed by SDS-PAGE and Coomassie staining. (D) On liposome GEF assay of Mon1wt and Mon1Δ1-50 containing trimer. Liposomes were preloaded with 150 nM prenylated Rab5 in the presence of 200 µM GTP and 1.5 mM Ethylenediaminetetraacetic acid (EDTA). The nucleotide was stabilized using 3 mM MgCl2. 250 nM Mant-GDP-loaded Rab7:GDI was added, and nucleotide exchange was triggered by adding 6.25 nM wild-type (blue) or mutant (green) GEF complex. The decrease in fluorescence was measured over time and normalized to fluorescence prior to GEF addition. (E) Comparison of fold-change in GEF activity of various Mon1 truncations. GEF assays were performed as in D, and kobs of each curve was determined as described in the Materials and Methods section. kobs values of mutants were normalized to the corresponding wild-type value in the respective experiment. Bar graphs represent average fold-change to the respective wild-type value, and dots represent individual changes from at least three experiments. (P value: *P < 0.05 and **P < 0.01, using a two-sample Student’s t test assuming equal variances). For kinetic constants of all GEF complexes, see SI Appendix, Table S3. (F) GEF activity of wild type and Mon1Δ1-100 containing trimer in solution. 2 µM nonprenylated Rab was incubated with increasing amounts of GEF. After baseline stabilization, nucleotide release was triggered by adding 0.1 mM GTP final. For details of data fitting and statistics, see the Materials and Methods section. The kcat/KM (M−1s−1) value for Mon1Δ1-100 containing trimer was normalized to the value of Mon1wt. Bar graphs represent average fold-change, and dots represent individual changes from three experiments. (P value: n.s. using a two-sample Student’s t test assuming equal variances). (G) Multiple sequence alignment of a hydrophobic patch in Mon1. The N-terminal regions of Mon1 from Drosophila melanogaster (D.m.), Saccharomyces cerevisiae (S.c.), Homo sapiens (H.s.), Mus musculus (M.m.), Caenorhabditis elegans (C.e.), and Schizosaccharomyces pombe (S.p.) were aligned using th Clustal omega web interface (26, 27). The hydrophobic patch is marked by a yellow box. Conservation was determined using Jalview (28). (H) Comparison of fold-change in GEF activity of Mon1I47, 48A. GEF assays were performed as in D, and kobs of each curve was determined as described in the Materials and Methods section. kobs values of mutants were normalized to the corresponding wild-type value in the respective experiment. Bar graphs represent average fold-change to the respective wild-type value, and dots represent individual changes from three experiments (P value: *P < 0.05, using a two-sample Student’s t test assuming equal variances). For kinetic constants of all GEF complexes, see SI Appendix, Table S3.
To address these questions, we investigated here a possible regulatory function of the mainly intrinsically disordered Mon1 N-terminal region. We show that this domain autoinhibits Mon1–Ccz1 and further identify a conserved Rab5 binding site in yeast and Drosophila Mon1 by structural modeling. Our results provide a framework on the regulation of GEFs, which thus control Rab transitions during organelle maturation.
Results
Mon1–Ccz1 Activity Is Autoinhibited.
We showed before that activity of the Rab7–GEF Mon1–Ccz1–Bulli is stimulated by Rab5 in a reconstituted system (10, 16), suggesting an order of events that promotes endosomal maturation (Fig. 1A) (1). In the past, we determined the structure of the dimeric Mon1–Ccz1 complex and identified the overall organization of the complex and a unique mechanism to drive nucleotide exchange of Rab7 (19, 20). However, in both structures, the N-terminal domain of Mon1 was not resolved. This missing part is predicted to be disordered in Drosophila melanogaster (D.m.) (Fig. 1B), Saccharomyces cerevisiae (S.c.), and human Mon1 (SI Appendix, Fig. S1 A and B). This feature is conserved and unique among TLD (tri-LD) subunits, suggesting a functional role of this region. We generated truncations of the N-terminal part and analyzed purified Mon1–Ccz1–Bulli complexes in vitro. All complexes were purified as a trimer (Fig. 1C and SI Appendix, Fig. S1 C and D).
To test for activity, we used an established in vitro GEF assay (10). In brief, Mon1–Ccz1–Bulli activity was determined by following the release of a fluorescently labeled GDP (Mant-GDP) from a preloaded Rab7-GDI complex in the presence of unlabeled GTP upon GEF addition. To mimic the membrane environment of the cell, this assay was conducted in the presence of liposomes, which carried prenylated Rab5-GTP that is required for efficient Rab7 GEF activity (10). We then tested the effect of Mon1 truncations on GEF activity. Compared to the wild-type complex, N-terminal truncations of the first 40, 50, or 100 residues resulted in a 1.5 to 3.5-fold increase in GEF activity (Fig. 1 D and E and SI Appendix, Table S3). This suggested that the N-terminal unstructured region inhibits Mon1–Ccz1. Surprisingly, GEF activity of the most active Mon1–Ccz1 mutant was like wild type in solution using nonprenylated, soluble Ypt7 as a substrate (Fig. 1F). Closer inspection of the region between residues 40 and 50 of D.m. Mon1 revealed a conserved hydrophobic motif in this region (Fig. 1G), A conservative I47, 48A mutant also resulted in an increase in Rab5-dependent Mon1–Ccz1–Bulli activity compared to wild type (Fig. 1H and SI Appendix, Fig. S1E), indicating that this hydrophobic patch regulates the availability of Mon1 and thus controls GEF complex activity.
The Regulatory Role of the Mon1 N-Terminal Region Is Conserved.
We next asked whether we could observe an effect of the Mon1 N-terminal truncation in Drosophila in vivo. In previous work, we analyzed the effect of Bulli deletions on the morphology and function of nephrocytes. Drosophila nephrocytes share histological and functional similarities with mammalian kidney podocytes, for example, a filtration apparatus and a complex endocytic system (16, 29). As the Drosophila GEF complex carrying Mon1∆100 had higher GEF activity than the wild type (Fig. 1E), we expected that expression of Mon1∆100 in Drosophila nephrocytes would generate a dominant effect on the morphology of the endocytic system. Therefore, a Drosophila line was generated expressing Mon1∆100, which was verified by western blot analysis (SI Appendix, Fig. S2A). As Bulli mutants had a strongly impaired ultrastructural morphology (16, 29), we initially analyzed nephrocytes by electron microscopy. The expression of Mon1∆100 had, however, little visible consequences on the histology of nephrocytes. Ultrastructural analyses revealed similar slit diaphragms, serving as filtration barriers, and a normal labyrinth channel system, representing the endocytic compartment. We also identified coated vesicles, early endosomes, and other typical organelles of nephrocytes (SI Appendix, Fig. S2B). The expression of Mon1∆100 also did not affect the endocytic activity of nephrocytes, as tested by uptake assays with Fluorescein isothiocyanate (FITC) albumin (SI Appendix, Fig. S2 C and D). These observations were not unexpected, as the Mon1∆100 supports Rab7 recruitment, and subtle defects in endolysosomal transport may not become apparent by these assays. However, the allele may nevertheless change the apparent transition from early to late endosomes.
To determine possible effects on the Rab transition in the endosomal system, we stained nephrocytes expressing Mon1∆100 for Rab5 and Rab7 and observed a clear decrease in the size of Rab5-positive endosomal structures (Fig. 2 A and B). However, changes in Rab7 staining were less obvious (SI Appendix, Fig. S2E), and also, the number of Rab5-positive structures did not show a significant change (SI Appendix, Fig. S2F). We thus conclude that the Mon1∆100 allele causes a shift in the balance from early to late endosomes and thus enhances the Rab5 to Rab7 transition in nephrocytes, while still allowing for endocytic transport. Since this observation is based on an overexpression of the alleles while having a wild-type copy expressed in the background, we turned toward the more amendable yeast system.
Fig. 2.

