

Abstract
The RNA-binding protein DDX5 is a polyfunctional regulator of gene expression, but its role in CD8+ T cell biology has not been extensively investigated. In this study, we demonstrate that deletion of DDX5 in murine CD8+ T cells reduced differentiation of terminal effector (TE), effector memory (TEM), and terminal effector memory (t-TEM) cells while increasing the generation of central memory (TCM) cells; forced expression of DDX5 elicited the opposite phenotype. DDX5-deficient CD8+ T cells exhibited increased expression of genes that promote TCM cell differentiation, including Tcf7 and Eomes. Together, these findings reveal a role for DDX5 in regulating the differentiation of effector and memory CD8+ T cell subsets in response to microbial infection.
Introduction
CD8+ T cells are a key component of the adaptive immune response. Activated CD8+ T cells differentiate into heterogeneous cell populations that provide efficient pathogen clearance and durable protection against reinfection. Terminal effector (TE) cells are responsible for mediating pathogen clearance through the production of inflammatory cytokines and cytolytic granules, whereas memory cells provide potent recall responses in the event of reinfection (1). The circulating memory cell population is comprised of several distinct subsets, including central memory (TCM), effector memory (TEM), and terminal effector memory (t-TEM) cells that each possess unique functional, migratory, and survival properties (2, 3, 4, 5). TCM cells express key transcription factors, such as Tcf7, Eomes, and Foxo1, that enable their stem cell-like capacity to expand, differentiate, and self-renew (6). TEM and t-TEM cells are characterized by potent effector function upon infectious rechallenge.
RNA-binding proteins are critical regulators of cellular differentiation due to their broad influence on RNA metabolism and gene regulation (7, 8). However, the understanding of their impact on T cell differentiation is still developing. We previously reported that the RNA-binding protein, Dead box protein 5 (DDX5), regulates the differentiation of an effector-like tissue-resident memory cell population (9). These findings, coupled with recent evidence suggesting a role for DDX5 in regulating the effector function of Th17 cells (10), raised the possibility that DDX5 might also regulate differentiation of circulating effector and memory CD8+ T cell subsets.
DDX5 is a member of the DEAD box family of RNA helicases that are named for a conserved DEAD (Asp-Glu-Ala-Asp) motif that is important for ATP binding, hydrolysis, and RNA helicase activities (11, 12). DDX5 has been shown to be crucial for cell development, proliferation, and organ maturation, and has been implicated in the tumorigenesis of many cancer types (13). DDX5 has been reported to influence nearly every modality of gene regulation, including chromatin organization and epigenetic remodeling, transcriptional co-activation and co-repression, and alternative splicing of RNA transcripts and RNA metabolism (11). Although DDX5 is a well-known polyfunctional regulator of gene expression, its role in CD8+ T cell biology has not been extensively explored.
In this study, we report the consequences of T cell-specific deletion of DDX5 in the differentiation and function of CD8+ T cells responding to lymphocytic choriomeningitis virus (LCMV) infection. DDX5 deficiency reduced the proportions of KLRG1hi CD127lo TE cells and increased the proportions of the KLRG1loCD127hi MP cell population at day 7 post-infection. Furthermore, DDX5 deficiency reduced the proportions of TEM and t-TEM cells, and increased the proportion of TCM cells at day 30 post-infection. DDX5-deficient CD8+ T cells exhibited decreased expression of genes associated with terminally differentiated effector cells, including Klrg1, Zeb2, and Cx3cr1, and increased expression of genes associated with TCM cell differentiation, including Tcf7 and Eomes. Ectopic expression of DDX5 increased the proportions of TEM and t-TEM cells and decreased the proportion of TCM cells, and was associated with reduced expression of Tcf7 and Eomes. Taken together, these findings suggest that DDX5 may regulate CD8+ T cell differentiation by virtue of repressing genes that promote the generation of TCM cells.
Materials and Methods
Mice
All mice were housed under specific pathogen–free conditions in an American Association of Laboratory Animal Care–approved facility at UCSD, and all procedures were approved by the UCSD Institutional Animal Care and Use Committee. C57BL6/J (CD45.1.2+ or CD45.2+) and P14 TCR transgenic (CD45.1+ or CD45.1.2+, maintained on a C57BL6/J background) mice were bred at UCSD or purchased from the Jackson Laboratories. Ddx5fl/fl mice were obtained from Dr. Frances Fuller-Pace’s laboratory (University of Dundee) and have been previously described (14). To generate congenically distinct P14 Ddx5fl/fl Cd4-Cre+ and P14 Ddx5fl/fl Cd4-Cre− mice, Ddx5fl/fl mice were crossed to P14 Cd4-Cre+ mice (either CD45.1+ or CD45.2+). All mice were used from 6 to 9 weeks of age, male mice were used as recipients, and male or female mice were used as donors in adoptive transfer experiments. No randomization of blinding was used in infection experiments.
CD8+ T cell isolation
For isolation of CD8+ T cells from the spleen and peripheral lymph nodes, tissues were collected and filtered through 70 μm cell strainers to yield a single-cell suspension before treatment with Red Blood Cell Lysing Buffer Hybri-Max (Sigma-Aldrich) for 5 minutes. CD8+ T cells were then isolated using the CD8a+ T Cell Isolation Kit and LS MACS Columns (Miltenyi Biotec) according to the manufacturer’s protocol.
Antibodies and flow cytometry
Surface proteins were stained for 10 minutes on ice in Hank’s Balanced Salt Solution (Corning) supplemented with 1% Fetal Bovine Serum (FBS) with the following antibodies: Vα2 (B20.1), CD8α (53–6.7), CD45.1 (A20), CD45.2 (104)), KLRG1 (2F1/KLRG1), CD127 (A7R34), CD27 (LG.3A10), CX3CR1 (SA011F11), CD62L (MEL-14), all purchased from BioLegend. Samples were then stained in Fixable Viability Dye eFluor780 (Thermo Fisher Scientific) at 1:1000 on ice for 10 min. Cells were then fixed in resuspended in eBio Fix/Perm (eBioscience) for 30 minutes at room temperature. After fixation, intracellular targets were stained in eBio Permeabilization Buffer (eBioscience) for 8 hours at 4°C with the following intracellular antibodies: TCF1/7 (C63D9), EOMES (Dan11mag), Ki67 (16A8), T-bet (4B10), and Granzyme B (GB11). For assessment of cytokine production, cells were cultured in the presence of LCMV GP33–41 peptide (GenScript) and Protein Transport Inhibitor Cocktail (Thermo Fisher Scientific) for 3 hours at 37°C. After cell surface and viability staining, cells were fixed and permeabilized using BD Cytofix/Cytoperm (BD Biosciences) for 30 min at room temperature before staining with anti–IFNy (XMG1.2), TNF-α (MP6-XT22), and IL-2 (JES6–5H4) antibodies (all from BioLegend) for 30 min on ice.
For DDX5 staining, after surface marker staining and fixation using eBiosciences Transcription Factor Fixation/Permeabilization Kit, isolated lymphocytes were stained with anti-DDX5 (JM52–30) 1:200–400 in 1X Permeabilization Buffer, at room temperature for 2 hours, followed by staining with Alexa Fluor® 488 Donkey anti-rabbit IgG (minimal x-reactivity) antibody (Biolegend), 1:100 at room temperature for 1–2 hours.
cDNA synthesis and qRT-PCR analysis
Total RNA was extracted with the RNeasy kit (QIAGEN) and reverse-transcribed using the iScript Select cDNA Synthesis Kit (Bio-Rad Laboratories). Real-time reverse transcription polymerase chain reaction (RT-PCR) was performed using iTaq Universal SYBR Green Supermix (Bio-Rad Laboratories). DDX5 expression data were normalized to Gapdh mRNA levels. hDdx5 primer sequences: Forward: ATGTCGGGTTATTCGAGTGACC; Reverse: ACTTCCTCCAAATCGAGGTGC. Gapdh primer sequences: Forward: TGAGTATGTCGTGGAGTCTAC; Reverse: TGGACTGTGGTCATGAGCC.
CD8+ T cell transfers and infection
For naïve CD8+ T cell transfers, splenocytes were collected from naïve congenically distinct Ddx5fl/fl-Cd4-Cre+ P14+ and Ddx5fl/fl-Cd4-Cre− P14+ mice. To quantify the number of Vα2+CD8+CD45.1+ cells, an aliquot of the isolated lymphocytes was stained with antibodies against Vα2, CD8α, and CD45.1, and analyzed using flow cytometry. Vα2+CD8+CD45.1+ cells (1 × 105) were adoptively transferred into CD45.2+ WT recipients 1 day before intraperitoneal (i.p.) infection with 2 × 105 plaque-forming units (PFU) of lymphocytic choriomeningitis virus (LCMV)-Armstrong. At days 7 and 30 p.i., donor mice were sacrificed, their spleens were harvested, and the donor cells were analyzed using flow cytometry.
For induced Ddx5 deletion, Vα2+CD8+CD45.1+ P14 cells from naïve congenically distinct Ddx5fl/fl-Ert2-Cre+ P14+ and Ddx5fl/fl-Ert2-Cre− P14+ mice were mixed 1:1 and adoptively transferred into CD45.2+ host mice before subsequent LCMV infection. To induce Ddx5 deletion, recipient mice were treated i.p. with 1 mg of tamoxifen diluted in sunflower oil on days 4–7 or 30–33 following infection.
