

ABSTRACT
The human immunodeficiency virus type 1 (HIV-1) envelope glycoprotein (Env) is incorporated into virions at the site of particle assembly on the plasma membrane (PM). The route taken by Env to reach the site of assembly and particle incorporation remains incompletely understood. Following initial delivery to the PM through the secretory pathway, Env is rapidly endocytosed, suggesting that recycling is required for particle incorporation. Endosomes marked by the small GTPase Rab14 have been previously shown to play a role in Env trafficking. Here, we examined the role of KIF16B, the molecular motor protein that directs outward movement of Rab14-dependent cargo, in Env trafficking. Env colocalized extensively with KIF16B+ endosomes at the cellular periphery, while expression of a motor-deficient mutant of KIF16B redistributed Env to a perinuclear location. The half-life of Env labeled at the cell surface was markedly reduced in the absence of KIF16B, while a normal half-life was restored through inhibition of lysosomal degradation. In the absence of KIF16B, Env expression on the surface of cells was reduced, leading to a reduction in Env incorporation into particles and a corresponding reduction in particle infectivity. HIV-1 replication in KIF16B knockout cells was substantially reduced compared to that in wild-type cells. These results indicated that KIF16B regulates an outward sorting step involved in Env trafficking, thereby limiting lysosomal degradation and enhancing particle incorporation.
IMPORTANCE The HIV-1 envelope glycoprotein is an essential component of HIV-1 particles. The cellular pathways that contribute to incorporation of envelope into particles are not fully understood. Here, we have identified KIF16B, a motor protein that directs movement from internal compartments toward the plasma membrane, as a host factor that prevents envelope degradation and enhances particle incorporation. This is the first host motor protein identified that contributes to HIV-1 envelope incorporation and replication.
KEYWORDS: HIV-1, KIF16B, kinesin, envelope glycoprotein, recycling, trafficking
INTRODUCTION
The HIV-1 Env protein plays critical roles in the viral life cycle, mediating receptor and coreceptor binding, fusion, and entry into target cells. Env is the principal neutralizing determinant on the surface of virions and has been a primary focus of vaccine design efforts since the onset of the HIV pandemic. While many aspects of Env structure and function are now understood, the determinants of particle incorporation of Env are not fully defined. HIV-1 Env is synthesized on endoplasmic reticulum (ER)-bound ribosomes as a precursor gp160 protein. Trimerization and initial glycosylation of Env take place in the ER, followed by additional glycan modifications and cleavage of gp160 into gp120 (SU) and gp41 (TM) subunits in the Golgi apparatus. After delivery to the plasma membrane (PM), the heterotrimeric Env complex is rapidly endocytosed into early endosomes (1–3). The trafficking events that occur following Env endocytosis have been poorly defined, and they are the subject of this report.
Clues to Env trafficking and particle incorporation have come from studies of the Env cytoplasmic tail (CT). Long CTs are a characteristic feature of most lentiviral Env proteins. HIV-1, HIV-2, and simian immunodeficiency virus have Env CTs of approximately 150 amino acids. The CT of HIV-1 Env contains multiple functional motifs that have been implicated in intracellular trafficking events (reviewed in references 4 and 5), suggesting that the CT interacts with host trafficking molecules to direct intracellular trafficking and particle incorporation of Env. In support of the critical role of the CT, Murakami and Freed showed that there are remarkable cell type-specific differences in incorporation of Env with a large deletion in the CT (6). While some cell lines, such as MT-4 and 293T cells, readily incorporate Env bearing a truncated CT, incorporation into virions in most T cell lines, in primary T lymphocytes, and in monocyte-derived macrophages requires an intact CT (6–9). HeLa cells demonstrate an intermediate phenotype, with a reduction but not complete loss of Env incorporation with a CT truncation (6). The cell type-dependent incorporation of Env with a truncated CT, together with evidence supporting trafficking motifs within the CT, suggests that specific host factors are required for incorporation of Env into particles.
We previously demonstrated that a Rab-related adaptor protein, Rab11-FIP1C (FIP1C), plays a role in Env incorporation, and we outlined a key role for recycling pathways in this process (10–12). The specific Rab GTPase involved in FIP1C-mediated recycling of Env was shown to be Rab14 (10). Additional evidence supporting a role for Rab14 comes from Hoffman and colleagues, who demonstrated that Env traffics to Rab14+ late endosomes and lysosomes (13). Recycling of Env to the PM was demonstrated in that study, and a potential role for Rab14 in regulating lysosomal degradation versus delivery to the PM was suggested. Given the accumulating evidence for Rab14 in the intracellular trafficking of Env, we sought to evaluate the role of the molecular motor protein that associates with Rab14, KIF16B.
KIF16B is a member of the kinesin-3 family of molecular motor proteins. KIF16B is a plus-end-directed molecular motor that regulates the steady-state distribution of Rab5+ early endosomes and plays a role in recycling of endocytosed cargo to the cell surface (14). A seminal study describing KIF16B function demonstrated that disruption of KIF16B led to redistribution of epidermal growth factor receptor (EGFR) and enhanced its degradation in the lysosome (14). KIF16B has also been implicated in anterograde transport of transferrin receptor and fibroblast growth factor receptor (FGFR), in contributing to major histocompatibility class I cross-presentation, and in directing apical transcytosis of cargo in epithelial cells (14–17). KIF16B binds directly to Rab14, and a Rab14-KIF16B complex has been shown to be essential for transport of FGFR and in mediating antigen cross-presentation by dendritic cells (15, 16). Given the role of Rab14 in HIV-1 Env trafficking, we hypothesized that KIF16B-mediated transport plays a role in anterograde trafficking of Env. We found that KIF16B directs Env to early endosomes located at the periphery of cells, while expression of a motor-deficient KIF16B molecule retained Env in perinuclear endosomal compartments. Measurement of the half-life of Env in KIF16B knockout (KO) cells revealed an enhanced rate of Env degradation that could be reversed using an inhibitor of lysosomal acidification. Knockout of KIF16B expression in a T-cell line diminished cell surface levels of Env incorporation, Env incorporation into particles, particle infectivity, and viral replication in a spreading infection. These studies implicated KIF16B in anterograde movement of Env away from degradation in the lysosome, allowing more efficient incorporation into HIV-1 particles.
RESULTS
Env localizes to KIF16B-positive compartments and is redistributed by dominant-negative KIF16B overexpression.
To determine if HIV-1 Env is found in KIF16B-positive endosomal compartments, we coexpressed the HIV-1 NL4-3 Env with KIF16B tagged with yellow fluorescent protein (YFP) in HeLa cells. KIF16B+ compartments were readily observed at the periphery of cells and demonstrated a striking colocalization with Env (Fig. 1A). Expression of KIF16B bearing a mutation in the nucleotide binding motif that disrupts its motor function, KIF16B-S109A-YFP, has been shown to act in a dominant-negative fashion to impair EGFR trafficking (14). Similar to what was seen with EGFR, HIV-1 Env was redistributed by expression of KIF16B-S109A-YFP to a perinuclear location (Fig. 1B). Expression of Env from intact NL4-3 proviral DNA together with wild-type KIF16B led to a peripheral distribution of Env, similar to that seen upon expression of Env alone (Fig. 1C). Expression of intact provirus with KIF16B-S109A-YFP also revealed relocalization of Env to a perinuclear location (Fig. 1D). To quantify the observed difference in subcellular localization, we used a mask of 30% the distance from the outer edge at the periphery of the cell to the stained nucleus and measured Env signal within this mask compared to total Env signal in the cell. Forty cells were examined for this analysis. The significant differences in peripheral localization shown in the representative images were confirmed quantitatively, as shown in Fig. 1E. Expression of either wild-type (WT) or S109A KIF16B resulted in a high degree of colocalization with Env, as measured by Pearson’s coefficient (Fig. 1F). We noted a somewhat higher degree of colocalization with KIF16B-S109A-YFP expression, which was statistically significant when Env alone was expressed (Fig. 1F). These results largely mirrored results reported for KIF16B relocalization of EGFR (14) and supported the hypothesis that KIF16B promotes anterograde transport of Env in early endosomal compartments.
FIG 1.

Env localizes to KIF16B-positive compartments and is redistributed by dominant-negative KIF16B. Approximate cell periphery outlines are shown as dashed lines. (A) Subcellular distribution of NL4-3 Env when coexpressed with KIF16B-WT-YFP in HeLa cells. Cells were fixed and immunolabeled with human neutralizing antibody 2G12 to stain HIV-1 gp120. Green, KIF16B; red, Env; rightmost image, overlay. (B) Distribution of NL4-3 Env when coexpressed with dominant KIF16B-S109A-YFP. Staining is as described for panel A. (C and D) Coexpression of proviral NL4-3 with WT and dominant-negative KIF16B, respectively. Staining for Env as described for panels A and B. (E) Percent Env signal at periphery of cells observed for each combination of coexpression shown in panels A to D. Signal at the periphery was the sum all Env signal within 30% of the distance from edge of the cell to the nucleus. Results are shown as means ± standard deviations from a total of 10 representative images. ns, not significant; ***, P < 0.0008; ****, P < 0.0001. P values were calculated using Student's unpaired t test using GraphPad Prism 9. (F) The degree of colocalization between KIF16B constructs and Env was measured by Pearson's correlation coefficient equation using Volocity 6.5.1 software after thresholding. Bars, 5 μm.
