

SUMMARY
Epstein-Barr virus (EBV) causes infectious mononucleosis, triggers multiple sclerosis, and is associated with 200,000 cancers/year. EBV colonizes the human B cell compartment and periodically reactivates, inducing expression of 80 viral proteins. However, much remains unknown about how EBV remodels host cells and dismantles key antiviral responses. We therefore created a map of EBV-host and EBV-EBV interactions in B cells undergoing EBV replication, uncovering conserved herpesvirus versus EBV-specific host cell targets. The EBV-encoded G-protein-coupled receptor BILF1 associated with MAVS and the UFM1 E3 ligase UFL1. Although UFMylation of 14-3-3 proteins drives RIG-I/MAVS signaling, BILF1-directed MAVS UFMylation instead triggered MAVS packaging into mitochondrial-derived vesicles and lysosomal proteolysis. In the absence of BILF1, EBV replication activated the NLRP3 inflammasome, which impaired viral replication and triggered pyroptosis. Our results provide a viral protein interaction network resource, reveal a UFM1-dependent pathway for selective degradation of mitochondrial cargo, and highlight BILF1 as a novel therapeutic target.
In brief
Epstein-Barr virus (EBV) contributes to ~ 2% of human cancers. Yiu et al. assemble an EBV protein interaction network in B cells undergoing viral replication, uncovering that EBV repurposes its G-protein-coupled receptor BILF1 to subvert MAVS-driven NLRP3 inflammasome activation. They highlight UFM1ylation in selective MAVS targeting for lysosomal proteolysis.
Graphical Abstract
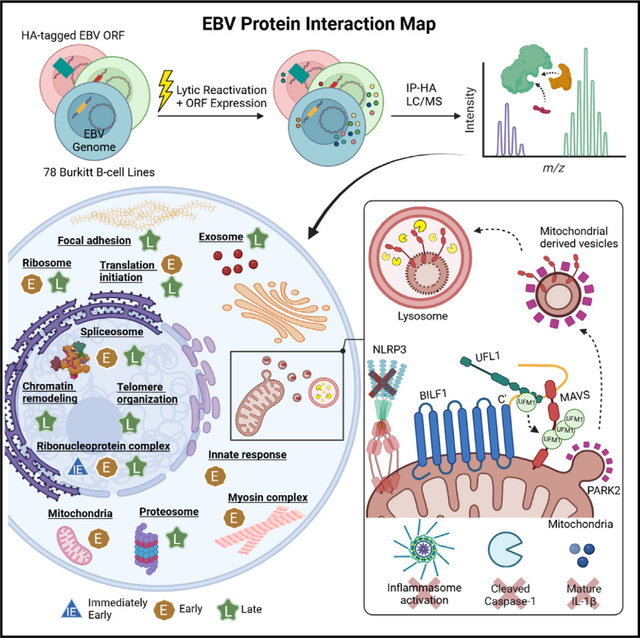
INTRODUCTION
The γ-herpesvirus Epstein-Barr virus (EBV) establishes lifelong infection in most people worldwide. EBV contributes to ~ 2% of human cancers, including endemic Burkitt lymphoma, Hodgkin lymphoma, and post-transplant lymphoproliferative diseases.1–5 EBV also contributes to nasopharyngeal carcinoma and ~ 10% of gastric cancers.6–8 EBV is a key trigger for multiple sclerosis.9 However, the function of many EBV-encoded proteins remains incompletely understood.
To achieve persistent infection, EBV employs a biphasic life-cycle, in which it alternates between latency and lytic replication. EBV latency programs express between one and nine viral oncogenes that support colonization of the memory B cell compartment. EBV reactivates, although memory B cells differentiate into plasma cells, highlighting the requirement to evade immune detection as EBV spreads to epithelial cells.10 Upon reactivation, 80 EBV proteins are expressed in a cascade of immediate early, early, and late genes leading to the synthesis of new viral progeny.5,11,12 Immediate early transcription activators BZLF1 and BRLF1 trigger expression of 40 early genes, which replicate EBV DNA and remodel host cells. ~35 late genes contribute to EBV virion assembly and secretion. Despite the label lytic replication, EBV+ B cells undergoing replication remain viable for a period of time as they secrete virion.5,11–13
Most EBV infections are asymptomatic, suggesting that EBV has evolved to modulate diverse aspects of host immunity. EBV encodes multiple proteins that mimic immune receptors, cytokines, or chemokines and subverts still other immune pathways to enter B cells.14,15 However, much remains to be learned about how EBV dismantles innate and adaptive immunities to hide pathogen-associated molecular patterns (PAMPs) produced during replication.
Although EBV latency proteins drive lymphoproliferative diseases,16,17 lytic cycle proteins are increasingly implicated in EBV pathogenesis, including lymphomagenesis.18,19 EBV lacking the lytic cycle inducing BZLF1 gene causes lymphomas at reduced frequencies in humanized mouse models.20 The lytic cycle BCL-2 homologs BHRF1 and BALF1 suppress apoptosis,21,22 whereas BGLF4, BGLF5, and BALF3 destabilize the host genome.23–25 The major EBV tegument protein BNRF1 not only destabilizes host SMC5/6 cohesins to support late gene expression26 but also causes host chromosome instability.27
Here, we assembled an EBV-B cell protein-protein interaction (PPI) network in B cells induced for EBV replication, an environment in which all EBV open reading frames (ORFs) are physiologically expressed. Cross-comparison with human cytomegalovirus (HCMV) and Kaposi sarcoma-associated herpesvirus (KSHV) interactomes28,29 revealed common targets. We leveraged the map to identify an unexpected mechanism by which EBV inhibits NLPR3 inflammasome activation. In addition to functioning as a viral G-protein-coupled receptor (GPCR), a subpopulation of EBV BILF1 traffics to mitochondria, where it drives mitochondrial antiviral-signaling protein (MAVS) UFMylation. This signal, together with Parkin ubiquitination, triggers MAVS selective dislocation from the mitochondrial outer membrane, incorporation into mitochondrial-derived vesicles (MDVs), and lysosomal degradation. Our study highlights a novel ubiquitinfold modifier 1 (UFM1)-dependent pathway for mitochondrial membrane cargo turnover, subverted by EBV to enable viral replication and inflammasome evasion.
RESULTS
EBV lytic cycle protein interaction map construction
To interrogate EBV PPIs in the lytic B cell environment, we generated 78 P3HR-1 Burkitt lymphoma cell lines, each expressing a doxycycline-inducible EBV ORF. Only EBV LF3 and BHLF1 were not included, due to their extremely high GC content. Lytic reactivation was driven by conditional immediate early BZLF1 and BRLF1 alleles fused to modified estrogen receptor 4-hydroxytamoxifen (4HT)-binding domains (referred to as ZHT and RHT).30 4HT addition triggers ZHT and RHT nuclear translocation and lytic reactivation. We recently used P3HR-1 ZHT/RHT cells for whole-cell EBV lytic cycle proteomic analysis, facilitating cross-comparison.31 EBV ORF expression was validated by anti-HA tag immunoblot and flow cytometry (Table S1).
EBV ORF expression and the viral lytic cycle were induced by the addition of doxycycline and 4HT for just 15 and 24 h, respectively. Anti-HA immunoprecipitation from whole-cell lysates followed by HA peptide elution was performed to isolate EBV protein complexes by liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Figure 1A). High-confidence PPIs were identified by CompPass.32–34 Data reported for each prey protein includes: (1) the number of peptide spectral matches (PSMs), averaged between technical replicates; (2) an entropy score, which compares the number of PSM between replicates to eliminate proteins that are not detected consistently; (3) a Z score, calculated in comparison to the average and standard deviation of PSMs observed across all IPs; and (4) a normalized WD (NWD) score. The NWD score addresses whether (1) the protein is detected across all IPs and (2) whether it is detected reproducibly among replicates.
Figure 1. EBV lytic cycle protein interaction map construction.

(A) EBV protein interaction map workflow. Very high-confidence interacting proteins (VHCIPs) were defined as prey meeting peptide spectral match (PSM) and entropy criteria with either an NWD ≥ 1.0 or Z score ≥ 4.0. High-confidence interacting proteins (HCIPs) were defined as prey meeting PSM and entropy criteria with either an NWD R 1.0 or Z score R 3.0 (Table S2).
(B and C) DAVID with default setting was applied to determine pathways enriched among all HCIPs, in comparison to all human proteins as background (full data, Table S3). Benjamini-Hochberg adjusted p values are shown in red with gradient scale to the top 16 unique pathway enriched (p < 0.01). Viral baits were assigned to their top unique enriched pathway with p < 0.05. Viral baits (blue squares), interacting proteins (circles, with viral proteins in light blue), enriched pathway members (red or green), and other host interactors not associated with the pathway (gray) are shown. Black solid lines indicate interactions identified in this study. See also Figure S1.
Using a stringent cutoff of either an NWD ≥ 1.0 or Z score ≥ 4.0, we identified 884 unique viral-host and 83 viral-viral very high-confidence interacting proteins (VHCIPs) (Table S2). Likewise, at a somewhat more relaxed entropy cutoff of either an NWD ≥ 1.0 or Z score ≥ 3.0, 1,398 viral-host and 90 viral-viral high-confidence interacting proteins (HCIPs) were identified. EBV baits retrieved 0–285 interactions, yielding a scale-free distribution similar to those reported for two human herpesvirus PPI networks28,29 (Figure S1A). Multiple well-characterized EBV-host interactions were identified, providing validation of the proteomic approach and raising confidence in the majority of high-confidence interactions that were not previously reported. For example, consistent with published reports, we identified high-confidence interactions between the viral pre-initiation complex TATT-binding protein BcRF1 and host RNA polymerase II,35 between EBV tegument protein BGLF2 and the cell cycle regulatory proteins GMIP and NEK936 (Table S2). Likewise, we captured multiple known high-confidence interactions between EBV proteins, such as between LF2 and the immediate early lytic protein BRLF1,37–39 between the viral helicase BBLF4 and primase BSLF140 (Figure 1B; Table S2), and between components of the tripartite terminase and viral pre-initiation complexes (Figure 1B; Table S2). However, most high-confidence EBV-EBV interactions in our dataset are not present in the Biogrid database, highlighting multiple EBV and host protein interaction hubs. For instance, three calcium/calmodulin-dependent protein kinase subunits were identified as VHCIPs of LF2, but not of any other EBV bait (Figure 1B; Table S2).
To gain further insights into host pathways enriched among viral bait interactors, we used database for annotation, visualization, and integrated discovery (DAVID) gene ontology (GO) analysis41,42 (Figures 1C and S1B–S1F; Table S3). DAVID indicated that EBV targeting of multiple host pathways not previously implicated in its replication, including the extracellular exosome (Figure 1C). EBV BALF4, which encodes the glycoprotein B (gB) fusogen conserved across herpesvirus cellular entry machinery, associated with exosome cargo (Figures 1B and 1C). Fascinatingly, 12 additional EBV lytic proteins also associated with exosome pathway components, suggesting potentially major exosome pathway roles in EBV egress (Figure 1C; Table S3). Likewise, DAVID highlighted EBV targeting of the mRNA splicing via the spliceosome pathway. Although EBV SM association with ribonucleoprotein complex proteins is well-characterized,43,44 five additional EBV lytic protein HCIPs were also enriched for ribonucleoprotein complexes, suggesting that EBV remodels this key pathway to a greater extent than has been appreciated (Figure 1C; Table S3). Glycyl-lysine isopeptide machinery was highly enriched within interactors of 13 EBV proteins, suggesting potentially major EBV roles in subverting ubiquitin, ISG15 and/or SUMO ligases (Figure 1C; Table S3).
Interactome analysis identifies herpesvirus-targeted host nodes
All herpesviruses use biphasic lifecycles and share the ability to establish persistent infection. To identify common herpesvirus host protein targets, we leveraged beta-herpesvirus HCMV and gamma-herpesvirus KSHV interactome datasets.28,29 Overlap between HCIP highlighted common biological targets of all three herpesviruses, which replicate across a wide range of host types (Figure 2A; Table S4). DAVID analysis of commonly interacting host HCIPs indicated enrichment of the promyelocytic leukaemia (PML) body, nuclear body, ubiquitin ligase complex, RNA phosphodiester bond hydrolysis, and the CCR4/NOT deadenylase complex, a major regulator of mRNA turnover46 (Figure 2A).
Figure 2. Systematic analysis of interactome data from three herpesviruses highlights common host targets.

(A) Overlap between EBV, HCMV,28 and KSHV29 HCIPs (see also Table S4). DAVID pathway enrichment analysis among HCIPs interacting with EBV, HCMV, and KSHV baits (default settings, against all human proteins as background) are shown at bottom, including a list of all commonly interacting CCR4-NOT complex components. Shown at upper right are representative terms from DAVID analysis of HCIPs interacting with EBV proteins but not HCMV or KSHV proteins, and a list of interacting components from the term “activation of innate immune response.” See Table S4 for full Venn diagram details.
(B) Network propagation identification of CORUM database human protein complexes that are closely associated with proteins from each herpesvirus. Large colored nodes represent each virus (HCMV, blue; EBV, red; KSHV, yellow). Edges connect to CORUM complexes closely associated with proteins from each virus. Edge thickness is proportional to the Z score observed, whereas CORUM nodes sizes are colored pale yellow and scaled according to the numbers of proteins in each complex.
(C and D) Interaction network depicting the CORUM CCR4-NOT and viral protein neighbors. Nodes represent proteins, whereas edges represent protein-protein interactions. Human proteins belonging to the CORUM CCR4-NOT complex (C) or RNA Pol II core complex (D) and edges among them are colored yellow. Viral proteins and their interactions are colored as in (B). Node size is scaled according to each node’s eigenvector centrality within the displayed subnetwork. Graph layouts were determined via gravity embedding.
(E) Immunoblot of 1% input and anti-HA complexes from P3HR-1 expressing BRRF1 or GFP cDNAs, representative of n = 3 experiments.
(F) Interaction of EBV proteins with components of the cellular degradation machinery. HCIP were searched against a database of E1, E2, E3 enzymes, cullins, deubiquitinases (DUBs), proteasome components (PSM), and ubiquitin-binding proteins (UBPs).45 Percentage of the total number of interactors for each protein from each of these categories is displayed. Values were clustered hierarchically (see also Table S5).
Next, we merged EBV, HCMV, and KSHV interactomes with the BioPlex 3.0 network of human protein interactions47 to derive a multi-species PPI network, viewable at https://wren.hms.harvard. edu/ViroPlex/. We then superimposed complexes from the CORUM database,48 a curated repository of experimentally characterized mammalian protein complexes onto the network. We used network propagation to quantify the proximity of each complex to each viral proteome. In addition to the CCR4/NOT deadenylase, CORUM analysis highlighted additional protein complexes targeted by all three human herpesviruses, including the DNA single-stranded binding RPA complex, and the RNA polymerase II complex (Figures 2B–2D). EBV early gene BRRF1 interacted with 7 of the 10 CCR4-NOT components, whereas KSHV ORF49 and HCMV UL72 associate with 6 and 10 CCR4-NOT components, respectively (Figure 2C). We validated the interaction between BRRF1 and CCR4/NOT complex members CNOT1 and CNOT10 by co-immunoprecipitation analysis (Figure 2E).
Ubiquitin ligase complexes were also highly enriched among HCIPs that commonly interacted with EBV-, KSHV-, and HCMV-encoded proteins (Figure 2A). We therefore more fully analyzed EBV PPIs with components of the ubiquitin-proteasome system. HCIPs were searched against a comprehensive database of E1, E2, and E3 enzymes, cullins, deubiquitinases (DUBs), proteasome components (PSM), and ubiquitin-binding proteins (UBPs).45 This revealed EBV association with cellular degradation machinery at multiple levels (Figure 2F; Table S5). This identified key “hubs” targeted by multiple herpes viruses.
BILF1 supports EBV late gene expression and blocks NLRP3 inflammasome activation
Much remains to be learned about how EBV evades immune responses to periodically reactivate. We were therefore intrigued that the EBV-encoded 7-transmembrane protein orphan GPCR BILF1 interacted with host factors involved in innate and adaptive immune responses, metabolism, and transport (Figure 3A). BILF1 is highly conserved across lymphocryptoviruses and is expressed early in the lytic cycle.49 Consistent with its roles in evasion of the human leukocyte antigen (HLA) presentation pathway,50,51 we identified high-confidence interactions between BILF1 and multiple HLA alleles. We also identified an association between BILF1 and the TAP-binding protein (TAPBP), which mediates an association between the TAP peptide transporter and class I molecules awaiting peptide cargo (Figure 3A). BILF1 may therefore subvert antigen presentation within the endoplasmic reticulum as well as at the plasma membrane (PM), perhaps together with BNLF2a, which blocks TAP.52
Figure 3. BILF1 inhibits NLRP3 inflammasome activation.

