

Abstract
Since the completion of the human genome project in 2003, extraordinary progress has been made in genome sequencing technologies, which has led to a decreased cost per megabase and an increase in the number and diversity of sequenced genomes. An astonishing complexity of genome architecture has been revealed, bringing these sequencing technologies to even greater advancements. Some approaches maximize the number of bases sequenced in the least amount of time, generating a wealth of data that can be used to understand increasingly complex phenotypes. Alternatively, other approaches now aim to sequence longer contiguous pieces of DNA, which are essential for resolving structurally complex regions. These and other strategies are providing researchers and clinicians a variety of tools to probe genomes in greater depth, leading to an enhanced understanding of how genome sequence variants underlie phenotype and disease.
Starting with the discovery of the structure of DNA1, great strides have been made in understanding the complexity and diversity of genomes in health and disease. A multitude of innovations in reagents and instrumentation supported the initiation of the Human Genome Project2. Its completion revealed the need for greater and more advanced technologies and data sets to answer the complex biological questions that arose; however, limited throughput and the high costs of sequencing remained major barriers. The release of the first truly high-throughput sequencing platform in the mid-2000s heralded a 50,000-fold drop in the cost of human genome sequencing since the Human Genome Project3 and led to the moniker: next-generation sequencing (NGS). Over the past decade, NGS technologies have continued to evolve — increasing capacity by a factor of 100–1,000 (REF. 4) — and have incorporated revolutionary innovations to tackle the complexities of genomes. These advances are providing read lengths as long as some entire genomes, they have brought the cost of sequencing a human genome down to around US$1,000 (as reported by Veritas Genomics)5, and they have enabled the use of sequencing as a clinical tool3,6.
Although exciting, these advancements are not without limitations. As new technologies emerge, existing problems are exacerbated or new problems arise. NGS platforms provide vast quantities of data, but the associated error rates (~0.1–15%) are higher and the read lengths generally shorter (35–700 bp for short-read approaches)7 than those of traditional Sanger sequencing platforms, requiring careful examination of the results, particularly for variant discovery and clinical applications. Although long-read sequencing overcomes the length limitation of other NGS approaches, it remains considerably more expensive and has lower throughput than other platforms, limiting the widespread adoption of this technology in favour of less-expensive approaches. Finally, NGS is also competing with alternative technologies that can carry out similar tasks, often at lower cost (BOX 1); it is not clear how these disparate approaches to genomics, medicine and research will interact in the years to come.
Box 1 |. Alternative genomic strategies.
There are other technologies that either compete with or complement next-generation sequencing (NGS). This section outlines these technologies and their relationship with NGS.
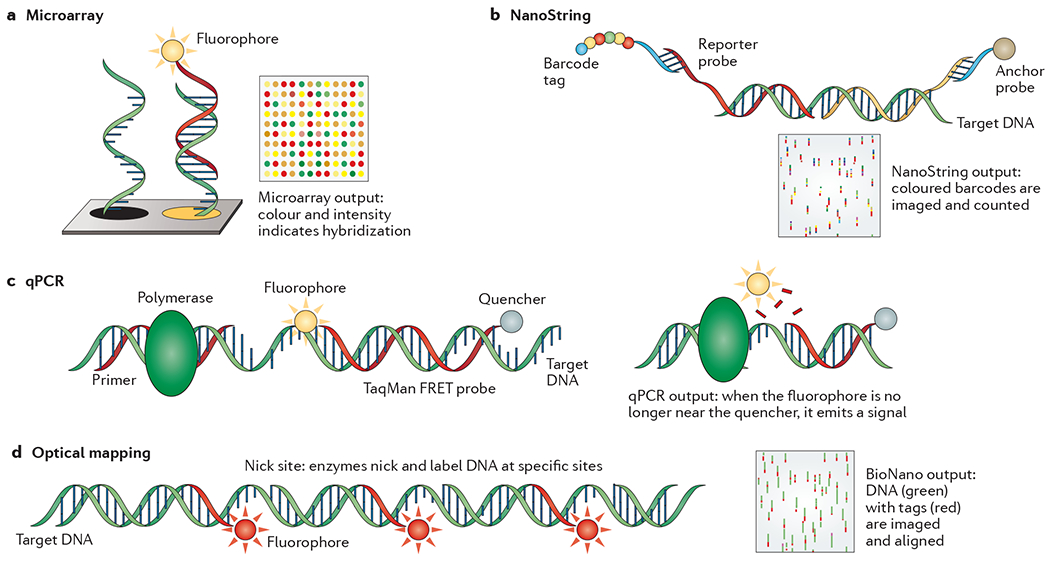
DNA microarrays
DNA microarrays have been used for genetic research since the early 1980s122 (see the figure, part a). In DNA microarrays, single-stranded DNA (ssDNA) probes are immobilized on a substrate in a discrete location with spots as small as 50 μm123. Target DNA is labelled with a fluorophore and hybridized to the array. The intensity of the signal is used to determine the number of bound molecules.