KIF16B fails to redistribute heterologous viral glycoproteins.
We considered the possibility that overexpression of KIF16B could divert early endosomes to the periphery of cells in a nonspecific manner that would be seen with other viral envelope glycoproteins. To address this possibility, we examined the effect of expression of WT versus S109A KIF16B on the distribution of three unrelated viral glycoproteins: influenza virus glycoprotein hemagglutinin (HA), Ebola virus glycoprotein (EBOV GP), and severe acute respiratory syndrome coronavirus 2 spike (SARS-CoV-2 S) protein. As noted previously, WT KIF16B was consistently found at the periphery of cells, while KIF16B-S109A localized near the cell nucleus. No redistribution of HA, EBOV GP, or SARS-CoV-2 S was seen for KIF16B versus KIF16B-S109A expression (Fig. 2A to F). Furthermore, in contrast to HIV-1 Env, these heterologous viral glycoproteins failed to colocalize significantly with KIF16B, as indicated by visual inspection and by determination of a slightly negative Pearson’s coefficient (see Fig. S1C in the supplemental material). These results demonstrated that HIV-1 Env colocalization with and redistribution by KIF16B is not widely generalizable to viral glycoproteins, at least as indicated by examination of a paramyxovirus, filovirus, or coronavirus envelope glycoprotein. Next, we examined the effects of expression of KIF1B, a closely related kinesin mediating plus-end-directed trafficking, or its dominant-negative (motor-deficient) form, KIF1B-Q98L (18), on HIV-1 Env distribution. KIF1B was found at the cellular periphery, while KIF1B-Q98L localized to perinuclear locations in a manner similar to that of KIF16B and KIF16B-S109A (Fig. S1A and B). However, distribution of HIV-1 Env did not appear to shift to the periphery or perinuclear region upon expression of KIF1B and KIF1B-Q98L, respectively (Fig. S1A and B). In contrast to results with KIF16B, Env was not significantly colocalized with either KIF1B protein (Fig. S1C, rightmost bars). Together, these results suggested that KIF16B plays a specific role in anterograde trafficking of HIV-1 Env.
FIG 2.
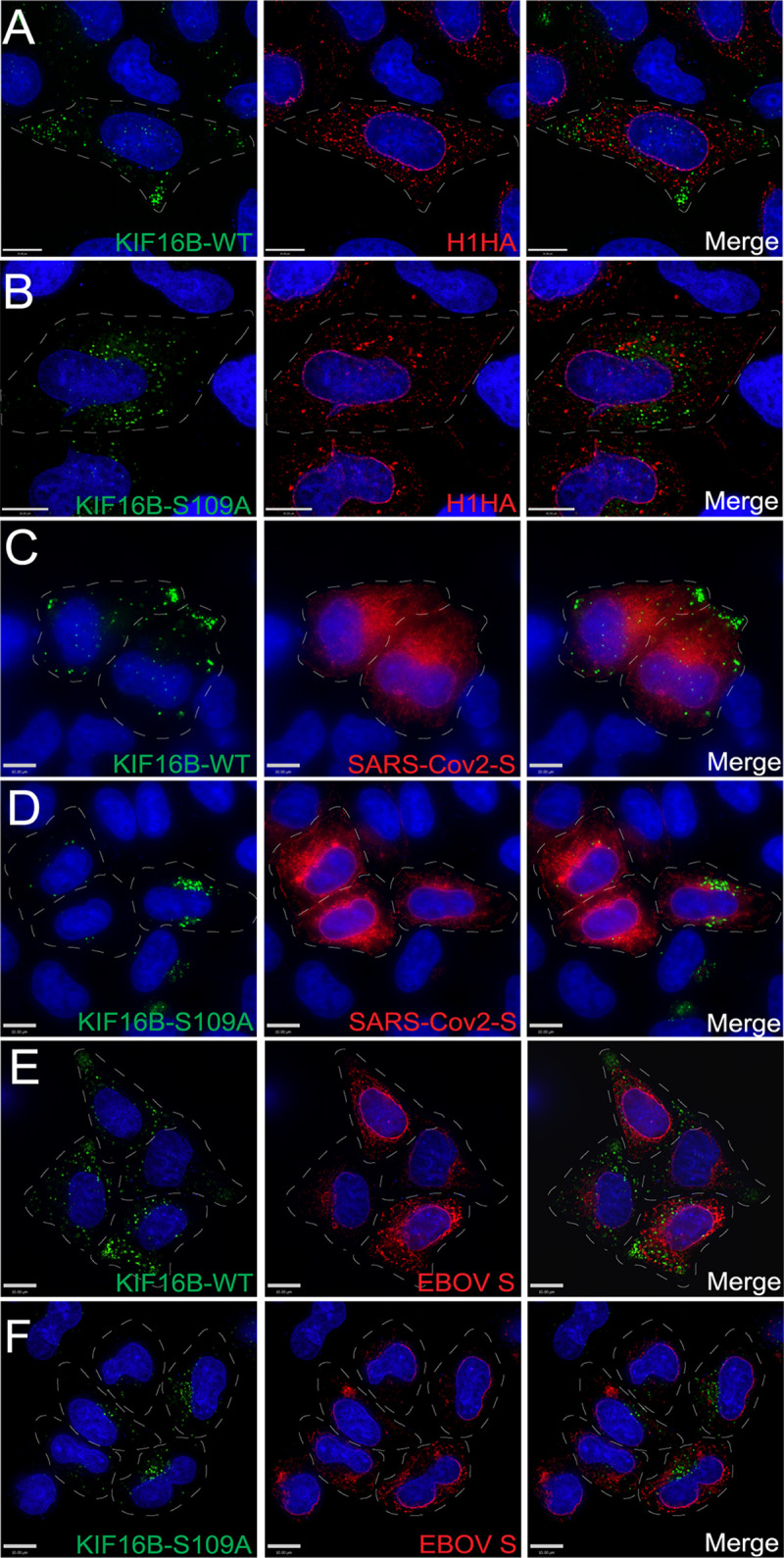
KIF16B and Env colocalization is not generalized to other viral envelope glycoproteins. Dashed lines indicate cell periphery. (A and B) Influenza HA glycoprotein was coexpressed with KIF16B-WT-YFP and KIF16B-S109A-YFP, respectively, in HeLa cells. Cells were fixed and immunolabeled with 11692-T54 antibody to stain HA. Green, KIF16B; red, HA; rightmost image, overlay. (C and D) SARS-Cov2-S protein was coexpressed with WT (C) or dominant-negative KIF16B (D). Cells were fixed and immunolabeled with anti-spike antibody 1A9 to stain S. Green, KIF16B; red, SARS-Cov2-S; rightmost image, overlay. (E and F) Coexpression of KIF16B-WT-YFP or KIF16B-S109A-YFP with Ebola GP. Cells were fixed and immunostained with 4F3 antibody specific to EBOV GP. Green, KIF16B; red, EBOV GP; rightmost image, overlay. Bars, 10 μm.
HIV-1 Env on the PM is endocytosed into KIF16B-positive endosomes.
The studies described above examined the total pool of Env in the cell and did not study the endocytosed pool of Env in isolation. In order to determine if Env is delivered into KIF16B-positive endosomes directly from the PM, we utilized a novel approach using fluorogen-activating protein (FAP) tagging (19). The FAP tag was placed within the V2 loop of the ectodomain of JR-FL Env, as illustrated in Fig. 3A, creating the FAP-Env construct. To validate this system for use in assessing Env incorporation, we examined particle incorporation of FAP-Env. FAP-Env was coexpressed with Env-deficient provirus and was efficiently incorporated into virions as indicated by Western blotting (Fig. S2). Combining expression of this construct (FAP-Env) with a fluorogen impermeant to the PM (MG-11p; αRED-np1 from Spectragenetics) allowed visualization of the fate of pulse-labeled Env present on the surface of cells. We first coexpressed FAP-Env with KIF16B WT in HeLa cells and fixed the cells after pulse-labeling them on ice to visualize the surface staining in the absence of endocytosis. Figure 3B demonstrates that the cell membrane-impermeable reagent fluoresced upon binding to Env on the cell surface and did not penetrate the PM. We next allowed internalization of FAP-Env for 15 min at 37°C after pulse-labeling, and we found that surface-labeled FAP-Env on the surface had accumulated into KIF16B-positive compartments (Fig. 3B). We also performed the same experiment with KIF16B S109A and found similar results (Fig. 3C). In order to follow the movement of this FAP-Env population expressed on the cell surface in real time, we pulse-labeled FAP-Env and recorded live-cell video of Env movement into HeLa cells expressing KIF16B or KIF16B-S109A. Pulse-labeled Env was endocytosed within a few minutes and demonstrated increasing colocalization with KIF16B+ endosomes at the periphery of cells, or with KIF16B-S109A+ endosomes in the perinuclear region of the cell (Fig. 3D and E; Videos S1 and S2). These results demonstrated that HIV-1 Env on the surface of the cell is rapidly endocytosed and thereafter accumulates in KIF16B+ endosomes.