(A) Cytoscape version 3.8.1 BILF1 interaction network. DAVID pathways enriched among BILF1 interactors are annotated in black. Sub-pathways are annotated either red or green with italicized characters. Host proteins associated with the main or sub-pathways are colored in red or green, remaining proteins are in gray.
(B) Immunoblot of WCL from P3HR-1 expressing the indicated sgRNA and uninduced or induced for lytic reactivation by 4HT (400 nM) for 24 h. Representative of n = 3 replicates.
(C) Mean ± SEM of plasma membrane (PM) gp350 levels in P3HR-1 expressing the indicated sgRNA, uninduced or 4HT-induced for reactivation for 24 h.
(D) Quantitative real-time PCR (qRT-PCR) of EBV intracellular genome copy number from P3HR-1 with the indicated sgRNA and 4HT-induced for 24 h. Mean ± SEM from n = 3 replicates.
(E) Immunoblot of 2.5% input and anti-HA immunopurified mitochondria from P3HR-1 stably expressing HA-OMP25, uninduced or 4HT-induced for 24 h. Representative of n = 2 replicates.
(F) Immunofluorescence analysis of NLRP3 and ASC in P3HR-1 expressing the indicated sgRNA, 4HT-induced for 24 h. Right: mean ± SEM percentage of cells with NLRP3/ASC specks from n = 3 replicates, as in (E), using data from 20 randomly selected panels of 200 nuclei, analyzed by ImageJ ComDet plugin.
(G) Immunoblot of ASC oligomerization from P3HR-1 expressing the indicated sgRNA, uninduced or 4HT-induced for 24 h. Representative of n = 2 replicates.
(H) Mean ± SEM from n = 3 replicates of caspase-1 activity normalized by live cell number from P3HR-1 expressing the indicated sgRNA and 4HT-induced for 24 h.
(I) Mean ± SEM from n = 3 replicates of caspase-1 activity normalized by live cell number from P3HR-1 cells expressing the indicated sgRNA, 4HT-induced ± the NLRP3 inhibitor MCC950 (10 μM) for 24 h.
(J) Mean ± SEM live cell percentages from n = 3 replicates of trypan blue staining of P3HR-1 expressing the indicated sgRNA and treated with 4HT, MCC950 (10 μM) or the caspase/pyroptosis inhibitor VX795 (10 μM) for 24 h.
(K) Mean ± SEM from n = 3 replicates of caspase-1 activity normalized by live cell number from P3HR-1 expressing the indicated cDNA and nigericin treated for 24 h.
Student’s t test was performed, with ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05. See also Figure S2.
Given the diversity of BILF1 host and viral protein interactions identified, we tested BILF1 knockout (KO) effects on EBV replication. Since an antibody against BILF1 is not available, high-frequency BILF1 CRISPR editing in latent P3HR-1 cells was validated by insertion or deletion (indel) sequencing and by immunoblot of exogenous V5-tagged BILF1 depletion (Figures S2A and S2B). A single-guide RNA (sgRNA) against EBV lytic gene BXLF1, which is not essential for replication,26,53 was performed as a control for EBV genome editing. BILF1 KO had little effect on EBV immediate early BZLF1 or early BMRF1 expression, but impaired late gene gp350 expression (Figures 3B, 3C, and S2C). BILF1 KO also reduced lytic EBV genome copy number and infectious virion production by approximately 75% and 50%, respectively (Figures 3D and S2D). Given BILF1 roles in support of both immune evasion and viral replication, we decided to pursue BILF1 function in depth.
Our proteomic analysis identified that BILF1 and BHRF1, but not other EBV proteins interacted with MAVS, a key pattern recognition receptor (PRR) (Figure 3A; Table S2). MAVS activates downstream interferon and inflammasome pathways in response to retinoic acid-inducible-I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5), typically in response to viral RNA PAMPs. Little has remained known about how EBV evades inflammasome pathways, which can exert potent antiviral effects. Therefore, given the proteomic signals that BILF1 associates with MAVS and other proteins potentially related to the inflammasome pathway activators (Figure 3A), we investigated potential BILF1 MAVS evasion roles. We first validated that a subpopulation of BILF1 interacts with MAVS and localizes to mitochondria. HA-tagged BILF1 co-immunoprecipitated MAVS from lysates of cells undergoing EBV lytic replication (Figure S2E). Similarly, we identified that a BILF1 subpopulation co-localized with the mitochondrial outer membrane protein TOMM20 (Figure S2F) and with immunopurified mitochondria (Figure 3E).
A key scaffold MAVS role in support of NLRP3 activation was reported.54 However, little has remained known about how EBV evades antiviral inflammasome pathways. As the EBV kinase BGLF4 suppresses interferon regulatory factor 3 (IRF3) responses downstream of MAVS,55 we hypothesized that BILF1 blocks MAVS to prevent NLRP3 inflammasome activation. To investigate this, we analyzed NLRP3 inflammasome assembly in control versus BILF1 KO B cells induced for reactivation. Although NLRP3 levels mildly increased upon EBV reactivation (Figure S2G), discrete foci of NLRP3 and the apoptosis-associated speck-like protein 2 (ASC2) co-localized, indicative of NLRP3 inflammasome activation (Figure 3F). We also observed the formation of high molecular weight ASC2 oligomers, indicative of NLRP3 inflammasome assembly (Figure 3G). Since the PRR absent in melanoma-2 (AIM2) assembles inflammasomes in response to murine cytomegalovirus DNA,56 we next tested EBV effects on AIM2. In contrast to effects on NLRP3, EBV reactivation did not induce co-localization between AIM2 and ASC, even with BILF1 KO (Figures S2H and S2I).
NLRP3 inflammasomes process IL-1β, IL-18, and gasdermin D (GSDMD) to induce pro-inflammatory signaling and pyroptosis cell death.57–59 We asked whether BILF1 restrains caspase-1 activation downstream of the NLRP3 inflammasome. Immunoblot analysis detected GSDMD and IL-1β caspase cleavage products in BILF1 KO, but not BXLF1 KO control P3HR-1 undergoing replication (Figure 3B). Similarly, caspase-1 activity was significantly increased in lytic BILF1 KO cells (Figure 3H), which was reduced by the NLRP3 inhibitor MCC95060 (Figure 3I). Similarly, treatment with either MCC95061 or the caspase-1/4 inhibitor VX76562,63 significantly increased live cell number in lytic BILF1 KO cells (Figure 3J).
Given that BILF1 associated with Na+/K+ transporting ATPase catalytic ATP1A1 and non-catalytic ATP1B3 subunits (Figure 3A), we tested whether BILF1 could block NLRP3 inflammasome assembly induced by the potassium ionophore nigericin. Constitutive expression of BILF1, but not BXLF1 cDNA, impaired NLRP3/ASC speck assembly, caspase-1 activity, and production of cleaved GSDMD and IL-1β in Akata cells (Figures 3K and S2J–S2L). Collectively, these results suggest that BILF1 subverts NLRP3 inflammasome activation otherwise triggered by EBV lytic replication.
BILF1 dislocates MAVS from the mitochondria to inhibit inflammasome activation
Subcellular mitochondrial or peroxisomal MAVS localization is crucial for NLRP3 inflammasome oligomerization and activation.54,64–66 In latent P3HR-1 and Akata B cells expressing BXLF1 or BILF1 sgRNAs, MAVS signal highly overlapped with the mitochondrial outer membrane translocase TOMM20 (Figure S3A). We hypothesized that BILF1 blocks inflammasome activation by interfering with NLPR3 recruitment to mitochondrial membrane MAVS foci. In support, minimal NLRP3 and TOMM20 co-localization was observed in 4HT-induced P3HR-1 expressing control BXLF1 sgRNA. However, BILF1 sgRNA expression resulted in a high degree of NLRP3 and TOMM20 signal overlap upon reactivation, suggesting that BILF1 interferes with NLRP3 mitochondrial recruitment, not previously reported in B lymphocytes (Figure 4A). Furthermore, CRISPR KO of both MAVS and BILF1 prevented NLRP3-ASC speck assembly and ASC oligomerization upon P3HR-1 lytic induction (Figures 4B and 4C). EBV lytic cycle-driven caspase-1 activity, IL-1β processing, and cell death were diminished in BILF1 KO cells by combined MAVS KO (Figures 4D–4F). Thus, despite emerging evidence that NLRP3 inflammasomes are assembled at the centrosome in macrophages,67,68 our studies highlight key mitochondrial membrane MAVS roles in NLRP3 inflammasome assembly at mitochondrial membranes in response to B cell EBV lytic induction but raise the question of how a viral 7TM GPCR can block MAVS/NLRP3 signaling.
Figure 4. BILF1 mediates MAVS dislocation from the mitochondria to inhibit NLRP3 inflammasome activation.

(A) Immunofluorescence analysis of NLRP3 and TOMM20 in P3HR-1 expressing the indicated sgRNA, 4HT-induced for 24 h. Right: mean ± SEM percentage of cells with NLRP3-TOMM20 co-localization from n = 3 replicates, using data from 10 randomly selected panels of 200 nuclei, analyzed by ImageJ ComDet plugin.
(B) Immunofluorescence analysis of NLRP3 and ASC speck formation in P3HR-1 expressing the indicated sgRNA and 4HT-induced for 24 h. Right: mean ± SEM percentage of cells with NLRP3/ASC specks from n = 3 replicates, using data from 20 randomly selected panels of 200 nuclei, analyzed by ImageJ ComDet plugin.
(C) Immunoblot of ASC oligomerization from P3HR-1 expressing the indicated sgRNA, uninduced, or 4HT-induced for 24 h. Representative of n = 2 replicates.
(D) Immunoblot of WCL from P3HR-1 expressing the indicated sgRNA, uninduced, or 4HT-induced for 24 h. # indicates low molecular weight bands immunoreactive with anti-MAVS antibody.
(E) Mean ± SEM from n = 3 replicates of caspase-1 activity normalized by live cell number from P3HR-1 expressing the indicated sgRNA, 4HT-induced for 24 h.
(F) Mean ± SEM from n = 3 replicates of trypan blue analysis of P3HR-1 expressing the indicated sgRNA, 4HT-induced for 24 h.
(G) Immunoblot of 293T transiently expressing the indicated cDNA. Representative of n = 2.
(H) Immunofluorescence analysis of MAVS and TOMM20 in P3HR-1 expressing the indicated sgRNA, 4HT-induced for 24 h. Right: mean ±SEM percentage of cells with delocalized MAVS from n = 3 replicates, using data from 20 randomly selected panels of 400 nuclei, analyzed by ImageJ ComDet plugin.
(I) Immunofluorescence analysis of MAVS and TOMM20 in P3HR-1, ± BILF1 cDNA induced by 5 mM doxycycline for 24 h. Right: mean ± SEM percentage of cells with delocalized MAVS from n = 3 replicates, using data from 30 randomly selected panels of 600 nuclei, analyzed by ImageJ ComDet plugin.
Student’s t test was performed, with ****p < 0.0001. ***p < 0.001. **p < 0.01. *p < 0.05. See also Figure S3.
We next characterized MAVS fate upon EBV lytic induction in control vs. BILF1 KO B cells. Interestingly, several low molecular weight polypeptides reactive with anti-MAVS antibody were evident upon lytic induction of P3HR-1 expressing BXLF1 control, but not BILF1 sgRNA (Figure 4D). MAVS sgRNA depleted these bands, even in cells co-expressing BILF1 sgRNA. The smaller MAVS species were recognized by a polyclonal antibody raised against full-length MAVS, but not by a monoclonal antibody against MAVS residues 34–96 (Figure S3B). Thus, BILF1 induces cleavage of the MAVS N terminus. To investigate this further, we expressed N-terminally EGFP-tagged and C-terminally HA-tagged MAVS (N′-EGFP-MAVS-C′-HA) with monomeric Cherry (mCherry)-tagged BILF1 or control BXLF1 in 293T cells. We note that mCherry-BILF1 migrates at a lower than predicted molecular weight, perhaps because of effects on BILF1 glycosylation. Expression of BILF1, but not BXLF1, resulted in appearance of MAVS fragments reactive with anti-HA antibody at ~ 70 kDa and just below 50 kDa (Figure 4G).
We next performed immunofluorescence microscopy on control versus BILF1 KO cells induced for lytic replication. In P3HR-1 expressing control BXLF1 sgRNA, 4HT treatment caused MAVS redistribution, with the formation of multiple puncta with limited TOMM20 overlap. However, MAVS remained highly TOMM20 co-localized in BILF1 KO P3HR-1 (Figure 4H). Similar results were obtained in Akata cells triggered for reactivation by immunoglobulin cross-linking (Figure S3C). MAVS puncta formation was also evident in lytic EBV+ gastric carcinoma AGS cells (Figure S3D). Therefore, immunoblot persistence of a population of full-length MAVS is likely due to a combination of MDV sequestration and MAVS re-synthesis. Alternatively, a subpopulation may be targeted, for example, multimerized MAVS.
BILF1 expression was sufficient to trigger MAVS dislocation (Figure 4I). We hypothesized that BILF1 might therefore perturb RIG-I-MAVS-driven IRF3 responses. Indeed, EBV lytic cycle-driven caspase-1 activity and cell death in BILF1 KO cells were significantly diminished by combined RIG-I KO (Figures S3E–S3G). IRF3 nuclear translocation and IRF3 puncta formation were observed in BILF1 KO, but not control cells (Figure S3H). EBV BHRF1 was reported to induce mitochondrial fission, induce mitophagy, and inhibit IRF3 nuclear translocation.69 Although our proteomic analysis identified BHRF1/MAVS association, neither BHRF1 KO nor BHRF1 cDNA expression altered MAVS localization (Figures S4A and S4B).
The BILF1 C-terminal tail recruits E3 ligase UFL1 to trigger MAVS K461 UFMylation
Proteomic analysis identified high-confidence interaction between BILF1 and host UFM1 specific ligase 1 (UFL1) (Figure 3A), an E3 ligase that mediates covalent attachment of the ubiquitinlike modifier UFM1 to target protein lysine residues in a process termed UFMylation.70 We validated this result by co-immunoprecipitation analysis (Figure S2E). Likewise, immunoprecipitation analysis identified that small populations of UFL1 and UFMylation pathway E2 ligase UFC1 were recruited to mitochondria in cells undergoing lytic replication (Figure 3E). Time-course analysis highlighted the appearance of a high molecular weight MAVS species migrating just above the full-length MAVS band, whose abundance was diminished by BILF1 KO (Figure S4C). We hypothesized that the high-molecular weight species was UFMylated MAVS and established control versus UFL1 KO P3HR-1. Although NLRP3 and ASC did not co-localize in 4HT-treated controls, NLRP3/ASC specks and ASC polymerization were evident in 4HT-treated UFL1 KO P3HR-1 (Figures 5A, S4D, and S4E). UFL1 KO also increased caspase-1 activity, GSDMD, and IL-1b processing in reactivated cells (Figures 5B and 5C). Likewise, UFL1 KO diminished the appearance of MAVS cleavage products (Figure S4F).
Figure 5. BILF1 triggers MAVS mitochondrial dislocation through UFMylation.

(A) Immunoblot of ASC oligomerization from P3HR-1 expressing the indicated sgRNA, uninduced or 4HT-induced for 24 h.
(B) Immunoblot analysis of WCL from P3HR-1 expressing the indicated sgRNA and 4HT-induced, as indicated.
(C) Mean ± SEM caspase-1 activity normalized by live cell number from n = 3 replicates of P3HR-1 expressing the indicated sgRNA and 4HT-treated for 24 h, as indicated.
(D) Immunoblot of 1% input and anti-FLAG-MAVS complexes from 293T co-transfected with FLAG-MAVS, BILF1, or BXLF1 cDNAs for 24 h, as indicated.
(E) PEAKS software identification of potential MAVS post-translational modification sites. Putative UFMylation sites were identified at lysines 362, 371, and 461. MAVS residues 321–500 are depicted. Black vertical lines represent individual peptide sequencing events.
(F) Immunoblot of anti-EGFP-MAVS immunopurified from 293T co-transfected with the indicated MAVS, BILF1, and UFM1 cDNAs for 24 h.