Microarrays are used in many applications. Single-nucleotide polymorphism (SNP) arrays identify common polymorphisms associated with disease and phenotypes, including cardiovascular disease124, cancer125–127, pathogens128,129, ethnicity130,131 and genome-wide association study (GWAS) analysis132,133. Additionally, lower-resolution arrays are used to identify structural variation, copy number variants (CNVs) and DNA–protein interactions134–137. Expression arrays measure expression levels by measuring the amount of gene-specific cDNA138.
Microarrays remain widely used in genomic research. They are used to identify SNPs at costs far below NGS routines. This is also true for expression studies, in which arrays inexpensively measure expression levels of thousands of genes. Variations in hybridization and normalization are problematic, leading some people to recommend RNA sequencing (RNA-seq) over gene expression microarrays139.
NanoString
Similar to microarrays, the nCounter Analysis System from NanoString relies on target-probe hybridization (see the figure, part b). Probes target a gene of interest; one probe is bound to a fluorophore ‘barcode’ and the other anchors the target for imaging. The number and type of each barcode is counted. NanoString is unique in that the probes are labelled molecules that are bound together in a discrete order, which can be changed to create hundreds of different labels.
nCounter applications are similar to those of microarrays and quantitative PCR (qPCR; see below), including gene expression analysis145,146, CNV and SNP detection147,148, and fusion gene detection149. This approach provides exceptionally high resolution, less than one copy per cell145, far below microarrays and approaching TaqMan in sensitivity. Unlike most NGS applications, neither template enrichment nor reverse transcription are required. Around 800 targets can be read at a time, far below either microarrays or NGS.
qPCR
Real-time qPCR utilizes the PCR reaction to detect targets of interest (see the figure, part c). Gene-specific primers are used and the target is detected either by the incorporation of a double-stranded DNA (dsDNA)-specific dye or by the release of a TaqMan FRET (fluorescence resonance energy transfer) probe through polymerase 5′–3′ exonuclease activity.
Developed in the early 1990s140, qPCR is widely used in both clinical and research settings for genotyping141, gene expression analysis142, CNV assays143 and pathogen detection144. qPCR is extremely rapid and robust, which is beneficial for point-of-care applications. Its high sensitivity and specificity make it the gold standard for clinical gene detection with several US Food and Drug Administration (FDA)-approved tests. The number of simultaneous targets that can be detected is in the hundreds rather than the thousands for microarrays and NGS. This method also requires primers and/or probes designed for specific targets.
Optical mapping
Optical mapping combines long-read technology with low-resolution sequencing (see the figure, part d). Originally a method for ordering restriction enzyme sites150 through digestion and size separation, this technology now uses fluorescent markers to tag particular sequences within DNA fragments that are up to ~1 Mb long. The results are imaged and aligned to each other, and/or a reference, to map the locations of the probes relative to each other.
A central application of this technology is the generation of genome maps that are used in de novo assembly and gap filling94,151. This technology can be used to detect structural variations that are up to tens of kilobases in length94,152. Haplotype blocks that are several hundred kilobases in size can also be resolved153.
Optical mapping can either be an alternative to NGS or a complementary approach. As an alternative, it provides a low-cost option for understanding structural and copy number variation, but it does not provide base-level resolution. As a complementary technology, optical mapping improves de novo genome assemblies by providing a long-range scaffold on which to align short-read data.
This Review evaluates various approaches used in NGS and how recent advancements in the field are changing the way genetic research is carried out. Details of each approach along with its benefits and drawbacks are discussed. Finally, various emerging applications within this field and its exciting future are explored.
Short-read NGS
Overview of clonal template generation approaches.
Short-read sequencing approaches fall under two broad categories: sequencing by ligation (SBL) and sequencing by synthesis (SBS). In SBL approaches, a probe sequence that is bound to a fluorophore hybridizes to a DNA fragment and is ligated to an adjacent oligonucleotide for imaging. The emission spectrum of the fluorophore indicates the identity of the base or bases complementary to specific positions within the probe. In SBS approaches, a polymerase is used and a signal, such as a fluorophore or a change in ionic concentration, identifies the incorporation of a nucleotide into an elongating strand. In most SBL and SBS approaches, DNA is clonally amplified on a solid surface. Having many thousands of identical copies of a DNA fragment in a defined area ensures that the signal can be distinguished from background noise. Massive parallelization is also facilitated by the creation of many millions of individual SBL or SBS reaction centres, each with its own clonal DNA template. A sequencing platform can collect information from many millions of reaction centres simultaneously, thus sequencing many millions of DNA molecules in parallel.
