

Key Points
Question
How prevalent is the NCSTN:c.671_682del variant in the ethnically Maltese patients with hidradenitis suppurativa (HS), and is it associated with a specific HS phenotype?
Findings
In this cross-sectional study of 113 patients with HS, 14 (12.39%) were found to be heterozygous for the NCSTN:c.671_682del variant. Patients with the variant were more likely to manifest HS earlier in atypical skin sites, such as the scalp, neck, torso, and antecubital fossae, and despite manifesting similar disease severity, patients with the variant were more likely to require treatment with biologic therapy.
Meaning
The results of this cross-sectional study suggest that monogenic variation in NCSTN may be associated with disease in a minority of patients with HS who manifest distinct phenotypic characteristics.
Abstract
Importance
Hidradenitis suppurativa (HS) is a complex trait that has a monogenic etiology in a subset of patients. Variation in genes that encode proteins of the γ secretase complex, particularly NCSTN, account for few patients who exhibit familial forms of HS. Thus far, extensive genotype-phenotype correlations have been lacking.
Objective
To establish the prevalence of the NCSTN:c.671_682del variant and explore potential genotype-phenotype associations in an ethnically Maltese HS cohort.
Design, Setting, and Participants
This cross-sectional study conducted from December 2021 to September 2022 included patients 18 years or older with a diagnosis of HS as defined by recurrent nodules, abscesses, and/or draining tunnels in typical (axilla, breast, groin, buttock, thighs, and inframammary folds) and less typical (scalp, ear pinnae, neck, arms, antecubital fossae) sites who were recruited from the sole national dermatology reference center servicing the Maltese archipelago. Clinical examination and targeted genetic analysis for an NCSTN deletion that was originally identified through whole-exome sequencing in a family with multigenerational disease were performed.
Exposure
Recruited patients were phenotyped and genotyped for the NCSTN:c.671_682del variant.
Main Outcome and Measures
To determine the prevalence of the NCSTN:c.671_682del variant and establish possible genotype-phenotype associations in the ethnically Maltese HS cohort.
Results
A total of 113 patients with HS (56 women [49.6%]) met the inclusion criteria and were enrolled in this study. The median age of disease onset was 18 years (range, 7-62 years), and the median International Hidradenitis Suppurativa Severity Score System score was 4.39 (range, 1.0-64.0). The NCSTN variant was identified in the heterozygous state in 14 patients (12.4%) from 5 unrelated, nonconsanguineous families of Maltese ethnicity. The variant was not identified in an ethnically matched reference genomic data set of disease-free individuals. Variant carriers manifested HS symptoms earlier and were more likely to exhibit a distinctive HS phenotype, which was characterized by involvement of the scalp, neck, torso, and antecubital fossae. Despite manifesting similar clinical disease severity, variant carriers were more likely to require treatment with adalimumab.
Conclusions and Relevance
The results of this cross-sectional study suggest that monogenic variation in NCSTN is associated with HS in a subset of patients who have a distinct, atypical phenotype.
This cross-sectional study examines the prevalence of the NCSTN:c.671_682del variant and potential genotype-phenotype associations in ethnically Maltese individuals with hidradenitis suppurativa.
Introduction
Hidradenitis suppurativa (HS) is a chronic, inflammatory condition of the pilosebaceous unit that is associated with genetic, lifestyle, and environmental factors. Hidradenitis suppurativa is a heritable trait,1,2 with up to 35% of patients having a family history.3 Variants in genes encoding the γ secretase complex (GSC) proteins (primarily NCSTN4) are implicated in most familial HS cases that have an underlying monogenic etiology. The GSC functions in Notch regulation, which plays a pivotal role in follicular differentiation and epidermal homeostasis.
Malta is a Mediterranean island whose population has a high prevalence of HS and its associated risk factors, particularly obesity.5 We previously described the NCSTN(NM_015331.3):c.671_682del (p.Val224_Thr227del) in-frame deletion, which was classified as a variant of uncertain significance (VUS) in a family of Maltese ethnicity with multigenerational HS.6 In this study, we aimed to establish its prevalence in the Maltese HS cohort and evaluate potential genotype-phenotype associations.