(G) Immunofluorescence analysis of wild type or K461R MAVS subcellular localization in 293T co-transfected with MAVS and BILF1 cDNAs for 24 h.
(H) Immunoblot of WCL from 293T expressing MAVS and BILF1 cDNA, nigericin stimulated for 24 h, as indicated.
(I) Immunoblot of 1% input and anti-HA immunopurified EGFP-MAVS and UFL1 from 293T transfected with BILF1 and EGFP-MAVS cDNAs for 24 h, as indicated.
(J) AlphaFold multimer model highlighting the predicted BILF1 and MAVS interaction domain and residues.
(K) Immunoblot of WCL from 293T expressing wild type, N- (ΔN) or C- (ΔC) terminal tail deletion mutant BILF1, nigericin stimulated for 24 h, as indicated.
(L) Schematic of BILF1 NLRP3 inflammasome inhibition.
Student’s t test was performed, with ***p < 0.001. *p < 0.05. See also Figures S4 and S5. Immunoblots are representative of n = 2.
We next investigated whether BILF1 expression was sufficient to trigger MAVS UFMylation. MAVS co-immunoprecipitated with UFL1 in cells expressing BILF1, but not BXLF1 (Figure S4G). Furthermore, BILF1 co-expression with C-terminal FLAG-tagged MAVS triggered the appearance of low molecular weight bands reactive with anti-FLAG antibody, suggestive of N-terminal MAVS cleavage. BILF1 expression also induced the formation of high molecular weight bands recognized by anti-UFM1 antibody on immunoblot analysis of purified FLAG-MAVS complexes (Figure 5D). Since samples were boiled prior to immunoprecipitation to disrupt protein complexes, this result is suggestive of covalent UFMylation.
To identify specific UFMylation site(s), we analyzed MAVS complexes immunopurified from BILF1+ vs. BXLF1+ cells by LC/MS. UFMylation signals were identified by the PEAKS algorithm at MAVS lysines K362/K371 and K461 only in BILF1+ cells.71 The absence of trypsin cleavage sites between K362 and K371 prevented assignment to just one of these lysines. We also used the CORE algorithm, which identified UFMylation signals only in BILF1+ cells at MAVS K461 (Figure 5E). To extend these MS results, we expressed MAVS lysine to arginine K362R/K371R and K461R point mutants together with BILF1. High-molecular-weight UFM1 signal of immunopurified K461R, but not K362R/K371R MAVS, suggested that BILF1 drives MAVS lysine 461 UFMylation (Figure 5F).
To test if UFMylation was necessary for MAVS mitochondrial dislocation, we knocked out the UFM1 E2 and E3 ligases, UFC1 and UFL1. Depletion of either impaired EBV MAVS dislocation (Figures S4H and S4I). Also suggestive of a key UFMylation role, BILF1 triggered dislocation of wild type and K362R/K371R MAVS, but not K461R in 293T (Figures 5G and S5A), and BILF1 blockade of nigericin NLRP3 inflammasome activation required UFL1 (Figures S5B and S5C). However, K461R MAVS expression precluded BILF1 NLRP3 antagonism (Figures 5H and S5D). We next asked if BILF1-mediated UFMylation impacted MAVS activation of downstream interferon responses. We overexpressed MAVS in 293T, which causes its oligomerization and activates downstream signaling.72 However, BILF1 blocked MAVS induction of the interferon-stimulated gene IFIT1 in a UFL1-dependent manner (Figure S5E).
Finally, we sought to identify the BILF1 region that associates with MAVS and UFL1. AlphaFold modeling73,74 suggested potential interactions between the BILF1 C-terminal tails, MAVS and UFL1 (Figures 5J and S5F). We generated BILF1 N- (ΔN) or C- (ΔC) terminal tail deletion mutants and performed co-immunoprecipitation analysis. ΔC-BILF1 had a reduced association with MAVS and UFL1 (Figure 5I) and diminished MAVS mitochondrial dislocation (Figure S5G). We note that BILF1 over-expression in 293T resulted in a low level of glycosylation, such that most BILF1 migrated at the predicted non-glycosylated molecular weight. DC-BILF1 also failed to inhibit nigericin NLRP3 activation (Figures 5K and S5H). By contrast, BILF1 C174A and K122A point mutants,75 which abolish GPCR signaling and MHC class I downregulation, did not affect MAVS dislocation or NLRP3 inflammasome inhibition (Figures S5I and S5J). These data suggest key BILF1 C-terminal tail roles in MAVS K461 UFMylation (Figure 5L).
BILF1 dislocates MAVS into MDVs for lysosomal proteolysis
To gain insights into MAVS disposition in BILF1+ cells, we performed confocal microscopy. 4HT triggered MAVS co-localization with the lysosomal marker LAMP1 in P3HR1 and AGS cells (Figures 6A–6C, S6A, and S6B). We therefore asked whether MAVS N-terminal cleavage fragments were enriched within immunopurified lysosomes.76 Immunoblots of cytosolic tubulin versus lysosomal LAMP1 controls indicated a high degree of lysosomal enrichment (Figure 6D). Importantly, the lysosomal fraction contained predominantly MAVS N-terminal cleavage products, whereas full-length and high-molecular-weight MAVS were instead more abundant in whole-cell lysate (WCL). We did not observe an enrichment of either UFC1 or UFL1 in lysosome fractions, suggesting that MAVS is delivered to lysosomes in the absence of other UFMylation pathway components (Figure 6E).
Figure 6. MAVS sequestration and lysosomal fusion events captured in real time.

(A) Left: immunofluorescence analysis of MAVS and LAMP1 in P3HR-1, uninduced, or 4HT-induced for 24 h. Right: fluorescence intensity line scanning of LAMP1 (red) and MAVS (green) in the white rectangle. * and # mark co-localization and non-co-localization, respectively.
(B) Mean ± SEM percentage of cells with MAVS-LAMP1 co-localization from n = 3 replicates, as in (A), using data from 12 randomly selected panels of 240 nuclei, analyzed by ImageJ ComDet plugin.
(C) Immunofluorescence analysis of MAVS and LAMP1 in EBV+ AGSiZ gastric carcinoma, lytic induced by doxycycline for 24 h. Right: mean ± SEM percentage of cells with MAVS-LAMP1 co-localization from n = 3 replicates, using data from 12 randomly selected panels of 240 nuclei, analyzed by ImageJ ComDet plugin.
(D) Immunoblot of 2.5% input and anti-HA immunopurified lysosomes from P3HR-1 HA-TMEM192+ cells, 4HT-induced for 24 h, as indicated. High and low molecular weight bands reactive with anti-MAVS antibodies are denoted by #. UFL1 is denoted by *. Representative of n = 2.
(E) Consecutive frames captured at 10 s intervals at 3.5 h post AGSiZ lytic induction by doxycycline. White arrows highlight a GFP-MAVS puncta (green) superimposed on BFP-labeled mitochondria (blue) and SIRylo-stained lysosomes (red). Consecutive frames show partial co-localization of the GFP-MAVS puncta and lysosome (red) signals at +10 and +20 s, and then loss of MAVS puncta signal at the site of lysosome overlap at +40 s. See also corresponding Video S1.
(F) Fluorescence intensity line scanning of lysosome SIRylo (red) and GFP-MAVS (green) signals at the white arrow marked puncta in (F) at +10, +20, and +40 s.
(G) Schematic illustration of UFMylated MAVS trafficking from mitochondria to lysosome via MDVs.
Student’s t test was performed, with ****p < 0.0001. See also Figure S6 and Video S1.
MDVs transport cargo generated by selective incorporation of mitochondrial content.77–81 Although these can include mitochondrial outer membrane proteins, MAVS incorporation into MDVs has not been described. Therefore, to investigate MAVS trafficking, we stably expressed N-terminally GFP-tagged MAVS and then transiently expressed mitochondrial-targeted Aequorea victoria blue fluorescence protein (BFP) in AGSiz cells. Lysosomes were imaged by live cell staining with the SiR lysosomal dye, which is activated by lysosomal cathepsins. Beginning at approximately 200 min post-induction, we observed GFP-MAVS signal budding off mitochondrial membranes (Figure 6E). MAVS-GFP signal then co-localized with SiR, with rapid subsequent loss of GFP signal, consistent with lysosomal destruction (Figures 6E and 6F; Video S1). These data support a model in which MAVS traffics via MDVs to lysosomes (Figure 6G).
BILF1 utilizes PARK2 for lysosomal MAVS dislocation
UFMylation is implicated in selective lysosomal autophagic turnover of endoplasmic reticulum contents.82 We hypothesized that BILF1/UFL1-driven UFMylation could serve to recruit PARK2 (also called PARKIN), an E3 ubiquitin ligase with MDV generation roles in response to mitochondrial stress.83,84 We established control and PARK2 KO P3HR-1 and as a further control, P3HR-1 knocked out for the mitophagy regulator Drp1. Dislocated MAVS puncta were evident in reactivated Drp1 KO cells but not PARK2 KO (Figures 7A and 7B). PARK2 KO also significantly increased levels of NLRP3/ASC specks, ASC oligomerization, caspase-1 activity, GSDMD and IL-1b cleavage (Figures 7C, 7D, and S6C), and MAVS fragments (Figure 7E), suggesting a key PARK2 role in MAVS lysosomal delivery.
Figure 7. BILF1 dislocates MAVS to mitochondria derived vesicles.

(A) Immunofluorescence analysis of MAVS and TOMM20 in P3HR-1 expressing the indicated PARK2 or Drp1 sgRNAs and 4HT-induced for 24 h.
(B) Mean ± SEM percentage of cells with dislocated MAVS from n = 3 replicates, as judged by appearance of MAVS puncta that did not overlap with TOMM20 signal as in (A), using data from 25 randomly selected panels of 500 nuclei, analyzed using ImageJ ComDet plugin.
(C) Mean ± SEM percentage of P3HR-1 with NLRP3/ASC specks from n = 3 replicates, using data from 20 randomly selected panels of 200 nuclei, analyzed by ImageJ ComDet plugin.
(D) Mean ± SEM caspase-1 activity normalized by live cell number from n = 3 replicates of P3HR-1 expressing the indicated sgRNA and 4HT-induced for 24 h, as indicated.
(E) Immunoblot of WCL from P3HR-1 expressing the indicated sgRNA and 4HT-induced for 24 h. # denotes low molecular weight bands immunoreactive with anti-MAVS antibody. Representative of n = 3.
(F) PAKR2 and TOMM20 immunofluorescence analysis in 293T transfected with BILF1 or BXLF1 cDNA for 24 h.
(G) Immunoblot of 1% input vs. anti-HA-MAVS immunopurified from wild type or UFL1-KO 293T transfected with MAVS and BILF1 cDNA for 24 h, as indicated. Representative of n = 2.
(H) Schematic of NLRP3 inflammasome subversion by BILF1. BILF1 recruits UFL1 to mediate MAVS UFMylation, which together with PARK2 triggers selective MAVS removal from the mitochondrial outer membrane, MDV packaging and delivery to lysosomes, preventing NLRP3 inflammasome activation and pyroptosis. Student’s t test was performed, with ****p < 0.0001. ***p < 0.001. ns > 0.05. See also Figure S6.
We hypothesized that PARK2 is recruited to mitochondria to ubiquitylate MAVS, perhaps following UFMylation. To investigate this, we stained PARK2 and TOMM20 in BILF1+ or BXLF1+ 293T. PARK2 co-localized with TOMM20 only with BILF1 expression (Figure 7F). To test if UFMylation was required, we co-expressed BILF1 and MAVS in control vs. UFL1 KO 293T. Immunoblot revealed high-molecular-weight ubiquitin conjugates of MAVS complexes immunopurified from control, but not UFL1 KO (Figure 7G).
Our proteomic analysis highlighted BILF1 association with the Ras-related proteins Rab2A and Rab7A (Figure 3A), which regulate intracellular vesicles transport. Rab2A localizes to autophagosomes,85 whereas Rab7A controls late endosome-lysosome fusion.86 Confocal analysis highlighted partial co-localization of dislocated MAVS puncta with Rab5A and Rab7A, but not Rab27B or the autophagy marker LC3B in 4HT-treated P3HR-1 (Figures S6D–S6G). This pattern suggests MAVS sorting to endosomes, independently of exosome or autophagy pathways. Collectively, our results support a model in which BILF1 triggers MAVS UFMylation, PARK2-dependent sorting to MDVs, and then lysosome degradation, highlighting a pathway for selective dislocation of mitochondrial membrane cargo (Figure 7H).
DISCUSSION
We leveraged the first lytic cycle EBV protein interaction map to identify high-confidence B cell and viral protein interactors of nearly all EBV ORFs, many of which have remained little studied. We also presented the Viroplex resource, in which EBV, KSHV, and HCMV interaction networks are presented for cross-comparison. This approach led to the observation that EBV BILF1 hijacks the UFMylation pathway to target MAVS for MDV trafficking and lysosomal degradation, highlighting a means to selectively degrade mitochondrial membrane cargo and prevent NLRP3 inflammasome activation.
In addition to characterized roles in the blockade of CXCR487 and MHC class I,50,51 we found that BILF1 targets MAVS to block NLRP3 inflammasomes and IRF3 nuclear translocation. These results raise the question of what MAVS and NLRP3 each sense to culminate in MAVS/NLRP3 association at mitochondrial membrane sites, necessitating BILF1 evasion. HSV and EBV replication unmask a host cell 5S rRNA pseudogene that binds and activates RIG-I.88 However, this 5S rRNA sensing pathway activated pro-inflammatory cytokine expression even in BILF1+ cells, suggesting an alternative stimulus may activate MAVS without BILF1.
EBV lytic replication produces multiple RNA species, including circular RNAs,89,90 which can activate RIG-I/MAVS.91 NLRP3 responds to stress induced by danger-associated molecular patterns,92 including alterations in levels of ATP, potassium, sodium, oxidized mitochondrial DNA, or mitochondrial reactive oxygen species. It is noteworthy that our proteomic analysis identified high-confidence interactions between BILF1 and MAVS and between BILF1 and PM transporters of sodium, potassium, glucose, and leucine, raising the possibility that BILF1 may perturb upstream aspects of NLRP3 activation. These proteomic signals were a major impetus to study BILF1 roles in inflammasome evasion.
In response to RNA viral infection, MAVS serves as a key mitochondrial docking site and driver of NLRP3 oligomerization.54,64,65 Whether this phenomenon also occurs in response to DNA virus infection has remained unknown. Our findings indicate that MAVS likewise plays key roles in NLRP3 recruitment to mitochondrial outer membranes and in NLRP3 inflammasome activation in response to EBV and therefore likely also to other DNA viruses. Although plasma cell differentiation is the major EBV reactivation trigger, high extracellular glucose can also trigger NLRP3 inflammasomes and EBV lytic reactivation.93 Once induced, BILF1 then blocks NLRP3, presumably to prevent pyroptosis, support viral replication, and dampen pro-inflammatory signaling.
Several herpesviruses activate NLRP inflammasomes, highlighting a key difference with EBV. KSHV ORF45 induces conformational changes that drive NLRP1 inflammasome assembly.94 This may be balanced by the KSHV ORF63 NLRP1 homolog, which inhibits NLRP1.95 However, we speculate that KSHV- and HCMV-encoded GPCRs may not target MAVS, given their low degree of C-terminal tail sequence similarity with BILF1.
A point mutation that disrupts BILF1 PM GPCR signaling did not abrogate MAVS dislocation, suggesting that BILF1 GPCR signaling and MAVS subversion are genetically separable. How BILF1 distributes to PM versus mitochondrial membrane sites remains unknown. Upon binding to kynurenic acid, host GPCR GPR35 traffics from the PM to the outer mitochondria membrane.96 Likewise, Rab5a, which partially co-localized with BILF1, translocates from early endosomes to mitochondrial membranes with oxidative stress.97
The signal transduction mediator 14-3-3ε is UFMylated in response to RNA virus infection, which supports RIG-I/MAVS association and their downstream activation of type I interferon responses.98 UFL1 is recruited to mitochondrial-associated ER membrane (MAM) upon RNA viral infection.98,99 Therefore, although not necessarily required for UFL1 recruitment MAM, we speculate that the BILF1 C-terminal tail evolved the ability to also recruit UFL1 to coordinate MAVS UFMylation. Thus, EBV converted a key antiviral UFMylation signal to subvert MAVS-driven NLRP3 inflammasomes.
