

Summary
The cardiac fibroblast interacts with an extracellular matrix (ECM), enabling myofibroblast maturation via a process called mechanosensing. Although in the aging male heart, ECM is stiffer than in the young mouse, myofibroblast development is impaired, as demonstrated in 2-D and 3-D experiments. In old male cardiac fibroblasts, we found a decrease in actin polymerization, α-smooth muscle actin (α-SMA), and Kindlin-2 expressions, the latter an effector of the mechanosensing. When Kindlin-2 levels were manipulated via siRNA interference, young fibroblasts developed an old-like fibroblast phenotype, whereas Kindlin-2 overexpression in old fibroblasts reversed the defective phenotype. Finally, inhibition of overactivated extracellular regulated kinases 1 and 2 (ERK1/2) in the old male fibroblasts rescued actin polymerization and α-SMA expression. Pathological ERK1/2 overactivation was also attenuated by Kindlin-2 overexpression. In contrast, old female cardiac fibroblasts retained an operant mechanosensing pathway. In conclusion, we identified defective components of the Kindlin/ERK/actin/α-SMA mechanosensing axis in aged male fibroblasts.
Subject areas: Cardiovascular medicine, Pathophysiology, Molecular microbiology
Graphical abstract
Highlights
-
•
Old male cardiac fibroblasts have defects in the ECM/Kindlin-2/ERK/αSMA pathway
-
•
Changes in ECM composition affect Kindlin-2 and αSMA expression
-
•
Loss of Kindlin-2 leads to ERK overactivation and actin depolymerization
-
•
ECM composition changes in aging female hearts but preserves mechanosensing
Cardiovascular medicine; Pathophysiology; Molecular microbiology.
Introduction
Extracellular matrix (ECM) is a complex meshwork of fibrillar and non-fibrillar collagens associated with growth factors and glycoproteins.1,2 Although all cardiac cells can synthesize different components of ECM, cardiac fibroblasts are the primary producers of fibrillar collagens, which determine ECM stiffness. Activated fibroblasts orchestrate the production and the remodeling of ECM.3,4 On the other hand, changes in ECM tension directly influence cardiac fibroblast phenotype and activation status via a mechanism called "mechanosensing."5,6,7 Therefore, active bidirectional crosstalk between ECM and the fibroblast translates ECM mechanical changes into intracellular pathways, activating a specific gene program. These changes may translate into phenotype switch (transition into myofibroblast) or ECM remodeling (degradation or synthesis).
One of the first steps in the mechanosensing pathway is the binding of integrins (cell surface receptors) to various ECM proteins. Integrin-based focal adhesions (a heterocomplex of proteins organized around the integrins) bridge ECM signals with the fibroblast’s actin cytoskeleton.8 After the integrin and intracellular pathways (PI3K, RhoGTpases, and Map Kinases) activation, actin in the cytoskeleton transitions from a monomeric (G-actin) into a polymerized (F-actin) form.9 Actin dynamics may translate to the expression of the cell differentiation program, and especially actin-related genes (essentially β-actin isoform and actin-binding proteins) via the nuclear translocation of transcription co-factors, such as myocardin-related transcription factor (MRTF) or Yes1 associated transcriptional regulator (YAP).10,11 The activation of MRTF and YAP results in cell differentiation (fibroblast-to-myofibroblast transition) and migration.12,13
In response to myocardial infarction (MI), fibroblasts synthesize collagens and other ECM proteins as a consequence of cardiomyocyte loss.3,14 As the newly formed scar becomes stiffer, it triggers the fibroblast-to-myofibroblast transition.15 The myofibroblast is a specialized type of cell that expresses α-smooth muscle actin (α-SMA).16,17,18 Myofibroblast differentiation can be triggered via biochemical signals (the TGF-β-dependent pathway) or mechanosensing.15,19,20,21 Myofibroblasts play an essential role in contracting the scar and preventing infarct expansion, but, on the other hand, they may contribute to adverse remodeling by producing collagens.
In the aging heart, ECM production, deposition, and crosslinking are exacerbated, leading to the development of invasive interstitial and perivascular fibrosis, a maladaptive process that affects the electrophysiological and mechanical properties of the myocardium.22,23,24,25 However, in an apparent paradox, the healing after MI (in aging male hearts) is impaired, with reduced myofibroblast number and inadequate scar, leading to infarct expansion and heart failure.26,27,28 The aging fibroblast becomes less capable of maturing into a myofibroblast due to uncoupling of the ECM/fibroblast crosstalk or impaired response to TGF-β.29,30,31,32,33 Increased infarct expansion and heart failure incidents after MI have been documented in older patients and experimental models in aged mice.27,28,34 Evidence suggests that changes in mechanosensing may be involved in the pathophysiological process of the aging heart,35 but there is a knowledge gap regarding the key effectors of the regulatory axis that may drive this defect.
In this paper, by using a combination of in vitro and ex vivo approaches, we identified the main defects in effectors of the fibroblast-to-ECM crosstalk in the old male heart. First, we found that fibroblasts from the old male expressed a reduced expression of α-SMA. Then, by a 2-D and 3-D matrix swapping strategy, we demonstrated that the ECM composition directly affected the fibroblast phenotype, especially its capacity to generate contractile stress fibers. We also demonstrated that the cardiac matrisome composition and architecture in the heart is affected by sex and age. Matrix composition also directly influenced the expression of Kindlin-2, the main adapter of integrin signaling that connects integrins to F-actin and transduces Map Kinase signaling downstream.36,37 Kindlin-2 deficiency in young cells recapitulated part of the old cell defects, while its overexpression in old cells promoted actin polymerization and α-SMA expression, favoring myofibroblast maturation. The improved maturation due to Kindlin-2 overexpression is even further amplified when Kindlin-2 overexpressing old cells were seeded on the young matrices. Finally, we demonstrated that extracellular regulated kinases 1 and 2 (ERK1/2) are overactivated (highly phosphorylated) in the fibroblasts isolated from the old mouse heart, but overexpression of Kindlin-2 can reduce ERK phosphorylation, rescuing actin polymerization and the downstream expression of α-SMA. In summary, we identified the ECM/Kindlin-2/ERK/actin axis as the main effector of the mechanosensing pathway that is impaired in the aging male heart.
Results
The aging phenotype of the male cardiac fibroblast is associated with a decreased expression of Kindlin-2
To better characterize the aging phenotype of the cardiac fibroblasts, cells from young and old male mice were cultured in standard tissue culture conditions on plastic dishes. These conditions favor the fibroblast-to-myofibroblast maturation via the mechanosensing pathway because of supra-physiological tensions exerted by rigid plastic.
We first focused on the main features of myofibroblast maturation, such as α-SMA expression and actin polymerization/depolymerization dynamics (polymerized F-actin versus monomeric G-actin).
Compared with young cells, fibroblasts cultured from the old male mouse heart have reduced α-SMA expression (0.32 ± 0.02 vs. 1.0 ± 0.14, p = 0.0025) (Figure 1A). In the old male fibroblasts, the F-to-G-actin ratio was also reduced almost by half when compared to the one observed in the young male cells (Figure 1B) (0.8 ± 0.02 in old cells vs. 1.80 ± 0.13 in young cells, p < 0.0001). We also examined these F- and G-actin fractions for other actin isoforms potentially expressed in fibroblasts and/or myofibroblasts such as β-cytosolic, ϒ-cytosolic, and α-skeletal actin (Figure S1A). Under our experimental conditions, the α-skeletal isoform was barely detectable. In the young male cells, β-cytosolic and ϒ-cytosolic were evenly distributed between F- and G-actin pools (Figure S1A). However, in the old male cells, the signal intensity for both these cytosolic isoforms seemed stronger in the G-actin pool (Figure S1A). Moreover, young male cardiac fibroblasts appear to be larger in shape and exhibit a complex F-actin network, while most of the old male fibroblasts are smaller and spindle-shaped or even round-shaped (more likely due to their lower adhesion to the surface) (Figure 1C). In addition, in old fibroblasts, the F-actin cytosolic staining was not evenly distributed, it appeared weak and mostly detectable near the plasma membrane (Figure 1C). Linked together, these observations suggested that the fibroblasts isolated from the old male heart failed to respond to a stiffer environment (glass or plastic), resulting in a combined dysregulated actin treadmill dynamics, favoring actin depolymerization and a defective α-SMA expression.
Figure 1.

The aging phenotype of the male cardiac fibroblast is associated with a decreased expression of Kindlin-2
(A) Representative Western blot depicting the difference in α-SMA protein level between the young and old male cardiac fibroblasts (upper panel) and the diagram representing the quantification from separated experiments (N = 5–6 per group) (lower panel).
(B) Representative Western blot for the actin (“F” depicting F-actin/Polymerized, and “G” for globular/monomeric). Actin quantity was estimated by densitometry for each sample, and the final F-to-G-actin ratio is shown in an individual-plotted diagram on the lower panel (N = 9–10 per group).
(C) Representative F-actin staining for young and old male cardiac fibroblasts seeded on glass, scale bar: 20 μm. N = 10 individual samples from 4 separate experiments.
(D) Representative Western blot depicting the difference in Kindlin-2 protein level in the lysate from young versus old cultured cardiac fibroblasts. Normalized densitometry was shown on the lower panel as an averaged value of 6–8 separated experiments.
(E) Staining for Kindlin-2 (green) with a DAPI nucleus counterstaining (blue) for young and old cardiac fibroblasts, scale bar: 20 μm.
(F–H) Representative staining for F-actin (F), α-SMA (G) and Kindlin-2/α-SMA (H) in cultured old male and old female cardiac fibroblasts, seeded on glass. Representative pictures from 3 to 4 biological repeats. Scale bar: 20 μm.
(I) Western blot depicting the F-/G-actin fractions in the lysate from old female versus old male cardiac fibroblasts. The diagram (lower panel) shows the relative comparison of the F-to-G-actin. N = 4.
(J) Western blot demonstrating the level of Kindlin-2 and α-SMA in the lysates from young and old female cardiac fibroblasts. Normalized densitometry is depicted as an individual-dotted diagram for both Kindlin-2 (lower left) and α-SMA (lower right). P-value was shown with an "N.S." below the bar, indicating a non-significant difference. M denotes a molecular weight marker; the corresponding molecular weight is indicated on the right side of the blot.
(K) Dot-plots depicting the correlation between the level of Kindlin-2 or F-/G-actin ratio (y axis) versus α-SMA (x axis). The regression line shows a positive correlation in male cells (left panel) but not in female cells (middle panel). There is also a positive correlation between the F-/G-actin ratio and the level of α-SMA (right panel). Statistics: All the data are shown as an averaged mean ± SEM. P-value was calculated as an Unpaired t-test with Welch’s correction. p value <0.05 is considered statistically significant.
Kindlin-2 is one the main adaptors of the mechanosensing axis, as it usually bridges the integrin subunits to the actin fibers, hence translating ECM biomechanical tension to the cytoskeleton. Since the old cardiac fibroblasts are less responsive to their environment, we hypothesized that the expression of Kindlin-2 might be affected by aging.
The Western blot analysis showed that Kindlin-2 was significantly reduced in the old cardiac fibroblast (0.26 ± 0.04 in old vs. 1.0 ± 0.09 in young male cells, p < 0.0001) (Figure 1D upper and lower panels). The distribution of Kindlin-2 was then determined by immunostaining in cardiac fibroblasts isolated from young and old male mice (Figure 1E). In the young cells, the Kindlin-2 signal was intense, and by contrast, in the old cardiac fibroblasts, Kindlin-2 was weaker and restricted to the perinuclear area (Figure 1E).
We previously reported that the age-related phenotype of the cardiac fibroblasts is dimorphic and linked to the sex-specific changes differentially determining the ECM composition between the old male versus the old female heart.38 We thus decided to assess the ability of the old female cardiac fibroblasts to transition into myofibroblasts. Contrary to the old male fibroblasts, the old female fibroblasts harbored a dense network of actin fibers that were strongly enriched in α-SMA (Figures 1F and 1G). Both Kindlin-2 and α-SMA signals were stronger in the old female cells than in the age-matched male cells (Figure 1H). Likewise, the old female cells had a higher yield of F-actin than G-actin (1.40 ± 0.12, p = 0.0006) and an overall higher content of actin than the old male cells (Figure 1I). Lastly, the levels of Kindlin-2 and α-SMA proteins were not significantly changed between the old and young female cardiac fibroblasts (0.84 ± 0.35 vs. 1.0 ± 0.05, p = 0.083 and 1.48 ± 0.21 vs. 1.0 ± 0.04, p = 0.1, respectively) (Figure 1J).
We also plotted α-SMA and Kindlin-2 respective protein levels, and we demonstrated that there was a strong correlation between them for the male (R2 = 0.75), but not for the female cardiac fibroblasts (R2 = 0.0009) (Figure 1K, left and middle panels). Similarly, we found a strong positive correlation between α-SMA protein level and F-/G-actin ratio values in the old male cells (R2 = 0.72) (Figure 1K, right panel). This demonstrates co-dependence between Kindlin-2 and α-SMA or Kindlin-2 and F-/G-actin expressions in the male cardiac fibroblasts.
Interestingly, the distribution pattern of the actin isoforms in the old female cells was also close to the one described for the young male cells (Figure S1B): α-SMA was mainly found in the F-actin fractions, while β-cytosolic and γ-cytosolic non-contractile isoforms were equally represented in both polymerized and depolymerized actin pools.
