

Abstract
Receptors for estrogen and progesterone frequently interact, via Cohesin/CTCF loop extrusion, at enhancers distal from regulated genes. Loss-of-function CTCF mutation in >20% of human endometrial tumors indicates its importance in uterine homeostasis. To better understand how CTCF-mediated enhancer-gene interactions impact endometrial development and function, the Ctcf gene was selectively deleted in female reproductive tissues of mice. Prepubertal Ctcfd/d uterine tissue exhibited a marked reduction in the number of uterine glands compared to those without Ctcf deletion (Ctcff/f mice). Post-pubertal Ctcfd/d uteri were hypoplastic with significant reduction in both the amount of the endometrial stroma and number of glands. Transcriptional profiling revealed increased expression of stem cell molecules Lif, EOMES and Lgr5, and enhanced inflammation pathways following Ctcf deletion. Analysis of the response of the uterus to steroid hormone stimulation showed that CTCF deletion affects a subset of progesterone responsive genes. This finding indicates 1) Progesterone mediated signaling remains functional following Ctcf deletion and 2) certain progesterone regulated genes are sensitive to Ctcf deletion, suggesting they depend on gene-enhancer interactions that require CTCF. The progesterone responsive genes altered by CTCF ablation included Ihh, Fst and Errfi1. CTCF-dependent progesterone responsive uterine genes enhance critical processes including anti-tumorigenesis, which is relevant to the known effectiveness of progesterone in inhibiting progression of early-stage endometrial tumors. Overall, our findings reveal that uterine Ctcf plays a key role in progesterone dependent expression of uterine genes underlying optimal post-pubertal uterine development.
Keywords: Chromatin, uterus, progesterone, estrogen, puberty
Graphical Abstract
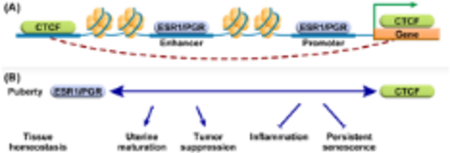
A. ESR1 and PGR interact with chromatin both at regions proximal to hormone responsive gene promoters and at distal enhancer regions. Cohesin/CTCF mediated loop extrusion facilitates interactions between distal enhancers and gene promoters. B. Deletion of uterine cell CTCF selectively perturbs post-pubertal progesterone regulation of tumor suppressive genes and leads to uterine hypoplasia, inflammation, and indications of persistent senescence. These observations suggest that interaction between hormone signaling, and CTCF-mediated chromatin structure is essential for pubertal uterine maturation and maintenance of biological integrity.
Introduction
Chromatin is organized in a manner that allows an ordered compacted structure to fit within the confines of the nucleus. Within the DNA strand, genes governing cellular identity, physiology and response to its environment are encoded and must be expressed by cell-specific spatial and temporally controlled mechanisms. Enhancer regions of DNA are often distal from regulated target genes, so that relative locations of genes and enhancers within the nucleus are arranged in ways that can facilitate or hinder their access by transcriptional mediators. One way these interactions are accomplished is by establishment of topologically associated domains (TADs) and enhancer-gene loops via Cohesin and CTCF mediated loop extrusion (1). Responses mediated by progesterone and estrogen receptors (PGR and ESR1) within the female reproductive tract serve as biological systems to study transcriptional mechanisms in vivo. PGR and ESR1 are hormonally induced transcriptional mediators that accumulate at enhancers and gene promoters to regulate genes critical for uterine tissue development, growth, and maturation in response to ovarian hormones beginning at puberty, and to establish and maintain pregnancy in reproductive-aged individuals (2, 3). Following the onset of estrous cycling at puberty, the mouse uterus, which is hypoplastic at birth, grows and acquires optimal responsiveness to estrogen and progesterone (4, 5). Perturbations in hormone signaling during this critical developmental window can lead to later life uterine dysfunction, including inability to establish pregnancy or increased susceptibility to endometrial cancer (6).
Previous work has indicated that ESR1 and PGR frequently interact with enhancers that are distal from responsive genes, such that the 3D structure of the chromatin is crucial for ESR1 and PGR mediated gene transcription (7–9). Accordingly, our studies endeavor to understand how chromatin structure impacts hormonally regulated enhancer-gene responses and resulting biological function. CTCF, which binds CTCF-motifs and anchors interacting ends of chromatin loops, acts as a tumor suppressor in multiple tissues (10, 11). Loss of function mutations are observed in >20% of uterine cancer (12–14). Similarly, cohesin subunits RAD21, SMC3 and SMC1A, which make up the cohesin ring that extrudes DNA in between CTCF-anchored chromatin loops, are altered in >8% of endometrial cancers (13, 14). This suggests that chromatin structure impacts uterine cell homeostasis. Since global deletion of Ctcf in mice is embryonic lethal, we used a conditional knockout approach to delete Ctcf in uterine cells using the PGR-Cre mouse, which expresses Cre-recombinase activity in uterine cells. Initially, recombination of loxP flanked alleles occurs most efficiently in epithelial cells and increases to also include stromal cells by 14–21 days after birth and the inner myometrial cell layer post pubertally (>28 days) (15). PgrCre and Ctcf-flox mice were inter-crossed examine Ctcf’s impact on uterine biology and hormone response.
Methods
Mice
All mouse studies were conducted under an Animal Study Protocol in accordance with the NIEHS Animal Care and Use Committee using the 2015 edition of the Public Health Service Policy on Humane Care and Use of Laboratory Animals. C57bl6 mice were purchased from Charles River Laboratories, Raleigh. Ctcff/f and Ctcfd/d mice were made by intercrossing Ctcf floxed mice (16) to PgrCre mice (15). Ihh19 KO mice were previously described (17). Ihh39 KO and Ihh 63 KO mice were made using CRISPR to target regions with PGR or CTCF ChIP peaks, respectively. Tail biopsies were sent to Transnexy Inc (Memphis, TN) for genotyping. For hormone responses in prepubertal mice, pnd 21 mice were injected with 125 ng estradiol (E2, Steraloids Inc., Newport RI), 0.5 mg progesterone (P4, Steraloids Inc.) or sesame oil vehicle (Sigma Chemicals, St Louis MO) and tissue was collected 6 hours later. For hormonal response in adult mice, animals were ovariectomized, and given 10–14 days to clear ovarian hormones. They were injected with1 mg P4 or sesame oil and tissue collected 1 or 6 hours later.
Ihh mice
Targeting of the Ihh −39 kb and −63 kb promoter peaks was done in C57BL/6J single cell embryos with CRISPR/Cas9-mediated genome editing. Single cell embryos from super-ovulated time mated C57BL/6J females were microinjected with 125 ng/ul Cas9 protein (EnCas9 from New England Biolabs, Ipswich, MA, USA) and 80 ng/ul synthesized sgRNA (Integrated DNA Technologies, Coralville, Iowa, USA). Microinjected single cell embryos were surgically transferred to pseudo-pregnant SWISS females. F0 offspring were screened by PCR amplicon sequencing. F0 mice carrying a mutation of interests were bred to C57BL/6J wildtype mice and resulting F1 offspring were re-sequenced using the same strategy as the F0 founders.
For the −39 kb peak, two Cas9 guides were used (Ihh39 SgD: CACAAGCCATGCTTTACTGCnGG and Ihh39 SgE: GGAGGGACATGAACAACCATnGG) and screened with long range PCR (Ihh39 Scr Fwd: GAGATTGGTACAGCTCCTCTCG, Rev: GGATTACTGCAGGGAAAAGCAC). Sequencing identified a guide to guide 928 bp excision that contained the entire predicted −39 kb peak (928 bp deletion: chr1:75,030,130–75,031,057 GRCm39/mm39).
For the −63 kb peak, two Cas9 guides were used (Ihh63 SgJ: ACCCACGGAGAAATTTACTCnGG and Ihh63 SgK: TCTATCTTGACAACCTACTTnGG) and screened with long range PCR (Ihh63 Scr Fwd: CCACTACCACAAGGGGGCTTAG, Rev: TGCCCACTGCTCACATGATTTA). Sequencing identified a founder carrying a large deletion of 1955 bp that contained the entire predicted −63 kb peak (1955 bp deletion: chr1:75,054,631–75,056,585 GRCm39/mm39). None of the Cas9 guides used had any predicted linked off-target mutations, and unlinked mutations were not screened. All alleles were crossed to C57BL/6J isogenic wildtype mice for at least three generations to clean up the genome from any unknown mutations. For all studies, wildtype littermates were used as the reference group to account for any unknown unlinked mutations.
Initial genotyping of the colony was done using the screening primer listed above. Once the colonies were established and germline transmission of the mutant allele was sequence confirmed, the colony were then transitioned to primer/probe genotyping at Transnetyx (Cordova, TN, USA). Primer/probe genotyping assay details for the Ihh-39 peak are Ihh39-WT (Fwd: GCATTAACAAATCACACAAGCGTACA, Rev: GCTAAAGTCCTCAATTCCCTGCA, Probe: CACAAGCCATGCTTTAC) and Ihh39-KO (Fwd: ACCCTGAGGGAGCAGCTATT, Rev: CACAGGAGCCAGACAAATGGA, Probe: ATAGCATACATTATAGCAATTTAT). Primer/probe genotyping assay details for the Ihh-63 peak are Ihh63-WT (Fwd: ACACTGGCTCCTGGACAAATT, Rev: GGGATGCTAAGGAACCAGACTTG, Probe: CTGCACCTCCACTTTC) and Ihh63-KO (Fwd: ACCACCACCACTACCACAAG, Rev: GCCTAGACTGACCTGGAACTCA, Probe: CTCCACTTGCCTCTGCC).
Histological Analysis
Tissues were fixed for 18–24h in 4% PFA (Electron Microscopy Inc), then transferred to 70% EtOH and embedded in paraffin. Five μm sections were cut and stained with hematoxylin and eosin (H&E). Histopathological evaluation was performed by a board-certified veterinary pathologist on whole slide images of scanned stained sections of uterine cross-sections. Whole slide image viewing software (Aperio eSlide Manager) was used to measure the longest dimension of the endometrial stroma (including the uterine lumen) and the longest perpendicular distance and averaged the two dimensions to obtain mean width of the uterine endometrial stroma (including lumen). Glands per section were counted.
RNA Isolation and Analysis
RNA was isolated from frozen uterine tissue as previously described using Trizol Reagent (Thermo Fisher, Waltham MA) (18). For RNAseq, RNA was further purified using the Zymo RNA clean up kit (Zymo Research, Irvine CA), with DNAse treatment, according to manufacturer’s instructions. For RT-PCR, cDNA was synthesized, and Real Time PCR performed as previously described (19). Primer sequences are listed in Table 1. For RNAseq, libraries were made and sequenced by the NIEHS Sequencing Core using the Illumina Tru-seq Stranded mRNA kit and Illumina NovaSeq instrument. For RNAseq, 4–8 replicates of each condition were used (Ctcff/f V n=8, Ctcff/f P n=4 Ctcfd/d V n=8, Ctcfd/d P n=4). Raw data were filtered to remove low-quality reads, mapped to mm10 using Tophat (20) and de-duplicated using Picard tools (2.18.15; “Picard Toolkit.” 2019. Broad Institute, GitHub Repository. https://broadinstitute.github.io/picard/ (accessed on 22 October 2018); Broad Institute, Cambridge, MA, USA). BAM files were imported into Partek Genomics Suite (Partek, Chesterfield MO) for further analysis. Gene sets were analyzed using Ingenuity Pathway Analysis (QIAGEN, Germantown, MD).