We suggest that BILF1 exploits an existing pathway, whereby UFMylation triggers the dislocation of mitochondrial outer membrane proteins to MDVs for lysosomal delivery. MDVs can act as mitochondrial stress responses, functioning prior to or in parallel with mitophagy.77–80 Although PARK2 poly-ubiquitination of mitofusins and voltage-dependent anion channels triggers autophagy,100 UFMylation may instead trigger selective removal of mitochondrial membrane proteins. Similarly, UFMylation of polypeptides stalled in the endoplasmic reticulum translocon supports their lysosomal degradation.101 PARK2 was essential for MAVS MDV packaging, suggesting that UFMylation and poly-ubiquitin chains may together guide its selective mitochondrial extraction and lysosomal delivery. Recently, UFMylation was implicated in CMV targeting of HLA molecules for proteasomal degradation.102 However, UFMylation was not detectable on HLA, suggesting a distinct mechanism.
GPCRs are targets of ~ 30% of FDA-approved drugs.103 The above studies highlight BILF1 as an intriguing therapeutic target. Antagonists of BILF1 association with MAVS or UFL1 or a small molecule BILF1 degrader compound, promises to re-sensitize cells undergoing lytic replication to pyroptosis. Furthermore, if used together with lytic reactivation strategies, BILF1 antagonists could trigger tumor cell death, although also inducing pro-inflammatory IL-1 and IL-18 responses to induce cellular anti-tumor immune responses.
In summary, a B cell protein EBV lytic cycle protein interaction map revealed a wide array of virus/host interactions. Cross-comparison with HCMV and KSHV proteomic datasets highlighted common and unique herpesvirus host targets. The EBV GPCR BILF1 utilizes its C-terminal tail to trigger MAVS K461 UFMylation, resulting in selective MAVS removal from mitochondrial membranes and routing via MDVs to lysosomes to prevent EBV activation of the NLRP3 inflammasome, which otherwise triggers pyroptosis and limits viral replication.
Limitations of the study
Our proteomic map used ORFs encoded by type I EBV, as it is more prevalent worldwide and is more transforming. However, a subset of results could differ with type II EBV strain ORFs. We used P3HR-1 cells with type II EBV, although a substantial portion of type I and II polymorphism resides within EBV latency proteins. Likewise, our proteomic studies were performed in B cells, and particular results may differ in epithelial cells. We used doxycycline to induce EBV ORFs, which can cause toxicity over long durations. However, we exposed cells for 15 h to minimize this concern. Attempts to remove sticky proteins by stringent filtering may have created false negative results when hosts or viral proteins truly interacted with multiple EBV baits. With regards to BILF1, studies were conducted in cell line models, since robust primary B cell systems for EBV lytic reactivation are not currently available. Key BILF1 results remain to be validated in EBV+ memory B cells triggered for lytic replication, a model for which does not presently exist.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Benjamin E. Gewurz (bgewurz@bwh.harvard.edu).
Materials availability
All reagents will be made available on request after completion of a Materials Transfer Agreement.
Data and code availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE104 partner repository (PRIDE: PXD041336) and made publicly available upon publication. All other raw data have been deposited at Mendeley and are publicly available as of the date of publication (Mendeley Data: https://doi.org/10.17632/zk884935vw.1). Microcopy data reported in this paper will be shared by the lead contact upon request. Figures were drawn with commercially available GraphPad, Biorender and Microsoft Powerpoint.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
HEK293T were cultured in DMEM supplemented with 10% FBS and 1% Pen/Strep. AGSiZ were cultured in F-12-Glutomax supplemented with 10% FBS, 1% Pen/Strep, 0.5 μg/ml puromycin and 0.5 μg/ml G418. P3HR-1-ZHT/RHT-Cas9+, EBV+ Akata-Cas9+, Daudi-Cas9+ cells and P3HR-1-TMEM192 cells were cultured in RPMI-1640 supplemented with 10% v/v FBS and 1% Pen/Strep. Cas9+ cells were maintained in 5 μg/ml blasticidin. P3HR-1-Z/R-HT were also maintained with 25 μg/ml G418 and 25 μg/ml hygromycin. All cells were incubated at 37°C with 5% CO2 and were routinely confirmed to be mycoplasma-negative. Cell lines were authenticated by STIR profiling. cDNA used in this study were cloned into the pLIX-402 vector. pLIX-402 uses a Tet ON TRE promoter to drive expression of the gene of interest with a C-terminal HA Tag. Stable cell lines were generated by lentiviral transduction and antibiotic selection with puromycin (pLIX-402). Cell lines were then maintained with 3.3 μg/ml puromycin.
METHOD DETAILS
Molecular cloning
Unless otherwise specified, all cloning experiments were performed by Gateway recombination. Briefly, 150 ng of the destination vector and donor vector containing the gene of interest were co-incubated with 1X LR Clonase Enzyme Mix (Invitrogen #11789–020) overnight at room temperature. The reaction mixture was then transformed into 50 μl of Stbl3 bacteria, spread on LB plates with corresponding antibiotics. MAVS and BILF1 mutants are generated either by standard restriction-ligation procedures or site-directed mutagenesis according to manufacturer’s protocol (NEB # E0554). Oligos used for mutagenesis are listed in Table S6.
Chemical compounds
Unless otherwise specified, EBV lytic induction in P3HR-1, Akata and AGSiZ cells were induced with 400 nM 4-Hydroxytamoxifen (4HT) (Sigma Aldrich) for 24h, 15 μg/ml anti-IgG crosslinking (Agilent) for 48h and 5 μg/ml doxycycline (Sigma Aldrich) for 24h, respectively. Inhibition of NLRP3 inflammasome in lytic-induced BILF1-depleted P3HR-1 cells was achieved by the addition of 10 μM MCC950 (InvivoGen) simultaneously with the EBV lytic inducer 4HT for 24h. NLRP3 inflammasome activation was induced by the potassium ionophore, Nigericin (InvivoGen) at 10 μM for 4h.
Sample preparation for LC/MS proteomics analysis
Samples were generated and analyzed in technical duplicate, using the method originally described in Huttlin et al.32,105 Whole cell lysates were prepared from 400 million P3HR-1 ZHT/RHT cells per replicate that were induced into the lytic cycle by 4HT (400 nM) and NaB (500 μM) for 24h, and that were also induced to express a HA-tagged bait by doxycycline (5 μg/ml) addition for the final 15hrs. Samples were prepared as previously described.28 Briefly, cells were lysed in (50 mM Tris-HCI pH 7.5, 300 mM NaCl, 0.5% v/v NP40, 1 mM DTT and Roche protease inhibitor cocktail). Samples were tumbled for 15 mins at 4°C and subjected to centrifugation at 16,000xg for 15 mins at 4°C. Lysates were then filtered through a 0.7 μm filter and incubated for 3h with immobilized mouse monoclonal anti-HA agarose resin (Sigma). Duplicates samples were combined and washed seven times with lysis buffer, followed by seven PBS washes. Immunopurified proteins were then eluted by addition of 200 μl of 250 μg/ml HA peptide in PBS at 37°C for 30 mins with agitation, followed by another identical peptide elution. Eluted material were then precipitated with 20% trichloroacetic acid (TCA), washed once with 10% TCA, washed three times with cold acetone and dried to completion, using a centrifugal evaporator. Samples were resuspended in digestion buffer (50 mM Tris-HCl pH 8.5, 10% acetonitrile (AcN), 1 mM DTT, 10 μg/ml trypsin (Promega) and incubated overnight at 37°C, with agitation. The reaction was quenched by 50% formic acid (FA), subjected to C18 solid-phase extraction, and vacuum-centrifuged to complete dryness. Samples were reconstituted in 4% AcN/5% FA and divided into technical duplicates prior to LC-MS/MS on an Orbitrap Lumos.
LC-MS Proteomic Analysis
Peptides for each sample were analyzed in technical duplicate, with the run order reversed from one batch of replicate analyses to the next to ensure that any carry-over was different in each case. Two washes were used between each sample to further minimize carry-over. Mass spectrometry data were acquired using an Orbitrap Fusion Lumos. An Ultimate 3000 RSLC nano UHPLC equipped with a 300 mm ID × 5 mm Acclaim PepMap m-Precolumn (Thermo Fisher Scientific) and a 75 μm ID × 75 cm 2 μm particle Acclaim PepMap RSLC analytical column was used. Loading solvent was 0.1% v/v FA, and the analytical solvents were (A) 0.1% v/v FA and (B) 80% v/v AcN + 0.1% v/v FA. All separations were carried out at 55C. Samples were loaded at 5 μl/min for 5 min in loading solvent before beginning the analytical gradient. The following gradient was used: 3–7% B over 3 min then 7–37% B over 54 min followed by a 4 min wash in 95% B and equilibration in 3% B for 15 min. The following settings were used: MS1, 350–1500 Thompsons (Th), 120,000 resolution, 2 × 105 automatic gain control (AGC) target, 50 ms maximum injection time. MS2, quadrupole isolation at an isolation width of m/z 0.7, higher-energy collisional dissociation (HCD) fragmentation (normalized collision energy (NCE) 34) with fragment ions scanning in the ion trap from m/z 120, 1 × 104 AGC target, 250 ms maximum injection time, with ions accumulated for all parallelizable times. The method excluded undetermined and very high charge states (≥25+). Dynamic exclusion was set to + /− 10 ppm for 25 s. MS2 fragmentation was triggered on precursors 5 × 103 counts and above. Two 45 min washes were included between every affinity purification-mass spectrometry (AP-MS) analysis, to minimize carry-over between samples. 1 μl transport solution (0.1% v/v trifluoroacetic acid) was injected, over the following gradient: 3–40% B over 29 min followed by a 3 min wash at 95% B and equilibration at 3% B for 10 min.
CompPASS identification of high confidence protein interactors
To identify interactors for each bait, replicate pairs were combined to attain a summary of proteins identified in both runs. Data reported for each protein in every IP in the dataset include: (a) the number of peptide spectrum matches (PSMs) averaged between technical replicates; (b) an entropy score, which compares the number of PSM between replicates to eliminate proteins that are not detected consistently; (c) a z-score, calculated in comparison to the average and standard deviation of PSMs observed across all IPs; and (d) an NWD score, which reflects (i) how frequently this protein was detected and (ii) whether it was detected reproducibly. NWD scores were calculated as described in34 using the fraction of runs in which a protein was observed, the observed number of PSMs, the average and standard deviation of PSMs observed for that protein across all IPs, and the number of replicates (1 or 2) containing the protein of interest. Protein interactors identified were filtered as described in the legend to Figure 1A. Specifically, stringent filters were applied to remove inconsistent and low-confidence protein identifications across all IPs and thus minimize both false protein identifications and associations.32 These included a minimum PSM score of 1.5 (i.e. ≥3 peptides per protein across both replicates) and an entropy score of ≥0.75. Very high confidence interacting proteins (VHCIPs) were defined as prey meeting PSM and entropy criteria with either an NWD≥1.0 or z-score≥4.0, and high confidence interacting proteins (HCIPs) were defined as prey meeting PSM and entropy criteria with either an NWD≥1.0 or z-score≥3.0.
NWD scores were normalized so that the top 2% earned scores of ≥ 1.0. Stringent filters were applied to remove inconsistent and low-confidence protein identifications across all IPs and thus minimize both false protein identifications and associations.32 These included a minimum PSM score of 1.5 (i.e. R3 peptides per protein across both replicates) and an entropy score of ≥0.75. Very high confidence interacting proteins (VHCIPs) were defined as prey meeting PSM and entropy criteria with either an NWD≥1.0 or z-scoreR4.0, and high confidence interacting proteins (HCIPs) were defined as prey meeting PSM and entropy criteria with either an NWD≥1.0 or z-score≥3.0 (Table S2). To facilitate global analysis of all data, HCIPs as opposed to VHCIPs were examined for the remainder of this study. Previous studies have estimated a 5% false discovery rate when employing a similar strategy with a top NWD score cutoff of 2%.33
CRIPSR analysis
CRISPR/Cas9 editing was performed as described.106 Briefly, sgRNAs were cloned into pLentiGuide-puro (Addgene plasmid #52963,107 pLenti-spBsmBI-sgRNA-Hygro (Addgene plasmid #62205108 or LentiGuide-zeo (Addgene plasmid #160091109 by restriction-ligation, and sequenced verified. Lentiviral transduction in 293T cells were performed as described previously.31 In brief, 293T cells were co-transfected with 500ng lentiviral plasmid, 400ng psPAX2 (a gift from Didier Trono, Addgene plasmid #12260) and 150ng VSV-G plasmids for packaging. Lentivirus produced were filtered with 0.45μm filter and transduced into P3HR-1-Z/R-HT-Cas9+ and Akata-EBV-Cas9+ cells. Transduced cells were selected for 1 week with 0.5 μg/ml puromycin or 2 weeks with 25 μg/ml hygromycin or 100 μg/ml zeocin. CRISPR KOs were verified by western blot analysis or amplicon sequencing (only when antibody is unavailable). sgRNA against genes used in this study are listed in Table S6.
Quantification of EBV copy number
Intracellular EBV genome copy # were quantified by qPCR analysis. For intracellular viral DNA extraction, total DNA from 1×106 cells were extracted by the Blood & Cell culture DNA mini kit (Qiagen). Extracted DNA were diluted to 10 ng/μl and were subjected to qPCR targeting the BALF5 gene. Serial dilutions of pHAGE-BALF5 plasmid at 25 ng/μl were used to generate the standard curve. Viral DNA copy number was calculated by substituting sample Cq values into the regression equation dictated by the standard curve. qPCR primer sequences are listed in Table S6.
