

Abstract
Trimethylation of lysine 36 on histone H3 (H3K36me3), an epigenetic mark associated with actively transcribed genes, plays an important role in multiple cellular processes, including transcription elongation, DNA methylation, DNA repair, etc. Aberrant expression and mutations of the main methyltransferase for H3K36me3, i.e., SET domain-containing 2 (SETD2), were shown to be associated with various cancers. Here, we performed targeted profiling of 154 epitranscriptomic reader, writer, and eraser (RWE) proteins using a scheduled liquid chromatography-parallel-reaction monitoring (LC-PRM) method coupled with the use of stable isotope-labeled (SIL) peptides as internal standards to investigate how H3K36me3 modulates the chromatin occupancies of epitranscriptomic RWE proteins. Our results showed consistent changes in chromatin occupancies of RWE proteins upon losses of H3K36me3 and H4K16ac and a role of H3K36me3 in recruiting METTL3 to chromatin following induction of DNA double-strand breaks. In addition, protein–protein interaction network and Kaplan–Meier survival analyses revealed the importance of METTL14 and TRMT11 in kidney cancer. Taken together, our work unveiled cross-talks between histone epigenetic marks (i.e., H3K36me3 and H4K16ac) and epitranscriptomic RWE proteins and uncovered the potential roles of these RWE proteins in H3K36me3-mediated biological processes.
Graphical Abstract
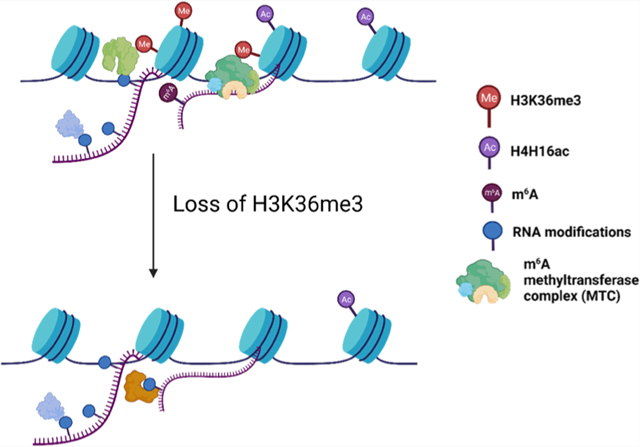
INTRODUCTION
Trimethylation of lysine 36 on histone H3 (H3K36me3) is an important epigenetic modification involved in multiple cellular processes.1–5 The presence of H3K36me3 on gene body is commonly associated with active transcription. In addition, H3K36me3 modulates a number of critical pathways. For instance, H3K36me3 can be recognized by the PWWP domain of DNMT3B,6 which recruits DNMT3B to spurious transcription start sites to ensure the fidelity of intragenic transcription initiation.7,8 H3K36me3 is also important for efficient alternative splicing in mammalian cells via chromatin-associated proteins, including MORF-related gene 15 (MRG15),9 lens epithelium-derived growth factor (LEDGF),10 and zinc finger MYND-type containing 11 (ZMYND11).11 Moreover, H3K36me3 constitutes an important signaling event in DNA repair, where it promotes homologous recombination (HR),12 nonhomologous end-joining (NHEJ),13 and mismatch repair (MMR).14
SET domain-containing 2 (SETD2) is the major methyltransferase catalyzing the formation of H3K36me3 from H3K36me2.5,15,16 Mutations and the ensuing loss of functions of SETD2 occur in a wide range of human cancers, suggesting the potential roles of H3K36me3 in preventing carcinogenesis and tumorigenesis.17,18 Diminished H3K36me3 also disrupts the patterns of other histone post-translational modifications (PTMs), where cross-talks between H3K36me3 and H3K79me2,19 H3K27me3,20–22 along with H4K16ac,16 have been identified.
Recent developments of the epitranscriptomics field led to the discovery of a number of epitranscriptomic RNA modifications, which modulate gene expression by altering the stabilities and translation efficiencies of mRNAs.23–26 Several recent studies documented intricate cross-talks between epitranscriptomic RNA modifications and epigenetic modifications. For instance, loss of N6-methyladenosine (m6A) on chromatin-associated regulatory RNAs (carRNAs) led to augmented stabilities of carRNAs, diminished H3K9me3, elevated chromatin accessibility, and active transcription in mouse embryonic stem cells.27 In addition, H3K36me3 was shown to guide the deposition of m6A in the transcriptome through recruiting methyltransferase-like 14 (METTL14) protein, a subunit of the major m6A methyltransferase complex (MTC).28 Apart from m6A, N1-methyladenosine (m1A), N7-methylguanosine (m7G), and pseudouridine (ψ) are abundant in mRNAs and assume important functions in multiple cellular pathways.26,29–31 The reader, writer, and eraser (RWE) proteins of these modified nucleosides are also actively involved in gene expression regulation.32,33 Owing to its association with active transcription, we reason that H3K36me3 may modulate the functions of other epitranscriptomic RWE proteins by regulating their chromatin occupancies. Hence, we set out to perform a comprehensive and quantitative assessment of the overall expression levels and chromatin occupancies of epitranscriptomic RWE proteins in HEK293T cells upon CRISPR-Cas9-mediated ablation of SETD2 gene to investigate the interplay between histone H3K36me3 and epitranscriptomic modulators.
EXPERIMENTAL SECTION
Cell Culture.
HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin. HEK293T cells with SETD2 and KAT5 genes being individually ablated with CRISPR/Cas9 were generated previously16 and maintained at 37 °C in a humidified chamber containing 5% CO2.
Stable Isotope-Labeled Peptides.
As described previously,34 47 crude synthetic stable isotope-labeled (SIL) peptides ([13C6,15N2 ]-Lys and [13C6, 15N4]-Arg) representing 44 RWE proteins were synthesized at 1.0 μmol scale and purified by Vivitide (Gardner, MA). The peptide purity was ~75%, and isotopic purity was approximately 99%. Each SIL peptide was reconstituted in an aqueous solution containing 15% acetonitrile and 0.1% formic acid. All SIL peptides were mixed as a stock solution for spiking into proteomic samples. The 47 stable isotope-labeled peptides were employed as internal standards for the corresponding peptides derived from endogenous epitranscriptomic RWE proteins. Peptides from endogenous epitranscriptomic RWE proteins lacking the corresponding stable isotope-labeled standards were grouped based on their retention times, where an appropriate stable isotope-labeled peptide with similar elution time was chosen as the surrogate standard for each group.
Cell Lysate Preparation.
HEK293T cells and the isogenic SETD2−/− and KAT5−/− cells were harvested and subsequently processed according to previously described procedures.34 Briefly, the cells were lysed on ice for 30 min with occasional vortexing using CelLytic M cell lysis reagent (Sigma) supplemented with 1% protease inhibitor cocktail and centrifuged at 16 000g for 30 min at 4 °C. The supernatants were collected, and the protein concentrations in the supernatants were determined using the Bradford assay.
Isolation of Chromatin Fraction.