Table 1:
RT-PCR Primer Sequences
| Gene | F | R |
|---|---|---|
| Ctcf | CAAACAGAACCAGCCAACAGC | GGCATCTGGCTCCTTTTTGAC |
| Pl7 | AGCTGGCCTTTGTCATCAGAA | GACGAAGGAGCTGCAGAACCT |
| Inhbb | GCTCATCGGCTGGAACGA | GCCCTCACAGTAGTTCCCGTAGT |
| Lif | GGCAACCTCATGAACCAGAT | TAGGCGCACATAGCTTTTCC |
| Igf1 | ACAGGCTATGGCTCCAGCAT | GCTCCGGAAGCAACACTCA |
| Ramp3 | GGAGCCACGTGTGACCTACTG | AGCCCACACTGGACACAGAAT |
| Ihh | CATCTTCAAGGACGAGGAGAACA | CATGACAGAGATGGCCAGTGA |
| Fst | TCTTCTGGCGTGCTTCTTGA | CCTCCGTTTCTTCCGAGATG |
| Errfi1 | GATGAGGCCGACAGTGAGGTA | TGAAGGCGCAGAGTCTTCTAAA |
| Npl | TCGCGGAGGAATGGGTTA | CGTTTAGTGCTCCCACGTGAA |
Western Blot
Uterine protein was isolated as previously described (21). 30 μg of protein was separated using a mini-PROTEAN TGX Precast Gel (Bio Rad, Hercules CA) and transferred to a nitrocellulose membrane using Bio Rad Trans-Blot Turbo. The membrane was blocked using 5% Blotto (Santa Cruz Biotech, Santa Cruz, CA) then probed overnight with Abcam anti CTCF (Abcam Cat# ab70303, RRID:AB_1209546) diluted 1:500 (21-day old samples) or Santa Cruz Biotechnology (Cat# sc-271474, RRID:AB_10649800) mouse monoclonal diluted1:250 (adult samples) in 5% Blotto. Proteins were normalized using anti b-ACTIN (Santa Cruz Biotechnology Cat# sc-1616, RRID:AB_630836) diluted 1:5000. Complexes were detected using LI-COR IR-labeled secondary antibodies (LI-COR Biosciences, Lincoln, NE) diluted 1:20,000 in 5% Blotto and scanned and quantified using a LI-COR Fc instrument and Image Studio Software.
Immunohistochemical Analysis
To detect CTCF, uterine tissue sections were de-paraffinized and antigen retrieval was done in a Biocare Decloaking instrument (Biocare Medical Pacheco, CA) for 5 minutes in Tris Antigen Unmasker pH9 (Vector Labs, Newark CA). Bethyl A300–543A Anti CTCF (Bethyl Cat# A300–543A, RRID:AB_2086903 Bethyl Laboratories Montgomery TX) was used at 1:500. F4/80, CASP3 and Ki67 IHC was done in the NIEHS Immunohistochemistry Core Lab. For F4/80 and CASP3 antigen retrieval was done in 1xEDTA Decloaker (Biocare Medical) and detected with BioLegend (San Diego CA) Anti-F4/80 (BioLegend Cat# 123102, RRID:AB_893506) diluted 1:500 or rabbit monoclonal anti-Caspase 3 (Cell Signaling Technology Cat# 9664s diluted 1:2000. For Ki67, antigen retrieval was done in 1x citrate buffer (Biocare) for 15 minutes and detected rabbit polyclonal anti-Ki67 (Abcam Cat# ab16667, RRID:AB_302459) at a 1:100 dilution.
CTCF ChIPseq
Five uteri from stock C57Bl/6 ovariectomized mice were frozen and pooled and shipped to Active Motif Inc (Carlsbad CA) for CTCF ChIPseq library preparation using Active Motif anti CTCF (Active Motif Cat# 61311, RRID:AB_2614975) and sequenced by the NIEHS Sequencing core. Raw ChIP-seq reads were processed and aligned to the mouse reference genome mm10 using Bowtie (parameters: -p 10 -q --phred33-quals -m 1 -v 2 -S) (22). The reads were de-duplicated, and peaks were called using MACS2 (23) (q-value cutoff 0.0001) relative to a pooled input sample from the same chromatin prep GEO GSE125972, sample GSM3587132) that was re-processed using the Bowtie pipeline. Peak Annotation and Visualization (PAVIS; (24)) was used to compare the locations of CTCF peaks relative to genes. Known motifs in CTCF peaks were identified using HOMER findMotifs (25). Partek list manager Venn diagram tool was used to overlap RNAseq DEG lists and genes <3 kb from CTCF peaks.
T-score computation
The Signature Analysis webtool was used to compute T-scores (26) between our 392 CTCF-dependent progesterone responsive gene signature (Table S6) and microarrays of 79 Stage I endometroid tumors (GSE17025) or 145 mixed stage endometroid tumors (GSE120490).
3C Assays
3C assays were conducted as described in (21) digesting with HindIII for the Ihh interactions and BglII for the Fst interactions. Primers used to amplify ligated interacting regions and control adjacent regions are shown in Table 2. Ligated region dCT values were calculated relative to control adjacent regions and ddCT relative to the mean dCT of the adult control samples. Negative control was DNA that was cut with restriction enzyme but not ligated.
Table 2.
3C Primers
| Product | F | R |
|---|---|---|
| Ihh TSS-63 | CATCCCTACTCCTATTTCCAATCT | AAGGGCACCTTGAATTCTCA |
| Ihh cont | CCTTCTTCTCTCTAGCCCAAAC | CCTCTGGAATTCACGACAGAAT |
| Fst TSS-350 | CTTTGGCAGAAGCAATGAAGTC | CACTTCCTTAGTCTACACGAGATA |
| Fst cont | TTCAGCTGCACCTTGGATAG | GGATGACAAGAGTCCTGGTAAC |
Results
PgrCre+Ctcf-floxed (Ctcfd/d) females were sterile, producing no pups after being continuously housed with fertile males for 100 days, whereas Ctcf-floxed (Ctcff/f) female littermates produced 4 litters each and a mean total of 35.8 pups (data not presented), indicating uterine Ctcf is essential for reproductive function. Gross morphological analysis of the uteri of Ctcfd/d mice revealed a hypoplastic uterus by 6 weeks of age, with smaller overall size and tissue weight (Fig 1A, Fig. S1A). Histological analysis showed the uterus has a reduced stroma layer and a decrease in the number of endometrial glands (Fig 1B,C, Fig. S1B). Additionally, stroma from older mice (>22 weeks) has indications of hyalinization and fibrosis (Fig S1B), suggesting inflammation and tissue remodeling (27). Examination of the uteri of 21 day old prepubertal females showed normal gross and histological morphology with the exception that fewer endometrial glands were present (Fig 1B,C). Ctcfd/d females ovulated after administration of exogenous gonadotrophins, exhibited normal ovarian histology and attained comparable serum progesterone and estradiol levels (data not presented).
Figure 1. Ctcf-null uteri develop post-pubertal hypoplasia.
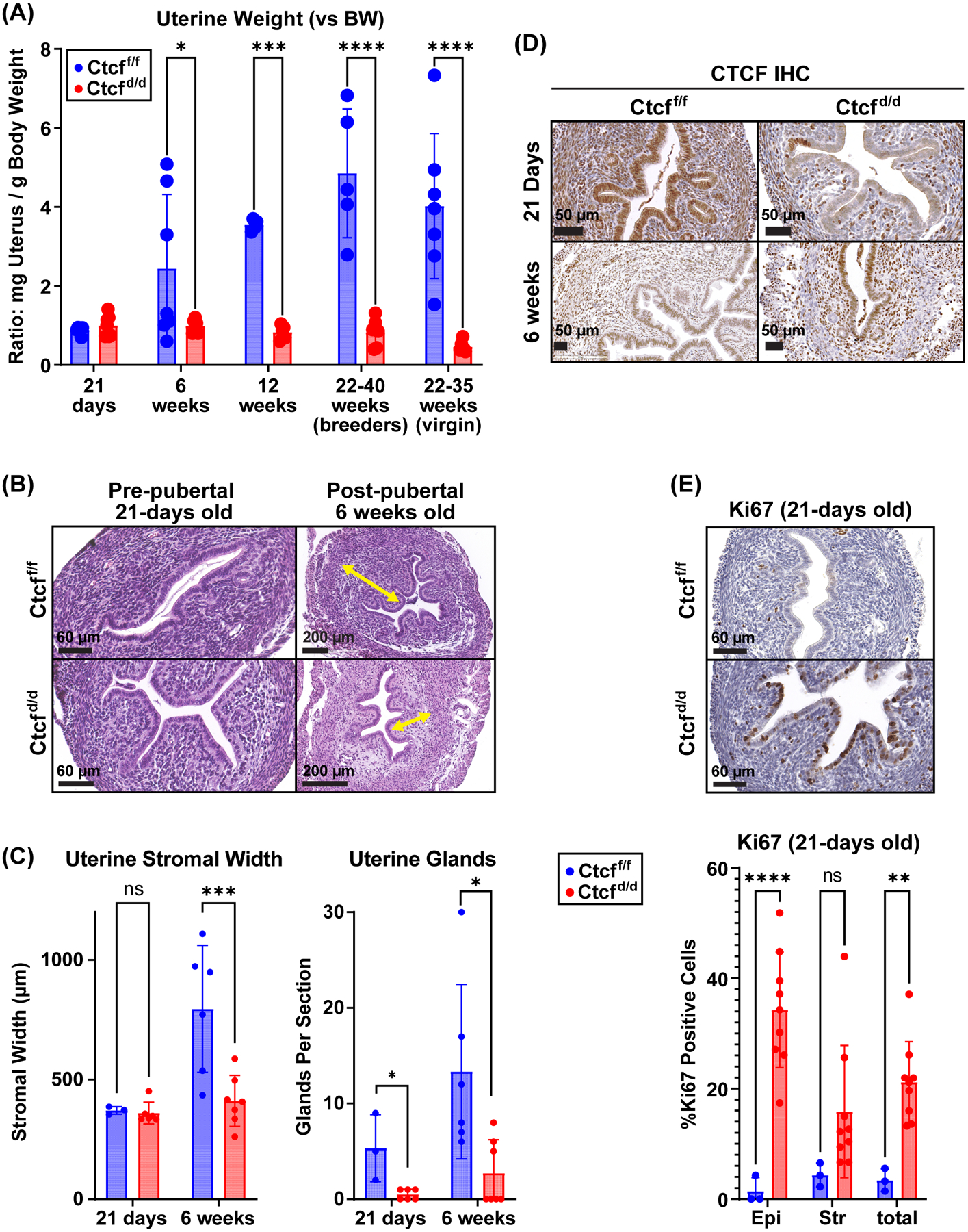
A. Uterine weights of uteri from Ctctf/f and Ctcfd/d mice (mg uterus normalized to g body weight); pre-pubertal (21 days) and post-pubertal (12 weeks and older). N=4 to 7 for each group. * p<0.05; ***p<0.001; ****p<0.0001 by 2 way ANOVA with uncorrected Fisher LSD post test.
