

Abstract
Post-translational modifications by small ubiquitin-like modifiers (SUMO) are dysregulated in many types of cancers. SUMO E1 enzyme has recently been suggested as a new immuno-oncology target. COH000 was recently identified as a highly specific allosteric covalent inhibitor of the SUMO E1. However, marked discrepancy was found between X-ray structure of the covalent COH000-bound SUMO E1 complex and available structure-activity-relationship (SAR) data of inhibitor analogs due to unresolved non-covalent protein-ligand interactions. Here, we have investigated non-covalent interactions between COH000 and SUMO E1 during the inhibitor dissociation through novel Ligand Gaussian accelerated Molecular Dynamics (LiGaMD) simulations. Our simulations have identified critical low-energy non-covalent binding intermediate conformation of COH000 that agreed excellently with published and new SAR data of the COH000 analogs, which were otherwise inconsistent with X-ray structure. Altogether, our biochemical experiments and LiGaMD simulations have uncovered a critical non-covalent binding intermediate during allosteric inhibition of the SUMO E1 complex.
Keywords: SUMO E1, allosteric inhibitor, Ligand Gaussian accelerated molecular dynamics, structure-activity-relationship, SUMOylation
Graphical Abstract
Since discovery of ubiquitin over 40 years ago, nearly 20 ubiquitin homologous protein modifiers have been discovered and are collectively called Ubl. The best characterized Ubl modifications include those by SUMO, Nedd8, and Atg8. All Ubl modifiers conjugate to other proteins or lipids through a cascade of reactions catalyzed by enzymes generally known as E1, E2, and E3 1. Briefly, a Ubl is first activated by E1—the activating enzyme—through ATP hydrolysis and then forms a thioester conjugate with E1. A Ubl is then transferred to an E2. Finally, a Ubl is transferred to target proteins, a step usually catalyzed by an E3 ligase. The modifications can be removed by deconjugation enzymes. Each Ubl has its corresponding set of E1, E2, E3, and deconjugation enzymes. Approximately 5% of the human proteome is involved in catalyzing the conjugation or removal of this type of post-translational modification (PTM).
The SUMO activating enzyme (SAE) has recently been recognized as a novel target for activating anti-tumor immunity. SUMOylation inhibition stimulates the production of type I interferons (IFNs) and IFNγ 2. This is highly significant because it has been known for nearly a decade that activation of type I IFN expression activates dendritic cells for immune rejection of tumors through antitumor CD8+ T cell responses 3. Besides immune effects, targeting SAE directly inhibits tumor cells 4. c-Myc was the first human oncogene to be identified more than 30 years ago 5 and is a driver in at least 70% of all human cancers 6, but has not been directly inhibited by an FDA approved drug. Genome-wide RNAi screens identified the genes encoding the SAE subunits (SAE1 and SAE2) to be those with the strongest synthetic lethal interactions with c-Myc7. We showed that inhibiting SAE activated the expression of the tumor suppressor miR-34 that targets the mRNA of c-Myc and other oncogenic pathways 8. Additionally, SAE is a target for inhibiting cancer cell stemness 9. The potentially wide therapeutic window of targeting SAE as a cancer therapeutic approach is supported by clinical trials of an ATP analog SAE inhibitor, TAK-981, developed by Takeda Pharmaceuticals.
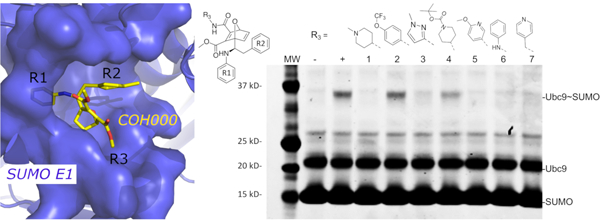
We have recently identified an allosteric, covalent mechanism to inhibit SAE that is potentially applicable to target other Ubl E1 enzymes because the covalent inhibitor binding site is highly conserved in this family of enzymes 10, 11 Our crystal structure of SAE in complex with an allosteric inhibitor, COH000, revealed a major conformational change including rotation of the catalytic Cys containing domain (CDD) and disruption of the ATP-binding site. This and other structural changes exposed a deep pocket that binds COH000 (Fig. 1A and 1B), which was hidden in all previously solved structures of Ubl E1s 12. Due to the sequence and structural similarities among the E1 enzymes, it is possible that this mechanism can be used to target other Ubl E1 enzymes.
Figure 1.
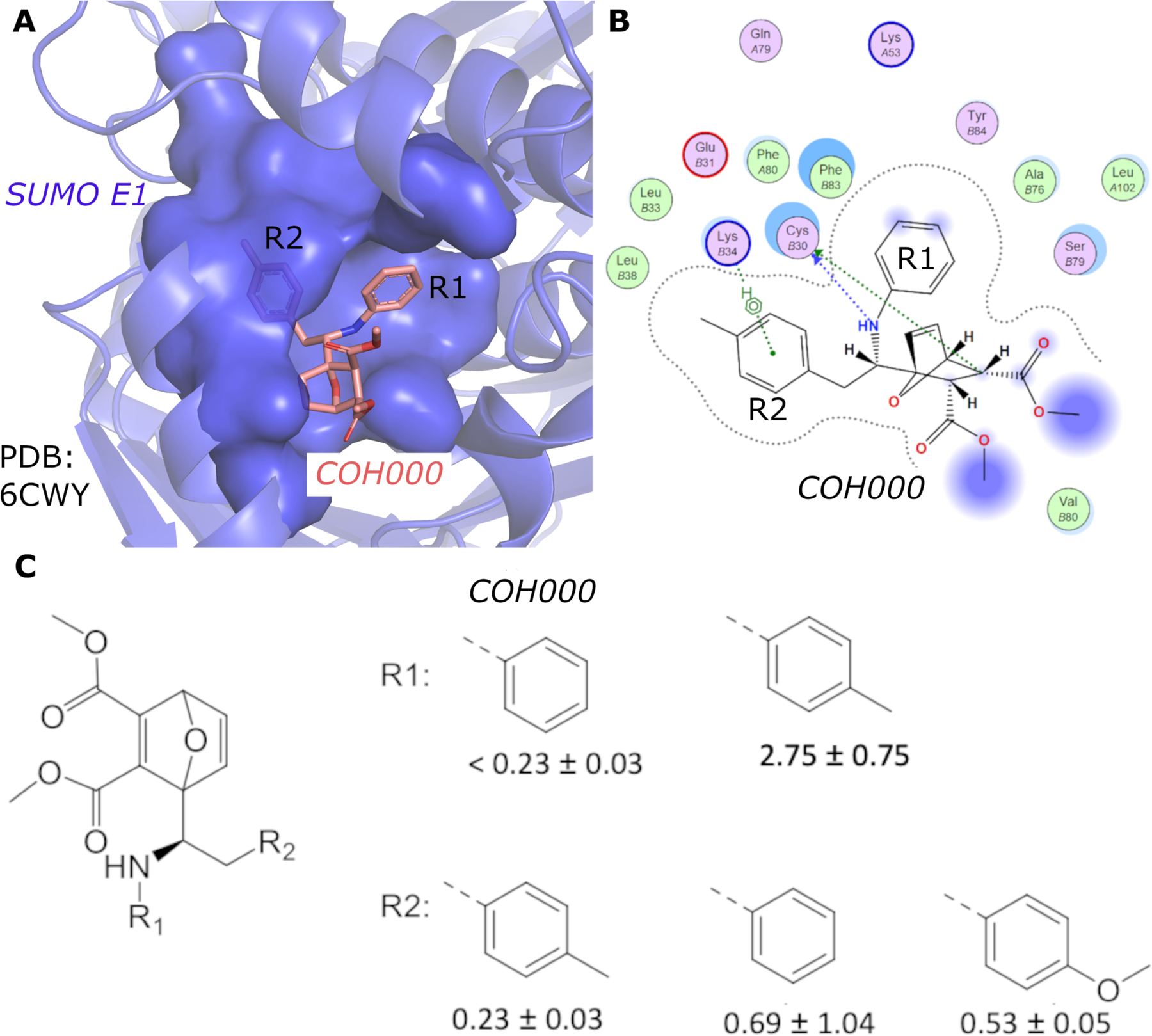