Since old female cardiac fibroblasts preserve their Kindlin-2 expression and they are able to mature into myofibroblasts, we focused our investigation on the male cardiac fibroblasts.
Cardiac ECM composition impacts on Kindlin-2 expression and distribution
Uncoated plastic or glass dishes promote supra-physiological conditions that may not recapitulate cardiac fibroblast phenotypic changes that occur in physiological or pathophysiological conditions.39,40,41 Therefore, we decided to perform ex vivo tridimensional (3-D) cell culture by seeding fibroblasts onto decellularized cardiac matrices and culturing the 3-D scaffolds in bioreactor vessels. Under these 3-D culture conditions, the fibroblasts adopt a more physiological phenotype, as they can infiltrate the cardiac tissue scaffold and respond to the natural 3-D organization and tension of the ECM. Cardiac fibroblasts were seeded on age-matched or age-unmatched (“matrix swapped”) 3-D matrices.
Three days after seeding, we found that the Kindlin-2 signal was intense and mainly cytosolic in the young cells cultured on young matrices, but weaker and mainly perinuclear in old fibroblasts seeded on old matrices (Figure 2A). By contrast, when matrices were swapped, the Kindlin-2 signal mislocation was corrected in the old cells cultured on young cardiac matrices, whereas the young fibroblasts grown on old matrices exhibited a partial mislocation of Kindlin-2 signal (Figure 2A). Old fibroblasts grown on young matrix exhibited an increased signal for α-SMA, compared with cells seeded on age-matched donor matrix, suggesting a better myofibroblast maturation. However, the young cells similarly exhibited a strong expression for α-SMA, regardless of the origin of the decellularized matrices, suggesting that they are able to correct the defect via an alternative pathway.
Figure 2.

The impact of cardiac ECM on Kindlin-2 expression
(A) Representative immunostaining of Kindlin-2 (red) and α-SMA (green) for tridimensional (3-D) culture of cardiac fibroblasts seeded on cardiac matrices from age-matched or un-matched mouse donors for 72 h. Representative pictures from 3 to 4 experiments per group (young vs. old) and two donors (young vs. old) per experiment. Note that the old male matrices tend to exhibit a more intense autofluorescence. Scale bar: 20 μm.
(B) Immunostaining for α-SMA (green) and F-actin (red) in old male cardiac fibroblasts seeded on glass (left), on matrix from old male cells (middle) or matrix from young male cells (right) (scale bar: 20 μm). Nuclei were counterstained with DAPI.
(C) Representative Western blot of Kindlin-2 expressed by cells seeded on age-matched and unmatched matrices in the 2-D experiment. The normalized dataset is shown in an individual-plotted diagram on the right panel (N = 5–6 per group).
(D) Representative Western blot showing the level of Kindlin-2 expressed by old male and old female fibroblasts seeded on sex-matched or unmatched matrices. The normalized dataset is shown in an individual-plotted diagram on the right panel (N = 4–5 per group). YM, OM, OF denote the young male, old male, and old female, respectively. M denotes a molecular weight marker; the corresponding molecular weight is indicated on the right side of the blot.
(E) Scanning electron microscopy pictures of decellularized 3-D matrices from young or old mouse male hearts (upper panel), young or old female hearts (middle panel) or decellularized 2-D matrices deposited by young or old male cardiac fibroblasts (lower panel). Red arrows indicate cleavage sites in the old male cardiac ECM, and cyan circles depict aggregates in the old female cardiac ECM. Scale bar: 2 μm. Pictures in enlarged frames (yellow squares) depict the presence of cleavage in the old male ECM and aggregates in the old female ECM. All the data are shown as an averaged mean ± SEM. For the experimental dataset involving 4 groups, Brown-Forsythe and Welch one-way ANOVA was used to test the significant difference among the four groups, to determine the significant difference among groups. p value <0.05 is considered statistically significant.
Then, we designed a two-dimensional (2-D) method for matrices deposited by cultured cells. In this protocol, 2-D matrices were directly deposited from “donor” cardiac fibroblasts, which were then removed, leaving the matrix.42
Cardiac fibroblasts from young or old male hearts were then seeded on age-matched or -unmatched 2-D matrices for 5 days. First, when seeded on glass coated with old 2-D matrices, the old fibroblasts exhibited a more regular shape, suggesting a better adhesion. Yet, the signal for F-actin and α-SMA remained especially weak (Figure 2B). By contrast, the same old cells cultured on young matrices exhibited a strongly noticeable F-actin network that was especially enriched in α-SMA (Figure 2B).
Then, while investigating the Kindlin-2 expression by Western blot, as previously shown for cells seeded on plastic (Figure 1D), the young fibroblasts on young matrices expressed higher levels of Kindlin-2, compared to the old cells seeded on 2D matrices secreted from old fibroblasts (Figure 2C). When matrices were swapped (young fibroblasts seeded on old matrices, and vice versa), Kindlin-2 expression decreased in young cardiac fibroblasts seeded on old matrices (Figure 2C). Likewise, when old cells were cultured on young matrices, Kindlin-2 protein levels significantly increased to reach averaged values that were expressed by young fibroblasts seeded on young matrices (Figure 2C).
Then, to ascertain the role of ECM composition on male fibroblast maturation, old male cells were compared with old female cells, both as matrix donors and recipients (Figure 2D). Male fibroblasts cultured on the old male matrix exhibited a trend to a lower yield of Kindlin-2 than the old females seeded on the old female matrix. In a similar fashion, once seeded on the old female matrix, Kindlin-2 expression was significantly increased in old male fibroblasts. However, the level of Kindlin-2 remained unaffected in the old female cells, regardless of the origin of the matrix (Figure 2D).
In a preceding study, we already demonstrated that ECM in the old male and female hearts differs in composition and ultrastructure organization.38 To identify the components of the ECM that could especially affect the old fibroblast phenotype, we performed mass spectrometry analysis from decellularized matrices directly prepared from mouse hearts (Figure S2A). We compared young vs. old male and young vs. old female cardiac ECMs, to decipher the changes in proteins and glycoproteins of the matrisome that are age-dependent and sex-specific to the ones that are aging-related but sex-independent.
As depicted on the Volcano plots, we identified an increase in Vitronectin as a main feature of the old cardiac ECM, both in male and female mice (Figure S2B, blue arrow). Then, our dataset indicated an age-related change in collagen composition that may be sex-specific. The old male matrix was indeed especially enriched in collagen VI (6a1, 6a2), Collagen IV (Col4a6), and two non-fibril-forming and atypical collagens. Interestingly, the level of Collagen XII also increased in the old male ECM, while it decreased in the old female ECMs (compared with sex-matched young mouse ECMs) (Figure S2B, white arrow). In addition to the collagens, we found that the old male ECM was enriched in Mfap4 (microfibril-associated protein 4), a protein usually located to the elastic fibers in the perivascular area of the heart, and depleted of von Willebrand factor C and EGF domain-containing protein (Vwce), a cytoplasmic protein potentially involved in β-catenin pathway. By contrast, the old female ECM had higher levels of matrix Gla protein (Mgp), peroxidasin homolog (Pxdn), but it was depleted in cartilage intermediate layer protein 2 (Cilp2).
Since these changes in collagen and glycoprotein composition may affect the ECM organization, we decided to investigate the ECM structure by scanning electron microscopy. We analyzed young vs. old male hearts (Figure 2E upper panel) and young vs. old female hearts (Figure 2E middle panels), aside with ECM secreted by young and old male cardiac fibroblasts (Figure 2E, lower panel). The ECM of the young male heart consisted of a network of thin fibrils connected with sparse thicker collagen fibers, whereas the old heart ECM was mainly characterized by the presence of many cleaved thin fibrils (Figure 2E, upper panel, red arrows). A similar cleavage pattern could be detected in the matrices generated in vitro by old male cardiac fibroblasts (Figure 2E, lower panel, red arrows). Regardless of age, the female ECM tends to be characterized by a denser network of thin fibrils that are more tightly connected together (Figure 2E, lower panel). The old female cardiac ECM did not strongly differ from the young female matrix, yet we did not observe any cleavage pattern as we repeatedly noticed in the cardiac ECM of the old male. By contrast, the old female cardiac ECM was characterized by the presence of amyloid-like aggregates along the fibers (Figure 2E, cyan circles). These findings support the concept of an impaired ECM-to-Kindlin-2 axis in old male cardiac fibroblasts.
Kindlin-2 can facilitate the old fibroblast-to-myofibroblast transition by promoting actin polymerization and α-SMA expression
To investigate the link between Kindlin-2 and the maturation profile of the cardiac fibroblasts, we then decided to manipulate Kindlin-2 expression. SiRNA interference was performed in young cardiac fibroblasts (“siK2”) to decrease the Kindlin-2 protein level, while old cells were transfected with a selective plasmid to promote Kindlin-2 overexpression (“K-OE”). At 72 h following transfection, siRNA transfected (siK2) young fibroblasts exhibited a drastic decrease in Kindlin-2 protein (0.30 ± 0.11 vs. 1.0 ± 0.031 in control scrambled RNA cells, p = 0.0025) (Figure 3A). Compared with control-transfected cells, these Kindlin-2 deficient cells were smaller in size. Their actin cytoskeleton was disorganized and characterized by a fewer number of F-actin fibers (Figure 3B). Interestingly, the remaining Kindlin-2 signal in siK2 cells was located in the perinuclear area (Figure 3B), as it is in the untreated old male cells (Figure 1D). When examining actin fractions prepared from siK2 transfected cells, an inversion of the F-to-G actin ratio was noticed in the siK2 fibroblasts, compared with control cells transfected with scrambled siRNA (Figure 3C) (0.65 ± 0.15 vs. 1.1 ± 0.076 in control scrambled cells, p = 0.024), and the actin expression in both fractions was also weaker in siK2 cells (Figure 3C). In the cells deficient for Kindlin-2, there was a modest decrease of β-cytoplasmic and ϒ-cytoplasmic in the F-actin fraction (Figure S1C).
Figure 3.

Kindlin-2 can facilitate the maturation of old fibroblasts by promoting actin polymerization and α-SMA expression
(A) Western-blot depicting a decrease in Kindlin-2 protein levels 72 h post-Kindlin-2 siRNA (siK2) transfection. Averaged densitometry was shown as an individual-plotted diagram (lower panel).
(B) Representative pictures of Kindlin-2 immunostaining and F-actin staining, scale bar: 30 μm.
(C) Representative Western blot of actin fractions in siK2- compared with control (scrambled)-transfected cells. The F-G actin ratio was calculated for each experimental sample and depicted in a mean ± SEM diagram (N = 5 per each experimental group).
(D) Staining of F-actin shown in false color using the OrientationJ plugin (ImageJ algorithm) depicting the changes in cell polarization and actin fiber orientation in control young male cells (left) and in Kindlin-2 deficient cells (right). Scale bar: 20 μm.
(E) Representative Western blot showing the increase in α-SMA protein levels in response to Kindlin-2 overexpression (K-OE). Densitometry (lower panel) is provided in an individual-plotted diagram for both Kindlin-2 and a-SMA (N = 3–5 per group). For each experiment, cells were transfected with either K-OE or CMV control).
(F) Immunostaining of Kindlin-2 in K-OE and CMV control clones (scale bar: 20 μm). Representative picture from 3 separate experiments.
(G) Representative Western blot of F-/G-actin fractions showing the enrichment of F-actin in K-OE lysates. The F-to-G actin ratio was calculated and plotted in a diagram (right panel N = 3–5 per group). M denotes a molecular weight marker; the corresponding molecular weight is indicated on the right side of the blot.
(H) Staining of F-actin shown in false color using the OrientationJ plugin (ImageJ algorithm) depicting the changes in cell polarization and actin fiber orientation in control old male cells (left) and in Kindlin-2 overexpressing cells (right).
(I) Staining for F-actin (red) and α-SMA (green) in K-OE cells seeded on glass, or in 2-D matrices, compared with old control cells seeded on glass (scale bar: 20 μm). Representative picture from 3 separate experiments. See also Figure S1. p value <0.05 is considered statistically significant.
In addition, the orientation of the F-actin fibers was assessed using OrientationJ plugin43 (Figure 3D). This indirect technique was developed to determine the overall cell orientation between either “polarized” (mono-color) or “not polarized” (multicolor) cells, which is highly dependent on ECM and the actin cytoskeleton and dynamics, as actin polymerization may promote optimal cell polarization. As demonstrated, mature young fibroblasts trended to harbor a unique monocolor “phenotype,” suggesting a specific polarization/direction, whereas siK2-transfected fibroblasts had several multicolor features, hence no specific direction (Figure 3D). This loss of directional orientation was also detected in old control cardiac fibroblasts (Figure 3H, “Control”).
These observations suggested that the loss of Kindlin-2 in young fibroblasts recapitulated some defects observed in the old male cardiac fibroblasts.