Loss of the disordered Mon1 N-terminal region affects localization of endosomal Rab GTPases and Rab5-dependent GEF activity. (A) Localization of endogenous Rab5 in Drosophila nephrocytes from 3rd instar larvae expressing wild type or Mon1∆100 under the control of the handC-GAL4 driver. Antibodies against Rab5 were used. Optical sections show the distribution of Rab5 in detail. Size bar: 10 µm. (B) Analysis of the mean size of Rab5 dots per cell from 15 cells from three animals for wild-type Mon1 and 13 cells from four animals for Mon1∆100. (P value ****< 0.0001 using an unpaired, two-sample Student’s t test). (C–F) Truncation of the Mon1 N-terminal affects Ypt7 and Vps21 localization. (C) Localization of mNEON-Ypt7 under its endogenous promotor in yeast cells in the presence of endogenously expressed wild type or Mon1Δ100 using fluorescence microscopy. Vacuoles were stained with FM4-64. Size bar, 2 µm, arrows show representative Ypt7 accumulations. (D) Quantification of Ypt7-positive dots in C. Cells (n ≥ 50) were counted from three independent experiments; (P value ***< 0.001 using a two-sample Student’s t test assuming equal variances). For image processing details, see the Materials and Methods section. (E) Localization of mCherry-tagged Vps21 in cells expressing mNeon-Ypt7 and endogenously expressed wild type or Mon1Δ100 using fluorescence microscopy. Size bar 2 µm, arrows show representative Vps21 accumulations. (F) Quantification of Vps21-positive dots in E. Cells (n ≥ 50) were counted from three independent experiments. (P value ***< 0.001 using a two-sample Student’s t test assuming equal variances). For image processing details, see the Materials and Methods section. (G) Effect on cell growth by Mon1 truncations. Strains endogenously expressing wild type or Mon1Δ100 were grown to the same OD600 in Yeast extract peptone dextrose (YPD) media and spotted in serial dilutions onto agar plates containing YPD or YPD supplemented with 4 mM ZnCl2. Plates were incubated for several days at 30 °C. (H) A hydrophobic patch mutation in the N-terminal part of Mon1 affects GEF activity. Liposomes were loaded with 150 nM prenylated Ypt10 in the presence of 200 µM GTP and 1.5 mM EDTA. The nucleotide was stabilized using 3 mM MgCl2. 250 nM Mant-GDP-loaded Ypt7:GDI was added, and the reaction was triggered by adding 12.5 nM wild-type (blue) or mutant complex expressing Mon1L95A,L96A (green). The decrease in fluorescence detected over time is normalized to initial fluorescence. (I) Comparison of fold-change in GEF activity of wild-type and Mon1L95A,L96A mutant GEF complexes on liposomes and in solution. For details of in solution GEF assay, see the Materials and Methods section. For the GEF assay on liposomes, kobs of each curve was determined as described in the Materials and Methods section. kobs values were normalized to the wild-type value. For in solution assays, kcat/Km values were determined and normalized to the wild-type value. Bar graphs represent average fold-change, and dots represent individual changes from three experiments. (P value: *P < 0.05 using a two-sample Student’s t test assuming equal variances).
Here, we deleted the first 100 residues in S.c. Mon1 (S.c. Mon1∆100), which correspond to the region including the hydrophobic patch, (Fig. 1G and SI Appendix, Fig. S2G), and analyzed the corresponding mutant for Ypt7 (yeast Rab7) and Vps21 (yeast Rab5) localization. In wild-type cells, endogenously expressed mNEON-Ypt7 localized to the vacuolar membrane and single dots. Cells expressing Mon1∆100 had round vacuoles, suggesting sufficient Ypt7 activation. However, Ypt7 now localized more prominently to several perivacuolar dots in addition to its localization to the vacuolar membrane, suggesting an endosomal localization (Fig. 2 C and D). We then analyzed the localization of the early endosomal Rab5-like Vps21 and observed that S.c. Mon1∆100 cells had significantly less Vps21-positive dots than wild-type cells (Fig. 2 E and F). This suggests that the balance between early Rab5/Vps21-positive endosomes and late endosomal Rab7/Ypt7-positive structures is altered in Mon1∆100-expressing cells. The localization of the Mon1–Ccz1 complex (SI Appendix, Fig. S3A) as well as the overall expression of both Rab GTPases was unaltered (SI Appendix, Fig. S3 B and C). To determine whether the truncation of Mon1 affects cellular physiology, we grew cells in sequential dilutions on plates containing 4 mM ZnCl2 (Fig. 2G). S.c. Mon1∆100 cells showed a slight growth defect in comparison to wild-type cells, indicating that S.c. Mon1∆100 cells had problems with ion detoxification due to a defect in the endolysosomal pathway. To determine whether S.c. Mon1∆100 cells had also defects in autophagy, we followed transport of the cytosolic Pho8∆60 reporter into the vacuole during nitrogen starvation (30) but detected no difference in Pho8 activity of mutant versus wild-type cells (SI Appendix, Fig. S3D).
We then asked whether the N-terminal truncation of S.c. Mon1 also resulted in higher Mon1–Ccz1 activity as observed for the truncated D.m. Mon1. However, the N-terminal deletion in Mon1 did not yield sufficiently clean purified Mon1–Ccz1 complex. We therefore tested whether a mutation in the corresponding hydrophobic motif present in the N-terminal domain (Fig. 1G; S.c. Mon1L95A,L96A) phenocopies the effect of the mutation in D.m. Mon1 (Fig. 1E). Introducing these mutations in Mon1 rescued the vacuolar morphology of a MON1 deletion strain and the GEF complex localized like wild-type Mon1–Ccz1 in vivo indicating stable complex formation (SI Appendix, Fig. S3E). It allowed purification of stable Mon1–Ccz1 (SI Appendix, Fig. S3F), which had an increased Rab5-dependent activity on liposomes when compared to wild type, but comparable GEF activity in solution (Fig. 2 H and I and SI Appendix, Table S3). However, this mutation in Mon1 did not impair growth of corresponding mutant strains on plates containing ZnCl2 (Fig. 3H), in agreement with the milder phenotype in vitro (Fig. 2H). Overall, these findings are in agreement with the altered endocytic system of Drosophila nephrocytes that express the more active D.m. Mon1∆100.
Fig. 3.