Immunoblot analysis
Immunoblot analyses were performed as described previously.26 In brief, whole cell lysates were separated by SDS-PAGE electrophoresis, transferred onto nitrocellulose membrane, blocked with 5% milk in TBST buffer for 1h and incubated with the corresponding primary antibodies at 4°C overnight. Blots were washed 3 times in TBST solution and were incubated with secondary antibodies for 1h at room temperature. Blots were then washed 3 times in TBST solution and were developed by incubating with ECL chemiluminescence. Images were captured by Licor Fc platform. All antibodies used in this study are listed in the key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-EBV ZEBRA mouse monoclonal antibody (BZ1) | Santa Cruz Biotechnology | cat# sc-53904; RRID: AB_783257 |
| Anti-EBV Ea-D mouse monoclonal antibody (0261) | Santa Cruz Biotechnology | cat# sc-58121; RRID: AB_631448 |
| Anti-gp350/220 mouse monoclonal antibody (OT6) | A gift from Jaap M Middeldorp | N/A |
| Anti-EBV BHRF1 rabbit polyclonal antibody | Thermo Fisher Scientific | cat# PA5-117549; RRID: AB_2902178 |
| Anti-GAPDH XP® rabbit monoclonal antibody (D16H11) | Cell Signaling Technology | cat# 5174; RRID: AB_10622025 |
| Anti-HA.11 tag mouse monoclonal antibody (16B12) | Biolegend | cat# 901513; RRID: AB_2820200 |
| Anti-NLRP3 rabbit polyclonal antibody | Proteintech | cat# 19771-1-AP; RRID: AB_10646484 |
| Anti-ASC/TMS1 rabbit monoclonal antibody (E1E3I) | Cell Signaling Technology | cat# 13833; RRID: AB_2798325 |
| Anti- ASC/TMS1 mouse monoclonal antibody | Proteintech | cat# 67494-1-Ig; RRID: AB_2882718 |
| Anti-AIM2 mouse monoclonal antibody | Biolegend | cat# 652802; RRID: AB_2561540 |
| Anti-TOMM20 mouse monoclonal antibody (F-10) | Santa Cruz Biotechnology | cat# sc-17764; RRID: AB_628381 |
| Anti-MAVS rabbit polyclonal antibody | Proteintech | cat# 14341-1-AP; RRID: AB_10548408 |
| Anti-MAVS mouse monoclonal antibody | Proteintech | cat# 66911-1-Ig; RRID: AB_2882238 |
| Anti-MAVS rabbit polyclonal antibody (Position: L34-Q96) | Thermo Fisher Scientific | cat# PA5-79636; RRID: AB_2746751 |
| Anti-RIG-I rabbit monoclonal antibody (D14G6) | Cell Signaling Technology | cat# 3743; RRID: AB_2269233 |
| Anti-GFP rabbit polyclonal antibody | Proteintech | cat# 50430-2-AP; RRID: AB_11042881 |
| Anti- mCherry rabbit monoclonal antibody (E5D8F) | Cell Signaling Technology | cat# 43590; RRID: AB_2799246 |
| Anti-UFM1 rabbit polyclonal antibody | Proteintech | cat# 15883-1-AP; RRID: AB_2878195 |
| Anti-UFL1 rabbit polyclonal antibody | Proteintech | cat# 26087-1-AP; RRID: AB_2880370 |
| Anti-UFC1 rabbit polyclonal antibody | Proteintech | cat# 15783-1-AP; RRID: AB_2213938 |
| Anti-IL-1β rabbit polyclonal antibody | Novus Biologicals | cat# NB600633; RRID: AB_577977 |
| Anti-GSDMD rabbit polyclonal antibody | Abcam | cat# ab155233; RRID: AB_2736999 |
| Anti-LAMP1 mouse monoclonal antibody (D4O1S) | Cell Signaling Technology | cat# 15665; RRID: AB_2798750 |
| Anti-PMP70 (ABCD3) mouse monoclonal antibody | Proteintech | cat# 66697-1-Ig; RRID: AB_2882050 |
| Anti-Rab5A mouse monoclonal antibody (E6N8S) | Cell Signaling Technology | cat# 46449; RRID: AB_2799303 |
| Anti-Rab7 mouse monoclonal antibody (E9O7E) | Cell Signaling Technology | cat# 95746; RRID: AB_2800252 |
| Anti-LC3B mouse monoclonal antibody (E5Q2K) | Cell Signaling Technology | cat# 83506; RRID: AB_2800018 |
| Anti-Rab27b mouse monoclonal antibody | Proteintech | cat# 66944-1-Ig; RRID: AB_2882268 |
| Anti-α-tubulin rabbit monoclonal antibody (11H10) | Cell Signaling Technology | cat# 2125; RRID: AB_2619646 |
| Anti-PARK2 mouse monoclonal antibody (Prk8) | Cell Signaling Technology | cat# 4211; RRID: AB_2159920 |
| Anti-Drp1 rabbit polyclonal antibody (N-terminal) | Proteintech | cat# 26187-1-AP; RRID: AB_2880417 |
| Anti-CNOT1 rabbit polyclonal antibody | Proteintech | cat# 14276-1-AP; RRID: AB_10888627 |
| Anti-CNOT10 rabbit polyclonal antibody | Proteintech | cat# 15938-1-AP; RRID: AB_2229678 |
| Anti-IRF3 rabbit polyclonal antibody | Proteintech | cat# 11312-1-AP; RRID: AB_2127004 |
| Anti-dsRNA mouse monoclonal antibody (clone rJ2) | Miilipore-Sigma | cat# MABE1134; RRID: AB_2819101 |
| Anti-Mouse IgG HRP-coupled secondary antibody | Cell Signaling Technology | cat# 7076; RRID: AB_330924 |
| Anti-Rabbit IgG HRP-coupled secondary antibody | Cell Signaling Technology | cat# 7074; RRID: AB_2099233 |
| Rabbit anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | cat# A-21239; RRID: AB_2535808 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | cat# A-11001; RRID: AB_143160 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | cat# A-21244; RRID: AB_2535812 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | cat# A-11008; RRID: AB_143165 |
| Anti-Human IgG rabbit polyclonal antibody (Gamma-Chains) | Agilent | cat# A042402; RRID: AB_578517 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Pierce™ Protein A/G Magnetic Beads | Thermo Fisher Scientific | 88803 |
| Pierce™ Anti-HA Magnetic Beads | Thermo Fisher Scientific | 88837 |
| T4 DNA ligase | New England Biolabs | M0202L |
| Doxycycline hyclate | Sigma-Aldrich | D9891-1G |
| (Z)-4-Hydroxytamoxifen | Sigma-Aldrich | H7904-25MG |
| NAE Inhibitor, MLN4924 | Sigma-Aldrich | 5.05477 |
| Bortezomib (PS-341) | APExBIO | A2614 |
| Sir-Lysosome Kit | Cytoskeleton Inc. | CY-SC012 |
| NLRP3 inhibitor, MCC950 | A gift from Jonathan Kagan; InvivoGen | MCC950 |
| Caspase-1/4 and Pyroptosis inhibitor, VX765 | InvivoGen | VX765 |
| Nigericin | InvivoGen | tlrl-nig |
| Lipofectamine™ 2000 Transfection Reagent | Thermo Fisher Scientific | 11668019 |
| Sodium butyrate, >98%, Alfa Aesar™ | Thermo Fisher Scientific | AAA1107922 |
| 2-NBDG (2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose) | Thermo Fisher Scientific | N13195 |
| NP40 | Sigma-Aldrich | 74385-1L |
| Sequencing Grade Modified Trypsin (Mass Spec Grade) (lyophilized) | Promega | V5111-5x20μg |
| HA Synthetic Peptide | Thermo Fisher Scientific | 26184-5mg |
| Monoclonal Anti-HA–Agarose antibody produced in mouse | Sigma-Aldrich | A2095-1ML |
| Formaldehyde solution | Sigma-Aldrich | F8775 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | 11697498001 |
| N-Ethylmaleimide (NEM) | Sigma-Aldrich | E3876 |
| Puromycin Dihydrochloride | Thermo Fisher Scientific | A1113803 |
| Hygromycin B | Millipore | 400052 |
| G418 Sulfate Solution (50 mg/mL) | GeminiBio | 400113 |
| Blasticidin | InvivoGen | ant-bl-5 |
| Zeocin™ Selection Reagent | Thermo Fisher Scientific | R25001 |
| TransIT®-LT1 Transfection Reagent | Mirus Bio | MIR 2306 |
| DSS (disuccinimidyl suberate) | Thermo Fisher Scientific | 21555 |
|
| ||
| Critical commercial assays | ||
|
| ||
| RNeasy Mini Kit | Qiagen | 74104 |
| QiAquick PCR Purification Kit | Qiagen | 28106 |
| QIAprep Spin Miniprep Kit | Qiagen | 27106 |
| DNeasy Blood& Tissue Kit | Qiagen | 69504 |
| QIAquick Gel Extraction Kit | Qiagen | 28704 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | 4367659 |
| Gateway™ LR Clonase™ II Enzyme Mix | Invitrogen | 11789-020 |
| Caspase-Glo® 1 Inflammasome Assay | Promega | G9951 |
| Dual-Luciferase® Reporter Assay System | Promega | E1910 |
|
| ||
| Deposited data | ||
|
| ||
| Mendeley dataset | https://doi.org/10.17632/zk884935vw.1 | |
| Mass spectrometry raw files and associated unmodified peptide and protein quantitation data | Data are available via ProteomeXchange PRIDE: PXD041336.104 | |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| EBV+ Burkitt lymphoma P3HR-1 ZHT | A gift from Eric Johannsen | N/A |
| EBV+ Burkitt lymphoma AKATA-Cas9 | Guo et al.106 | N/A |
| EBV+ Burkitt lymphoma Daudi-Cas9 | Ma et al., 2017 | N/A |
| HEK293T | ATCC | CRL-3216 |
| EBV+ Gastric carcinoma AGSiZ | A gift from Sankar Swaminathan | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT-TMEM192 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT-OMP25 | This study | N/A |
| HEK293T sgUFL1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT sgBILF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT sgBXL1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT sgBHRF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT sgBILF1/MAVS | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT sgBILF1/RIG-I | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT sgPARK2 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT sgDrp1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT sgUFL1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT sgUFC1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-Zta | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BRLF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BDLF3.5 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BALF5 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BGLF3.5 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BNLF2b | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BKRF3 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BVLF1.5 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BLLF3 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BRRF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BFLF2 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BGLF3 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-LF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-SM | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BALF1/0 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BFRF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-LF2 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BGLF4 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BGLF5 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BFLF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BXLF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BALF3 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BBLF2/3 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BcRF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BBLF4 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BSLF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BALF2 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BNRF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BBLF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BLRF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BFRF0.5 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BKRF2 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BLLF2 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BLRF2 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BCRF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BFRF3 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BKRF4 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BSRF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BZLF2 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BDLF3 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BILF2 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BXRF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BBRF2 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BDLF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BILF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BGLF2 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BdRF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BMRF2 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BORF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BTRF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BBRF3 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BDLF2 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BGLF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BRRF2 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BVRF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BVRF2 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BBRF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-B-GD-RF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BXLF2 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BALF4 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-gp350/220 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BOLF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BcLF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BHRF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BARF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BaRF1 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BFRF2 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BORF2 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BPLF1 1–1000 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BPLF1 501–1500 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BPLF1 1001–2000 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BPLF1 1501–2500 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BPLF1 2001–3000 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BPLF1 3001–3149 | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BNLF2a | This study | N/A |
| EBV+ Burkitt lymphoma P3HR-1 ZHT pLIX402-BDLF4 | This study | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| CRISPR sgRNAs are listed in Table S6 | This paper | N/A |
| qPCR primers for EBV copy number quantification are listed in Table S6 | This paper | N/A |
| Primers used for amplicon sequencing on BILF1 gene are listed in Table S6 | This paper | N/A |
| gBlocks and oligos used for molecular cloning of MAVS and BILF1 mutants are listed in Table S6 | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pLentiGuide-Puro | A gift from Feng Zhang (Addgene plasmid # 52963; http://n2t.net/addgene:52963)107 | RRID:Addgene_52963 |
| pLenti SpBsmBI sgRNA Hygro | A gift from Rene Maehr (Addgene plasmid # 62205; http://n2t.net/addgene:62205)108 | RRID:Addgene_62205 |
| LentiGuide-zeo | A gift from Rizwan Haq (Addgene plasmid # 160091; http://n2t.net/addgene:160091)109 | RRID:Addgene_160091 |
| pLX-TRC313 | Broad Institute | N/A |
| pLX-402 | Broad Institute | N/A |
| pDEST-CMV-N-EGFP | A gift from Robin Ketteler (Addgene plasmid # 122842; http://n2t.net/addgene:122842)110 | RRID:Addgene_122842 |
| pDEST-CMV-N-mCherry | A gift from Robin Ketteler (Addgene plasmid # 123215; http://n2t.net/addgene:123215)110 | RRID:Addgene_123215) |
| pHAGE-C-HA-FLAG | A gift from James Decaprio | N/A |
| Mito-BFP | A gift from Gia Voeltz (Addgene plasmid # 49151; http://n2t.net/addgene:49151)111 | RRID:Addgene_49151 |
| pLJC6-3XHA-TMEM192 | A gift from David Sabatini (Addgene plasmid # 104434; http://n2t.net/addgene:104434)112 | RRID:Addgene_104434 |
| pMXs-3XHA-EGFP-OMP25 | A gift from David Sabatini (Addgene plasmid # 83356; http://n2t.net/addgene:83356)113 | RRID:Addgene_83356 |
| pDONR221-MAVS | DNASU | HsCD00296475 |
| pENTR223-UFM1 | DNASU | HsCD00504493 |
| pENTR-Zta | A gift from Eric Johannsen | N/A |
| pENTR-BRLF1 | Eric Johannsen | N/A |
| pENTR-BDLF3.5 | Eric Johannsen | N/A |
| pENTR-BALF5 | Eric Johannsen | N/A |
| pENTR-BGLF3.5 | Eric Johannsen | N/A |
| pENTR-BNLF2b | Eric Johannsen | N/A |
| pENTR-BKRF3 | Eric Johannsen | N/A |
| pENTR-BVLF1.5 | Eric Johannsen | N/A |
| pENTR-BLLF3 | Eric Johannsen | N/A |
| pENTR-BRRF1 | Eric Johannsen | N/A |
| pENTR-BFLF2 | Eric Johannsen | N/A |
| pENTR-BGLF3 | Eric Johannsen | N/A |
| pENTR-LF1 | Eric Johannsen | N/A |
| pENTR-SM | Eric Johannsen | N/A |
| pENTR-BALF1/0 | Eric Johannsen | N/A |
| pENTR-BFRF1 | Eric Johannsen | N/A |
| pENTR-LF2 | Eric Johannsen | N/A |
| pENTR-BGLF4 | Eric Johannsen | N/A |
| pENTR-BGLF5 | Eric Johannsen | N/A |
| pENTR-BFLF1 | Eric Johannsen | N/A |
| pENTR-BXLF1 | Eric Johannsen | N/A |
| pENTR-BALF3 | Eric Johannsen | N/A |
| pENTR-BBLF2/3 | Eric Johannsen | N/A |
| pENTR-BcRF1 | Eric Johannsen | N/A |
| pENTR-BBLF4 | Eric Johannsen | N/A |
| pENTR-BSLF1 | Eric Johannsen | N/A |
| pENTR-BALF2 | Eric Johannsen | N/A |
| pENTR-BNRF1 | Eric Johannsen | N/A |
| pENTR-BBLF1 | Eric Johannsen | N/A |
| pENTR-BLRF1 | Eric Johannsen | N/A |
| pENTR-BFRF0.5 | Eric Johannsen | N/A |
| pENTR-BKRF2 | Eric Johannsen | N/A |
| pENTR-BLLF2 | Eric Johannsen | N/A |
| pENTR-BLRF2 | Eric Johannsen | N/A |