Then the reverse approach was performed by transfecting old male cardiac fibroblasts with a selective Kindlin-2 overexpressing plasmid (K-OE). Control cells were prepared at the same time and transfected with CMV-neomycin empty plasmid (CMV) instead. First, we found that the increase in Kindlin-2 was associated with a modest but significant increase in α-SMA protein level (1.36 ± 0.017 vs. 1.0 ± 0.05 in control cells, p = 0.0192) (Figure 3E). The increase in Kindlin-2 expression was also clearly discernable by immunostaining (Figure 3F). The signal was stronger and distributed in the cytosol, compared with a perinuclear stain usually observed in old male cells (Figure 3F). In addition, the F-to-G-actin ratio was also significantly higher in K-OE cells (1.14 ± 0.06 vs. 0.74 ± 0.04 in control cells, p = 0.0012), indicating that actin polymerization increased in response to Kindlin-2 overexpression (Figure 3G). The increased polymerization applies to all the expressed actin isoforms (α-SMA, and β- and ϒ-cytoplasmic), but α-SMA was especially enriched in the F-actin fraction (Figure S1D). By using the OrientationJ plugin, we could also determine that the polarization of the K-OE fibroblasts also improved compared with their age-matched controls (Figure 3H).
K-OE and control clones were then used for the 2-D matrix-swap experiment (using non-manipulated young and old males as matrix donors). In accordance with our previous observations (Figures 1C, 1D, and 2E), control old fibroblasts poorly adhered to the glass, which affected their morphology (Figure 3I). Under these conditions, only a few α-SMA+ cells could be detected, and most of these cells also had a weak F-actin signal that barely formed discernible actin fibers. Compared with this observation, the α-SMA signal was weak and sporadically distributed in control cells seeded on the old matrix, but stronger yet sparse in response to the young matrix (Figure 3I). By contrast, K-OE cells adhered more firmly and developed a denser F-actin network, regardless of the composition of the support (glass versus old or young 2D-matrix). KOE cells exhibited a strong signal for both F-actin and α-SMA once seeded on the young matrix (Figure 3G). However, interestingly, when K-OE cells were cultured on an old donor matrix, both F-actin and α-SMA signals were slightly weaker. This observation suggests negative feedback from the ECM that could antagonize the benefits of Kindlin-2 overexpression and/or affect actin treadmill dynamics.
The aging-associated deleterious activation of the MAP kinase ERK can be rescued by Kindlin-2 overexpression in the old cells
The aging process is associated with profound disturbances of intracellular signaling pathways. The MAP Kinases ERK1/2 represent the main effector of cell signaling that can be activated in response to mechanical stress44,45 and activated ERK1/2 can indirectly promote actin depolymerization.46,47,48 In this study, we accumulated evidence that actin polymerization is impaired in the old cardiac fibroblasts, and so is Kindlin-2 distribution and expression. Therefore, we hypothesized that the defective ECM/Kindlin-2/actin axis in the old male cardiac fibroblasts might imply an exacerbated activation of ERKs.
The level of phosphorylated ERK was investigated by Western blot using lysates obtained from cultured young and old cardiac fibroblasts. The old male fibroblasts were characterized by a significant (almost 3-fold) increased P-ERK/ERK ratio when compared with young male fibroblasts, while the P-ERK/ERK ratio was low and unchanged in young vs. old female cells (Figure 4A). To ascertain the involvement of this overactivation of ERK1/2 in the phenotype of the old cardiac fibroblasts, we treated old male cardiac fibroblasts with 5 μM Temuterkib (LY3214996, an ERK1/2 specific inhibitor) for 3 h or 72 h, to measure the impact on actin polymerization rate and the resulting potential changes on the level of α-SMA and Kindlin-2 expressions. First, we noticed that 3 h of ERK inhibition promoted actin polymerization as the F-to-G-actin ratio significantly increased, compared to control DMSO-treated old cells (0.77 ± 0.04 vs. 0.46 ± 0.11 in DMSO control cells) (Figure 4B). But, we also noticed the overall level of actin tends to be higher in cells treated with Temuterkib, both in F- and G-actin fractions. This may thus suggest a faster increase in actin expression that may underestimate the net impact on actin polymerization. After a 72 h-long culture with the inhibitor, the α-SMA protein level was also significantly increased in the old male fibroblasts treated with the ERK1/2 inhibitor (1.77 ± 0.15 vs. 1.0 ± 0.15) (Figure 4C). Interestingly, this induction of α-SMA expression was equivalent to the one induced by Kindlin-2 overexpression in K-OE cells (Figure 3D). Yet, the Kindlin-2 protein level itself was not significantly affected by ERK inhibition (Figure 4C). Both of these observations were confirmed by the immunofluorescence staining of α-SMA and Kindlin-2 on old treated cells (Figures 4D and 4E). There is no direct transcriptional feedback between ERK activation and Kindlin-2 expression. Furthermore, we noticed that the incubation with Temuterkib promoted Kindlin-2 relocation to the plasma membrane next to the extremity of the F-actin fibers (Figure 4E, middle panel).
Figure 4.

The aging-associated deleterious activation of ERK can be rescued by Kindlin-2 overexpression in the old cells
(A) Representative Western blot showing the difference in phosphorylated ERK1/2 (P-ERK) versus total ERK1/2 protein levels in the lysate from old and young male cardiac fibroblasts. Densitometry was presented on an individual-plotted diagram (N = 5–6 per group).
(B) Representative Western blot depicting changes in F- and G-actin fraction enrichment in the cells cultured with 5 μM Temuterkib (ERK1/2 specific inhibitor, also known as LY3214996, “ERK-LY”) or DMSO (control, “-”) for 3 h. The F-to-G-actin ratio was calculated (lower panel) (N = 3–5 per group).
(C–E). Representative Western blot showing the change in α-SMA and Kindlin-2 in the lysate of old male cells treated for 72 h with 5 μM LY3214996 (ERK-LY, “+”) or DMSO (control, “-“). For each protein, the normalized densitometry of each individual value is plotted in a diagram (lower panel) (N = 4–6 per group, “ERKinh” depicts cells treated with 5μM Temuterkib). Staining for F-actin (red)/α-SMA (green) (D.) and Kindlin-2 (green)/F-actin (red) (E.) on cells treated with 5 μM Temuterkib (ERK inhibitor) or DMSO (control) for 72 h. In E., arrows indicate the location of Kindlin-2 at the plasma membrane, and “N” its perinuclear location. Scale bar: 20 μm. Pictures in enlarged frames (yellow square) depicted the changes in Kindlin-2 localization between control and Temuterkib-treated cells. White arrows indicate focal adhesion localization, while “N” depicts nuclear/perinuclear localization of Kindlin-2 (Scale bar: 5 μm).
(F) Representative Western blot depicting the level of phosphorylated ERK1/2 (P-ERK) and ERK1/2 in the lysate of pre-selected clones overexpressing Kindlin-2 (“K-OE + ”) or a control plasmid “K-OE -“). The P-ERK to ERK ratio was calculated and shown in an individual-plotted diagram (N = 6 per group). M denotes a molecular weight marker; the corresponding molecular weight is indicated on the right side of the blot.All the data are shown as an averaged mean ± SEM. Statistical significance was challenged by an unpaired t-test with Welch’s correction. p value <0.05 is considered statistically significant.
This last dataset suggested that ERK acts downstream of Kindlin-2 and actin, affecting actin polymerization, which promotes α-SMA expression. Interestingly, the level of P-ERK was significantly decreased in old cells overexpressing Kindlin-2 (0.38 ± 0.02 vs. 1.0 ± 0.13 in CMV control cells) (Figure 4F), while total ERK trended to increase (2.01 ± 0.19 vs. 1.0 ± 0.07 in CMV control cells, p = 0.24). Kindlin-2 deficiency may thus favor ERK overactivation in old cardiac fibroblasts.
Discussion
Mechanosensing is a complex regulatory axis that promotes the cell response to ECM tension. It coordinates tissue growth, homeostasis, and integrity throughout the whole lifetime, since the earliest steps of embryonic development.49 Therefore, defective ECM-to-cells crosstalks may also be involved in pathophysiological processes. In the myocardium, mechanical forces affect both the cells and the matrix.50,51 While the cardiomyocyte contraction generates a combination of active and passive forces to the ECM,52 the fibroblasts ensure the optimal tuning of the ECM’s properties.53,54 The biochemical signature of ECM can be modified to respond to any stress that could impair tissue integrity.
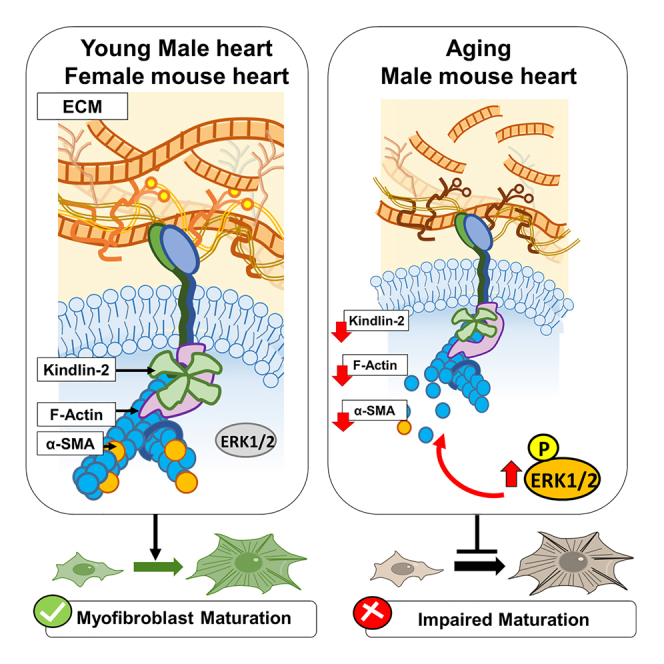
Yet, although suspected to be pivotal, the impact of mechanosensing on the aging phenotype of the heart remains unclear. In addition, while sex-specific differences are extensively reported in the pathogenesis of the aging heart, little is known regarding the effectors that contribute to this dimorphism. Here we described an ECM/Kindlin-2/actin/ERK1/2 axis, as a main part of the mechanosensing that is defective in the old cardiac fibroblast phenotype, impairing their maturation into myofibroblast (Figure 5).
Figure 5.

The ECM/Kindlin-2/ERKs/actin axis in the mechanosensing-dependent maturation of the cardiac fibroblasts
p value <0.05 is considered statistically significant.
Extracellular matrix
In this study, we demonstrated that the origin of a 2-D or 3-D matrix may impact the fibroblast-to-myofibroblast maturation. Previously, we already determined that the protein and glycoprotein composition of the aging heart differ between the old male and the old female heart, which may affect the fibroblast phenotype.38 In this newly generated mass spectrometry analysis, we identified age- and sex-specific ECM changes that may explain the effect of matrix on cardiac fibroblasts.
Interestingly, the old male cardiac ECM is characterized by a specific enrichment in atypical collagens (collagen IV, VI, and XII) that usually plays a critical role both in mechanotransduction and ECM remodeling. As a main component of the basement membrane, collagen IV is at the interface between the fibril-forming collagens of the ECM and the focal adhesions. Noteworthy, Integrins α1β1 have a higher affinity for collagen IV, and this facilitates ERK activation downstream.55 On the other hand, collagen XII and collagen VI are both reported to bridge collagens I and III, as well as collagen IV (directly or indirectly). In addition, evidence from previous studies also demonstrated that they colocalize and have synergetic functions in tissue remodeling, during development, or in response to injury.56 Collagen XII expression seems to be especially sensitive to mechanical tension,57 and its preferential location in elastic tissue (such as a tendon) suggests that it can either facilitate or absorb the ECM mechanical tension.58 Consequently, in the old male cardiac ECM, the enrichment of these collagens, and especially collagen IV and XII, may disconnect the ECM signaling to the cardiac fibroblasts downstream while potentially facilitating injury-like ECM remodeling dynamics.
In addition to collagens, our matrisomal analysis also identified Mfap4 in the old male matrix. Mfap4 is reported to be especially expressed in cardiac fibroblasts, and it is involved in TGF-β signaling, and FAK and ERK activation.58,59 It is thus surprising to find its enrichment within the old male ECM, in which both the TGF-β pathway and integrin signaling seem to be impaired,31 suggesting a potential compensatory mechanism. Our dataset also detected a depletion in Vwce in the old male cardiac ECM. Since Vwce can be a regulator of the β-catenin pathway, and that it can bind to TIMP3, a tissue inhibitor of metalloproteases,59,60 both of these findings suggest the possibility of a regulatory feedback or an adaptive role of Vwce in the mechanosensing regulation. Yet, this protein has been barely studied in cardiac fibroblasts, so its function should be the subject of further investigation.
Therefore, the aging process may favor injury-like remodeling and lesser maturation in the old male cardiac ECM, which may disconnect the fibroblast to the ECM, impairing the mechanosensing signaling downstream. By contrast, collagen crosslinking of the old female ECM may reinforce the ECM-to-cell connection, hence facilitating fibroblast activation and maturation. This interpretation is also in accordance with our observations in scanning electron microscopy.
Kindlin
Kindlin-2 is a member of the FERM family and the main one expressed in the heart.61,62 At the focal adhesion, Kindlin-2 mainly works as an adaptor for the integrins, as it can bridge the subunits to the actin cytoskeleton, which favors (and even reinforces) their downstream signaling.63,64,65 As suggested by both total and conditional knockout mouse models, Kindlin-2 expression is essential in the myocardium from the earliest stages of cardiogenesis.66,67 The postnatal cardiomyocyte-specific loss of Kindlin-2 causes severe fibrosis, followed by the development of heart failure.68 There is, thus, a strong correlation between cardiac homeostasis and Kindlin-2 expression. While its involvement in cardiac fibroblast-to-myofibroblast maturation has been suggested by several studies,69,70 how Kindlin-2 is affected in the aging heart remained unknown.