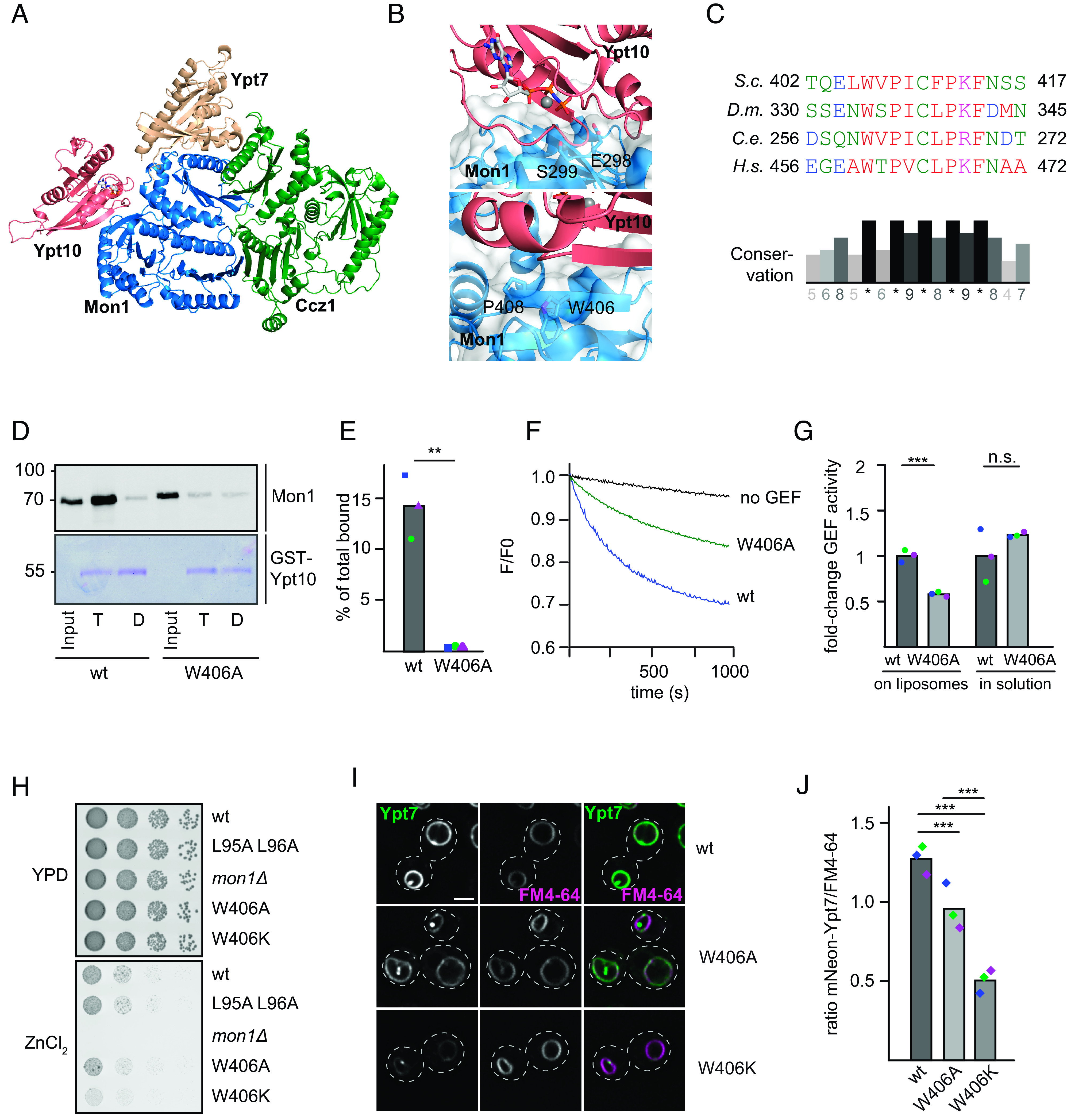
Identification of the Rab5-binding site in the Mon1–Ccz1 complex. (A) Identification of the putative Rab5-binding site in Mon1. Composite model of the S.c. Mon1 (blue)-Ccz1 (green)-Ypt7 (beige)-Ypt10 (pink) complex based on an Alphafold 2 prediction (31, 32) and the crystal structure of the catalytic core complex (PDB ID: 5LDD) (19, 32, 33). (B) Close-up view of the Mon1–Ypt10 binding interface. Colors are as in A. (C) Multiple sequence alignment of Mon1 β-strand in the modeled Rab5 binding region. Mon1 sequences from S.c., D.m., Caenorhabditis elegans (C.e.), and Homo sapiens (H.s.) were aligned using the Clustal omega web interface. Conservation was determined using Jalview. (D) Effect of the Mon1W406A mutant on Rab5 binding. 75 µg purified GST-Ypt10 was loaded with GTP (T) or GDP (D) and incubated with 25 µg of either wild-type or Mon1W406A GEF complex. Elution of bound GEF was performed with EDTA. 20% of the eluate was analyzed together with 1% input by western blotting using an anti-Mon1 antibody. 2% GST-Ypt10 was stained with Coomassie as loading control (E) Quantification of bound GEF complex to Ypt10-GTP. The band intensity of Mon1 signal in elution fraction was measured using Fiji software and compared to input signal. (P value **< 0.01 using a two-sample Student’s t test assuming equal variances). (F) Effect of the Mon1W406 mutation on Rab5-dependent GEF activity. 250 nM Mant-GDP-loaded Ypt7:GDI was added, and Rab activation was measured by the fluorescence decrease over time. Liposomes were loaded with 150 nM prenylated Ypt10 using 200 µM GTP and 1.5 mM EDTA. The nucleotide was stabilized using 3 mM MgCl2. The reaction was triggered by adding 25 nM GEF complex. The decrease in fluorescence was normalized to fluorescence prior to GEF addition. (G) Comparison of fold-change in GEF activity of Mon1W406A to Mon1wt complex on liposomes and in solution. For details of in solution GEF assay, see Materials and Methods. For GEF assay on liposomes, kobs of each curve was determined as described in the Materials and Methods section. kobs values of mutant were normalized to the wild-type GEF complex value. For in solution assays, kcat/Km values were normalized to the wild-type value. Bar graphs represent average fold-change, and dots represent individual values from three experiments. (P value ***< 0.001, using a two-sample Student’s t test assuming equal variances). (H) Growth assay. Indicated Mon1 variants in a mon1 deletion background were grown to the same OD600 in YPD media and then spotted in serial dilutions onto agar plates containing YPD or YPD supplemented with 4 mM ZnCl2. Plates were incubated for several days at 30 °C. (I) Localization of Ypt7 in wild-type and mutant strains. Plasmids encoding Mon1wt, Mon1W406A, or Mon1W406K variants were expressed under their endogenous promoter in a mon1 deletion strain. Vacuoles were stained with FM4-64. Images were deconvolved (SOftWoRx software 5.5). Size bar, 2 µm. (J) Quantification of the ratio between FM4-64 and mNeon-Ypt7 mean intensity signal on the vacuolar rim. Mean intensity signals were determined by a line profile across the vacuole. For details of image processing and quantification, see the Materials and Methods section. Cells (n ≥ 50) were counted from three independent experiments. (P value ***< 0.001 using a one-way ANOVA).
Taken together, these results suggest a regulatory role of the intrinsically disordered Mon1 N-terminal region including a short, conserved hydrophobic motif. The hyperactivity of Mon1∆100 is only observed in the Rab5-dependent GEF assay on liposomes, suggesting that the N-terminal region either affects Rab5 binding to Mon1–Ccz1 or access of the substrate Ypt7 to the active site in a membrane context.
Rab5 Interacts with a Conserved Region of Mon1.
To further elucidate the Rab5-dependent regulation of Mon1–Ccz1 GEF activity, we searched for interaction sites between Rab5 and Mon1– Ccz1 using AlphaFold2. As we observed the strongest interaction between the Rab5-like Ypt10 and S.c. Mon1–Ccz1 in the past (10), we used these proteins as templates for our modeling. Using AlphaFold2-Multimer, we generated a model for the S.c. Mon1–Ccz1 complex bound to Ypt7 (nucleotide-free) and Ypt10 (GTP-bound) (Fig.3A and SI Appendix, Fig. S4 A–C). In these models, Ypt10 was consistently positioned to interact with a β-sheet in LD2 of Mon1 with high confidence (S4C). A similar binding mode is predicted for D.m. Rab5 to D.m. Mon1 (SI Appendix, Fig. S4 D and E) and observed for the interaction of Rsg1 to the Fuzzy subunit of CPLANE (SI Appendix, Fig. S4F), further supporting the validity of the proposed interaction site. This region is next to the so-called elbow-loop of Mon1, which is involved in Mon1–Ccz1 GEF function and its interaction with Ypt7 (19). The loop residues E298 and S299 of Mon1 are predicted to interact with the P-loop and switch regions of Ypt10 (Fig. 3 B, Upper).