B. H&E stained uterine tissue sections from prepubertal (21-days old) and post-pubertal (6 weeks old) Ctcff/f and Ctcfd/d mice. Yellow arrows indicate stromal thickness. Scale bars indicate 60 or 200 μm.
C. Stromal width and number of glands in uterine tissue. N=3 to 6 for each group. * p<0.05; p<0.001; by 2 way ANOVA with uncorrected Fisher LSD post test.
D. IHC for CTCF protein in uterine sections from prepubertal (21 days) and post pubertal (6 weeks) Ctcff/f and Ctcfd/d mice.
E. Analysis of Ki67 proliferative marker in 21-day old Ctcff/f and Ctcfd/d mice. Ki67 positive cells are brown. The % Ki67 positive cells were quantified in the luminal epithelium (epi) and the stroma (str) cells. **p<0.01; ****p<0.0001 by 2 way ANOVA with uncorrected Fisher LSD post test.
Analysis of the efficiency of CTCF deletion in the uterus revealed initial ablation with later reappearance of CTCF positive cells. Immunostaining for CTCF protein demonstrated that at 21 days old, most epithelial and stromal cells of Ctcfd/d uteri lacked CTCF protein (Fig 1D) and that most epithelial and stromal cells in the 6 week old Ctcfd/d uterine tissue expressed CTCF (Fig 1D). RT-PCR of RNA from 21 day old uterine samples indicated Ctcf transcript was decreased by 60% in Ctcfd/d vs Ctcff/f, whereas by 6 weeks old, the decrease was more modest (16% of Ctcff/f; Fig S1C). Western blot revealed CTCF protein of 21-day old Ctcfd/d uterus was decreased to 46% of the level in Ctcff/f, whereas the level of CTCF protein in 6-week old Ctcfd/d uterus was the same as in Ctcff/f (Fig S1D). Although the deletion of CTCF in the uteri was not sustained, the re-appearance of CTCF positive cells in post-pubertal uterine tissue did not restore the uterine size or stroma compartment (Fig 1A, B,C Fig S1B). Overall, our observations indicate that pubertal uterine growth and maturation relies on CTCF-dependent processes and suggests that lack of CTCF prevents growth or survival of uterine cells resulting in uterine hypoplasia and CTCF expression in remaining cells. To discover CTCF dependent mechanisms that contribute to the observed phenotype, we utilized prepubertal Ctcff/f and Ctcfd/d uterine tissue as it is more comparable in terms of size and cellular composition and the Ctcf deletion is more prominent at this developmental stage.
First, we evaluated the uterine tissue for indications of altered proliferation or apoptosis using immunostaining for the proliferative marker, Ki67 and the apoptotic enzyme Cleaved Caspase 3 (CASP3). Few Ki67 positive cells were detected in Ctcff/f uteri (Fig 1E), in contrast many Ki67 positive cells, mainly in the luminal epithelium, were present in Ctcfd/d samples (Fig 1E), indicating that deletion of CTCF increased cellular proliferation. No CASP3 positive cells were seen in Ctcff/f samples, whereas few (1–2) CASP3 positive cells were seen in Ctcfd/d sections (Fig S1E). The observation of increased proliferation and minimally increased apoptosis is unexpected, considering the appearance of hypoplasia in post-pubertal uteri.
Next, we compared the transcriptomes of Ctcff/f and Ctcfd/d uterine tissue, revealing 1257 differentially expressed genes (DEG; 846 increased, 411 decreased; 2- fold, FDR<0.05; Table S1) following Ctcf deletion. Increased expression of the majority of DEG suggests a role for CTCF in repression of uterine genes, potentially via maintenance of loops. Ingenuity Pathway Analysis (IPA) revealed, among others, CTCF as a decreased pathway (Fig 2A; Table S2), consistent with the deletion of this gene. The DEGs that IPA used to derive this finding include decreased expression of several protocadherin (Pcdh) genes (Fig. S2A and B), mostly the “clustered” b and g Pcdhs (cPcdh).
Figure 2. Transcriptional profile indicates prepubertal Ctcf deletion leads to an inflammatory response.
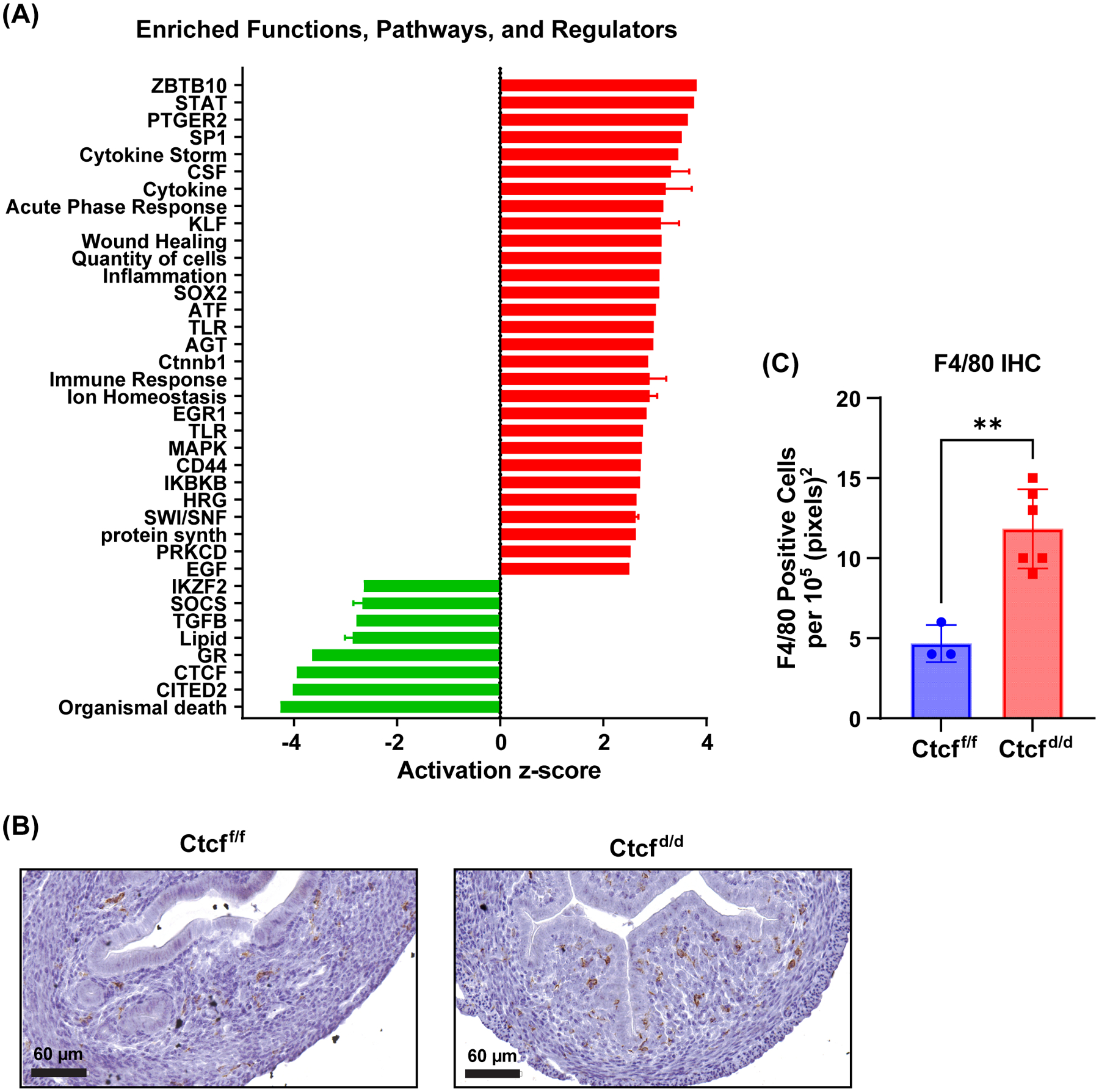
A. Summary of functions and pathways enriched in prepubertal uterine Ctcfd/d vs. Ctcff/f gene set. Activation z-score indicates degree and direction of impact on the pathway (activation (red) or inhibition (green)).
B. IHC for F4/80 reveals increased signal (brown) in uterine stroma of Ctcfd/d indicative of monocyte infiltration.
C. F4/80 positive cells per 105 (pixels)2 n=3–5 t-test **p<0.01
The DEG also correlate with activation of inflammation and cytokine signaling pathways (Fig 2A; Table S2), including STAT signaling, cytokine storm, acute phase response and inflammation together with inhibition of glucocorticoid receptor (GR) signaling. GR signaling is anti-inflammatory (28), thus decreased activity aligns with increased inflammation. We examined uterine tissue for expression of the monocyte protein, F4/80, to evaluate the inflammatory response finding. More F4/80 positive cells were detected in sections from Ctcfd/d uterine tissue than in sections from Ctcff/f (Fig 2B and C), consistent with increased inflammation and tissue remodeling as a result of Ctcf deletion. Inflammation, growth arrest, proliferation, resistance to apoptosis and senescence-associated secretory phenotype (SASP) are potential indicators of senescence. Senescent cells are a feature of normal tissue development (29), however, altering senescent processes can lead to chronic inflammation and fibrosis (29). Genes encoding several indicators of senescence, including the anti-apoptotic BCL2 apoptosis regulator, Bcl2, indicator of growth arrest, p21 (cyclin dependent kinase inhibitor 1A, Cdkn1a), indicators of SASP, beta-galactosidase (Glb1) and several metalloproteinases (Mmps) are increased in Ctcfd/d (Fig S2C and D). This suggests altered senescence signals in Ctcfd/d might contribute to the clearence of CTCF-null uterine cells and development of uterine hypoplasia and fibrosis that we observed (Fig 1 and S1). The transcriptome was also consistent with decreased TGFβ signaling (Fig 2A), a pathway essential for uterine development, function and homeostasis (30). We also analyzed the Ctcf-dependent genes’ most significantly impacted upstream regulators that were classified as ligand-dependent nuclear receptors or transcriptional regulators to look at potential effects via these factors (overlap p value<0.05, absolute value of activation Z-score >1.2; Table S2). The inhibited regulators included GR and CTCF as well as chromatin and transcriptional mediators such as CITED2, HDAC4, SMARCA5. Increased cytokine signaling (IRFs, STAT1) and increased activity via several transcription factors (SP1, AP1, KLF 4 and 6) as well as increased CTNNB1, EGR1, and CEBP activity are apparent (Table S2).To find potential CTCF targets, we analyzed CTCF ChIP-seq from uterine tissue of ovariectomized adult mice (Fig S3). Most CTCF peaks were enriched at and within 10 kb of annotated transcripts (Fig S3A). CTCF or CTCFL motifs were most significantly enriched, with bHLH, Nanog, NF-1, bZIP/AP-1 and HOX motifs less significantly enriched (Fig S3B). Based on a study indicating a role for promoter-proximal CTCF in promotion of enhancer driven gene activation (31), we filtered the DEG to focus on the 439 genes with TSS <3kb from a CTCF ChIPseq peak in adult uterine tissue (Fig 3A,Table S3; 167 genes decreased, 272 increased following Ctcf deletion). Analysis of significantly enriched functions and signaling pathways of the putative CTCF target genes confirmed the decreased activity of CTCF, and increased activity of cytokine and inflammation associated components previously observed using all DEG (Figs 2A and 3B; Table S4), as well as inhibition of the anti-inflammatory nuclear receptor, NR3C1 (glucocorticoid receptor). Notably, within the putative CTCF targets, stem cell factors, Aldehyde dehydrogenase family 1, subfamily A3 (Aldh1a3), Leukemia inhibitory factor (Lif), Eomesodesmin (Eomes), and Leucine rich repeat containing G protein coupled receptor 5 (Lgr5), were expressed at increased levels in Ctcfd/d uterine RNA (Fig 3A), which correlates with indications of CTCF disruption in some uterine tumors (32, 33).