By using ex vivo and in vitro approaches, our study demonstrated that Kindlin-2 is strongly sensitive to ECM composition. The fibroblasts of the old male heart have a decreased expression of Kindlin-2 compared with the young male cells. By seeding these cells on age-unmatched matrices (in a matrix swap experiment), Kindlin-2 expression significantly increased in the old cells seeded on young matrices, but it decreased in young cells cultured on old matrices.
Artificial manipulation of Kindlin-2 (siK2 young cells versus K-OE old cells) also highlighted how Kindlin-2 regulation determines the fibroblast maturation capacity. The loss of Kindlin-2 in siK2-transfected young male cells can recapitulate part of the defect of the old male cells, while constitutive Kindlin-2 overexpression in old cells can rescue actin polymerization, favoring α-SMA expression and their later response to a stiffer matrix. Interestingly, this defect was only observed in the old male hearts/fibroblasts since old female fibroblasts can mature into myofibroblasts, their Kindlin-2 protein level was unaffected by age, and the female matrix can modestly increase Kindlin-2 expression in the old male fibroblasts. Thus, our datasets indicate that Kindlin-2 represents a critical gauge of the mechanosensing regulatory signaling affecting cardiac fibroblast phenotype.
The net impact of Kindlin-2 on the myofibroblast program, and therefore on α-SMA expression, is more likely linked to a downstream impact on transcriptional factors. Among them, Serum Response Factor (SRF) is known to be involved in cardiac fibrosis.71 SRF activity depends on two different families of co-activators that overlap but can antagonize each other72,73: Ternary complex factor that promotes cell proliferation and signaling (TGF-β, TNF-α, and mTORC); and the MRTF that is mainly involved in cell differentiation and maturation. Interestingly, the ternary complex factor (TCF) family is mainly ERK-activated,74,75 while the activity of the MRTF family is actin-regulated and repressed by G-actin.13,76 In this paper, we demonstrated that the cytosol of the old male fibroblasts is significantly enriched in both G-actin and overactivated ERK1/2, and these changes can be reversed by Kindlin-2 overexpression. These observations and others from the literature might thus suggest a critical role of Kindlin-2 in the myofibroblast program, beyond its function in the focal adhesion.
In a previous study, we identified Gli1 as a potential orchestrator of cardiac fibroblast phenotype.38 Briefly, we found that the AMP-activated protein kinase (AMPK)-dependent regulation of Gli1 expression is dimorphic, the female cells responding to AMPK agonist by a decrease in Gli1 protein and pro-fibrotic genes, but this was not the case in the male cells.38 Noteworthy, Gli1 is a transcriptional co-activator that is reported both as a target gene and a repressor of the Kindlin-2/β-catenin complex.77 In the old male cardiac fibroblasts, the maintenance of Gli1 activation may be involved that its repression of Kindlin-2 expression is preserved or exacerbated. Although some evidence may suggest an involvement of a Hedgehog/cAMP feedback, the potential relationship between Gli1 and Kindlin-2 in the old cardiac fibroblasts should be worth clarifying.
Actins
In quiescent fibroblasts, actin is localized mainly cortically, which preserves the overall cell shape; but it can be located at the podocytes or filopodia for optimal cell polarity and motility.78,79 Within mature myofibroblasts, actin represents the main component of the stress fibers that could contract the ECM.17 There are different actin isoforms that can polymerize into microfilaments. Distinct actin proteins can be found in F- and G-actin pools. As actin isoforms differ from each other by only a few amino acids,80 they usually harbor some functional redundancy. In addition, the expression of the actin-coding genes is inter-connected in such a way that a transcriptional change affecting one isoform (loss or gain of function) would affect the mRNA levels of the other isoform.81,82,83,84
Some suggested that α-SMA could be dispensable to fibroblast-to-myofibroblast maturation, as cells could compensate for the loss of Acta2 (α-SMA) by the expression of other actin isoforms, such as α-skeletal actin (Acta1).84 In our study, α-skeletal actin was not detected in the F- or G-actin fractions. However, here we were working under different conditions, aside from drastic stress such as genetic manipulation or in vivo injury, which may not promote the acute expression of Acta1.
Here we highlight the fact that regular actin stress fibers are a heterogeneous assemblage of three different actins: α-SMA, β-cytoplasmic, and ϒ-cytoplasmic (to a lesser extent). In the old male cells, F-actin depolymerization affected all three isoforms, but with a relatively better persistence of β-cytoplasmic actin. On phalloidin stainings performed on old male cells, F-actin is mainly detected at the periphery of the cell, just below the plasma membrane, which hence more likely corresponds to the cortical actin that is made of β-cytoplasmic actin. This may partly explain why the old male cardiac fibroblasts are able to maintain a spindle shape, smaller but still characteristic of fibroblasts, despite this significant loss of polymerized actin.
The manipulation of Kindlin-2 strongly affects the polymerization rate of all the isoforms, as well as the shape of the cells and their ability to mature into myofibroblasts. However, while Kindlin-2 overexpression may facilitate the old cells to transit into myofibroblasts, seeding the K-OE cells on old matrix trends to decrease the α-SMA immunostaining signal, which suggests that the ECM/Kindlin-2/F-actin/α-SMA axis is especially dynamic.
ERKs overactivation in the cardiac fibroblasts
ERK1/2 are effectors of the MAP Kinase pathway. They integrate the signaling from extracellular growth factors receptors, ECM, and inner compartments.45 The role of ERK in cell migration and proliferation has also been extensively studied for many years, including in various models of fibrosis.85,86,87 In this work, we found that ERK1/2 overactivation is directly responsible for the exacerbated actin depolymerization observed in the old male cardiac fibroblasts. This is in accordance with several studies that linked mechanical stress to ERK1/2 activation and actin depolymerization. In a striking manner, we found that Kindlin-2 overexpression is able to strongly decrease ERK1/2 phosphorylation in the old cells. It is notably reported that oxidative stress-triggered ERK activation is Kindlin-dependent,88 but the repressive activity of Kindlin-2 on ERK (as we showed in this study) was not clearly documented before. In the literature, there is no evidence of direct interaction between Kindlin-2 and ERK subunits. However, they both interact at the focal adhesion core through actin-binding proteins. In a model of retinal microglia vasculature, the loss of the Kindlin-3 isoform causes ERK1/2 mislocation, leading to its ectopic and over-phosphorylation.89 Our findings indicated that the loss of Kindlin-2 may indeed cause disorganization of the actin fibers, which would cause ERK activation in a similar fashion. Consequently, ERK activation may be more directly linked to the actin cytoskeleton and mechanosensation, the latter depending on Kindlin-2. Linked together, findings from our team and others reinforce the concept of more complex crosstalk between ECM and fibroblasts in which Kindlin-2/ERK coordination could play a crucial role in the mechanosensing-dependent signaling.
Lastly, another study suggests that cardiac fibrogenesis may be an ERK-dependent and SMAD3-independent process that is regulated upstream by glycogen synthase kinase 3 α (GSK3 α subunit).90 Interestingly, in pancreatic β-cells, the loss of Kindlin-2 favors GSK3-β activation, making GSK3 a potential substrate of Kindlin-2.91 The potential mechanism linking the decrease of Kindlin-2 expression with GSK3 and ERK1/2 activations will be examined in future studies.
Old female cardiac fibroblast
Unlike the male cells, female cells are able to bypass the ECM signal and maintain Kindlin-2 expression, which favors the maturation of fibroblasts into α-SMA-positive myofibroblasts. Noteworthy, the young female heart is characterized by better resistance to deleterious stress, such as myocardial infarction or pressure overload.92,93 However, paradoxically, females do have a trend to develop more severe cardiac fibrosis than males, especially during aging.94,95,96,97 It is possible that less-known characteristics of the female fibroblasts play an active role in these specific features: in fact, as suggested by our in vitro study, female fibroblast maturation is not affected by aging, which may thus facilitate scar formation after MI. But, with time, the accumulation of these activated fibroblasts may lead to more extensive fibrosis in the old female heart. In our study, K-OE cells exhibited a similar increase in α-SMA regardless of the composition of the matrix. Linked together, these findings once again highlight differences between male and female cells and responses to stress.
In conclusion, Kindlin-2 represents the main gauge of the mechanosensing downstream of the ECM and may be a pivotal orchestrator of the myofibroblast maturation program in the old male cardiac fibroblasts. In the aging male heart, strong negative feedback originating from the ECM decrease Kindlin-2 and exacerbate ERK1/2 activation leading to actin depolymerization and a decreased α-SMA expression. By contrast, the Kindlin-actin-α-SMA pathway is fully operant in the old female cells.
Limitations of the study
Mass spectrometry has identified several changes in the matrisome that point to age and sex differences. Although partial validation of these findings has been published by us before,38 further studies should follow to fully address the links between differentially expressed ECM proteins and the mechanosensing axis. In addition, while we demonstrated the respective impact of ECM on Kindlin-2 and Kindlin-2 on intracellular mechanosensing, the understanding of the mechanism may require further investigation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Kindlin-2 rabbit polyclonal | Thermofisher | RRID:AB_2262660 |
| FITC Conjugated anti-α-smooth muscle actin mouse monoclonal | MilliporeSigma | RRID:AB_476977 |
| Phalloidin–Tetramethylrhodamine B isothiocyanate | MilliporeSigma | RRID:AB_2315148 |
| Anti-α-smooth muscle actin mouse monoclonal (unconjugated) | MilliporeSigma | RRID:AB_262054 |
| Anti-P-ERK1/2 Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) rabbit polyclonal |
Cell Signaling | RRID:AB_331772 |
| Anti-ERK1/2 mouse monoclonal | Cell Signaling | RRID:AB_390780 |
| Anti-β-actin rabbit | Cell Signaling | RRID:AB_330288 |
| Anti-β-actin rabbit polyclonal | Proteintech | RRID:AB_2687938 |
| Anti-ϒ-actin rabbit polyclonal | Proteintech | RRID:AB_2223507 |
| Anti-α-skeletal actin mouse polyclonal | Proteintech | RRID:AB_2273608 |
| Anti-pan Actin Antibody | Novus Biologicals | RRID:AB_2222878 |
| IRDye® 800CW anti-Rabbit antibody | Li-Cor | RRID:AB_2651127 |
| IRDye® 800CW anti-Mouse antibody | Li-Cor | RRID: AB_2687825 |
| IRDye® 680RD anti-Rabbit antibody | Li-Cor | RRID: AB_10954442 |
| IRDye® 680RD anti-Mouse antibody | Li-Cor | RRID: AB_2651128 |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase type IV | Worthington | LS004188 |
| Dispase II | MilliporeSigma | D4818-2MG |
| DNAse | MilliporeSigma | D45131VL |
| PBS Ca2+/Mg2+ | ThermoFisher | 14190144 |
| PBS without Ca2+/Mg2+ | ThermoFisher | 14190250 |
| Antibiotic Antimycotic 100X | ThermoFisher | 15240062 |
| Mycoplasma removal agent (MRA) | Biorad | BUF035 |
| Temuterkib (Synonyms: LY3214996) | MedChem Express | HY-101494 |
| DMEM/F-12 | Gibco | 11320033 |
| Fetal Bovine Serum | Gibco | 10437–028 |
| TrypLExpress | Gibco | 12563011 |
| 0.05% trypsin-0.02% EDTA | Gibco | 25300054 |
| Lipofectamine RNAimax | ThermoFisher | 13778030 |
| FuGENE® HD Transfection Reagent | Promega | E2311 |
| OptiMEM Reduced Serum medium | ThermoFisher | 51985034 |
| Selective Antibiotic Geneticin™ (G418 Sulfate) | ThermoFisher | 10131035 |
| Tannic acid | ThermoFisher | 403040 |
| PIPES | Goldbio | P-281-100 |
| cytochalasin D | MilliporeSigma | C8273 |
| Intercept Blocking Buffer | LI-COR Biosciences | 927–60001 |
| Intercept® T20 (TBS) Antibody Diluent | LI-COR Biosciences | 927–65001 |
| Revert 700 nm Total Protein staining | LI-COR Biosciences | 926–11021 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | 11836170001 |
| PhosSTOP™ | Roche | 4906845001 |
| Critical commercial assays | ||
| SlowFade™ Diamond Antifade Mountant with DAPI | Molecular Probes | S36964 |
| BCA assay Protein concentration | ThermoFisher | 23227 |
| Deposited data | ||
| Original dataset, on Mendeley | This paper | |
| Mass Spectrometry Database | This paper | Data are available via ProteomeXchange (http://www.ebi.ac.uk/pride) with identifier PXD031223 |
| Mass Spectrometry Database | This paper and Angelini et al.38 | Data are available via ProteomeXchange (http://www.ebi.ac.uk/pride) with identifier PXD041660 |
| Mendeley Repository Database | This paper | https://doi.org/10.17632/4brzy98vw9.1 |
| Experimental models: Cell lines | ||
| Primary adult mouse cardiac fibroblasts | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57bl6/J - young male and female (3–4 months old) | Inner source | |
| C57bl6/J mice old male and old female (24–30 months old) | National Institute of Aging | |
| Oligonucleotides | ||
| Fermt2 Mouse siRNA Oligo duplexes | Origene | SR419055 |
| Trilencer-27 Universal Scrambled Negative Control siRNA Duplex | Origene | SR30005 |
| Fermt2 Mouse Untagged clone plasmid | Origene | MC202155 |
| PCMV6-Kan/Neo Untagged Cloning Vector | Origene | PCMV6KN |
| Software and algorithms | ||
| ImageJ | Schneider et al.105 | https://imagej.nih.gov/ij/download.html |
| OrientationJ plugin | Püspöki et al.43 | http://bigwww.epfl.ch/demo/orientationj/ |
| Image Studio Lite (version 5.2) | Li-COR Biosciences | Discontinued. Replaced by Empiria-Studio: https://www.licor.com/bio/empiria-studio/ |
| Prism version 9.2.0 | GraphPad | https://www.graphpad.com/ |
| Blender version 3.3 | Blender | https://www.blender.org/ |
Resource availability
Lead contact
For further information and requests for resources, please contact Dr. Katarzyna A. Cieslik (cieslik@bcm.edu).