Methods
Study Population
We conducted a single-center, cross-sectional study from December 2021 to September 2022 at the sole national reference dermatology center in Malta (population: 550 000 individuals). Ethical approval was granted by the University of Malta. Patients provided written consent for participation in this study and genetic analysis.
Enrollment was restricted to adult patients (age ≥18 years) of Maltese ethnicity. Consanguinity was excluded by clinical questionnaire. A diagnosis of HS was made based on the Dessau definition: “recurrent, painful or purulent, deep-seated lesions occurring more than twice every six months, located in the groin, axilla, perineum, buttocks or sub mammary folds.”7
Severity of HS was scored according to the Hurley Severity Scoring System8 and the International Hidradenitis Suppurativa Severity Scoring System.9 Baseline demographic characteristics, clinical parameters, and anthropometric measurements were captured at enrollment. Blood samples were taken after a 12-hour fast.
Targeted Genetic Analysis
We screened the HS cohort for the NCSTN(NM_015331.3):c.671_682delTCATCAGCACTG variant by polymerase chain reaction amplification-restriction fragment length polymorphism and subsequently confirmed genotypes by Sanger sequencing, as described in the eMethods in Supplement 1. Segregation studies were conducted.
Controls
An anonymized in-house whole-exome sequencing data set was used to screen for variants of potential clinical significance within NCSTN in an ethnically matched reference population unselected for HS. The data set comprised 320 clinically characterized, unrelated individuals of third-generation Maltese ethnicity (age, 17-42 years; 154 women [48%]) who were recruited through convenience and random sampling from the general population. No participants with known HS at recruitment were present in this cohort.
Bioinformatics
Image analyses were performed with the default parameters of the Illumina RTA pipeline. The raw FASTQ files were mapped and aligned to the human reference genome (University of California, Santa Cruz hg19; National Center for Biotechnology Information build 37) using the Burrows-Wheeler transformation algorithm, and duplicated reads were removed using Picard, version 3.0.0. The quality of sequencing data was checked using FastQC (Babraham Bioinformatics), and Cutadapt, version 4.4, was used to remove adaptor sequences. The Genome Analysis Toolkit Unified Genotyper was used to call single-nucleotide variations and indels in variant call files. The annotation of identified genetic variants and a prediction of their effects was done through SnpEff, version 5.1. VarAFT, version 2.1.5, was used to compute coverage statistics, and genetic variants with a read depth of less than 20 times and a quality mapping score of less than 30 were filtered out. On-target NCSTN sequencing depth and NCSTN exon 6 depth in the control reference population were assessed; summary sequencing metrics are tabulated in eTable 1 in Supplement 1. The aggregate whole-exome sequencing data set from the control reference population was interrogated for the presence of protein-altering, nonsynonymous variants in NCSTN.
Variant pathogenicity was classified based on a combination of computational, population frequency and functional data according to guidelines from the American College of Medical Genetics and Genomics and Association for Molecular Pathology criteria.10 Variant interpretation was conducted using VarSome. A principal components analysis of the sequencing data was performed using Locating Ancestry from Sequence Reads11 to estimate the genetic ancestry of the control reference cohort in association with the human genetic diversity panel.
Statistical Analysis
The characteristics of the study cohort were summarized using descriptive statistics. The normality of continuous variables was assessed by the Shapiro-Wilk and Kolmogorov-Smirnoff tests. All parameters exhibited a skewed non-normal distribution, and nonparametric statistics with medians and interquartile ranges are presented. To compare differences in quantitative variables between 2 categories, the independent samples Mann-Whitney U test was applied. The χ2 test was used to compare categorical variables. All analyses were performed using IBM SPSS, version 26. P < .05 was considered statistically significant.
Results
Study Population
A total of 113 patients with HS (80.5% unrelated) from 98 unrelated families of Maltese ethnicity participated in this study. The salient clinical and biochemical characteristics of the study cohort are presented in the Table. Most patients had obesity (median body mass index [calculated as weight in kilograms divided by height in meters squared] of 31) and were active or ex-smokers (72 [64.0%]). The median age of disease onset was 18 years (range, 7-62 years), with a diagnostic delay of 7 years (range, 0-59 years). Forty-five patients (39.8%) reported a family history, and 52 (46.4%) had Hurley I disease.