(A) Crystal structure of SUMO E1 (cartoon) in complex with the COH000 allosteric inhibitor (sticks) (PDB: 6CWY). The protein residues of SUMO E1 surrounding COH000 are represented in surface. (B) 2D LigPlot represention of the binding pocket residues of SUMO E1 that interact with COH000. Blue filled circles on COH000 represent solvent accessibility with larger circles representing greater solvent accessibility. Polar and non-polar residues are colored in pink and green circles, respectively. Acidic and basic residues have a red and blue circle border, respectively. (C) Discrepancy between the crystal structure and the structure-activity-relationship of the compounds obtained. Specifically, the R1 group does not tolerate a small change from a benzyl to a methyl benzyl group, although the methyl addition site is partially solvent accessible and has the space to accommodate the addition of a methyl group in the crystal structure. In contrast, variations of the R2 group that is deeply buried in a hydrophobic pocket and is not solvent accessible in the crystal structure tolerates a variety of changes, including removal of the methyl group or replacement of methyl with a methoxy.
While revealing an exciting allosteric inhibition mechanism for SAE, the crystal structure is not consistent with the structure-activity-relationship of the compounds obtained 10. As shown in Fig. 1, the aniline, which binds to an exposed shallow pocket according to the crystal structure, is not tolerable to changes; addition of a methyl group to the aniline resulted in > 15-fold reduction in inhibition potency. However, the tolyl group, which is deeply buried in a hydrophobic pocket, can accommodate various substitutions, such as methoxy group, without significant changes in inhibitory potency (Fig. 1C). Removal of the methyl group also did not significantly affect inhibition potency (Fig. 1C). The mechanism of a covalent inhibitor involves specific non-covalent interaction to recruit a small molecule inhibitor followed by covalent adduct formation. Therefore, the inhibitory potency depends on the non-covalent binding affinity (KI) and the rate of covalent adduct formation (kinact). The KI/kinact ratio, therefore, correlates to the 50% inhibition concentration (IC50) of a covalent inhibitor 13. Because these analogs contain the same a-b unsaturated carbonyl as the electrophilic acceptor for covalent adduct formation, it is likely that the intrinsic covalent adduct formation rates (kinact) are similar. Therefore, non-covalent interaction (KI) is likely reflected by the observed SAR. However, such transient non-covalent interaction is not visible by the crystal structure.
Molecular Dynamics (MD) has proven useful in simulations of biomolecular dynamics. Gaussian accelerated molecular dynamics (GaMD) is an unconstrained enhanced sampling technique for large biomolecular simulations14. The system energy barriers are reduced by adding a harmonic boost potential to smooth the potential energy surface of biomolecules14. Moreover, proper energetic reweighting of GaMD simulations is achieved through cumulant expansion to the 2nd order (“Gaussian approximation”). GaMD does not require predefined reaction coordinates or collective variables to study complex biological processes such as ligand binding to proteins. Compared to conventional MD (cMD), GaMD could speed up biomolecular simulations by orders of magnitude. GaMD simulations have been successfully demonstrated on ligand binding, protein folding, GPCR activation, and the protein-membrane, protein-protein and protein-nucleic acid interactions15. GaMD has been further developed to new algorithms such as Ligand GaMD (LiGaMD)16, Peptide GaMD (Pep-GaMD)17 and Protein-Protein interaction-GaMD (PPI-GaMD)18, which have greatly expanded the abilities to simulate biomolecular binding processes. Microsecond LiGaMD, Pep-GaMD and PPI-GaMD simulations could successfully capture repetitive binding and dissociation of small-molecule ligands16, highly flexible peptides17 and proteins18, which could further aid in ligand/peptide/protein binding thermodynamics and kinetics calculations.
In this study, we have performed all-atom simulations using LiGaMD to understand the mechanism of non-covalent inhibitor interaction with SUMO E1 after removing the covalent adduct. We generated a computational model using the X-ray crystal structure of the SUMO E1 in complex with the allosteric inhibitor COH000 (PDB: 6CWY)19 (Fig. 2A). Homology modeling was performed using SWISS-MODEL20 to add the missing regions including residues 179–204 in chain C and 54–68, 225–237, 291–312, 333–341 in chain D of the protein, respectively. The thioester bond between COH000 and Cys30 was removed using PyMOL21 (Fig. 2C–D). The simulation system was set up by solvating the complex in 0.15M NaCl solution and ten independent 1μs LiGaMD simulations were performed for analysis (See SI for details). Multiple dissociation events of the COH000 inhibitor were captured in the LiGaMD simulations that helped identify an important low-energy non-covalent bound conformation of COH000. The non-covalent bound conformation was in excellent agreement with the SAR experimental data. This will greatly facilitate rational design of new covalent and non-covalent allosteric inhibitors targeting the SUMO E1.