Materials availability
This study did not generate new unique reagents (plasmids and siRNA are available from Origene). There are restrictions to the availability of the primary adult mouse cardiac fibroblasts because of their limited passage number for amplification.
Experimental model
Mice
Young (3-4 month-old) male and female C57BL/6J mice were obtained from Jackson Laboratory, and 21-month old female and male C57BL/6J mice were provided by the National Institute on Aging. All the mice were housed at the Center of Comparative Medicine of Baylor College of Medicine, under a 12 h day/night cycle, with food and water provided ad libitum. Young mice were used after one-week of acclimation time, and old mice were aged until they reached 24–30 months old. At the time of tissue collection, mice were placed under deep gas anesthesia (5% isoflurane) and directly euthanized by cervical dislocation. The absence of a toe reflex was verified before proceeding to a necropsy.
For histology, a slice of the heart corresponding to the mid-myocardium area was directly placed in Zinc-Tris fixative [0.1 M Tris-HCl pH 7.8, 0.05% calcium acetate (CH3COO)2Ca, 0.5% zinc acetate (CH3COO)2Zn, 0.5% zinc chloride ZnCl2], dehydrated, and embedded in paraffin.
All animals were treated following the NIH Guide for the Care and Use of Laboratory Animals (DHHS publication (NIH) 85-23, revised 1996) and approved by the Baylor College of Medicine Institutional Animal Care and Use Committee. Baylor College of Medicine is certified by USDA (Animal Welfare Act, certification number 74-R-0018).
Adult mouse cardiac fibroblasts
Fibroblasts were isolated from young (3–4 months old) and old (24–30 months old) male and female mouse hearts. The isolation method was performed, as described below (see STAR methods details). Cells were cultured in complete growth medium [DMEM/F-12 1:1, 10% fetal bovine serum, Antibiotic/Antimycotic 1X (ThermoFisher # 15240062)] in a 5% CO2 wet incubator at 37°C. The cultures are regularly tested for mycoplasma contamination using the LookOut Mycoplasma PCR Detection Kit (Millipore-Sigma # MP0035), and the results were negative. Cells prepared from old animals were also carefully checked for phenotype changing.
A unique identification number is provided to each cell line prepared, based on the following nomenclature “Mouse age (in months-old). Mouse ID_Sex”. For instance, 30.80_M refers to the cells from the 30 months-old mouse #80, which is a male. Fibroblasts are stored in a dedicated storage place in a liquid nitrogen tank, and the location of each vial is recorded in a folder.
Methods details
Isolation and culture of adult mouse cardiac fibroblasts
Cardiac fibroblasts were prepared as previously described.38 Briefly, the slice of the myocardium (mid-myocardium to apical) was cut into 1 mm3 –sized pieces and incubated in a digestion buffer containing 2 mg/mL Collagenase type IV (Worthington Cat#LS004188), 2 mg/mL Dispase (Sigma Cat# D4818-2MG) and DNAse (14.8 U/mL, prepared from Millipore-Sigma #D45131VL) in PBS Ca2+/Mg2+ (ThermoFisher #14190144). The supernatant was passed through a Falcon 40 μm strainer (Fisher-Scientific #08-771-1) and mixed with an ice-cold quenching buffer [2% fetal bovine serum (from HyClone #SH30071.03), 14.8 U/mL DNAse (prepared from Millipore-Sigma #D45131VL) in PBS without Ca2+/Mg2+ (ThermoFisher #14190250)]. The final cell suspension was spun down at 300 g to pellet the cells, washed once with sterile D-PBS, then resuspended in the complete growth medium [DMEM/F-12 1:1, 10% fetal bovine serum, Antibiotic/Antimycotic 1X (ThermoFisher # 15240062)]. After isolation, cells were left seeding undisturbed for 48 h, then were thoroughly washed twice with sterile D-PBS.
To prevent mycoplasma contamination, medium was supplemented with 0.5 μg/mL of Mycoplasma removal agent (Biorad #BUF035) for the first 2 weeks following the isolation.
Cell culture
Fibroblasts are cultured in a complete growth medium in a 5% CO2 wet incubator at 37°C. Once the cells reach no further than 70% confluency, passaging is performed by rinsing the plate once with PBS without Ca2+ and Mg2+ then applying TrypLExpress (Gibco # 12563011) for 5 min at 37°C. The cells are then pelleted by 300 g spin down for 5 min. The supernatant is discarded and the cell pellet resuspended in a volume of growth medium, according to the experimental need and/or the passaging dilution.
Before the experiment, primary fibroblasts were synchronized in serum-limited medium [DMEM/F-12, FBS 1%, antibiotics-antimycotics 1%]. Cells were dissociated with TrypLExpress (Gibco # 12563011).
For 3D culture: 5x105 cells were seeded on 3D matrices for 2 h at 37°C (without rotation). The assembled cells/matrices were placed into the biovessels and the motorized rotating system (Synthecon) within a tissue culture incubator. The rotation speed (rpm) was adjusted so that the tissue pieces remained in freefall. 3D cell culture was conducted for 72 h, with a medium change after 48 h.
Matrices invaded by the cells were then fixed, dehydrated and processed for histology.
Two-dimensional (2-D) matrices preparation from cultured cardiac fibroblasts
To prepare 2-D matrices from "donor" cells, experiment was performed following the protocol from Castaldo et al., 201342 with minor adjustments. Primary cardiac fibroblasts were seeded at 30–50% confluency. Depending on the later use of the matrices, cells were placed onto wells of a 6-well plate or in 4-well chamber slides. Twenty-four hours after maximal confluency was reached, cells were washed twice with sterile D-PBS and then placed in a decellularization buffer [20 mM NH4OH and 0.5% Triton in D-PBS] and for 10 min. The supernatant was gently discarded. The wells and chambers were rinsed once with D-PBS, then the plates were incubated with 7.4 U/mL of DNAse (in D-PBS) for 10–20 min at 37°C. The matrices were gently rinsed twice with D-PBS and stored at 4°C until ready to use.
Tri-dimensional (3-D) matrices preparation from mouse hearts
Cardiac 3-D Matrices were prepared as previously described.98 The heart was sliced in 1-2 mm thick cross sections, and the pieces were immediately snap-frozen in liquid nitrogen. After thawing, the tissue was soaked for 24 h with sterile water in a rocker at 4°C. Tissues were incubated in 0.05% trypsin-0.02% EDTA (Gibco #25300054) for 1 h at 37°C, and then placed in decellularization buffer (2% Triton X-100 and 0.8% ammonium hydroxide in sterile deionized water) for 24 h in a rocker at 4°C. The samples were rinsed in water for 48 h on a rocker at 4°C. Samples were finally incubated in 7.4 U/ml of DNase (Millipore Sigma #D4513) in D-PBS for 1 h at 37°C.
Cell transfection (siRNA interference, overexpressing plasmid, and clone selection)
To induce Kindlin-2 knockdown, young cardiac fibroblasts were transfected with 27-mer siRNAs (Origene) by using Lipofectamine RNAimax (ThermoFisher #13778030) following suppliers' recommendations. Briefly, cells were passaged and left growing overnight (10–12 h), so they reached 50–60% confluency on the day of the experiment. Then, Fermt2 Mouse siRNA Oligo duplexes (Origene #SR419055) were combined with Lipofectamine RNAimax in OptiMEM Reduced Serum medium (ThermoFisher #51985034) for 5 min at room temperature (final concentration 10 nM). These complexes were added directly into the culture growth medium, and incubation was performed for 72 h. Negative control cells were transfected with 10 nM of Trilencer-27 Universal Scrambled Negative Control siRNA Duplex (Origene cat#SR30005).
To ensure Kindlin-2 overexpression, old cardiac fibroblasts were transfected with 2 μg of Fermt2 Mouse Untagged clone plasmid (Origene #MC202155) by using FuGene transfection reagent. The complexes were assembled in OptiMEM Reduced Serum medium (ThermoFisher #51985034) and incubated for 10 min at room temperature (final concentration 10 nM). Control cells were transfected with 2 μg of PCMV6-Kan/Neo Untagged Cloning Vector (Origene #PCMV6KN), following the same protocol.
Clone selection was conducted for both sub-cultured Kindlin-2 overexpressing (K-OE) and transfection control cells by using 300 μg/mL of Selective Antibiotic Geneticin (G418 Sulfate) (ThermoFisher #10131035). Due to the lower growth rate of old cardiac fibroblasts (compared with regular cell lines undergoing transfection), the selection was extended for up to 2–3 weeks.
Scanning electron microscopy
The matrices from both mouse hearts and fibroblasts were prepared for scanning electron microscopy. Fibroblasts were grown on EM grids (Ted Pella) and after 5 days in culture cells were removed (see 2-D matrices preparation). Then matrices were fixed as we described below.
For visualization of the myocardial ECM, the samples were prepared as previously described.99 Briefly, a 1 mm3 piece of heart (cut at left ventricular mid-myocardium area) was fixed in 4% glutaraldehyde for 1 h, rinsed with distilled H2O and decellularized in 10% (m/vol) aqueous NaOH for 6 days. Next, the tissue was again washed with water, then stained with 1% (m/vol) tannic acid (ThermoFisher #403040) for 4 h. After a 12 h-long washing, matrices first underwent osmication and dehydration and then were dried and coated with 5 nm of platinum using an argon ion beam sputter coater. Image acquisition was performed using an FEI Nova NanoSEM 230 at the Houston Methodist Electron Core Microscopy.
Immunostaining
Immunostaining was conducted on cells cultured in chamber slides, we used 2% PFA for 10 min as a fixative. Cells were then permeabilized with Triton X-100 0.5% in PBS for 10 min, and blocking was performed for 60 min in PBS Ca2+/Mg2+ buffer containing 0.1% NaN3, 0.1% Tween 20, and 1% Bovine Serum Albumin. Incubation was conducted overnight (12–16 h) with the primary antibodies targeting the protein of interest (dilutions and references are provided in the table below). Washing steps were performed in Triton X-100 0.1% in PBS thrice for 10 min and staining with a secondary antibody was performed if needed.
Slides were all mounted with coverslips using SlowFade Diamond Antifade Mountant with DAPI (Molecular Probes #S36964).
Picture acquisition was performed using an Olympus Provis-AX70 microscope and a QImaging Retina 2000R camera (x10 and x20 objective lens) on QCapture Pro 6.0 software. RGB stacking and channel merging (from greyscale originals) was performed using ImageJ 1.52a version. The experimenter was blinded during the image acquisition.
| Antibody/reagent | Specie | Provider | Cat# | RRID | Dilution |
|---|---|---|---|---|---|
| Anti-Kindlin-2 | Polyclonal rabbit | Thermofisher | 11453-1-AP | RRID:AB_2262660 | 1:200 |
| Conjugated anti-α-SMA | Mouse monoclonal (conjugated) | MilliporeSigma | F3777 | RRID:AB_476977 | 1:200 |
| Phalloidin–Tetramethylrhodamine B isothiocyanate | N/A | MilliporeSigma | P1951 | RRID:AB_2315148 | 1:200 |
Actin fractionation
Cells were lysed directly in 100 μL of actin stabilization buffer [0.1M PIPES(pH 6.9), 1 mM MgSO4, 1 mM EGTA, 1 mM ATP, 30% glycerol, 5% DMSO, 1% Triton X-100, pH 7–8, containing cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail (Roche #11836170001) and PhosSTOP (Roche #4906845001)]. The homogenate was incubated for 15 min on ice. The lysate was then centrifuged at 300 g at 4°C to remove insoluble particles.
The F-actin and G-actin of the cell lysates were separated by centrifugation at 16,000 g for 45 min at 4°C. After centrifugation, the supernatant (G-actin) was collected into a fresh tube, while the pellet (insoluble F-actin) was resuspended in an ice-cold actin depolymerization buffer [0.1M PIPES, 1 mM MgSO4, CaCl2 0.01M, pH 7–8, containing 1 mM cytochalasin D (Sigma-Aldrich #C8273) and proteases/phosphatase inhibitors cocktail (cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail #11836170001 and PhosSTOP #4906845001, both from Roche)]. The resuspended pellet was incubated on ice for 15 min to make it soluble for Western blot.
Cell lysate
Cells were washed twice with ice-cold D-PBS and directly homogenized in a cell lysis buffer [HEPES 10 mM, KCl 10 mM, EDTA 0.1 mM, and NP-40 0.5%] containing 1X antiproteases/antiphosphatases (cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail #11836170001 and PhosSTOP #4906845001, both from Roche).