There are several different strategies used to generate clonal template populations: bead-based, solid-state and DNA nanoball generation (FIG. 1). The first step of DNA template generation is fragmentation of the sample DNA, followed by ligation to a common adaptor set for clonal amplification and sequencing. For bead-based preparations, one adaptor is complementary to an oligonucleotide fragment that is immobilized on a bead (FIG. 1a). Using emulsion PCR (emPCR)8, the DNA template is amplified such that as many as one million clonal DNA fragments are immobilized on a single bead9. These beads can be distributed onto a glass surface10 or arrayed on a PicoTiterPlate (Roche Diagnostics)11. Solid-state amplification12 eschews the use of emPCR in favour of amplification directly on a slide13 (FIG. 1b,c). In this approach, forward and reverse primers are covalently bound to the slide surface, either randomly or on a patterned slide. These primers provide complementary ends to which single-stranded DNA (ssDNA) templates can bind. Precise control over template concentration enables the amplification of templates into localized, non-overlapping clonal clusters, thus maintaining spatial integrity. Recently, several NGS platforms have utilized patterned flow cells. By defining precisely where primers are bound to the slide, more DNA templates can be spatially resolved, enabling higher densities of reaction centre clusters and increasing sequencing throughput.
Figure 1 |. Template amplification strategies.
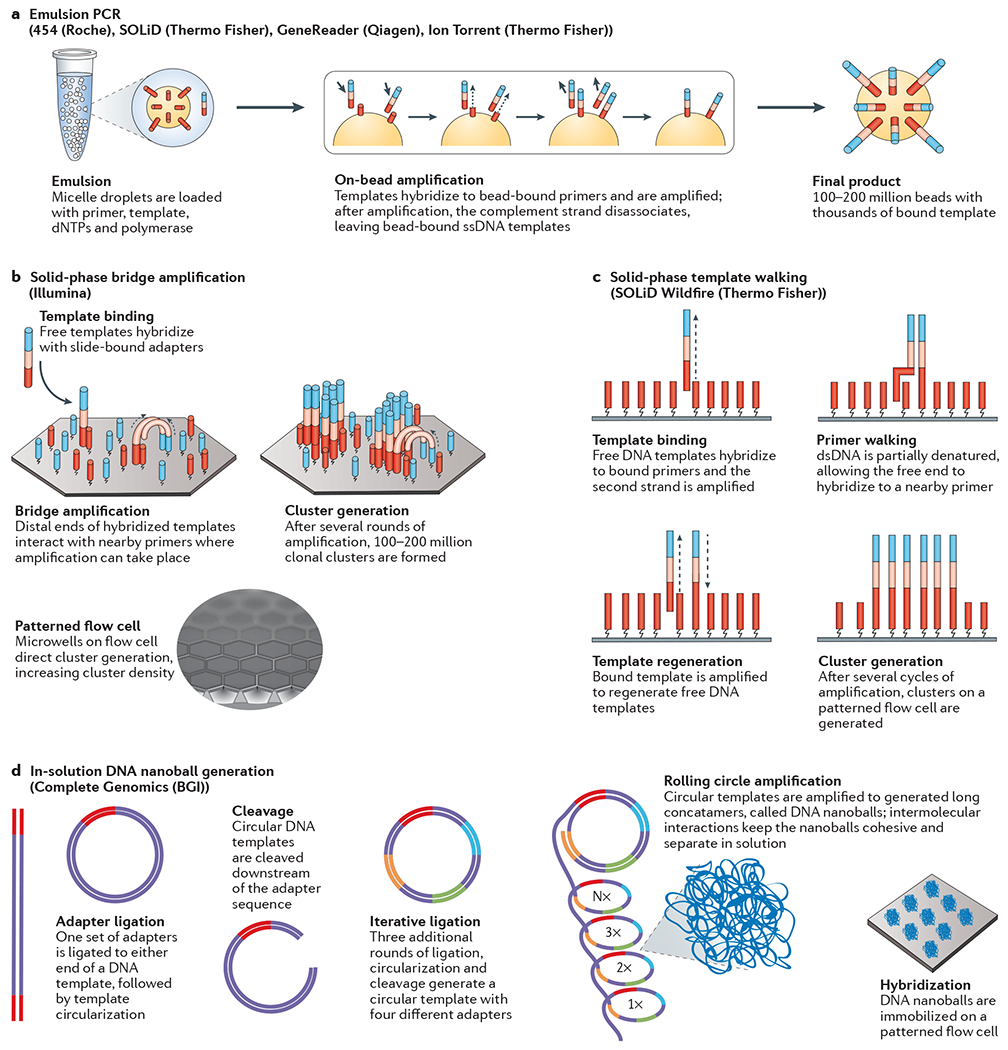
a | In emulsion PCR, fragmented DNA templates are ligated to adapter sequences and are captured in an aqueous droplet (micelle) along with a bead covered with complementary adapters, deoxynucleotides (dNTPs), primers and DNA polymerase. PCR is carried out within the micelle, covering each bead with thousands of copies of the same DNA sequence. b | In solid-phase bridge amplification, fragmented DNA is ligated to adapter sequences and bound to a primer immobilized on a solid support, such as a patterned flow cell. The free end can interact with other nearby primers, forming a bridge structure. PCR is used to create a second strand from the immobilized primers, and unbound DNA is removed. c | In solid-phase template walking154, fragmented DNA is ligated to adapters and bound to a complementary primer attached to a solid support. PCR is used to generate a second strand. The now double-stranded template is partially denatured, allowing the free end of the original template to drift and bind to another nearby primer sequence. Reverse primers are used to initiate strand displacement to generate additional free templates, each of which can bind to a new primer. d | In DNA nanoball generation, DNA is fragmented and ligated to the first of four adapter sequences. The template is amplified, circularized and cleaved with a type II endonuclease. A second set of adapters is added, followed by amplification, circularization and cleavage. This process is repeated for the remaining two adapters. The final product is a circular template with four adapters, each separated by a template sequence. Library molecules undergo a rolling circle amplification step, generating a large mass of concatamers called DNA nanoballs, which are then deposited on a flow cell. Parts a and b are adapted from REF. 18, Nature Publishing Group.