FIG 3.

Env on the plasma membrane is endocytosed into KIF16B-positive compartments. (A) FAP-Env construct shown as a cartoon. The FAP tag was placed on the V2 loop of the Env ectodomain. This tag is inherently nonfluorescent on the far left. In the middle, the addition of a nonpermeable dye is shown. On the far right, the FAP tag becomes fluorescent, allowing visualization of a pulsed population of surface Env. (B) FAP-Env coexpressed with KIF16B-WT-YFP in HeLa cells. Left image, FAP-Env was pulse-labeled for 10 min on ice. Cells were then fixed and imaged immediately to show population of Env on plasma membrane surface. Right image, cells were pulsed as before and then were returned to a 37°C incubator for 15 min to allow internalization, and then fixed and imaged to show FAP-Env endocytosed into KIF16B-positive compartments. (C) FAP-Env construct coexpressed with KIF16B-S109A-YFP in HeLa cells. Left image is FAP-Env collecting on surface as before. Right image is the cell shown after 15 min of internalization as before. (D and E) Frames from live-cell imaging video of FAP-Env coexpressed with KIF16B-WT-YFP (D) or KIF16B-S109A-YFP (E). Time at T =-0 min shows the cell before pulse. Subsequent time points show FAP-Env collecting in KIF16B-positive compartments. Dotted lines are a visual aid to represent periphery of the cell body. Bars, 10 μm.
Deletion of KIF16B leads to enhanced degradation of Env and accumulation within the lysosome.
It has been established that KIF16B-mediated transport can divert cellular cargo from default pathways that lead to lysosomal degradation, while depletion of KIF16B enhances their degradation (14, 16). We therefore hypothesized that a loss of normal anterograde trafficking of Env mediated by KIF16B could result in enhanced Env degradation. To evaluate this possibility, we utilized CRISPR/Cas9 technology to knock out KIF16B in both HeLa cells and H9 T cells, followed by characterization of isolated cell clones. Figure S3 shows characterization of one HeLa and one H9 clone used for subsequent studies; the results were consistent with the findings from several clones examined for each cell type. Note the complete absence of KIF16B by Western blotting in Fig. S3A. We then utilized the KIF16B KO HeLa cell clones together with pulse-labeling of Env using the FAP tag technique described above. FAP-Env was expressed in WT or KIF16B KO cells, and the kinetics of disappearance of total cellular FAP-Env signal was measured over time. These experiments were controlled for the effects of photobleaching on FAP-Env signal, as outlined in Materials and Methods. Remarkably, the FAP-Env signal degraded at a considerably higher rate in KIF16B KO cells than in WT cells (Fig. 4A versus Fig. 4B; Video S3). The half-life of endocytosed Env measured in WT HeLa cells by this method was 159.5 min (95% confidence interval [CI], 145.4 to 175.6), while in the absence of KIF16B Env was more rapidly degraded, with a half-life of 56.9 min (95% CI, 54.1 to 59.8). Furthermore, when we incubated KIF16B KO cells with the lysosomal inhibitor bafilomyacin-A1 (Baf-A1), the degradation rate in KIF16B KO cells returned to wild-type levels (Fig. 4 A, B, C, and E; Video S3). To corroborate the observation that KIF16B knockout leads to enhanced degradation of Env as measured by degradation of FAP-tagged Env, we performed surface pulse-labeling with N-hydroxysuccinimide–biotin in WT and KIF16B KO HeLa cells on ice, followed by quenching of remaining biotin and pulldown of internalized biotinylated proteins with streptavidin beads at multiple time points after incubation at 37°C. Detection of internalized Env was then performed using quantitative Western blotting. Figure 4D shows Western blot examples representative of results from five individual experiments for each experimental arm. Compared with WT cells, pulse-labeled Env in KIF16B KO cells was more rapidly degraded (Fig. 4D, compare WT to KO blots). The addition of Baf-A1 reduced the degradation to essentially WT levels (Fig. 4D and F). Using repeated experiments, we generated degradation curves for pulsed Env in WT, KO, and KO + Baf-A1 cells that were similar, but not identical, to the curves derived from FAP-Env labeling (Fig. 4F, compared to Fig. 4E). The Env half-life measured by this method was 154.1 min for WT cells, 32.1 min for KO cells, and 94.9 min for Baf-A1-treated KO cells. While we noted that the results were not identical between the two methods employed, they were in good general agreement and established that KIF16B KO leads to more rapid Env degradation that can be rescued by Baf-A1. Together, these results clearly showed that KIF16B limits lysosomal degradation of Env, likely through promoting anterograde trafficking.
FIG 4.

KIF16B KO leads to enhanced degradation of endocytosed Env in the lysosome. (A, B, and C) Frames from live-cell movies for Fap-Env expressed in WT HeLa cells with vehicle DMSO (A), KIF16B KO HeLa cells with DMSO (B), or KIF16B KO HeLa cells with 100 μM Baf-A1 (C). Representative frames are shown for each hour over the time course. One cell in each series of images is outlined with a dashed line. (B) Western blots show biotinylated Env from surface-labeled cells, with lysates chased over the time course, pulled down with streptavidin beads, and blotted for Env. Labels indicate WT, KIF16B KO, and KIF16B KO plus Baf-A1 HeLa cells. (C) Quantification of signal intensity of each time point over the FAP-Env imaging experiment normalized to the first frame. (D) Quantification of the Western blot signal for each time point for the biotin-streptavidin pulse-chase experiment. Error bars represent SD from 5 independent experiments. Fit curves for both panels C and D were generated using the Prism 9 nonlinear regression one-phase decay algorithm. Bars, 60 μm.
Given results above with the lysosomal inhibitor Baf-A1, we sought to determine if the absence of KIF16B results in a shift in Env distribution into the lysosomal compartment. Results of immunofluorescence staining were striking. WT HeLa cells revealed punctate lysosomes with a moderate degree of Env colocalization, while intracellular Env was clearly distinguished outside the lysosome (marked by LAMP1 staining) (Fig. 5A and D). In KIF16B KO cells, Env colocalization with LAMP1 was dramatically higher (Fig. 5B and E). Baf-A1 treatment of KIF16B KO cells did not result in significant differences in colocalization of Env and LAMP1 compared with untreated KO cells (Fig. 5C and F, compare to Fig. 5B and E). The visual differences in LAMP1 colocalization noted above in KIF16B KO cells were confirmed to be significant when quantified by Pearson’s correlation coefficient (Fig. 5G). Note that LAMP1 expression as indicated by total fluorescence intensity per square micrometer was modestly increased in KIF16B KO cells, although this difference did not reach statistical significance (Fig. 5H). This increase may reflect the effect of early endosomes that are unable to move in an anterograde fashion in the absence of KIF16B and instead act to expand the lysosomal compartment. Taken together, these results from imaging and pulse-chase analysis suggest that deletion of KIF16B increases the rate of Env degradation due to loss of anterograde movement normally conferred by KIF16B, resulting in shunting to the lysosome for degradation.
FIG 5.

KIF16B KO shunts Env to lysosomal compartments. (A) HIV-1 NL4-3 Env expressed in WT HeLa cells. Magnification, ×60. Bar, 10 μm. (B) NL4-3 Env expressed in KIF16B KO HeLa cells. Magnification, ×60. Bar = 10 μm. (C) NL4-3 Env expressed in KIF16B KO HeLa cells with Baf-A1 added for 3 h prior to imaging. Magnification, ×60. Bar, 10 μm. (D) HIV-1 NL4-3 Env expressed in WT HeLa cells. Magnification, ×20. Bar, 35 μm. (E) NL4-3 Env expressed in KIF16B KO HeLa cells. Magnification, ×20. Bar, 35 μm. (F) NL4-3 Env expressed in KIF16B KO HeLa cells with Baf-A1 added 3 h prior to imaging. Magnification, ×20. Bar, 35 μm. For all imaging, cells were fixed and immunolabeled for Env with 2G12 antibody and for endogenous LAMP1 with H4A3 antibody. (G) Pearson’s correlation coefficient showing the degree of colocalization between Env and LAMP1 positive compartments between WT, KIF16B KO cells, and WT cells with Baf-A1 in panels A, B, and C, respectively. Pearson's correlation coefficient equation used Volocity 6.5.1 software after manual thresholding. One-way analysis of variance performed using Prism 9 to obtain significance values for difference in colocalization. ****, P < 0.0001. (H) Comparison of LAMP1 fluorescence intensity in WT and KIF16B KO cells. Ten individual cells for each cell type were selected to quantify total signal (per micrometer squared), and mean values ± SD. An unpaired t test was utilized to analyze significance (P = 0.182; nonsignificant [ns]).
KIF16B deletion leads to defects in Env incorporation, particle infectivity, and viral replication.