Figure 2.
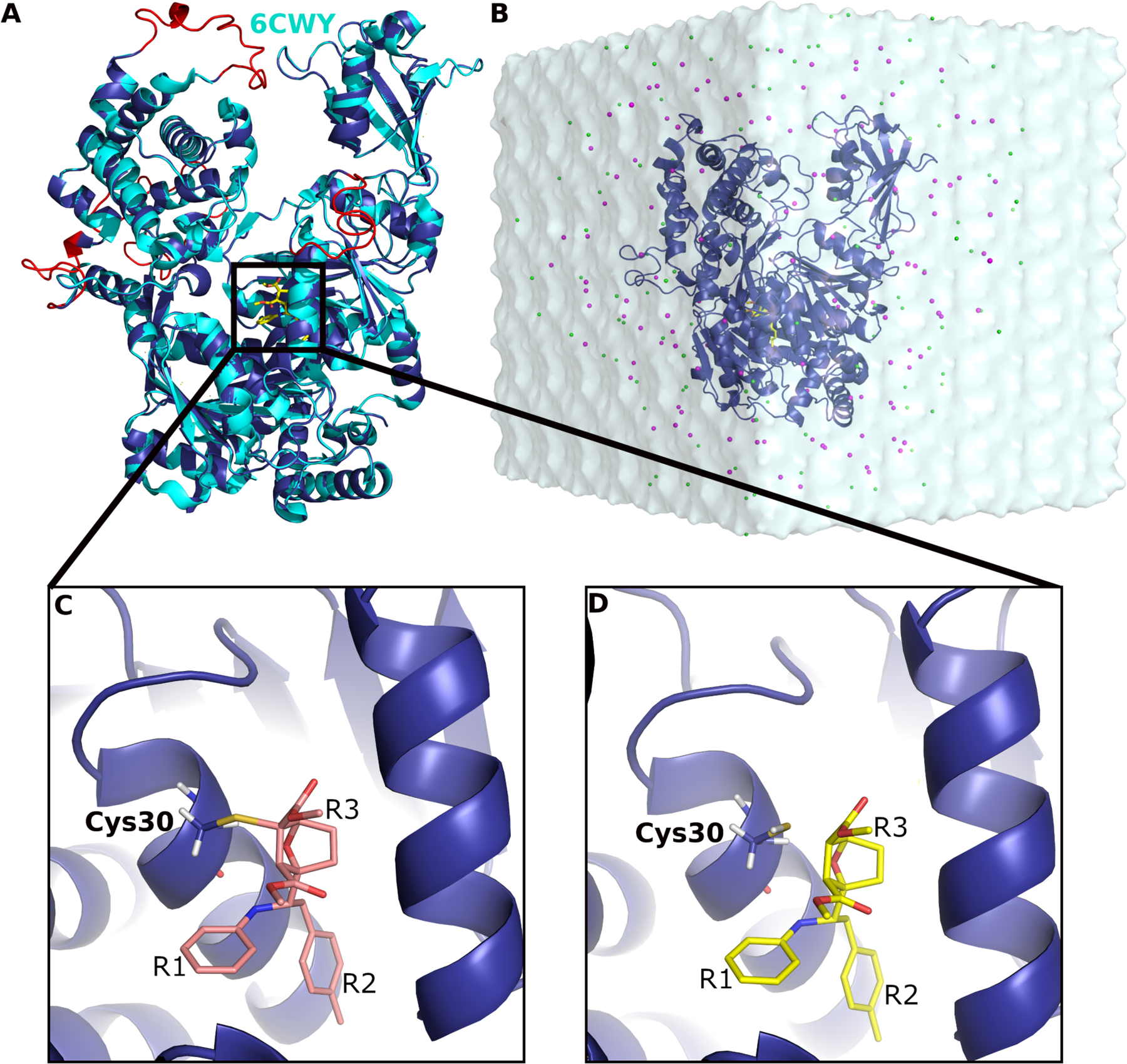
(A) Computational model of SUMO E1 (blue cartoon) aligned to the X-ray structure (cyan cartoon) in complex with COH000 (yellow sticks). The missing regions added through homology modeling are highlighted in red. (B) LiGaMD simulation input system with COH000 (yellow sticks) bound to SUMO-E1 (blue cartoon) that was solvated in 0.15 M NaCl (magenta and green beads) solution. (C) A covalent bond was formed between COH000 and Cys30 in SUMO E1 in the X-ray structure. (D) The protein-ligand covalent bond was broken for LiGaMD simulations of non-covalent COH000 interacting with the SUMO E1. The R1, R2 and R3 groups are indicated in (C) and (D).
Dissociation of the COH000 allosteric inhibitor was observed in ten independent 1μs LiGaMD simulations (Fig. 3A). The root-mean square deviation (RMSD) of COH000 relative to the X-ray crystal structure became greater than 30 Å within ~500 ns of all simulations except “Sim9”, in which the ligand dissociated completely into bulk solvent after ~750 ns. Ten LiGaMD simulations were combined for energetic reweighting to calculate the original free energy profiles. RMSD of COH000 relative to the starting X-ray structure was used as one reaction coordinate. A previous study suggested that the g1/g2 region undergoes conformational changes during the thioester bond formation22. It appeared flexible with atomic coordinates missing in the X-ray crystal structure of COH000-bound SUMO E119. Missing residues in this region were added using homology modeling for our LiGaMD simulations. Hence, the RMSD of the g1/g2 region relative to the simulation starting structure was used as the second reaction coordinate. The COH000 RMSD ranged from 0 Å to ~110 Å (Fig. 3A), and the g1/g2 RMSD ranged from ~5 Å to ~20 Å (Fig. S2). Two distinct low-energy wells “Non-Covalent Bound” (”NC-B”) and “Unbound” were identified in the reweighted free energy profile (Fig. 3B). In the “Unbound” state, the COH000 and g1/g2 RMSDs relative to the starting structure were >~30 Å and ~10–18 Å, respectively. In the “NC-B” state, the COH000 and g1/g2 RMSDs relative to the starting structure were ~ 5 Å and ~12 Å, respectively.
