

Summary
Diagnostic error can be defined as deviation from a gold standard diagnosis, typically defined in terms of expert opinion, although sometimes in terms of unexpected events that might occur in follow-up (such as progression and death from disease). Although diagnostic error does exist for melanoma, deviations from gold standard diagnosis, certainly among appropriately trained and experienced practitioners, are likely to be the result of uncertainty and lack of specific criteria, and differences of opinion, rather than lack of diagnostic skills. In this review, the concept of diagnostic error will be considered in relation to diagnostic uncertainty, and the concept of overdiagnosis in melanoma will be presented and discussed.
Keywords: Melanoma, dermatopathology, surgical pathology, diagnostic uncertainty, overdiagnosis, standardised classification tool
INTRODUCTION
Histopathology is often regarded as the gold standard for diagnosis of cancer. However, if cancer is defined as a malignant tumour that has the potential to cause the death of patients in the absence of effective therapy, then the only true modality for definition of cancer is long-term follow-up (which would have to be without effective treatment). The histopathological diagnosis of cancer can be predictive of mortality, but usually only in a probabilistic sense. In melanoma in particular, the cure rate for surgical excision of lesions diagnosed as localised primary ‘malignant melanoma’ is over 90%. The remaining 7–10% of patients who die of the disease do so despite complete excision of the local tumour, perhaps because of the existence of dormant metastases beyond the local site, which later become activated,1 or alternatively, because of progressive angiotropic extravascular migration of tumour cells to local, regional, and possibly distant sites.2,3 Of course, it is desirable to recognise lesions that have the potential to cause mortality with high sensitivity, to eliminate, as far as possible, those cases that have risk of progression. This quest for sensitivity inevitably results in loss of specificity, so that many lesions diagnosed as melanoma will not cause death to patients after therapy, and in many cases likely would not have caused death even in the absence of therapy. This phenomenon has been termed ‘overdiagnosis’ and is not the same as diagnostic error.4,5 Diagnostic error can be defined as deviation from a gold standard diagnosis, typically defined in terms of expert opinion, although sometimes in terms of unexpected events that might occur in follow-up (such as progression and death from disease). Although diagnostic error does exist for melanoma, deviations from gold standard diagnosis, certainly among appropriately trained and experienced practitioners, are likely to be the result of uncertainty and lack of specific criteria, and differences of opinion, rather than lack of diagnostic skills.6 In this review, the concept of diagnostic error will be considered in relation to diagnostic uncertainty, and the concept of overdiagnosis in melanoma will be presented and discussed.
DIAGNOSTIC ERROR AND UNCERTAINTY
A diagnostic error in pathology may be defined as an ‘incorrect’ diagnosis,7 or perhaps more specifically as a deviation from a ‘gold standard’ diagnosis.8 Defining errors in the diagnosis of melanoma is problematic given the difficulty of defining a true gold standard for melanoma. Since pathology is often regarded as the gold standard for melanoma diagnosis, this definition may be regarded as self-referential, except in the cases where unexpected malignant behaviour of a lesion previously diagnosed as benign reveals the true potential of the tumour. These latter examples of diagnostic error will not be extensively discussed in this review. Reviews of medicolegal cases have provided some insight into these uncommon situations.9
In general, since pathological diagnosis aims to be predictive rather than to simply recapitulate the past behaviour of lesions, and since biological outcomes are not available at the time of diagnosis for most patients and in most studies, expert panels have been utilised to provide a ‘ground truth’ or ‘gold standard’ diagnosis. In a series of studies over the past decade, the M-Path group has studied diagnostic error and uncertainty in the diagnosis of melanocytic skin lesions.10 The M-Path study recruited community pathologists to review cases that were derived from a particular dermatopathology laboratory. The cases were selected to provide a full spectrum of diagnoses from benign through atypical to fully evolved malignant melanoma. The ‘atypical’ group was purposely over-represented because this was perceived to be an area in which there was particular difficulty of specificity and, perhaps, accuracy of diagnosis. The 240 cases that were selected were all reviewed independently by three expert diagnosticians with long records of research and publication in the field. Following independent review, individual diagnoses were presented to the three members of the panel around a multi-headed microscope and then discussed using a modified Delphi approach.11 Consensus was reached by discussion and was not always easy to achieve. For some cases, it was necessary to agree that the diagnosis was uncertain, with the lesions being interpreted as either ‘superficial atypical melanocytic proliferation of uncertain significance’ or ‘melanocytic tumour of uncertain malignant potential’ for lesions that either lacked, or had a tumourigenic (i.e., mass forming) dermal component, respectively.12 Although there was full agreement for many lesions, in some there was complete disagreement with each of the three experts providing a different diagnosis for the same case during their independent review. Thus, great diagnostic uncertainty became apparent early in the study, complicating the concept of ‘diagnostic error.’
To manage the very diverse terminology used by the different dermatopathologists, the M-Path study team developed a histology reporting form that was given to the study participants, who were practitioners in both academic and private practices. This histology reporting form was conceived as a thesaurus of terms encompassing most or all possible terms that pathologists might use for melanocytic proliferations. The concept of melanocytic lesions of uncertain potential, where the observer cannot decide whether the lesion is clearly benign or malignant, was incorporated. The major classes included ‘wholly benign’ lesions without atypia, lesions with atypia considered to have potential significance either as precursor lesions or as risk factors for future development of melanoma, melanoma in situ, and invasive melanoma. The ‘atypical’ category included dysplastic naevi, atypical spitzoid tumours, and melanocytomas (a term that refers, in general, to lesions that have atypical histopathological features with more than a single pathogenic mutation, and an expected, albeit generally small, increased risk of neoplastic progression).13 In the melanoma category the multiple World Health Organization subtypes were distinguished,14 and the American Joint Committee on Cancer (AJCC) stages separating the low stage AJCC T1a melanomas where metastasis is very rare, and the T1b or greater melanomas where the risk of metastasis becomes more appreciable and continues to increase with further progression.15
These lesions were placed into MPATH-Dx (Melanocytic Pathology Assessment Tool and Hierarchy for Diagnosis) categories I through V. An update of this MPATH-Dx classification scheme (MPATH-Dx 2.0) has been developed, with four rather than five categories, in an effort to simplify the classification scheme, and to reduce uncertainty and misclassification.16 In both the original and the updated MPATH-Dx scheme, the Category I represents lesions such as banal naevi or those with low grade dysplasia, having ‘no apparent risk for continued local proliferation and adverse outcome’, for which no further treatment is required. Category II in the current version consists of lesions with ‘low risk for progression’ such as high-grade dysplastic naevi and melanoma in situ, for which consideration of narrow but complete re-excision is recommended. Category III consists of lesions with ‘relatively low probability of local tumour progression and greater need for intervention’, and includes stage T1a invasive melanomas, which in general may have capacity for local progression if not excised but usually not for metastasis. Category IV comprises ‘lesions at greater risk for regional and or distant metastasis’ and this includes T1b or greater stage melanomas, which are lesions with potential competence for metastasis and for which wide local excision is recommended, along with sentinel node staging. These treatment recommendations are presented as being for consideration only, and in the case of melanomas it is emphasised that national guidelines should take precedence. These guidelines exist for only some of the benign lesions with atypia,17 and the MPATH-Dx guidelines are presented as being potentially helpful for practitioners, especially those who lack the more specific expertise of specialists in the field.