Table. Salient Clinical and Biochemical Characteristics of the Study Cohort as Stratified by the NCSTN p.Val224_Thr227del Genotypea.
| Characteristic | Overall (n = 113) | NCSTN deletion absent (n = 99) | NCSTN p.Val224_Thr227del (n = 14) | P value |
|---|---|---|---|---|
| Sex, No. (%) | ||||
| Female | 56 (49.6) | 47 (48.0) | 8 (57.1) | .58 |
| Male | 57 (50.4) | 52 (52.0) | 6 (42.9) | |
| Age of disease onset, y | 18 (9) | 18 (10) | 12 (7) | .001 |
| Diagnostic delay, y | 7 (15) | 7 (12) | 21 (34) | .001 |
| Current/previous smoker, No. (%) | 72 (64.0) | 64 (65.0) | 8 (57.1) | .52 |
| Prescribed biologic therapy, No. (%) | 17 (15.0) | 12 (12.1) | 5 (35.7) | .04 |
| History of HS surgery, No. (%) | 57 (50.4) | 48 (48.5) | 9 (64.3) | .21 |
| Pilonidal sinus disease, No. (%) | 45 (39.8) | 38 (38.4) | 7 (50.0) | .56 |
| Acne keloidalis nuchae, No. (%) | 7 (6.2) | 5 (5.1) | 2 (14.3) | .21 |
| Hurley severity (mild/moderate/severe), No. (%) | 52 (46.4)/44 (38.4)/17 (15.2) | 45 (45.0)/37 (37.4)/17 (17.2) | 5 (35.7)/6 (42.9)/3 (21.4) | .32 |
| IHS4 score | 4 (6) | 4 (5) | 6 (9) | .52 |
| Systolic BP, mm Hg | 132 (22) | 133 (21) | 127 (38) | .40 |
| Diastolic BP, mm Hg | 79 (15) | 80 (16) | 76 (17) | .22 |
| BMI | 31 (10.8) | 32 (11.5) | 25.5 (8) | .01 |
| Fasting plasma glucose level, mg/dL | 91.17 (15.67) | 90.90 (15.31) | 95.14 (16.75) | .58 |
| HbA1c, % of total blood | 5.6 (0.6) | 5.3 (0.7) | 5.4 (0.3) | .67 |
| C-reactive protein, mg/L | 4 (7.6) | 3.9 (7.2) | 4.7 (8.6) | .98 |
| Erythrocyte sedimentation rate, mm/h | 14.5 (23) | 13 (22) | 29 (17) | .01 |
| Neutrophil:lymphocyte | 2.33 (0.99) | 2.32 (0.98) | 2.36 (0.78) | .60 |
| Platelet:lymphocyte | 135.29 (73.5) | 133.32 (61.74) | 181.29 (99.99) | .03 |
Abbreviations: BMI, body mass index (calculated as weight in kilograms divided by height in meters squared); HbA1c, hemoglobin A1c; HS, hidradenitis suppurativa; IHS4, International Hidradenitis Suppurativa Severity Score System.
SI conversion factors: To convert HbA1c to the proportion of total hemoglobin, multiply by 0.01; for glucose to mmol/L, multiply by 0.0555.
Quantitative variables are presented as medians (IQR), and categorical variables as percentages. At baseline, variant carriers were leaner, demonstrated a lower age of disease onset, greater diagnostic delay, and a higher likelihood of biological therapy. Deletion carriers exhibited a significantly higher platelet:lymphocyte ratio and erythrocyte sedimentation rate values. Quantitative variables are compared using the Mann-Whitney U test and categorical variables using the χ2 test.
Genetic Analysis and Genotype-Phenotype Associations
The NCSTN:c.671_682del in-frame deletion was identified in the heterozygous state in 14 patients (12.4%), representing 5 nonconsanguineous families (Figure 1). No genealogical link between kindreds could be ascertained. The variant segregates with HS in an autosomal dominant pattern and was absent in the ethnically matched control group. No NCSTN variant of likely clinical significance was identified in the reference cohort that was unselected for HS. The 14 patients carrying the NCSTN deletion comprised 31.1% of patients with a family history of HS in a first-degree relative.
Figure 1. Pedigrees and Controls.