Figure 3.
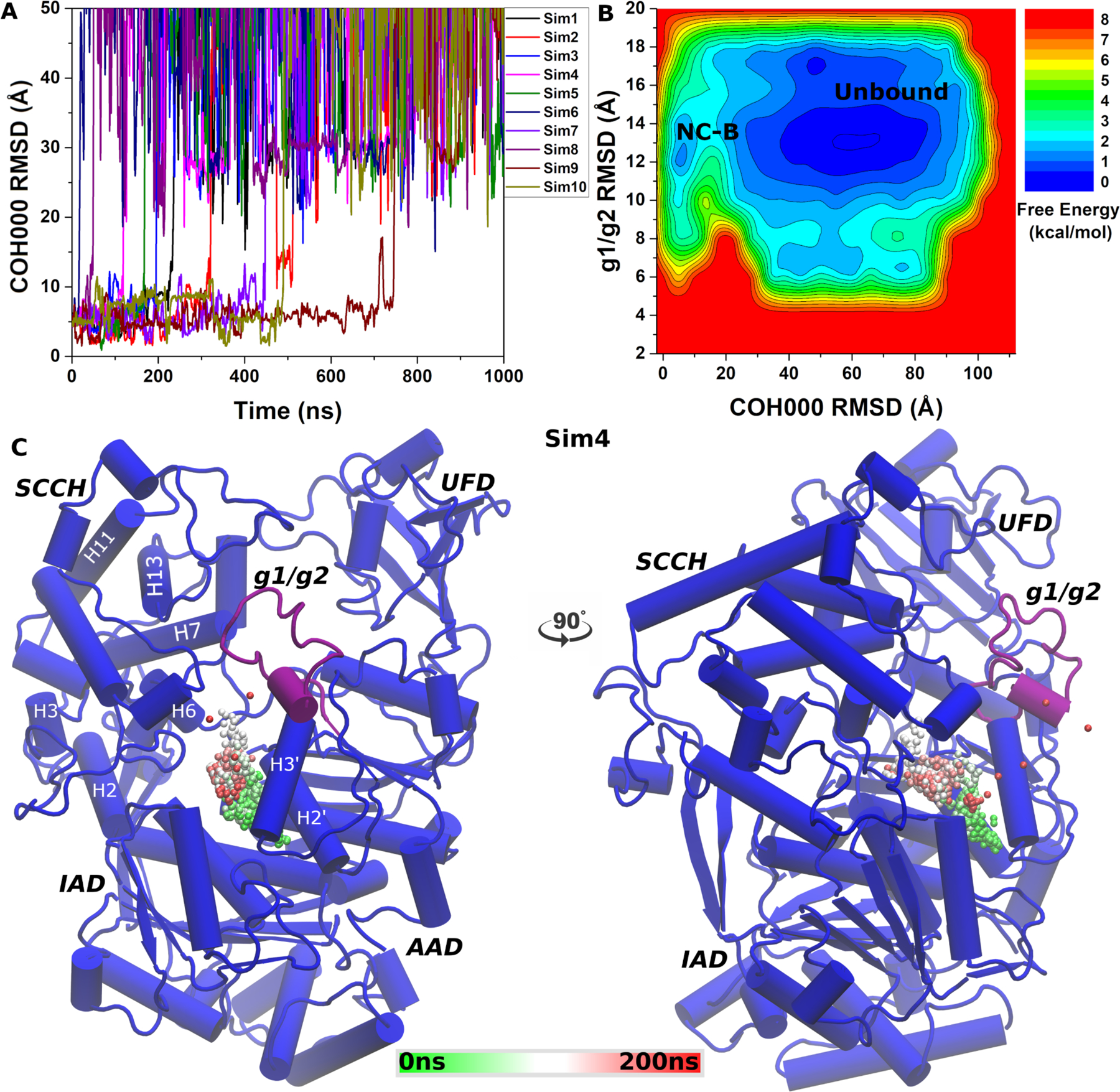
(A) Time course of COH000 RMSD relative to the X-ray conformation observed in 10 independent LiGaMD simulations. (B) Free energy profiles calculated regarding the COH000 RMSD relative to the X-ray conformation and g1/g2 RMSD relative to the starting computational model. The LiGaMD simulations sampled two low-energy conformations labeled “Non-covalent Bound” (NC-B) and “Unbound”. (C) Representative COH000 dissociation pathway from the allosteric binding pocket to the bulk solvent observed in “Sim3”. The dissociation pathway observed in the other LiGaMD simulations are shown in Figure S1.
With ligand dissociation observed in all the ten LiGaMD simulations, a representative dissociation pathway of the COH000 allosteric inhibitor obtained from “Sim4” was highlighted in Fig. 3C. Within the first ~150 ns, starting from the deeply buried binding pocket near the H2’ and H3’ helices of the active adenylation domain (AAD), the ligand gradually moved towards the inactive adenylation domain (IAD) and the second catalytic cysteine half-domain (SCCH) interface. The ligand then completely dissociated through the opening between the AAD, IAD and SCCH domains into the bulk solvent near ~200 ns. Similar dissociation pathway was observed in the other 9 LiGaMD trajectories as shown in Fig. S1A–S1I.
The g1/g2 region appeared unstructured in the homology model obtained using SWISS-MODEL as highlighted in Fig. 2A. In LiGaMD simulations, significant conformational changes were observed in the g1/g2 region during ligand dissociation. Structural clustering was performed using hierarchical agglomerative algorithm on the simulation snapshots of the g1/g2 region with all ten LiGaMD trajectories combined to obtain it’s top 20 representative conformations. The clusters were further energetically reweighted and ranked according to their free energy values as shown in Fig. 4A. The g1/g2 RMSD relative to the simulation starting structure in the structural clusters ranged from ~4 Å to ~19 Å. Folding of g1/g2 into a helical conformation was captured. However, the present LiGaMD simulations did not sample the completely bound conformation of g1/g2 in the allosteric pocket as observed in the X-ray structure of ligand-free SUMO E1 (PDB: 6CWZ). This suggested that longer time scale simulations would be required to sample the completely bound conformation of g1/g2 in SUMO E1 after ligand dissociation.
Figure 4.