Data from the original M-PATH-Dx study reflecting observer accuracy and reproducibility were published in the British Medical Journal.18 Pathologists’ interpretations of the 240 lesions were condensed into the five classes in the original scheme. Reproducibility was assessed by intra- and inter-observer concordance, and accuracy defined by concordance with the consensus reference diagnosis reached by the panel of three expert diagnosticians. Two additional reference diagnoses were explored including an ‘experienced participant reference diagnosis’ based on the most frequent classification (mode diagnosis) of each case by board-certified and/or dermatopathology fellowship-trained participants, who comprised 74 of the 187 study pathologists. The third reference standard was defined by the mode diagnosis of each case by all participating pathologists. The results were similar using the three different standards. In this study, pathologists interpreted the same cases in two separate phases separated by a wash-out period, thus data are available on reproducibility (i.e., intra-observer agreement of a pathologist). Among pathologists who diagnosed a case in class I or class V during phase 1 interpretations, they gave the same diagnosis in phase 2 with an intra-observer agreement of 76.7% and 82.6%, respectively. In contrast, the intra-observer agreement was considerably lower for cases interpreted as class II (35.2%), class III (59.5%) and class IV (63.2%). Inter-observer agreement rates (i.e., agreement among different observers) were lower but with similar trends.
Examination of the individual case level data and categories indicated, in summary, that the diagnosis of ‘wholly benign’ (non-atypical) naevi was reasonably reproducible, as was the diagnosis of those melanomas that had substantial risk for metastasis (AJCC stage T1b and greater). The reproducibility of diagnoses was objectively poor for lesions in the category of having ‘atypia’ and these lesions included not only dysplastic naevi, but also melanoma in situ and T1a melanoma. While the results for dysplastic naevi were perhaps not surprising, the poor reproducibility of diagnoses of melanoma in situ and T1a invasive melanoma were somewhat unexpected, potentially calling into question the validity of the distinctions among severe melanocytic dysplasia, melanoma in situ, and at least a subset of T1a invasive melanomas.
Based on the M-Path study results, it was estimated that at a United States population level, 82% of melanocytic skin biopsy diagnoses would have the diagnosis verified if reviewed by a consensus reference panel of experienced pathologists, with 8% of cases being over-interpreted by the initial pathologist and 9% under-interpreted. It was concluded that diagnoses spanning moderately dysplastic naevi to early-stage invasive melanoma (T1a melanomas) were neither reproducible nor accurate. The research group suggested that a standardised classification system acknowledging uncertainty in pathology reports was justified, along with developing tools such as molecular markers to support visual criteria.14 However, the available molecular markers, in general, suffer from the same problem as traditional criteria in that they are based on the problematic ‘gold standard’ histopathological diagnoses and thus on expert opinion.
Considering the low intra- and inter-observer reproducibility for the diagnosis of melanocytic skin tumours, and the lack of a true gold standard, it seems that the term ‘error’ is not appropriately applied to this variability, at least in general. Rather, these results indicate the existence of substantial diagnostic uncertainty in these categories of melanocytic proliferations, likely a consequence of criteria that are inadequate to make the distinctions proposed, and/or on an unrealistic assessment of the validity of the distinctions being made. For example, the distinction between banal naevus and mildly dysplastic naevus and that between severe junctional and dermal dysplasia and low risk subsets of T1a melanoma may be of no consequence and might even be artificial, given that the behaviour of these pairs of lesions may be similar.19 In some instances, this uncertainty results from competing diagnostic systems where different groups of pathologists have applied different criteria (or different weighting of criteria) to various diagnostic entities. In other instances, the uncertainty results from the difficulty of setting thresholds for multiple parameters that are part of the process of distinguishing between diagnostic subsets. In any case, the existence of diagnostic uncertainty suggests that efforts should be applied to resolving the uncertainty. In other tumour systems, similar demonstrations of diagnostic variability have led to changes in the nomenclature of lesions, designed to simplify the diagnostic task without harming patients. These changes will be discussed later in this essay, in the context of overdiagnosis. Additional promising efforts to resolve uncertainty include attempts to develop better criteria including the use of more fundamental molecular markers, and such efforts have proliferated in recent years, with variable efficacy, as briefly discussed in the next section.
ADJUNCT DIAGNOSTIC CRITERIA
Adjunct criteria for the diagnosis of melanocytic proliferations include immunohistochemistry (IHC) directed in general at protein and/or carbohydrate antigens, and genomic studies directed at genomic DNA or mRNA expression.20,21 Studies of these criteria have provided substantial assistance to the diagnostic process, which was not available to the observers in the M-Path study described above. IHC studies as adjuncts for diagnosis include markers that can help to evaluate the distribution of cells (relevant to the diagnosis of malignancy) such as S100 and SOX10,22,23 and markers that have differential expression in benign versus malignant lesions, such as Mart-1,24 HMB45, the tumour suppressor p16,25 and more recently the cancer testis antigen PRAME.26 All these markers, and others, are used as adjuncts to histopathological diagnosis, and likely have improved specificity in certain subsets. However, use of these IHC stains is quite variable and their impact on diagnostic accuracy and relationship to outcome has not been adequately studied.27
Genomic studies that are now available have included comparative genomic hybridisation and fluorescence in situ hybridisation,28 which address copy number variation that is more common in malignant than in benign tumours, mRNA profiling studies which have addressed profiles developed in preliminary studies for their predictive diagnostic ability in diagnostic settings,29 and DNA and RNA sequencing (next generation sequencing) performed to reveal mutations and translocations (gene fusions).30 Of note, many of these studies have used ‘expert opinion’ as the gold standard for evaluating the diagnostic algorithm.