Figure 3. Putative CTCF target genes include stem cell factors.
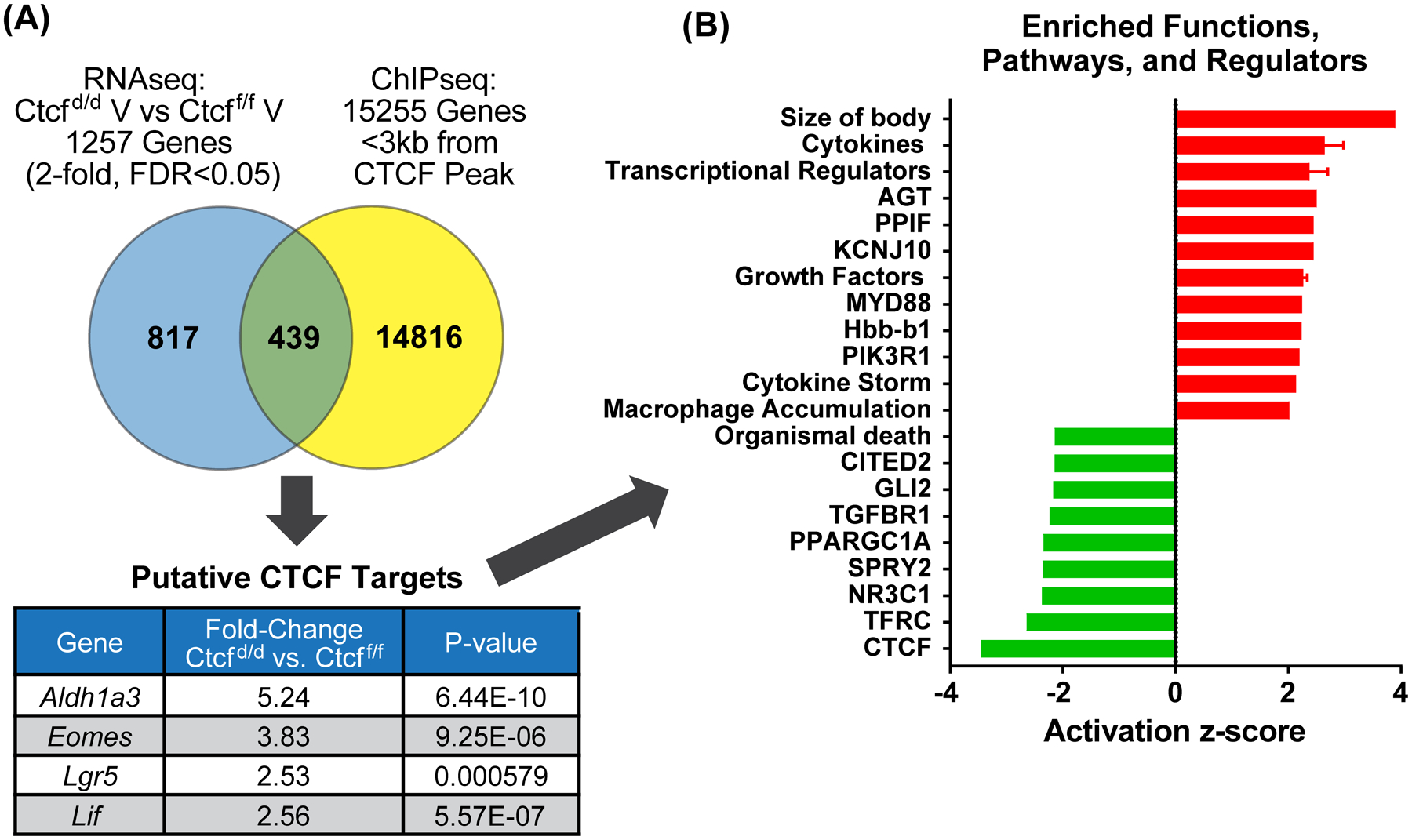
A. Venn diagram comparing prepubertal uterine Ctcfd/d vs. Ctcff/f gene set to genes <3kb from a CTCF ChIPseq peak. Values for four stem cell genes are listed in table.
B. Summary of functions and pathways enriched in the putative CTCF target gene set (439 genes).
The primary uterine stimulus at the onset of puberty is as the result of ovarian secretion of estrogen and progesterone with the initiation of estrus cycling. Using RT-PCR, we evaluated panels of estrogen or progesterone responsive uterine transcripts after administering either hormone to 21-day old Ctcff/f and Ctcfd/d females. Comparable induction of estrogen response was observed (Fig. 4A), whereas some but not all progesterone responsive genes assayed were sensitive to Ctcf deletion (Fig. 4B). Indian hedgehog (Ihh) exhibited decreased basal transcription, but was increased by P, P response of Follistatin (Fst) was lost, P response of ERBB receptor feedback inhibitor 1 (Errfi1) was reduced, and N-acetylneuraminate pyruvate lyase (Npl) was comparably increased by P. RNAseq was used to compare the transcriptomes of progesterone treated Ctcff/f and Ctcfd/d uteri to comprehensively assess the CTCF dependence. Similar to the genes examined by RT-PCR (Figs 4B), subsets of progesterone response patterns are indicated (Fig 4C,D, Fig S4, Table S5). One subset consists of CTCF independent genes, which are regulated by progesterone in both Ctcff/f and Ctcfd/d (Fig S4C, Table S8), and a second subset consists of CTCF dependent genes, which have attenuated response to progesterone in Ctcfd/d (Fig 4D, Fig S4B). The maintenance of comparable progesterone response for the gene in the first subset indicates that progesterone signaling is intact and functioning in uterine cells. The CTCF dependence of a subset of, but not all, progesterone responsive genes, is consistent with a role for CTCF in mediating promoter-enhancer interactions, which would be expected to vary from gene to gene based on the location of each enhancer relative to its target gene and each one’s need for interaction.
Figure 4. Ctcf deficiency causes progesterone insensitivity in a subset of uterine genes.
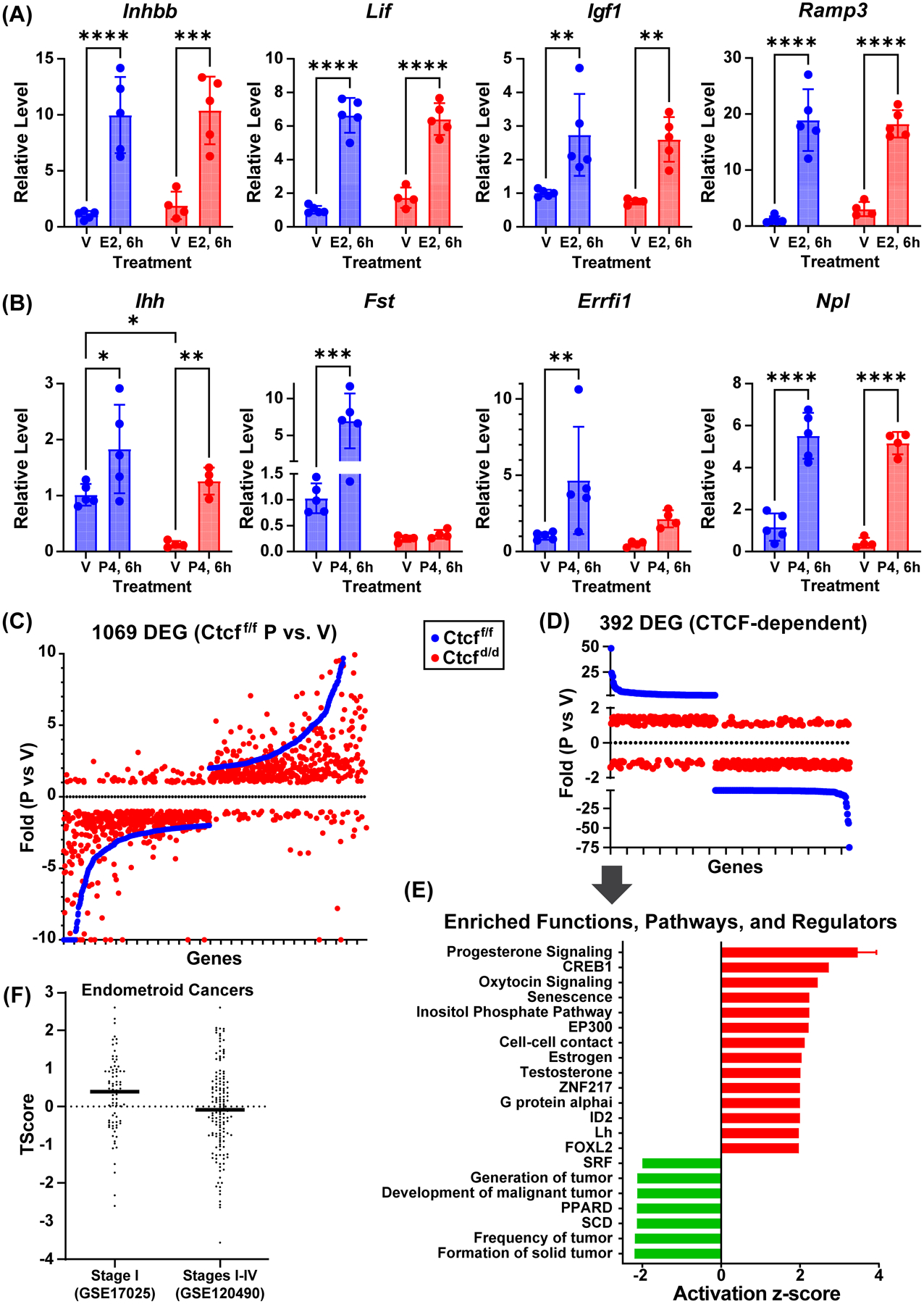
A. RT-PCR of estrogen responsive genes from prepubertal (21 days) Ctcff/f and Ctcfd/d uterine RNA. Mice were treated for 6 hours (6h) with sesame oil vehicle (V) or estrogen (E2). All genes were normalized to Rpl7. N=5 *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001 by 2 way ANOVA with uncorrected Fisher LSD post test.