The Complete Genomics technology used by the Beijing Genomics Institute (BGI) is currently the only approach that achieves template enrichment in solution. In this case, DNA undergoes an iterative ligation, circularization and cleavage process to create a circular template, with four distinct adaptor regions. Through the process of rolling circle amplification (RCA), up to 20 billion discrete DNA nanoballs are generated (FIG. 1d). The nanoball mixture is then distributed onto a patterned slide surface containing features that allow a single nanoball to associate with each location14.
Sequencing by ligation (SOLiD and Complete Genomics).
Fundamentally, SBL approaches involve the hybridization and ligation15 of labelled probe and anchor sequences to a DNA strand. The probes encode one or two known bases (one-base-encoded probes or two-base-encoded probes) and a series of degenerate or universal bases, driving complementary binding between the probe and template, whereas the anchor fragment encodes a known sequence that is complementary to an adapter sequence and provides a site to initiate ligation. After ligation, the template is imaged and the known base or bases in the probe are identified16. A new cycle begins after complete removal of the anchor–probe complex or through cleavage to remove the fluorophore and to regenerate the ligation site.
The SOLiD platform utilizes two-base-encoded probes, in which each fluorometric signal represents a dinucleotide17. Consequently, the raw output is not directly associated with the incorporation of a known nucleotide. Because the 16 possible dinucleotide combinations cannot be individually associated with spectrally resolvable fluorophores, four fluorescent signals are used, each representing a subset of four dinucleotide combinations. Thus, each ligation signal represents one of several possible dinucleotides, leading to the term colour-space (rather than base-space), which must be deconvoluted during data analysis. The SOLiD sequencing procedure is composed of a series of probe–anchor binding, ligation, imaging and cleavage cycles to elongate the complementary strand (FIG. 2a). Over the course of the cycles, single-nucleotide offsets are introduced to ensure every base in the template strand is sequenced.
Figure 2 |. Sequencing by ligation methods.
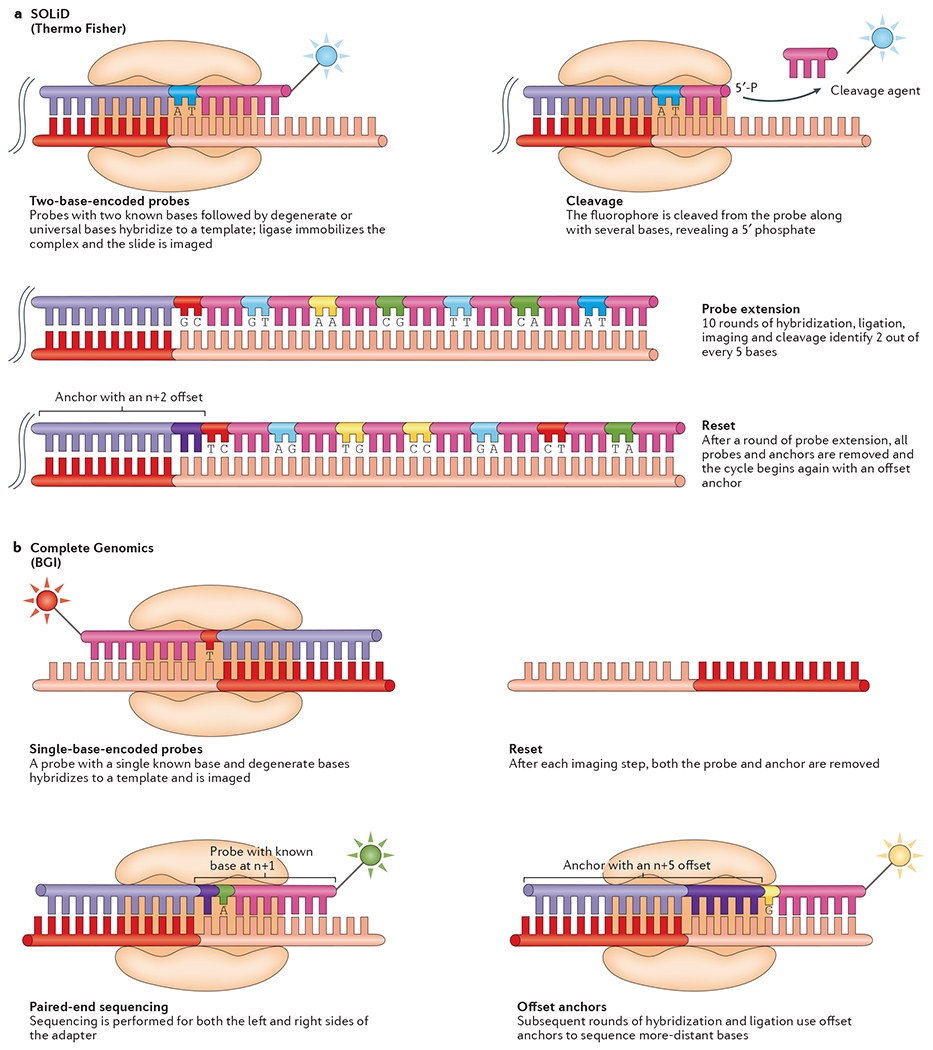
a | SOLiD sequencing. Following cluster generation or bead deposition onto a slide, fragments are sequenced by ligation, in which a fluorophore-labelled two-base-encoded probe, which is composed of known nucleotides in the first and second positions (dark blue), followed by degenerate or universal bases (pink), is added to the DNA library. The two-base probe is ligated onto an anchor (light purple) that is complementary to an adapter (red), and the slide is imaged to identify the first two bases in each fragment. Unextended strands are capped by unlabelled probes or phosphatase to maintain cycle synchronization. Finally, the terminal degenerate bases and the fluorophore are cleaved off the probe, leaving a 5 bp extended fragment. The process is repeated ten times until two out of every five bases are identified. At this point, the entire strand is reset by removing all of the ligated probes and the process of probe binding, ligation, imaging