Few if any of these studies reviewed above have been evaluated against the gold standard of prediction of outcome, and none with sufficient statistical power, for example, to suggest that they could be used as a replacement for diagnostic histopathology in primary diagnosis of malignancy, as a ‘black box’ diagnostic tool or algorithm. These criteria and methods are not the focus of this review but suffice it to say that they have not yet revolutionised the diagnosis of melanoma, although undoubtedly having improved the process at the margins. Artificial intelligence (AI) is another modality that has promise in diagnosis that has not yet been realised in practice.31
OVERDIAGNOSIS
Overdiagnosis is the diagnosis of a tumour as malignant that would not have caused the death of the patient, even if it had not been excised.32 ‘Overdiagnosis’ is not the same as ‘erroneous diagnosis’, in the sense that the lesions that are overdiagnosed meet current diagnostic criteria to be called cancer.
Overdiagnosis, as presented in seminal studies by Welch and colleagues,33–35 occurs in settings where there is an undiagnosed pool of cancer cases in the community, or a pool of cases that look like cancer but are not, coupled with efforts aiming for early diagnosis, as is the situation for melanoma and several other cancers. Compelling evidence for the pervasive existence of overdiagnosis has been provided in several databases and in several tumour types.36 For example, in the SEER population-based cancer database, as noted by others using earlier data,28 there has been a dramatic rise in the incidence of invasive melanoma, without a corresponding increase in mortality (Fig. 1). In a study using trends among black and white patients in the US, it was estimated that about 60% of melanomas in white men and women were overdiagnosed in 2014,35 while in Australia an estimated 54% of melanomas (invasive melanoma 15%) in women and 58% (invasive 22%) in men were overdiagnosed in 2012.37
Fig. 1.
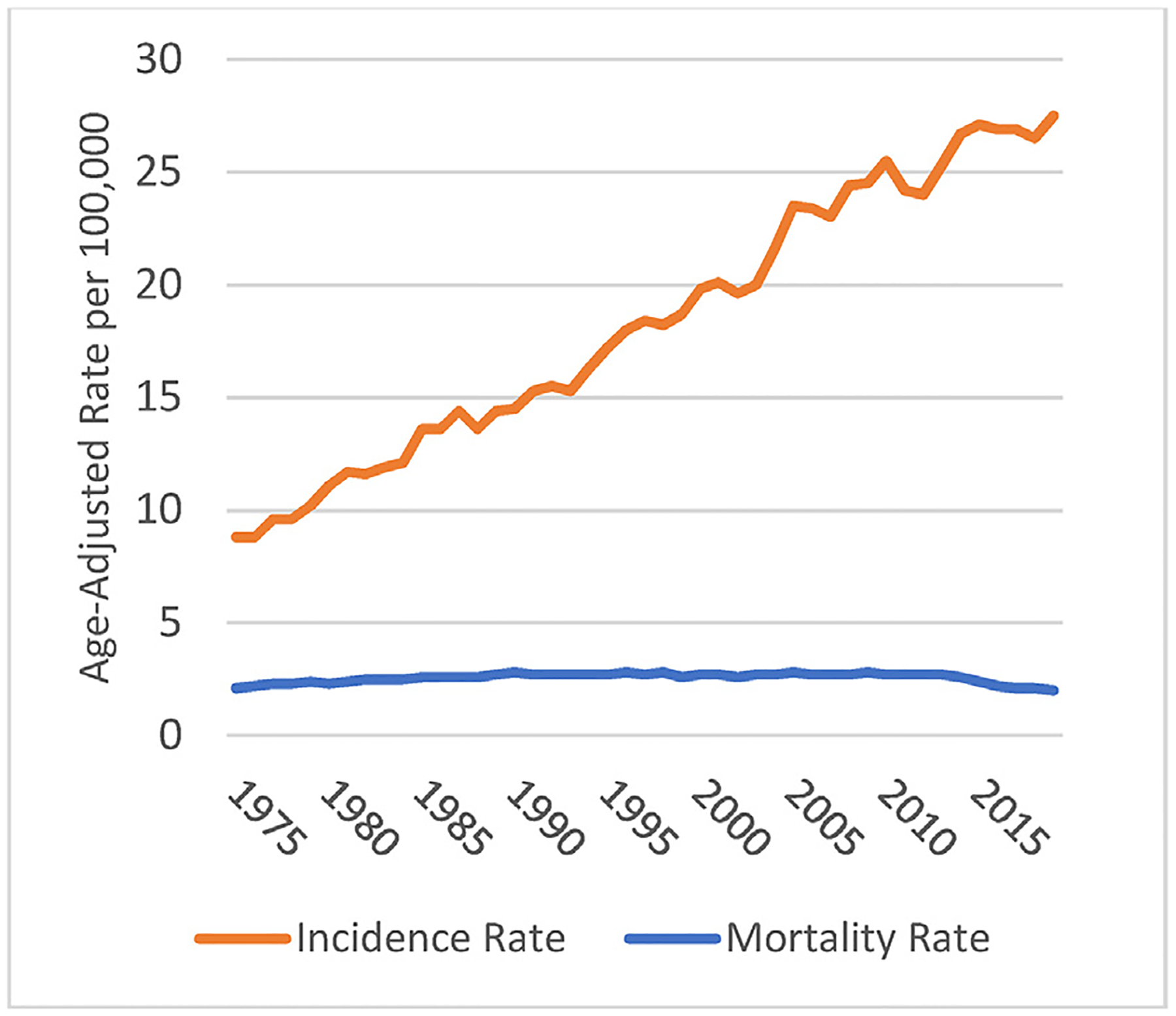
SEER age-adjusted incidence and mortality rates for melanoma of the skin, 1975–2019. Rates are per 100,000 and age-adjusted to the 2000 US Standard Population (19 age groups) (M. Eguchi). Source for incidence rate: Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov) SEER*Stat Database: Incidence -; SEER Research Data, 8 Registries, Nov 2021 Sub (1975–2019) - Linked To County Attributes - Time Dependent (1990–2019) Income/Rurality, 1969–2020 Counties, National Cancer Institute, DCCPS, Surveillance Research Program, released April 2022, based on the November 2021 submission. Source for mortality rate: Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov) SEER*Stat Database: Mortality - All COD, Aggregated Total US (1969–2020) <Katrina/Rita Population Adjustment>, National Cancer Institute, DCCPS, Surveillance Research Program, released June 2022. Underlying mortality data provided by NCHS (www.cdc.gov/nchs).