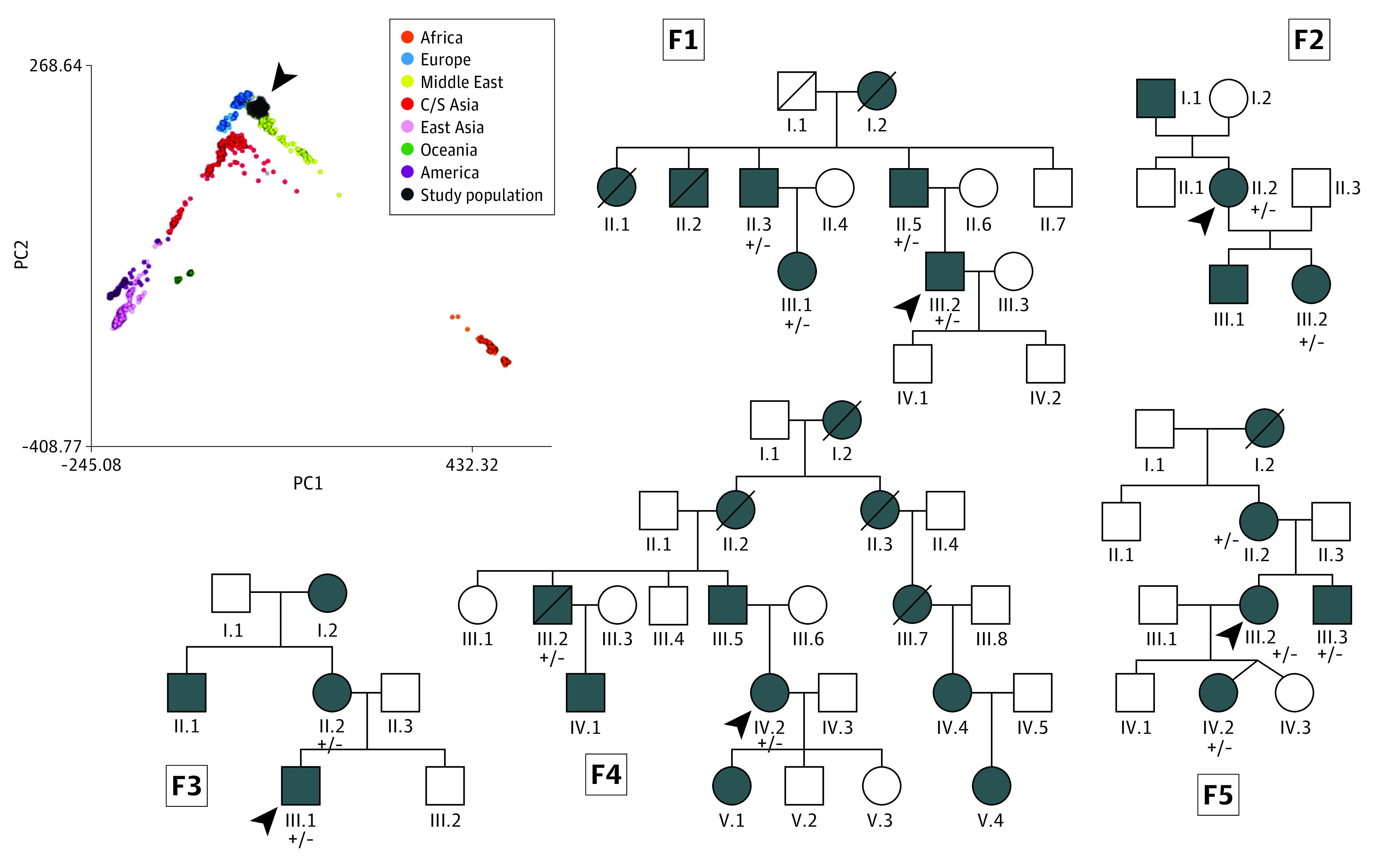
The pedigrees of the 5 unrelated, nonconsanguineous families in whom the variant was identified (F1-F5). The probands are indicated by the arrowheads. Patients are represented by shaded symbols. Heterozygous alleles in the genotyped patients are labeled +/−. The inset panel represents the reference principal component (PC) analysis coordinates for samples from the Human Genome Diversity Project reference panel. The Maltese genomic data set is shown superimposed in black and indicated by the arrowhead, mapping close to the European and Middle Eastern clusters and suggesting a shared genetic ancestry between the 2 populations. C/S indicates central/south.