We next examined the effect of KIF16B depletion on Env trafficking and incorporation into particles. We first assessed surface levels of Env by flow cytometry after infecting WT or KIF16B KO H9 cells with NL4-3 virus. We note that H9 cells are nonpermissive for incorporation of Env in the absence of the CT, similar to primary T cells and macrophages, whereas HeLa cells are considered “semipermissive,” allowing Env incorporation in the absence of the CT (6). Thus, H9 cells were chosen both to reflect CT-dependent Env incorporation and to facilitate analysis of spreading infection in culture. Surface expression levels of Env were consistently diminished in KIF16B KO cells compared with WT cells (Fig. 6A and B), consistent with diminished anterograde movement of Env. Total cellular Env measured by flow cytometry was also diminished, but to a somewhat lesser degree than cell surface Env (Fig. 6B). This result was consistent with the enhanced lysosomal degradation of endocytosed Env shown in prior analyses. Viruses were then purified from supernatants of infected WT and KIF16B KO H9 cells, in order to examine levels of Env incorporation into particles. Viral particle Env content was significantly reduced in KIF16B KO cells compared to WT H9 (>50% reduction from wild-type levels, as quantified by LI-COR Image Studio software, n = 5) (Fig. 6C). The leftmost blot in Fig. 6C shows particle Env content normalized by equal p24 content loading of the lanes. When cell lysates were examined following multiple rounds of replication in WT or KIF16B KO cells and normalized for total protein, there was a reduction of all viral proteins, reflecting diminished replication (Fig. 6C, cell lysate). Notably, however, production of gp160 in KIF16B KO cells was not diminished, as indicated by Western blotting of lysates from infected cells examined after a single round of replication (Fig. 6C, rightmost blot). This suggested that the reduced levels of Env seen on the PM and on particles derived from KIF16B KO cells were not due to an Env production defect. To evaluate the significance of the diminished particle incorporation of Env induced by KIF16B KO, we next examined particle infectivity and viral replication. Consistent with the observed reduction of Env on particles, the infectivity of particles released from KIF16B KO cells was consistently reduced (Fig. 6D). We noted that the reduction in Env incorporation shown in Fig. 6C and the reduction in particle infectivity in Fig. 6D were consistent but are incomplete, suggesting that although KIF16B alters the level of Env incorporation, it is not absolutely required for incorporation of Env into particles. To determine the significance of this level of Env reduction on viral replication, we infected WT H9 and KIF16B KO cells and measured viral replication in a spreading infection. Infection of KIF16B KO H9 cells resulted in a significant delay in replication and much lower overall levels of particle production compared with WT H9 cells (Fig. 6E). We concluded from these results that the anterograde movement of Env by KIF16B plays a role in Env incorporation and viral replication. Loss of KIF16B results in lower levels of cell surface Env and a corresponding loss of Env incorporation into particles.
FIG 6.

KIF16B depletion leads to reduction of Env on the cell surface, particle incorporation, infectivity, and viral replication. (A) Flow plots for cell surface staining (nonpermeabilized) and total Env content (permeabilized) in WT H9 and KIF16B KO H9 cells. Cells were fixed and stained for Env using anti-gp120 2G12 antibody. (B) Quantification of flow cytometry mean fluorescent intensity of Env expression on surface and total cell, averaged across 5 independent experiments. Unpaired t test was performed on Prism 9 to for significance values between surface versus total. **, P < 0.005. (C) WT and KIF16B H9 cells were infected with VSV-G-pseudotyped NL4-3. Shown are Western blots of Env and Gag content for particles in viral supernatant (left) and cell lysates (middle) collected on day 5 following infection. Note that loading was normalized for p24 content (supernatants) or for total cellular protein (cell lysates). Rightmost blot, single-round infection of WT and KIF16B KO H9 cells infected with VSV-G-pseudotyped NL4-3 at an MOI of 0.5 and harvested for cell lysate analysis after 24 h. Uninfected WT and KO cell lysates were run on the same blot as controls. Blotting was performed with 2G12 antibody against HIV-1 gp160 and ACTN05 (Invitrogen) against actin as a loading control. (D) Infectivity of viral particles released from WT or KIF16B KO H9 cells was determined from 3 independent experiments evaluated using TZM-bl indicator cells. (E) WT (black line) and KIF16B KO (green line) cells were infected with VSV-G-pseudotyped NL4-3 virus. Viral growth was monitored over time by detection of p24 in the culture supernatant.
KIF16B depletion effects in semipermissive HeLa cells and role of the Env CT.
HeLa cells allow incorporation of Env into particles in the absence of the CT, as outlined above. To define the dependence of Env incorporation on KIF16B in HeLa cells, we transfected NL4-3 or NL4-3 CT144 (lacking most of the CT) into WT HeLa or KIF16B KO HeLa cells and examined Env incorporation into particles. Incorporation of full-length Env as judged by Western blotting was diminished in KO cells (Fig. 7A, top, KO lanes), but the difference did not reach statistical significance (Fig. 7B) (P = 0.174 for NL4-3). As expected, CT144 Env was also incorporated into particles (Fig. 7A, bottom blots). No difference in Env incorporation was observed between WT and KIF16B KO cells (Fig. 7A and B) (P = 0.711). Incorporation of WT Env was therefore more significantly diminished by KIF16B KO in nonpermissive H9 cells than in HeLa cells (compare Fig. 6C to Fig. 7A). Next, we asked if KIF16B S109A resulted in sequestration of Env in perinuclear KIF16B+ endosomes in the absence of the CT. CT144 Env was present on the PM and on intracellular membranes in a relatively diffuse distribution, and we did note some colocalization of intracellular CT144 Env with YFP-KIF16B-S109A signal (Fig. 7C, CT144 images). The concentration of Env overlying KIF16B signal appeared visually more prominent, however, for WT Env (Fig. 7C, compare WT and CT144 images). This impression was verified by Pearson’s correlation, with a mean of 0.401 for WT versus 0.228 for CT144 (Fig. 7D) (P < 0.001). We concluded that the redistribution of Env to perinuclear KIF16B+ compartments by KIF16B-S109A is more marked in the presence of the CT, suggesting a CT-dependent effect of KIF16B trafficking.
FIG 7.

Semipermissive HeLa cells demonstrated reduced dependence on KIF16B. (A) HeLa cells and KIF16B KO HeLa cells were transfected with NL4-3 provirus (top) or NL4-3 CT144 provirus (bottom), and cell lysates and viral pellets were analyed by Western blotting. Molecular mass markers are shown on the left. (B) Fluorescence signals for gp41 and p24 in cells and supernatants from three independent experiments were measured, with the gp41/p24 ratio normalized to a setting of 1.0 for NL4-3 in WT HeLa cells. Differences between Env particle content in particles derived from WT or KIF16B KO cells were analyzed by an unpaired t test and found to be nonsignificant. Shown are mean values + standard deviations. (C) WT and CT144 versions of JR-FL Env were coexpressed in HeLa cells with GFP-KIF16B-S109A. Bars: top = 7 μm; bottom = 10 μm. (D) Pearson’s correlation coefficient values and mean representing colocalizaton of Env and YFP-KIF16B-S109A. Analysis was by unpaired t test; ***, P < 0.001.
DISCUSSION
Molecular motor proteins are essential to the normal function of eukaryotic cells. The cytoplasm of cells is dense, crowded with proteins, vesicles, nucleotides, and organelles. Free diffusion of large molecules within the cytoplasm is restricted and cannot be relied upon to move these cellular materials with the efficiency and accuracy required for proper cell function. Instead, the cell must use specialized molecular motors. These motors—kinesins, dyneins, and myosins—use ATP to power movement of cargo along cytoskeletal structures. Kinesins and dyneins utilize microtubule networks as their pathways for transporting cellular cargo. With some exceptions, kinesins move in an anterograde direction along microtubules. Dyneins, in contrast, move in a retrograde direction toward the minus end of microtubules often localized at the microtubule-organizing center. Kinesins and dyneins are most commonly structured with an N-terminal motor head that is responsible for powering processive, step-like movement of motor subunits, attached via a middle stalk region to a C-terminal tail composed of light and heavy chains that bind the cellular cargo (20–23).
Viruses hijack host cellular pathways in order to perform essential steps in their life cycle, including coopting molecular motor-driven intracellular transport (24–27). HIV-1 has been shown to utilize motor proteins at several steps of replication (27, 28). Following entry into a cell, the conical core of HIV-1 has been shown to bind to dynein and to use the KIF5 motor protein to successfully traffic toward the nucleus (29, 30). Depletion of the KIF5B motor prevents nuclear entry and infection of HIV-1 in a CA-dependent manner (31). In studies of outward trafficking of Gag, KIF4 has been shown to directly interact with Gag in both yeast two-hybrid and coimmunoprecipitation assays (32, 33). Additionally, disruption of KIF4 slowed progression of Gag through trafficking intermediates, and depletion of KIF4 increased degradation of Gag that was reversed upon reintroduction of KIF4 (34). However, no motor protein has previously been shown to interact with the HIV-1 Env protein or participate in its trafficking within host cells. Because Env that reaches the cell surface through the secretory pathway is rapidly endocytosed and must subsequently traffic in an anterograde manner to return to the PM for particle incorporation, we hypothesized in this study that HIV-1 would also likely use a kinesin or kinesins for outward trafficking of Env. Several pieces of evidence implicate Rab14 as an important outward trafficking factor for Env. Our group has shown that Rab14 mediates FIP1C-dependent recycling of Env and plays a role in Env incorporation (10). Hoffman and colleagues similarly found that Env colocalizes to Rab14+ endosomal compartments in T-cell lines and unexpectedly found that these compartments shared some characteristics with late endosomes and lysosomes (13). KIF16B is an effector of Rab14-mediated trafficking that binds directly to Rab14 (15). In this study, we demonstrated that KIF16B plays a role in Env trafficking and in the ability of HIV-1 to form fully infectious particles. Similar to other reported cargo of KIF16B, such as EGFR, Env colocalized strongly with KIF16B at the periphery of cells. Motor mutant dominant-negative KIF16B redistributed Env to a perinuclear position, entirely consistent with what was seen for EGFR. Video microscopy of pulse-labeled Env expressed on the surface of cells in this study indicated that Env on the PM is endocytosed into KIF16B-positive compartments. In the absence of KIF16B, Env was shifted to the lysosome, and the half-life of Env that had been endocytosed from the PM was significantly reduced. Taken together, these studies strongly suggest that KIF16B mediates anterograde movement of Env and limits the pool of Env that is shunted to the lysosome for degradation. To our knowledge, this is the first evidence for a motor protein contributing to Env trafficking and particle incorporation.