Western blot
Migration and transfer were performed under denaturing conditions. The nitrocellulose membranes were blocked for 1 h with Intercept Blocking Buffer at room temperature (LI-COR Biosciences #927–60001) and blotted with the primary antibodies diluted in Intercept T20 (TBS) Antibody Diluent (Li-Cor #927–65001) for 12 h (at 4°C) (details of antibody dilution provided in the table below). Secondary antibodies (anti-rabbit IgG 800 nm (Cat# 926–32211, RRID:AB_621843) and anti-mouse IgG 680 nm (Cat# 926–68072, RRID:AB_10953628), both from LI-COR Biosciences) were diluted at 1:20,000 and incubated for 1 h. Washing steps were performed at room temperature using PBS-Tween-0.1% or PBS. Infrared detection was performed using an Odyssey Imager (LI-COR Biosciences). Quantification was done by densitometry using Image Studio Lite (version 5.2) and normalized to the signal for total protein staining (performed before blocking using Revert 700 nm Total Protein staining from LI-COR Biosciences (Cat# 926–11021)).
| Antibody/reagent | Specie | Provider | Cat# | RRID | Dilution |
|---|---|---|---|---|---|
| Anti-Kindlin-2 | Polyclonal rabbit | Thermofisher | 11453-1-AP | RRID:AB_2262660 | 1:1000 |
| Anti-α-smooth muscle actin | Mouse monoclonal | MilliporeSigma | F3777 | RRID:AB_262054 | 1:3000 |
| Anti-P-ERK1/2 Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) |
Rabbit | Cell Signaling | #4376 | RRID:AB_331772 | 1:1000 |
| Anti-ERK1/2 | Mouse | Cell Signaling | 4696 | RRID:AB_390780 | 1:2000 |
| Anti-β-actin | Rabbit | Cell Signaling | 4967 | RRID:AB_330288 | 1:3000 |
| Anti-β-actin | Rabbit | Proteintech | 66009-1-Ig | RRID:AB_2687938 | 1:3000 |
| Anti-ϒ-actin | Mouse | Proteintech | 11227-1-AP | RRID:AB_2223507 | 1:3000 |
| Anti-α-skeletal actin | Mouse | Proteintech | 17521-1-AP | RRID:AB_2273608 | 1:3000 |
| IRDye® 800CW anti-Rabbit antibody | Goat IgG Secondary | Li-Cor | 926–32211 | RRID:AB_2651127 | 1:10,000 |
| IRDye® 800CW anti-Mouse antibody | Goat IgG Secondary | Li-Cor | 925–32210 | RRID: AB_2687825 | 1:20,000 |
| IRDye® 680RD anti-Rabbit antibody | Donkey IgG Secondary | Li-Cor | 926–68073 | RRID: AB_10954442 | 1:10,000 |
| IRDye® 680RD anti-Mouse antibody | Goat IgG Secondary | Li-Cor | 925–68070 | RRID: AB_2651128 | 1:20,000 |
Mass spectrometry
We followed the detailed protocol published by Angelini et al.38 Briefly, the decellularized hearts were digested using LysC and Trypsin. Peptides were subjected to C18 cleanup using a C18 disk plug (3M Empore C18) and dried in a speed vac. Then, LC-MS/MS analysis was carried out using an EASY-nLC 1200 system (Thermo Fisher Scientific, San Jose, CA) coupled to Orbitrap Lumos ETD mass spectrometer (Thermo Fisher Scientific, San Jose, CA). Obtained MS/MS spectra were searched against target-decoy Mus musculus NCBI RefSeq protein database in the Proteome Discoverer (PD2.1, Thermo Fisher) with Mascot algorithm (Mascot 2.4, Matrix Science100). The peptides identified from the mascot result file were validated by Percolator101 and were thresholded at a 5% false discovery rate (FDR). The gene product inference and iBAQ-based quantification was carried out using the gpGrouper algorithm.102 The median normalized iBAQ values were used for data analysis.103 The differentially expressed proteins were calculated using the moderated t-test to calculate p values and log2 fold changes in the R package limma. The FDR corrected p value was calculated using the Benjamini-Hochberg procedure.104
Microfilament orientation
Microfilament orientation was determined using OrientationJ plugin in ImageJ, based on the method described by Püspöki et al.43 The gradient-based estimating algorithm generates multicolor pictures from original grayscale pictures of phalloidin-TRIC stainings, one color representing one orientation degree (between 0° and 180°) of the F-actin filaments.
Figure preparation
Immunostaining and Western blot files were processed using ImageJ.105 For the graphical abstract, the first layout was made on 3D software (Blender version 3.3) using a WACOM digital tablet and Powerpoint.
Quantification and statistical analysis
Statistics
Data are shown as a mean ± standard error of the mean (SEM). For the comparison of two groups, an unpaired Student’s t-test was performed, followed by Welch’s correction. When the statistics analysis involved more than two groups, one-way ANOVA was performed, followed by a post hoc Brown-Forsythe and a Welch analysis. Statistics were run on GraphPad Version 9.2.0. Statistical details of the experiments (including the replicates and what n number represents) can be found in the legends of the figures, while means ± SEM and P-value are also indicated within the text. On each diagram of the figures, p value is indicated, and asterisk depicted the degree of significance (∗p < 0.05; ∗∗ 0.05 > p > 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001).
Information about replicates, randomization and blinding methods
For each experiment, the n number indicated in the caption of the figures represent the number of experimental replicates. To comply with ethical refinement (i-e: limited use of animals), and the slow growth state of the primary old cells, a cell line is routinely used to perform multiple tests once they reach the exponential growth state, but each cell line is only used once per each experimental test (e-g: Western blot for Kindlin-2 and F-G actin fractions).
When sex- and/or age-specific effects are to be estimated, young and old cells are placed in culture, synchronized and treated at the same time, in such a way that young male, young female, old male and old female cells reach the overall same passage number and be compared in a specific experiment point.
Each cell line is assigned an unique ID based on original mouse ID. Table in the repository document indicates the ID of each cell line for each experiments shown in the figures.
At the time of processing samples, samples received a unique but neutral identification number (for instance “K” for Kindlin-2 manipulation, “MX” for a matrix swap experiment, “LY” for ERK inhibitor, for both controls and treated samples. Numbers are the order of collection, with no specific organization by treatment). When possible, Western blots were run by third-party. For immunostaining, slide identification is masked and revealed only after picture analysis. However, the change of phenotype in ERK1/2 inhibitor treated cells, si-K2 cells and K-OE cells on matrix swap is so obvious that the experimenter is aware about the group composition while analyzing the pictures.
As an attempt to remain objective, data are regularly blind-shared to ascertain the robustness of the experimenters’ observations.
Spontaneous transformation may happen stochastically in culture, and especially while working with primary cells from aged animals. The risk of dedifferentiation increases with the passage number and freeze/thaw stress. For this reason, a cell line is removed from any experimental studies once it reaches passage 10. It can be eventually excluded, regardless of its passage number, when uncontrolled growth and/or changes in cell shape (glioblastoma-like) are observed. These cells usually acquired mutations and tumorigenic adaptive capacities that make them non-representative from their original group.
Acknowledgments
The authors thank Drs. Mark Entman and George Taffet for critical reading of the manuscript and Sharon Malinowski for her editorial assistance. We also thank the Electron Microscopy Core at Houston Methodist Hospital for providing access to the scanning electron microscope, and Huie Wang for her technical assistance. We also thank Isaac Karim for his help in data analysis of cardiac fibroblasts and 3-D matrices preparation, and Sharonya Shetty for her help in cell culture and Western blot preparation.
The project was supported by National Institute on Aging (AG059599), the Medallion Foundation, and the Hankamer Foundation. The following grants supported work done in Mass Spectrometry Proteomics Core at Baylor College of Medicine the Dan L. Duncan Comprehensive Cancer Center Award (P30 CA125123), CPRIT Core Facility Awards (RP170005 and RP210227), Intellectual and Developmental Disabilities Research Center Award (P50 HD103555), and NIH High-End Instrument Award (S10 OD026804, Orbitrap Exploris 480).
Authors contributions
Conceptualization, Methodology, Investigation, Formal analysis, and Resources: A.A., J.T., A.S., A.M., and K.C.; Writing – Original draft: A.A., K.C.; Writing – Review & editing: A.A., K.C., J.T., A.S., and A.M.; Visualization: A.A.; Supervision: K.C.; Funding acquisition: K.C. and A.M.
Declaration of interests
The authors declare no competing interest.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: July 4, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107283.
Supplemental information
Data and code availability
-
1.Data
- The Matrisomal Mass Spectrometry dataset has been deposited at Proteome Xchange (PRIDE). It is publicly available online at the time of publication with the identifiers PXD041660 and PXD031223 via ProteomeXchange. Accession numbers and links are also listed in the key resources table.
- All original files generated in this paper have been deposited at Mendeley and are available as of the date of publication. DOI is listed in the key resource table.
-
2.Code
- This paper does not report the original code.
-
3.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Pizzo A.M., Kokini K., Vaughn L.C., Waisner B.Z., Voytik-Harbin S.L. Extracellular matrix (ECM) microstructural composition regulates local cell-ECM biomechanics and fundamental fibroblast behavior: a multidimensional perspective. J. Appl. Physiol. 2005;98:1909–1921. doi: 10.1152/japplphysiol.01137.2004. [DOI] [PubMed] [Google Scholar]
- 2.de Haas H.J., Arbustini E., Fuster V., Kramer C.M., Narula J. Molecular imaging of the cardiac extracellular matrix. Circ. Res. 2014;114:903–915. doi: 10.1161/CIRCRESAHA.113.302680. [DOI] [PubMed] [Google Scholar]
- 3.Hortells L., Johansen A.K.Z., Yutzey K.E. Cardiac fibroblasts and the extracellular matrix in regenerative and nonregenerative hearts. J. Cardiovasc. Dev. Dis. 2019;6:29. doi: 10.3390/jcdd6030029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeLeon-Pennell K.Y., Barker T.H., Lindsey M.L. Fibroblasts: The arbiters of extracellular matrix remodeling. Matrix Biol. 2020;91–92:1–7. doi: 10.1016/j.matbio.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Avery D., Govindaraju P., Jacob M., Todd L., Monslow J., Puré E. Extracellular matrix directs phenotypic heterogeneity of activated fibroblasts. Matrix Biol. 2018;67:90–106. doi: 10.1016/j.matbio.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Atance J., Yost M.J., Carver W. Influence of the extracellular matrix on the regulation of cardiac fibroblast behavior by mechanical stretch. J. Cell. Physiol. 2004;200:377–386. doi: 10.1002/jcp.20034. [DOI] [PubMed] [Google Scholar]
- 7.Banerjee I., Yekkala K., Borg T.K., Baudino T.A. Dynamic interactions between myocytes, fibroblasts, and extracellular matrix. Ann. N. Y. Acad. Sci. 2006;1080:76–84. doi: 10.1196/annals.1380.007. [DOI] [PubMed] [Google Scholar]
- 8.Green H.J., Brown N.H. Integrin intracellular machinery in action. Exp. Cell Res. 2019;378:226–231. doi: 10.1016/j.yexcr.2019.03.011. [DOI] [PubMed] [Google Scholar]
- 9.Romero S., Le Clainche C., Gautreau A.M. Actin polymerization downstream of integrins: signaling pathways and mechanotransduction. Biochem. J. 2020;477:1–21. doi: 10.1042/BCJ20170719. [DOI] [PubMed] [Google Scholar]
- 10.Seo J., Kim J. Regulation of hippo signaling by actin remodeling. BMB Rep. 2018;51:151–156. doi: 10.5483/bmbrep.2018.51.3.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morita T., Hayashi K. G-actin sequestering protein thymosin-beta4 regulates the activity of myocardin-related transcription factor. Biochem. Biophys. Res. Commun. 2013;437:331–335. doi: 10.1016/j.bbrc.2013.06.069. [DOI] [PubMed] [Google Scholar]
- 12.Perestrelo A.R., Silva A.C., Oliver-De La Cruz J., Martino F., Horváth V., Caluori G., Polanský O., Vinarský V., Azzato G., de Marco G., et al. Multiscale analysis of extracellular matrix remodeling in the failing heart. Circ. Res. 2021;128:24–38. doi: 10.1161/CIRCRESAHA.120.317685. [DOI] [PubMed] [Google Scholar]
- 13.Small E.M. The actin-MRTF-SRF gene regulatory axis and myofibroblast differentiation. J. Cardiovasc. Transl. Res. 2012;5:794–804. doi: 10.1007/s12265-012-9397-0. [DOI] [PubMed] [Google Scholar]