| pENTR-BCRF1 | Eric Johannsen | N/A |
| pENTR-BFRF3 | Eric Johannsen | N/A |
| pENTR-BKRF4 | Eric Johannsen | N/A |
| pENTR-BSRF1 | Eric Johannsen | N/A |
| pENTR-BZLF2 | Eric Johannsen | N/A |
| pENTR-BDLF3 | Eric Johannsen | N/A |
| pENTR-BILF2 | Eric Johannsen | N/A |
| pENTR-BXRF1 | Eric Johannsen | N/A |
| pENTR-BBRF2 | Eric Johannsen | N/A |
| pENTR-BDLF1 | Eric Johannsen | N/A |
| pENTR-BILF1 | Eric Johannsen | N/A |
| pENTR-BGLF2 | Eric Johannsen | N/A |
| pENTR-BdRF1 | Eric Johannsen | N/A |
| pENTR-BMRF2 | Eric Johannsen | N/A |
| pENTR-BORF1 | Eric Johannsen | N/A |
| pENTR-BTRF1 | Eric Johannsen | N/A |
| pENTR-BBRF3 | Eric Johannsen | N/A |
| pENTR-BDLF2 | Eric Johannsen | N/A |
| pENTR-BGLF1 | Eric Johannsen | N/A |
| pENTR-BRRF2 | Eric Johannsen | N/A |
| pENTR-BVRF1 | Eric Johannsen | N/A |
| pENTR-BVRF2 | Eric Johannsen | N/A |
| pENTR-BBRF1 | Eric Johannsen | N/A |
| pENTR-B-GD-RF1 | Eric Johannsen | N/A |
| pENTR-BXLF2 | Eric Johannsen | N/A |
| pENTR-BALF4 | Eric Johannsen | N/A |
| pENTR-gp350/220 | Eric Johannsen | N/A |
| pENTR-BOLF1 | Eric Johannsen | N/A |
| pENTR-BcLF1 | Eric Johannsen | N/A |
| pENTR-BALF1 | Eric Johannsen | N/A |
| pENTR-BHRF1 | Eric Johannsen | N/A |
| pENTR-BARF1 | Eric Johannsen | N/A |
| pENTR-BaRF1 | Eric Johannsen | N/A |
| pENTR-BFRF2 | Eric Johannsen | N/A |
| pENTR-BORF2 | Eric Johannsen | N/A |
| pENTR-BPLF1 1–1000 | Eric Johannsen | N/A |
| pENTR-BPLF1 501–1500 | Eric Johannsen | N/A |
| pENTR-BPLF1 1001–2000 | Eric Johannsen | N/A |
| pENTR-BPLF1 1501–2500 | Eric Johannsen | N/A |
| pENTR-BPLF1 2001–3000 | Eric Johannsen | N/A |
| pENTR-BPLF1 3001–3149 | Eric Johannsen | N/A |
| pENTR-BNLF2a | Eric Johannsen | N/A |
| pENTR-BDLF4 | Eric Johannsen | N/A |
| pDEST-CMV-N-EGFP BILF1 | This study | N/A |
| pDEST-CMV-N-EGFP-BXLF1 | This study | N/A |
| pDEST-CMV-N-EGFP-MAVS (wildtype) | This study | N/A |
| pDEST-CMV-N-EGFP-MAVS (K362R/K371R) | This study | N/A |
| pDEST-CMV-N-EGFP-MAVS (K461R) | This study | N/A |
| pLIX402-Zta | This study | N/A |
| pLIX402-BRLF1 | This study | N/A |
| pLIX402-BDLF3.5 | This study | N/A |
| pLIX402-BALF5 | This study | N/A |
| pLIX402-BGLF3.5 | This study | N/A |
| pLIX402-BNLF2b | This study | N/A |
| pLIX402-BKRF3 | This study | N/A |
| pLIX402-BVLF1.5 | This study | N/A |
| pLIX402-BLLF3 | This study | N/A |
| pLIX402-BRRF1 | This study | N/A |
| pLIX402-BFLF2 | This study | N/A |
| pLIX402-BGLF3 | This study | N/A |
| pLIX402-LF1 | This study | N/A |
| pLIX402-SM | This study | N/A |
| pLIX402-BALF1/0 | This study | N/A |
| pLIX402-BFRF1 | This study | N/A |
| pLIX402-LF2 | This study | N/A |
| pLIX402-BGLF4 | This study | N/A |
| pLIX402-BGLF5 | This study | N/A |
| pLIX402-BFLF1 | This study | N/A |
| pLIX402-BXLF1 | This study | N/A |
| pLIX402-BALF3 | This study | N/A |
| pLIX402-BBLF2/3 | This study | N/A |
| pLIX402-BcRF1 | This study | N/A |
| pLIX402-BBLF4 | This study | N/A |
| pLIX402-BSLF1 | This study | N/A |
| pLIX402-BALF2 | This study | N/A |
| pLIX402-BNRF1 | This study | N/A |
| pLIX402-BBLF1 | This study | N/A |
| pLIX402-BLRF1 | This study | N/A |
| pLIX402-BFRF0.5 | This study | N/A |
| pLIX402-BKRF2 | This study | N/A |
| pLIX402-BLLF2 | This study | N/A |
| pLIX402-BLRF2 | This study | N/A |
| pLIX402-BCRF1 | This study | N/A |
| pLIX402-BFRF3 | This study | N/A |
| pLIX402-BKRF4 | This study | N/A |
| pLIX402-BSRF1 | This study | N/A |
| pLIX402-BZLF2 | This study | N/A |
| pLIX402-BDLF3 | This study | N/A |
| pLIX402-BILF2 | This study | N/A |
| pLIX402-BXRF1 | This study | N/A |
| pLIX402-BBRF2 | This study | N/A |
| pLIX402-BDLF1 | This study | N/A |
| pLIX402-BILF1 | This study | N/A |
| pLIX402-BGLF2 | This study | N/A |
| pLIX402-BdRF1 | This study | N/A |
| pLIX402-BMRF2 | This study | N/A |
| pLIX402-BORF1 | This study | N/A |
| pLIX402-BTRF1 | This study | N/A |
| pLIX402-BBRF3 | This study | N/A |
| pLIX402-BDLF2 | This study | N/A |
| pLIX402-BGLF1 | This study | N/A |
| pLIX402-BRRF2 | This study | N/A |
| pLIX402-BVRF1 | This study | N/A |
| pLIX402-BVRF2 | This study | N/A |
| pLIX402-BBRF1 | This study | N/A |
| pLIX402-B-GD-RF1 | This study | N/A |
| pLIX402-BXLF2 | This study | N/A |
| pLIX402-BALF4 | This study | N/A |
| pLIX402-gp350/220 | This study | N/A |
| pLIX402-BOLF1 | This study | N/A |
| pLIX402-BcLF1 | This study | N/A |
| pLIX402-BHRF1 | This study | N/A |
| pLIX402-BARF1 | This study | N/A |
| pLIX402-BaRF1 | This study | N/A |
| pLIX402-BFRF2 | This study | N/A |
| pLIX402-BORF2 | This study | N/A |
| pLIX402-BPLF1 1–1000 | This study | N/A |
| pLIX402-BPLF1 501–1500 | This study | N/A |
| pLIX402-BPLF1 1001–2000 | This study | N/A |
| pLIX402-BPLF1 1501–2500 | This study | N/A |
| pLIX402-BPLF1 2001–3000 | This study | N/A |
| pLIX402-BPLF1 3001–3149 | This study | N/A |
| pLIX402-BNLF2a | This study | N/A |
| pLIX402-BDLF4 | This study | N/A |
| PHAGE-3X FLAG-HA MAVS | This study | N/A |
| PHAGE-3X FLAG-HA BILF1 | This study | N/A |
| PHAGE-3X FLAG-HA BXLF1 | This study | N/A |
| PHAGE-3X FLAG-HA UFM1 | This study | N/A |
| pDEST-CMV-N-mCherry BXLF1 | This study | N/A |
| pDEST-CMV-N-mCherry BILF1 (wildtype) | This study | N/A |
| pDEST-CMV-N-mCherry BILF1 (ΔN) | This study | N/A |
| pDEST-CMV-N-mCherry BILF1 (ΔC) | This study | N/A |
| pDEST-CMV-N-mCherry BILF1 (C174A) | This study | N/A |
| pDEST-CMV-N-mCherry BILF1 (K122A) | This study | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Database for Annotation, Visualization and Integrated Discovery (DAVID) | Laboratory of Human Retrovirology and Immunoinformatics | https://david.ncifcrf.gov/home.jsp |
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Flowjo X | Flowjo LLC. | https://www.flowjo.com/ |
| Biorender | Biorender | https://biorender.com/ |
| ImageJ | ImageJ | https://imagej.nih.gov/ij/ |
| ImageJ- ComDet | ImageJ | https://imagej.net/imagej-wiki-static/Spots_colocalization_(ComDet) |
| Zeiss Zen Lite (Blue) | Zeiss | https://www.zeiss.com/microscopy/int/products/microscope-software/zen-lite.html |
| Arivis Vision4D | Arivis | https://imaging.arivis.com/en/imaging-science/arivis-vision4d |
| AlphaFold Collab | Highly accurate protein structure prediction with AlphaFold73 | https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebooks/AlphaFold.ipynb#scrollTo=pc5-mbsX9PZC |
| Robetta Server | Protein structure prediction service114 | https://robetta.bakerlab.org/ |
| The PyMOL Molecular Graphics System, Version 2.0 | Schrödinger, LLC | http://www.pymol.org/pymol |
| R | R Core Team116 | https://www.R-project.org/ |
| R-shiny | shiny: Web Application Framework for R. R package version 1.7.2.9000117 | https://shiny.rstudio.com/ |
| PEAKS X Pro | Bioinformatics Solutions Inc. | https://www.bioinfor.com/peaks-xpro/ |
|
| ||
| Other | ||
|
| ||
| Standard Fetal Bovine Serum, Qualified, USDA-Approved Regions | Thermo Fisher Scientific | 10437028 |
| RPMI 1640 Medium | Life Technologies | 11875085 |
| DMEM, high glucose, pyruvate | Life Technologies | 11995081 |
| Ham’s F-12 Nutrient Mix, GlutaMAX™ Supplement | Thermo Fisher Scientific | 31765035 |
Flow cytometry analysis
Cells were washed once with cold PBS supplemented with 2% v/v fetal bovine serum (FBS). Cells were then incubated with Cy5-conjugaged anti-gp350 antibody (1:1000) in 2% FBS v/v, PBS for 30 mins at 4°C. Cells were pelleted, washed twice, resuspended in 2% FBS v/v, PBS into flow cytometry-compatible tubes and processed immediately. Flow cytometric data was acquired with a BD FACSCalibur instrument and analysis was performed with FlowJo V10.
Immunofluorescence analysis
Cells dried on glass slides were fixed with 4% paraformaldehyde/PBS solution for 10 mins, permeabilized with 0.5% Triton X-100/PBS for 5 mins and blocked with 1% BSA/PBS for 1h at room temperature. Subsequently, cells were incubated with a cocktail of primary antibodies at 1:100 against NLRP3, ASC, TOMM20, MAVS, LAMP1, AIM2, Rab5A, Rab7, Rab27b and LC3B in blocking solution for 1h at 37°C. Cells were then washed twice with PBS and incubated with a cocktail of secondary antibodies at 1:1000 in PBS for 1h at 37°C in the dark. Finally, cells were washed twice with PBS and were stained/mounted overnight with ProLong™ Gold Antifade Mountant with DAPI. Image acquisition and analysis was performed with Zeiss LSM 800 instrument and with Zeiss Zen Lite (Blue) software, respectively. Arivis Vision4D from ZEISS ZEN lite (blue edition) was used for 3D reconstruction in Figures S5 and S6. Image J was used to score the % of colocalization of MAVS-TOMM20, NLRP3-ASC, NLRP3-AIM2 and MAVS-LAMP1 in P3HR-1 cells, using the ImageJ “ComDet” plugin.
Live cell imaging
MAVS cDNA (DNASU#HsCD00296475) was cloned into pDEST-CMV-N-EGFP plasmid (a gift from Robin Ketteler, Addgene plasmid #122842110 by Gateway cloning to produce pDEST-CMV-N-EGFP-MAVS plasmid. To visualize the mitochondria, the mito-BFP plasmid (a gift from Gia Voeltz, Addgene plasmid #49151111 was used. Mito-BFP expresses BFP fused to an N-terminal mitochondrial targeting signal sequence obtained from COX4 amino acids 1–21. AGSiZ cells were plated in glass bottom 35 mm dish (ibidi #81158) at 0.4 million/ml. The next day, the plated cells were replenished with fresh medium at least 1h prior to co-transfection by pDEST-CMV-N-EGFP-MAVS and mito-BFP plasmids using Lipofectamine 2000 (Thermo) according to manufacturer’s instruction. Transfection medium was replaced by fresh medium after 6h and cells were incubated for an addition of 18h before live cell imaging. EBV lytic cycle was induced with 10 μg/ml Doxycycline and active lysosome was stained by 200 nM SiR-lysosome (#CY-SC012, Cytoskeleton, Inc.) during the course of experiment. At 200 mins post-lytic induction, image acquisition was performed with Zeiss LSM 800 instrument at an interval of 10 s for 15 mins and Zeiss Zen Lite (Blue) software was used for image analysis.
Co-immunoprecipitation analysis
Expression of either BILF1-WT, BILF1-ΔN or BILF1-ΔC were induced by the addition of 5 mg/ml doxycycline, 5 nM of Bortezomib was added to preserve UFMylation and 400 nM 4HT was used to induce the lytic cycle in P3HR-1 cells. 100 million cells were harvested and was lysed in ice cold lysis buffer (1% v/v NP40, 150 mM Tris, 300 mM NaCl in dH2O) supplemented with 1X cOmplete™ EDTA-free protease inhibitor cocktail (Sigma), 1 mM Na3VO4 and 1 mM NaF for 1h at 4°C with rotation. Lysed cells were pelleted, and lysates were incubated with anti-HA tag magnetic beads (Pierce, Thermo) at 4°C overnight. Beads were washed with lysis buffer for four times and were eluted using 1X SDS loading buffer incubated for 10 mins at 95°C. Proteins were separated by SDS-PAGE gel and transferred to nitrocellulose membranes. Subsequent procedures were similar to that mentioned in “Immunoblot analysis”
UFMylation co-immunoprecipitation analysis
Expression of MAVS was induced by addition of 5 μg/ml doxycycline. 100 million cells were harvested and lysed in ice cold lysis buffer (1% v/v NP40, 50 mM Tris, 150 mM NaCl, 0.5% w/v sodium deoxycholate, 10% glycerol in dH2O) supplemented with 1X cOmplete™ EDTA-free protease inhibitor cocktail (Sigma), 1 mM Na3VO4, 1 mM NaF and 20 mM N-Ethylmaleimide (NEM, Sigma-Aldrich) for 1h at 4°C with rotation. Lysed cells were pelleted, and lysates were boiled at 100°C for 5 mins and then re-pelleted. Lysates were then precleared with 0.2 μg anti-mouse IgG isotype antibodies and protein A/G magnetic beads (Pierce, Thermo) for 30 mins at 4°C with rotation. Precleared lysates were then incubated with anti-MAVS antibody for 1 h at 4°C. Protein A/G magnetic beads were then added to the immunocomplex and were incubated overnight at 4°C. Beads were washed with cold lysis-boil buffer for four times and were eluted using 1X SDS loading buffer incubated for 10 mins at 95°C. Proteins were separated by SDS-PAGE gel and transferred to nitrocellulose membranes. Subsequent procedures were similar to that mentioned in “Immunoblot analysis”
Sample preparation and LC-MS/MS for post-translational modification analysis
Samples were reduced, alkylated and digested in-gel using trypsin. The resulting peptides were dried and re-suspended 20 μl 3% MeCN/0.1% TFA.
Mass spectrometry data was acquired using a Q Exactive Plus coupled to an Ultimate 3000 RSLC nano UHPLC equipped with a 100 μm ID × 2 cm Acclaim PepMap Precolumn (Thermo Fisher Scientific) and a 50 μm ID × 50 cm, 2 μm particle Acclaim PepMap RSLC analytical column. Loading solvent was 0.1% FA with analytical solvents A: 0.1% FA and B: 80% MeCN + 0.1% FA. Samples were loaded at 5 μl/minute loading solvent for 5 minutes before beginning the analytical gradient. The analytical gradient was 10–40% B over 42 minutes rising to 95% B by 45 minutes followed by a 4 minute wash at 95% B and equilibration at 3% solvent B. Columns were held at 40°C. Data was acquired in a DDA fashion with the following settings: MS1: 400–1500 Th, 70,000 resolution, 1×105 AGC target, 250 ms maximum injection time. MS2: Quadrupole isolation at an isolation width of m/z 3.0, HCD fragmentation (NCE 30). Dynamic exclusion was set for 30 s. MS2 fragmentation was trigged on precursors 3.2 ×104 counts and above.
Raw files were processed using PEAKS X Pro (Bioinformatics Solutions Inc.). Data was searched against a human Uniprot Database (downloaded 12/1/21) and a database of common contaminants. The data was searched using the following variable PTMs: carbamidomethylation on cysteine residues, oxidation on methionine residues and UFMylation on lysine residues. In Figure *, peptides identified by de novo sequencing in PEAKS software are not shown.
Lysosome-immunoprecipitation and mitochondria-immunoprecipitation
Lysosome-immunoprecipitation or mitochondria-immunoprecipitation was performed as described previously.112,113 In brief, 35 million P3HR-1- stably expressing HA-TMEM192 or HA-OMP25 cells were harvested and washed twice with cold PBS. The cells were resuspended in 1 ml of cold KPBS (136 mM KCl, 10 mM KH2PO4, pH7.25) and were then centrifuged at 1000 xg for 2 mins at 4°C. Pelleted cells were resuspended in 500 μl of ice cold KPBS and were homogenized on ice with a 1 ml homogenizer. 2.5% of the homogenate was reserved as input and the remaining fraction was centrifuged at max speed for 15 mins at 4°C. The lysosome or mitochondria containing supernatant fraction was then incubated with 150 μl of anti-HA magnetic beads (Pierce, Thermo) prewashed with KPBS for 15 mins at 4°C with rotation. Beads were washed with cold KPBS for three times and were eluted using 1X SDS loading buffer incubated for 10 mins at 95°C. Subsequent procedures were similar to that mentioned in “Immunoblot analysis”
Caspase-1 activity measurement
Caspase-1 activity in this study was measured using Caspase-Glo® 1 Inflammasome Assay from Promega (#G9951) according to manufacturer’s instructions. In brief, cells were mixed with Caspase-Glo® 1 reagent at a 1:1 ratio and incubated for 1h at room temperature. Luminescence signal was measured using a Molecular Devices microplate reader. Final caspase-1 activities presented were determined by normalizing the signal against live cell number, as determined by trypan blue vital dye exclusion.
ASC polymerization
ASS polymerization was performed as described previously. Briefly, 3 million cells were harvested and washed with cold PBS once. The pellet was homogenized with 100 μl 1% NP-40 and was centrifuged at max speed for 5 mins at 4°C. Supernatant was collected, mixed with 4X SDS loading buffer and boiled for 10 mins. 1mM DSS (disuccinimidyl suberate) in PBS was added to the pellet and incubated in shaking incubators for 15 mins at 37°C. DSS solution is removed followed by centrifugation at max speed for 5 mins. The pellet was resuspended in 1X SDS loading buffer and boiled for 10 mins. Subsequent procedures were similar to that mentioned in “Immunoblot analysis”
Structural prediction
Structural prediction for BILF1-MAVS and BILF1-UFL1 was performed with AlphaFold Collab- Multimer73 and Robetta server,114 respectively.Structure visualization was performed with PyMOL version 2.0.