The chromatin fractions of parental HEK293T cells and the isogenic SETD2−/− and KAT5−/− cells were prepared following the previously reported protocol.16 The cells were lysed by incubating in cytoplasmic lysis buffer (10 mM Tris-HCl, Ph 8.0, 0.34 M sucrose, 3 mM CaCl2, 2 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 0.5% Nonidet P-40, and 1% protease inhibitor cocktail) for 30 min on ice, and the intact nuclei were subsequently pelleted by centrifugation at 5000 rpm for 2 min. Nuclei were lysed with nuclear lysis buffer (20 mM HEPES, pH 7.9, 1.5 mM MgCl2, 1 mM EDTA, 150 mM KCl, 0.1% Nonidet P-40, 1 mM DTT, 10% glycerol, and 1% protease inhibitor mixture) by homogenization and incubated on ice for 5 min. After centrifugation at 13 000 rpm for 30 min, the chromatin-enriched pellet fraction was incubated in a chromatin isolation buffer, which contained 20 mM HEPES (pH 7.9), 1.5 mM MgCl2, 150 mM KCl, 10% glycerol, 1% protease inhibitor mixture, and 0.15 unit/μL benzonase (Sigma), on a thermomixer at 4 °C with interval vortexing (on: 20 s and off: 9 min 40 s) at 1200 rpm for 2 h. Debris was then removed by centrifugation at 5000 rpm for 2 min, and the supernatant was collected as the chromatin fraction. Protein concentrations in chromatin fractions were determined by Bradford assay (Bio-Rad).
Sample Preparation for Liquid Chromatography-Parallel-Reaction Monitoring (LC-PRM) Analysis.
The samples were prepared following the filter-aided sample preparation (FASP) protocol,35 where 40 μg of protein samples were denatured twice with 8 M urea in 50 mM NH4HCO3 using the polyethersulfone (PES) membrane centrifugal filter units (with a 30 kDa molecular weight cutoff, VWR), and the sample tubes were centrifuged at 10 000g for 30 min at room temperature. The denatured samples were reduced with 20 mM dithiothreitol (DTT) at 37 °C for 1 h and alkylated with 55 mM iodoacetamide (IAA) at room temperature in the dark for 30 min, followed by washing with 50 mM NH4HCO3 three times. The samples were digested with MS-grade trypsin (Pierce) at a 1:40 ratio (trypsin/protein, by mass) in 50 mM NH4HCO3 at 37 °C for 18 h. The tryptic peptides were collected by centrifugation, dried in a Speed-vac, desalted using Pierce C18 pipette tips (Thermo Scientific), and reconstituted in 0.1% formic acid. To the mixture was subsequently added a mixture of SIL peptides at a final concentration of 2 fmol/μL each.
LC-PRM Analysis.
LC-PRM analyses were conducted on a Q Exactive Plus quadrupole-Orbitrap mass spectrometer coupled with a Dionex UltiMate 3000 RSLCnano UPLC system. A trapping column (150 μm ID, 40 mm) packed in-house with 5 μm Reprosil-Pur C18-AQ resin (Dr. Maisch GmbH HPLC) and an analytical column (75 μm ID, 200 mm) packed in-house with 3 μm Reprosil-Pur C18-AQ resin (Dr. Maisch GmbH HPLC) were utilized to trap and resolve peptides, respectively. Formic acid (0.1%, v/v) in water and formic acid (0.1%, v/v) in acetonitrile/H2O (80:20, v/v) were used as mobile phases A and B, respectively. A linear gradient of 6–43% B in 125 min was employed, and the flow rate was 300 nL/min. The voltage for electrospray was 1.8 kV, and the capillary temperature was 320 °C. The precursor ions were isolated in the quadrupole at an isolation width of 1.6 m/z and fragmented in the HCD collision cell at a normalized collision energy (NCE) of 28. MS/MS were acquired at a resolution of 17 500, where the automated gain control target was 1 × 105 and the maximum accumulation time was 50 ms. After calibrating the retention times for precursor ions using the tryptic peptides of bovine serum albumin (BSA) as references, MS/MS for the precursor ions on the inclusion lists were acquired in the scheduled PRM mode with a 10 min retention time window.
The raw LC-PRM data were imported to Skyline v21.236 for processing. The acquired MS/MS of each precursor ion was compared with that in the spectral library, where similarity was gauged by dot product (dotp) value.37 A dotp value of >0.7 and 4–6 fragment ions eluting at the same retention time were considered a positive identification. Additional data processing was conducted in Excel, and the results are shown in Table S1. In brief, the ratio of each peptide representing a specific RWE protein was calculated based on a two-step normalization: (1) the peak area of an endogenous peptide was normalized to that of its corresponding SIL peptide or a surrogate standard with a similar retention time; (2) the ratio was further normalized against the ratio of the sum of peak areas for all light peptides over the sum of peak areas for all heavy peptides in each LC-PRM run. The relative ratios of the peptides in SETD2−/− and KAT5−/− samples were then normalized against the corresponding ratios in parental HEK293T samples. The relative ratios of the epitranscriptomic RWE proteins in one biological replicate were calculated from the mean of the relative ratios of the peptides derived from the same protein. The final ratios of the RWE proteins were averaged from the quantification results of three independent biological replicates.
RESULTS AND DISCUSSION
The major objective of the present study is to explore how H3K36me3 modulates the chromatin occupancies of epitranscriptomic RWE proteins. To this end, we first employed liquid chromatography-parallel-reaction monitoring (LC-PRM) analysis, in conjunction with the use of synthetic stable isotope-labeled (SIL) peptides,34 to examine the relative expression of 154 epitranscriptomic RWE proteins in HEK293T and the isogenic SETD2−/− cells (Figure 1A). Li et al.16 showed that knockout of SETD2 gene in HEK293T cells not only abolished H3K36me3 but also significantly diminished H4K16ac as a consequence of the cross-talk between the two histone epigenetic marks. In particular, upon the induction of DNA double-strand breaks (DSBs), H3K36me3 recruits histone acetyltransferase KAT5 via LEDGF for the deposition of H4K16ac, thereby augmenting chromatin accessibility and facilitating DNA repair.16 Hence, we also monitored the relative expression levels of the 154 epitranscriptomic RWE proteins in the isogenic KAT5−/− cells for comparison (Figure 1A).
Figure 1.