The proposed interface contains a highly conserved motif in LD2 of Mon1 with a hydrophobic residue (Fig. 3C) that corresponds to the surface-exposed W406 in S.c. Mon1 (Fig. 3 B, Lower). Expression and purification of a corresponding Mon1W406A-Ccz1 complex resulted in a stable preparation (SI Appendix, Fig. S4G). We then asked whether the mutant complex was still able to interact with the Rab5-like Ypt10. We therefore bound GTP- or GDP-loaded GST-Ypt10 on glutathione sepharose and added either purified wild-type or Mon1W406A complex. Whereas wild-type Mon1–Ccz1 showed robust binding to Ypt10-GTP as shown before (10), almost no Ypt10 binding was observed for the mutant complex, suggesting that the mutation strongly impaired Ypt10 binding (Fig. 3 D and E). In agreement, the purified Mon1W406A-Ccz1 complex had half the Rab5-dependent GEF activity toward Ypt7 on membranes (Fig. 3 F and G and SI Appendix, Table S3). Importantly, GEF activity was unperturbed in solution (Fig. 3G and SI Appendix, Table S3). This suggests that Rab5 binding results in activation of Mon1–Ccz1 on membranes.
We wondered whether deficient Rab5 binding of Mon1W406A would affect Mon1–Ccz1 function in vivo. To address this, we transformed a MON1 deletion strain with plasmids either carrying MON1 or MON1W406A. Both alleles rescued the growth defect of the MON1 deletion strain on medium containing ZnCl2 (Fig. 3H). We then analyzed vacuolar morphology and localization of mNEON-tagged Ypt7 and observed an almost wild-type-like localization of Ypt7 and normal vacuole morphology in Mon1W406A cells (Fig. 3 I and J). However, mutation of W406 into a basic lysine residue in Mon1 resulted in far more dramatic phenotypes. Cells expressing Mon1W406K had a strong decrease in vacuolar rim signal (Fig. 3 I and J), consistent with a growth deficiency in vivo (Fig. 3H). The overall expression of Ypt7 was not affected in these strains (SI Appendix, Fig. S4H) This suggests problems with correct spatiotemporal activation of Ypt7, possibly due to insufficient Mon1–Ccz1 activation by S.c. Rab5.
To test whether the Rab5-binding site in Mon1 is conserved among species (Fig. 3 B and C), we went back to the Drosophila system. The structural analysis of the D.m. Mon1–Ccz1–Bulli complex (34) suggested a similar binding interface for D.m. Rab5 as identified for S.c. Mon1 (SI Appendix, Fig.S4 D and E). We therefore generated a corresponding Mon1W334A mutation in the trimeric Mon1–Ccz1–Bulli complex and obtained a stable complex (Fig. 4A). Strikingly, this complex showed reduced activity in the Rab5-dependent GEF-assay (Fig. 4 B and C and SI Appendix, Table S3) but had normal GEF activity in solution toward Rab7 (Fig. 4C and SI Appendix, Table S3).
Fig. 4.
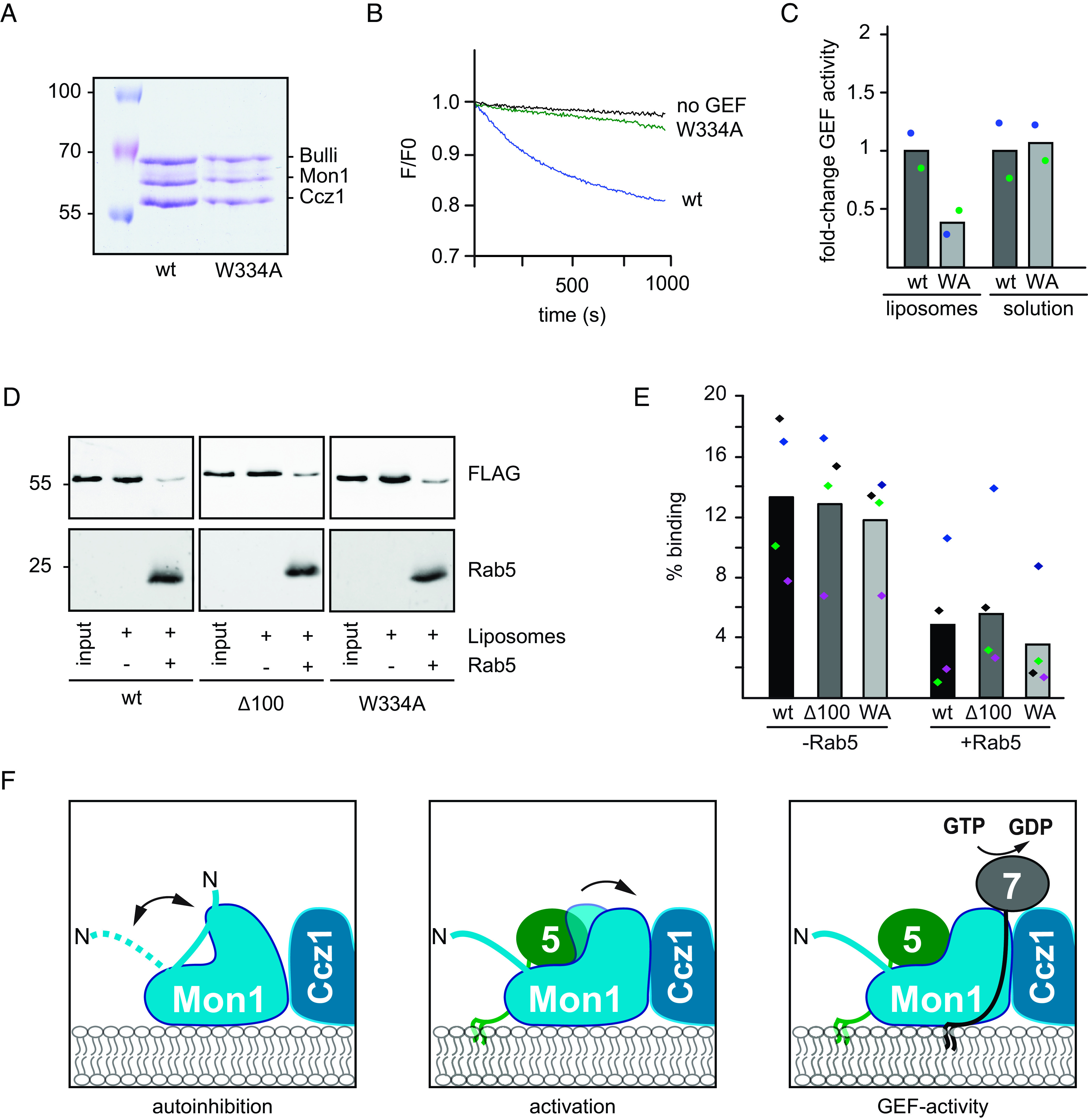
The Rab5-binding site in Mon1 is conserved. (A) Analysis of the Drosophila Mon1–Ccz1–Bulli complex with a Mon1W334A mutation. Stability of wild-type and Mon1W334A trimeric GEF complexes was analyzed by SDS-PAGE and Coomassie staining. (B) GEF activity of the wild-type and Mon1W334A trimeric GEF complex. Rab activation was measured by fluorescence decrease over time. Liposomes were loaded with 150 nM prenylated Rab5 in the presence of 200 µM GTP and 1.5 mM EDTA; bound nucleotide was stabilized with 3 mM MgCl2. 250 nM Mant-GDP-loaded Rab7:GDI complex was added, and the reaction was triggered by adding 6.25 nM of the corresponding GEF complex. The decrease in fluorescence was normalized to fluorescence prior to GEF addition. (C) Fold-change in GEF activity of Mon1W334A compared to wild-type complex on liposomes and in solution. For details, see Materials and Methods. Bar graphs represent average fold-change, and dots represent individual values of two experiments normalized to corresponding wild-type value. (D) Membrane association of wild-type and mutant GEF complexes. 333 µM liposomes were preloaded with 150 nM pRab5:Rab escort protein (REP) as in Fig. 4B. As control, the Rab was omitted. Liposomes were preincubated for 8 min at 30 °C; then, 12.5 nM GEF complex was added, and incubation was continued for 15 min. Liposomes were sedimented at 20,000 g for 20 min. 100% of the pellet was loaded together with 10% input on SDS gels and analyzed with anti-FLAG antibody. (E) Quantification of C. Band intensities were quantified using Fiji, and binding in the absence and presence of Rab5 was normalized to the input value. Bar graphs represent average binding, and dots represent individual values from four experiments. (F) Working model. The unstructured N-terminal part of Mon1 is folding back to the core of Mon1 resulting in autoinhibition of GEF activity. Rab5 (green) binding to a conserved site activates Mon1–Ccz1 (blue) and drives nucleotide exchange of Ypt7. For details, see text.