The Table presents the clinical data of the study population as stratified by genotype. Carriers of the NCSTN variant were more likely to experience HS lesions that involved the scalp, neck, torso, breasts, infra-abdominal fold, and antecubital fossa (Figure 2). Patients with the deletion were leaner (median body mass index, 25.5 vs 32.0; P = .01) and experienced an earlier disease onset (median, 12 years vs 18 years; P = .001) but a longer diagnostic delay (median, 21 years vs 7 years; P = .001). Despite no differences in disease severity, deletion carriers exhibited a higher baseline inflammatory burden and were more likely to have been prescribed biologic therapy with adalimumab (35.7% vs 12.1%; P = .04).
Figure 2. Characteristic Clinical Phenotype of Patients Who Were Heterozygous for the NCSTN Deletion Variant.
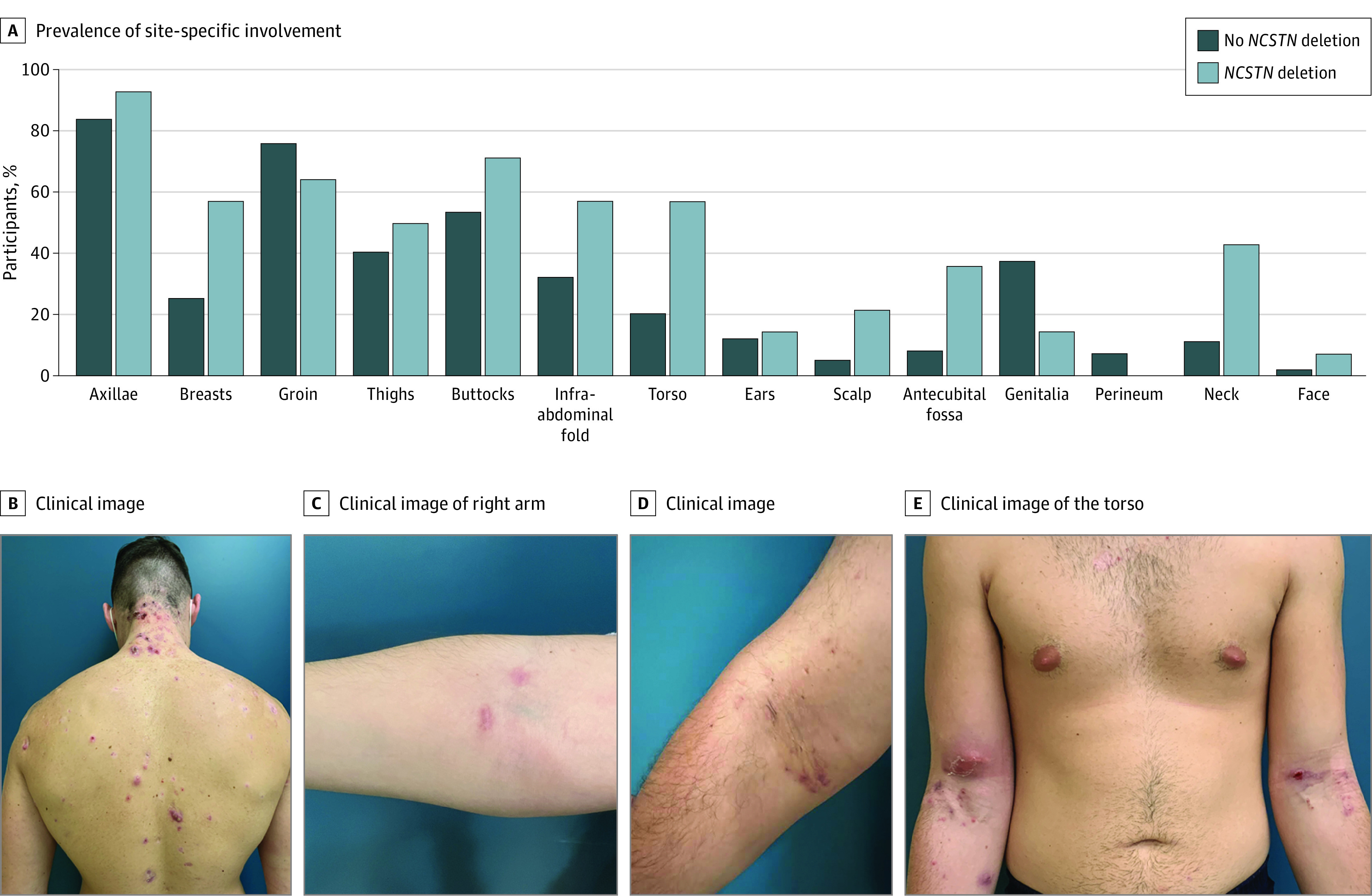
A, Prevalence of various sites of cutaneous involvement according to genotype. Carriers of the deletion were more likely to have disease that affected the scalp, neck, breasts, and antecubital fossa. B, A patient with hidradenitis suppurativa who was heterozygous for the NCSTN deletion variant demonstrated inflamed nodules and abscess in the nuchal area and torso. C-E, Antecubital involvement in increasing degrees of severity.