For forced DDX5 (pMThy1.1_hDDX5_FL) experiments, retroviral particles were generated using platinum E cells grown in 10 cm plates with full selection media (DMEM 10% FBS (v/v), 2 mM L-Glutamine, 100 U/mL Penicillin-Streptomycin, 1 ug/mL puromycin and 10 ug/mL blasticidin). Eighteen hours before transfection, selection media was replaced with antibiotic-free media (DMEM, 10% FBS (v/v), 2 mM L-Glutamine). For each 10 cm plate, 10 μg of each construct and 5 μg pCL-Eco helper plasmids were mixed in Opti-MEM (ThermoFisher) to a volume of 700 μl. This was combined with 45 μl TransIT-LT1 Reagent (Mirus) and 655 μl Opti-MEM for 20 minutes at room temperature. The mixture was then added dropwise to each 10 cm plate. Twelve hours later, media was replaced with fresh antibiotic-free media supplemented with cholesterol 30uM and Viralboost 1:500 (Alstem Cell Advancements). The viral supernatant was subsequently harvested at 48 and 72 hours. Retroviral supernatant was filtered through a 0.45 μm syringe filter and stored at −80°C. Naive WT P14 CD8+ T cells were plated at a density of 1×106 cells/mL in 24 well plates pre-coated with 5 μg/ml each of anti-CD3 (clone 3C11) and anti-CD28 (clone 37.51) with T cell media supplemented with IL-2 (100 U/ml). The cells were then centrifuged for 90 minutes at 2000 rpm. Retroviral supernatant was removed and replaced with stored T cell media and incubated at 37°C for 2 hours before adoptive transfer.
For co-transfer experiments, cells of each construct were mixed at a 1:1 ratio and a total of 5×105 donor cells/mouse was adoptively transferred into CD45.2+ male recipient mice. One hour later, mice were infected with 2×105 PFU LCMV. Seven and 30 days after infection, mice were euthanized, spleens were harvested, and the donor cells were analyzed by flow cytometry.
For ex vivo re-stimulation assays, P14 CD8+ T cells were plated in a U-bottom 96-well plate at 10–15× 106 cells/well in the presence of 1 ng/μl LCMV GP33–41 peptide (Genscript) and 1X Protein Transport Inhibitor Cocktail (eBioScience) for >3 h at 37°C. Cells were then fixed and permeabilized using BD Cytofix/Cytoperm (BD Biosciences) and stained for IFNγ (XMG1.2), TNF-α (MP6-XT22), and IL-2 (JES6–5H4) antibodies, all purchased from BioLegend, for 30 min on ice. Flow cytometry was performed on a Novocyte (ACEA Biosciences). FACS sorting of cells was done on a FACSAria Fusion or FACSAria2 (BD Biosciences). FlowJo software (BD Biosciences) was used for analysis of flow cytometry data.
CITE-sequencing and analysis
WT CD45.1 and Ddx5fl/flCd4-Cre+ CD8+ CD45.1.2 P14 cells were adoptively co-transferred into congenically distinct hosts. The host mice were infected with 2×105 PFU LCMV 24 hours after transfer and sacrificed on days 4, 7, and 30 post-infection. Circulating lymphocytes were isolated from the spleen and resuspended with antibody staining buffer containing TruStain FcX™ PLUS (anti-mouse CD16/32; Biolegend). Cells were incubated with TotalSeq-A antibody panel (Supplemental Table 1) and fluorescently labeled antibodies targeting CD8, Vα2, CD45.1, CD45.2 for 30 min at 4°C. Stained cells were washed with antibody staining buffer three times before sorting for P14 CD8+ T cells of each genotype. About 10,000 cells per sample were loaded into Single Cell G chips (10x Genomics) and partitioned into Gel Bead In-Emulsions in a Chromium Controller (10x Genomics). Single-cell RNA libraries were prepared according to the 10x Genomics Chromium Next GEM Single Cell 3’ v3.1 (Dual Index) User Guide and sequenced on a HiSeq 4000 (Illumina).
Reads from CITE-seq were aligned to mm10 using Cellranger (v.6.0.1). The Unique Molecular Identifier count matrix was used for downstream analysis using Seurat (v4.1.1). In R, samples were subsampled to 2000 cells per sample. Cells were further filtered to exclude cells with < 300 genes per cell and <5% mitochondrial genes. Standard parameters were used for normalization, filtering, PCA reduction, and UMAP creation. The samples were clustered with Louvain clustering using the FindClusters function with a resolution of 1. Differential expression analysis was completed between clusters and conditions using the FindMarkers function. Data are available at Gene Expression Omnibus under GSE230028.
Results
DDX5 may regulate CD8+ T cell proliferation and apoptosis
To assess phenotypic changes in circulating CD8+ T cell populations resulting from deletion of DDX5, we adoptively co-transferred P14 CD8+ T cells, which express a transgenic T cell receptor (TCR) that recognizes the immunodominant epitope of lymphocytic choriomeningitis (LCMV), from mice with a T cell-specific deletion of Ddx5 (Ddx5fl/fl Cd4-Cre+, “Ddx5cKO”) and congenically distinct control mice (Ddx5fl/fl Cd4-Cre−,“WT”) into CD45.2+ recipient mice. Relatively high numbers of P14 cells were transferred (2 × 105 CD8+ P14 cells total), which may alter the kinetics and phenotype of the ensuing CD8 T cell response, representing a limitation of the present study (15). One day after cell transfer, recipient mice were infected with the Armstrong strain of LCMV. At days 7 and 30 p.i., lymphocytes were isolated from the spleens of recipient mice and donor cells were analyzed by flow cytometry (Fig. 1A). DDX5-deficient CD8+ T cells were reduced compared to their control counterparts at days 7 and 30 post-infection (p.i.), suggesting a possible role for DDX5 in cellular proliferation and/or survival (Fig. 1B). Further, the altered frequencies of DDX5-deficient CD8+ T cells compared to control cells remained relatively consistent through the duration of acute infection (Fig. 1C, 1D). Consistent with the notion of DDX5 repressing cellular proliferation, DDX5-deficient CD8+ T cells expressed higher levels of Ki-67, a marker of cellular proliferation (Fig. 1E, 1F), compared to control cells. To further elucidate the role of DDX5 in proliferation, we stained WT and DDX5-deficient CD8+ T cells with the fluorescent dye CFSE, which dilutes as cells divide, allowing for efficient tracing of cell proliferation patterns. CFSE-labeled WT and DDX5-deficient cells were mixed at a 1:1 ratio and adoptively transferred into congenically distinct recipients. Forty-eight hours after LCMV infection, recipient mice were sacrificed, and donor cells were analyzed by flow cytometry. DDX5-deficient cells exhibited greater CFSE dilution than control cells (Fig. 1G, 1H; Supplemental Fig. 1A, 1B). Lastly, we observed that DDX5-deficient CD8+ T cells expressed higher levels of Annexin V, a membrane-bound apoptotic marker at 48 hours p.i. (Fig. 1I, 1J). Having shown that conditional knock-out of Ddx5 accelerated proliferation and led to an early death phenotype, we next investigated whether deletion of DDX5 after the initial proliferative burst of early infection also affected CD8+ T cell survival. Importantly, inducible deletion of Ddx5 during the peak of infection (days 4–7 p.i.) did not result in a numerical deficit in DDX5-deficient cells at days 9 and 30 p.i. (Supplemental Fig. 1C, 1D), suggesting that the DDX5 may only play a role in proliferation and survival during the first few days of infection. Taken together, these results indicate that DDX5 may have an important role in inhibiting apoptosis, leading to the observed decrease of DDX5-deficient cells on days 7 and 30 post-infection.
FIGURE 1.