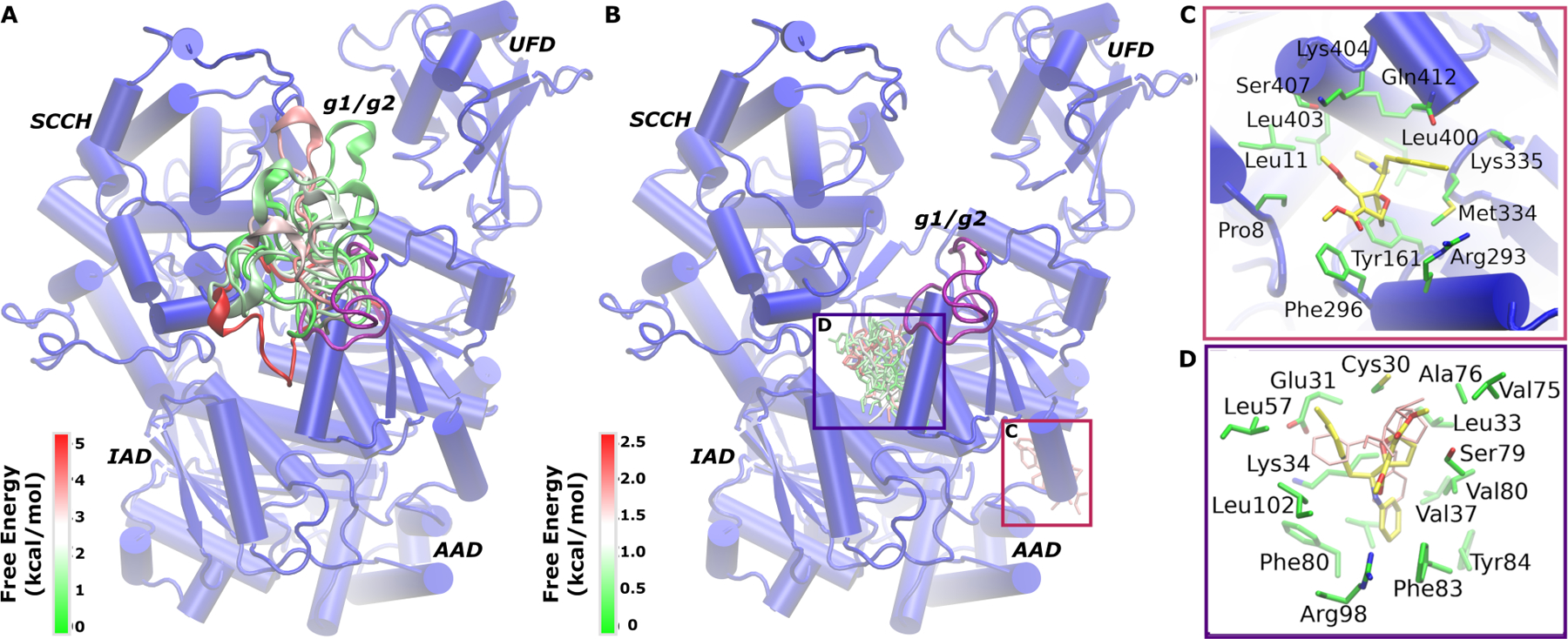
(A) Different conformations of the g1/g2 obtained through structural clustering of LiGaMD simulations colored by the reweighted free energy values in a green(0 kcal/mol)-white-red5 kcal/mol) color scale. (B) Top 30 structural clusters of the COH000 ligand obtained from LiGaMD simulations colored by the reweighted free energy values in a green(0 kcal/mol)-white-red(2.5 kcal/mol) color scale. The SUMO-E1 is shown in blue cartoon and the protein domains are labeled. (C-D) Two representative low-energy conformations of COH000 (yellow sticks) extracted from structural clusters in (B) with the surrounding protein residues highlighted in green sticks. COH000 conformation in (D) corresponded to the “Non-covalent Bound” (NC-B) state in Fig. 3B. The X-ray structure conformation of COH000 is shown in pink sticks.
Structural clustering of the COH000 ligand snapshots was performed using hierarchical agglomerative clustering method with RMSD cutoff of 3.2 Å. We observed two densely populated clusters of COH000 in the allosteric binding pocket and on the protein surface near the AAD domain, respectively (Fig. 4B). We further ranked the clusters according to their reweighted free energy values and highlighted representative conformations of the top 30 clusters (Fig. 4B). The lowest-energy cluster in each binding region of the ligand was extracted and the protein-ligand interactions were examined. Fig. 4C highlights the intermediate ligand binding conformation on the protein surface of the AAD domain. In this intermediate binding state, the ligand interacted with non-polar protein residues including Met334, Leu400, Ser407, Leu403, Leu11, Pro8, Phe296 and Tyr161 in a hydrophobic pocket. The hydrophobic interactions between the protein and ligand were consistent with the hydrophobic nature of the ligand. Polar residues such as Lys404, Lys335, Arg293 and Gln412 were also located within 4 Å of COH000 in the pocket. However, the side chains of these polar residues appeared facing away from the hydrophobic pocket and did not form direct interactions with the ligand.
The top-ranked binding conformation of COH000 was identified in the allosteric binding pocket of SUMO E1 (Fig. 4D). It corresponded to the “NC-B” (Non-Covalent Bound) low-energy state in the free energy profile (Fig. 3B). Compared to the X-ray crystallographic conformation, the R1 and R2 groups in the NC-B state of COH000 flipped in the allosteric pocket of SUMO E1 (Fig. 1C). COH000 in the NC-B conformation formed hydrophobic interactions with surrounding non-polar protein residues including Glu31, Lys34, Leu38, Val37, Leu102, Phe83, Val80, Leu33, Phe80, Cys30, Ser79 and Ala76, similar to those in the X-ray crystal structure. In addition, two distinct residues, Arg98 and Tyr84, formed new interactions with the ligand in the NC-B conformation.
The new simulation predicted non-covalent bound conformation of COH000 was in excellent agreement with the previously described SAR experimental data 10 (Fig. 1). In the NC-B conformation, COH000 positioned the R1 group in a buried pocket while the R2 group is partially solvent exposed (Fig. 5A and 5B). Accordingly, addition of a methyl group to the R1 group in COH000 reduced the inhibitory potency by more than 10 folds (Fig. 1C) 10. The partial solvent exposure of the R2 group in the NC-B conformation was highly consistent with SAR data that replacement of this group with the phenyl or methoxy phenyl group did not significantly change the inhibitory potency 10.
Figure 5.