These observations could be explained by a lack of either recruitment or activation of Mon1–Ccz1 by Rab5. To address this, we tested for membrane association of the GEF complex in either the presence or absence of Rab5. Importantly, for Mon1∆100 and Mon1W334A, similar protein amounts were present in the membrane fraction compared to wild type. However, in the presence of prenylated Rab5 on these membranes, the overall amount of GEF complex decreased comparably in all cases (Fig. 4 D and E). This is in agreement with observations obtained earlier with the yeast Mon1–Ccz1 complex (10). Artificial tethering of the GEF complex via a His-tag to DOGS-NTA containing liposomes did not show any significant change in the overall response of the mutant Mon1–Ccz1 complex to the presence of Rab5 on membranes (SI Appendix, Fig. S5 A and B). Artificial tethering of Mon1–Ccz1 to DOGS-NTA liposomes was comparable and, importantly, independent of Rab5 (SI Appendix, Fig. S5 C and D). These results indicate that the effect of Rab5 on Mon1–Ccz1 seems to be rather in activating than recruiting the complex to membranes. However, dependent on the membrane environment, Rab5 also shows a recruiting effect (21).
Taken together, our results identify a conserved motif containing a hydrophobic tryptophan residue in Mon1 being involved in Rab5 binding and consequently in recruitment and activation of Mon1–Ccz1 (Figs. 3B and 4I).
Discussion
Within this study, we focused on the regulation of the Mon1–Ccz1 complex. We here identify two conserved regions in Mon1, controlling the activity of the GEF complex on membranes. First, we show that deletion of the intrinsically disordered N-terminal part of Mon1 results in a two- to three-fold more active GEF complex on membranes. This N-terminal part may thus function as an autoinhibitory loop for the complex. It is nevertheless required for Mon1–Ccz1 function in vivo as yeast cells as well as Drosophila nephrocytes lacking the Mon1 N-terminal have an altered Rab5 and Rab7 distribution along the endocytic pathway and physiological deficits. Second, we identify W406 as a key residue in Mon1 involved in Rab5 binding. Mutation of this residue results in poor binding to the Rab5-like Ypt10 in yeast, and strongly reduced Rab5-dependent GEF activity, both for the yeast and Drosophila Mon1–Ccz1 complex. Finally, we find a strong overlap of the identified site involved in Rab5 binding with the position of the noncanonical GTPase Rsg1 of the homologous CPLANE complex (18), suggesting that the position of small regulatory GTPases to TLD-GEF complexes may be conserved.
The Mon1–Ccz1 complex has a key role in the transition from early to late endosomes (1). On endosomes, the GEF complex arrives at the same time as Rab7, which is accompanied by a sharp Rab5 to Rab7 transition (35, 36), and is later released from lysosomes (37). A similar release is observed from yeast vacuoles (38). Furthermore, Mon1–Ccz1 is required for Rab7 activation on autophagosomes (39, 40). Its targeting requires specific cues such as the recently identified amphipathic helix in Ccz1, which may recognize packaging defects within the membrane of autophagosomes (21). It is thus not unexpected that the complex is regulated at multiple sites. One of these is the N-terminal part of Mon1. Its removal results in a hyperactive complex and a defective endosomal system. This part of Mon1 is predicted to be intrinsically disordered but also heavily phosphorylated (www.yeastgenome.org). Phosphorylation by one or multiple kinases or the accumulation of PI(3)P and Rab5 on endosomes may differentially control Mon1–Ccz1 activity (Fig. 4D). One possibility is that the Mon1 N-terminal controls Rab5 binding. On membranes without established Rab5 domains, the N-terminal region would then restrict Rab5 binding to the Mon1 subunit resulting in an autoinhibited GEF. Increasing local concentrations of Rab5 on growing endosomes, potentially accompanied by increasing PI(3)P concentrations produced by the Rab5 effector Vps34 kinase complex II (8), would then act as maturation signal. Under these conditions, Rab5 binding and consequently Rab7 GEF activation could then take place. This activation would result in a sharp and timely regulated Rab5-to-Rab7 transition. The N-terminal region could also directly block the active site of the GEF complex, and Rab5 binding then in turn would allow structural rearrangements and release of the active site. However, we speculate that this region controls the access of Rab7 to Mon1–Ccz1, likely by restricting binding of the Rab7 HVD to the complex. According to our model, Rab7 would partition out of the GDI complex onto membranes, where it eventually encounters the GEF. A first interaction may require binding of the Rab7 HVD to the complex. If the N-terminal domain of Mon1 in an autoinhibited state would prevent binding of the HVD, Rab7 may not get efficient access to the active site.
Such a control of Rab access to the active site would be in part reminiscent of the gating mechanism of the TRAPP complexes (41, 42). These complexes share the same active site, but have distinct subunits in TRAPPIII and TRAPPII, which determine specificity. Here, the length of the HVD of Ypt1 targets it specifically to TRAPPIII, whereas the larger TRAPPII complex can only activate Ypt32 with its longer HVD (41, 42). It is likely that the interactions of the Rabs with the respective TRAPP complex are further regulated by interaction of the HVD with the core of the TRAPP complex. Future experiments will have to reveal whether the Rab7-HVD is involved in interaction with Mon1–Ccz1 and how phosphorylation specifically controls Mon1–Ccz1 functions.
Multiple studies identified Rab cascades, where one Rab is required for the recruitment of the downstream GEF (1, 4, 22), yet mechanistic insight on how this works has been lacking. Here, we identify a region in Mon1 that can explain how Rab5 may not only recruit but activate the Mon1–Ccz1 complex. Mutation of the conserved tryptophan residue within Mon1 strongly impairs binding to the Rab5-like Ypt10 and interferes with GEF activity of the yeast and Drosophila complex on membranes. The identified motif in Mon1 lies in a β-sheet of LD2 and in proximity to the elbow loop implicated in regulating the active site (19). This elbow loop was not resolved in the structure of the Mon1–Ccz1 complex in the absence of Ypt7 site (19, 20), suggesting that this region is involved in Ypt7 binding and activation. It is thus possible that Rab5 binding stabilizes the active site and thus both recruits and activates the Mon1–Ccz1 complex as revealed in our in vitro assays (10, 21). Curiously, the noncanonical small GTPase Rsg1 can bind to the tri-longin CPLANE complex and here occupies the same site on the Fuzzy subunit, where Rab5 binds Mon1 (18). We thus speculate that Rsg1 may also activate the CPLANE complex.
Our data reveal that the targeting and activation mechanism of the Rab7 GEF is conserved. However, we still understand very little of the precise regulation. Our recent analysis of the lipid and protein requirements of the Mon1–Ccz1 complex showed that targeting to autophagosomes requires an amphipathic helix in Ccz1 and Atg8, whereas targeting to endosomes relies on negative charges and Rab5. Regulation may occur by distinct posttranslational modifications, likely phosphorylation by kinases such as the casein kinase Yck3 (10, 38). Other kinases may affect Mon1–Ccz1 in autophagy. One obvious regulatory site is the N-terminal segment of Mon1. Here, phosphorylation and dephosphorylation may occur by multiple kinases and phosphatases, depending on the organelle Mon1–Ccz1 is targeted to. As organelle maturation depends on the spatiotemporal activation of the GEFs, it is crucial to understand the regions involved in targeting and regulation. Our analysis of Mon1–Ccz1 may thus help to understand the underlying mechanisms guiding Rab exchange and organelle maturation.