The effect of KIF16B KO on particle incorporation of Env was more pronounced in H9 cells than in HeLa cells, suggesting that the role of KIF16B is more prominent in those cell types that require the Env CT for incorporation (nonpermissive cell types). Dominant-negative S109A also had less of an effect on the subcellular distribution of CT144 Env than on WT Env. Together, these results support a model in which CT-directed endocytosis and trafficking leads to subsequent degradation or recycling as shown in Fig. 8. According to this model, Env is transported through the secretory pathway to the PM and subsequently endocytosed to the early or recycling compartments, following which Env can be directed in an anterograde fashion by KIF16B or can follow a default pathway leading to fusion with the lysosome (Fig. 8A). In the absence of KIF16B, a larger pool of endocytosed Env is shunted to the lysosome, as indicated by the relative sizes of the arrows in Fig. 8B. The fraction of the endocytosed pool of Env that is directed to the periphery and is subsequently available for incorporation into particles is diminished in the absence of KIF16B, resulting in diminished particle incorporation, particle infectivity, and reduced replication. Figure 8 for simplicity does not include other potential trafficking routes of Env, such as retrograde trafficking to the Golgi and the potential for alternative routes for Env to recycle to the PM.
FIG 8.

(A and B) Model of Env Trafficking with (A) and without (B) KIF16B. (A) In a wild-type cell, Env is produced in the rough endoplasmic reticulum and follows the secretory pathway to the cell surface. Env is then endocytosed into early endosomes (EEs; path 1 in the figure), which later may sort to the endosomal recycling compartment (ERC) for recycling. This compartment is depicted as “EE/ERC.” A fraction of Env in EEs can progress to fuse with lysosomes, leading to lysosomal degradation in path 2. In path 3, Env is directed in an anterograde manner to the PM, where it is available for incorporation into viral particles. Path 3 is the relevant pathway that is partially dependent upon KIF16B. (B) In a cell with KIF16B KO, a diminished fraction of Env is delivered to the PM from the EE/ERC via path 3. This results in an increase in the amount of Env shunted to the lysosome (path 2, illustrated as a broader arrow) and enhanced degradation of Env. The change in the distribution of endocytosed Env is shown graphically as fewer trimers on the budding and mature virion, as well as fewer trimers on the PM and more trimers in the lysosome. The figure was created with BioRender.
Four models for Env incorporation into HIV particles have been proposed by the Freed laboratory (5). Passive incorporation at the PM is not likely to be the correct model in nonpermissive cell types, including primary T cells and macrophages, as Env with a full-length CT is excluded in these cell types. This model remains a potential in permissive cell lines that do not require the CT (4). The identification of host factors, such as Rab11-FIP1C, Rab14, and KIF16B, that influence particle incorporation favors the Gag-Env cotargeting model, whereby Env is directed to specific microdomains on the PM that are enriched in Gag molecules and serve as sites of particle budding. At present, there is no evidence favoring an indirect interaction model with an adaptor or linker connecting Gag to Env. The direct Gag-Env interaction model has been supported by two biochemical studies (35, 36) and by evidence that maturation is required before Env can mediate membrane fusion, while CT truncation relieves the constraint on fusion posed by the immature Gag lattice (9, 37, 38). Trimerization of MA has been shown to be required for Env incorporation, while some mutations in MA that result in Env exclusion from particles can be rescued by compensatory mutations at the trimer interface (39, 40). Furthermore, MA trimerization-impaired mutants can be rescued by substitutions that simultaneously restore Env incorporation (41). This has added substantially to the evidence favoring an interaction of the CT with MA in the form of MA trimers and supports a model whereby the structural integrity of the MA lattice is an important determinant of Env incorporation (41). Results in the present study showing a role for kinesin-mediated trafficking of Env do not negate the importance of structural integrity of the MA lattice for particle incorporation of Env. Trafficking to the site of the developing Gag lattice mediated by host factor interactions with the CT may be a necessary step preceding a direct Gag-Env interaction. We expect that a defect in anterograde trafficking and increase in Env degradation in the lysosome would diminish the delivery of Env to sites of assembly and reduce the potential interaction of Env with the Gag lattice.
This study provides new information regarding the half-lives of Env trimers following endocytosis from the PM. A prior study employing autoradiographic pulse-labeling of total cellular Env indicated that cellular gp160 and gp120 were degraded over a period of hours and that inhibition of lysosomal acidification prevented gp160 degradation (42). Degradation of gp160 has been shown to not only occur in the lysosome, but also within the biosynthetic pathway within the ER or Golgi compartments (43, 44). However, these studies labeled all Env populations within the cell, whereas the current study utilized two methods of cell surface pulse-labeling followed by chase periods to measure the half-lives of Env trimers that had reached the plasma membrane. We found that endocytosed Env had a half-life of 150 to 160 min and that the absence of KIF16B promoted much more rapid degradation (57 or 32 min, measured by FAP-Env degradation or surface biotinylation, respectively). An important role for lysosomal degradation of endocytosed Env is suggested by the reversal of the enhanced rates of degradation in KIF16B KO cells by Baf-A1. A model in which endocytosed Env is shunted to the lysosome for degradation in the absence of KIF16B is also supported by the increased appearance of Env in lysosomal compartments in KIF16B KO cells.
An important caveat for our conclusions is that a complete KIF16B KO resulted in an incomplete reduction of Env incorporation and particle infectivity in H9 cells. Env incorporation and particle infectivity were diminished by approximately 50%. This suggested that kinesins other than KIF16B may also be involved in trafficking Env to the PM, or that there are multiple pathways whereby Env may be recycled to the surface. Indeed, it is known that successful movement of cargo within cells is often accomplished by groups of motors rather than just one (45). Retrograde trafficking of Env from early endosomes to the Golgi may play a role in Env incorporation into particles, as depletion of components of the retromer complex modulated Env distribution and particle incorporation (46). However, the identification of KIF16B as a regulator of Env trafficking and degradation defines another component of the host machinery regulating Env incorporation. Kinesins have been proposed as potential therapeutic targets in some areas of medicine, such as cancer and chronic pain (47–49). Although targeting KIF16B function globally would be disruptive to many cell processes, it is possible that a more directed approach disrupting KIF16B-mediated Env trafficking can be developed. In conclusion, we identified KIF16B as an anterograde motor involved in Env trafficking and particle incorporation. Future studies will be needed to fully elucidate the complex pathways taken by Env to reach the site of particle assembly and enable particle incorporation.
MATERIALS AND METHODS
Cells and plasmids.
HeLa cells were obtained from the American Type Culture Collection (ATCC; CRM-CCL-2). TZM-bl cells were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, from John C. Kappes, Xiaoyun Wu, and Tranzyme, Inc. The H9 T cell line was obtained from ATCC (HTB-176), and cells were maintained in RPMI medium 1640 supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, and antibiotics. HeLa and TZM-bl cells were maintained in Dulbecco's modified Eagle medium containing 10% FBS and 2 mM penicillin-streptomycin. pNL4-3 proviral plasmid was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, from Malcolm Martin. Env expression plasmid pIIINL4Env was a gift kindly provided by Eric Freed (HIV DRP, NCI, NIH) (6). The influenza A vaccine strain of A/Brisbane/02/2018 (H1N1) HA sequence was used to construct a human codon-optimized gene, which was synthesized and cloned into the BamHI and EcoRI sites of pcDNA3.1+ by GenScript. The SARS-CoV-2 spike gene (Wuhan) was synthesized by Genscript for Biao He (University of Georgia) and gifted to our lab. The human codon-optimized full-length SARS-CoV-2 spike gene was subcloned into the KpnI and BamHI sites of the plasmid pcDNA5TO/Puro. pcDNA5/TO-puro was constructed from backbone pCDNA5/TO (Thermo Fisher Scientific) as previously described (50). The EBOV S protein plasmid pcDNA5/TO-puro EBOV GP has been previously described (51). Plasmid pcDNA5/TO JR-FLoptgp160 expresses a codon-optimized JR-FL env gene in the pcDNA5/TO vector backbone (Thermo Fisher Scientific, Waltham, MA) as previously described (12). Overlap-extension PCR was used to create novel XhoI and PstI sites within the exposed portion of the V2 loop of JR-FL. The fluorogen-activated peptide (FAP) sequence from pMFAP-alpha2 (Spectragenetics, Pittsburgh, PA) was PCR amplified as an XhoI-PstI fragment and ligated into pcDNA5/TO JR-FLoptgp160, placing the FAP peptide in-frame within the V2 loop. A complete cloning schema with primers is available upon request. The WT KIF16B-YFP and dominant KIF16B-S109A-YFP were kindly provided by Sebastian Hoepfner (14). Expression plasmids for Flag-KIF1Bβ and Flag-KIF1Bβ Q98L were kindly provided by Susanne Schlisio and have been described by Fell et al. (18).