and cleavage is repeated four times, each with an n + 1, n + 2, n + 3 or n + 4 offset anchor. b | Complete Genomics. DNA is sequenced using the combinatorial probe–anchor ligation (cPAL) approach. After DNA nanoball deposition, an anchor complementary to one of four adapter sequences and a fluorophore-labelled probe are bound to each nanoball. The probe is degenerate at all but the first position. The anchor and probe are then ligated into position and imaged to identify the first base on either the 3′ or the 5′ side of the anchor. Next, the probe–anchor complex is removed and the process begins again with the same anchor but a different probe with the known base at the n + 1 position. This is repeated until five bases from the 3′ end of the anchor and five bases from the 5′ end of the anchor are identified. Another round of hybridization occurs, this time using anchors with a five-base offset identifying an additional five bases on either side of the anchor. Finally, this whole process is repeated for each of the remaining three adapter sequences in the nanoball, generating 100 bp paired-end reads.
Complete Genomics performs DNA sequencing using combinatorial probe–anchor ligation (cPAL)14 or combinatorial probe–anchor synthesis (cPAS; see the BGISEQ-500 website). In cPAL (FIG. 2b), an anchor sequence (complementary to one of the four adaptor sequences) and a probe hybridize to a DNA nanoball at several locations. In each cycle, the hybridizing probe is a member of a pool of one-base-encoded probes, in which each probe contains a known base in a constant position and a corresponding fluorophore. After imaging, the entire probe–anchor complex is removed and a new probe–anchor combination is hybridized. Each subsequent cycle utilizes a probe set with the known base in the n + 1 position. Further cycles in the process also use adaptors of variable lengths and chemistries, allowing sequencing to occur upstream and downstream of the adaptor sequence. The cPAS approach is a modification of cPAL intended to increase read lengths of Complete Genomics’ chemistry; however, at present, details about the approach are limited.
Sequencing-by-synthesis categories.
SBS is a term used to describe numerous DNA-polymerase-dependent methods in the literature, but it does not delineate the different mechanisms involved in SBS approaches. For this article, SBS approaches will be classified either as cyclic reversible termination (CRT) or as single-nucleotide addition (SNA)18.
Sequencing by synthesis: CRT (Illumina, Qiagen).
CRT approaches are defined by their use of terminator molecules that are similar to those used in Sanger sequencing, in which the ribose 3′-OH group is blocked, thus preventing elongation19,20. To begin the process, a DNA template is primed by a sequence that is complementary to an adapter region, which will initiate polymerase binding to this double-stranded DNA (dsDNA) region. During each cycle, a mixture of all four individually labelled and 3′-blocked deoxynucleotides (dNTPs) are added. After the incorporation of a single dNTP to each elongating complementary strand, unbound dNTPs are removed and the surface is imaged to identify which dNTP was incorporated at each cluster. The fluorophore and blocking group can then be removed and a new cycle can begin.