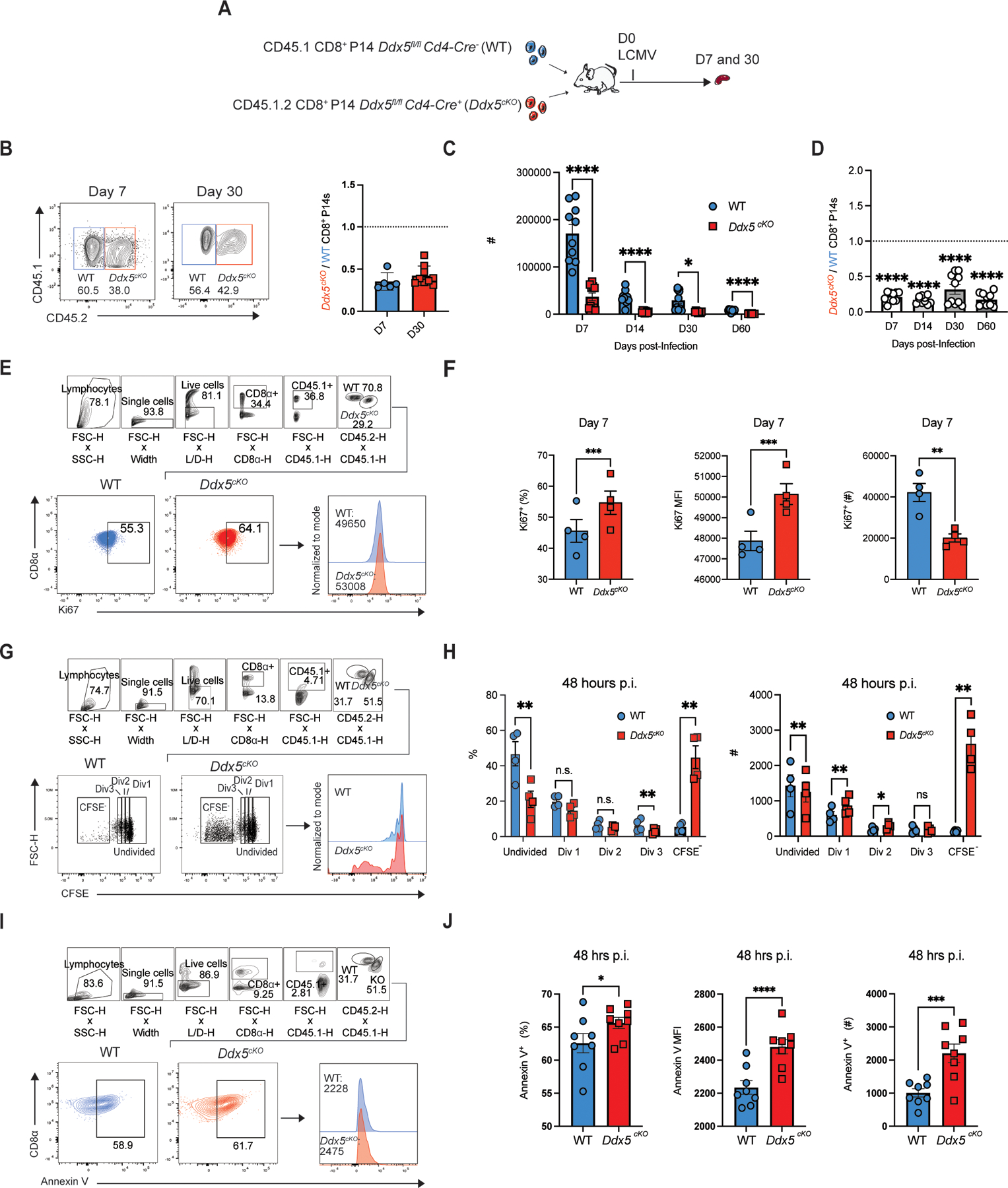
DDX5 may regulate CD8+ T cell proliferation and apoptosis. (A) Experimental set-up. Congenically distinct Ddx5fl/fl Cd4-Cre+ (Ddx5cKO) cells and Ddx5fl/fl CD4-Cre− (WT) CD8+ P14 T cells were mixed at a 1:1 ratio and adoptively transferred i.v. into CD45.2+ hosts. One day after transfer, recipient mice were infected with LCMV, sacrificed at days 7 and 30 p.i., and donor cells analyzed by flow cytometry. (B) Representative flow cytometry plot showing the proportions of WT vs Ddx5cKO cells (left) and ratio of the absolute numbers of Ddx5cKO to WT CD8+ P14 cells at days 7 and 30 p.i., normalized to the input ratio (right). (C) Absolute numbers of WT vs. Ddx5cKO CD8+ P14 cells at days 7, 14, 30 and 60 post-infection. (D) Ratio of Ddx5cKO to WT CD8+ P14 cells at days 7, 14, 30 and 60 post-infection, normalized to the input ratio. (E-F) Representative flow cytometry plots and histograms showing the frequency, absolute numbers, and MFI of Ki67+ WT vs, Ddx5cKO CD8+ P14 cells at day 7 post-infection. Gates were determined by Ki67 fluorescence minus one (FMO). (G-H) CFSE proliferation analysis of WT and Ddx5cKO CD8+ T cells at 48 hours p.i., represented as (G) flow cytometry plots (left), histograms (right). (H) Frequency and absolute numbers of CFSE-labeled undivided, Division 1 (Div 1), Division 2 (Div 2), Division 3 (Div 3), and CFSE− WT vs. Ddx5cKO CD8+ P14 cells. (I-J) Representative flow plots and histograms and showing the frequency, absolute numbers, and MFI of Annexin V+ WT and Ddx5cKO CD8+ P14 cells at 48 hours post-infection. All data are from one representative experiment out of two independent experiments with n = 5 to 10 per group: ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (Student’s paired two-tailed t test). Graphs indicate mean ± SEM, symbols represent individual mice.
DDX5 regulates CD8+ TE, TEM, and t-TEM cell differentiation
We next examined the consequences of T cell-specific deletion of Ddx5 (Ddx5fl/fl Cd4-Cre+, “Ddx5cKO”) on circulating effector and memory cell populations (Fig. 1A). Ddx5 deletion resulted in reduced proportions and absolute numbers of KLRG1hiCD127lo TE-phenotype cells and increased proportions of KLRG1loCD127hi MP-phenotype cells at day 7 p.i. (Fig. 2A, Supplemental Fig. 1E). Furthermore, DDX5 deletion resulted in increased proportions of CD62LhiCD127hi TCM cells at day 30 post-infection (Fig. 2B; Supplemental Fig. 1F). Conversely, Ddx5cKO memory cells displayed reduced proportions and absolute numbers of CD62LloCD127hi TEM cells and CD62LloCD127lo t-TEM cells at day 30 post-infection (Fig. 2B, 2C). Moreover, compared to control cells, DDX5-deficient memory cells exhibited increased expression of CD62L at day 30 p.i. (Supplemental Fig. 2A). Consistent with the observed increase in TCM cell differentiation, CD127 (Il7r) and CD122 (Il15rb) expression were both increased in the absence of DDX5, suggesting that these cells may be better able to compete for IL-7 and IL-15 (Supplemental Fig. 2B, 2C). Analyses of functional parameters demonstrated that compared to control cells, DDX5-deficient CD8+ T cells displayed equivalent frequencies of IFNγ+TNF+ cells, whereas the frequency of IL-2+ DDX5-deficient CD8+ T cells was increased compared to controls, upon ex vivo restimulation with GP33–41 peptide (Fig. 2D, 2E). Moreover, IFNγ, TNF, IL-2 expression and IFNγ+ IL-2+ co-expression were unaffected in Ddx5cKO cells, compared to controls (Supplemental Fig. 2D-G). Importantly, analysis of pre-transfer WT and Ddx5cKO cells showed similar frequencies of naïve CD44loCD62Lhi CD8+ T cells (Supplemental Fig. 3A). In addition, naïve pre-transfer WT and Ddx5cKO CD8+ T cells exhibited similar expression of the transcription factor TCF1, which is expressed by all naïve CD8+ T cells, suggesting that the phenotypes observed in Ddx5cKO cells were not a consequence of altered thymic selection or homeostasis (Supplemental Fig. 3B, 3C). Taken together, these data demonstrate that deletion of DDX5 changes the composition of the antigen-experienced CD8+ T cell pool.
FIGURE 2.