Variant Reclassification and Genetic Ancestry
The NCSTN:c.671_682del variant describes a 12–base pair in-frame deletion in exon 6, translating into loss of valine 224, isoleucine 225, serine 226, and threonine 227 residues (eFigure 3 in Supplement 1). The deletion is absent in gnomAd, TOPMed, GenomeAsia, GME Variome, and Iranome and does not overlap with structural variation from the gnomAD and Database of Genomic Variants data sets. The deleted residues are in a conserved region that is highly intolerant for substitutions (eFigure 3 in Supplement 1). The analysis results suggest variant segregation in multiple affected family members, lending support for reclassification from VUS to likely pathogenic according to the American College of Medical Genetics and Genomics and Association for Molecular Pathology criteria. A principal components analysis of the in-house ethnically matched reference genomic data set mapped the Maltese cohort as close to the European and Middle Eastern data points, suggesting a shared genetic ancestry between the 2 populations (Figure 1) and reinforcing the uniqueness of the Maltese population.12
Discussion
This cross-sectional study identified an NCSTN in-frame deletion in multiple individuals from 5 unrelated families; thus, it potentially presents a unique opportunity for describing genotype-phenotype associations. To our knowledge, this report represents the first description of a unique variant segregating in more than 2 families that exhibit familial HS.
The distinct HS phenotype reported, together with the phenomenon of genetic determinism, could partially account for the longer diagnostic delay, despite earlier disease onset.13 Consistent with a recent review,14 deletion carriers were more likely to require treatment with biologic therapy, notwithstanding having similar disease severity as that of patients without the variant.
This study did not identify any homozygotes for the NCSTN variant. Based on Hardy-Weinberg equilibrium analysis, 1 homozygote for the deletion was expected. When the gnomAD reference data set was filtered for either putative loss-of-function NCSTN variants or in-frame insertions/deletions, none were biallelic. When missense NCSTN variants in gnomAD were selected (n = 358) only 5 (1.3%) were biallelic, and none involved residues implicated in the deletion. Relevantly, the NCSTN locus is loss of function–intolerant (pLI) in gnomAD (pLI >0.9; observed/expected ratio, 0.1; 95% CI, 0.05-0.23). Such observations may suggest that the lower-than-expected frequency of homozygous individuals reflects lower fitness in homozygotes. This is supported by data from the Mouse Genome Database, which shows that NCSTN homozygous variant embryos exhibit morphological defects of the somites, yolk sac vasculature, neural tube, and pericardial sacs and fail to thrive.15
Direct comparison with other HS genetic studies was limited by differences in population ethnicity, study methods, and the HS diagnostic criteria used. To date, 8 studies have screened HS cohorts for GSC variation, with extensive differences in the reported prevalence of NCSTN variants.16,17 The findings of these studies (eTable 1 in Supplement 1) highlight research gaps, including the exclusion of racial and ethnic minority groups and unstandardized methods of patient prioritization. While phenotype-driven approaches are key to identifying causal variation, prioritizing patients with multigenerational or early-onset disease skews toward the detection of high-penetrance variants that represent the low-hanging fruit of genomic discovery. Thus, lower penetrance variants, which are not associated with familial segregation or predispose to later onset or milder disease, may be missed. In contrast to other complex traits, the contribution of noncoding or regulatory genomic variants and polygenic risk scores to HS is unknown. The GSC bears a high burden of rare deleterious variants in the gnomAD reference data set4 that can be associated with late-onset disease and confound variant classification.18 Thus, rare or novel nonsynonymous variants are often classified as VUS, limiting their interpretation and clinical actionability.