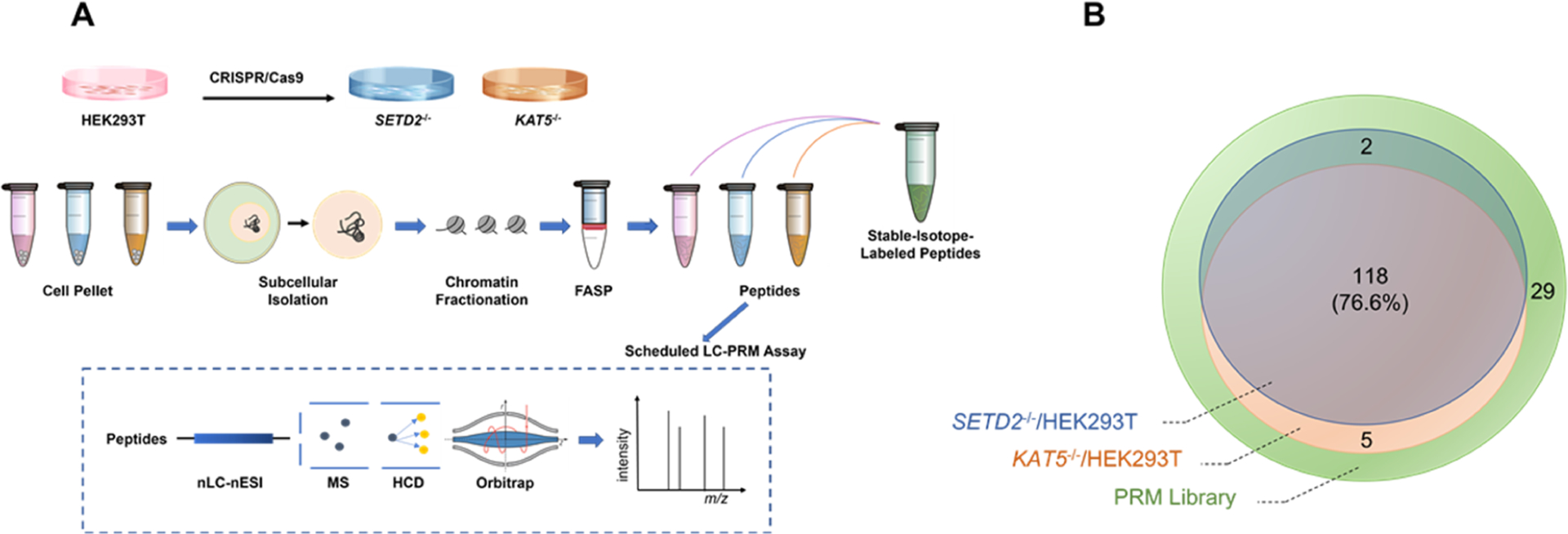
Overview of the LC-PRM method for examining the differential expression and chromatin occupancy of epitranscriptomic RWE proteins in HEK293T and the isogenic SETD2−/− and KAT5−/− cells. (A) LC-PRM workflow for targeted profiling of alterations in epitranscriptomic RWE proteins in the whole-cell lysate and chromatin fraction of HEK293T cells upon genetic ablation of SETD2 and KAT5. Confluent cells were collected and processed with the FASP protocol. Targeted profiling of epitranscriptomic RWE proteins was performed using LC-PRM analysis with SIL peptides as internal or surrogate standards. (B) Venn diagram depicting the numbers of quantified RWE proteins in SETD2−/− and KAT5−/− cells vs parental HEK293T cells, compared to those deposited in the PRM library.
Overall, 228 and 236 tryptic peptides, representing 120 and 123 epitranscriptomic RWE proteins, were quantified in SETD2−/− and KAT5−/− cells relative to parental HEK293T cells, respectively. Among them, 34 and 36 out of 228 and 236 tryptic peptides were quantified with their SIL peptides, respectively. The remaining peptides were quantified with surrogate SIL peptides with similar elution times as the target peptides. A total of 118 common RWE proteins were detected in both knockout cell lines, accounting for 76.6% of the proteins in the PRM library (Figure 1B).
Pearson’s correlation analysis (R = 0.7522, p < 0.0001) showed a significant positive correlation between the relative expression levels of RWE proteins in SETD2−/− and KAT5−/− cells (Figure 2A). This result is in line with the previous observation that SETD2-mediated H3K36me3 promotes KAT5-catalyzed H4K16ac.16 RWE proteins in the knockout cells with relative expression levels being greater than 1.50- or less than 0.67-fold in comparison with parental HEK293T cells were considered up- or downregulated, respectively. A Venn diagram revealed that 23 and 26 RWE proteins were up- and downregulated, respectively, in both SETD2−/− and KAT5−/− cells (Figure 2B). By imposing an additional criterion of p < 0.05, we found that 2 and 14 RWE proteins were significantly up- and downregulated, respectively, in both knockout cell lines (Figure 2D).
Figure 2.
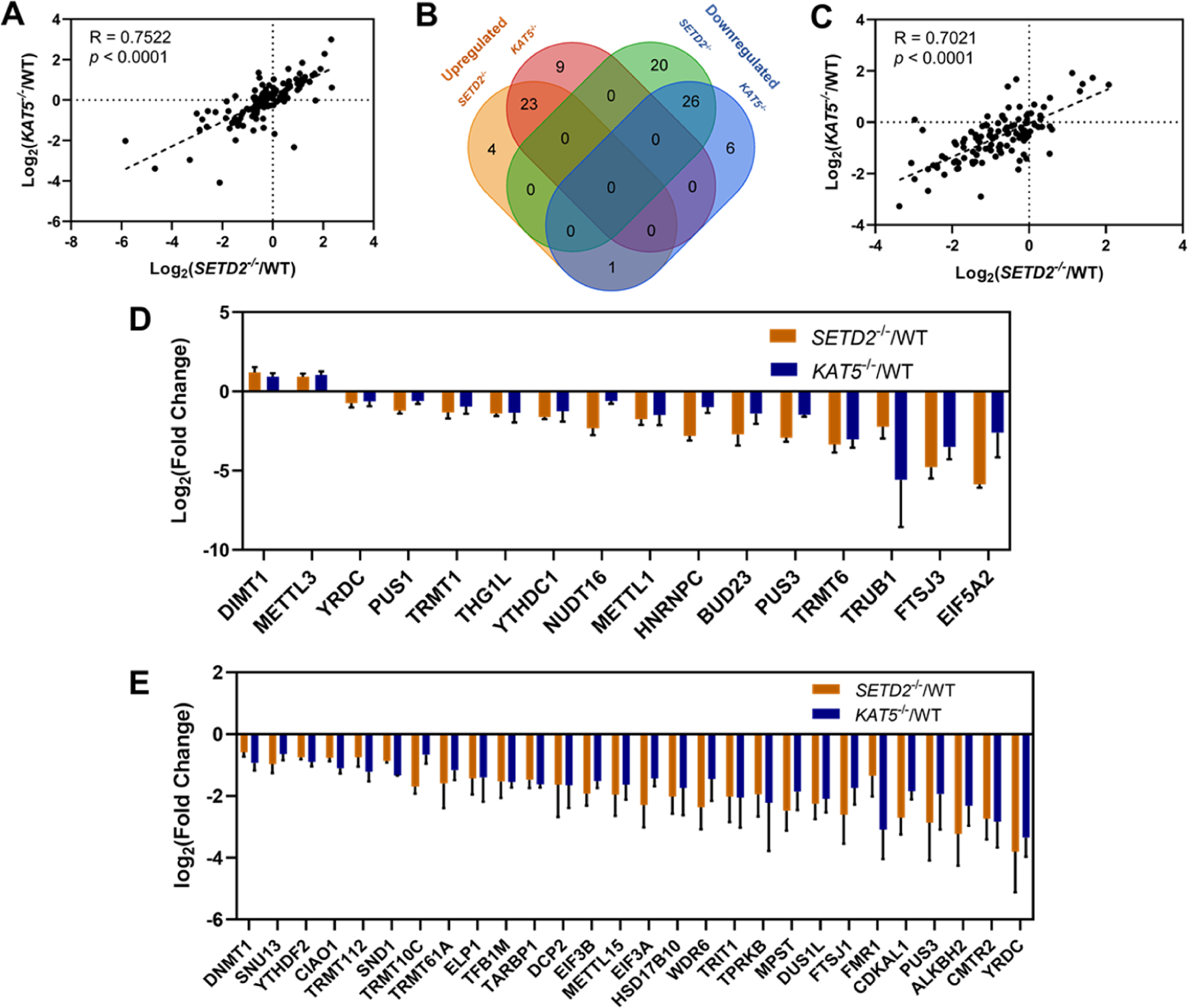
Comparison of the LC-PRM quantification results of RWE proteins in SETD2−/− (n = 3) and KAT5−/− (n = 3) cells relative to parental HEK293T cells (WT, n = 3). (A) Pearson’s correlation analysis displaying the Log2-transformed expression fold changes of the 118 epitranscriptomic RWE proteins in SETD2−/− (n = 3) and KAT5−/− (n = 3) cells relative to parental HEK293T cells (n = 3). (B) Venn diagram depicting the numbers of up- and downregulated RWE proteins in SETD2−/− and KAT5−/− cells relative to parental HEK293T cells. Proteins with expression fold changes being greater than 1.5 and less than 0.67 are considered up- and downregulated, respectively. (C) Pearson’s correlation analysis displaying the Log2-transformed fold changes of the 119 epitranscriptomic RWE proteins in the chromatin fractions of SETD2−/− (n = 3) and KAT5−/− (n = 3) cells relative to parental HEK293T cells (n = 3). (D) Bar chart showing the RWE proteins with significantly increased and decreased expressions in both SETD2−/− (n = 3) and KAT5−/− (n = 3) cells (expression fold changes: > 1.5 or < 0.67; p < 0.05). (E) Bar chart showing the RWE proteins significantly up- and downregulated in chromatin fractions of both SETD2−/− (n = 3) and KAT5−/− (n = 3) cells (expression fold changes: > 1.5 or < 0.67; p < 0.05).