The Illumina CRT system (FIG. 3a) accounts for the largest market share for sequencing instruments compared to other platforms21. Illumina’s suite of instruments for short-read sequencing range from small, low-throughput benchtop units to large ultra-high-throughput instruments dedicated to population-level whole-genome sequencing (WGS). dNTP identification is achieved through total internal reflection fluorescence (TIRF) microscopy using either two or four laser channels. In most Illumina platforms, each dNTP is bound to a single fluorophore that is specific to that base type and requires four different imaging channels, whereas the NextSeq and Mini-Seq systems use a two-fluorophore system.
Figure 3 |. Sequencing by synthesis: cyclic reversible termination approaches.
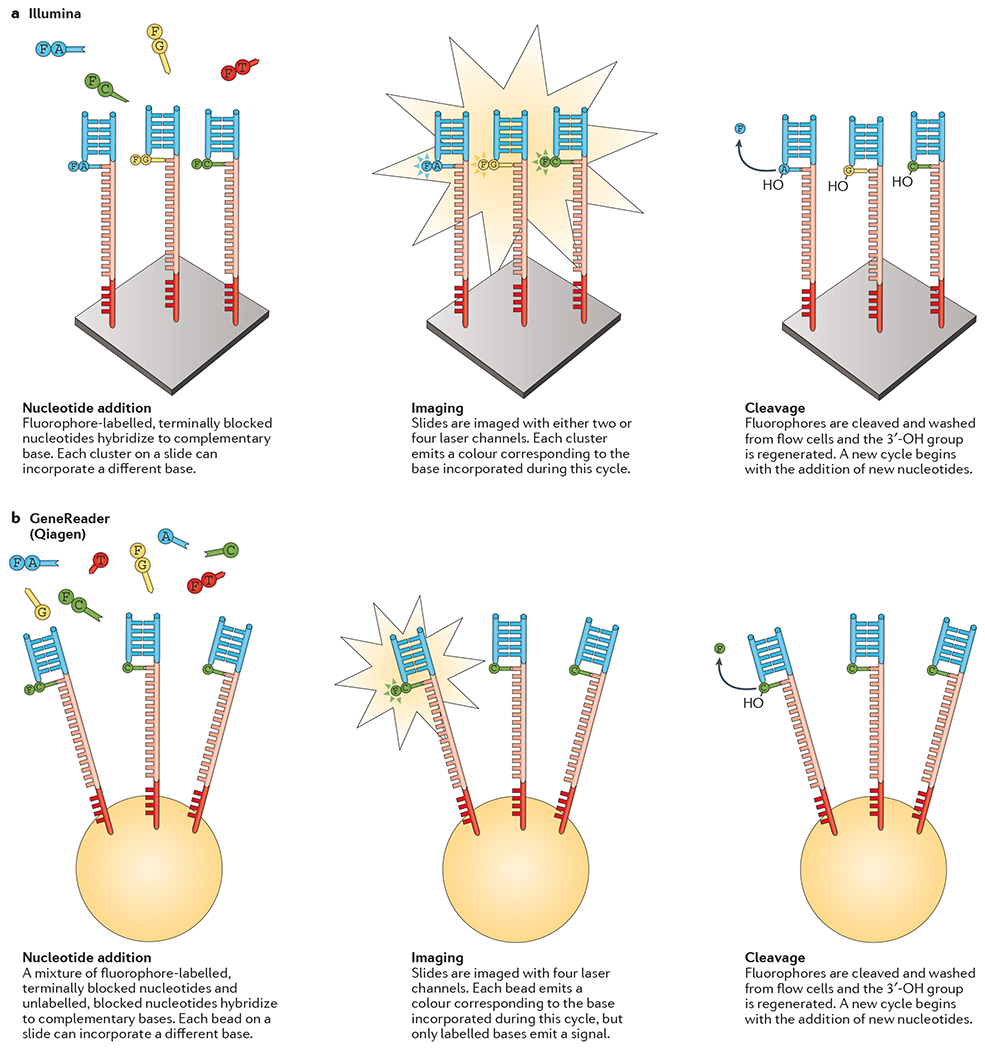
a | Illumina. After solid-phase template enrichment, a mixture of primers, DNA polymerase and modified nucleotides are added to the flow cell. Each nucleotide is blocked by a 3′-O-azidomethyl group and is labelled with a base-specific, cleavable fluorophore (F). During each cycle, fragments in each cluster will incorporate just one nucleotide as the blocked 3′ group prevents additional incorporations. After base incorporation, unincorporated bases are washed away and the slide is imaged by total internal reflection fluorescence (TIRF) microscopy using either two or four laser channels; the colour (or the lack or mixing of colours in the two-channel system used by NextSeq) identifies which base was incorporated in each cluster. The dye is then cleaved and the 3′-OH is regenerated with the reducing agent tris(2-carboxyethyl)phosphine (TCEP). The cycle of nucleotide addition, elongation and cleavage can then begin again. b | Qiagen. After bead-based template enrichment, a mixture of primers, DNA polymerase and modified nucleotides are added to the flow cell. Each nucleotide is blocked by a 3′-O-allyl group and some of the bases are labelled with a base-specific, cleavable fluorophore. After base incorporation, unincorporated bases are washed away and the slide is imaged by TIRF using four laser channels. The dye is then cleaved and the 3′-OH is regenerated with the reducing agent mixture of palladium and P(PhSO3Na)3 (TPPTS).