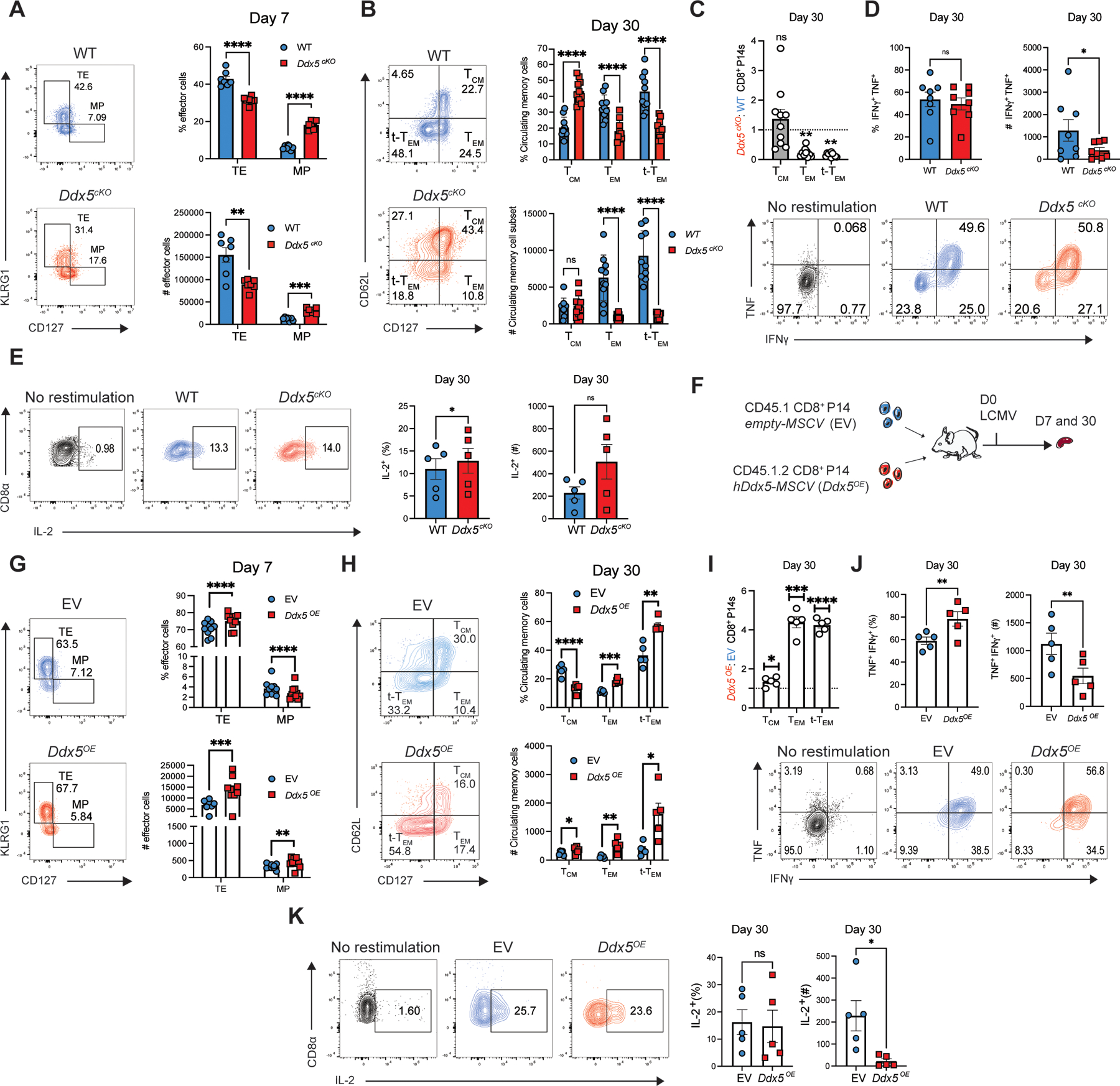
DDX5 regulates CD8+ TE, TEM, and t-TEM cell differentiation. (A) Representative flow cytometry plots (left) and quantification (right) of WT and Ddx5cKO TE- and MP-phenotype cells. (B) Representative flow cytometry plots (left) and quantification (right) of WT and Ddx5cKO TCM, TEM, t-TEM cells at day 30 p.i. (C) Ratio of Ddx5cKO and WT TCM-, TEM-, t-TEM-phenotype CD8+ P14 cells at day 30 post-infection. (D) Representative flow cytometry plots (bottom) and quantification (top) of TNF+IFNy+ WT and Ddx5cKO cells isolated at day 30 p.i. and restimulated ex vivo in the presence of GP33–41 peptide for 3 hours. (E) Representative flow plots (left) and quantification (right) of WT and Ddx5cKO IL-2+ CD8+ T cells after ex vivo restimulation at day 30 p.i. (F) Experimental set-up. Congenically distinct CD8+ T cells were activated and transduced with empty vector (EV) or forced Ddx5 expression (Ddx5OE) retroviral constructs. Transduced cells were mixed at a 1:1 ratio and adoptively transferred into CD45.2+ recipients. Recipient mice were infected with LCMV and sacrificed at days 7 and 30 p.i. (G) Representative flow cytometry plots (left) and quantification (right) of EV and Ddx5OE TE- and MP-phenotype cells at day 7 p.i. (H) Representative flow cytometry plots (left) and quantification (right) of EV and Ddx5OE TCM, TEM, t-TEM cells at day 30 p.i. (I) Ratio of Ddx5OE and WT TCM-, TEM-, t-TEM-phenotype CD8+ P14 cells at day 30 post-infection. (J) Representative flow plots (left) and quantification (right) of EV and Ddx5OE IFN-γ+ and TNF+ CD8+ T cells after ex vivo restimulation at day 30 p.i. (I) Representative flow plots (left) and quantification (right) of EV and Ddx5OE IL-2+ CD8+ T cells after ex vivo restimulation at day 30 p.i. All data are from one representative experiment out of two independent experiments with n = 5–10 per group; ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; student’s paired two-tailed t test (A,B,D,E,G,H,J,K), one sample t test (C and I). Graphs indicate mean ± SEM, symbols represent individual mice.
Since DDX5 deletion appeared to inhibit terminal effector cell differentiation and promote the generation of TCM cells, we next assessed the functional consequences of ectopic expression of DDX5. We isolated and activated congenically distinct CD8+ P14 T cells, retrovirally transduced the cells with empty vector (EV) or DDX5 ectopic-expression (Ddx5OE) constructs, mixed them at a 1:1 ratio, and adoptively co-transferred the cells into CD45.2+ mice (Fig. 2F). Recipient mice were sacrificed at days 7 and 30 p.i. and donor cells were analyzed using flow cytometry. In contrast to decreased proportions of TE cells and increased proportions of TCM cells resulting from Ddx5 deletion, ectopic expression resulted in increased proportions and absolute numbers of TE cells and reduced proportions of MP cells at day 7 p.i., along with decreased proportions of TCM cells and increased proportions and absolute numbers of TEM and t-TEM cells at day 30 post-infection (Fig. 2G, 2H). Furthermore, Ddx5OE memory cells exhibited reduced CD62L and CD127, while CD122 expression was unaffected (Supplemental Fig. 2H-J). In contrast to conditional knock-out of Ddx5, a greater proportion of Ddx5OE cells expressed TNF and IFNγ compared to EV controls, and TNF expression was increased among Ddx5OE cells (Fig. 2K, Supplemental Fig. 2K, 2L). Additionally, while the proportions of IL-2-producing or IL-2 and IFNγ – co-producing memory cells were not affected by ectopic expression of Ddx5, the expression of IL-2 was enhanced in Ddx5cKO CD8+ T cells (Fig. 2L, Supplemental Fig. 2M, 2N). Taken together, these findings indicate that DDX5 promotes the differentiation of effector-like subsets of CD8+ T cells.
DDX5 regulates the maintenance of established CD8+ t-TEM cells
Having shown that DDX5 plays a role in the generation of memory cells, we next investigated whether DDX5 also plays a role in established memory cells. We therefore used an inducible Cre recombinase model in which the deletion of Ddx5 is temporally regulated by administration of tamoxifen. Congenically distinct Ddx5fl/fl Ert2-Cre− (WT) and Ddx5fl/fl Ert2-Cre+ (Ddx5iKO) and CD8+ P14 T cells were adoptively co-transferred into CD45.2+ recipients and infected with LCMV 24 hours later. Recipient mice received tamoxifen on days 30–33 p.i. and donor cells were analyzed at day 40 p.i. by flow cytometry (Fig. 3A). A reduction in DDX5 protein and mRNA expression was confirmed by flow cytometry and RT-qPCR of sorted WT and Ddx5iKO CD8+ T cells (Supplemental Fig. 3D-F). Induced deletion of Ddx5 in established circulating memory cells resulted in increased absolute numbers of total DDX5-deficient memory cells relative to control cells (Fig. 3B). Among memory T cell subsets, Ddx5 deletion resulted in increased proportions and absolute numbers of TCM cells and reduced proportions of t-TEM cells (Fig. 3C). Furthermore, compared to control cells, reduced proportions of DDX5-deficient CD8+ T cells could produce IFNγ and TNF (Fig. 3D). Notably, the production of IFNγ and TNF were largely similar among Ddx5iKO and control cells among the circulating memory cell subsets, except for reduced expression of IFNγ in the Ddx5iKO TCM cell population (Fig. 3E, 3F). The frequency of IL-2 producing Ddx5iKO memory cells was also reduced compared to WT control cells, while IL-2 production was only significantly decreased within the t-TEM cell population and increased in the TCM cell population (Fig. 3H). Together, these findings suggest a role for DDX5 in promoting the maintenance of t-TEM cells.
FIGURE 3.