Considering that histone PTMs could influence the recruitment of RWE proteins to chromatin directly, we performed subcellular fractionation and implemented targeted LC-PRM analyses for the 154 epitranscriptomic RWE proteins in the chromatin fraction. In this vein, Western blot analysis confirmed the successful subcellular fractionation (Figure S1). In total, 253 and 263 tryptic peptides, representing 123 and 121 epitranscriptomic RWE proteins, were quantified in the chromatin fractions from SETD2−/− and KAT5−/− cells, respectively. Among them, 39 tryptic peptides derived from 37 RWE proteins were quantified with their SIL peptides in the two knockout cell lines. In addition, 119 common RWE proteins were detected in the chromatin fractions of the two knockout cell lines, and they represent 78% of RWE proteins in the PRM library (Figure S2a). Although the proteomic results of chromatin fraction and whole-cell lysate yielded similar coverage of RWE proteins in the PRM library, chromatin fractions offered better peptide coverage, indicating enrichment of these RWE proteins in the chromatin fraction relative to the whole-cell lysate.
We again observed a significant positive correlation between the chromatin-associated RWE proteins in SETD2−/− and KAT5−/− cells based on Pearson’s correlation analysis (R = 0.7021, p < 0.0001, Figure 2C). The scatter plots of Pearson’s correlation analysis also showed that the expression levels of RWE proteins, i.e., in the whole-cell lysates, showed similar numbers of up- (fold change > 1.5) and downregulated (fold change < 1.5) proteins, whereas a large number of RWE proteins exhibited diminished occupancy in the chromatin fraction. Consistently, the Venn diagram showed that 5 and 47 RWE proteins in the chromatin fractions were up- and downregulated, respectively, in both knockout cell lines (Figure S2b). By employing p < 0.05 as a cutoff, 28 common RWE proteins exhibited significantly diminished presence in the chromatin fractions of the two knockout cell lines (Figure 2E). The substantially attenuated levels of RWE proteins in the chromatin fractions of SETD2−/− and KAT5−/− cells suggested the roles of H3K36me3 and H4K16ac in the recruitment of these epitranscriptomic RWE proteins to chromatin.
The diminished levels of epitranscriptomic RWE proteins in the chromatin fraction may also arise from their decreased expression. To illustrate the correlation between expression levels of RWE proteins and their chromatin occupancy, we plotted the Log2-transformed fold changes of RWE proteins in the whole-cell lysate vs chromatin fraction of the two knockout cell lines separately. A total of 106 RWE proteins were commonly quantified in the whole-cell lysates and chromatin fractions in the two knockout cell lines (Figure 3A,B). Downregulated chromatin-associated RWE proteins were grouped based on their expression levels (Figures 3C,D and S3). In SETD2−/− cells, 13, 23, and 27 RWE proteins downregulated in the chromatin fraction were upregulated, unchanged, and downregulated, respectively, in overall expression levels. Additionally, 11, 23, and 18 RWE proteins downregulated in chromatin fraction were upregulated, unchanged, and downregulated, respectively, in their overall expression levels in KAT5−/− cells.
Figure 3.
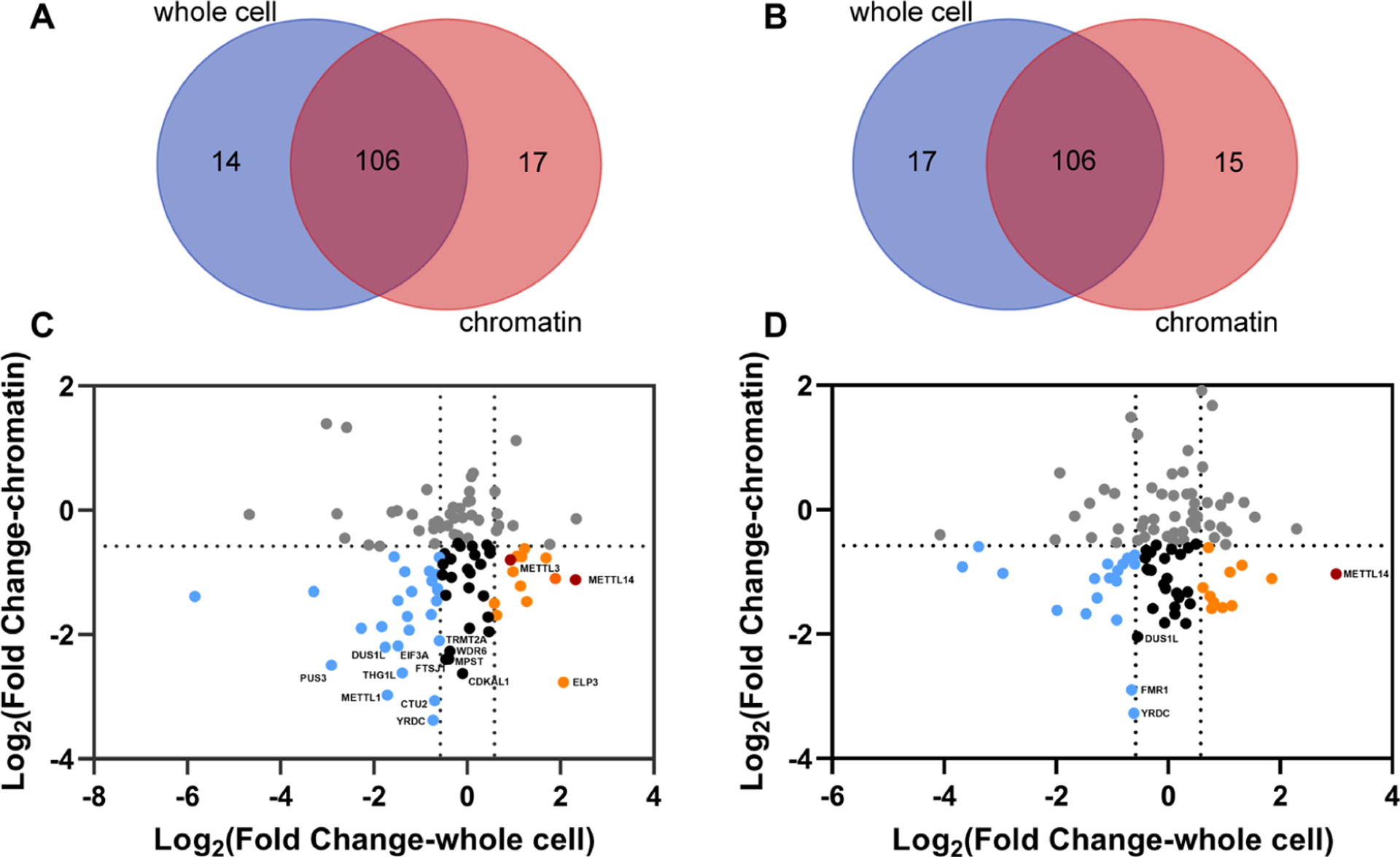
Comparison of the LC-PRM quantification results of the RWE proteins in the chromatin fractions and whole-cell lysates of SETD2−/− (n = 3) and KAT5−/− (n = 3) cells relative to parental HEK293T cells. (A, B) Venn diagrams depicting the numbers of quantified RWE proteins in chromatin fractions and whole-cell lysates of SETD2−/− (A) and KAT5−/− (B) cells relative to parental HEK293T cells. (C, D) Scatter plots showing the Log2-transformed fold changes in overall expression and chromatin occupancy of RWE proteins in SETD2−/− (C) and KAT5−/− (D) cells. RWE proteins downregulated in the chromatin fraction but upregulated and unchanged in whole-cell lysate are labeled in orange and black, respectively. RWE proteins downregulated in both chromatin fraction and whole-cell lysate are labeled in blue.