This study’s findings should be interpreted in the context of population genetics. Contemporary Maltese individuals have inherited a genetic legacy derived from the various peoples that settled, conquered, or conducted trade during the course of the archipelago’s history. The collective genetic profile reported in this article suggests evidence of an island genetic isolate that is distinct from the mainland European average.19 Notwithstanding the phenotypic overlap in HS clinical characteristics with other populations, this study provides preliminary evidence for rare community-specific disease variation in HS. Because HS is late onset and is not adversely associated with reproductive fitness, a founder effect in a dominant form is biologically plausible, especially in an isolated island population.20 Alternatively, the described variant may have arisen independently.
Limitations
This study does not provide a comprehensive assessment of deleterious variant burden in known or putative HS loci. Furthermore, the HS diagnostic criteria that were used may exclude patients who have rare phenotypes, such as exclusively facial HS. This study lacks functional evidence linking the deletion to disease pathophysiology and is limited by the absence of sequencing data that enable assessment of shared ancestral haplotypes among variant carriers.
Conclusions
The results of this cross-sectional study suggest that monogenic variation in NCSTN is associated with HS in a subset of patients who have a distinct, atypical phenotype. Patients harboring the NCSTN:c.671_682del variant experience early-onset HS, which is more likely to require treatment with biological therapy. Standardizing pathways for deep phenotyping is likely to narrow the diagnostic gap and enhance our understanding of HS.
eMethods.
eTable 1. Summary sequencing metrics for NCSTN NM_015331 and NCSTN exon 6 in the control population
eFigure 1. Representative RFLP results
eFigure 2. Sanger traces
eFigure 3. The NCSTN (NM_015331.3):c.671_682delTCATCAGCACTG variant
eTable 2. A summary of studies investigating the prevalence of GSC variation in cohorts
eReferences.
Data sharing statement
References
- 1.Kjaersgaard Andersen R, Clemmensen SB, Larsen LA, et al. Evidence of gene-gene interaction in hidradenitis suppurativa: a nationwide registry study of Danish twins. Br J Dermatol. 2022;186(1):78-85. doi: 10.1111/bjd.20654 [DOI] [PubMed] [Google Scholar]
- 2.van Straalen KR, Prens EP, Willemsen G, Boomsma DI, van der Zee HH. Contribution of genetics to the susceptibility to hidradenitis suppurativa in a large, cross-sectional Dutch twin cohort. JAMA Dermatol. 2020;156(12):1359-1362. doi: 10.1001/jamadermatol.2020.3630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schrader AMR, Deckers IE, van der Zee HH, Boer J, Prens EP. Hidradenitis suppurativa: a retrospective study of 846 Dutch patients to identify factors associated with disease severity. J Am Acad Dermatol. 2014;71(3):460-467. doi: 10.1016/j.jaad.2014.04.001 [DOI] [PubMed] [Google Scholar]
- 4.Mintoff D, Pace NP, Borg I. Interpreting the spectrum of gamma-secretase complex missense variation in the context of hidradenitis suppurativa—an in-silico study. Front Genet. 2022;13:962449. doi: 10.3389/fgene.2022.962449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mintoff D, Camilleri L, Aquilina S, Boffa MJ, Clark E, Scerri L. Prevalence of hidradenitis suppurativa in Malta: comparison with established epidemiological data. Clin Exp Dermatol. 2020;45(6):758-759. doi: 10.1111/ced.14271 [DOI] [PubMed] [Google Scholar]
- 6.Mintoff D, Pace NP, Bauer P, Borg I. A novel c.671_682del NCSTN variant in a family with hidradenitis suppurativa: a pilot study. Clin Exp Dermatol. 2021;46(7):1306-1308. doi: 10.1111/ced.14677 [DOI] [PubMed] [Google Scholar]