This finding indicates, without question, that a large fraction of the melanomas that are currently diagnosed would not cause the death of patients, even if they had not been excised. The reason for this conclusion can be explained in terms of the efficacy of efforts at ‘early diagnosis’. If the additional melanoma diagnoses were catching and successfully treating 100% of precursor lesions in the community, then we would expect increases in diagnoses to be followed by corresponding decreases in mortality. If additional melanoma diagnoses represented greater incidence of melanoma in the community, then we would expect a corresponding increase in mortality unless the increase in incidence happened to correspond to advances in treatment. In reality, mortality rates are flat, indicating that a large fraction of melanomas that are currently diagnosed would not cause the death of patients, even if they had not been excised. In other words, they are overdiagnosed. In contrast, the efficacy of early diagnosis is undoubtedly much less than 100%, and requires large randomised controlled trials to reliably measure. This would indicate that for every lesion diagnosed as a ‘melanoma’ and contributing to the rising frequency of diagnosis, there must be dozens or even hundreds of exactly similar lesions in the community, remaining undiagnosed and untreated, and yet failing to contribute to any rising mortality from the disease. That being said, there are instances of demonstrable progression of intermediate and even benign lesions, but the incidence of such progression must be very low in relation to the enormous pool of (very low risk) potential precursors and simulants of melanoma.
MINIMAL RISK MELANOMA AND MELANOCYTIC NEOPLASMS OF LOW MALIGNANT POTENTIAL (MNLMP)
Given the incontrovertible evidence of overdiagnosis in melanoma, it is appropriate to consider what might be the nature of the lesions that are being diagnosed as invasive melanoma yet not contributing to mortality. Possible candidates include various forms of ‘pseudomelanoma’ including some that have been described and no doubt others that have yet to be described.38 The published experience of clinical groups that review outside slides prior to treatment in their institutions, where there is generally only a small subset of cases with disagreement that results in a change in diagnosis (although changes in management are not uncommon),39 suggests that there is not a very large number of such lesions being (recognisably) misdiagnosed as melanoma in the community. There has been recent evidence presented that some examples of lesions that are truly Spitz tumours may be indistinguishable from more usual melanomas (without sophisticated genomic testing) and these might form a significant subset of pseudomelanomas.36 The number of such lesions is presently unknown but more of them will be discovered as comprehensive genomic testing becomes more pervasive in case management. However, at present the most prominent group of cases that can explain the overdiagnosis phenomenon, albeit not completely, is likely to be found in the group of T1a melanomas which enjoy a very good, but not perfect, survival rate after local excision without any additional forms of therapy.1
To explore the possibility that T1a melanomas include a subset of overdiagnosed cases, we have recently studied these lesions, looking for prognostic indicators that can distinguish a group of cases with survival rates approaching or equaling 100%. Such cases could help explain the overdiagnosis phenomenon. In this study, the 2020 submission of the SEER Cancer registry comprised of 18 population-based registries covering approximately 28% of the US population was used, including patients with survival follow-up through December 2018.40 Similar methods have been used by others in melanoma, and in other cancers (e.g., breast, prostate, and thyroid).5 The SEER database for melanoma includes Breslow thickness, ulceration, Clark level, stage of disease and mitotic rate. Because mitotic rate was included in the 7th edition of the AJCC system but not in the previous or subsequent editions, we included cases from patients diagnosed in 2010 and 2011, years when the 7th edition criteria applied and allowed for evaluating 7-year survival. The sample was randomly divided into training (67%) and testing (33%) sets. Logistic regression models and CART (classification and regression tree) or ‘tree’ models were developed. The initial models developed in the training set were applied to the testing set. We identified two CART models and one regression model of particular interest. One of the CART models is illustrated in Fig. 2; 638 patients in the training set and 331 in the test set who were aged ≤43 and had Clark level II melanomas enjoyed an observed 100% survival. Despite the identification of this relatively small subset of patients with perfect survival, in general the goal of identifying a large group of patients with 100% survival was not met in this study. Nevertheless, the survival experience of patients identified in these models is indicative of a very low risk group of patients for whom the suitability of the term ‘cancer’ could be questioned. However, it would be desirable to identify criteria in addition to those in the SEER database that might lead to identification of more cases with excellent survival, that might account for a greater fraction of the observed phenomenon of overdiagnosis illustrated in Fig. 1.
Fig. 2.