In 2012, Qiagen acquired the Intelligent BioSystems CRT platform, which was commercialized and relaunched in 2015 as the GeneReader22 (FIG. 3b). Unlike other systems, this platform is intended to be an all-in-one NGS platform, from sample preparation to analysis. To accomplish this, the GeneReader system is bundled with the QIAcube sample preparation system and the Qiagen Clinical Insight platform for variant analysis. The GeneReader uses virtually the same approach as that used by Illumina; however, it does not aim to ensure that each template incorporates a fluorophore-labelled dNTP23. Rather, GeneReader aims to ensure that just enough labelled dNTPs are incorporated to achieve identification.
Sequencing by synthesis: SNA (454, Ion Torrent).
Unlike CRT, SNA approaches rely on a single signal to mark the incorporation of a dNTP into an elongating strand. As a consequence, each of the four nucleotides must be added iteratively to a sequencing reaction to ensure only one dNTP is responsible for the signal. Furthermore, this does not require the dNTPs to be blocked, as the absence of the next nucleotide in the sequencing reaction prevents elongation. The exception to this is homopolymer regions where identical dNTPs are added, with sequence identification relying on a proportional increase in the signal as multiple dNTPs are incorporated.
The first NGS instrument developed was the 454 pyrosequencing24 device. This SNA system distributes template-bound beads into a PicoTiterPlate along with beads containing an enzyme cocktail. As a dNTP is incorporated into a strand, an enzymatic cascade occurs, resulting in a bioluminescence signal. Each burst of light, detected by a charge-coupled device (CCD) camera, can be attributed to the incorporation of one or more identical dNTPs at a particular bead (FIG. 4a).
Figure 4 |. Sequencing by synthesis: single-nucleotide addition approaches.
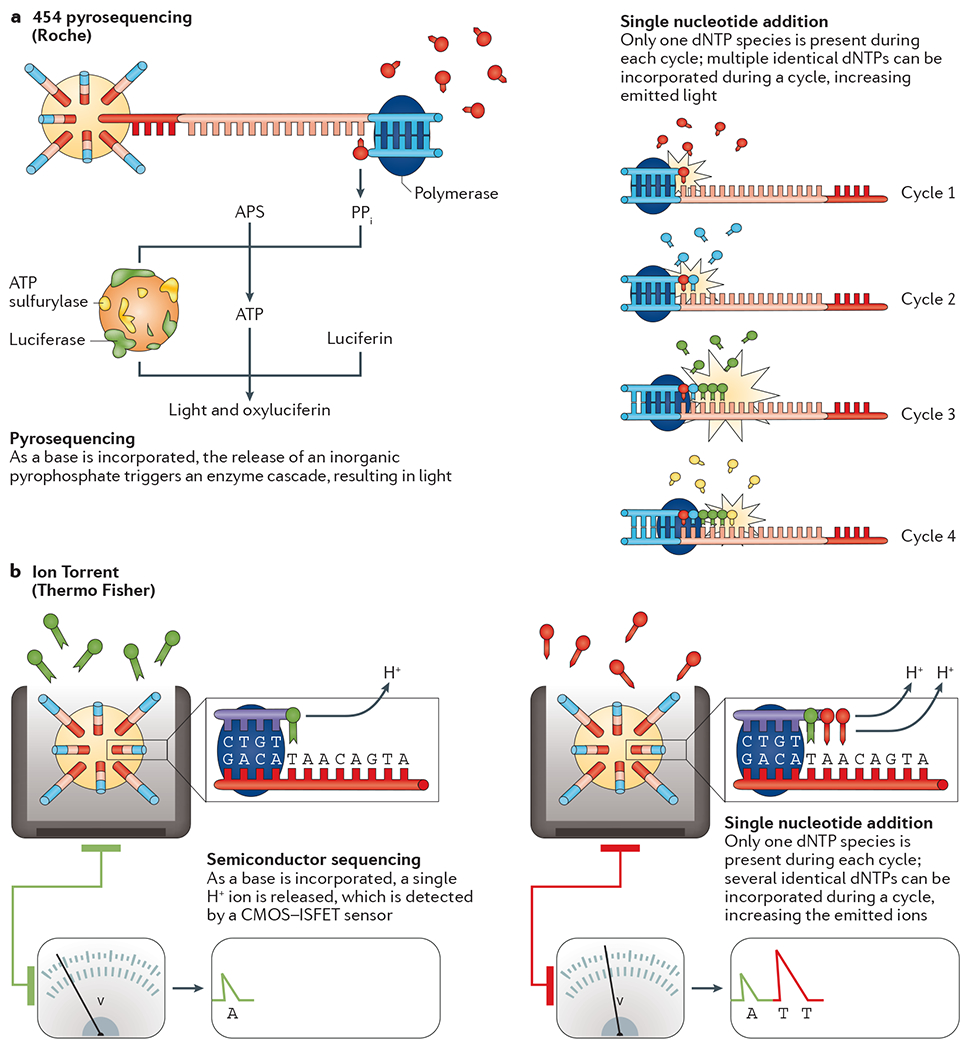
a | 454 pyrosequencing. After bead-based template enrichment, the beads are arrayed onto a microtitre plate along with primers and different beads that contain an enzyme cocktail. During the first cycle, a single nucleotide species is added to the plate and each complementary base is incorporated into a newly synthesized strand by a DNA polymerase. The by-product of this reaction is a pyrophosphate molecule (PPi). The PPi molecule, along with ATP sulfurylase, transforms adenosine 5′ phosphosulfate (APS) into ATP. ATP, in turn, is a cofactor for the conversion of luciferin to oxyluciferin by luciferase, for which the by-product is light. Finally, apyrase is used to degrade any unincorporated bases and the next base is added to the wells. Each burst of light, detected by a charge-coupled device (CCD) camera, can be attributed to the incorporation of one or more bases at a particular bead. b | Ion Torrent. After bead-based template enrichment, beads