Viral infectivity and multiround replication assays.
H9 T cells were infected with vesicular stomatitis virus (VSV) G-pseudotyped NL4-3 virus at a multiplicity of infection (MOI) of 0.2. Cells and supernatants were harvested 4 to 5 days after infection for analysis by Western blotting, p24 antigen quantitation, and infectivity assays. Infectivity of cell culture supernatants was measured using TZM-bl indicator cells following p24 normalization as previously described (52). Replications assays were performed by taking aliquots of the supernatant after infection every 3 days for 21 days. Supernatant was then used in a p24 enzyme-linked immunosorbent assay (ELISA) to graph the amount of virus produced over the course of viral growth and replication. Murine anti-p24 capture antibody 183-H12-5C (CA183) was obtained from Bruce Chesebro and Kathy Wehrly through the NIH AIDS Research and Reference Reagent Program, and the p24 ELISA was performed as previously described (53).
Western blotting, antibodies, and immunostaining.
For analysis of viral protein content, viruses were pelleted from cell culture supernatants through a 20% sucrose cushion at 35,000 × g for 3 h at 4°C; cells were harvested and pelleted at 400 × g. Virus pellets and cell lysates were dissolved in radioimmunoprecipitation assay (RIPA) buffer (Thermo Scientific) containing protease inhibitors. Cell lysates for blotting KIF16B were prepared with a hypotonic lysis buffer consisting of 10 mM HEPES, 1.5 mM MgCL2, 10 mM KCl in phosphate-buffered saline (PBS) on ice for 15 min, vortexed with 10% IGEPAL CA630 (Sigma) to break open cells, and then centrifuged to pellet nuclei. Virus pellets and cell lysates were separated on 4% to 12% polyacrylamide gels, transferred to nitrocellulose, and subjected to Western blotting using the antibodies outlined below. Goat polyclonal antibody AHP2204, from Bio-Rad AbD Serotec (Oxford, United Kingdom; catalog number D7324), was used for detecting HIV-1 gp120 for Western blotting. Recombinant human monoclonal antibody 2F5 from Polymun Scientific (Klosterneuburg, Austria) was used to detect HIV-1 gp41 in Western blots. Actin was detected with Invitrogen actin antibody ACTN05. KIF16B antibody used was polycolonal mouse from Abnova (H00055614-B01P). Mouse anti-p24 monoclonal CA-183 (provided by Bruce Chesebro and Kathy Wehrly through the NIH AIDS Research and Reference Reagent Program) was used to detect p24 and Pr55Gag in Western blotting and for ELISA to measure HIV-1 Gag in virus-containing culture supernatant and virus pellets; the capture ELISA was performed as previously described (54). IRDye goat anti-mouse, IRDye rabbit anti-goat, and IRDye goat anti-rabbit secondary antibodies from LiCor Biosciences (Lincoln, NE) were used for Western blotting. All blots were acquired and analyzed using the LiCor Odyssey infrared detection system. The following antibodies were utilized in immunofluorescence assays: human anti-gp120 antibody IgG1 2G12 from Polymun scientific (catalog number A002), monoclonal anti-Flag antibody produced in rabbit from Sigma-Aldrich (F2555), rabbit polyclonal anti-HA1 antibody from Sino Biological (Beijing, China; catalog number 11692-T54), mouse monoclonal SARS-CoV/SARS-CoV-2 (COVID-19) spike antibody from Genetex (Irvine, CA; catalog number 1A9), mouse anti-EBOV GP monoclonal antibody 4F3 from IBT Bioservices (Rockville, MD), and anti-LAMP1 mouse monoclonal antibody from BD Biosciences (Franklin Lakes, NJ; catalog number H4A3).
Env degradation assays.
In order to determine degradation rates of Env microscopically using our FAP-tagged Env, we plated 105 HeLa WT or KIF16B KO HeLa cells in wells of an Ibidi 4-well μ-Slide plate (Ibidi) and then transfected overnight with the FAP-Env construct. In one of the KO cell wells, a 100 μM concentration of Bafilomycin A1 (Baf-A1, Invivogen) was employed while the WT and other KO cell wells received an equal amount of dimethyl sulfoxide (DMSO). Three hours later, live cell imaging was conducted following a 10-min pulse-label incubation with FAP reagent αRED-np1 (Spectragenetics) in 5% CO2 at 37°C chamber on a Widefield Nikon Ti2 inverted SpectraX system (Cincinnati Children’s Confocal Core). Data from these movies were extracted with Volocity imaging software (Quorum Technologies) to acquire the signal intensity of each region of interest drawn around individual cells analyzed in the field of view over the time course. The averaged normalized signal intensity was then calculated using the method outlined in Alber et al. to attain the degradation curve and half-life of the FAP-tagged Env protein (55). For protein analysis of Env degradation by Western blotting, we plated 1.75 million WT or KIF16B KO HeLa cells in 10-cm dishes (5 dishes for WT, 10 dishes for KO). The next morning, 3 μg of pIII-Env and 250 ng of psv72-Tat protein were cotransfected into each plate. Following 24 h of incubation, 100 μM Baf-A1 was added to 5 of the KO dishes, while the other KO and WT dishes received an equal amount of DMSO. Three hours later, a 1 mM solution of biotin-PBS was applied to the cells and allowed to label surface proteins for 15 min. This labeling reaction was quenched with 1× Tris-buffered saline solution for 5 min. Cells were then washed with PBS and returned to normal media. Cell plates were then put on ice and in a 4°C cold room to stop endocytosis at 0, 30, 60, 90, and 120 min after biotin labeling and were then subsequently scraped and harvested for cell lysates in RIPA buffer. Cell lysates were then immunoprecipitated overnight with 60 μL of Pierce high-capacity NeutrAvidin agarose (Thermo Fischer). Beads were then collected in Pierce spin columns (Thermo Fischer), and protein was then eluted off the beads at 95°C in 1× Laemmli buffer with reducing agent. After a 2,000 × g spin for 3 min, equal amounts of elution were run for each time point on a 4 to 12% SDS-PAGE gel and then analyzed by immunoblotting for gp120.
Image acquisition and analysis.
HeLa cells were plated in MatTek 35-mm poly-d-lysine-treated dishes (Brooke Knapp MatTek) overnight and were then transfected with the indicated constructs using jetPRIME (Polyplus) per the manufacturer’s instructions. After a 24 h of incubation, cells were rinsed with PBS three times and fixed in 4% paraformaldehyde for 10 min at room temperature. Cells were washed with PBS three times with mild agitation after fixation and then permeabilized for 10 min with 0.2% Triton X-100, followed by washing with PBS. Dako blocking solution was applied to cells for 30 min under gentle shaking conditions. Cells were then incubated with the appropriate primary antibody, detailed above, diluted in Dako (Agilent) antibody diluent to 1:500. Species-appropriate Alex Fluor (Thermo Fischer) secondary antibody was diluted in Dako antibody diluent to 1:1,000. Images were acquired on a widefield DeltaVision Elite high-resolution microscope (GE Healthcare). The percentage of total Env signal at the periphery of cells was calculated using measurements from Volocity 6.5.1 imaging analysis software. Briefly, regions of interest were drawn around the entire cell, around 30% of the distance from the edge of the cell signal to the nucleus (inner 70% of total cell area), and around the nucleus. Peripheral signal was calculated by subtracting the middle two regions from the sum of signal from the total cell region, giving a percentage of peripheral signal as all signal in the last 30% of the cell body toward the edge. Quantitation of colocalization was performed using Volocity software features by derivation of Pearson's correlation coefficient after manual thresholding.
Flow cytometry for HIV-1 Env expression on the cell surface.
For cell surface staining, approximately 5 million H9 cells were infected overnight with 1,000 ng p24 of VSV-G-pseudotyped NL4.3. The next day, cells were washed once with PBS and plated in a 6-well plate in RPMI for 48 h. Infected cells were stained for viability with Zombie violet dye (BioLegend) diluted 1:500 in PBS, followed by fixation with 4% paraformaldehyde. Cells were blocked with Dako protein block and then stained for cell surface Env with 2G12 anti-gp120 directly conjugated to allophycocyanin (Abcam catalog number ab201807) and diluted 1:100 in Dako antibody diluent (10 μg/mL final concentration) for 2 h. 2G12 was washed out twice with PBS followed by permeabilization with 0.2% Triton X-100. Lastly, cells were stained for total infected cells using p24 antibody, KC-57 fluorescein isothiocyanate diluted 1:100 in Dako antibody diluent. Cells were washed twice with PBS and then resuspended in magnetic activated cell sorting (Miltenyi Biotec) buffer and analyzed using the BD FACS Canto II.