B. RT-PCR of progesterone responsive genes from prepubertal (21 days) Ctcff/f and Ctcfd/d uterine RNA. Mice were treated for 6 hours (6h) with sesame oil vehicle (V) or progesterone (P4). All genes were normalized to RPL7. N=4–5 *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001 by 2 way ANOVA with uncorrected Fisher LSD post test.
C. Scatter plot of genes that are progesterone responsive in Ctcff/f uterus (blue) in rank order of fold change along X axis together with fold changes of same genes in Ctcfd/d uterine RNA samples (red).
D. Scatter plot illustrating Ctcff/f progesterone responsive uterine genes (blue) that are progesterone insensitive in Ctcfd/d (red; absolute value fold change <1.5).
E. Summary of functions and pathways enriched in Ctcff/f progesterone responsive uterine genes that lack response in Ctcfd/d.
F. T-Scores calculated for CTCF-dependent progesterone target genes relative to transcriptomes from Stage I endometroid cancer (GSE17025) or from Stage I to Stage IV endometroid cancer (GSE120490).
Because the biological outcome of post-pubertal uterine hypoplasia indicates that pubertal growth and maturation is CTCF dependent, we evaluated what progesterone is doing in CTCF-intact uterus that is missing in CTCF-deleted tissue. We analyzed pathways enriched in the progesterone responsive CTCF-dependent gene signature (392 genes Table S6) of Ctcf-intact mice (Figs 4D and 4E, Table S7). These progesterone regulated genes were enriched for functions associated with inhibition of tumorigenesis and increased cell-to-cell contact (Fig 4E), consistent with CTCF’s role as a tumor suppressor, and suggests that CTCF works with PGR mediated signaling in this capacity. The values for the 13 genes used by IPA to derive the finding of increased cell-to-cell contact are shown in Fig S5A. Analysis of cBioPortal datasets (13, 14) revealed that inactivating mutations are found in endometrial cancer datasets for six of the 13 genes (Cadherin 11 (Cdh11), Claudin 2 (Cldn2), Follistatin (Fst), Leucine rich repeat transmembrane neuronal 3 (Lrrtm3), Muscle, skeletal, receptor tyrosine kinase (Musk), Tumor necrosis factor (ligand) superfamily, member 11 (Tnfsf11), consistent with decreased cell-cell contact, whereas progesterone treatment increases their expression in the mouse uterus (Fig S5A), correlating with stabilized contact between cells.
Progesterone can inhibit the progression of earlier stage endometrial cancers (33, 34), with progesterone resistance seen as the cancer progresses to later stages. We computed “T-scores” (26) representing projected activity of our CTCF-dependent progesterone responsive gene signature in comparison to two reposited endometrial cancer microarray datasets. The first included Stage I (early) endometroid tumors (GSE17025), the second was a mixture of Stages I, II, III and IV endometrioid tumors (GSE120490). The T-score for the early-stage selective dataset in relation to our gene signature was higher than the dataset containing multiple stages (Fig 4F), consistent with our signature more closely representing progesterone-sensitive early-stage endometrial tumors.
We also note increased activity of not only progesterone signaling, but also testosterone and estrogen signaling in CTCF-dependent progesterone responsive genes (Fig 4E). Estrogen signaling is involved in uterine pubertal maturation, evidenced by uterine hypoplasia of Esr1-null mice (3). Testosterone signaling within the uterus is less studied, but has clinical relevance for individuals with polycystic ovarian syndrome, who are exposed to increased levels of serum testosterone (35). Additionally, we observed inhibition of SRF signaling, which has also been observed after myometrial deletion of PGR (36). Oxytocin signaling, which is involved in parturition (37), is increased as well. The CTCF-dependent progesterone responsive gene signature is enriched for activation of senescence, implying roles for senescent cells in tissue homeostasis.
The impact of CTCF-ablation on expression of Indian hedgehog (Ihh) was complex, with CTCF-dependent basal expression and CTCF-independent progesterone response (Fig. 4B). IHH is a uterine mediator of endometrial function, as demonstrated by decreased fertility after its deletion in mouse uterus (38). Previous work has identified a progesterone dependent uterine enhancer 19kb 5’ of the Ihh gene (17), and further work has revealed an additional PGR ChIP peak 39 kb 5’ of the Ihh gene (19). We examined CTCF ChIPseq peaks as well as HiC loop calls to assess potential distal regulatory mechanisms (Fig 5A). We observed a loop anchor and CTCF peak 63kb 5’ of Ihh that interacts with a CTCF peak and loop anchor 12kb 5’ of Ihh as well as within the Ihh gene. The CTCF motif in the 63kb 5’ CTCF peak is convergent with the CTCF motifs in both the CTCF peaks 12kb 5’ and in the Ihh gene. To determine the potential contributions of the 39 kb 5’ PR binding site or the 63 kb 5’ CTCF binding region, we utilized CRISPR-Cas9 deletion to remove each of these. We confirmed our previous observation that deletion of the 19kb 5’ enhancer (Ihh 19 KO) decreased basal Ihh expression and prevented progesterone induction (Fig. 5B). It is intriguing that this enhancer does not include a CTCF peak, but that its deletion recapitulates the lowered basal expression seen after Ctcf deletion, suggesting that CTCF dependent interactions are involved in activity of this enhancer. Deletion of the 39kb 5’ PR binding site (Ihh39KO) resulted in normal basal expression, but attenuated progesterone response (Fig. 5B). To evaluate the interdependence of the two progesterone dependent enhancers, progesterone response in compound 19 kb+39 kb heterozygotes (Ihh19het39het) was examined, revealing an intermediate impact of normal basal expression together with progesterone induction of Ihh that is further decreased relative to the 39kb enhancer deletion (Fig. 5B). Deletion of the CTCF binding region 63kb 5’ of Ihh (Ihh63KO) decreased Ihh progesterone response (Fig. 5C). We utilized 3C to confirm the interaction between the Ihh gene and 63kb 5’ region (Fig. 5A, 5D).The interaction was detected in post-pubertal (“control”) uterine tissue as well as in prepubertal Ctcff/f and Ctcfd/d tissue (Figs 5D), indicating the distal 63kb region can interact with Ihh and potentially contributes to Ihh regulation by progesterone by facilitating interaction with intervening PR-binding enhancers 19kb and 39 kb upstream. However, the qualitative nature of a 3C assay means our positive result does not preclude the possibility that Ctcf deletion alters the interaction. As we have previously reported (19), analysis of PGR and ESR1 ChIPseq and HiC data from human endometrium and epithelial organoids in regions near IHH reveals ESR1 and PGR binding at 5’ enhancers 20 kb and 70 kb from IHH, which resemble the mouse 19 kb and 39 kb 5’ enhancers (Fig S6A and Fig 5A). Additionally, interaction between IHH and an enhancer 100 kb 5’ of IHH is similar to the interaction between the mouse Ihh gene and a 63 kb distal region (Fig S6A and Fig 5A). This suggests conserved mechanisms of hormonal regulation. Conservation analysis indicates that these enhancer and interacting regions contain stretches of homology with mouse DNA (Fig S6A).
Figure 5. Chromatin structures and hormone dependent enhancers mediate uterine Ihh regulation.
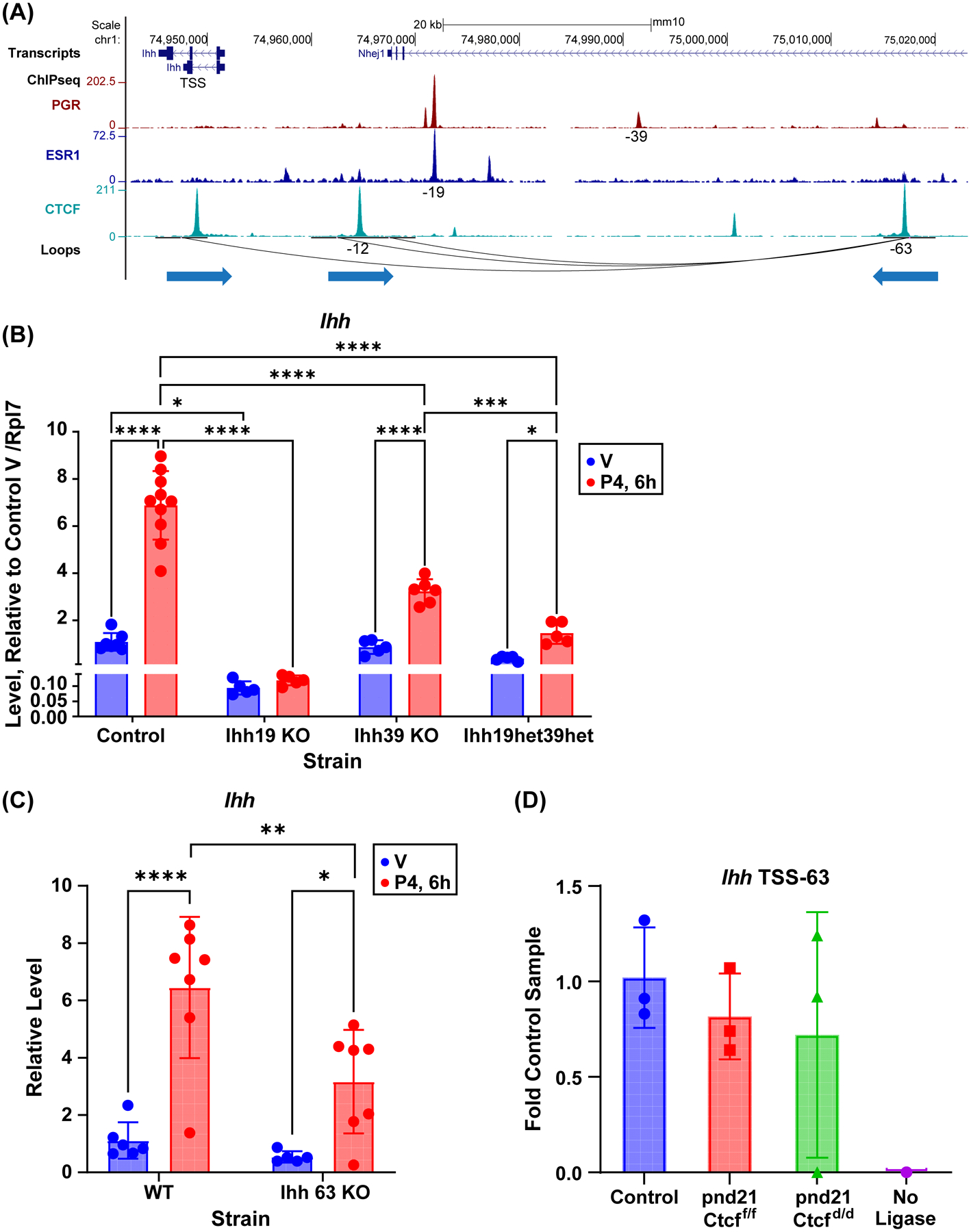
A. PGR, ESR1 and CTCF ChIPseq and loops near Ihh gene. Both ESR1 and PGR interact with an enhancer 19 kb 5’ (–19) of the Ihh gene. PGR interacts with a second enhancer 39 kb 5’ of Ihh (–39). Interactions with Ihh occur via CTCF bound loop anchors at the Ihh gene and 63 kb 5’ (–63). An interaction between regions 12 kb 5’ of Ihh (–12) and the −63 region are also observed. The direction of teach CTCF motif is indicated by arrow under the loop anchors.