To identify those proteins whose chromatin localizations are enabled by the histone epigenetic marks, we focused on those RWE proteins displaying attenuated levels in the chromatin fraction but exhibiting no change or augmented levels in the whole-cell lysate after genetic ablation of SETD2. In this vein, Royal family domains, e.g., chromodomain, chromobarrel, MBT, PWWP, Tudor, and TTD, can interact with histone methylation marks and are considered reader domains for histone methylation.38,39 Staphylococcal nuclease and Tudor domain-containing 1 (SND1) protein was shown to be a reader protein for histone arginine methylation.40 Meanwhile, SND1 was identified as a reader protein of m6A in the Kaposi’s sarcoma-associated herpesvirus (KSHV) ORF50 RNA transcripts.41,42 We found that genetic ablation of SETD2 did not alter the expression level of SND1 but led to its diminished presence in the chromatin fraction, indicating that H3K36me3 may enable the recruitment of SND1 to chromatin through its Tudor domain.
Our results also revealed diminished chromatin occupancies of METTL3 and METTL14, whereas their overall expressions were elevated in SETD2−/− cells (Figures S3 and S4). In this vein, our Western blot results confirmed a diminished chromatin occupancy of METTL3 protein in SETD2−/− cells, and RT-qPCR results revealed no appreciable change in mRNA expression level of METTL3 gene upon genetic ablation of SETD2 (Figure S5), suggesting the involvement of a post-transcriptional mechanism in modulating the expression level of METTL3 protein in SETD2-depleted cells. Attenuated recruitment of METTL14 to chromatin upon loss of H3K36me3 is consistent with the previous finding that H3K36me3 guides the deposition of m6A through its interaction with METTL14.28 METTL3, which forms a complex with METTL14 along with several other proteins and constitutes the catalytic subunit of the major m6A MTC, is involved in multiple cellular processes.43 Increasing lines of evidence illustrated the active participation of METTL3, METTL14, and the ensuing RNA m6A methylation in DNA repair.44–47 In addition, H3K36me3 is a significant chromatin signal for DNA damage repair.3 Hence, we reason that METTL3 and METTL14′s chromatin occupancies could be regulated by H3K36me3 during DNA repair. Therefore, we next asked how the chromatin occupancy of METTL3 is affected by the loss of H3K36me3 following DNA damage induction.
Li et al.16 showed that DNA DSB-inducing agents stimulated H3K36me3 in cultured human cells in a time-dependent manner. Consistent with this previous finding, we observed that, upon neocarzinostatin (NCS) treatment, H3K36me3 level in HEK293T cells first increased, peaked at 30 min, and then returned to the initial level at 4-h following treatment with 100 ng/mL NCS (Figure 4A,C). As expected, H3K36me3 was not detectable in SETD2−/− cells (Figures 4A and S6). Quantification results from the Western blot showed that chromatin-bound METTL3 first increased, peaked at 60 min, and then decreased in HEK293T cells (Figures 4B,D and S7). The observation that the increase in chromatin occupancy of METTL3 trails that of H3K36me3 is consistent with the notion that H3K36me3 promotes the chromatin recruitment of METTL3. In keeping with the PRM results, we observed that the levels of chromatin-bound METTL3 were significantly lower in SETD2−/− cells than those in HEK293T cells throughout the entire time course following NCS exposure. Western blot analysis of METTL3 in the whole-cell lysate did not reveal any apparent alterations in the expression levels of the protein following NCS treatment in HEK293T or the isogenic SETD2−/− cells (Figure S8). Zhang et al.45 showed previously that METTL3 is involved in HR-mediated repair. In this vein, upon DNA DSB induction, activated METTL3 is localized to DNA damage sites and installs m6A on nascent RNA transcribed from DNA damage loci, which subsequently recruits YTHDC1, RAD51, and BRCA1 to promote HR repair.45 The above results suggest that SETD2-mediated H3K36me3 contributes to the recruitment of METTL3 to chromatin during DNA DSB repair.
Figure 4.