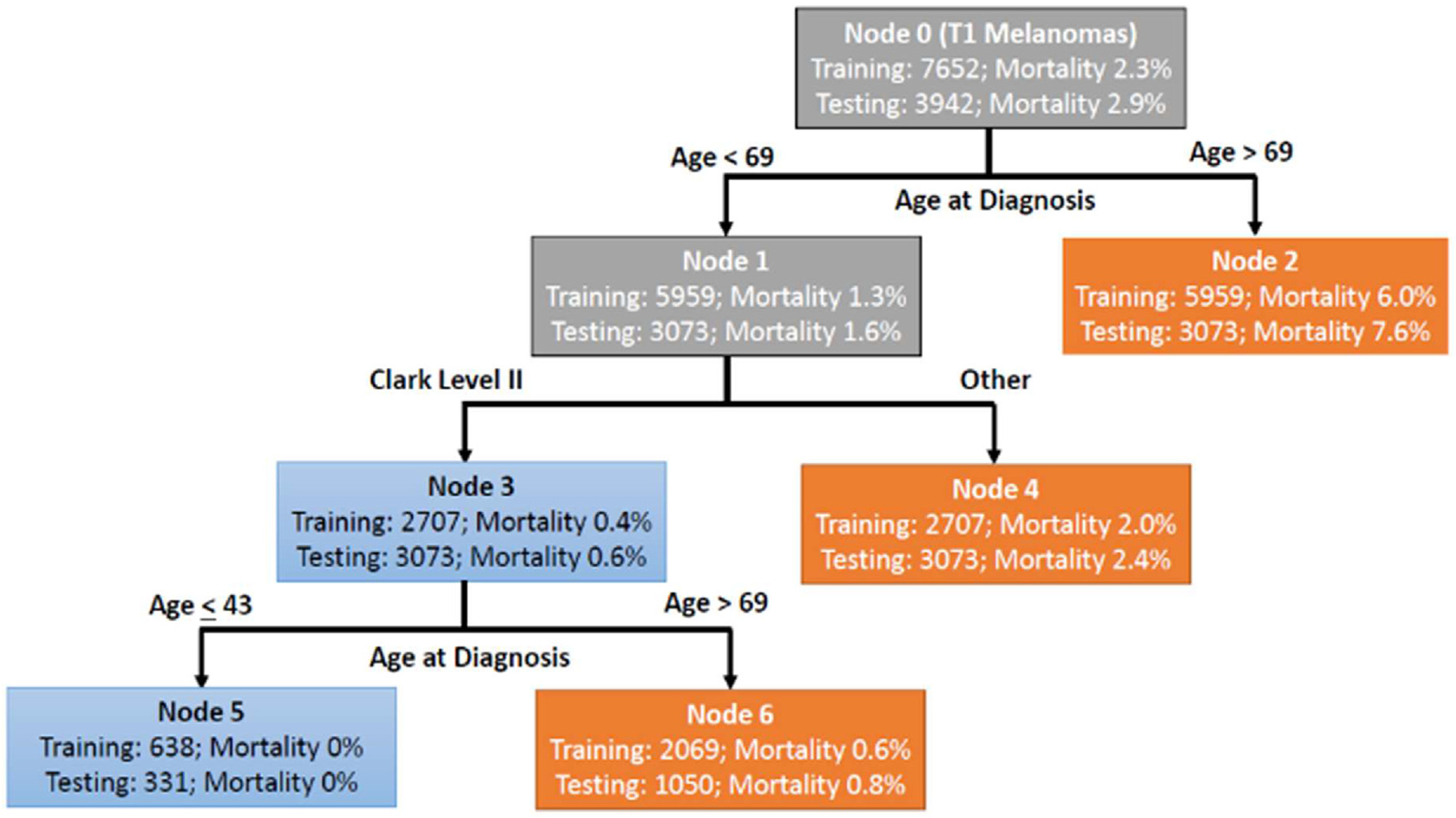
Prognostic model for Stage 1 melanoma. The importance of Clark level II. The diagram displays the number of patients in each node and the proportion of patients in the node who died. Blue leaves indicate subsets of patients classified as at low risk of death and orange leaves indicate subset of patients classified as relatively higher risk of death. These models were constructed in the training dataset weighting patients who died within 7 years 160:1 compared to patients that survived. Data from Eguchi et al.40
CRITERIA FOR MINIMAL RISK MELANOMA
Among the potential criteria for minimal risk melanoma, one could include molecular testing that, up to now, has not been evaluated in large databases because of the lack of stand-ardisation and generalisability of such testing, at least at this early stage of the evolution of molecular diagnosis. There is one criterion, however, that can be evaluated by routine histopathological evaluation of tumours, and that is the property of ‘vertical growth phase’ or ‘tumourigenicity’ in melanomas, first described by Clark in 1967.41,42 The term vertical growth phase (VGP) is based on the concept of tumour evolution from a stage in which it spreads in the skin along the radii of an imperfect circle, called the radial growth phase (RGP), which is at risk of developing a focal area where tumour cells proliferate not only in or near the epidermis but also in the dermis itself, forming an expansile mass or ‘tumour’. This mass tends to expand the lesion in a vertical direction, hence the term VGP, which like RGP was originally based on evaluation of clinical photographs. In contrast, the RGP tends to present as a horizontal proliferation (HGP) in tissue sections while the VGP tends to be a more or less circular structure that has a ‘radius’ (Fig. 3). The VGP or ‘tumourigenic’ melanomas have potential competence for metastasis, which can be explained by the idea that a metastasis represents an expansile tumour in a remote site and the consideration that melanomas that lack the capacity to form such tumours at the primary site (i.e., RGP melanomas) would lack such capacity in distant sites as well.43
Fig. 3.
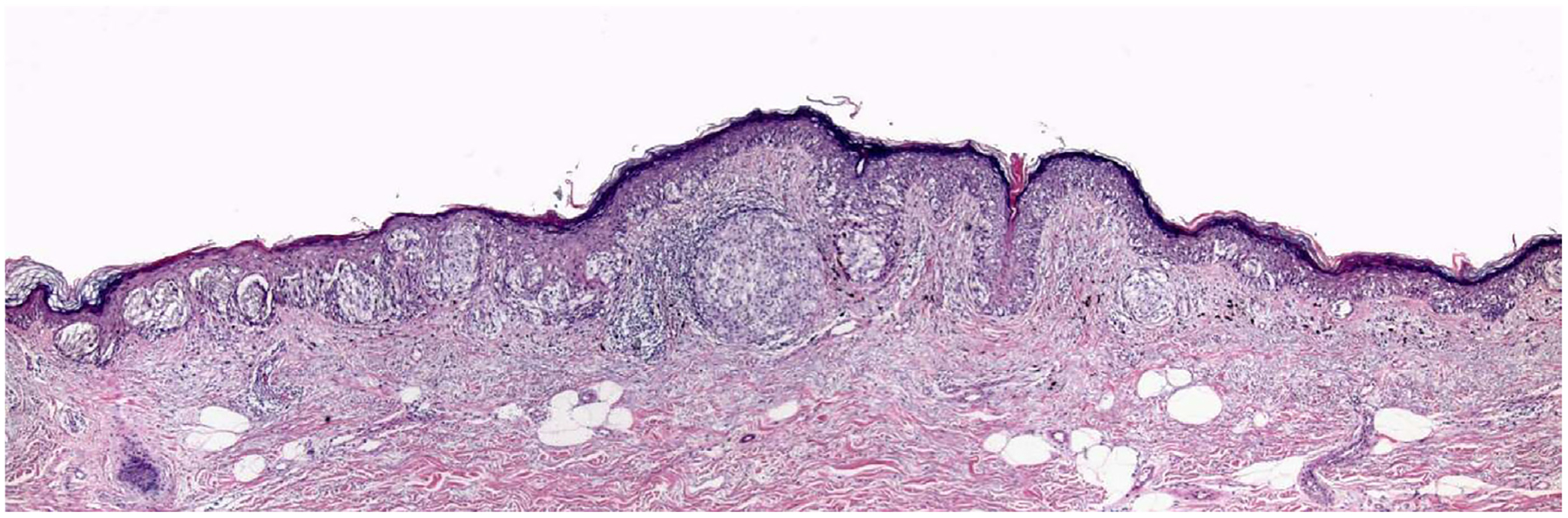
A thin melanoma with an early Clark level II vertical growth phase papule. Despite a Breslow thickness of considerably less than 1 mm, this T1a Clark level II melanoma has a cluster of cells in the dermis that is larger than the clusters of cells in the epidermis, consistent with its having capacity for growth as a mass lesion in the dermis, and increased potential for metastasis, illustrating that while most Clark level II melanomas will lack VGP, there are a few where the tumour cells do not fill and expand the papillary dermis and thus do not qualify as level III.