B. RT-PCR of Ihh in ovariectomized adult uterine RNA from mice with deletion of enhancers −19 (Ihh19 KO) or −39 (Ihh39 KO), mice that are heterozygous for deletion of one copy each of the −19 and −39 enhancers (Ihh19het39het), and control littermates. Mice were treated for 6 hours (6h) with sesame oil vehicle (V) or progesterone (P4). N=5–10 *p<0.05; ***p<0.001; ****p<0.0001 by 2 way ANOVA with uncorrected Fisher LSD post test.
C. RT-PCR of Ihh in ovariectomized adult uterine RNA from mice with deletion of a CTCF site at −63 (Ihh 63 KO) or their control littermates (WT). Mice were treated for 6 hours (6h) with sesame oil vehicle (V) or progesterone (P4). N=5–7 *p<0.05; **p<0.01; ****p<0.0001 by 2 way ANOVA with uncorrected Fisher LSD post test.
D. 3C PCR to detect ligation between HindIII fragments at the Ihh TSS and the −63 CTCF binding loop anchor (IHH TSS-63) in adult mice (control) or prepubertal (pnd21) Ctcff/f and Ctcfd/d uterine DNA.
Progesterone induction of Fst was completely Ctcf dependent (Fig. 4B). Fst is crucial to uterine function (39), as its deletion impairs the progesterone-dependent decidual response. Multiple potential PGR-binding enhancers 3’ (270, 190, 145, 88 and 55 kb) of the Fst gene are within a large >350kb interacting region with CTCF peaks at flanking loop anchors (Fig 6A). The interaction between the distal 350 kb region and the Fst gene was confirmed using 3C (Fig 6B), indicating distal interaction might facilitate Fst expression by bringing PR-binding regions closer to the gene TSS. We examined PGR and ESR1 ChIPseq from human endometrium and HiC data from epithelial organoids in regions flanking the human FST gene and observe several potential PGR or ESR1 binding enhancers (80 kb, 400–450 kb, 680 kb) between the FST gene and a CTCF-binding loop end 730 kb 3’ of the gene (Fig S6B). As with the IHH gene, there are regions of conservation with mouse DNA that correspond to these potential regulatory regions, suggesting that both IHH and FST have conserved mechanisms of hormonal regulation in the uterus.
Figure 6. Chromatin structures and PGR binding enhancers interacting with Fst gene.
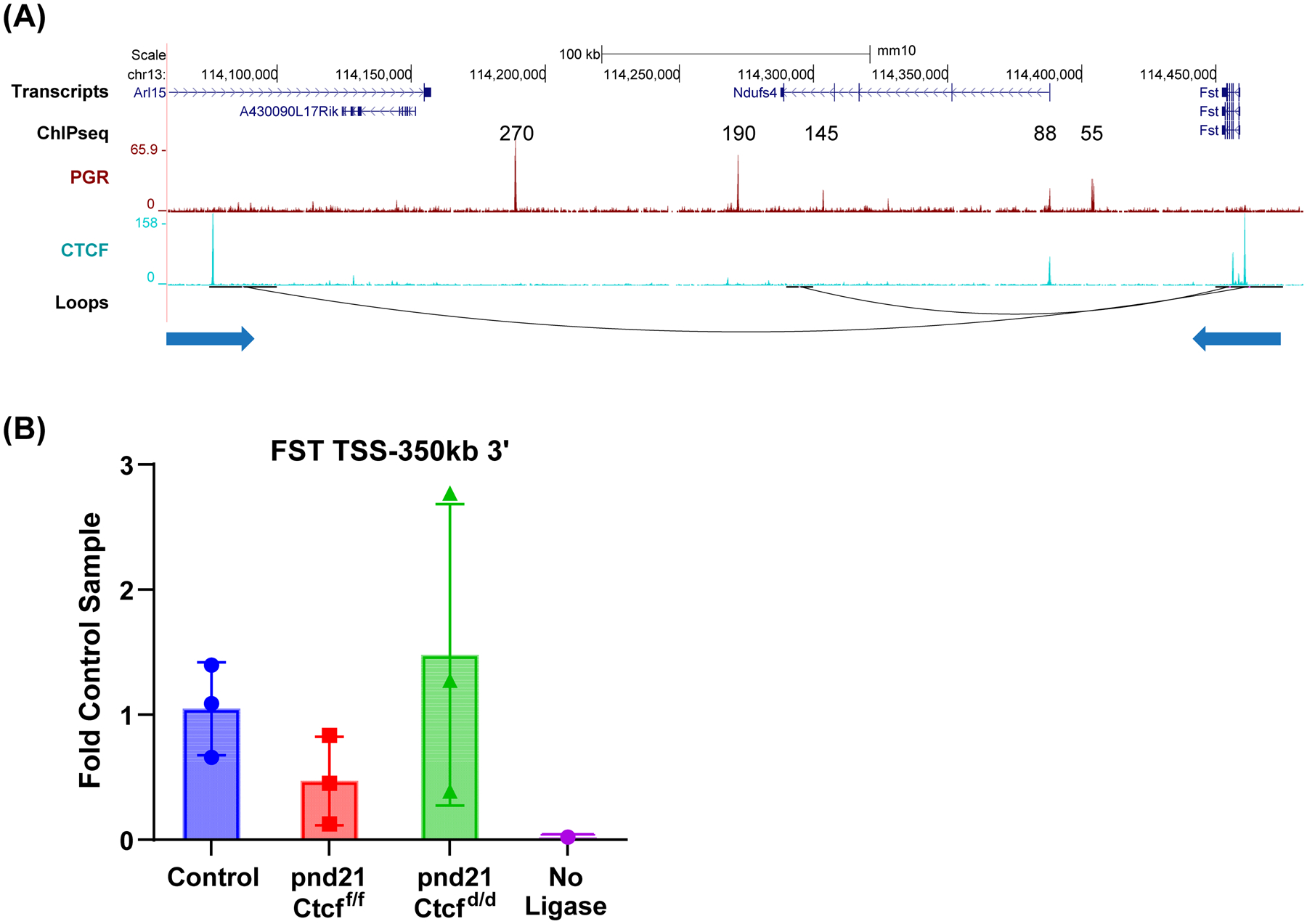
A. PGR and CTCF ChIPseq and loops near the Fst gene. Interactions with Fst occur via CTCF bound loop anchors at the Fst gene and 350 kb 3’ of Fst in the Arl15 gene. The direction of each CTCF motif is indicated by the arrow underneath the loop anchor.
B. 3C PCR to detect ligation between BamHI fragments at the Fst TSS and the 350kb 3’ CTCF binding loop anchor (FST TSS-350 3’) in adult mice (control) or prepubertal (pnd21) Ctcff/f and Ctcfd/d uterine DNA.
Discussion
Puberty is a critical and sensitive developmental window for establishment of optimal reproductive tract physiological response later in life. It is not surprising that this corresponds to the appearance of the most dramatic physiologic impact of Ctcf deletion on uterine cells, as the onset of ovarian estrogen and progesterone production initiates rapid coordinated growth and development of uterine tissue. Inhibition of cellular proliferation and survival is a hallmark of systems lacking Ctcf (10, 40). We confirmed deletion of Ctcf in terms of transcript and protein expression in our pre-pubertal samples but observed CTCF expression at levels near to those of Ctcff/f in Ctcfd/d post-pubertal samples (Fig 1D, S1C,D). Taken together with the hypoplastic appearance of the uterine tissue (Figs 1, S1), this likely reflects selective growth or survival of CTCF positive cells through the pubertal period. We did observe elevation in Ki67 positive cells (Fig 1E), however the proliferating cells included CTCF-null cells (Fig 1D). Additionally, we observed minimal indications of apoptosis in Ctcfd/d tissue (Fig S1E). However, our observations are suggestive of an impact on cellular senescence. Several studies have observed impacts of senescence on both normal and pathological uterine biology. For example, a subpopulation of endometrial stromal cells undergo senescence and secrete endometrial receptivity mediators (41). Conversely, endometrial decidual cell senescence has been associated with implantation failure (42, 43) and recurrent pregnancy loss (44). Expression of genes encoding indicators of SASP and senescence were increased in our Ctcfd/d samples (Fig S2C and D). Additionally, Ctcf deletion increased F4/80+ cells (Fig 2B), which have been shown to facilitate clearance of senescent cells during postpartum uterine remodeling in mice (45). Multiple inflammation associated genes, including CD68, and multiple mediators of chemokine signaling are elevated in the CTCF associated gene set (Fig S2A), as was also observed in a study of Ctcf deletion in brain (46). Increased expression of metalloproteinases, indicative of SASP, following Ctcf deletion was notable (Fig S2C and D). Increased MMP7 and MMP12 expression has been observed in endometrial adenocarcinomas (47, 48). Elevated MMP7 was associated with poor prognosis for disease free survival in endometrial cancer (47) and MMP12 is expressed in more invasive stage III endometrial tumors, suggesting a role in the invasion (48). Transient SASP and occasional senescent cells are expected in developing tissue, while chronic SASP with inflammation and increased prevalence of senescent cells is detrimental, often causing fibrosis (29). We propose that enrichment of senescence activity in the CTCF-dependent progesterone responsive gene signature (Fig 4E) represents a transient aspect of normal tissue homeostasis, whereas chronic SASP and senescence as a result of Ctcf deletion (Fig 2, S2C,D) contributes to the uterine hypoplasia and fibrosis. Since the floxed region of the CTCF is large, spans >35 Kb, this would decrease the efficiency of Cre action and there may be a small fraction of the cells that escape recombination. Their emergence may be slow and hindered by the senescence/inflammation/tissue remodeling phenotype. This selected cell population may have a phenotype that does not reflect the normal CTCF expressing cells which makes it incapable of rescuing the hypoplasia phenotype.
Other investigators have faced challenges when utilizing the Cre-lox system to disrupt Ctcf and chromatin structure, including studies in cardiomyocytes. Even after >80% decrease of CTCF using a cardiomyocyte specific Cre, residual CTCF was maintained in chromatin and 3-dimentional structures were not impacted (49). Other approaches that used auxin-inducible degrons to rapidly and more completely deplete CTCF or Cohesin to <1% have shown the relative impacts of CTCF and Cohesin in chromatin structure, where Cohesin depletion effectively disrupts looping, but CTCF deletion maintains looping that has become disordered (50). In our study, because some cells retained CTCF expression, and because of the observation that loops can remain even after complete CTCF deletion (50), it is not surprising that interactions in specific chromatin regions we tested (Ihh TSS-63kb and Fst TSS-350kb; Figs 5D and 6B) could still be detected after Ctcf deletion (Figs 5D, 6B). The 3C assays used to detect interactions are not quantitative, so that our positive result indicates the presence of interaction, as might be anticipated from the presence of CTCF-positive cells in the tissue but cannot rule out disruption in interactions in CTCF-null cells. Alternatively, CTCF may regulate affected genes as a promoter proximal transcription factor, rather than through its role in chromatin dynamics. Here, for our gene regulation studies, we focused our analyses on pre-pubertal uterine samples, in which CTCF was the most effectively deleted.