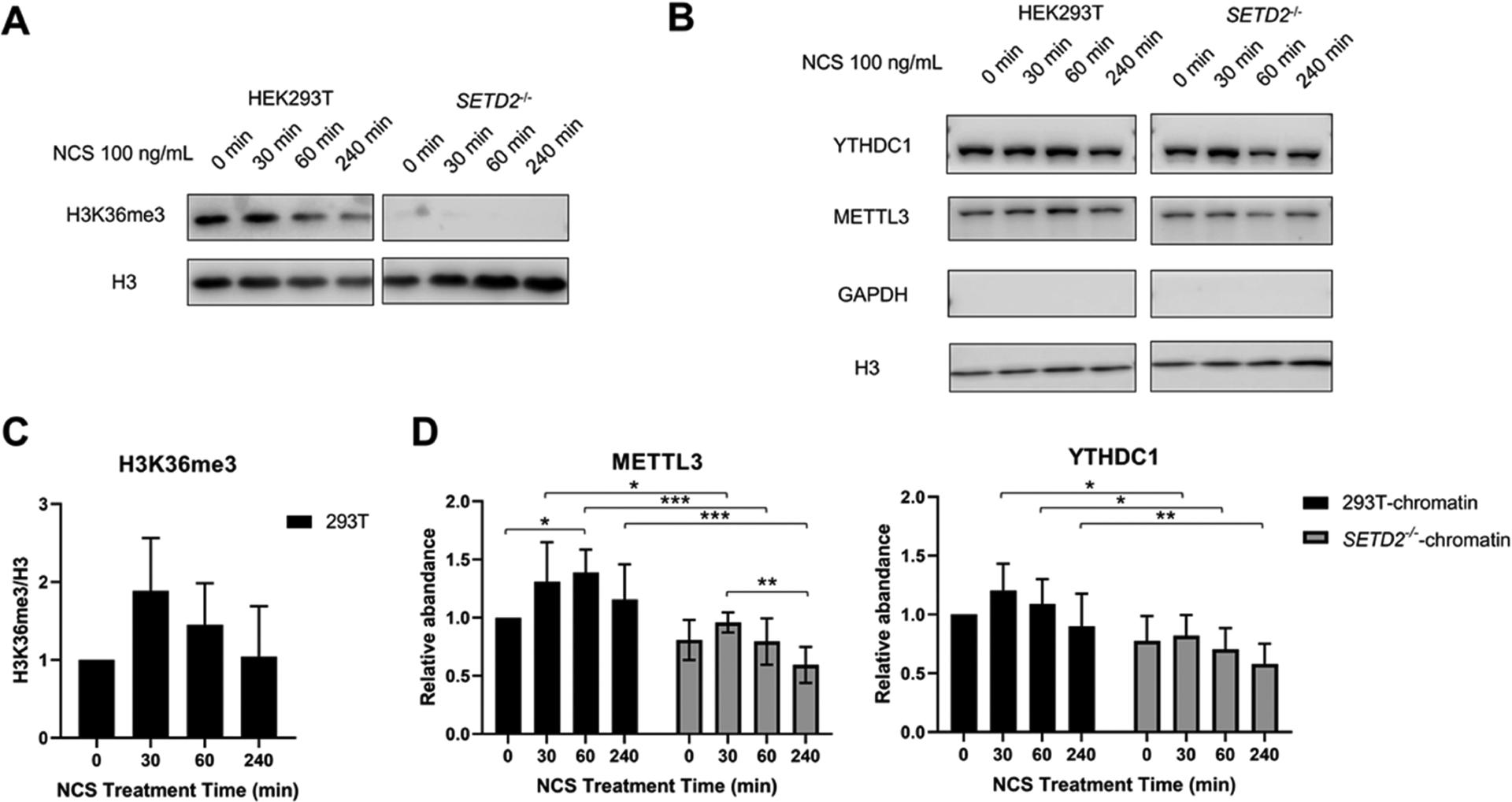
Western blot quantification results of METTL3 and YTHDC1 proteins in HEK293T and the isogenic SETD2−/− cells at different time points following treatment with 100 ng/mL NCS. (A) Representative western blot result of H3K36me3 in core histones extracted from HEK293T and the isogenic SETD2−/− cells during the treatment of 100 mg/mL NCS. Histone H3 was employed as a loading control. (B) Representative Western blot result of METTL3 and YTHDC1 proteins in the chromatin fractions of HEK293T and the isogenic SETD2−/− cells at different time points following treatment with 100 mg/mL NCS. Histone H3 was employed as a loading control. The lack of GAPDH signal indicates the lack of contamination of cytoplasmic proteins in the chromatin fraction. (C) Western blot quantification results displaying the NCS-induced changes in H3K36me3 levels in HEK293T cells. (D) Western blot quantification results showing the NCS-induced changes in the levels of METTL3 and YTHDC1 proteins in the chromatin fractions of HEK293T and the isogenic SETD2−/− cells. The quantification results displayed in panels (C, D) represent the mean and S.D. of results obtained from five independent biological replicates conducted on 5 separate days. The p values were calculated using nonparametric one-way ANOVA with further adjustment by the Benjamini−Hochberg FDR method for the comparison among different NCS treatment times in HEK293T and the isogenic SETD2−/− cells. #, p > 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
We also monitored the chromatin occupancy and expression level of YTHDC1, a known nuclear m6A reader protein, in HEK293T cells and the isogenic SETD2−/− cells following NCS treatment (Figures 4B,D, S7, and S8). Our results revealed that the chromatin occupancy of YTHDC1 in HEK293T cells increased first and then started to decrease after 30 min following NCS treatment. Moreover, the chromatin occupancy of YTHDC1 in SETD2−/− cells is significantly lower than that in HEK293T cells following NCS exposure. This is in line with the aforementioned findings made for METTL3, suggesting that increased chromatin occupancy of the METTL3 methyltransferase complex and the ensuing elevated m6A deposition in carRNAs promote the chromatin localization of the YTHDC1, the nuclear m6A reader protein.
We next performed protein–protein interaction (PPI) network analysis to reveal the relationship among the up- and downregulated epitranscriptomic RWE proteins after knockout of SETD2 gene. Our results showed that METTL14 and TRMT11 are the hub proteins with the highest interconnectivity to other nodes in the up- and downregulated RWE PPI network (Figure S9). Upregulation of METTL14 and downregulation of TRMT11 in SETD2−/− cells are reflected in the extracted-ion chromatograms of the representative tryptic peptides of the two proteins and their corresponding SIL or surrogate standard peptides (Figure S10a,c). SETD2 is one of the top mutated genes in clear-cell renal cell carcinoma48 (ccRCC) and is associated with tumor progression.49 Mutations in SETD2 gene or lower expression of SETD2 protein are accompanied with poor prognosis in ccRCC patients.50,51 Those RWE proteins substantially altered after SETD2 deletion could also be involved in ccRCC progression. Kaplan–Meier survival analysis for kidney cancer cases in the Cancer Genome Atlas (TCGA) showed that higher levels of METTL14 expression or lower levels of TRMT11 expression are significantly associated with better prognosis in kidney renal clear-cell carcinoma (KIRC, Figure S10b,d), suggesting potential roles of METTL14 and TRMT11 during ccRCC disease progression.
In summary, by using a scheduled LC-PRM-based targeted proteomic method coupled with SIL standard peptides, we examined quantitatively the chromatin occupancies and overall expression levels of 154 epitranscriptomics RWE proteins in HEK293T and the isogenic SETD2−/− and KAT5−/− cells. The quantification results for the RWE proteins exhibited positive correlations in SETD2−/− and KAT5−/− cells with respect to their overall expression levels and chromatin occupancies, indicating that H3K36me3 and H4K16ac may coregulate epitranscriptomic RWE proteins in human cells. We found that H3K36me3 modulates the chromatin occupancies of METTL3 and METTL14, the core components of the major m6A methyltransferase complex, and revealed the implication of this modulation in response to DNA DSB induction in cells. High mutation rate of SETD2 is associated with multiple cancers, especially ccRCC.17 PPI network analysis indicated the importance of METTL14 among the upregulated proteins and TRMT11 among the downregulated proteins after knockout of SETD2 protein. Kaplan–Meier survival analysis for kidney cancers in TCGA suggested the potential regulatory roles of METTL14 and TRMT11 in ccRCC disease. Together, our results revealed the cross-talks between epitranscriptomic RWE proteins and H3K36me3 and their potential regulatory roles in DNA repair and ccRCC disease.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (R35 ES031707).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.3c01552.
Western blot results of METTL3, METTL14, and YTHDC1; PPI network analysis; and LC-PRM quantification results of epitranscriptomic RWE proteins in HEK293T and the isogenic SETD2−/− and KAT5−/− cells (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.3c01552
The authors declare no competing financial interest.
Contributor Information
Jiekai Yin, Environmental Toxicology Graduate Program, University of California, Riverside, California 92521-0403, United States.
Tianyu F. Qi, Environmental Toxicology Graduate Program, University of California, Riverside, California 92521-0403, United States
Lin Li, Deparment of Chemistry, University of California, Riverside, California 92521-0403, United States.
Yinsheng Wang, Environmental Toxicology Graduate Program, University of California, Riverside, California 92521-0403, United States;.