- 14.Daseke M.J., 2nd, Tenkorang M.A., Chalise U., Konfrst S.R., Lindsey M.L. Cardiac fibroblast activation during myocardial infarction wound healing: Fibroblast polarization after MI. Matrix Biol. 2020;91–92:109–116. doi: 10.1016/j.matbio.2020.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang X., Yang N., Fiore V.F., Barker T.H., Sun Y., Morris S.W., Ding Q., Thannickal V.J., Zhou Y. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am. J. Respir. Cell Mol. Biol. 2012;47:340–348. doi: 10.1165/rcmb.2012-0050OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinz B., Celetta G., Tomasek J.J., Gabbiani G., Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell. 2001;12:2730–2741. doi: 10.1091/mbc.12.9.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sandbo N., Dulin N. Actin cytoskeleton in myofibroblast differentiation: ultrastructure defining form and driving function. Transl. Res. 2011;158:181–196. doi: 10.1016/j.trsl.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shinde A.V., Humeres C., Frangogiannis N.G. The role of alpha-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim. Biophys. Acta, Mol. Basis Dis. 2017;1863:298–309. doi: 10.1016/j.bbadis.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hinz B., McCulloch C.A., Coelho N.M. Mechanical regulation of myofibroblast phenoconversion and collagen contraction. Exp. Cell Res. 2019;379:119–128. doi: 10.1016/j.yexcr.2019.03.027. [DOI] [PubMed] [Google Scholar]
- 20.MacKenna D., Summerour S.R., Villarreal F.J. Role of mechanical factors in modulating cardiac fibroblast function and extracellular matrix synthesis. Cardiovasc. Res. 2000;46:257–263. doi: 10.1016/s0008-6363(00)00030-4. [DOI] [PubMed] [Google Scholar]
- 21.van Putten S., Shafieyan Y., Hinz B. Mechanical control of cardiac myofibroblasts. J. Mol. Cell. Cardiol. 2016;93:133–142. doi: 10.1016/j.yjmcc.2015.11.025. [DOI] [PubMed] [Google Scholar]
- 22.Anderson R., Richardson G.D., Passos J.F. Mechanisms driving the ageing heart. Exp. Gerontol. 2018;109:5–15. doi: 10.1016/j.exger.2017.10.015. [DOI] [PubMed] [Google Scholar]
- 23.Anisimov S.V., Boheler K.R. Aging-associated changes in cardiac gene expression: large scale transcriptome analysis. Adv. Gerontol. 2003;11:67–75. [PubMed] [Google Scholar]
- 24.Annoni G., Luvarà G., Arosio B., Gagliano N., Fiordaliso F., Santambrogio D., Jeremic G., Mircoli L., Latini R., Vergani C., Masson S. Age-dependent expression of fibrosis-related genes and collagen deposition in the rat myocardium. Mech. Ageing Dev. 1998;101:57–72. doi: 10.1016/s0047-6374(97)00165-6. [DOI] [PubMed] [Google Scholar]
- 25.Grilo G.A., Shaver P.R., Stoffel H.J., Morrow C.A., Johnson O.T., Iyer R.P., de Castro Brás L.E. Age- and sex-dependent differences in extracellular matrix metabolism associate with cardiac functional and structural changes. J. Mol. Cell. Cardiol. 2020;139:62–74. doi: 10.1016/j.yjmcc.2020.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gould K.E., Taffet G.E., Michael L.H., Christie R.M., Konkol D.L., Pocius J.S., Zachariah J.P., Chaupin D.F., Daniel S.L., Sandusky G.E., Jr., et al. Heart failure and greater infarct expansion in middle-aged mice: a relevant model for postinfarction failure. Am. J. Physiol. Heart Circ. Physiol. 2002;282:H615–H621. doi: 10.1152/ajpheart.00206.2001. [DOI] [PubMed] [Google Scholar]
- 27.Bujak M., Kweon H.J., Chatila K., Li N., Taffet G., Frangogiannis N.G. Aging-related defects are associated with adverse cardiac remodeling in a mouse model of reperfused myocardial infarction. J. Am. Coll. Cardiol. 2008;51:1384–1392. doi: 10.1016/j.jacc.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ezekowitz J.A., Kaul P., Bakal J.A., Armstrong P.W., Welsh R.C., McAlister F.A. Declining in-hospital mortality and increasing heart failure incidence in elderly patients with first myocardial infarction. J. Am. Coll. Cardiol. 2009;53:13–20. doi: 10.1016/j.jacc.2008.08.067. [DOI] [PubMed] [Google Scholar]
- 29.Trial J., Cieslik K.A. Changes in cardiac resident fibroblast physiology and phenotype in aging. Am. J. Physiol. Heart Circ. Physiol. 2018;315:H745–H755. doi: 10.1152/ajpheart.00237.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trial J., Heredia C.P., Taffet G.E., Entman M.L., Cieslik K.A. Dissecting the role of myeloid and mesenchymal fibroblasts in age-dependent cardiac fibrosis. Basic Res. Cardiol. 2017;112:34. doi: 10.1007/s00395-017-0623-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cieslik K.A., Trial J., Entman M.L. Defective myofibroblast formation from mesenchymal stem cells in the aging murine heart rescue by activation of the AMPK pathway. Am. J. Pathol. 2011;179:1792–1806. doi: 10.1016/j.ajpath.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lindsey M.L., Goshorn D.K., Squires C.E., Escobar G.P., Hendrick J.W., Mingoia J.T., Sweterlitsch S.E., Spinale F.G. Age-dependent changes in myocardial matrix metalloproteinase/tissue inhibitor of metalloproteinase profiles and fibroblast function. Cardiovasc. Res. 2005;66:410–419. doi: 10.1016/j.cardiores.2004.11.029. [DOI] [PubMed] [Google Scholar]
- 33.Trial J., Entman M.L., Cieslik K.A. Mesenchymal stem cell-derived inflammatory fibroblasts mediate interstitial fibrosis in the aging heart. J. Mol. Cell. Cardiol. 2016;91:28–34. doi: 10.1016/j.yjmcc.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Curcio F., Gerundo G., Sasso G., Panicara V., Liguori I., Testa G., Della-Morte D., Gargiulo G., Galizia G., Ungar A., et al. Type 2 myocardial infarction: is it a geriatric syndrome? Aging Clin. Exp. Res. 2020;32:759–768. doi: 10.1007/s40520-019-01452-8. [DOI] [PubMed] [Google Scholar]
- 35.Angelini A., Trial J., Ortiz-Urbina J., Cieslik K.A. Mechanosensing dysregulation in the fibroblast: A hallmark of the aging heart. Ageing Res. Rev. 2020;63:101150. doi: 10.1016/j.arr.2020.101150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Plow E.F., Qin J., Byzova T. Kindling the flame of integrin activation and function with kindlins. Curr. Opin. Hematol. 2009;16:323–328. doi: 10.1097/MOH.0b013e32832ea389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yasuda-Yamahara M., Rogg M., Frimmel J., Trachte P., Helmstaedter M., Schroder P., Schiffer M., Schell C., Huber T.B. FERMT2 links cortical actin structures, plasma membrane tension and focal adhesion function to stabilize podocyte morphology. Matrix Biol. 2018;68–69:263–279. doi: 10.1016/j.matbio.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 38.Angelini A., Ortiz-Urbina J., Trial J., Reddy A.K., Malovannaya A., Jain A., Entman M.L., Taffet G.E., Cieslik K.A. Sex-specific phenotypes in the aging mouse heart and consequences for chronic fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2022;323:H285–H300. doi: 10.1152/ajpheart.00078.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang K., Xu X., Yang B., Cook B., Ramos H., Krishnan N.M.A., Smedskjaer M.M., Hoover C., Bauchy M. Predicting the young's modulus of silicate glasses using high-throughput molecular dynamics simulations and machine learning. Sci. Rep. 2019;9:8739. doi: 10.1038/s41598-019-45344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mih J.D., Sharif A.S., Liu F., Marinkovic A., Symer M.M., Tschumperlin D.J. A multiwell platform for studying stiffness-dependent cell biology. PLoS One. 2011;6:e19929. doi: 10.1371/journal.pone.0019929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woodley J.P., Lambert D.W., Asencio I.O. Understanding fibroblast behavior in 3D biomaterials. Tissue Eng. Part B Rev. 2022;28:569–578. doi: 10.1089/ten.TEB.2021.0010. [DOI] [PubMed] [Google Scholar]
- 42.Castaldo C., Di Meglio F., Miraglia R., Sacco A.M., Romano V., Bancone C., Della Corte A., Montagnani S., Nurzynska D. Cardiac fibroblast-derived extracellular matrix (biomatrix) as a model for the studies of cardiac primitive cell biological properties in normal and pathological adult human heart. BioMed Res. Int. 2013;2013:352370. doi: 10.1155/2013/352370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Püspöki Z., Storath M., Sage D., Unser M. Transforms and operators for directional bioimage analysis: a survey. Adv. Anat. Embryol. Cell Biol. 2016;219:69–93. doi: 10.1007/978-3-319-28549-8_3. [DOI] [PubMed] [Google Scholar]
- 44.De Belly H., Stubb A., Yanagida A., Labouesse C., Jones P.H., Paluch E.K., Chalut K.J. Membrane tension gates erk-mediated regulation of pluripotent cell fate. Cell Stem Cell. 2021;28:273–284.e6. doi: 10.1016/j.stem.2020.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang J.M., Bhattacharya S., West-Foyle H., Hung C.F., Wu T.C., Iglesias P.A., Huang C.H. Integrating chemical and mechanical signals through dynamic coupling between cellular protrusions and pulsed ERK activation. Nat. Commun. 2018;9:4673. doi: 10.1038/s41467-018-07150-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chatzifrangkeskou M., Yadin D., Marais T., Chardonnet S., Cohen-Tannoudji M., Mougenot N., Schmitt A., Crasto S., Di Pasquale E., Macquart C., et al. Cofilin-1 phosphorylation catalyzed by ERK1/2 alters cardiac actin dynamics in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 2018;27:3060–3078. doi: 10.1093/hmg/ddy215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fringer J., Grinnell F. Fibroblast quiescence in floating or released collagen matrices: contribution of the ERK signaling pathway and actin cytoskeletal organization. J. Biol. Chem. 2001;276:31047–31052. doi: 10.1074/jbc.M101898200. [DOI] [PubMed] [Google Scholar]
- 48.Shrestha D., Choi D., Song K. Actin dysfunction induces cell cycle delay at G2/M with sustained ERK and RSK activation in IMR-90 normal human fibroblasts. Mol. Cells. 2018;41:436–443. doi: 10.14348/molcells.2018.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haack T., Abdelilah-Seyfried S. The force within: endocardial development, mechanotransduction and signalling during cardiac morphogenesis. Development. 2016;143:373–386. doi: 10.1242/dev.131425. [DOI] [PubMed] [Google Scholar]
- 50.Howard C.M., Baudino T.A. Dynamic cell-cell and cell-ECM interactions in the heart. J. Mol. Cell. Cardiol. 2014;70:19–26. doi: 10.1016/j.yjmcc.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 51.Knöll R., Hoshijima M., Chien K. Cardiac mechanotransduction and implications for heart disease. J. Mol. Med. 2003;81:750–756. doi: 10.1007/s00109-003-0488-x. [DOI] [PubMed] [Google Scholar]
- 52.Samarel A.M. Costameres, focal adhesions, and cardiomyocyte mechanotransduction. Am. J. Physiol. Heart Circ. Physiol. 2005;289:H2291–H2301. doi: 10.1152/ajpheart.00749.2005. [DOI] [PubMed] [Google Scholar]
- 53.Sullivan K.E., Black L.D. The role of cardiac fibroblasts in extracellular matrix-mediated signaling during normal and pathological cardiac development. J. Biomech. Eng. 2013;135:71001. doi: 10.1115/1.4024349. [DOI] [PubMed] [Google Scholar]
- 54.Kihara T., Ito J., Miyake J. Measurement of biomolecular diffusion in extracellular matrix condensed by fibroblasts using fluorescence correlation spectroscopy. PLoS One. 2013;8:e82382. doi: 10.1371/journal.pone.0082382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanders M.A., Basson M.D. Collagen IV regulates Caco-2 migration and ERK activation via alpha1beta1- and alpha2beta1-integrin-dependent Src kinase activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004;286:G547–G557. doi: 10.1152/ajpgi.00262.2003. [DOI] [PubMed] [Google Scholar]
- 56.Marro J., Pfefferli C., de Preux Charles A.S., Bise T., Jaźwińska A. Collagen XII contributes to epicardial and connective tissues in the zebrafish heart during ontogenesis and regeneration. PLoS One. 2016;11:e0165497. doi: 10.1371/journal.pone.0165497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arai K., Nagashima Y., Takemoto T., Nishiyama T. Mechanical strain increases expression of type XII collagen in murine osteoblastic MC3T3-E1 cells. Cell Struct. Funct. 2008;33:203–210. doi: 10.1247/csf.08025. [DOI] [PubMed] [Google Scholar]
- 58.Izu Y., Adams S.M., Connizzo B.K., Beason D.P., Soslowsky L.J., Koch M., Birk D.E. Collagen XII mediated cellular and extracellular mechanisms regulate establishment of tendon structure and function. Matrix Biol. 2021;95:52–67. doi: 10.1016/j.matbio.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kanaan R., Medlej-Hashim M., Jounblat R., Pilecki B., Sorensen G.L. Microfibrillar-associated protein 4 in health and disease. Matrix Biol. 2022;111:1–25. doi: 10.1016/j.matbio.2022.05.008. [DOI] [PubMed] [Google Scholar]