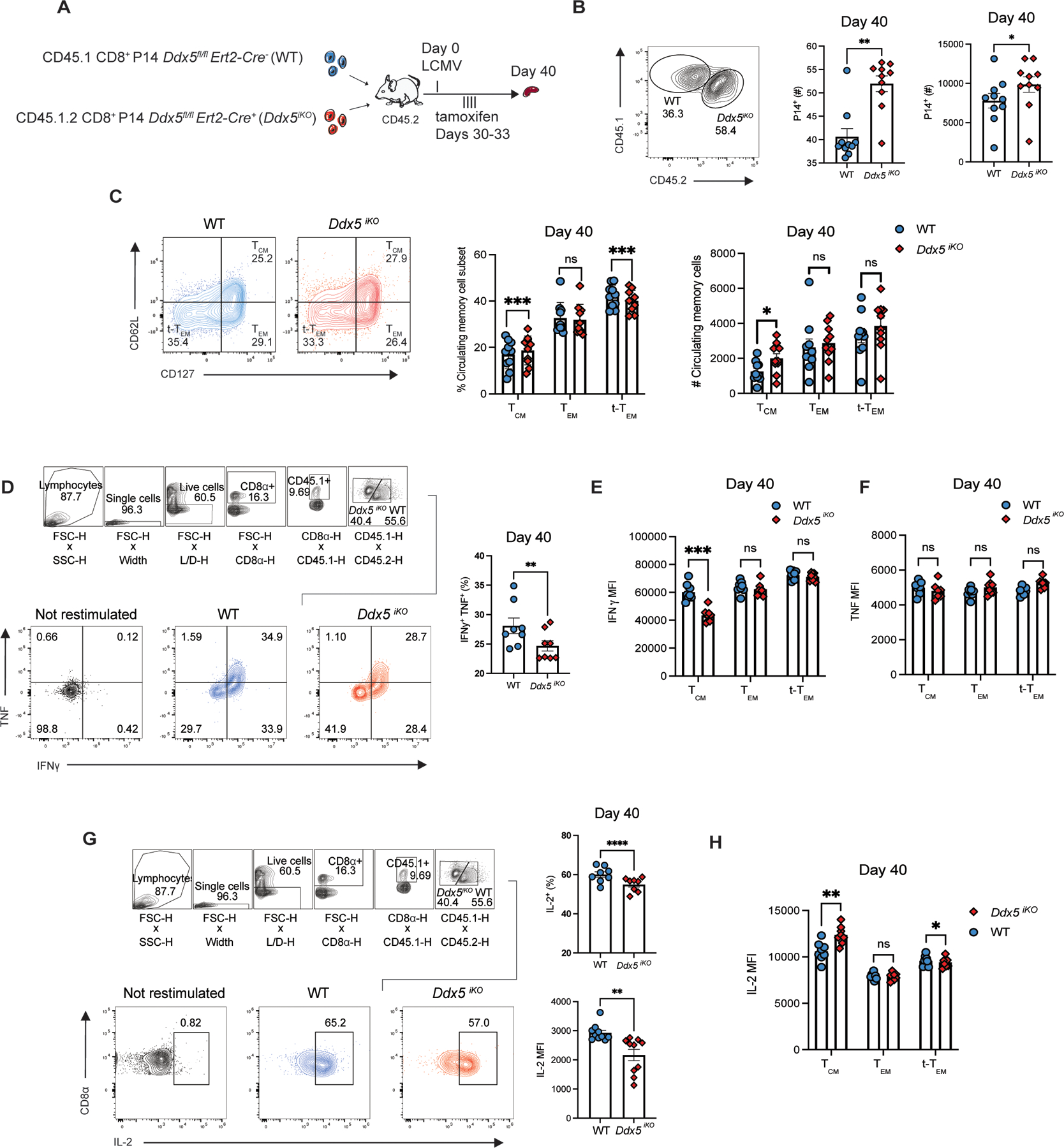
DDX5 regulates the maintenance of established CD8+ t-TEM cells. (A) Experimental set-up. Congenically distinct Ddx5fl/fl Ert2-Cre− (WT ) and Ddx5fl/fl Ert2-Cre+ (Ddx5iKO ) CD8+ P14 T cells were mixed at a 1:1 ratio and adoptively transferred into CD45.2+ recipients intravenously. One day post-transfer, recipient mice were infected with LCMV. At days 30–33 p.i., mice received 1μg tamoxifen i.p. to induce the deletion of Ddx5. Recipient mice were sacrificed at day 40 p.i. and donor cells were analyzed by flow cytometry. (B) Representative flow cytometry plots (left) and quantification (right) of WT and Ddx5iKO CD8+ P14 cells at day 40 p.i. (C) Representative flow cytometry plots (left) and quantification (right) of WT and Ddx5iKO TCM, TEM, and t-TEM cells at day 40 p.i. (D) Representative flow plots (left) and quantification (right) of WT and Ddx5iKO IFN-γ+ and TNF+ CD8+ P14 cells after ex vivo restimulation at day 30 p.i. (E-F) IFNγ and TNF median fluorescence intensity in WT and Ddx5iKO TCM, TEM, and t-TEM cells at day 40 p.i. (G) Representative flow plots (left) and quantification (right) of WT and Ddx5iKO IL-2+ CD8+ T cells after ex vivo restimulation at day 40 p.i. (H) IL-2 median fluorescence intensity of WT and Ddx5iKO TCM, TEM, and t-TEM cells at day 40 p.i. All data are from one representative experiment out of two independent experiments with n = 5 to 10 per group; ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (Student’s paired two-tailed t test). Graphs indicate mean ± SEM, symbols represent individual mice.
Genes associated with TCM cell differentiation are upregulated in the absence of DDX5
To investigate potential mechanisms by which DDX5 regulates CD8+ T cell differentiation, we employed Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-seq), which allows for measurements of the transcriptome and selected proteins in the same single-cells (16). We adoptively co-transferred control WT and Ddx5cKO CD8+ P14 T cells into congenically distinct recipient mice that were infected with LCMV one day after transfer (Fig. 4A). At days 4, 7, and 30 days p.i., splenic CD8+ P14 T cells were isolated by FACS and processed for CITE-seq using the 10x Genomics platform. Antibodies targeting cell surface molecules, some of which have previously implicated in CD8+ T cell activation and differentiation, were selected for inclusion in the CITE-seq antibody panel (Supplemental Table 1).
FIGURE 4.
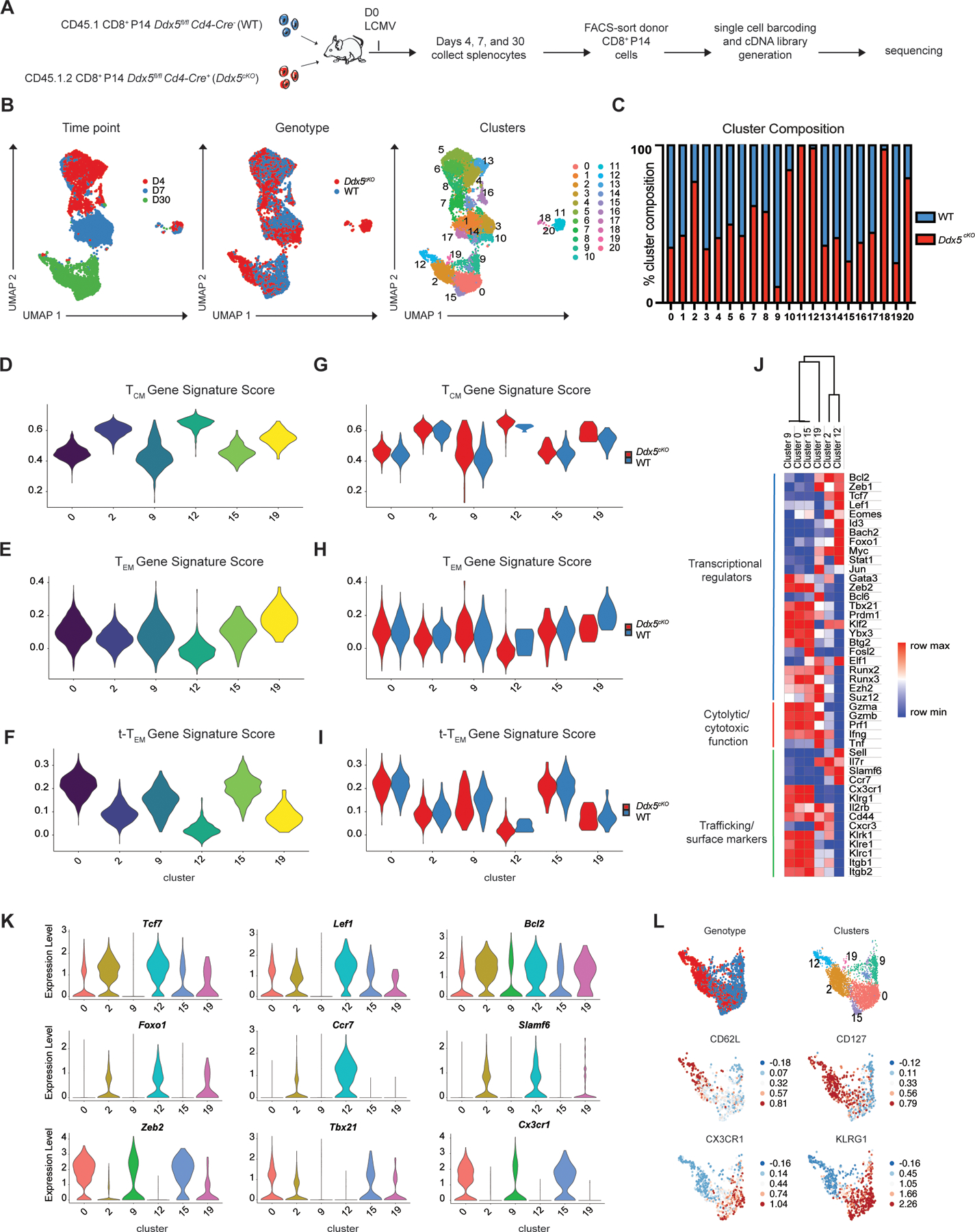
Genes associated with TCM cell differentiation are upregulated in the absence of DDX5. (A) Experimental set-up. Congenically distinct Ddx5fl/fl Cd4-Cre− (WT) and Ddx5fl/fl Cd4-Cre+ (Ddx5cKO) CD8+ P14 cells were mixed and adoptively transferred into CD45.2+ recipients. One day after transfer, recipient mice were infected with LCMV. Mice were sacrificed at days 4, 7, and 30 p.i. and stained with a TotalSeq-A antibody panel (Supplemental Table 1) before FACS-sorting and processing for CITE-seq. (B) UMAP analyses of WT and Ddx5cKO CD8+ T cells on days 4, 7, and 30 p.i. (C) Proportions of WT (red) and Ddx5cKO (blue) cells within each cluster. (D-F) TCM, TEM, and t-TEM gene signature scores applied to Day 30 clusters, represented as violin plots. (G-I) TCM, TEM, and t-TEM gene signature scores applied to WT and Ddx5cKO cells within each Day 30 cluster and represented as violin plots. (J) Heatmap representing the expression of selected genes among cells from each of the Day 30 clusters. (K) Quantification of expression level of selected genes among cells from each of the Day 30 clusters, represented as violin plots. (L) Quantification of selected proteins by Day 30 cells, represented as relative expression plots superimposed on individual cells in the UMAP.