Analysis of the impact of Ctcf deletion on the uterine transcriptome confirmed enrichment for decreased CTCF activity (Figs 2A and 3B; Tables S2 and S4). Genes by which IPA derived this finding included protocadherin (Pcdh) genes (Fig. S2A and B), mostly the “clustered” b and g Pcdhs (cPcdh) previously reported to be decreased in the brain after deletion of Ctcf (51–53). cPdch are members of the cadherin family of cell adhesion molecules (54, 55) that establish specific inter-neuron connections and are most highly expressed in the central nervous system (54, 56). Though they are expressed in non-neural tissues as well, those functions are less well studied. In the mouse, Pcdha1 to Pcdha12, Pcdhb1 to Pcdhb22, and Pcdhga1 to Pchga12, Pchgb1 to Pchgb8, and Pcdhgc3 to Pcdhgc5 are arrayed in 3 clusters along Chromosome 18 (52, 56). Based on our RNAseq datasets, multiple Pcdhb and Pcdhg genes are well expressed in uterine tissue, whereas reads mapped to Pcdha genes are found at <1% of the levels of Pcdhb or Pcdhg genes (Table S9). Studies in neural cells have shown that Pcdhb and Pcdhg genes are regulated via CTCF-mediated interactions with an enhancer at the 3’ side of the Pcdhg gene cluster (52) and CTCF sites at each cPcdh gene (52). Examining the chromatin of the mouse uterus reveals similar interaction with an enhancer 3’ of the Pcghg cluster (Fig S2E) that could underlie the decreased expression of 13 Pchdb genes and 4 Pcdhg genes we observe after CTCF deletion (Fig S2B). Based on our HiC analysis, the mouse uterine Pcdha cluster does not interact with the Pcdhb and Pcdhg clusters or their enhancer (Fig S2E). Intriguingly, promoter hypermethylation and decreased expression of cPcdh members is observed in several cancers, including breast and endometrial cancer (57, 58), suggesting one mechanism for CTCF’s tumor suppressor role (59, 60) via maintenance of Pcdh mediated cell adhesion.
The primary outcome of Ctcf deletion was loss of progesterone response of a subset of uterine genes. Analysis of the progesterone mediated signals impacted by these genes highlighted decreased tumorigenesis signals (Table S7). Progesterone is effective in management of endometrial hyperplasia and cancer, with earlier stage cancers having better response, likely due to loss of PGR during progression to more advanced stages (34). It is possible that CTCF-dependent progesterone sensitivity of this subset of uterine genes is contributing to the observed impact of CTCF loss of function mutations seen in endometrial cancer (59, 60). Further, we observed elevated levels of several stem cell associated factors after Ctcf deletion, which suggests retention of undifferentiated stem cells as precursors to cancer. One study has analyzed genomic features and classified endometrial cancers into four molecular subgroups (POLE ultramutated, microsatellite instability hypermutated, copy-number low, and copy-number high) (32). CTCF mutations were mostly associated with the first 3 subgroups, (32). Phosphatase and tensin homolog (PTEN), the most frequently mutated gene in endometrial cancers, was also mostly associated with the same 3 categories as CTCF, whereas TP53 mutation occurred mostly in the copy number high group (32). Uterine cystic hyperplasia and leiomyoma was previously described in tissue from mice with global heterozygous deletion of Ctcf examined 24 months after DMBA exposure (60). We did not observe any indications of hyperplasia or cancers in our Ctcfd/d or Ctcfd/+ uterine samples (not shown), however our mice were not exposed to DMBA and all of our studies utilized mice no older than 8 months. Additionally, uterine cystic hyperplasia, as was observed in the Kemp study, is frequently observed in aging mice (60, 61). Deletion of Pten in the mouse uterus results in hyperplasia and cancer (62, 63), quite rapidly in Ptend/d and more slowly after heterozygous deletion (Ptend/+). Breeding Pten-flox mice with Ctcfd/d mice, resulting in mice heterozygous for both Pten and Ctcf (Ptend/+Ctcfd/+), did not impact the progression of hyperplasia or the appearance of cancer in comparison to Ptend/+ females (data not presented).
Our study reveals that Ctcf plays a key role in P dependent expression of uterine genes underlying optimal post pubertal uterine development, so that Ctcf deletion leads to impaired pubertal maturation resulting in development of uterine hypoplasia, inflammation and hyalinization. Although previous work in vitro has demonstrated interaction between CTCF, ESR1 and PGR activities in breast cancer derived cell lines (64–67), ours is the first observation indicating a role for CTCF in mediating hormone-driven biological processes in vivo in a tissue. Ultimately, dysregulation of hormone response at puberty leads to loss of CTCF-null cells, resulting in endometrial tissue that is not favorable to pregnancy, emphasizing a key role for CTCF in uterine homeostasis.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported [in part] by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences, project Z1AES103311-01. NIEHS Sequencing Core Lab, NIEHS Histology Core Lab, NIEHS, Immunohistochemistry Core Lab, NIEHS Knockout Mouse Core Lab. We greatly appreciate the bioinformatics advice and training provided by Tianyuan Wang. We appreciate the suggestions and feedback provided by Drs. Kenneth S. Korach and Jackson Hoffman.
Footnotes
Conflict of Interest Statement
The authors declare no conflicts of interest
Data Availability
The data that support the findings of this study are available in the methods and/or supplementary material of this article or are openly available in GEO. Previously unpublished data is reposited (RNAseq, CTCF ChIPseq) at GEO (GSE227517 and GSE227519, respectively). Other data used includes mouse HiC (GSE125972 and GSE147843) and organoid HiC (GSE200807), endometrial (ER GSE200807; PR GSE132713) and organoid PR and ER (GSE200807) ChIP, mouse ER (GSE56501), PR (GSE34927) ChIP), H3K27Ac (GSE125972).
References
- 1.Sikorska N, and Sexton T (2020) Defining Functionally Relevant Spatial Chromatin Domains: It is a TAD Complicated. Journal of Molecular Biology 432, 653–664 [DOI] [PubMed] [Google Scholar]
- 2.DeMayo FJ, and Lydon JP (2020) 90 YEARS OF PROGESTERONE: New insights into progesterone receptor signaling in the endometrium required for embryo implantation. Journal of molecular endocrinology 65, T1–T14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hewitt SC, and Korach KS (2018) Estrogen Receptors: New Directions in the New Millennium. Endocrine reviews 39, 664–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stewart CA, Fisher SJ, Wang Y, Stewart MD, Hewitt SC, Rodriguez KF, Korach KS, and Behringer RR (2011) Uterine gland formation in mice is a continuous process, requiring the ovary after puberty, but not after parturition. Biol Reprod 85, 954–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hewitt SC, Carmona M, Foley KG, Donoghue LJ, Lierz SL, Winuthayanon W, and Korach KS (2020) Peri- and Postpubertal Estrogen Exposures of Female Mice Optimize Uterine Responses Later in Life. Endocrinology 161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iguchi T, Sato T, Nakajima T, Miyagawa S, and Takasugi N (2021) New frontiers of developmental endocrinology opened by researchers connecting irreversible effects of sex hormones on developing organs. Differentiation 118, 4–23 [DOI] [PubMed] [Google Scholar]
- 7.Hewitt SC, Grimm SA, Wu SP, DeMayo FJ, and Korach KS (2020) Estrogen receptor alpha (ERalpha)-binding super-enhancers drive key mediators that control uterine estrogen responses in mice. The Journal of biological chemistry 295, 8387–8400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mazur EC, Vasquez YM, Li X, Kommagani R, Jiang L, Chen R, Lanz RB, Kovanci E, Gibbons WE, and DeMayo FJ (2015) Progesterone receptor transcriptome and cistrome in decidualized human endometrial stromal cells. Endocrinology 156, 2239–2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rubel CA, Lanz RB, Kommagani R, Franco HL, Lydon JP, and DeMayo FJ (2012) Research resource: Genome-wide profiling of progesterone receptor binding in the mouse uterus. Molecular endocrinology (Baltimore, Md 26, 1428–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bailey CG, Metierre C, Feng Y, Baidya K, Filippova GN, Loukinov DI, Lobanenkov VV, Semaan C, and Rasko JEJ (2018) CTCF Expression is Essential for Somatic Cell Viability and Protection Against Cancer. International Journal of Molecular Sciences 19, 3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Segueni J, and Noordermeer D (2022) CTCF: A misguided jack-of-all-trades in cancer cells. Comput Struct Biotechnol J 20, 2685–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Debaugny RE, and Skok JA (2020) CTCF and CTCFL in cancer. Curr Opin Genet Dev 61, 44–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, and Schultz N (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, and Schultz N (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soyal SM, Mukherjee A, Lee KY, Li J, Li H, DeMayo FJ, and Lydon JP (2005) Cre-mediated recombination in cell lineages that express the progesterone receptor. Genesis 41, 58–66 [DOI] [PubMed] [Google Scholar]
- 16.Heath H, Ribeiro de Almeida C, Sleutels F, Dingjan G, van de Nobelen S, Jonkers I, Ling KW, Gribnau J, Renkawitz R, Grosveld F, Hendriks RW, and Galjart N (2008) CTCF regulates cell cycle progression of alphabeta T cells in the thymus. Embo J 27, 2839–2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Li X, Wang T, Wu SP, Jeong JW, Kim TH, Young SL, Lessey BA, Lanz RB, Lydon JP, and DeMayo FJ (2018) SOX17 regulates uterine epithelial-stromal cross-talk acting via a distal enhancer upstream of Ihh. Nat Commun 9, 4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hewitt SC, Deroo BJ, Hansen K, Collins J, Grissom S, Afshari CA, and Korach KS (2003) Estrogen receptor-dependent genomic responses in the uterus mirror the biphasic physiological response to estrogen. Molecular endocrinology (Baltimore, Md: 17, 2070–2083 [DOI] [PubMed] [Google Scholar]
- 19.Hewitt SC, Wu SP, Wang T, Young SL, Spencer TE, and DeMayo FJ (2022) Progesterone Signaling in Endometrial Epithelial Organoids. Cells 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trapnell C, Pachter L, and Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hewitt SC, Lierz SL, Garcia M, Hamilton KJ, Gruzdev A, Grimm SA, Lydon JP, Demayo FJ, and Korach KS (2019) A distal super enhancer mediates estrogen-dependent mouse uterine-specific gene transcription of Igf1 (insulin-like growth factor 1). The Journal of biological chemistry 294, 9746–9759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langmead B, Trapnell C, Pop M, and Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008) Model-based analysis of ChIP-Seq (MACS). Genome biology 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang W, Loganantharaj R, Schroeder B, Fargo D, and Li L (2013) PAVIS: a tool for Peak Annotation and Visualization. Bioinformatics 29, 3097–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Bushel PR, Lin L, Day K, Wang T, DeMayo FJ, Wu SP, and Li JL (2021) Structural Equation Modeling of In silico Perturbations. Front Genet 12, 727532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Y, Li M, Zhang J, and Wang S (2023) Unveiling uterine aging: Much more to learn. Ageing Research Reviews 86, 101879. [DOI] [PubMed] [Google Scholar]