Data Availability Statement
All of the LC–MS/MS raw files and Skyline PRM library were deposited to the ProteomeXchange Consortium via the PRIDE52 partner repository with the data set identifier (PXD041343).
REFERENCES
- (1).Wagner EJ; Carpenter PB Nat. Rev. Mol. Cell Biol 2012, 13, 115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Zaghi M; Broccoli V; Sessa A Front. Genet 2020, 10, No. 1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Sun Z; Zhang Y; Jia J; Fang Y; Tang Y; Wu H; Fang D Cell Biosci. 2020, 10, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Xiao C; Fan T; Tian H; Zheng Y; Zhou Z; Li S; Li C; He J Clin. Epigenet 2021, 13, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sharda A; Humphrey TC DNA Repair 2022, 119, No. 103407. [DOI] [PubMed] [Google Scholar]
- (6).Rondelet G; Dal Maso T; Willems L; Wouters JJ Struct. Biol 2016, 194, 357–367. [DOI] [PubMed] [Google Scholar]
- (7).Baubec T; Colombo DF; Wirbelauer C; Schmidt J; Burger L; Krebs AR; Akalin A; Schübeler D Nature 2015, 520, 243–247. [DOI] [PubMed] [Google Scholar]
- (8).Neri F; Rapelli S; Krepelova A; Incarnato D; Parlato C; Basile G; Maldotti M; Anselmi F; Oliviero S Nature 2017, 543, 72–77. [DOI] [PubMed] [Google Scholar]
- (9).Luco RF; Pan Q; Tominaga K; Blencowe BJ; Pereira-Smith OM; Misteli T Science 2010, 327, 996–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Pradeepa MM; Sutherland HG; Ule J; Grimes GR; Bickmore WA PLoS Genet. 2012, 8, No. e1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Guo R; Zheng L; Park JW; Lv R; Chen H; Jiao F; Xu W; Mu S; Wen H; Qiu J; Wang Z; Yang P; Wu F; Hui J; Fu X; Shi X; Shi YG; Xing Y; Lan F; Shi Y Mol. Cell 2014, 56, 298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Pfister SX; Ahrabi S; Zalmas L-P; Sarkar S; Aymard F; Bachrati CZ; Helleday T; Legube G; La Thangue NB; Porter ACG; Humphrey TC Cell Rep. 2014, 7, 2006–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chang CF; Chu PC; Wu PY; Yu MY; Lee JY; Tsai MD; Chang MS Cell Death Dis. 2015, 6, e1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Li F; Mao G; Tong D; Huang J; Gu L; Yang W; Li G-M Cell 2013, 153, 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Edmunds JW; Mahadevan LC; Clayton AL EMBO J. 2008, 27, 406–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Li L; Wang YJ Biol. Chem 2017, 292, 11951–11959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lu M; Zhao B; Liu M; Wu L; Li Y; Zhai Y; Shen X NPJ Precis. Oncol 2021, 5, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Xie Y; Sahin M; Sinha S; Wang Y; Nargund AM; Lyu Y; Han S; Dong Y; Hsieh JJ; Leslie CS; Cheng EH Nat. Cancer 2022, 3, 188–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bu J; Chen A; Yan X; He F; Dong Y; Zhou Y; He J; Zhan D; Lin P; Hayashi Y; Sun Y; Zhang Y; Xiao Z; Grimes HL; Wang QF; Huang G Leukemia 2018, 32, 890–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yuan W; Xu M; Huang C; Liu N; Chen S; Zhu BJ Biol. Chem 2011, 286, 7983–7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Schmitges FW; Prusty AB; Faty M; Stützer A; Lingaraju GM; Aiwazian J; Sack R; Hess D; Li L; Zhou S; Bunker RD; Wirth U; Bouwmeester T; Bauer A; Ly-Hartig N; Zhao K; Chan H; Gu J; Gut H; Fischle W; Müller J; Thomä NH Mol. Cell 2011, 42, 330–341. [DOI] [PubMed] [Google Scholar]
- (22).Streubel G; Watson A; Jammula SG; Scelfo A; Fitzpatrick DJ; Oliviero G; McCole R; Conway E; Glancy E; Negri GL; Dillon E; Wynne K; Pasini D; Krogan NJ; Bracken AP; Cagney G Mol. Cell 2018, 70, 371–379.e5. [DOI] [PubMed] [Google Scholar]
- (23).Wang X; Lu Z; Gomez A; Hon GC; Yue Y; Han D; Fu Y; Parisien M; Dai Q; Jia G; Ren B; Pan T; He C Nature 2014, 505, 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wang X; Zhao BS; Roundtree Ian A.; Lu Z; Han D; Ma H; Weng X; Chen K; Shi H; He C Cell 2015, 161, 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Arango D; Sturgill D; Alhusaini N; Dillman AA; Sweet TJ; Hanson G; Hosogane M; Sinclair WR; Nanan KK; Mandler MD; Fox SD; Zengeya TT; Andresson T; Meier JL; Coller J; Oberdoerffer S Cell 2018, 175, 1872–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Roundtree IA; Evans ME; Pan T; He C Cell 2017, 169, 1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Liu J; Dou X; Chen C; Chen C; Liu C; Xu MM; Zhao S; Shen B; Gao Y; Han D; He C Science 2020, 367, 580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Huang H; Weng H; Zhou K; Wu T; Zhao BS; Sun M; Chen Z; Deng X; Xiao G; Auer F; Klemm L; Wu H; Zuo Z; Qin X; Dong Y; Zhou Y; Qin H; Tao S; Du J; Liu J; Lu Z; Yin H; Mesquita A; Yuan CL; Hu Y-C; Sun W; Su R; Dong L; Shen C; Li C; Qing Y; Jiang X; Wu X; Sun M; Guan J-L; Qu L; Wei M; Müschen M; Huang G; He C; Yang J; Chen J Nature 2019, 567, 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Frye M; Harada BT; Behm M; He C Science 2018, 361, 1346–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Delaunay S; Frye M Nat. Cell Biol 2019, 21, 552–559. [DOI] [PubMed] [Google Scholar]
- (31).Barbieri I; Kouzarides T Nat. Rev. Cancer 2020, 20, 303–322. [DOI] [PubMed] [Google Scholar]
- (32).Shi H; Wei J; He C Mol. Cell 2019, 74, 640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Boriack-Sjodin PA; Ribich S; Copeland RA Nat. Rev. Drug Discovery 2018, 17, 435–453. [DOI] [PubMed] [Google Scholar]
- (34).Qi TF; Liu X; Tang F; Yin J; Yu K; Wang Y Anal. Chem 2022, 94, 12559–12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wiśniewski JR; Zougman A; Nagaraj N; Mann M Nat. Methods 2009, 6, 359–362. [DOI] [PubMed] [Google Scholar]