CRISPR KO strategy.
CRISPR-Cas9 technology was used to knockout KIF16B in both HeLa and H9 cell lines. Using the Crispor.tefor.net open-access website and algorithm designed to find areas of the genome to target with guide RNAs (gRNAs), we targeted exon 13 of KIF16B using the sequence from NCBI (Gene ID 55614, Assembly GCF_000001405.40, Chr 20, location NC_000020.11) and found the stretch of DNA with the highest score to target for deletion, CTCACGGATAAGTTTGACGT. We ordered sense (CACCGCTCACGGATAAGTTTGACGT) and antisense (AAACACGTCAAACTTATCCGTGAGC) gRNA oligonucleotides plus protospacer adjacent motif sequence from Integrated DNA Technologies to clone into the lentiCRISPR v2, a gift from Feng Zhang (Addgene plasmid 52961; http://n2t.net/addgene:52961; RRID:Addgene_52961) using the target guide sequence protocol from Sanjana Lab (http://sanjanalab.org/library/protocol_lentiOligo.pdf) (56). After transformation of MAX Efficiency Stbl2 competent cells (Thermo Fischer) and DNA harvest, we packaged the plasmid into a lentivirus using sPAX2 and PMGD2 and transduced our HeLa and H9 cell lines. Puromycin selection and limiting dilution were used to obtain clones knocked out for KIF16B (Fig. S1).
ACKNOWLEDGMENTS
This work was supported by R01AI150486.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Flow cytometry was performed using the Cincinnati Children’s Hospital Medical Center (CCHMC) Flow Cytometry Core. Live-cell imaging of was conducted in the Confocal Imaging Center at CCHMC. DNA sequencing was performed at the DNA Sequencing Core at CCHMC.
Footnotes
Supplemental material is available online only.
Contributor Information
Paul Spearman, Email: paul.spearman@cchmc.org.
Guido Silvestri, Emory University.
REFERENCES
- 1.Egan MA, Carruth LM, Rowell JF, Yu X, Siliciano RF. 1996. Human immunodeficiency virus type 1 envelope protein endocytosis mediated by a highly conserved intrinsic internalization signal in the cytoplasmic domain of gp41 is suppressed in the presence of the Pr55gag precursor protein. J Virol 70:6547–6556. PMC190695. doi: 10.1128/JVI.70.10.6547-6556.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ohno H, Aguilar RC, Fournier MC, Hennecke S, Cosson P, Bonifacino JS. 1997. Interaction of endocytic signals from the HIV-1 envelope glycoprotein complex with members of the adaptor medium chain family. Virology 238:305–315. doi: 10.1006/viro.1997.8839. [DOI] [PubMed] [Google Scholar]
- 3.Rowell JF, Stanhope PE, Siliciano RF. 1995. Endocytosis of endogenously synthesized HIV-1 envelope protein. Mechanism and role in processing for association with class II MHC. J Immunol 155:473–488. doi: 10.4049/jimmunol.155.1.473. [DOI] [PubMed] [Google Scholar]
- 4.Anokhin B, Spearman P. 2022. Viral and host factors regulating HIV-1 envelope protein trafficking and particle incorporation. Viruses 14:1729. doi: 10.3390/v14081729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Checkley MA, Luttge BG, Freed EO. 2011. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J Mol Biol 410:582–608. PMC3139147. doi: 10.1016/j.jmb.2011.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murakami T, Freed EO. 2000. The long cytoplasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc Natl Acad Sci USA 97:343–348. doi: 10.1073/pnas.97.1.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akari H, Fukumori T, Adachi A. 2000. Cell-dependent requirement of human immunodeficiency virus type 1 gp41 cytoplasmic tail for Env incorporation into virions. J Virol 74:4891–4893. PMC112014. doi: 10.1128/jvi.74.10.4891-4893.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dubay JW, Roberts SJ, Hahn BH, Hunter E. 1992. Truncation of the human immunodeficiency virus type 1 transmembrane glycoprotein cytoplasmic domain blocks virus infectivity. J Virol 66:6616–6625. PMC240157. doi: 10.1128/JVI.66.11.6616-6625.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang J, Aiken C. 2007. Maturation-dependent human immunodeficiency virus type 1 particle fusion requires a carboxyl-terminal region of the gp41 cytoplasmic tail. J Virol 81:9999–10008. PMC2045384. doi: 10.1128/JVI.00592-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qi M, Williams JA, Chu H, Chen X, Wang J-J, Ding L, Akhirome E, Wen X, Lapierre LA, Goldenring JR, Spearman P. 2013. Rab11-FIP1C and Rab14 direct plasma membrane sorting and particle incorporation of the HIV-1 envelope glycoprotein complex. PLoS Pathog 9:e1003278. doi: 10.1371/journal.ppat.1003278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qi M, Chu H, Chen X, Choi J, Wen X, Hammonds J, Ding L, Hunter E, Spearman P. 2015. A tyrosine-based motif in the HIV-1 envelope glycoprotein tail mediates cell-type– and Rab11-FIP1C–dependent incorporation into virions. Proc Natl Acad Sci USA 112:7575–7580. doi: 10.1073/pnas.1504174112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirschman J, Qi M, Ding L, Hammonds J, Dienger-Stambaugh K, Wang J-J, Lapierre LA, Goldenring JR, Spearman P. 2018. HIV-1 envelope glycoprotein trafficking through the endosomal recycling compartment is required for particle incorporation. J Virol 92. doi: 10.1128/JVI.01893-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffman HK, Aguilar RS, Clark AR, Groves NS, Pezeshkian N, Bruns MM, van Engelenburg SB. 2022. Endocytosed HIV-1 envelope glycoprotein traffics to Rab14(+) late endosomes and lysosomes to regulate surface levels in T-cell lines. J Virol 96:e0076722. PMC9327703. doi: 10.1128/jvi.00767-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoepfner S, Severin F, Cabezas A, Habermann B, Runge A, Gillooly D, Stenmark H, Zerial M. 2005. Modulation of receptor recycling and degradation by the endosomal kinesin KIF16B. Cell 121:437–450. doi: 10.1016/j.cell.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 15.Ueno H, Huang X, Tanaka Y, Hirokawa N. 2011. KIF16B/Rab14 molecular motor complex is critical for early embryonic development by transporting FGF receptor. Dev Cell 20:60–71. doi: 10.1016/j.devcel.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 16.Weimershaus M, Mauvais FX, Saveanu L, Adiko C, Babdor J, Abramova A, Montealegre S, Lawand M, Evnouchidou I, Huber KJ, Chadt A, Zwick M, Vargas P, Dussiot M, Lennon-Dumenil AM, Brocker T, Al-Hasani H, van Endert P. 2018. Innate immune signals induce anterograde endosome transport promoting MHC class I cross-presentation. Cell Rep 24:3568–3581. doi: 10.1016/j.celrep.2018.08.041. [DOI] [PubMed] [Google Scholar]
- 17.Perez Bay AE, Schreiner R, Mazzoni F, Carvajal-Gonzalez JM, Gravotta D, Perret E, Lehmann Mantaras G, Zhu YS, Rodriguez-Boulan EJ. 2013. The kinesin KIF16B mediates apical transcytosis of transferrin receptor in AP-1B-deficient epithelia. EMBO J 32:2125–2139. PMC3730227. doi: 10.1038/emboj.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fell SM, Li S, Wallis K, Kock A, Surova O, Rraklli V, Höfig CS, Li W, Mittag J, Henriksson MA, Kenchappa RS, Holmberg J, Kogner P, Schlisio S. 2017. Neuroblast differentiation during development and in neuroblastoma requires KIF1Bβ-mediated transport of TRKA. Genes Dev 31:1036–1053. doi: 10.1101/gad.297077.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szent-Gyorgyi C, Schmidt BF, Creeger Y, Fisher GW, Zakel KL, Adler S, Fitzpatrick JA, Woolford CA, Yan Q, Vasilev KV, Berget PB, Bruchez MP, Jarvik JW, Waggoner A. 2008. Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat Biotechnol 26:235–240. doi: 10.1038/nbt1368. [DOI] [PubMed] [Google Scholar]
- 20.Hirokawa N, Noda Y, Tanaka Y, Niwa S. 2009. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol 10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- 21.Verhey KJ, Hammond JW. 2009. Traffic control: regulation of kinesin motors. Nat Rev Mol Cell Biol 10:765–777. doi: 10.1038/nrm2782. [DOI] [PubMed] [Google Scholar]
- 22.Gennerich A, Vale RD. 2009. Walking the walk: how kinesin and dynein coordinate their steps. Curr Opin Cell Biol 21:59–67. doi: 10.1016/j.ceb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kardon JR, Vale RD. 2009. Regulators of the cytoplasmic dynein motor. Nat Rev Mol Cell Biol 10:854–865. doi: 10.1038/nrm2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greber UF, Way M. 2006. A superhighway to virus infection. Cell 124:741–754. doi: 10.1016/j.cell.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 25.Döhner K, Nagel CH, Sodeik B. 2005. Viral stop-and-go along microtubules: taking a ride with dynein and kinesins. Trends Microbiol 13:320–327. doi: 10.1016/j.tim.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 26.Radtke K, Döhner K, Sodeik B. 2006. Viral interactions with the cytoskeleton: a hitchhiker's guide to the cell. Cell Microbiol 8:387–400. doi: 10.1111/j.1462-5822.2005.00679.x. [DOI] [PubMed] [Google Scholar]