- 28.Vandevyver S, Dejager L, Tuckermann J, and Libert C (2013) New Insights into the Anti-inflammatory Mechanisms of Glucocorticoids: An Emerging Role for Glucocorticoid-Receptor-Mediated Transactivation. Endocrinology 154, 993–1007 [DOI] [PubMed] [Google Scholar]
- 29.Paramos-de-Carvalho D, Jacinto A, and Saúde L (2021) The right time for senescence. eLife 10, e72449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Q (2014) Transforming growth factor β signaling in uterine development and function. Journal of Animal Science and Biotechnology 5, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kubo N, Ishii H, Xiong X, Bianco S, Meitinger F, Hu R, Hocker JD, Conte M, Gorkin D, Yu M, Li B, Dixon JR, Hu M, Nicodemi M, Zhao H, and Ren B (2021) Promoter-proximal CTCF binding promotes distal enhancer-dependent gene activation. Nature Structural & Molecular Biology 28, 152–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cancer Genome Atlas Research, N., Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, Yau C, Laird PW, Ding L, Zhang W, Mills GB, Kucherlapati R, Mardis ER, and Levine DA (2013) Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Makker V, MacKay H, Ray-Coquard I, Levine DA, Westin SN, Aoki D, and Oaknin A (2021) Endometrial cancer. Nat. Rev. Dis. Primers 7, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang S, Thiel KW, and Leslie KK (2011) Progesterone: the ultimate endometrial tumor suppressor. Trends in endocrinology and metabolism: TEM 22, 145–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheehan MT (2004) Polycystic ovarian syndrome: diagnosis and management. Clin Med Res 2, 13–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu S-P, Wang T, Yao Z-C, Peavey MC, Li X, Zhou L, Larina IV, and DeMayo FJ (2022) Myometrial progesterone receptor determines a transcription program for uterine remodeling and contractions during pregnancy. PNAS Nexus 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kimura T, Ogita K, Kumasawa K, Tomimatsu T, and Tsutsui T (2013) Molecular analysis of parturition via oxytocin receptor expression. Taiwan J Obstet Gynecol 52, 165–170 [DOI] [PubMed] [Google Scholar]
- 38.Lee K, Jeong J, Kwak I, Yu CT, Lanske B, Soegiarto DW, Toftgard R, Tsai MJ, Tsai S, Lydon JP, and DeMayo FJ (2006) Indian hedgehog is a major mediator of progesterone signaling in the mouse uterus. Nature genetics 38, 1204–1209 [DOI] [PubMed] [Google Scholar]
- 39.Fullerton PT Jr., Monsivais D, Kommagani R, and Matzuk MM (2017) Follistatin is critical for mouse uterine receptivity and decidualization. Proc Natl Acad Sci U S A 114, E4772–E4781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohlsson R, Lobanenkov V, and Klenova E (2010) Does CTCF mediate between nuclear organization and gene expression? BioEssays : news and reviews in molecular, cellular and developmental biology 32, 37–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brighton PJ, Maruyama Y, Fishwick K, Vrljicak P, Tewary S, Fujihara R, Muter J, Lucas ES, Yamada T, Woods L, Lucciola R, Hou Lee Y, Takeda S, Ott S, Hemberger M, Quenby S, and Brosens JJ (2017) Clearance of senescent decidual cells by uterine natural killer cells in cycling human endometrium. eLife 6, e31274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deryabin P, Griukova A, Nikolsky N, and Borodkina A (2020) The link between endometrial stromal cell senescence and decidualization in female fertility: the art of balance. Cellular and Molecular Life Sciences 77, 1357–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deryabin PI, and Borodkina AV (2022) Stromal cell senescence contributes to impaired endometrial decidualization and defective interaction with trophoblast cells. Hum Reprod 37, 1505–1524 [DOI] [PubMed] [Google Scholar]
- 44.Lucas ES, Vrljicak P, Muter J, Diniz-da-Costa MM, Brighton PJ, Kong CS, Lipecki J, Fishwick KJ, Odendaal J, Ewington LJ, Quenby S, Ott S, and Brosens JJ (2020) Recurrent pregnancy loss is associated with a pro-senescent decidual response during the peri-implantation window. Commun Biol 3, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Egashira M, Hirota Y, Shimizu-Hirota R, Saito-Fujita T, Haraguchi H, Matsumoto L, Matsuo M, Hiraoka T, Tanaka T, Akaeda S, Takehisa C, Saito-Kanatani M, Maeda K. i., Fujii T, and Osuga Y (2017) F4/80+ Macrophages Contribute to Clearance of Senescent Cells in the Mouse Postpartum Uterus. Endocrinology 158, 2344–2353 [DOI] [PubMed] [Google Scholar]
- 46.McGill BE, Barve RA, Maloney SE, Strickland A, Rensing N, Wang PL, Wong M, Head R, Wozniak DF, and Milbrandt J (2018) Abnormal Microglia and Enhanced Inflammation-Related Gene Transcription in Mice with Conditional Deletion of Ctcf in Camk2a-Cre-Expressing Neurons. The Journal of Neuroscience 38, 200–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Misugi F, Sumi T, Okamoto E, Nobeyama H, Hattori K, Yoshida H, Matsumoto Y, Yasui T, Honda K, and Ishiko O (2005) Expression of matrix metalloproteinases and tissue inhibitors of metalloproteinase in uterine endometrial carcinoma and a correlation between expression of matrix metalloproteinase-7 and prognosis. Int J Mol Med 16, 541–546 [PubMed] [Google Scholar]
- 48.Yang X, Dong Y, Zhao J, Sun H, Deng Y, Fan J, and Yan Q (2007) Increased expression of human macrophage metalloelastase (MMP-12) is associated with the invasion of endometrial adenocarcinoma. Pathology - Research and Practice 203, 499–505 [DOI] [PubMed] [Google Scholar]
- 49.Foo RS, Anene-Nzelu CG, Rosa-Garrido M, and Vondriska TM (2019) Dissecting Chromatin Architecture for Novel Cardiovascular Disease Targets. Circulation 140, 446–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cummings CT, and Rowley MJ (2022) Implications of Dosage Deficiencies in CTCF and Cohesin on Genome Organization, Gene Expression, and Human Neurodevelopment. Genes 13, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elbert A, Vogt D, Watson A, Levy M, Jiang Y, Brûlé E, Rowland ME, Rubenstein J, and Bérubé NG (2019) CTCF Governs the Identity and Migration of MGE-Derived Cortical Interneurons. The Journal of Neuroscience 39, 177–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirayama T, Tarusawa E, Yoshimura Y, Galjart N, and Yagi T (2012) CTCF is required for neural development and stochastic expression of clustered Pcdh genes in neurons. Cell reports 2, 345–357 [DOI] [PubMed] [Google Scholar]
- 53.Kim S, Yu N-K, Shim K-W, Kim J. i., Kim H, Han DH, Choi JE, Lee S-W, Choi DI, Kim MW, Lee D-S, Lee K, Galjart N, Lee Y-S, Lee J-H, and Kaang B-K (2018) Remote Memory and Cortical Synaptic Plasticity Require Neuronal CCCTC-Binding Factor (CTCF). The Journal of Neuroscience 38, 5042–5052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pancho A, Aerts T, Mitsogiannis MD, and Seuntjens E (2020) Protocadherins at the Crossroad of Signaling Pathways. Frontiers in Molecular Neuroscience 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen WV, and Maniatis T (2013) Clustered protocadherins. Development 140, 3297–3302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Flaherty E, and Maniatis T (2020) The role of clustered protocadherins in neurodevelopment and neuropsychiatric diseases. Curr Opin Genet Dev 65, 144–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Novak P, Jensen T, Oshiro MM, Watts GS, Kim CJ, and Futscher BW (2008) Agglomerative Epigenetic Aberrations Are a Common Event in Human Breast Cancer. Cancer research 68, 8616–8625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Men C, Chai H, Song X, Li Y, Du H, and Ren Q (2017) Identification of DNA methylation associated gene signatures in endometrial cancer via integrated analysis of DNA methylation and gene expression systematically. J Gynecol Oncol 28, e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marshall AD, Bailey CG, Champ K, Vellozzi M, O’Young P, Metierre C, Feng Y, Thoeng A, Richards AM, Schmitz U, Biro M, Jayasinghe R, Ding L, Anderson L, Mardis ER, and Rasko JEJ (2017) CTCF genetic alterations in endometrial carcinoma are pro-tumorigenic. Oncogene 36, 4100–4110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kemp CJ, Moore JM, Moser R, Bernard B, Teater M, Smith LE, Rabaia NA, Gurley KE, Guinney J, Busch SE, Shaknovich R, Lobanenkov VV, Liggitt D, Shmulevich I, Melnick A, and Filippova GN (2014) CTCF haploinsufficiency destabilizes DNA methylation and predisposes to cancer. Cell reports 7, 1020–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Creasy D (2012) Reproduction of the rat, mouse, dog, non-human primate and minipig. In Background Lesions in Laboratory Animals (McInnes EF, and Mann P, eds) pp. 101–122, W.B. Saunders, Saint Louis [Google Scholar]
- 62.Joshi A, and Ellenson LH (2017) PI3K/PTEN/AKT Genetic Mouse Models of Endometrial Carcinoma. Adv Exp Med Biol 943, 261–273 [DOI] [PubMed] [Google Scholar]
- 63.Daikoku T, Hirota Y, Tranguch S, Joshi AR, DeMayo FJ, Lydon JP, Ellenson LH, and Dey SK (2008) Conditional loss of uterine Pten unfailingly and rapidly induces endometrial cancer in mice. Cancer research 68, 5619–5627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oh S, Oh C, and Yoo KH (2017) Functional roles of CTCF in breast cancer. BMB Rep. 50, 445–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ross-Innes CS, Brown GD, and Carroll JS (2011) A co-ordinated interaction between CTCF and ER in breast cancer cells. BMC Genomics 12, 593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Korkmaz G, Manber Z, Lopes R, Prekovic S, Schuurman K, Kim Y, Teunissen H, Flach K, Wit E. d., Galli GG, Zwart W, Elkon R, and Agami R (2019) A CRISPR-Cas9 screen identifies essential CTCF anchor sites for estrogen receptor-driven breast cancer cell proliferation. Nucleic acids research 47, 9557–9572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Le Dily F, Vidal E, Cuartero Y, Quilez J, Nacht AS, Vicent GP, Carbonell-Caballero J, Sharma P, Villanueva-Canas JL, Ferrari R, De Llobet LI, Verde G, Wright RHG, and Beato M (2019) Hormone-control regions mediate steroid receptor-dependent genome organization. Genome Res 29, 29–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the methods and/or supplementary material of this article or are openly available in GEO. Previously unpublished data is reposited (RNAseq, CTCF ChIPseq) at GEO (GSE227517 and GSE227519, respectively). Other data used includes mouse HiC (GSE125972 and GSE147843) and organoid HiC (GSE200807), endometrial (ER GSE200807; PR GSE132713) and organoid PR and ER (GSE200807) ChIP, mouse ER (GSE56501), PR (GSE34927) ChIP), H3K27Ac (GSE125972).