- 7.Zouboulis CC, Del Marmol V, Mrowietz U, Prens EP, Tzellos T, Jemec GBE. Hidradenitis suppurativa/acne inversa: criteria for diagnosis, severity assessment, classification and disease evaluation. Dermatology. 2015;231(2):184-190. doi: 10.1159/000431175 [DOI] [PubMed] [Google Scholar]
- 8.Hurley H. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa, and familial benign pemphigus: surgical approach. in Roenigk RK, Roenigk HH, eds. Dermatologic Surgery: Principles and Practice. Marcel Dekker; 1989:729-739. [Google Scholar]
- 9.Zouboulis CC, Tzellos T, Kyrgidis A, et al. ; European Hidradenitis Suppurativa Foundation Investigator Group . Development and validation of the International Hidradenitis Suppurativa Severity Score System (IHS4), a novel dynamic scoring system to assess HS severity. Br J Dermatol. 2017;177(5):1401-1409. doi: 10.1111/bjd.15748 [DOI] [PubMed] [Google Scholar]
- 10.Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taliun D, Chothani SP, Schönherr S, et al. LASER server: ancestry tracing with genotypes or sequence reads. Bioinformatics. 2017;33(13):2056-2058. doi: 10.1093/bioinformatics/btx075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarno S, Boattini A, Pagani L, et al. Ancient and recent admixture layers in Sicily and Southern Italy trace multiple migration routes along the Mediterranean. Sci Rep. 2017;7(1):1984. doi: 10.1038/s41598-017-01802-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harden KP. Genetic determinism, essentialism and reductionism: semantic clarity for contested science. Nat Rev Genet. 2023;24(3):197-204. doi: 10.1038/s41576-022-00537-x [DOI] [PubMed] [Google Scholar]
- 14.Mintoff D, Pace NP, Borg I. Management of patients with hidradenitis suppurativa having underlying genetic variation: a systematic review and a call for precision medicine. Clin Exp Dermatol. 2023;48(2):67-72. doi: 10.1093/ced/llac045 [DOI] [PubMed] [Google Scholar]
- 15.Blake JA, Richardson JE, Bult CJ, Kadin JA, Eppig JT, Group TMGD; Mouse Genome Database Group . MGD: the Mouse Genome Database. Nucleic Acids Res. 2003;31(1):193-195. doi: 10.1093/nar/gkg047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marzano AV, Genovese G, Moltrasio C, et al. Whole-exome sequencing in 10 unrelated patients with syndromic hidradenitis suppurativa: a preliminary step for a genotype-phenotype correlation. Dermatology. 2022;238(5):860-869. doi: 10.1159/000521263 [DOI] [PubMed] [Google Scholar]
- 17.Theut Riis P, Loft IC, Yazdanyar S, et al. Full exome sequencing of 11 families with Hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2021;35(5):1203-1211. doi: 10.1111/jdv.17095 [DOI] [PubMed] [Google Scholar]
- 18.Federici G, Soddu S. Variants of uncertain significance in the era of high-throughput genome sequencing: a lesson from breast and ovary cancers. J Exp Clin Cancer Res. 2020;39(1):46. doi: 10.1186/s13046-020-01554-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilbert E, Shanmugam A, Cavalleri GL. Revealing the recent demographic history of Europe via haplotype sharing in the UK Biobank. Proc Natl Acad Sci U S A. 2022;119(25):e2119281119. doi: 10.1073/pnas.2119281119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Charoute H, Bakhchane A, Benrahma H, et al. Mediterranean Founder Mutation Database (MFMD): taking advantage from founder mutations in genetics diagnosis, genetic diversity and migration history of the Mediterranean population. Hum Mutat. 2015;36(11):E2441-E2453. doi: 10.1002/humu.22835 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods.
eTable 1. Summary sequencing metrics for NCSTN NM_015331 and NCSTN exon 6 in the control population
eFigure 1. Representative RFLP results
eFigure 2. Sanger traces
eFigure 3. The NCSTN (NM_015331.3):c.671_682delTCATCAGCACTG variant
eTable 2. A summary of studies investigating the prevalence of GSC variation in cohorts
eReferences.
Data sharing statement