Materials and Methods
All methods are listed in detail in SI Appendix.
Strains and Plasmids.
Strains used in this study are listed in SI Appendix, Table S1. Plasmids used in this study are listed in SI Appendix, Table S2.
Expression and Purification of Rab GTPases and Prenylation Machinery Components.
GST–Rabs and the components of the prenylation machinery were expressed and purified as before (SI Appendix, Table S2) (10). In brief, Escherichia coli BL21 Rosetta cells were transformed with plasmid DNA and grown in Lysogeny broth (LB) media. Protein expression was induced using 0.25 mM IPTG for 14 h at 16 °C. Cells were lysed using the microfluidizer (Microfluidics, Westwood, MA, USA). Rab GTPases were either purified as glutathione S-transferase (GST) fusion constructs or as Tag-free version. S.c. GST-PreSc-Gdi1 was eluted by PreScission protease (PreSc) cleavage. Bet4 His-TEV-Bet2, Mrs6-His, and His-Sumo-D.m. GDI were eluted with 300 mM imidazole in elution buffer. Proteins eluted with imidazole or glutathione were dialyzed twice against buffer (50 mM HEPES–NaOH, pH 7.5, 150 mM NaCl, and 1.5 mM MgCl2). To remove the His-Tag from His-Sumo-D.m. GDI, the dialyzed protein was incubated with SUMO protease. Protein purity and efficiency were analyzed by SDS gel electrophoresis.
Expression and Purification of Drosophila GEF Complexes in Sf21 Cells.
Drosophila GEF complexes were expressed and purified as described (10).
Tandem-Affinity Purification.
Purification of yeast Mon1–Ccz1 was essentially performed as described (43).
In Vitro Prenylation of Yeast and Drosophila Rab GTPases.
Prenylated yeast and Drosophila Rab–REP and Rab–GDI complexes were generated as described (10, 44). For generation of Drosophila Rab7-GDI, yeast prenylation machinery and Drosophila GDI were used.
In Solution and Membrane-Associated Fluorescent Nucleotide Exchange Assays.
In solution nucleotide exchange factor assays were performed as described (19) with minor changes. Purified Rab7 was loaded with MANT-GDP (Jena Bioscience, Germany). 2 µM Rab was incubated with varying amounts of GEF in a SpectraMax M3 Multi-Mode Microplate Reader (Molecular Devices, San Jose, CA, USA); nucleotide exchange was triggered by 0.1 mM GTP (for D.m. GEFs) or 1 mM GTP (S.c. GEFs). MANT-GDP fluorescence was monitored over time (excitation: 355 nm/emission: 448 nm). Data were fitted individually against a first-order exponential decay using OriginPro9 software (OriginLab Corporation, Northampton, USA).
The liposome-based GEF assays were performed as described (10) with liposomes extruded to 400 nm using a polycarbonate filter and a hand extruder (Avanti Polar Lipids, Inc., Alabaster, AL, USA). Liposomes were decorated with prenylated recruiter GTPase. The reaction was transferred to a half micro cuvette 109.004F-QS (Hellma, Müllheim, Germany). 250 nM Rab7/Ypt7-GDI was added, and the cuvette was filled up to 800 µL with prenylation buffer omitting the volume of the GEF. The reaction was measured in a fluorimeter (Jasco, Gross-Umstadt, Germany) at 30 °C. After baseline stabilization, the indicated concentration of GEF was added to start the reaction. GEF assays with artificial recruitment of His-tagged GEF complexes to liposomes using His-tagged Bulli-MC1 were performed essentially as above. 3 mol % DOGS-NTA (18:1 18:1) was included into liposomes and corrected for by adjustment of DLPC amounts. The measurements were performed in a SpectraMax iD3 Multi-Mode Microplate Reader (Molecular Devices, San Jose, CA, USA). All kinetic parameters of used GEF complexes are listed in SI Appendix, Table S3.
Liposome Sedimentation Assay.
Membrane association of GEF complexes was analyzed via liposome sedimentation. Liposomes containing the lipid composition used for the nucleotide exchange assays including 0.5 % ATTO550 (AD 550, ATTO-TEC, Siegen, Germany) were loaded with recruiter GTPase. As control, Rab GTPases were omitted. After preincubation at 30 °C, 12.5 nM GEF complex was added and incubated for 15 min. Liposomes were sedimented at 20,000 g for 20 min at 4 °C. The presence of GEF was detected using Sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and western blot with an anti-FLAG M2 antibody (1:1,000, F3165, Merck, Darmstadt, Germany) and a fluorescence-coupled secondary antibody (1:10,000, SA-35521, Thermo Fisher Scientific, Dreieich, Germany). Band intensity was analyzed using Fiji software (NIH, Bethesda, MD, USA) and normalized to the respective input.
GST-Rab Pull Downs.
75 µg GST-tagged Rab GTPase was loaded with a nucleotide (Sigma-Aldrich, Taufkirchen, Germany) in the presence of 20 mM EDTA and 50 mM HEPES–NaOH, pH 7.4, at 30 °C for 30 min. The nucleotide was stabilized using 25 mM MgCl2. Protein was incubated for 1 h at 4 °C with 30 µl GSH sepharose (Cytiva, Freiburg im Breisgau, Germany). 25 µg Mon1–Ccz1, 7 mg/mL BSA, and 1 mM nucleotide were added, and the tube was filled up to 300 µL with pull-down buffer (50 mM HEPES–NaOH, pH 7.4, 150 mM NaCl, 1 mM MgCl2, 5% (v/v) glycerol, and 0.1% (v/v) Triton X-100) and incubated for 1.5 h at 4 °C. Beads were washed 3× using pull-down buffer. Bound protein was eluted using 300 µL elution buffer supplemented with EDTA. The eluted fraction was analyzed by SDS-PAGE and western blot.
Pho8Δ60 Assay.
The assay was essentially performed as described (45). Yeast expressing a genetically truncated version of the PHO8 gene (Pho8Δ60) were grown until the logarithmic phase. Cells were washed and incubated in starvation media (0.17% yeast nitrogen base without amino acids or ammonium sulfate and 2% glucose) for indicated times. Cells were harvested, washed in H2O, and lysed. Pho8 activity was monitored in the presence of 1.25 mM p-nitrophenyl phosphate. The colorimetric reaction was measured in a SpectraMax M3 Multi-Mode Microplate Reader (Molecular Devices, San Jose, CA, USA) at OD400. The slopes of measured curves were normalized to WT cells under nutrient-rich conditions. Significance was determined using a one-way ANOVA with a Tukey post hoc test [OriginPro9 software (OriginLab Corporation, Northampton, USA)].
Analysis of Yeast and Drosophila Protein Expression.
To test protein expression in yeast cells, 4 optical density (OD) units were harvested and resuspended in buffer (0.2 M NaOH and 30 mM β-mercaptoethanol). Proteins were precipitated and analyzed via SDS-PAGE and western blotting. Protein expression of Mon1::hemagglutini (HA) and Mon1∆100::HA in transgenic flies was verified by western blot of whole cell lysate.
Fluorescence Microscopy of Yeast Cells.
For yeast fluorescence microscopy, cells were grown overnight in synthetic media containing 2% (w/v) glucose and essential amino acids (SDC+all). Cells were grown to the logarithmic phase. Vacuoles were stained with FM4-64 (Thermo Fisher Scientific, Dreieich, Germany). 1 OD equivalent of cells was resuspended in 50 µL SDC+all containing 30 µM FM4-64 and incubated at 30 °C for 20 min. Cells were washed twice and incubated with 500 µL SDC+all for 45 min at 30 °C. Cells were imaged on an Olympus IX-71 inverted microscope (DeltaVision Elite, GE Healthcare, Solingen, Germany). For details, see SI Appendix.
Growth Test.