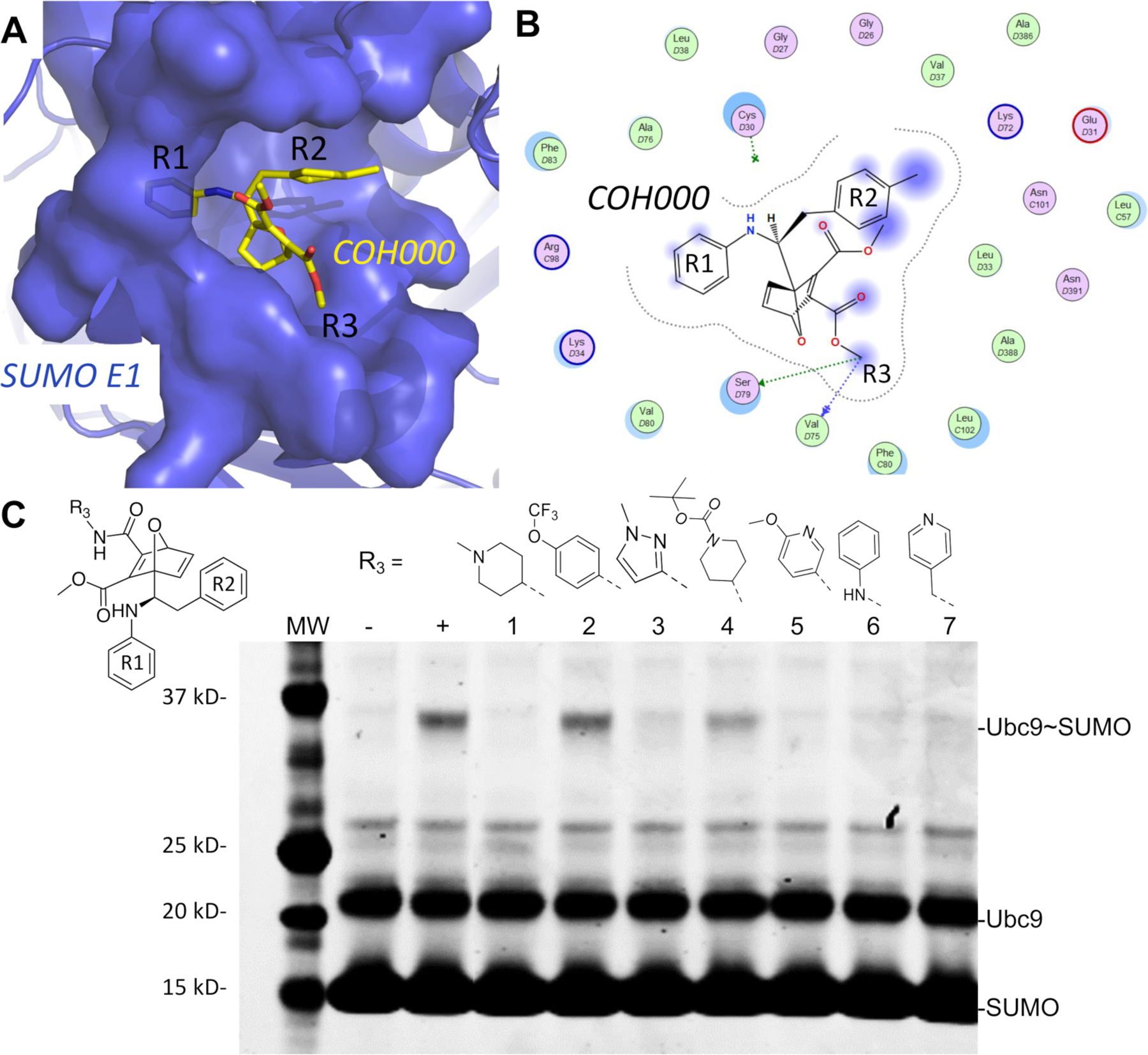
The lowest energy non-covalent bound conformation of the SUMO E1-COH000 complex identfied from LiGaMD simulations were supported by SAR of the inhibitor analogs. (A) The Non-covalent Bound” (NC-B) conformation of COH000 in the SUMO E1 protein pocket. (B) 2D LigPlot representation of the binding pocket residues of SUMO E1 that interact with COH000 as observed in Fig. 4D. Blue filled circles on COH000 represent solvent accessibility with larger circles representing greater solvent accessibility. Polar and non-polar residues are colored in pink and green circles, respectively. Acidic and basic residues have a red and blue circle border, respectively. (C) Analogs with larger R3 groups exhibit lower inhibitory effects, which is consistent with the steric restriction of the region interacting with this site of the compound as shown in (A). “−“ and “+” represent Ubc9-SUMO thioester formation catalyzed by E1, in the absence and presence of ATP, respectively, as controls. All reactions with compounds were conducted in the presence of ATP and at the same compound molar concentration, and lack of Ubc9-SUMO thioester formation bands indicates inhibition and the presence of Ubc9-SUMO thioester formation bands indicates lack of inhibition.
Furthermore, additional SAR on the R3 group of COH000 validated the NC-B conformation of COH000 predicted from the LiGaMD simulations (Fig. 5C). The R3 group in the NC-B conformation was located in a partially buried pocket (Fig. 5A and 5B), unlike the fully solvent accessible conformation shown in the crystal structure (Fig. 1A–B). It is conceivable that large groups of R3 could result in steric clashes to prevent the formation of this non-covalent bound complex and thus prevented the covalent adduct formation for the enzyme inhibition. We compared the inhibitory activities of a set of R3 analogs side-by-side in the same experiment. The SUMO-Ub9 conjugation formation assay catalyzed by SAE was used. The substrates, SUMO E1 and Ubc9, were prepared as a master mix along with ATP that was added to each SAE – inhibitor mixture to minimize experimental variations. Formation of the SUMO-Ubc9 conjugate was used to determine the inhibitory potency. Less SUMO-Ubc9 conjugate formation indicated higher inhibitory potency. The impact of chemical modifications in the R3 group on the inhibitory potency (Fig. 5C) would not be anticipated from the crystal structure of the covalent complex (Fig. 2). The molecular weights of the R3 groups were used to estimate their sizes as a first approximation. There appeared to be size dependence in potency. For example, compounds 3 and 5 were both bulkier and larger than groups in the other compounds and exhibited reduced inhibitory potency. From compounds 1 to 7, the molecular weights of the R3s are 98, 161, 81, 184, 108, 79 and 92 Da, correspondingly. The two compounds that had the poorest inhibition are compound 2 and 4 that have R3 groups of 161 Da and 184 Da. The molecular weights are statistically significantly different between compounds that nearly fully inhibited reaction and did not, with p value of 0.0007 by unpaired t test (Fig. S3). The new SAR data further supported the non-covalent bound conformation of COH000 predicted from the LiGaMD simulations. Taken together, the SAR of R1, R2 and R3 groups collectively supported the non-covalent binding intermediate identified in this study.