Multiple studies have demonstrated that the VGP cells differ from those of the RGP cells in many properties related to tumourigenicity.44–49 In addition, there have been studies that have demonstrated the prognostic significance of the presence or absence of VGP. In one such study of patients prospectively diagnosed and followed in a pigmented lesion clinic, the melanoma disease-free survival of 161 patients whose tumours lacked vertical growth phase, followed for a minimum of 10 years, and a median of 13.7 years, was literally 100%.50,51 There have been rare anecdotal exceptions to this finding published in the literature, but nevertheless the lack of tumourigenic or mitogenic VGP is a property in melanoma that is highly correlated with favorable outcomes, and the histopathological diagnosis is reasonably reproducible. McDermott et al. found that ‘although overall agreement for the growth phase is moderate, agreement between experienced observers is good’ (k=0.68), and further stated ‘in fact, agreement for the growth phase among this group was equal to the agreement for Breslow thickness’.52 Lefevre et al. found ‘a very high level of agreement for growth phase diagnosis’ (k=0.86) and stated that ‘vertical growth phase is the only statistically significant prognostic factor for thin level II cutaneous SSM’.53 The importance of Clark level II (or absence of level III or greater) in identifying patients with ‘minimal risk’ melanomas (Fig. 2) can be explained by its being quite highly, but not perfectly, correlated with absence of VGP (see Fig. 3).
These considerations suggest that vertical growth phase should be reinstated in the AJCC protocol and recorded in the SEER database. In the meantime, we suggest that the provision of optimal care for patients with T1 melanoma should include the incorporation of VGP in diagnostic protocols and management planning. The diagnosis of VGP is obvious in thicker melanomas and may not be necessary to record. Criteria for the diagnosis of VGP in a thin melanoma are listed in Table 1.
Table 1.
Minimal criteria for VGP in a melanoma
| • Presence of a cluster of cells in the dermis that is larger than the largest cluster in the epidermis (‘tumourigenic VGP’) and/or •Presence of any dermal lesional cell mitoses (‘mitogenic VGP’) |
CONCLUSION
In sum, the studies reviewed above have established the existence of a category of lesions currently called melanoma that present minimal risk of mortality, and the existence of overdiagnosis indicates the biological plausibility of the idea that there is a substantial subset of ‘melanomas’ in which the mortality rate due to the melanoma is literally zero. Identification of this subset is currently imperfect, but one can list a set of criteria for identification of a subset of lesions that we have proposed could be called ‘melanocytic neoplasms of low malignant potential’ (MNLMP). Use of this nomenclature could reduce the perceived need for surgery and other interventions in melanocytic proliferations and reduce anxiety in the patient population. Similar terminology has been used in other tumour systems such as the bladder,54 and in melanoma by others, most notably in the concept of ‘melanocytic intraepithelial neoplasia’ (MIN) of Cook et al.43,55 A limitation of this MIN terminology is the fact that RGP melanomas can be invasive, thus no longer entirely intraepithelial and yet they lack capacity for metastasis. These lesions have also been called ‘microinvasive’ melanomas, and this term can be regarded as an acceptable synonym for ‘RGP confined invasive melanoma’. The criteria that we propose as providing the best possible definition of MNLMP tumours, emphasising specificity over sensitivity, are listed in Table 2.
Table 2.
Criteria for melanocytic neoplasms of low malignant potential (MNLMP)
These histopathological criteria are all amenable to study with routine light microscopy, and all of them have been subjected to studies that indicate a reasonable degree of reproducibility.52,53,56,57
To establish a diagnostic category of MNLMP, or perhaps in the future to identify categories of lesions presently called melanoma for which the diagnosis should be revised to benign (e.g., severely dysplastic naevi or even wholly benign naevi), additional studies and consideration by consensus groups will be essential. Nevertheless, using present knowledge and available techniques, it is clearly possible to identify a subset of melanoma cases for which the mortality is exceedingly low and, in some subsets, approaches or equals zero. Confirmation of the prognostic utility of excluding tumourigenic vertical growth phase in lesions under consideration for a diagnosis of melanoma should be studied in expanded prospective data sets.
For the present, we recommend use of the criteria presented in Table 2, and recording of these criteria in national databases, to support expanding the evidence base for identifying the large group of cases that are currently over-diagnosed as melanomas, but that in reality represent benign neoplasms of melanocytes, better considered in the category of melanocytic naevi, or perhaps, in some cases, in the intermediate category of melanocytomas. Funding for studies designed to address these issues should receive higher priority than is presently the case.
Conflicts of interest and sources of funding:
Supported by the National Cancer Institute (R01 CA201376). The funding agency had no role in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the article for publication. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors state that there are no conflicts of interest to disclose.