Yeast cells were incubated in YPD media overnight at 30 °C. In the morning, cultures were diluted and grown to the logarithmic phase at 30 °C. Cells were diluted to OD600 = 0.25 in YPD and spotted in 1:10 serial dilutions onto control and selection plates.
Fly Stocks.
Fly stocks were obtained from the Drosophila stock center (Bloomington): da-GAL4 (RRID:BDSC_55850) and w1118 (RRID:BDSC_5905). handC-GAL4 was previously generated by us (46). The UAS-Mon1::HA line was obtained from T. Klein, Düsseldorf, Germany (47). Fly husbandry was carried out as described previously (48).
Generation of the Transgenic UAS-Mon1∆100::HA Line.
To generate a Mon1∆100::HA construct, a Mon1∆100-cDNA was used as PCR template [Source DNA clone IP03303 from the Drosophila Genomics Resource Center (DGRC, Bloomington, IN, USA)]. The Mon1∆100 construct was fused to an HA-tag and was cloned into plasmid for yeast/E. coli/Drosophila (pYED) (49). The expression plasmid was injected into RRID:BDSC_24749 for integration at 86Fb on the 3rd Chromosome (50). A commercial service was used for establishing transgenic fly lines (BestGene, Chino Hills, CA, USA).
FITC-Albumin Uptake.
FITC-albumin uptake was performed in pericardial nephrocytes (16). Briefly, 3rd instar larvae, expressing Mon1::HA or Mon1∆100::HA in nephrocytes using handC-GAL4 as a driver, were anesthetized and fixed ventral side upward on Sylgard 184 silicone elastomer plates. Preparation buffer was replaced by artificial hemolymph buffer containing 0.2 mg/mL FITC-albumin (A9771, albumin–fluorescein isothiocyanate conjugate, MW: 66 kDa; Sigma-Aldrich, Taufkirchen, Germany) for 5 min or 10 min. Uptake was stopped by fixation in 8% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). Tissues were embedded in Fluoromount-G mounting medium containing DAPI (Thermo Fisher, Waltham, MA, USA). Uptake efficiency was quantified by imaging respective nephrocytes (LSM800, Zeiss, Jena, Germany). The mean pixel-intensity measurement function (Fiji software) was used to quantify uptake efficiency (51). The mean pixel intensity was calculated in relation to the perimeter of the cell. An unpaired, two-tailed Student’s t test was used (GraphPad Prism 9, Boston, MA, USA).
Immunohistochemistry.
3rd instar larvae expressing handC driven Mon1::HA and Mon1∆100::HA were dissected in PBS and fixed with 4% PFA in PBS. After washing, specimens were permeabilized with 1% Triton X-100 in PBS, followed by washing with BBT (0.1% BSA and 0.1% Tween-20 in PBS). Specimens were incubated for 30 min in blocking solution (1% BSA and 0.1% Tween-20 in PBS) followed by antibody staining (rabbit anti-Rab5, 1:250, Abcam Cat# ab31261, RRID:AB_882240, Cambridge, United Kingdom; mouse anti-Rab7, 1:10, Developmental Studies Hybridoma Bank Cat# Rab7, RRID:AB_2722471, University of Iowa, IA, USA) (52). Afterwards they were rinsed with BBT, blocked with blocking solution, and incubated with secondary antibodies (anti-rabbit Alexa Fluor 488, 1:200, Jackson ImmunoResearch Laboratories, Inc, Ely, United Kingdom, Code Number: 111-545-006); anti-mouse Cy3, 1:200, Dianova GmbH, Eching, Germany). Samples were embedded in Fluoromount-G mounting medium containing DAPI (Thermo Fisher, Waltham, MA, USA). Confocal images were captured with an LSM800 (Zeiss, Jena, Germany). For details, see SI Appendix.
Measurement of Rab5 Structures in Nephrocytes.
To analyze the size and number of Rab5-positive vesicles, the “Intermodes” auto threshold (Fiji) was used to highlight Rab5 dots above the set threshold. Two sections of each cell (one from the periphery and one from the center of the cell) were analyzed using the “analyzed particle” function. The mean size of Rab5 dots per cell and number per 100 µm2 cell area was calculated. An unpaired, two-tailed Student’s t test was used (GraphPad Prism 9, Boston, MA, USA).
Transmission Electron Microscopy (TEM).
Briefly, specimens were prepared in PBS and subsequently fixed (2% glutaraldehyde (Sigma-Aldrich, Taufkirchen, Germany)/4% PFA [Merck, Darmstadt, Germany) in 0.05 M cacodylate buffer pH 7.4]. Specimens were postfixed for 2 h at RT [1% osmium tetroxide in 0.05 M cacodylate buffer pH 7.4 (Sciences Services, München, Germany)] and dehydrated stepwise in a graded ethanol series followed by 100% acetone. Specimens were embedded in Epon 812 and polymerized for 48 h at 60 °C. Ultrathin sections (70 nm) were cut on an ultramicrotome (UC6 and UC7 Leica, Wetzlar, Germany) and mounted on formvar-coated copper slot grids. Sections were stained for 30 min in 2% uranyl acetate (Sciences Services, München, Germany) and 20 min in 3% lead citrate (Roth, Karlsruhe, Germany). A detailed protocol for processing nephrocytes for TEM analysis can be found elsewhere (29). All samples were analyzed at 80 kV with a Zeiss 902 and Zeiss LEO912 and at 200 kV with a Jeol JEM2100-Plus transmission electron microscope (Zeiss, Jena, Germany; Jeol, Tokyo, Japan).
Modeling of the Yeast Mon1–Ccz1 Complex Bound to Ypt7 and Ypt10.
The protein complex model was generated using AlphaFold2-Multimer (31). The switch regions of nucleotide-free Ypt7 were manually edited based on the crystal structure of the catalytic MC1-Ypt7 core complex (19), and GTP and Mg2+ were added to the Ypt10 nucleotide-binding pocket. Only regions that were modeled with high confidence are shown in the figures.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Kerstin Etzold, Angela Perz, and Kathrin Auffarth for expert technical assistance. This work was supported by a grant from the Deutsche Forschungsgemeinschaft to C.U. and A.P. (UN111/9-2; PA517/12-2) and by the Sonderforschungsbereich 944 and Sonderforschungsbereich 1557 (to D.K., A.P., and C.U.). We also acknowledge the support of the Open Access Publishing Fund of Osnabrück University.
Author contributions
A.-C.B., A.M., A.P., C.U., and L.L. designed research; A.-C.B., M.J., and J.-H.S. performed research; A.-C.B., M.J., D.K., A.P., C.U., and L.L. analyzed data; J.-H.S. performed initial structural modeling; A.M. supervised J.-H.S.; and A.-C.B., D.K., A.P., C.U., and L.L. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Christian Ungermann, Email: cu@uos.de.