Uniform Manifold Approximation and Projection (UMAP) analyses revealed that CD8+ T cells clustered distinctly at all time points post-infection (Fig. 4B), indicating that T cells exhibit distinct gene expression patterns during differentiation, as previously demonstrated (16, 9, 17). Notably, WT and Ddx5cKO CD8+ T cells clustered distinctly at days 7 and 30 p.i. (Fig. 4B). Ddx5cKO CD8+ T cells exhibited increased expression of Sell and Il7r, and decreased expression of genes associated with terminal effector differentiation, such as Zeb2, Klrg1, and Bhlhe40, consistent with the observed bias toward TCM cell differentiation in the absence of DDX5 (Supplemental Fig. 4A). Moreover, pathway analyses revealed an enrichment of genes regulating cytokine production and cytotoxicity in WT cells relative to Ddx5cKOcells, as well as of processes related to ribosome biogenesis and cytoplasmic translation, consistent with DDX5’s known involvement in ribosome assembly and function (19) (Supplemental Fig. 4B). We next evaluated circulating memory cell heterogeneity at day 30 post-infection. Clusters 0, 9, 15, and 19 were primarily comprised of day 30 p.i. WT CD8+ T cells, whereas clusters 2 and 12 were primarily comprised of Ddx5cKOCD8+ T cells (Fig. 4C). Comparative gene ontology analyses among the six day 30 clusters revealed that the Ddx5cKO clusters were more similar to each other than to the WT clusters (Supplemental Fig. 4C). Application of TCM, TEM, and t-TEM transcriptional signatures to WT and DDX5-deficient memory cells revealed that the four WT-predominant clusters (0, 9, 15, and 19) tended to score more highly for the TEM, and t-TEM transcriptional signatures, whereas the two Ddx5cKO-predominant clusters (2, 12) tended to score more highly for the TCM gene signature (Fig. 4D-4F). Furthermore, application of these transcriptional signatures to WT and DDX5-deficient memory cells within each cluster demonstrated that WT cells scored higher for the TEM and t-TEM gene signatures, while DDX5-deficient cells scored higher for the TCM gene signature (Fig. 4G-4I). Analyses focused on specific genes revealed that compared to the four WT clusters (0, 9, 15, 19), the two Ddx5cKO clusters (2, 12) exhibited increased expression of transcriptional regulators associated with TCM cells, including Bcl2, Tcf7, Lef1, Eomes, Foxo1, and Bach2 (Fig. 4J, 4K). In contrast, the two Ddx5cKO clusters (2, 12) exhibited reduced expression of genes associated with effector cell differentiation and function, including Zeb2, Tbx21, Gzma, Gzmb, Prf1, Cx3cr1, and Klrg1, compared to the WT clusters (0, 9, 15, 19) (Fig. 4J, 4K). Using protein expression derived from the CITE-seq protein panel, we observed that the WT clusters (0, 9, 15, and 19) tended to express higher levels of molecules associated with terminal effector cells, including CX3CR1 and KLRG1, whereas the two Ddx5cKO clusters (2, 12) tended to express higher levels of molecules associated with TCM cells, such as CD62L and CD127 (Fig. 4L, Supplemental Fig. 4D). Together these findings raised the possibility that DDX5 functions to repress the expression of TCM cell-associated genes to promote the differentiation of effector-like subsets of CD8+ T cells.
DDX5-deficient t-TEM cells exhibit increased TCF1 and Eomes protein expression
The CITE-seq analyses indicated that a major consequence of DDX5 deletion was increased expression of genes that promote TCM cell generation. We therefore sought to confirm some of the key transcriptional findings with independent flow cytometry experiments. Indeed, greater proportions of DDX5-deficient TE and MP cells expressed TCF1 and DDX5-deficient TE cells displayed increased expression of TCF1 compared to control cells at day 7 p.i. (Fig. 5A, 5B); conversely, ectopic expression of Ddx5 resulted in decreased proportions of TE and MP cells expressing TCF1 and reductions of TCF1 expression in the Ddx5OE TE and MP cell populations (Fig. 5C, 5D). Analyses of TCF1 expression among DDX5-deficient circulating memory CD8+ T cell subsets revealed increased expression of TCF1 at day 30 post-infection (Fig. 5E, Supplemental Fig. 4E). The observed increase in TCF1 expression among circulating memory CD8+ T cells was also evident within the TCM, TEM, and t-TEM cell populations and DDX5-deficient CD8+ T cells displayed increased proportions of TCM, TEM, and t-TEM cells expressing TCF1 (Fig. 5F, Supplemental Fig. 4F, 4G). Conversely, forced expression of DDX5 resulted in decreased expression of TCF1 among circulating memory CD8+ T cells (Fig. 5G), which was significant within the TCM and TEM cell subsets (Fig. 5H). Importantly, forced expression of Ddx5 also resulted in decreased proportions of TCM, TEM, and t-TEM cells expressing TCF1 (Supplemental Fig. 4H, 4I). Similarly, Eomes expression was increased in DDX5-deficient CD8+ T cells as well as within the TCM and t-TEM cell populations (Fig. 5I, 5J); moreover, increased proportions of DDX5-deficient t-TEM cells expressed Eomes (Supplemental Fig. 4J-L). Conversely, ectopic expression of Ddx5 resulted in reduced proportions of cells expressing Eomes in the total circulating memory CD8+ T cell pool and among TCM, TEM, and t-TEM cell subsets (Fig. 5K, 5L). Moreover, ectopic expression of Ddx5 resulted in reduced proportions of Eomeshi TCM, TEM, and t-TEM cells (Supplemental Fig. 4M, 4N). Furthermore, DDX5-deficient CD8+ T cells exhibited increased co-expression of Eomes and TCF1 (Supplemental Fig. 4O). Together, these data suggest that DDX5 may promote the differentiation and maintenance of t-TEM cells by repressing genes associated with generation of TCM cells.
FIGURE 5.
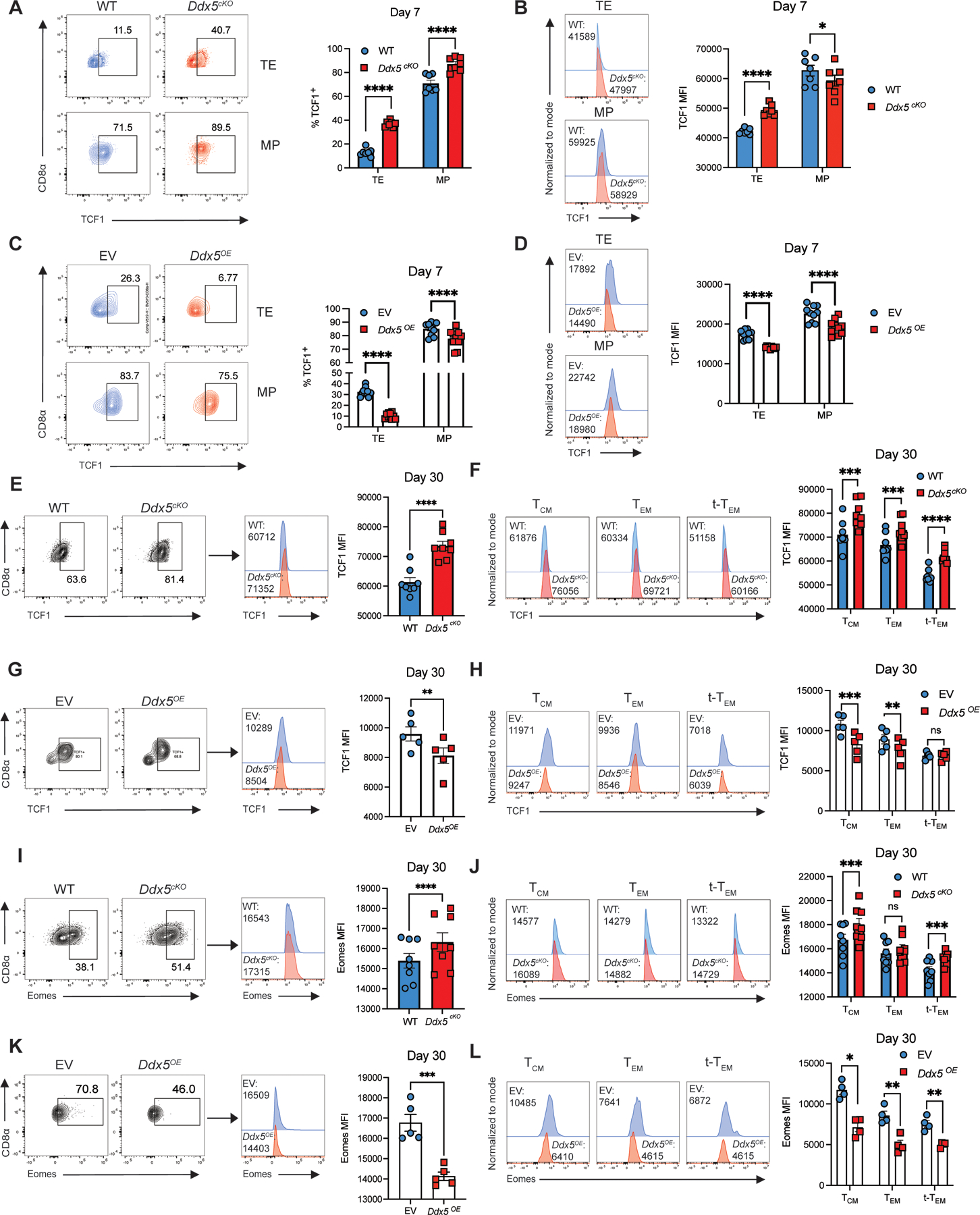
DDX5-deficient t-TEM cells exhibit increased TCF1 and Eomes protein expression. (A) Representative flow cytometry plots (left) and quantification (right) of proportions of WT and Ddx5cKO TE and MP CD8+ T cells expressing TCF1 at day 7 p.i. (B) Representative histograms (left) and median fluorescence intensity (right) of TCF1 expression in WT and Ddx5cKO TE and MP cells at day 7 p.i. (C) Representative flow cytometry plots (left) and quantification (right) of proportions of WT and Ddx5OE TE and MP CD8+ T cells expressing TCF1 at day 7 p.i. (D) Representative histograms (left) and median fluorescence intensity (right) of TCF1 expression in WT and Ddx5OE TE and MP cells at day 7 p.i. (E) Representative flow cytometry plots (left) and histograms (middle) and quantification (right) of TCF1 MFI in WT and Ddx5cKO CD8+ P14 cells at day 30 p.i. (F) Representative histograms (left) and quantification (right) of proportions of WT and Ddx5cKO TCM, TEM, t-TEM cells expressing TCF1. (G) Representative flow cytometry plots (left), histograms (middle), and quantification (right) of TCF1 MFI in WT and Ddx5OE CD8+ P14 cells at day 30 p.i. (H) Representative histograms (left) and quantification (right) of proportions of WT and Ddx5OE TCM, TEM, t-TEM cells expressing TCF1. (I) Representative flow cytometry plots (left) and histograms (middle) and quantification (right) of Eomes MFI in WT and Ddx5cKO CD8+ P14 cells at day 30 p.i. (J) Representative histograms (left) and quantification (right) of proportions of WT and Ddx5cKO TCM, TEM, t-TEM cells expressing Eomes.(K) Representative flow cytometry plots (left), histograms (middle), and quantification (right) of Eomes MFI in WT and Ddx5OE CD8+ P14 cells at day 30 p.i. (L) Representative histograms (left) and quantification (right) of proportions of WT and Ddx5OE TCM, TEM, t-TEM cells expressing Eomes. All data are from one representative experiment out of two independent experiments with n = 5 to 10 per group; ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (Student’s paired two-tailed t test). Graphs indicate mean ± SEM, symbols represent individual mice.