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise indicated, all bar and line graphs represent the arithmetic mean of three independent experiments (n = 3), with error bars denoting SEM. Significance between the control and experimental groups, or indicated pairs of groups, was assessed using the unpaired Student’s t-test in the GraphPad Prism 7 software. P values correlate with symbols as follows, unless otherwise indicated: ns = not significant, p > 0.05; *p % 0.05; **p % 0.01; ***p % 0.001; ****p % 0.0001.
Network Assembly
To facilitate cross-species comparisons, we combined our EBV interactions with published interaction networks from human Cytomegalovirus and Kaposi’s Sarcoma Associated Herpesvirus,28,29 as well as human protein-protein interactions from the BioPlex project.47 For this analysis, interactions of human proteins observed in both 293T and HCT116 cell lines were merged. Across all viral interaction datasets, human preys were mapped to Entrez Gene ID’s and then merged with the combined BioPlex network to create a single composite interaction network. Interactions among virus proteins in each species were included as well. Depending on the analysis, EBV interactions were filtered using either stringent or relaxed criteria prior to incorporation.
Pathway and cluster analysis
The Database for Annotation, Visualization and Integrated Discovery (DAVID) 2021 version41 was used to determine pathway enrichment for Figures 1B and 1C and Table S3. Indicated subsets of human HCIPs were searched against a background of all human proteins, using default settings.
To identify baits that interact with host E1s, E2s, E3s, cullins, deubiquitinases (DUB), proteasome components (PSM) and ubiquitin-binding proteins (UBP), HCIPs were searched against a comprehensive list provided in https://elifesciences.org/articles/40009#s4.45 To supplement this list with E3s that ligate other moieties in addition to ubiquitin (such as SUMO, UFM), HCIPs that included the term ‘E3’ in their protein name were additionally searched.
Hierarchical centroid clustering based on uncentered Pearson correlation was performed using Cluster 3.0 (Stanford University) and visualised using Java Treeview (http://jtreeview.sourceforge.net).
Proximity Scoring of CORUM Complexes
An approach based on network propagation115 was used to quantify the proximity of human proteins and protein complexes to proteins belonging to each virus. Each viral proteome was scored separately via network propagation. To begin, each viral protein was assigned a starting weight of 1.0 with all other proteins assigned a weight of zero. Forty iterations of a Random Walk were then performed while allowing restarts at each step with probability 0.5. The final weights assigned to each protein were then returned. This procedure was then repeated across 100 additional randomized versions of the combined human-virus protein-protein interaction network. Network randomization preserved bait and prey identities as described previously.47
To identify protein complexes that were associated with each virus, “core” complexes from the CORUM database48 were downloaded and scored individually. To score a complex, its constituent proteins were identified in the network and their network propagation scores were extracted and summed. Any complex members not found in the interaction network were ignored in this analysis. This process was then repeated across all 100 randomized networks and the resulting scores were used to determine averages and standard deviations that could be used to convert the complex’s summed propagation score into a Z-score. To consider a complex associated with a particular virus, we required a minimum Z-score of 4 and a minimum summed propagation score of 0.04. Identical criteria were used for all three viruses and were found to correspond to approximately a 5% FDR based on scoring of each virus against an additional randomized network. Scores are provided in Table S4.
For purposes of plotting (see Figure 2B), CORUM complexes were displayed if their Z-scores observed across all three cell lines summed to 20 or greater. This was simply done to ensure that the resulting figure was legible. Plots of individual complexes (Figures 2C and 2D) were generated by extracting CORUM complex members and all viral proteins within a graph distance of 2 or less of at least one complex member. Network layouts were calculated via gravity embedding, and unconnected nodes were manually removed.
Network analyses and plotting were performed using Mathematica 13.1 (Wolfram Research).
Interaction Network Web Browser
A custom viewer for our combined viral-human protein interaction network is available at https://wren.hms.harvard.edu/ViroPlex/. It was implemented using R116 as well as R-Shiny.117 Our viewer includes interactions for all three viral interactomes – including both stringent and relaxed filtering of EBV interactions – as well as human protein-protein interactions drawn from BioPlex. To improve performance, only those human protein-protein interactions within max distance of 2 are displayed in the viewer.
Supplementary Material
Highlights.
First EBV interactome identifies key EBV-host and EBV-EBV interactions in lytic B cells
Common host protein nodes targeted by multiple human herpesviruses identified
EBV BILF1 drives MAVS lysine 461 UFMylation to block NLRP3 inflammasome activation
UFM1 and Parkin trigger selective degradation of mitochondrial membrane cargo
ACKNOWLEDGMENTS
We thank Eric Johannsen for EBV cDNA vectors and for helpful discussions. We thank Jaap Middeldorp for the anti-gp350 monoclonal antibody OT6; Michael Calderwood for the split luciferase plasmids; Adam Maynard and Naama Kanarek for the UFC1 sgRNA vector; and Megan Orzalli, Pascal Devant, and Jonathan Kagan for discussion and the NLRP3 inhibitor. This work was supported by NIH R01s AI137337, AI164709, CA228700 and U01 CA275301 and a Burroughs Wellcome Career Award in Medical Sciences to B.E.G., by HG010730, and by NIH R01 HG010730. M.P.W. is supported by the Medical Research Council (MR/W025647/1), Addenbrooke’s Charitable Trust, the Wellcome Trust Institutional Strategic Support Fund (204845/Z/16/Z) and the Cambridge Biomedical Research Centre, UK. For the purpose of open access, the author has applied a CC-BY public copyright license to any Author Accepted Manuscript version arising from this submission.
Footnotes
DECLARATION OF INTERESTS
B.E.G. receives support from an Abbvie-Harvard grant for research unrelated to these studies.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2023.05.018.
REFERENCES
- 1.Parkin DM (2006). The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 118, 3030–3044. 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- 2.Zur Hausen H, and de Villiers EM (2015). Reprint of: cancer “causation” by infections–individual contributions and synergistic networks. Semin. Oncol. 42, 207–222. 10.1053/j.seminoncol.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 3.Farrell PJ (2019). Epstein-Barr virus and cancer. Annu. Rev. Pathol. 14, 29–53. 10.1146/annurev-pathmechdis-012418-013023. [DOI] [PubMed] [Google Scholar]
- 4.Shannon-Lowe C, Rickinson AB, and Bell AI (2017). Epstein-Barr virus-associated lymphomas. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372. 20160271. 10.1098/rstb.2016.0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gewurz BE, Longnecker RM, and Cohen JI (2021). Epstein-Barr Virus. Fields Virology, Seventh Edition (Wolters Kluwer Health Adis (ESP)). [Google Scholar]
- 6.Young LS, and Dawson CW (2014). Epstein-Barr virus and nasopharyngeal carcinoma. Chin. J. Cancer 33, 581–590. 10.5732/cjc.014.10197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wen KW, Wang L, Menke JR, and Damania B (2022). Cancers associated with human gammaherpesviruses. FEBS Journal 289, 7631–7669. 10.1111/febs.16206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raab-Traub N (2015). Nasopharyngeal carcinoma: an evolving role for the Epstein-Barr virus. Curr. Top. Microbiol. Immunol. 390, 339–363. 10.1007/978-3-319-22822-8_14. [DOI] [PubMed] [Google Scholar]
- 9.Soldan SS, and Lieberman PM (2023). Epstein-Barr virus and multiple sclerosis. Nat. Rev. Microbiol. 21, 51–64. 10.1038/s41579-022-00770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thorley-Lawson DA (2015). EBV persistence–introducing the virus. Curr. Top. Microbiol. Immunol. 390, 151–209. 10.1007/978-3-319-22822-8_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hammerschmidt W, and Sugden B (2013). Replication of Epstein-Barr viral DNA. Cold Spring Harb. Perspect. Biol. 5, a013029. 10.1101/cshperspect.a013029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murata T, Sugimoto A, Inagaki T, Yanagi Y, Watanabe T, Sato Y, and Kimura H (2021). Molecular basis of Epstein-Barr virus latency establishment and lytic reactivation. Viruses 13, 2344. 10.3390/v13122344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan J, Cahir-McFarland E, Zhao B, and Kieff E (2006). Virus and cell RNAs expressed during Epstein-Barr virus replication. J. Virol. 80, 2548–2565. 10.1128/JVI.80.5.2548-2565.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Münz C (2019). Latency and lytic replication in Epstein-Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 17, 691–700. 10.1038/s41579-019-0249-7. [DOI] [PubMed] [Google Scholar]
- 15.Albanese M, Tagawa T, and Hammerschmidt W (2022). Strategies of Epstein-Barr virus to evade innate antiviral immunity of its human host. Front. Microbiol. 13, 955603. 10.3389/fmicb.2022.955603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang MS, and Kieff E (2015). Epstein-Barr virus latent genes. Exp. Mol. Med. 47, e131. 10.1038/emm.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pei Y, Wong JH, and Robertson ES (2020). Herpesvirus epigenetic reprogramming and oncogenesis. Annu. Rev. Virol. 7, 309–331. 10.1146/annurev-virology-020420-014025. [DOI] [PubMed] [Google Scholar]
- 18.Rosemarie Q, and Sugden B (2020). Epstein-Barr virus: how its lytic phase contributes to oncogenesis. Microorganisms 8, 1824. 10.3390/microorganisms8111824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frappier L (2021). Epstein-Barr virus: current questions and challenges. Tumour Virus Res. 12, 200218. 10.1016/j.tvr.2021.200218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma SD, Hegde S, Young KH, Sullivan R, Rajesh D, Zhou Y, Jankowska-Gan E, Burlingham WJ, Sun X, Gulley ML, et al. (2011). A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 85, 165–177. 10.1128/JVI.01512-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bellows DS, Howell M, Pearson C, Hazlewood SA, and Hardwick JM (2002). Epstein-Barr virus BALF1 is a BCL-2-like antagonist of the herpesvirus antiapoptotic BCL-2 proteins. J. Virol. 76, 2469–2479. 10.1128/jvi.76.5.2469-2479.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fitzsimmons L, Cartlidge R, Chang C, Sejic N, Galbraith LCA, Suraweera CD, Croom-Carter D, Dewson G, Tierney RJ, Bell AI, et al. (2020). EBV BCL-2 homologue BHRF1 drives chemoresistance and lymphomagenesis by inhibiting multiple cellular pro-apoptotic proteins. Cell Death Differ. 27, 1554–1568. 10.1038/s41418-019-0435-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang YH, Lee CP, Su MT, Wang JT, Chen JY, Lin SF, Tsai CH, Hsieh MJ, Takada K, and Chen MR (2012). Epstein-Barr virus BGLF4 kinase retards cellular S-phase progression and induces chromosomal abnormality. PLoS One 7, e39217. 10.1371/journal.pone.0039217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu CC, Liu MT, Chang YT, Fang CY, Chou SP, Liao HW, Kuo KL, Hsu SL, Chen YR, Wang PW, et al. (2010). Epstein-Barr virus DNase (BGLF5) induces genomic instability in human epithelial cells. Nucleic Acids Res. 38, 1932–1949. 10.1093/nar/gkp1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiu SH, Wu CC, Fang CY, Yu SL, Hsu HY, Chow YH, and Chen JY (2014). Epstein-Barr virus BALF3 mediates genomic instability and progressive malignancy in nasopharyngeal carcinoma. Oncotarget 5, 8583–8601. 10.18632/oncotarget.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yiu SPT, Guo R, Zerbe C, Weekes MP, and Gewurz BE (2022). Epstein-Barr virus BNRF1 destabilizes SMC5/6 cohesin complexes to evade its restriction of replication compartments. Cell Rep. 38, 110411. 10.1016/j.celrep.2022.110411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shumilov A, Tsai MH, Schlosser YT, Kratz AS, Bernhardt K, Fink S, Mizani T, Lin X, Jauch A, Mautner J, et al. (2017). Epstein-Barr virus particles induce centrosome amplification and chromosomal instability. Nat. Commun. 8, 14257. 10.1038/ncomms14257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nobre LV, Nightingale K, Ravenhill BJ, Antrobus R, Soday L, Nichols J, Davies JA, Seirafian S, Wang EC, Davison AJ, et al. (2019). Human cytomegalovirus interactome analysis identifies degradation hubs, domain associations and viral protein functions. eLife 8, e49894. 10.7554/eLife.49894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davis ZH, Verschueren E, Jang GM, Kleffman K, Johnson JR, Park J, Von Dollen J, Maher MC, Johnson T, Newton W, et al. (2015). Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol. Cell 57, 349–360. 10.1016/j.molcel.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johannsen E, Luftig M, Chase MR, Weicksel S, Cahir-McFarland E, Illanes D, Sarracino D, and Kieff E (2004). Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. USA 101, 16286–16291. 10.1073/pnas.0407320101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ersing I, Nobre L, Wang LW, Soday L, Ma Y, Paulo JA, Narita Y, Ashbaugh CW, Jiang C, Grayson NE, et al. (2017). A temporal proteomic map of Epstein-Barr virus lytic replication in B cells. Cell Rep. 19, 1479–1493. 10.1016/j.celrep.2017.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, et al. (2015). The BioPlex network: A systematic exploration of the human interactome. Cell 162, 425–440. 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sowa ME, Bennett EJ, Gygi SP, and Harper JW (2009). Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403. 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Behrends C, Sowa ME, Gygi SP, and Harper JW (2010). Network organization of the human autophagy system. Nature 466, 68–76. 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aubry V, Mure F, Mariamé B, Deschamps T, Wyrwicz LS, Manet E, and Gruffat H (2014). Epstein-Barr virus late gene transcription depends on the assembly of a virus-specific preinitiation complex. J. Virol. 88, 12825–12838. 10.1128/JVI.02139-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paladino P, Marcon E, Greenblatt J, and Frappier L (2014). Identification of herpesvirus proteins that contribute to G1/S arrest. J. Virol. 88, 4480–4492. 10.1128/JVI.00059-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calderwood MA, Holthaus AM, and Johannsen E (2008). The Epstein-Barr virus LF2 protein inhibits viral replication. J. Virol. 82, 8509–8519. 10.1128/JVI.00315-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Calderwood MA, Venkatesan K, Xing L, Chase MR, Vazquez A, Holthaus AM, Ewence AE, Li N, Hirozane-Kishikawa T, Hill DE, et al. (2007). Epstein-Barr virus and virus human protein interaction maps. Proc. Natl. Acad. Sci. USA 104, 7606–7611. 10.1073/pnas.0702332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heilmann AM, Calderwood MA, and Johannsen E (2010). Epstein-Barr virus LF2 protein regulates viral replication by altering Rta subcellular localization. J. Virol. 84, 9920–9931. 10.1128/JVI.00573-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yokoyama N, Fujii K, Hirata M, Tamai K, Kiyono T, Kuzushima K, Nishiyama Y, Fujita M, and Tsurumi T (1999). Assembly of the Epstein-Barr virus BBLF4, BSLF1 and BBLF2/3 proteins and their interactive properties. J. Gen. Virol. 80, 2879–2887. 10.1099/0022-1317-80-11-2879. [DOI] [PubMed] [Google Scholar]