Lars Langemeyer, Email: lars.langemeyer@uos.de.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Borchers A.-C., Langemeyer L., Ungermann C., Who’s in control? Principles of Rab GTPase activation in endolysosomal membrane trafficking and beyond. J. Cell Biol. 220, e202105120 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Romano R., et al. , Alteration of the late endocytic pathway in Charcot–Marie–Tooth type 2B disease. Cell Mol. Life Sci. 78, 351–372 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goody R. S., Müller M. P., Wu Y.-W., Mechanisms of action of Rab proteins, key regulators of intracellular vesicular transport. Biol. Chem. 398, 565–575 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Barr F. A., Rab GTPases and membrane identity: Causal or inconsequential? J. Cell Biol. 202, 191–199 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bezeljak U., Loya H., Kaczmarek B., Saunders T. E., Loose M., Stochastic activation and bistability in a Rab GTPase regulatory network. Proc. Natl. Acad. Sci. U.S.A. 117, 6540–6549 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cezanne A., Lauer J., Solomatina A., Sbalzarini I. F., Zerial M., A non-linear system patterns Rab5 GTPase on the membrane. Elife 9, e54434 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kümmel D., Ungermann C., Principles of membrane tethering and fusion in endosome and lysosome biogenesis. Curr. Opin. Cell Biol. 29, 61–66 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Tremel S., et al. , Structural basis for VPS34 kinase activation by Rab1 and Rab5 on membranes. Nat. Commun. 12, 1564 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kinchen J. M., Ravichandran K. S., Identification of two evolutionarily conserved genes regulating processing of engulfed apoptotic cells. Nature 464, 778–782 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langemeyer L., et al. , A conserved and regulated mechanism drives endosomal Rab transition. Elife 9, e56090 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui Y., et al. , Activation of the Rab7 GTPase by the Mon1–CCZ1 complex is essential for PVC-to-vacuole trafficking and plant growth in Arabidopsis. Plant Cell 26, 2080–2097 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh M. K., et al. , Protein delivery to vacuole requires SAND protein-dependent Rab GTPase conversion for MVB-vacuole fusion. Curr. Biol. 24, 1383–1389 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Wang C.-W., Stromhaug P. E., Shima J., Klionsky D. J., The Ccz1-Mon1 protein complex is required for the late step of multiple vacuole delivery pathways. J. Biol. Chem. 277, 47917–47927 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaites L. P., Paulo J. A., Huttlin E. L., Harper J. W., Systematic analysis of human cells lacking ATG8 proteins uncovers roles for GABARAPs and the CCZ1/MON1 regulator C18orf8/RMC1 in macroautophagic and selective autophagic flux. Mol. Cell. Biol. 38, e00392-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van den Boomen D. J. H., et al. , A trimeric Rab7 GEF controls NPC1-dependent lysosomal cholesterol export. Nat. Commun. 11, 5559 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dehnen L., et al. , A trimeric metazoan Rab7 GEF complex is crucial for endocytosis and scavenger function. J. Cell Sci. 133, jcs247080 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Gerondopoulos A., Langemeyer L., Liang J.-R., Linford A., Barr F. A., BLOC-3 mutated in Hermansky-Pudlak syndrome is a Rab32/38 guanine nucleotide exchange factor. Curr. Biol. 22, 2135–2139 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langousis G., et al. , Structure of the ciliogenesis-associated CPLANE complex. Sci. Adv. 8, eabn0832 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kiontke S., et al. , Architecture and mechanism of the late endosomal Rab7-like Ypt7 guanine nucleotide exchange factor complex Mon1–Ccz1. Nat. Commun. 8, 14034 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klink B. U., et al. , Structure of the Mon1–Ccz1 complex reveals molecular basis of membrane binding for Rab7 activation. Proc. Natl. Acad. Sci. U.S.A. 119, e2121494119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herrmann E., Langemeyer L., Auffarth K., Ungermann C., Kümmel D., Targeting of the Mon1–Ccz1 Rab guanine nucleotide exchange factor to distinct organelles by a synergistic protein and lipid code. J. Biol. Chem. 299, 102915 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hutagalung A. H., Novick P. J., Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 91, 119–149 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.del Conte-Zerial P., et al. , Membrane identity and GTPase cascades regulated by toggle and cut-out switches. Mol. Syst. Biol. 4, 206 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erdős G., Dosztányi Z., Analyzing protein disorder with IUPred2A. Curr. Protoc. Bioinf. 70, e99 (2020). [DOI] [PubMed] [Google Scholar]
- 25.Mészáros B., Erdős G., Dosztányi Z., IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 46, W329–W337 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sievers F., et al. , Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539–539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goujon M., et al. , A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38, W695–W699 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waterhouse A. M., Procter J. B., Martin D. M. A., Clamp M., Barton G. J., Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Psathaki O.-E., Dehnen L., Hartley P. S., Paululat A., Drosophila pericardial nephrocyte ultrastructure changes during ageing. Mech. Ageing Dev. 173, 9–20 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Duarte P. V., et al. , The yeast LYST homolog Bph1 is a Rab5 effector and prevents Atg8 lipidation at endosomes. J. Cell Sci. 135, jcs259421 (2022). [DOI] [PubMed] [Google Scholar]
- 31.Evans R., et al. , Protein complex prediction with AlphaFold-multimer. biorxiv [Preprint] (2022). 10.1101/2021.10.04.463034 (Accessed 3 November 2022). [DOI]
- 32.Jumper J., et al. , Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varadi M., et al. , AlphaFold protein structure database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herrmann E., et al. , Structure of the metazoan Rab7 GEF complex Mon1–Ccz1–Bulli. Proc. Natl. Acad. Sci. U.S.A. 120, e2301908120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rink J., Ghigo E., Kalaidzidis Y., Zerial M., Rab conversion as a mechanism of progression from early to late endosomes. Cell 122, 735–749 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Poteryaev D., Datta S., Ackema K., Zerial M., Spang A., Identification of the switch in early-to-late endosome transition. Cell 141, 497–508 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Yasuda S., et al. , Mon1–Ccz1 activates Rab7 only on late endosomes and dissociates from the lysosome in mammalian cells. J. Cell Sci. 129, 329–340 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Lawrence G., et al. , Dynamic association of the PI3P-interacting Mon1–Ccz1 GEF with vacuoles is controlled through its phosphorylation by the type 1 casein kinase Yck3. Mol. Biol. Cell 25, 1608–1619 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao J., Langemeyer L., Kümmel D., Reggiori F., Ungermann C., Molecular mechanism to target the endosomal Mon1–Ccz1 GEF complex to the pre-autophagosomal structure. Elife 7, e31145 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hegedűs K., et al. , The Ccz1-Mon1–Rab7 module and Rab5 control distinct steps of autophagy. Mol. Biol. Cell 27, 3132–3142 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas L. L., van der Vegt S. A., Fromme J. C., A steric gating mechanism dictates the substrate specificity of a Rab–GEF. Dev. Cell 48, 100–114.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bagde S. R., Fromme J. C., Structure of a TRAPPII-Rab11 activation intermediate reveals GTPase substrate selection mechanisms. Sci. Adv. 8, eabn7446 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nordmann M., et al. , The Mon1–Ccz1 complex is the GEF of the late endosomal Rab7 homolog Ypt7. Curr. Biol. 20, 1654–1659 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Langemeyer L., Perz A., Kümmel D., Ungermann C., A guanine nucleotide exchange factor (GEF) limits Rab GTPase–driven membrane fusion. J. Biol. Chem. 293, 731–739 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guimaraes R. S., Delorme-Axford E., Klionsky D. J., Reggiori F., Assays for the biochemical and ultrastructural measurement of selective and nonselective types of autophagy in the yeast Saccharomyces cerevisiae. Methods 75, 141–150 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Sellin J., Albrecht S., Kölsch V., Paululat A., Dynamics of heart differentiation, visualized utilizing heart enhancer elements of the Drosophila melanogaster bHLH transcription factor Hand. Gene. Exp. Patterns 6, 360–375 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Yousefian J., et al. , Dmon1 controls recruitment of Rab7 to maturing endosomes in Drosophila. J. Cell Sci. 126, 1583–1594 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang S., et al. , GBF1 (Gartenzwerg)-dependent secretion is required for Drosophila tubulogenesis. J. Cell Sci. 125, 461–472 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Paululat A., Heinisch J. J., New yeast/E. coli/Drosophila triple shuttle vectors for efficient generation of Drosophila P element transformation constructs. Gene 511, 300–305 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Bischof J., Maeda R. K., Hediger M., Karch F., Basler K., An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc. Natl. Acad. Sci. U.S.A. 104, 3312–3317 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schindelin J., et al. , Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Riedel F., Gillingham A. K., Rosa-Ferreira C., Galindo A., Munro S., An antibody toolkit for the study of membrane traffic in Drosophila melanogaster. Biol. Open 5, 987–992 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.