References
- 1.Landow SM, Gjelsvik A, Weinstock MA. Mortality burden and prognosis of thin melanomas overall and by subcategory of thickness, SEER registry data, 1992–2013. J Am Acad Dermatol 2017; 76: 258–63. [DOI] [PubMed] [Google Scholar]
- 2.Lugassy C, Kleinman H, Vermeulen P, Barnhill R. Angiotropism, pericytic mimicry and extravascular migratory metastasis: an embryogenesis-derived program of tumor spread. Angiogenesis 2020; 23: 27–41. [DOI] [PubMed] [Google Scholar]
- 3.Massagué J, Ganesh K. Metastasis-initiating cells and ecosystems. Cancer Discov 2021; 11: 971–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swerlick RA, Chen S. The melanoma epidemic. Is increased surveillance the solution or the problem? Arch Dermatol 1996; 132: 881–4. [DOI] [PubMed] [Google Scholar]
- 5.Welch HG, Mazer BL, Adamson AS. The rapid rise in cutaneous melanoma diagnoses. N Engl J Med 2021; 384: 72–9. [DOI] [PubMed] [Google Scholar]
- 6.Jafry MA, Peacock S, Radick AC, et al. Pathologists’ agreement on treatment suggestions for melanocytic skin lesions. J Am Acad Dermatol 2020; 82: 1435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newman-Toker DE. A unified conceptual model for diagnostic errors: underdiagnosis, overdiagnosis, and misdiagnosis. Diagnosis (Berl) 2014; 1: 43–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Foucar E Error identification: a surgical pathology dilemma. Am J Surg Pathol 1998; 22: 1–5. [DOI] [PubMed] [Google Scholar]
- 9.Kornstein MJ, Byrne SP. The medicolegal aspect of error in pathology: a search of jury verdicts and settlements. Arch Pathol Lab Med 2007; 131: 615–8. [DOI] [PubMed] [Google Scholar]
- 10.Piepkorn MW, Barnhill RL, Elder DE, et al. The MPATH-Dx reporting schema for melanocytic proliferations and melanoma. J Am Acad Dermatol 2014; 70: 131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carney PA, Reisch LM, Piepkorn MW, et al. Achieving consensus for the histopathologic diagnosis of melanocytic lesions: use of the modified Delphi method. J Cutan Pathol 2016; 43: 830–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elder DE, Xu X. The approach to the patient with a difficult melanocytic lesion. Pathology 2004; 36: 428–34. [DOI] [PubMed] [Google Scholar]
- 13.Yeh I. New and evolving concepts of melanocytic nevi and melanocytomas. Mod Pathol 2020; 33 (Suppl 1): 1–14. [DOI] [PubMed] [Google Scholar]
- 14.Elder DE, Bastian BC, Cree IA, Massi D, Scolyer RA. The 2018 World Health Organization classification of cutaneous, mucosal, and uveal melanoma: detailed analysis of 9 distinct subtypes defined by their evolutionary pathway. Arch Pathol Lab Med 2020; 144: 500–22. [DOI] [PubMed] [Google Scholar]
- 15.Gershenwald JE, Scolyer RA. Melanoma staging: American Joint committee on cancer (AJCC) 8th edition and beyond. Ann Surg Oncol 2018; 25: 2105–10. [DOI] [PubMed] [Google Scholar]
- 16.Barnhill RL, Elder DE, Piepkorn MW, et al. Revision of the Melanocytic Pathology Assessment Tool and Hierarchy for Diagnosis version 2.0 classification schema for melanocytic lesions: a consensus statement. JAMA Netw Open 2023; 6: e2250613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de la Fouchardiere A, Blokx W, van Kempen LC, et al. ESP, EORTC, and EURACAN Expert Opinion: practical recommendations for the pathological diagnosis and clinical management of intermediate melanocytic tumors and rare related melanoma variants. Virchows Arch 2021; 479: 3–11. [DOI] [PubMed] [Google Scholar]
- 18.Elmore JG, Barnhill RL, Elder DE, et al. Pathologists’ diagnosis of invasive melanoma and melanocytic proliferations: observer accuracy and reproducibility study. BMJ 2017; 357: j2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foucar E Do pathologists play dice? Uncertainty and early histopathological diagnosis of common malignancies. Histopathology 1997; 31: 495–502. [DOI] [PubMed] [Google Scholar]
- 20.Lee JJ, Lian CG. Molecular testing for cutaneous melanoma: an update and review. Arch Pathol Lab Med 2019; 143: 811–20. [DOI] [PubMed] [Google Scholar]
- 21.Wick MR. Cutaneous melanoma: a current overview. Semin Diagn Pathol 2016; 33: 225–41. [DOI] [PubMed] [Google Scholar]
- 22.De Vries TJ, Smeets M, de Graaf R, et al. Expression of gp100, MART-1, tyrosinase, and S100 in paraffin-embedded primary melanomas and locoregional, lymph node, and visceral metastases: implications for diagnosis and immunotherapy. A study conducted by the EORTC Melanoma Cooperative Group. J Pathol 2001; 193: 13–20. [DOI] [PubMed] [Google Scholar]
- 23.Mohamed A, Gonzalez RS, Lawson D, Wang J, Cohen C. SOX10 expression in malignant melanoma, carcinoma, and normal tissues. Appl Immunohistochem Mol Morphol 2012; 21: 506–10. [DOI] [PubMed] [Google Scholar]
- 24.Busam KJ, Chen YT, Old LJ, et al. Expression of melan-A (MART1) in benign melanocytic nevi and primary cutaneous malignant melanoma. Am J Surg Pathol 1998; 22: 976–82. [DOI] [PubMed] [Google Scholar]
- 25.Redon S, Guibourg B, Talagas M, Marcorelles P, Uguen A. A diagnostic algorithm combining immunohistochemistry and molecular cytogenetics to diagnose challenging melanocytic tumors. Appl Immunohistochem Mol Morphol 2018; 26: 714–20. [DOI] [PubMed] [Google Scholar]
- 26.Lezcano C, Jungbluth AA, Nehal KS, Hollmann TJ, Busam KJ. PRAME expression in melanocytic tumors. Am J Surg Pathol 2018; 42: 1456–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.May CJ, Piepkorn MW, Knezevich SR, et al. Factors associated with use of immunohistochemical markers in the histopathological diagnosis of cutaneous melanocytic lesions. J Cutan Pathol 2020; 47: 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagarajan P, Tetzlaff MT, Curry JL, Prieto VG. Use of new techniques in addition to IHC applied to the diagnosis of melanocytic lesions, with emphasis on CGH, FISH, and mass spectrometry. Actas Dermosifiliogr 2017; 108: 17–30. [DOI] [PubMed] [Google Scholar]