SAE serves as a novel therapeutic target for activating anti-tumor immunity. The crystal structure of the covalent allosteric inhibitor COH000 targeting the SAE10, 11 revealed a novel allosteric ligand-binding pocket that is highly conserved among the E1 family of enzymes and large scale conformational changes (Fig. 1A and 1B), which were not identified in any previously resolved structures of Ubl E1s 12. However, the crystal structure and the SAR of the compounds obtained lacked consistency10. Covalent inhibition mechanism involves specific non-covalent protein-ligand interaction that is essential to covalent adduct formation. We hypothesized that the non-covalent interaction prior to covalent adduct formation is the determinant of SAR, but such non-covalent transient intermediate has not been observed. To validate this hypothesis, in this work we have applied novel LiGaMD simulations to investigate the transient non-covalent ligand interactions in combination with additional SAR studies. These studies have revealed a non-covalent intermediate representing a “flip” of the ligand binding pose in comparison to the X-ray covalent complex.
Ten independent LiGaMD simulations successfully captured complete dissociation of COH000 from the allosteric binding pocket in SUMO E1. It is likely that the association and dissociation pathways involve the same low-energy intermediate. Energetic reweighting of LiGaMD simulations identified important low-energy conformational states of the system. Accurate free energy calculations of large biomolecular systems, such as the SUMO E1-COH000 complex, were exceedingly difficult even through enhanced sampling, hence, it is important to note that the free energy profiles presented in the study were not converged. Nevertheless, multiple dissociation events along with structural clustering of COH000 identified an important low-energy conformation that agreed excellently with the SAR experimental data.
LiGaMD simulations demonstrated the previously unknown transition of the g1/g2 helices between ligand-bound and ligand-free conformational states of SUMO E1 crystal structures. The previously solved apo structure of SUMO E1, i.e., the adenylation active structure, showed a well-ordered structure of the g1/g2 region of the AAD domain that occupied the deeply buried allosteric pocket11. During covalent binding of COH000, the g1/g2 is completely disordered which exposes the binding pocket and Cys30 for thioester bond formation. This suggested that the g1/g2 may undergo a large conformational transition during COH000 binding to the cryptic binding pocket that was previously unobserved. LiGaMD simulations illustrated that the g1/g2 sampled a vast degree of conformational space that was not shown by the previous crystal structures. Starting from a disordered loop exposed to the solvent, LiGaMD showed that the g1/g2 was able to form helical conformations at varying positions. The g1/g2 was extremely dynamic with RMSD calculated up to ~19 Å relative to its starting structure in the course of ligand dissociation. Thus, LiGaMD simulations have captured the large conformational transition of the g1/g2 region in SUMO E1 during ligand dissociation, although the g1/g2 had not reached the completely bound conformation in the protein allosteric pocket.
One unexpected finding of the LiGaMD simulations was the flip of the R1 and R2 groups of COH000 in the lowest energy non-covalent bound conformation compared with the covalent X-ray crystal structure. The predicted non-covalent bound pose of COH000 agreed well with the previously reported SAR and new SAR data described in this study, including the SAR of the R1, R2 and R3 groups (Fig. 1C and 5C). In contrast, the crystal structure of the covalent complex showed discrepancies with the SAR of these chemical groups as described in Introduction and Results. Therefore, the SAR experimental data supported the LiGaMD simulation predicted non-covalent bound conformation of the COH000 inhibitor, which apparently flipped to form covalent adduct in the allosteric binding pocket of SUMO E1.
In conclusion, with novel LiGaMD simulations of the COH000 allosteric inhibitor dissociation from the cryptic binding pocket of SUMO E1, we have not only revealed the ligand dissociation pathway, but more importantly resolved the discrepancies between the previously solved crystal structure of covalent COH000-bound SUMO E1 and the SAR experiments. Our new findings have provided important mechanistic insights into recognition of the allosteric inhibitor by SUMO E1 that is potently applicable to facilitate rational mechanism-based design of new covalent and non-covalent allosteric inhibitors of SAE and other Ubl E1 enzymes.
Supplementary Material
ACKNOWLEDGEMENTS
This work used supercomputing resources with allocation award TG-MCB180049 through the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS), and project M2874 through the National Energy Research Scientific Computing Center (NERSC), which is a U.S. Department of Energy Office of Science User Facility operated under Contract No. DE-AC02–05CH11231, and the Research Computing Cluster at the University of Kansas. This work was supported by the startup funding in the College of Liberal Arts and Sciences at the University of Kansas (to Y.M.) and 5R01CA265410 (to Y.C.) and R44CA189499 (to S.X.O).
Footnotes
SUPPORTING INFORMATION
Materials and methods, Gaussian accelerated molecular dynamics (GaMD), energetic reweighting of GaMD for Free Energy Calculations, ligand Gaussian accelerated molecular dynamics (LiGaMD), system setup, simulation protocol, simulation analysis, LiGaMD simulation input parameters, Figures S1–S3 and Compound synthesis.
Competing Interest Statement
Y.C. reports equity ownership and Board of Director with Suvalent Therapeutics and consulting fees from Suvalent Therapeutics, Inc. outside the submitted work.