- 27.Ward BM. 2011. The taking of the cytoskeleton one two three: how viruses utilize the cytoskeleton during egress. Virology 411:244–250. doi: 10.1016/j.virol.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brandenburg B, Zhuang X. 2007. Virus trafficking: learning from single-virus tracking. Nat Rev Microbiol 5:197–208. doi: 10.1038/nrmicro1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carnes SK, Zhou J, Aiken C. 2018. HIV-1 engages a dynein-dynactin-BICD2 complex for infection and transport to the nucleus. J Virol 92. doi: 10.1128/JVI.00358-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malikov V, da Silva ES, Jovasevic V, Bennett G, de Souza Aranha Vieira DA, Schulte B, Diaz-Griffero F, Walsh D, Naghavi MH. 2015. HIV-1 capsids bind and exploit the kinesin-1 adaptor FEZ1 for inward movement to the nucleus. Nat Commun 6:6660. doi: 10.1038/ncomms7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dharan A, Talley S, Tripathi A, Mamede JI, Majetschak M, Hope TJ, Campbell EM. 2016. KIF5B and Nup358 cooperatively mediate the nuclear import of HIV-1 during infection. PLoS Pathog 12:e1005700. doi: 10.1371/journal.ppat.1005700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaudin RL, Cunha De Alencar B, Arhel N, Benaroch P. 2013. HIV trafficking in host cells: motors wanted! Trends Cell Biol 23:652–662. doi: 10.1016/j.tcb.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 33.Tang Y, Winkler U, Freed EO, Torrey TA, Kim W, Li H, Goff SP, Morse HC. 1999. Cellular motor protein KIF-4 associates with retroviral Gag. J Virol 73:10508–10513. doi: 10.1128/JVI.73.12.10508-10513.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez NW, Xue X, Berro RG, Kreitzer G, Resh MD. 2008. Kinesin KIF4 regulates intracellular trafficking and stability of the human immunodeficiency virus type 1 Gag polyprotein. J Virol 82:9937–9950. doi: 10.1128/JVI.00819-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alfadhli A, Staubus AO, Tedbury PR, Novikova M, Freed EO, Barklis E. 2019. Analysis of HIV-1 matrix-envelope cytoplasmic tail interactions. J Virol 93. doi: 10.1128/JVI.01079-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cosson P. 1996. Direct interaction between the envelope and matrix proteins of HIV-1. EMBO J 15:5783–5788. doi: 10.1002/j.1460-2075.1996.tb00964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wyma DJ, Jiang J, Shi J, Zhou J, Lineberger JE, Miller MD, Aiken C. 2004. Coupling of human immunodeficiency virus type 1 fusion to virion maturation: a novel role of the gp41 cytoplasmic tail. J Virol 78:3429–3435. doi: 10.1128/jvi.78.7.3429-3435.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murakami T, Ablan S, Freed EO, Tanaka Y. 2004. Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J Virol 78:1026–1031. doi: 10.1128/jvi.78.2.1026-1031.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tedbury PR, Novikova M, Ablan SD, Freed EO. 2016. Biochemical evidence of a role for matrix trimerization in HIV-1 envelope glycoprotein incorporation. Proc Natl Acad Sci USA 113:E182–E190. PMC4720328. doi: 10.1073/pnas.1516618113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tedbury PR, Mercredi PY, Gaines CR, Summers MF, Freed EO. 2015. Elucidating the mechanism by which compensatory mutations rescue an HIV-1 matrix mutant defective for gag membrane targeting and envelope glycoprotein incorporation. J Mol Biol 427:1413–1427. PMC4844178. doi: 10.1016/j.jmb.2015.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tedbury PR, Novikova M, Alfadhli A, Hikichi Y, Kagiampakis I, KewalRamani VN, Barklis E, Freed EO. 2019. HIV-1 matrix trimerization-impaired mutants are rescued by matrix substitutions that enhance envelope glycoprotein incorporation. J Virol 94 PMC6912099. doi: 10.1128/JVI.01526-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willey RL, Bonifacino JS, Potts BJ, Martin MA, Klausner RD. 1988. Biosynthesis, cleavage, and degradation of the human immunodeficiency virus 1 envelope glycoprotein gp160. Proc Natl Acad Sci USA 85:9580–9584. PMC282803. doi: 10.1073/pnas.85.24.9580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Courageot J, Fenouillet E, Bastiani P, Miquelis R. 1999. Intracellular degradation of the HIV-1 envelope glycoprotein. Evidence for, and some characteristics of, an endoplasmic reticulum degradation pathway. Eur J Biochem 260:482–489. doi: 10.1046/j.1432-1327.1999.00193.x. [DOI] [PubMed] [Google Scholar]
- 44.Kimura T, Nishikawa M, Fujisawa J. 1996. Uncleaved env gp160 of human immunodeficiency virus type 1 is degraded within the Golgi apparatus but not lysosomes in COS-1 cells. FEBS Lett 390:15–20. doi: 10.1016/0014-5793(96)00614-x. [DOI] [PubMed] [Google Scholar]
- 45.Gross SP, Vershinin M, Shubeita GT. 2007. Cargo transport: two motors are sometimes better than one. Curr Biol 17:R478–R486. doi: 10.1016/j.cub.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 46.Groppelli E, Len AC, Granger LA, Jolly C. 2014. Retromer regulates HIV-1 envelope glycoprotein trafficking and incorporation into virions. PLoS Pathog 10:e1004518. PMC4231165. doi: 10.1371/journal.ppat.1004518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shantanu PA, Sharma D, Sharma M, Vaidya S, Sharma K, Kalia K, Tao YX, Shard A, Tiwari V. 2019. Kinesins: motor proteins as novel target for the treatment of chronic pain. Mol Neurobiol 56:3854–3864. doi: 10.1007/s12035-018-1327-y. [DOI] [PubMed] [Google Scholar]
- 48.Huszar D, Theoclitou ME, Skolnik J, Herbst R. 2009. Kinesin motor proteins as targets for cancer therapy. Cancer Metastasis Rev 28:197–208. doi: 10.1007/s10555-009-9185-8. [DOI] [PubMed] [Google Scholar]
- 49.Liu X, Gong H, Huang K. 2013. Oncogenic role of kinesin proteins and targeting kinesin therapy. Cancer Sci 104:651–656. doi: 10.1111/cas.12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singh K, Marasini B, Chen X, Ding L, Wang JJ, Xiao P, Villinger F, Spearman P. 2020. A bivalent, spherical virus-like particle vaccine enhances breadth of immune responses against pathogenic Ebola viruses in rhesus macaques. J Virol 94 PMC7163155. doi: 10.1128/JVI.01884-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singh K, Marasini B, Chen X, Spearman P. 2018. A novel Ebola virus antibody-dependent cell-mediated cytotoxicity (Ebola ADCC) assay. J Immunol Methods 460:10–16. doi: 10.1016/j.jim.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hammonds J, Chen X, Fouts T, DeVico A, Montefiori D, Spearman P. 2005. Induction of neutralizing antibodies against human immunodeficiency virus type 1 primary isolates by Gag-Env pseudovirion immunization. J Virol 79:14804–14814. PMC1287556. doi: 10.1128/JVI.79.23.14804-14814.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rai MA, Hammonds J, Pujato M, Mayhew C, Roskin K, Spearman P. 2020. Comparative analysis of human microglial models for studies of HIV replication and pathogenesis. Retrovirology 17:35. PMC7678224. doi: 10.1186/s12977-020-00544-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hammonds JE, Beeman N, Ding L, Takushi S, Francis AC, Wang JJ, Melikyan GB, Spearman P. 2017. Siglec-1 initiates formation of the virus-containing compartment and enhances macrophage-to-T cell transmission of HIV-1. PLoS Pathog 13:e1006181. PMC5298340. doi: 10.1371/journal.ppat.1006181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alber AB, Suter DM. 2018. Single-cell quantification of protein degradation rates by time-lapse fluorescence microscopy in adherent cell culture. JoVE PMC5912353. doi: 10.3791/56604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanjana NE, Shalem O, Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11:783–784. PMC4486245. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Download jvi.00255-23-s0002.mp4, MP4 file, 0.4 MB (364.4KB, mp4)
Video S2. Download jvi.00255-23-s0003.mp4, MP4 file, 0.1 MB (153.4KB, mp4)
Video S3. Download jvi.00255-23-s0004.mp4, MP4 file, 0.4 MB (431.8KB, mp4)
Fig. S1 to S3. Download jvi.00255-23-s0001.docx, DOCX file, 14.3 MB (14.3MB, docx)
Legends of Videos S1 to S3. Download jvi.00255-23-s0005.docx, DOCX file, 0.01 MB (12.7KB, docx)