DISCUSSION
RNA-binding proteins are important regulators of cellular differentiation due to their unique ability to operate at multiple levels of gene regulation, but their role in immune cell biology has not been extensively explored (19, 20). Here, we identify the RNA-binding protein DDX5 as a regulator of effector and circulating memory CD8+ T cell differentiation in response to microbial infection.
While DDX5 has been implicated in nearly every aspect of genetic regulation from chromatin organization to RNA metabolism and suggested to broadly regulate cell differentiation, the role of DDX5 in T cells during acute infection is only beginning to be explored (21, 22, 23). We show here that DDX5-deficient CD8+ T cells exhibited accelerated proliferation and reduced survival compared to controls, implicating DDX5 in the regulation of CD8+ T cell proliferation. However, the substantial CFSE dilution was accompanied by a small difference in the proportion of Ki67+ cells, suggesting that factors other than cell cycle entry might be contributing to the observed phenotype. Furthermore, consistent with our previously reported role for DDX5 in regulating the differentiation of an effector-like subset of tissue-resident memory cells (9), we demonstrate that deletion of DDX5 resulted in a reduction in the proportions of TE, TEM, and t-TEM cells, along with increased proportions of MP and TCM cells. In established memory cells, Ddx5 deletion promoted maintenance of the TCM population while simultaneously limiting the t-TEM population. Some observed differences between WT and Ddx5iKO cells were modest, representing an important limitation of the present study. Further, DDX5-deficient CD8+ T cells exhibited increased expression of key genes known to be involved in TCM cell differentiation, such as Tcf7 and Eomes. Together, these findings raise the possibility that DDX5 regulates differentiation of effector-like CD8+ T cell subsets by virtue of repressing genes that promote TCM cell generation.
There are several potential mechanisms by which DDX5 may repress expression of genes associated with TCM cells. Multiple TCF1 isoforms have been identified in T cells (24, 25); the long isoforms possess a beta-catenin binding domain and are dispensable for TE cell differentiation, but are necessary for the generation of TCM cells (26). Our data suggest that DDX5 may repress TCF1 expression, raising the possibility that DDX5 could regulate the differentiation of TE and t-TEM cells through alternative splicing of TCF1 and shifting the composition of the TCF1 isoform pool to favor the short isoforms (27–29); such a possibility will be investigated in future studies. Furthermore, prior studies have described the intrinsic histone-modifying capabilities of TCF1 and its homolog, LEF1 (30), suggesting the possibility that repression of TCF1 expression by DDX5 may shift the epigenetic landscape to favor expression of genes associated with TE cell differentiation. Another potential mechanism by which DDX5 could repress expression of genes associated with TCM cells is through an association with the Polycomb Repressive Complex 2 (PRC2), thereby mediating the subsequent silencing of memory genes through deposition of repressive H3K27me3 marks. Indeed, recent reports have posited a role for long non-coding RNAs (lncRNAs) in enabling targeted PRC2-mediated gene silencing (27, 28). Recently published global RNA interactions with DNA by deep sequencing (GRID-seq) data revealed that the lncRNA Malat1 interacts at genomic loci associated with H3K27me3 deposition in TE cells; furthermore, shRNA-mediated knockdown of Malat1 inhibited TE and t-TEM cell differentiation (32). Coupled with the well-established capacity of DDX5 to bind lncRNAs, these findings raise the possibility that lncRNAs, such as Malat1, may guide DDX5/PRC2 activity for sequence-specific silencing of TCM cell-associated transcripts in TE and t-TEM cells. Future studies exploring the putative relationships between DDX5, PRC2, and lncRNAs may reveal previously unknown mechanisms of sequence-specific PRC2-directed CD8+ T cell differentiation.
The possibility of DDX5-mediated repression of the TCM cell-associated transcriptional program may be especially pertinent in the context of chronic infections and cancer. In the current study, deletion of DDX5 appeared to de-repress Tcf7 expression, potentially allowing for the preferential differentiation of TCM cells. The ability of TCF1 to preserve cellular plasticity has recently garnered a lot of attention, as many groups have shown that this property is essential for maintaining a stem-like population of CD8+ T cells that may play a critical role in chronic infections and cancer (29, 30, 31, 32). Furthermore, high expression of DDX5 has been correlated with aggressive tumor features, including treatment resistance, and has been shown to be predictive of poor clinical outcomes in many cancer types (33, (38). Together, these insights suggest that targeting DDX5 in cancer may inhibit DDX5-driven tumorigenesis and drug-resistance within the tumor, as well as potentially skewing the immune response towards TCF1-expressing stem-like CD8+ T cells. Future studies evaluating the efficacy of DDX5-deficient CD8+ T cells in controlling tumor growth may provide support for DDX5 as a target for cancer therapy.
Overall, our study has identified a role for DDX5 in promoting the differentiation of TE, TEM, and t-TEM cells, potentially through repression of TCM cell-associated gene expression. These findings advance our understanding of the role of RNA-binding proteins in regulating CD8+ T cell differentiation and serve as the basis for future studies aimed at improving CD8+ T cell responses in infection and cancer.
Supplementary Material
KEY POINTS.
The RNA-binding protein DDX5 regulates CD8+ T cell differentiation.
DDX5 represses the generation of central memory CD8+ T (TCM) cells.
DDX5-deficient T cells exhibit increased expression of TCM-associated genes.
Acknowledgments
CITE-seq using the 10x Genomics platform was performed at the UCSD IGM Genomics Center and supported by NIH grants P30KC063491, P30CA023100, and S10OD026929. This work was supported by the NIDDK-funded San Diego Digestive Diseases Research Center (P30DK120515) and funded by grants from the NIH: AI132122, AI123202, AI129973, BX005106, and CX002396 (J.T.C.); and GM124494 (W.J.M.H.).