- 60.Fan D., Kassiri Z. Biology of tissue inhibitor of metalloproteinase 3 (timp3), and its therapeutic implications in cardiovascular pathology. Front. Physiol. 2020;11:661. doi: 10.3389/fphys.2020.00661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhan J., Yang M., Chi X., Zhang J., Pei X., Ren C., Guo Y., Liu W., Zhang H. Kindlin-2 expression in adult tissues correlates with their embryonic origins. Sci. China Life Sci. 2014;57:690–697. doi: 10.1007/s11427-014-4676-4. [DOI] [PubMed] [Google Scholar]
- 62.Chen C., Manso A.M., Ross R.S. Talin and kindlin as integrin-activating proteins: focus on the heart. Pediatr. Cardiol. 2019;40:1401–1409. doi: 10.1007/s00246-019-02167-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bledzka K., Bialkowska K., Sossey-Alaoui K., Vaynberg J., Pluskota E., Qin J., Plow E.F. Kindlin-2 directly binds actin and regulates integrin outside-in signaling. J. Cell Biol. 2016;213:97–108. doi: 10.1083/jcb.201501006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lu F., Zhu L., Bromberger T., Yang J., Yang Q., Liu J., Plow E.F., Moser M., Qin J. Mechanism of integrin activation by talin and its cooperation with kindlin. Nat. Commun. 2022;13:2362. doi: 10.1038/s41467-022-30117-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu Z., Gao J., Hong J., Ma Y.Q. Integrity of kindlin-2 FERM subdomains is required for supporting integrin activation. Biochem. Biophys. Res. Commun. 2013;434:382–387. doi: 10.1016/j.bbrc.2013.03.086. [DOI] [PubMed] [Google Scholar]
- 66.Zhang Z., Mu Y., Zhang J., Zhou Y., Cattaneo P., Veevers J., Peter A.K., Manso A.M., Knowlton K.U., Zhou X., et al. Kindlin-2 is essential for preserving integrity of the developing heart and preventing ventricular rupture. Circulation. 2019;139:1554–1556. doi: 10.1161/CIRCULATIONAHA.118.038383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Montanez E., Ussar S., Schifferer M., Bösl M., Zent R., Moser M., Fässler R. Kindlin-2 controls bidirectional signaling of integrins. Genes Dev. 2008;22:1325–1330. doi: 10.1101/gad.469408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Z., Mu Y., Veevers J., Peter A.K., Manso A.M., Bradford W.H., Dalton N.D., Peterson K.L., Knowlton K.U., Ross R.S., et al. Postnatal loss of kindlin-2 leads to progressive heart failure. Circ. Heart Fail. 2016;9 doi: 10.1161/CIRCHEARTFAILURE.116.003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Godbout E., Son D.O., Hume S., Boo S., Sarrazy V., Clement S., Kapus A., Wehrle-Haller B., Bruckner-Tuderman L., Has C., Hinz B. Kindlin-2 mediates mechanical activation of cardiac myofibroblasts. Cells. 2020;9 doi: 10.3390/cells9122702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.He Y., Esser P., Schacht V., Bruckner-Tuderman L., Has C. Role of kindlin-2 in fibroblast functions: implications for wound healing. J. Invest. Dermatol. 2011;131:245–256. doi: 10.1038/jid.2010.273. [DOI] [PubMed] [Google Scholar]
- 71.Angelini A., Li Z., Mericskay M., Decaux J.F. Regulation of connective tissue growth factor and cardiac fibrosis by an SRF/MicroRNA-133a axis. PLoS One. 2015;10 doi: 10.1371/journal.pone.0139858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miano J.M. Serum response factor: toggling between disparate programs of gene expression. J. Mol. Cell. Cardiol. 2003;35:577–593. doi: 10.1016/s0022-2828(03)00110-x. [DOI] [PubMed] [Google Scholar]
- 73.Wang Z., Wang D.Z., Hockemeyer D., McAnally J., Nordheim A., Olson E.N. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature. 2004;428:185–189. doi: 10.1038/nature02382. [DOI] [PubMed] [Google Scholar]
- 74.Esnault C., Gualdrini F., Horswell S., Kelly G., Stewart A., East P., Matthews N., Treisman R. ERK-Induced activation of tcf family of srf cofactors initiates a chromatin modification cascade associated with transcription. Mol. Cell. 2017;65:1081–1095.e5. doi: 10.1016/j.molcel.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tresini M., Lorenzini A., Frisoni L., Allen R.G., Cristofalo V.J. Lack of Elk-1 phosphorylation and dysregulation of the extracellular regulated kinase signaling pathway in senescent human fibroblast. Exp. Cell Res. 2001;269:287–300. doi: 10.1006/excr.2001.5334. [DOI] [PubMed] [Google Scholar]
- 76.Posern G., Sotiropoulos A., Treisman R. Mutant actins demonstrate a role for unpolymerized actin in control of transcription by serum response factor. Mol. Biol. Cell. 2002;13:4167–4178. doi: 10.1091/mbc.02-05-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gao J., Khan A.A., Shimokawa T., Zhan J., Strömblad S., Fang W., Zhang H. A feedback regulation between kindlin-2 and GLI1 in prostate cancer cells. FEBS Lett. 2013;587:631–638. doi: 10.1016/j.febslet.2012.12.028. [DOI] [PubMed] [Google Scholar]
- 78.Safiejko-Mroczka B., Bell P.B., Jr. Reorganization of the actin cytoskeleton in the protruding lamellae of human fibroblasts. Cell Motil Cytoskeleton. 2001;50:13–32. doi: 10.1002/cm.1038. [DOI] [PubMed] [Google Scholar]
- 79.Bunnell T.M., Burbach B.J., Shimizu Y., Ervasti J.M. beta-Actin specifically controls cell growth, migration, and the G-actin pool. Mol. Biol. Cell. 2011;22:4047–4058. doi: 10.1091/mbc.E11-06-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vandekerckhove J., Weber K. At least six different actins are expressed in a higher mammal: an analysis based on the amino acid sequence of the amino-terminal tryptic peptide. J. Mol. Biol. 1978;126:783–802. doi: 10.1016/0022-2836(78)90020-7. [DOI] [PubMed] [Google Scholar]
- 81.Tondeleir D., Lambrechts A., Müller M., Jonckheere V., Doll T., Vandamme D., Bakkali K., Waterschoot D., Lemaistre M., Debeir O., et al. Cells lacking beta-actin are genetically reprogrammed and maintain conditional migratory capacity. Mol. Cell. Proteomics. 2012;11:255–271. doi: 10.1074/mcp.M111.015099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sztal T.E., McKaige E.A., Williams C., Ruparelia A.A., Bryson-Richardson R.J. Genetic compensation triggered by actin mutation prevents the muscle damage caused by loss of actin protein. PLoS Genet. 2018;14 doi: 10.1371/journal.pgen.1007212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Angelini A., Gorey M.A., Dumont F., Mougenot N., Chatzifrangkeskou M., Muchir A., Li Z., Mericskay M., Decaux J.F. Cardioprotective effects of alpha-cardiac actin on oxidative stress in a dilated cardiomyopathy mouse model. FASEB J. 2020;34:2987–3005. doi: 10.1096/fj.201902389R. [DOI] [PubMed] [Google Scholar]
- 84.Li Y., Li C., Liu Q., Wang L., Bao A.X., Jung J.P., Dodlapati S., Sun J., Gao P., Zhang X., et al. Loss of Acta2 in cardiac fibroblasts does not prevent the myofibroblast differentiation or affect the cardiac repair after myocardial infarction. J. Mol. Cell. Cardiol. 2022;171:117–132. doi: 10.1016/j.yjmcc.2022.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Foglia B., Cannito S., Bocca C., Parola M., Novo E. ERK pathway in activated, myofibroblast-like, hepatic stellate cells: a critical signaling crossroad sustaining liver fibrosis. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20112700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen H., Chen H., Liang J., Gu X., Zhou J., Xie C., Lv X., Wang R., Li Q., Mao Z., et al. TGF-beta1/IL-11/MEK/ERK signaling mediates senescence-associated pulmonary fibrosis in a stress-induced premature senescence model of Bmi-1 deficiency. Exp. Mol. Med. 2020;52:130–151. doi: 10.1038/s12276-019-0371-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weng C.H., Li Y.J., Wu H.H., Liu S.H., Hsu H.H., Chen Y.C., Yang C.W., Chu P.H., Tian Y.C. Interleukin-17A induces renal fibrosis through the ERK and smad signaling pathways. Biomed. Pharmacother. 2020;123 doi: 10.1016/j.biopha.2019.109741. [DOI] [PubMed] [Google Scholar]
- 88.Emmert H., Patel H., Brunton V.G. Kindlin-1 protects cells from oxidative damage through activation of ERK signalling. Free Radic. Biol. Med. 2017;108:896–903. doi: 10.1016/j.freeradbiomed.2017.05.013. [DOI] [PubMed] [Google Scholar]
- 89.Dudiki T., Meller J., Mahajan G., Liu H., Zhevlakova I., Stefl S., Witherow C., Podrez E., Kothapalli C.R., Byzova T.V. Microglia control vascular architecture via a TGFbeta1 dependent paracrine mechanism linked to tissue mechanics. Nat. Commun. 2020;11:986. doi: 10.1038/s41467-020-14787-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Umbarkar P., Tousif S., Singh A.P., Anderson J.C., Zhang Q., Tallquist M.D., Woodgett J., Lal H. Fibroblast GSK-3alpha promotes fibrosis via RAF-MEK-ERK pathway in the injured heart. Circ. Res. 2022;131:620–636. doi: 10.1161/CIRCRESAHA.122.321431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhu K., Lai Y., Cao H., Bai X., Liu C., Yan Q., Ma L., Chen D., Kanaporis G., Wang J., et al. Kindlin-2 modulates MafA and beta-catenin expression to regulate beta-cell function and mass in mice. Nat. Commun. 2020;11:484. doi: 10.1038/s41467-019-14186-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mehta L.S., Beckie T.M., DeVon H.A., Grines C.L., Krumholz H.M., Johnson M.N., Lindley K.J., Vaccarino V., Wang T.Y., Watson K.E., et al. Acute myocardial infarction in women: a scientific statement from the american heart association. Circulation. 2016;133:916–947. doi: 10.1161/CIR.0000000000000351. [DOI] [PubMed] [Google Scholar]
- 93.Chen Q., Williams R., Healy C.L., Wright C.D., Wu S.C., O'Connell T.D. An association between gene expression and better survival in female mice following myocardial infarction. J. Mol. Cell. Cardiol. 2010;49:801–811. doi: 10.1016/j.yjmcc.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fazal L., Azibani F., Vodovar N., Cohen Solal A., Delcayre C., Samuel J.L. Effects of biological sex on the pathophysiology of the heart. Br. J. Pharmacol. 2014;171:555–566. doi: 10.1111/bph.12279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dworatzek E., Baczko I., Kararigas G. Effects of aging on cardiac extracellular matrix in men and women. Proteomics. Clin. Appl. 2016;10:84–91. doi: 10.1002/prca.201500031. [DOI] [PubMed] [Google Scholar]
- 96.Achkar A., Saliba Y., Fares N. Differential gender-dependent patterns of cardiac fibrosis and fibroblast phenotypes in aging mice. Oxid. Med. Cell. Longev. 2020;2020 doi: 10.1155/2020/8282157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Trial J., Diaz Lankenau R., Angelini A., Tovar Perez J.E., Taffet G.E., Entman M.L., Cieslik K.A. Treatment with a DC-SIGN ligand reduces macrophage polarization and diastolic dysfunction in the aging female but not male mouse hearts. Geroscience. 2021;43:881–899. doi: 10.1007/s11357-020-00255-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sheridan W.S., Duffy G.P., Murphy B.P. Mechanical characterization of a customized decellularized scaffold for vascular tissue engineering. J. Mech. Behav. Biomed. Mater. 2012;8:58–70. doi: 10.1016/j.jmbbm.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 99.Stephenson M.K., Lenihan S., Covarrubias R., Huttinger R.M., Gumina R.J., Sawyer D.B., Galindo C.L. Scanning electron microscopy of macerated tissue to visualize the extracellular matrix. J. Vis. Exp. 2016 doi: 10.3791/54005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brosch M., Yu L., Hubbard T., Choudhary J. Accurate and sensitive peptide identification with mascot percolator. J. Proteome Res. 2009;8:3176–3181. doi: 10.1021/pr800982s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Anderson D.C., Li W., Payan D.G., Noble W.S. A new algorithm for the evaluation of shotgun peptide sequencing in proteomics: support vector machine classification of peptide MS/MS spectra and SEQUEST scores. J. Proteome Res. 2003;2:137–146. doi: 10.1021/pr0255654. [DOI] [PubMed] [Google Scholar]
- 102.Saltzman A.B., Leng M., Bhatt B., Singh P., Chan D.W., Dobrolecki L., Chandrasekaran H., Choi J.M., Jain A., Jung S.Y., et al. gpGrouper: A peptide grouping algorithm for gene-centric inference and quantitation of bottom-up proteomics data. Mol. Cell. Proteomics. 2018;17:2270–2283. doi: 10.1074/mcp.TIR118.000850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- 105.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to imageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
1.Data
- The Matrisomal Mass Spectrometry dataset has been deposited at Proteome Xchange (PRIDE). It is publicly available online at the time of publication with the identifiers PXD041660 and PXD031223 via ProteomeXchange. Accession numbers and links are also listed in the key resources table.
- All original files generated in this paper have been deposited at Mendeley and are available as of the date of publication. DOI is listed in the key resource table.
-
2.Code
- This paper does not report the original code.
-
3.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.