REFERENCES
- (1).Sarge KD; Park-Sarge OK Sumoylation and human disease pathogenesis. Trends in biochemical sciences 2009, 34 (4), 200–205. Yeh ET SUMOylation and De-SUMOylation: wrestling with life’s processes. The Journal of biological chemistry 2009, 284 (13), 8223–8227. Hay RT SUMO: a history of modification. Molecular cell 2005, 18 (1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Decque A; Joffre O; Magalhaes JG; Cossec JC; Blecher-Gonen R; Lapaquette P; Silvin A; Manel N; Joubert PE; Seeler JS; et al. Sumoylation coordinates the repression of inflammatory and anti-viral gene-expression programs during innate sensing. Nat Immunol 2016, 17 (2), 140–149. DOI: 10.1038/ni.3342. Crowl JT; Stetson DB SUMO2 and SUMO3 redundantly prevent a noncanonical type I interferon response. Proceedings of the National Academy of Sciences of the United States of America 2018, 115 (26), 6798–6803. DOI: . [DOI] [PubMed] [Google Scholar]
- (3).Fuertes MB; Kacha AK; Kline J; Woo SR; Kranz DM; Murphy KM; Gajewski TF Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med 2011, 208 (10), 2005–2016. DOI: 10.1084/jem.20101159. Diamond MS; Kinder M; Matsushita H; Mashayekhi M; Dunn GP; Archambault JM; Lee H; Arthur CD; White JM; Kalinke U; et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 2011, 208 (10), 1989–2003. DOI: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).He X; Riceberg J; Soucy T; Koenig E; Minissale J; Gallery M; Bernard H; Yang X; Liao H; Rabino C; et al. Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor. Nat Chem Biol 2017, 13 (11), 1164–1171. DOI: 10.1038/nchembio.2463. [DOI] [PubMed] [Google Scholar]
- (5).Hayward WS; Neel BG; Astrin SM Activation of a cellular onc gene by promoter insertion in ALV-induced lymphoid leukosis. Nature 1981, 290 (5806), 475–480, Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S. [DOI] [PubMed] [Google Scholar]
- (6).Dang CV MYC on the path to cancer. Cell 2012, 149 (1), 22–35, Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't Review. DOI: 10.1016/j.cell.2012.03.003. Vita M; Henriksson M The Myc oncoprotein as a therapeutic target for human cancer. Semin Cancer Biol 2006, 16 (4), 318–330. DOI: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Luo J; Emanuele MJ; Li D; Creighton CJ; Schlabach MR; Westbrook TF; Wong KK; Elledge SJ A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009, 137 (5), 835–848. DOI: 10.1016/j.cell.2009.05.006. Kessler JD; Kahle KT; Sun T; Meerbrey KL; Schlabach MR; Schmitt EM; Skinner SO; Xu Q; Li MZ; Hartman ZC; et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 2012, 335 (6066), 348–353, Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, Non-P.H.S. DOI: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Li YJ; Du L; Aldana-Masangkay G; Wang X; Urak R; Forman SJ; Rosen ST; Chen Y Regulation of miR-34b/c-targeted gene expression program by SUMOylation. Nucleic Acids Res 2018, 46 (14), 7108–7123. DOI: 10.1093/nar/gky484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Du L; Li YJ; Fakih M; Wiatrek RL; Duldulao M; Chen Z; Chu P; Garcia-Aguilar J; Chen Y Role of SUMO activating enzyme in cancer stem cell maintenance and self-renewal. Nature communications 2016, 7, 12326. DOI: 10.1038/ncomms12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Li YJ; Du L; Wang J; Vega R; Lee TD; Miao Y; Aldana-Masangkay G; Samuels ER; Li B; Ouyang SX; et al. Allosteric Inhibition of Ubiquitin-like Modifications by a Class of Inhibitor of SUMO-Activating Enzyme. Cell Chem Biol 2019, 26 (2), 278–288 e276. DOI: 10.1016/j.chembiol.2018.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lv Z; Yuan L; Atkison JH; Williams KM; Vega R; Sessions EH; Divlianska DB; Davies C; Chen Y; Olsen SK Molecular mechanism of a covalent allosteric inhibitor of SUMO E1 activating enzyme. Nature communications 2018, 9 (1), 5145. DOI: 10.1038/s41467-018-07015-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lois LM; Lima CD Structures of the SUMO E1 provide mechanistic insights into SUMO activation and E2 recruitment to E1. The EMBO journal 2005, 24 (3), 439–451. Olsen SK; Capili AD; Lu X; Tan DS; Lima CD Active site remodelling accompanies thioester bond formation in the SUMO E1. Nature 2010, 463 (7283), 906–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Miyahisa I; Sameshima T; Hixon MS Rapid Determination of the Specificity Constant of Irreversible Inhibitors (kinact/KI) by Means of an Endpoint Competition Assay. Angew Chem Int Ed Engl 2015, 54 (47), 14099–14102. DOI: 10.1002/anie.201505800. [DOI] [PubMed] [Google Scholar]
- (14).Miao Y; Feher VA; McCammon JA Gaussian accelerated molecular dynamics: Unconstrained enhanced sampling and free energy calculation. Journal of chemical theory and computation 2015, 11 (8), 3584–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wang J; Arantes PR; Bhattarai A; Hsu RV; Pawnikar S; Huang Y. m. M.; Palermo G; Miao Y. Gaussian accelerated molecular dynamics: Principles and applications. Wiley Interdisciplinary Reviews: Computational Molecular Science 2021, 11 (5), e1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Miao Y; Bhattarai A; Wang J Ligand Gaussian accelerated molecular dynamics (LiGaMD): Characterization of ligand binding thermodynamics and kinetics. J Chem Theory Comput 2020, 16 (9), 5526–5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wang J; Miao Y Peptide Gaussian accelerated molecular dynamics (Pep-GaMD): Enhanced sampling and free energy and kinetics calculations of peptide binding. The Journal of Chemical Physics 2020, 153 (15), 154109. DOI: 10.1063/5.0021399 (acccessed 2020/10/22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wang J; Miao Y Protein–Protein Interaction-Gaussian Accelerated Molecular Dynamics (PPI-GaMD): Characterization of Protein Binding Thermodynamics and Kinetics. Journal of Chemical Theory and Computation 2022, 18 (3), 1275–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lv Z; Yuan L; Atkison JH; Williams KM; Vega R; Sessions EH; Divlianska DB; Davies C; Chen Y; Olsen SK Molecular mechanism of a covalent allosteric inhibitor of SUMO E1 activating enzyme. Nature communications 2018, 9 (1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Waterhouse A; Bertoni M; Bienert S; Studer G; Tauriello G; Gumienny R; Heer FT; de Beer TAP; Rempfer C; Bordoli L SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic acids research 2018, 46 (W1), W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).DeLano WL Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr 2002, 40 (1), 82–92. [Google Scholar]
- (22).Olsen SK; Capili AD; Lu X; Tan DS; Lima CD Active site remodelling accompanies thioester bond formation in the SUMO E1. Nature 2010, 463 (7283), 906–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.