- (36).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Bioinformatics 2010, 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kawahara R; Bollinger JG; Rivera C; Ribeiro ACP; Brandão TB; Leme AFP; MacCoss MJ Proteomics 2016, 16, 159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Maurer-Stroh S; Dickens NJ; Hughes-Davies L; Kouzarides T; Eisenhaber F; Ponting CP Trends Biochem. Sci 2003, 28, 69–74. [DOI] [PubMed] [Google Scholar]
- (39).Musselman CA; Lalonde M-E; Côté J; Kutateladze TG Nat. Struct. Mol. Biol 2012, 19, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Liu K; Chen C; Guo Y; Lam R; Bian C; Xu C; Zhao DY; Jin J; MacKenzie F; Pawson T; Min J Proc. Natl. Acad. Sci. U.S.A 2010, 107, 18398–18403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Baquero-Perez B; Antanaviciute A; Yonchev ID; Carr IM; Wilson SA; Whitehouse A eLife 2019, 8, No. e47261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Röder K; Barker AM; Whitehouse A; Pasquali S PLoS Comput. Biol 2022, 18, No. e1010150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Liu J; Yue Y; Han D; Wang X; Fu Y; Zhang L; Jia G; Yu M; Lu Z; Deng X; Dai Q; Chen W; He C Nat. Chem. Biol 2014, 10, 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Xiang Y; Laurent B; Hsu C-H; Nachtergaele S; Lu Z; Sheng W; Xu C; Chen H; Ouyang J; Wang S; Ling D; Hsu P-H; Zou L; Jambhekar A; He C; Shi Y Nature 2017, 543, 573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zhang C; Chen L; Peng D; Jiang A; He Y; Zeng Y; Xie C; Zhou H; Luo X; Liu H; Chen L; Ren J; Wang W; Zhao Y Mol. Cell 2020, 79, 425–442. [DOI] [PubMed] [Google Scholar]
- (46).Yang Z; Yang S; Cui Y-H; Wei J; Shah P; Park G; Cui X; He C; He Y-Y Proc. Natl. Acad. Sci. U.S.A 2021, 118, No. e2025948118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Li E; Xia M; Du Y; Long K; Ji F; Pan F; He L; Hu Z; Guo Z eLife 2022, 11, No. e75231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Creighton CJ; Morgan M; Gunaratne PH; Wheeler DA; Gibbs RA; Gordon Robertson A; Chu A; Beroukhim R; Cibulskis K; Signoretti S; Vandin Hsin-Ta Wu F; Raphael BJ; Verhaak RGW; Tamboli P; Torres-Garcia W; Akbani R; Weinstein JN; Reuter V; Hsieh JJ; Rose Brannon A; Ari Hakimi A; Jacobsen A; Ciriello G; Reva B; Ricketts CJ; Marston Linehan W; Stuart JM; Kimryn Rathmell W; Shen H; Laird PW; Muzny D; Davis C; Morgan M; Xi L; Chang K; Kakkar N; Treviño LR; Benton S; Reid JG; Morton D; Doddapaneni H; Han Y; Lewis L; Dinh H; Kovar C; Zhu Y; Santibanez J; Wang M; Hale W; Kalra D; Creighton CJ; Wheeler DA; Gibbs RA; Getz G; Cibulskis K; Lawrence MS; Sougnez C; Carter SL; Sivachenko A; Lichtenstein L; Stewart C; Voet D; Fisher S; Gabriel SB; Lander E; Beroukhim R; Schumacher SE; Tabak B; Saksena G; Onofrio RC; Carter SL; Cherniack AD; Gentry J; Ardlie K; Sougnez C; Getz G; Gabriel SB; Meyerson M; Gordon Robertson A; Chu A; Chun H-JE; Mungall AJ; Sipahimalani P; Stoll D; Ally A; Balasundaram M; Butterfield YSN; Carlsen R; Carter C; Chuah E; Coope RJN; Dhalla N; Gorski S; Guin R; Hirst C; Hirst M; Holt RA; Lebovitz C; Lee D; Li HI; Mayo M; Moore RA; Pleasance E; Plettner P; Schein JE; Shafiei A; Slobodan JR; Tam A; Thiessen N; Varhol RJ; Wye N; Zhao Y; Birol I; Jones SJM; Marra MA; Auman JT; Tan D; Jones CD; Hoadley KA; Mieczkowski PA; Mose LE; Jefferys SR; Topal MD; Liquori C; Turman YJ; Shi Y; Waring S; Buda E; Walsh J; Wu J; Bodenheimer T; Hoyle AP; Simons JV; Soloway MG; Balu S; Parker JS; Neil Hayes D; Perou CM; Kucherlapati R; Park P; Shen H; Triche T Jr; Weisenberger DJ; Lai PH; Bootwalla MS; Maglinte DT; Mahurkar S; Berman BP; Van Den Berg DJ; Cope L; Baylin SB; Laird PW; Creighton CJ; Wheeler DA; Getz G; Noble MS; DiCara D; Zhang H; Cho J; Heiman DI; Gehlenborg N; Voet D; Mallard W; Lin P; Frazer S; Stojanov P; Liu Y; Zhou L; Kim J; Lawrence MS; Chin L; Vandin F; Wu H-T; Raphael BJ; Benz C; Yau C; Reynolds SM; Shmulevich I; Verhaak RGW; Torres-Garcia W; Vegesna R; Kim H; Zhang W; Cogdell D; Jonasch E; Ding Z; Lu Y; Akbani R; Zhang N; Unruh AK; Casasent TD; Wakefield C; Tsavachidou D; Chin L; Mills GB; Weinstein JN; Jacobsen A; Rose Brannon A; Ciriello G; Schultz N; Ari Hakimi A; Reva B; Antipin Y; Gao J; Cerami E; Gross B; Arman Aksoy B; Sinha R; Weinhold N; Onur Sumer S; Taylor BS; Shen R; Ostrovnaya I; Hsieh JJ; Berger MF; Ladanyi M; Sander C; Fei SS; Stout A; Spellman PT; Rubin DL; Liu TT; Stuart JM; Ng S; Paull EO; Carlin D; Goldstein T; Waltman P; Ellrott K; Zhu J; Haussler D; Gunaratne PH; Xiao W; Shelton C; Gardner J; Penny R; Sherman M; Mallery D; Morris S; Paulauskis J; Burnett K; Shelton T; Signoretti S; Kaelin WG; Choueiri T; Atkins MB; Penny R; Burnett K; Mallery D; Curley E; Tickoo S; Reuter V; Kimryn Rathmell W; Thorne L; Boice L; Huang M; Fisher JC; Marston Linehan W; Vocke CD; Peterson J; Worrell R; Merino MJ Nature 2013, 499, 43–49.23792563 [Google Scholar]
- (49).Ho TH; Park IY; Zhao H; Tong P; Champion MD; Yan H; Monzon FA; Hoang A; Tamboli P; Parker AS; Joseph RW; Qiao W; Dykema K; Tannir NM; Castle EP; Nunez-Nateras R; Teh BT; Wang J; Walker CL; Hung MC; Jonasch E Oncogene 2016, 35, 1565–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Hakimi AA; Ostrovnaya I; Reva B; Schultz N; Chen Y-B; Gonen M; Liu H; Takeda S; Voss MH; Tickoo SK; Reuter VE; Russo P; Cheng EH; Sander C; Motzer RJ; Hsieh JJ Clin. Cancer Res 2013, 19, 3259–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Li L; Miao W; Huang M; Williams P; Wang Y Mol. Cell Proteomics 2019, 18, 437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Perez-Riverol Y; Bai J; Bandla C; García-Seisdedos D; Hewapathirana S; Kamatchinathan S; Kundu DJ; Prakash A; Frericks-Zipper A; Eisenacher M; Walzer M; Wang S; Brazma A; Vizcaíno JA Nucleic Acids Res. 2022, 50, D543–D552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the LC–MS/MS raw files and Skyline PRM library were deposited to the ProteomeXchange Consortium via the PRIDE52 partner repository with the data set identifier (PXD041343).