REFERENCES
- 1.Chang JT, Wherry EJ, and Goldrath AW. 2014. Molecular regulation of effector and memory T cell differentiation. Nat. Immunol 15: 1104–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jameson SC, and Masopust D. 2018. Understanding Subset Diversity in T Cell Memory. Immunity 48: 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sallusto F, Lenig D, Förster R, Lipp M, and Lanzavecchia A. 1999. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401: 708–712. [DOI] [PubMed] [Google Scholar]
- 4.Hamann D, Baars PA, Rep MH, Hooibrink B, Kerkhof-Garde SR, Klein MR, and van Lier RA. 1997. Phenotypic and functional separation of memory and effector human CD8+ T cells. J. Exp. Med 186: 1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milner JJ, Nguyen H, Omilusik K, Reina-Campos M, Tsai M, Toma C, Delpoux A, Boland BS, Hedrick SM, Chang JT, and Goldrath AW. 2020. Delineation of a molecularly distinct terminally differentiated memory CD8 T cell population. Proc. Natl. Acad. Sci 117: 25667–25678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou X, Yu S, Zhao D-M, Harty JT, Badovinac VP, and Xue H-H. 2010. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 33: 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li D, Liu J, Yang X, Zhou C, Guo J, Wu C, Qin Y, Guo L, He J, Yu S, Liu H, Wang X, Wu F, Kuang J, Hutchins AP, Chen J, and Pei D. 2017. Chromatin Accessibility Dynamics during iPSC Reprogramming. Cell Stem Cell 21: 819–833.e6. [DOI] [PubMed] [Google Scholar]
- 8.Shigunov P, and Dallagiovanna B. 2015. Stem Cell Ribonomics: RNA-Binding Proteins and Gene Networks in Stem Cell Differentiation. Front. Mol. Biosci 2. [DOI] [PMC free article] [PubMed]
- 9.Kurd NS, He Z, Louis TL, Milner JJ, Omilusik KD, Jin W, Tsai MS, Widjaja CE, Kanbar JN, Olvera JG, Tysl T, Quezada LK, Boland BS, Huang WJ, Murre C, Goldrath AW, Yeo GW, and Chang JT. 2020. Early precursors and molecular determinants of tissue-resident memory CD8+ T lymphocytes revealed by single-cell RNA sequencing. Sci. Immunol 5: eaaz6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma S, Zhou B, Abbasi N, Luo C, Yeo GW, Fu X, and Huang WJM. 2019. DDX5 regulates chromatin lncRNAs to direct T cell programs. J. Immunol 202: 125.20–125.20. [Google Scholar]
- 11.Giraud G, Terrone S, and Bourgeois CF. 2018. Functions of DEAD box RNA helicases DDX5 and DDX17 in chromatin organization and transcriptional regulation. BMB Rep 51: 613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xing Z, Wang S, and Tran EJ. 2017. Characterization of the mammalian DEAD-box protein DDX5 reveals functional conservation with S. cerevisiae ortholog Dbp2 in transcriptional control and glucose metabolism. RNA 23: 1125–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Panchbhai N, Turaga RC, Sharma M, Satyanarayana G, and Liu Z-R. 2021. P68 RNA Helicase facilitates Breast Cancer progression by promoting Proliferation and Migration via PDGFR-β/AR axis. J. Cancer 12: 6543–6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bates GJ, Nicol SM, Wilson BJ, Jacobs A-MF, Bourdon J-C, Wardrop J, Gregory DJ, Lane DP, Perkins ND, and Fuller-Pace FV. 2005. The DEAD box protein p68: a novel transcriptional coactivator of the p53 tumour suppressor. EMBO J 24: 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marzo AL, Klonowski KD, Bon AL, Borrow P, Tough DF, and Lefrançois L. 2005. Initial T cell frequency dictates memory CD8+ T cell lineage commitment. Nat. Immunol 6: 793–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, and Smibert P. 2017. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14: 865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kakaradov B, Arsenio J, Widjaja CE, He Z, Aigner S, Metz PJ, Yu B, Wehrens EJ, Lopez J, Kim SH, Zuniga EI, Goldrath AW, Chang JT, and Yeo GW. 2017. Early transcriptional and epigenetic regulation of CD8+ T cell differentiation revealed by single-cell RNA sequencing. Nat. Immunol 18: 422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Milner JJ, Toma C, He Z, Kurd NS, Nguyen QP, McDonald B, Quezada L, Widjaja CE, Witherden DA, Crowl JT, Shaw LA, Yeo GW, Chang JT, Omilusik KD, and Goldrath AW. 2020. Heterogenous Populations of Tissue-Resident CD8+ T Cells Are Generated in Response to Infection and Malignancy. Immunity 52: 808–824.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin R, Straub AU, Doebele C, and Bohnsack MT. 2013. DExD/H-box RNA helicases in ribosome biogenesis. RNA Biol 10: 4–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turner M, and Díaz-Muñoz MD. 2018. RNA-binding proteins control gene expression and cell fate in the immune system. Nat. Immunol 19: 120–129. [DOI] [PubMed] [Google Scholar]
- 21.Petkau G, Mitchell TJ, Chakraborty K, Bell SE, D′Angeli V, Matheson L, Turner DJ, Saveliev A, Gizlenci O, Salerno F, Katsikis PD, and Turner M 2022. The timing of differentiation and potency of CD8 effector function is set by RNA binding proteins. Nat. Commun 13: 2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yao H, Brick K, Evrard Y, Xiao T, Camerini-Otero RD, and Felsenfeld G. 2010. Mediation of CTCF transcriptional insulation by DEAD-box RNA-binding protein p68 and steroid receptor RNA activator SRA. Genes Dev 24: 2543–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dardenne E,; Polay Espinoza M, Fattet L, Germann S, Lambert M-P, Neil H, Zonta E, Mortada H, Gratadou L, Deygas M, Chakrama FZ, Samaan S, Desmet F-O, Tranchevent L-C, Dutertre M, Rimokh R, Bourgeois CF, and Auboeuf D. 2014. RNA Helicases DDX5 and DDX17 Dynamically Orchestrate Transcription, miRNA, and Splicing Programs in Cell Differentiation. Cell Rep 7: 1900–1913. [DOI] [PubMed] [Google Scholar]
- 24.Fuller-Pace FV 2013. The DEAD box proteins DDX5 (p68) and DDX17 (p72): multi-tasking transcriptional regulators. Biochim. Biophys. Acta 1829: 756–763. [DOI] [PubMed] [Google Scholar]
- 25.Steinke FC, Yu S, Zhou X, He B, Yang W, Zhou B, Kawamoto H, Zhu J, Tan K, and Xue H-H. 2014. TCF-1 and LEF-1 act upstream of Th-POK to promote the CD4(+) T cell fate and interact with Runx3 to silence Cd4 in CD8(+) T cells. Nat. Immunol 15: 646–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu Z, Xing S, Shan Q, Gullicksrud JA, Bair TB, Du Y, Liu C, and Xue H-H. 2017. Cutting Edge: β-Catenin–Interacting Tcf1 Isoforms Are Essential for Thymocyte Survival but Dispensable for Thymic Maturation Transitions. J. Immunol 198: 3404–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dardenne E, Pierredon S, Driouch K, Gratadou L, Lacroix-Triki M, Espinoza MP, Zonta E, Germann S, Mortada H, Villemin J-P, Dutertre M, Lidereau R, Vagner S, and Auboeuf D. 2012. Splicing switch of an epigenetic regulator by RNA helicases promotes tumor-cell invasiveness. Nat. Struct. Mol. Biol 19: 1139–1146. [DOI] [PubMed] [Google Scholar]
- 28.Germann S, Gratadou L, Zonta E, Dardenne E, Gaudineau B, Fougère M, Samaan S, Dutertre M, Jauliac S, and Auboeuf D. 2012. Dual role of the ddx5/ddx17 RNA helicases in the control of the pro-migratory NFAT5 transcription factor. Oncogene 31: 4536–4549. [DOI] [PubMed] [Google Scholar]
- 29.Guil S, Gattoni R, Carrascal M, Abián J, Stévenin J, and Bach-Elias M. 2003. Roles of hnRNP A1, SR Proteins, and p68 Helicase in c-H-ras Alternative Splicing Regulation. Mol. Cell. Biol 23: 2927–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xing S, Li F, Zeng Z, Zhao Y, Yu S, Shan Q, Li Y, Phillips FC, Maina PK, Qi HH, Liu C, Zhu J, Pope RM, Musselman CA, Zeng C, Peng W, and Xue H-H. 2016. Tcf1 and Lef1 transcription factors establish CD8(+) T cell identity through intrinsic HDAC activity. Nat. Immunol 17: 695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fiorenzano A, Pascale E, Patriarca EJ, Minchiotti G, and Fico A. 2019. LncRNAs and PRC2: Coupled Partners in Embryonic Stem Cells. Epigenomes 3: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanbar JN, Ma S, Kim ES, Kurd NS, Tsai MS, Tysl T, Widjaja CE, Limary AE, Yee B, He Z, Hao Y, Fu X-D, Yeo GW, Huang WJ, and Chang JT. 2022. The long noncoding RNA Malat1 regulates CD8+ T cell differentiation by mediating epigenetic repression. J. Exp. Med 219: e20211756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, Pradervand S, Thimme R, Zehn D, and Held W. 2016. T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 45: 415–427. [DOI] [PubMed] [Google Scholar]
- 34.Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, Anderson SM, Handon R, Kenyon E, Elkahloun A, Wu W, Lang PA, Gattinoni L, McGavern DB, and Schwartzberg PL. 2016. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol. 1: eaai8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, Sharpe AH, Freeman GJ, Germain RN, Nakaya HI, Xue H-H, and Ahmed R. 2016. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537: 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, Johnson J, Staupe RP, Bengsch B, Xu C, Yu S, Kurachi M, Herati RS, Vella LA, Baxter AE, Wu JE, Khan O, Beltra J-C, Giles JR, Stelekati E, McLane LM, Lau CW, Yang X, Berger SL, Vahedi G, Ji H, and Wherry EJ. 2019. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity 51: 840–855.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le TK, Cherif C, Omabe K, Paris C, Lannes F, Audebert S, Baudelet E, Hamimed M, Barbolosi D, Finetti P, Bastide C, Fazli L, Gleave M, Bertucci F, Taïeb D, and Rocchi P. 2022. DDX5 mRNA-targeting antisense oligonucleotide as a new promising therapeutic in combating castration-resistant prostate cancer. Mol. Ther. [DOI] [PMC free article] [PubMed]
- 38.Abbasi N, Long T, Li Y, Yee BA, Cho BS, Hernandez JE, Ma E, Patel PR, Sahoo D, Sayed IM, Varki N, Das S, Ghosh P, Yeo GW, and Huang WJM. 2020. DDX5 promotes oncogene C3 and FABP1 expressions and drives intestinal inflammation and tumorigenesis. Life Sci. Alliance 3. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.