- 41.Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using David bioinformatics resources. Nat. Protoc. 4, 44–57. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 42.Sherman BT, Hao M, Qiu J, Jiao X, Baseler MW, Lane HC, Imamichi T, and Chang W (2022). David: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221. 10.1093/nar/gkac194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Verma D, and Swaminathan S (2008). Epstein-Barr virus SM protein functions as an alternative splicing factor. J. Virol. 82, 7180–7188. 10.1128/JVI.00344-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verma D, Bais S, Gaillard M, and Swaminathan S (2010). Epstein-Barr virus SM protein utilizes cellular splicing factor SRp20 to mediate alternative splicing. J. Virol. 84, 11781–11789. 10.1128/JVI.01359-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Menzies SA, Volkmar N, van den Boomen DJ, Timms RT, Dickson AS, Nathan JA, and Lehner PJ (2018). The sterol-responsive RNF145 E3 ubiquitin ligase mediates the degradation of HMG-CoA reductase together with gp78 and Hrd1. eLife 7, e40009. 10.7554/eLife.40009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chalabi Hagkarim N, and Grand RJ (2020). The regulatory properties of the Ccr4-not complex. Cells 9, 2379. 10.3390/cells9112379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huttlin EL, Bruckner RJ, Navarrete-Perea J, Cannon JR, Baltier K, Gebreab F, Gygi MP, Thornock A, Zarraga G, Tam S, et al. (2021). Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 184, 3022–3040.e28. 10.1016/j.cell.2021.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giurgiu M, Reinhard J, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, and Ruepp A (2019). Corum: the comprehensive resource of mammalian protein complexes-2019. Nucleic Acids Res. 47, D559–D563. 10.1093/nar/gky973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spiess K, Fares S, Sparre-Ulrich AH, Hilgenberg E, Jarvis MA, Ehlers B, and Rosenkilde MM (2015). Identification and functional comparison of seven-transmembrane G-protein-coupled BILF1 receptors in recently discovered nonhuman primate lymphocryptoviruses. J. Virol. 89, 2253–2267. 10.1128/JVI.02716-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Griffin BD, Gram AM, Mulder A, Van Leeuwen D, Claas FH, Wang F, Ressing ME, and Wiertz E (2013). EBV BILF1 evolved to downregulate cell surface display of a wide range of HLA class I molecules through their cytoplasmic tail. J. Immunol. 190, 1672–1684. 10.4049/jimmunol.1102462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zuo J, Quinn LL, Tamblyn J, Thomas WA, Feederle R, Delecluse HJ, Hislop AD, and Rowe M (2011). The Epstein-Barr virus-encoded BILF1 protein modulates immune recognition of endogenously processed antigen by targeting major histocompatibility complex class I molecules trafficking on both the exocytic and endocytic pathways. J. Virol. 85, 1604–1614. 10.1128/JVI.01608-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Horst D, Burrows SR, Gatherer D, van Wilgenburg B, Bell MJ, Boer IG, Ressing ME, and Wiertz EJ (2012). Epstein-Barr virus isolates retain their capacity to evade T cell immunity through BNLF2a despite extensive sequence variation. J. Virol. 86, 572–577. 10.1128/JVI.05151-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meng Q, Hagemeier SR, Fingeroth JD, Gershburg E, Pagano JS, and Kenney SC (2010). The Epstein-Barr virus (EBV)-encoded protein kinase, EBV-PK, but not the thymidine kinase (EBV-TK), is required for ganciclovir and acyclovir inhibition of lytic viral production. J. Virol. 84, 4534–4542. 10.1128/JVI.02487-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Subramanian N, Natarajan K, Clatworthy MR, Wang Z, and Germain RN (2013). The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153, 348–361. 10.1016/j.cell.2013.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang JT, Doong SL, Teng SC, Lee CP, Tsai CH, and Chen MR (2009). Epstein-Barr virus BGLF4 kinase suppresses the interferon regulatory factor 3 signaling pathway. J. Virol. 83, 1856–1869. 10.1128/JVI.01099-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, et al. (2010). The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402. 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brubaker SW, Bonham KS, Zanoni I, and Kagan JC (2015). Innate immune pattern recognition: a cell biological perspective. Annu. Rev. Immunol. 33, 257–290. 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Broz P, and Dixit VM (2016). Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420. 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 59.Sharma D, and Kanneganti TD (2016). The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J. Cell Biol. 213, 617–629. 10.1083/jcb.201602089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, et al. (2015). A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21, 248–255. 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu D, Chen Y, Sun Y, Gao Q, Li H, Yang Z, Wang Y, Jiang X, and Yu B (2020). Target of MCC950 in inhibition of NLRP3 inflammasome activation: a literature review. Inflammation 43, 17–23. 10.1007/s10753-019-01098-8. [DOI] [PubMed] [Google Scholar]
- 62.Jin Y, Liu Y, Xu L, Xu J, Xiong Y, Peng Y, Ding K, Zheng S, Yang N, Zhang Z, et al. (2022). Novel role for caspase 1 inhibitor VX765 in suppressing NLRP3 inflammasome assembly and atherosclerosis via promoting mitophagy and efferocytosis. Cell Death Dis. 13, 512. 10.1038/s41419-022-04966-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li Y, Niu X, Xu H, Li Q, Meng L, He M, Zhang J, Zhang Z, and Zhang Z (2020). VX-765 attenuates atherosclerosis in ApoE deficient mice by modulating VSMCs pyroptosis. Exp. Cell Res. 389, 111847. 10.1016/j.yexcr.2020.111847. [DOI] [PubMed] [Google Scholar]
- 64.Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes-Alnemri T, Yu JW, and Alnemri ES (2013). The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J. Immunol. 191, 4358–4366. 10.4049/jimmunol.1301170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Franchi L, Eigenbrod T, Muñoz-Planillo R, Ozkurede U, Kim YG, Arindam C, Gale M Jr., Silverman RH, Colonna M, Akira S, et al. (2014). Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J. Immunol. 193, 4214–4222. 10.4049/jimmunol.1400582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, et al. (2010). Peroxisomes are signaling platforms for antiviral innate immunity. Cell 141, 668–681. 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W, Deng Q, Orning P, Alam HB, Maliga Z, et al. (2020). HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science 369. 10.1126/science.aas8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu D, Zhang Z, Jiang X, Du Y, Zhang S, and Yang XD (2022). Inflammasome meets centrosome: understanding the emerging role of centrosome in controlling inflammasome activation. Front. Immunol. 13, 826106. 10.3389/fimmu.2022.826106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vilmen G, Glon D, Siracusano G, Lussignol M, Shao Z, Hernandez E, Perdiz D, Quignon F, Mouna L, Poüs C, et al. (2021). BHRF1, a BCL2 viral homolog, disturbs mitochondrial dynamics and stimulates mitophagy to dampen type I IFN induction. Autophagy 17, 1296–1315. 10.1080/15548627.2020.1758416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tatsumi K, Sou YS, Tada N, Nakamura E, Iemura S, Natsume T, Kang SH, Chung CH, Kasahara M, Kominami E, et al. (2010). A novel type of E3 ligase for the Ufm1 conjugation system. J. Biol. Chem. 285, 5417–5427. 10.1074/jbc.M109.036814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xin L, Qiao R, Chen X, Tran H, Pan S, Rabinoviz S, Bian H, He X, Morse B, Shan B, et al. (2022). A streamlined platform for analyzing tera-scale DDA and DIA mass spectrometry data enables highly sensitive immunopeptidomics. Nat. Commun. 13, 3108. 10.1038/s41467-022-30867-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seth RB, Sun L, Ea CK, and Chen ZJ (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682. 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 73.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, and Steinegger M (2022). ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682. 10.1038/s41592-022-01488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fares S, Spiess K, Olesen ETB, Zuo J, Jackson S, Kledal TN, Wills MR, and Rosenkilde MM (2019). Distinct roles of extracellular domains in the Epstein-Barr virus-encoded BILF1 receptor for signaling and major histocompatibility complex Class I downregulation. mBio 10, e01707–e01718. 10.1128/mBio.01707-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Abu-Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH, Freinkman E, and Sabatini DM (2017). Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813. 10.1126/science.aan6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, and McBride HM (2012). A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 22, 135–141. 10.1016/j.cub.2011.11.057. [DOI] [PubMed] [Google Scholar]
- 78.Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, and McBride HM (2012). Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One 7, e52830. 10.1371/journal.pone.0052830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cadete VJ, Deschênes S, Cuillerier A, Brisebois F, Sugiura A, Vincent A, Turnbull D, Picard M, McBride HM, and Burelle Y (2016). Formation of mitochondrial-derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J. Physiol. 594, 5343–5362. 10.1113/JP272703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Towers CG, Wodetzki DK, Thorburn J, Smith KR, Caino MC, and Thorburn A (2021). Mitochondrial-derived vesicles compensate for loss of LC3-mediated mitophagy. Dev. Cell 56, 2029–2042.e5. 10.1016/j.devcel.2021.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sugiura A, McLelland GL, Fon EA, and McBride HM (2014). A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 33, 2142–2156. 10.15252/embj.201488104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liang JR, Lingeman E, Luong T, Ahmed S, Muhar M, Nguyen T, Olzmann JA, and Corn JE (2020). A genome-wide ER-phagy screen highlights key roles of mitochondrial metabolism and ER-resident UFMylation. Cell 180, 1160–1177.e20. 10.1016/j.cell.2020.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Todkar K, Chikhi L, Desjardins V, El-Mortada F, Pépin G, and Germain M (2021). Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat. Commun. 12, 1971. 10.1038/s41467-021-21984-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Heo JM, Harper NJ, Paulo JA, Li M, Xu Q, Coughlin M, Elledge SJ, and Harper JW (2019). Integrated proteogenetic analysis reveals the landscape of a mitochondrial-autophagosome synapse during PARK2-dependent mitophagy. Sci. Adv. 5, eaay4624. 10.1126/sciadv.aay4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ding X, Jiang X, Tian R, Zhao P, Li L, Wang X, Chen S, Zhu Y,Mei M, Bao S, et al. (2019). RAB2 regulates the formation of autophagosome and autolysosome in mammalian cells. Autophagy 15, 1774–1786. 10.1080/15548627.2019.1596478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cantalupo G, Alifano P, Roberti V, Bruni CB, and Bucci C (2001). Rab-interacting lysosomal protein (RILP): the Rab7 effector required for transport to lysosomes. EMBO J. 20, 683–693. 10.1093/emboj/20.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nijmeijer S, Leurs R, Smit MJ, and Vischer HF (2010). The Epstein-Barr virus-encoded G protein-coupled receptor BILF1 hetero-oligomerizes with human CXCR4, scavenges Galphai proteins, and constitutively impairs CXCR4 functioning. J. Biol. Chem. 285, 29632–29641. 10.1074/jbc.M110.115618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chiang JJ, Sparrer KMJ, van Gent M, Lässig C, Huang T, Osterrieder N, Hopfner KP, and Gack MU (2018). Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat. Immunol. 19, 53–62. 10.1038/s41590-017-0005-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.O’Grady T, Cao S, Strong MJ, Concha M, Wang X, Splinter Bondurant S, Adams M, Baddoo M, Srivastav SK, Lin Z, et al. (2014). Global bidirectional transcription of the Epstein-Barr virus genome during reactivation. J. Virol. 88, 1604–1616. 10.1128/JVI.02989-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ungerleider N, Concha M, Lin Z, Roberts C, Wang X, Cao S, Baddoo M, Moss WN, Yu Y, Seddon M, et al. (2018). The Epstein Barr virus circRNAome. PLoS Pathog. 14, e1007206. 10.1371/journal.ppat.1007206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen YG, Chen R, Ahmad S, Verma R, Kasturi SP, Amaya L, Broughton JP, Kim J, Cadena C, Pulendran B, et al. (2019). N6-methyladenosine modification controls circular RNA immunity. Mol. Cell 76, 96–109.e9. 10.1016/j.molcel.2019.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Swanson KV, Deng M, and Ting JP (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489. 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Burton EM, Goldbach-Mansky R, and Bhaduri-McIntosh S (2020). A promiscuous inflammasome sparks replication of a common tumor virus. Proc. Natl. Acad. Sci. USA 117, 1722–1730. 10.1073/pnas.1919133117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang X, Zhou J, Liu C, Qu Y, Wang W, Xiao MZX, Zhu F, Liu Z, and Liang Q (2022). KSHV-encoded ORF45 activates human NLRP1 inflammasome. Nat. Immunol. 23, 916–926. 10.1038/s41590-022-01199-x. [DOI] [PubMed] [Google Scholar]
- 95.Gregory SM, Davis BK, West JA, Taxman DJ, Matsuzawa S, Reed JC, Ting JP, and Damania B (2011). Discovery of a viral NLR homolog that inhibits the inflammasome. Science 331, 330–334. 10.1126/science.1199478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wyant GA, Yu W, Doulamis IP, Nomoto RS, Saeed MY, Duignan T, McCully JD, and Kaelin WG Jr. (2022). Mitochondrial remodeling and ischemic protection by G protein-coupled receptor 35 agonists. Science 377, 621–629. 10.1126/science.abm1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hsu F, Spannl S, Ferguson C, Hyman AA, Parton RG, and Zerial M (2018). Rab5 and Alsin regulate stress-activated cytoprotective signaling on mitochondria. eLife 7, e32282. 10.7554/eLife.32282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Snider DL, Park M, Murphy KA, Beachboard DC, and Horner SM (2022). Signaling from the RNA sensor RIG-I is regulated by ufmylation. Proc. Natl. Acad. Sci. USA 119, e2119531119. 10.1073/pnas.2119531119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Horner SM, Wilkins C, Badil S, Iskarpatyoti J, and Gale M Jr. (2015). Proteomic analysis of mitochondrial-associated ER membranes (MAM) during RNA virus infection reveals dynamic changes in protein and organelle trafficking. PLOS One 10, e0117963. 10.1371/journal.pone.0117963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.McLelland GL, Soubannier V, Chen CX, McBride HM, and Fon EA (2014). Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 33, 282–295. 10.1002/embj.201385902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang L, Xu Y, Rogers H, Saidi L, Noguchi CT, Li H, Yewdell JW, Guydosh NR, and Ye Y (2020). UFMylation of RPL26 links translocation-associated quality control to endoplasmic reticulum protein homeostasis. Cell Res. 30, 5–20. 10.1038/s41422-019-0236-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schuren ABC, Boer IGJ, Bouma EM, Van de Weijer ML, Costa AI, Hubel P, Pichlmair A, Lebbink RJ, and Wiertz EJHJ (2021). The UFM1 pathway impacts HCMV US2-mediated degradation of HLA Class I. Molecules 26, 287. 10.3390/molecules26020287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, and Gloriam DE (2017). Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842. 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Perez-Riverol Y, Bai J, Bandla C, García-Seisdedos D, Hewapathirana S, Kamatchinathan S, Kundu DJ, Prakash A, Frericks-Zipper A, Eisenacher M, et al. (2022). The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552. 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. 10.1038/nature22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Guo R, Jiang C, Zhang Y, Govande A, Trudeau SJ, Chen F, Fry CJ, Puri R, Wolinsky E, Schineller M, et al. (2020). MYC controls the Epstein-Barr virus lytic switch. Mol. Cell 78, 653–669.e8. 10.1016/j.molcel.2020.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pham H, Kearns NA, and Maehr R (2016). Transcriptional regulation with CRISPR/Cas9 effectors in mammalian cells. Methods Mol. Biol. 1358, 43–57. 10.1007/978-1-4939-3067-8_3. [DOI] [PubMed] [Google Scholar]
- 109.Gstalder C, Liu D, Miao D, Lutterbach B, DeVine AL, Lin C, Shettigar M, Pancholi P, Buchbinder EI, Carter SL, et al. (2020). Inactivation of Fbxw7 impairs dsRNA sensing and confers resistance to PD-1 blockade. Cancer Discov. 10, 1296–1311. 10.1158/2159-8290.CD-19-1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Agrotis A, Pengo N, Burden JJ, and Ketteler R (2019). Redundancy of human ATG4 protease isoforms in autophagy and LC3/GABARAP processing revealed in cells. Autophagy 15, 976–997. 10.1080/15548627.2019.1569925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, and Voeltz GK (2011). ER tubules mark sites of mitochondrial division. Science 334, 358–362. 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wyant GA, Abu-Remaileh M, Wolfson RL, Chen WW, Freinkman E, Danai LV, Vander Heiden MG, and Sabatini DM (2017). mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell 171, 642–654.e12. 10.1016/j.cell.2017.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen WW, Freinkman E, Wang T, Birsoy K, and Sabatini DM (2016). Absolute quantification of matrix metabolites reveals the dynamics of mitochondrial metabolism. Cell 166, 1324–1337.e11. 10.1016/j.cell.2016.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kim DE, Chivian D, and Baker D (2004). Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32, W526–W531. 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cowen L, Ideker T, Raphael BJ, and Sharan R (2017). Network propagation: a universal amplifier of genetic associations. Nat. Rev. Genet. 18, 551–562. 10.1038/nrg.2017.38. [DOI] [PubMed] [Google Scholar]
- 116.R Core Team (2021). R: a Language and Environment for Statistical Computing (R Foundation for Statistical Computing).
- 117.Winston Chang JC, Allaire JJ, Sievert C, Schloerke B, Xie Y, Allen J, McPherson J, Dipert A, and Borges B (2022). Shiny: Web Application Framework for R. https://shiny.posit.co/r/reference/shiny/latest/shiny-package.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE104 partner repository (PRIDE: PXD041336) and made publicly available upon publication. All other raw data have been deposited at Mendeley and are publicly available as of the date of publication (Mendeley Data: https://doi.org/10.17632/zk884935vw.1). Microcopy data reported in this paper will be shared by the lead contact upon request. Figures were drawn with commercially available GraphPad, Biorender and Microsoft Powerpoint.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.