- 29.Clarke LE, Flake DD, Busam K, et al. An independent validation of a gene expression signature to differentiate malignant melanoma from benign melanocytic nevi. Cancer 2017; 123: 617–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roth A, Lampley N 3rd, Boutko A, et al. Next-generation sequencing improves agreement and accuracy in the diagnosis of Spitz and spitzoid melanocytic lesions. J Cutan Pathol 2022; 49: 868–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomsen K, Iversen L, Titlestad TL, Winther O. Systematic review of machine learning for diagnosis and prognosis in dermatology. J Dermatolog Treat 2020; 31: 496–510. [DOI] [PubMed] [Google Scholar]
- 32.Carter SM, Barratt A. What is overdiagnosis and why should we take it seriously in cancer screening? Public Health Res Pract 2017; 27: 2731722. [DOI] [PubMed] [Google Scholar]
- 33.Welch HG, Black WC. Overdiagnosis in cancer. J Natl Cancer Inst 2010; 102: 605–13. [DOI] [PubMed] [Google Scholar]
- 34.Kurtansky NR, Dusza SW, Halpern AC, et al. An epidemiologic analysis of melanoma overdiagnosis in the United States, 1975–2017. J Invest Dermatol 2022; 142: 1804–1811.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adamson AS, Suarez EA, Welch HG. Estimating overdiagnosis of melanoma using trends among black and white patients in the US. JAMA Dermatol 2022; 158: 426–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raghavan SS, Peternel S, Mully TW, et al. Spitz melanoma is a distinct subset of spitzoid melanoma. Mod Pathol 2020; 33: 1122–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glasziou PP, Jones MA, Pathirana T, Barratt AL, Bell KJ. Estimating the magnitude of cancer overdiagnosis in Australia. Med J Aust 2020; 212: 163–8. Erratum in: Med J Aust 2020; 212: 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cook MG. Benign melanocytic lesions mimicking melanomas. Pathology 2004; 36: 414–8. [DOI] [PubMed] [Google Scholar]
- 39.Patrawala S, Maley A, Greskovich C, et al. Discordance of histopathologic parameters in cutaneous melanoma: clinical implications. J Am Acad Dermatol 2016; 74: 75–80. [DOI] [PubMed] [Google Scholar]
- 40.Eguchi MM, Elder DE, Barnhill RL, et al. Prognostic modeling of cutaneous melanoma stage I patients using cancer registry data identifies subsets with very-low melanoma mortality. Cancer 2023; 129: 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clark WHJ, From L, Bernardino EA, Mihm MCJ. The histogenesis and biologic behavior of primary human malignant melanomas of the skin. Cancer Res 1969; 29: 705–27. [PubMed] [Google Scholar]
- 42.Clark WHJ. A classification of malignant melanoma in man correlated with histogenesis and biologic behavior. In: Montagna W, Hu F, editors. Advances in the biology of the skin volume VIII. New York: Pergamon Press, 1967; 621–47. [Google Scholar]
- 43.Elder D. Tumor progression, early diagnosis and prognosis of melanoma. Acta Oncol 1999; 38: 535–47. [DOI] [PubMed] [Google Scholar]
- 44.Herlyn M, Balaban G, Bennicelli J, et al. Primary melanoma cells of the vertical growth phase: similarities to metastatic cells. J Natl Cancer Inst 1985; 74: 283–9. [PubMed] [Google Scholar]
- 45.Herlyn M, Thurin J, Balaban G, et al. Characteristics of cultured human melanocytes isolated from different stages of tumor progression. Cancer Res 1985; 45: 5670–6. [PubMed] [Google Scholar]
- 46.Balaban GB, Herlyn M, Clark WHJ, Nowell PC. Karyotypic evolution in human malignant melanoma. Cancer Genet Cytogenet 1986; 19: 113–22. [DOI] [PubMed] [Google Scholar]
- 47.Chi HI, Otsuka F, Ishibashi Y. Topographical differences in cellular DNA content between the radial growth phase and the vertical growth phase of superficial spreading melanoma. Nippon Hifuka Gakkai Zasshi 1989; 99: 493–7. [PubMed] [Google Scholar]
- 48.Elder DE, Rodeck U, Thurin J, et al. Antigenic profile of tumor progression stages in human melanocytic nevi and melanomas. Cancer Res 1989; 49: 5091–6. [PubMed] [Google Scholar]
- 49.Albelda SM, Mette SA, Elder DE, et al. Integrin distribution in malignant melanoma: association of the beta 3 subunit with tumor progression. Cancer Res 1990; 50: 6757–64. [PubMed] [Google Scholar]
- 50.Elder DE, Guerry DI, Epstein MN, et al. Invasive malignant melanomas lacking competence for metastasis. Am J Dermatopathol 1984; 6: 55–62. [PubMed] [Google Scholar]
- 51.Guerry DIV, Synnestvedt M, Elder DE, Schultz D. Lessons from tumor progression: the invasive radial growth phase of melanoma is common, incapable of metastasis, and indolent. J Invest Dermatol 1993; 100: 342. –5S. [DOI] [PubMed] [Google Scholar]
- 52.McDermott NC, Hayes DP, al-Sader MH, et al. Identification of vertical growth phase in malignant melanoma. A study of interobserver agreement. Am J Clin Pathol 1998; 110: 753–7. [DOI] [PubMed] [Google Scholar]
- 53.Lefevre M, Vergier B, Balme B, et al. Relevance of vertical growth pattern in thin level II cutaneous superficial spreading melanomas. Am J Surg Pathol 2003; 27: 717–24. [DOI] [PubMed] [Google Scholar]
- 54.Compérat E, Oszwald A, Wasinger G, Shariat S, Amin M. Update on flat and papillary urothelial lesions: genitourinary Pathology Society consensus recommendations. Surg Pathol Clin 2022; 15: 629–40. [DOI] [PubMed] [Google Scholar]
- 55.Cook MG, Clarke TJ, Humphreys S, et al. The evaluation of diagnostic and prognostic criteria and the terminology of thin cutaneous melanoma by the CRC Melanoma Pathology Panel. Histopathology 1996; 28: 497–512. [DOI] [PubMed] [Google Scholar]
- 56.Scolyer RA, Shaw HM, Thompson JF, et al. Interobserver reproducibility of histopathologic prognostic variables in primary cutaneous melanomas. Am J Surg Pathol 2003; 27: 1571–6. [DOI] [PubMed] [Google Scholar]
- 57.Scolyer RA, Rawson RV, Gershenwald JE, Ferguson PM, Prieto VG. Melanoma pathology reporting and staging. Mod Pathol 2020; 33 (Suppl 1): 15–24. [DOI] [PubMed] [Google Scholar]
- 58.Guitart J, Lowe L, Piepkorn M, et al. Histological characteristics of metastasizing thin melanomas: a case-control study of 43 cases. Arch Dermatol 2002; 138: 603–8. [DOI] [PubMed] [Google Scholar]