are carefully arrayed into a microtitre plate where one bead occupies a single reaction well. Nucleotide species are added to the wells one at a time and a standard elongation reaction is performed. As each base is incorporated, a single H+ ion is generated as a by-product. The H+ release results in a 0.02 unit change in pH, detected by an integrated complementary metal-oxide semiconductor (CMOS) and an ion-sensitive field-effect transistor (ISFET) device. After the introduction of a single nucleotide species, the unincorporated bases are washed away and the next is added. Part a is adapted from REF. 18, Nature Publishing Group.
The Ion Torrent was the first NGS platform without optical sensing25. Rather than using an enzymatic cascade to generate a signal, the Ion Torrent platform detects the H+ ions that are released as each dNTP is incorporated. The resulting change in pH is detected by an integrated complementary metal-oxide-semiconductor (CMOS) and an ion-sensitive field-effect transistor (ISFET) (FIG. 4b). The pH change detected by the sensor is imperfectly proportional to the number of nucleotides detected, allowing for limited accuracy in measuring homopolymer lengths.
Comparison of short-read platforms.
Individual short-read sequencing platforms vary with respect to throughput, cost, error profile and read structure (TABLE 1). Despite the existence of several NGS technology providers, NGS research is increasingly being conducted within the Illumina suite of instruments21. Although this implies high confidence in their data, it also raises concerns about systemic biases derived from using a single sequencing approach26–28. As a consequence, new approaches are being developed and researchers increasingly have the choice to integrate multiple sequencing methods with complementary strengths.
Table 1 |.
Summary of NGS platforms
| Platform | Read length (bp) | Throughput | Reads | Runtime | Error profile | Instrument cost (US$) | Cost per Gb (US$, approx.) |
|---|---|---|---|---|---|---|---|
| Sequencing by ligation | |||||||
| SOLiD 5500 Wildfire | 50 (SE) | 80 Gb | ~700 M* | 6 d* | ≤0.1%, AT bias‡ | NA§ | $130‡ |
| 75 (SE) | 120 Gb | ||||||
| 50 (SE)* | 160 Gb* | ||||||
| SOLiD 5500 xl | 50 (SE) | 160 Gb | ~1.4 B* | 10 d* | ≤0.1%, AT bias‡ | $251,000‡ | $70‡ |
| 75 (SE) | 240 Gb | ||||||
| 50 (SE)* | 320 Gb* | ||||||
| BGISEQ-500 FCS155 | 50–100 (SE/PE)* | 8–40 Gb* | NA|| | 24 h* | ≤0.1%, AT bias‡ | $250 (REF. 155) | NA|| |
| BGISEQ-500 FCL155 | 50–100 (SE/PE)* | 40–200 Gb* | NA|| | 24 h* | ≤0.1%, AT bias‡ | $250,000 (REF. 155) | NA|| |
| Sequencing by synthesis: CRT | |||||||
| Illumina MiniSeq Mid output | 150 (SE)* | 2.1–2.4 Gb* | 14–16 M* | 17 h* | <1%, substitution‡ | $50,000 (REF. 118) | $200–300 (REF. 118) |
| Illumina MiniSeq High output | 75 (SE) | 1.6–1.8 Gb | 22–25 M (SE)* | 7 h | <1%, substitution‡ | $50,000 (REF. 118) | $200–300 (REF. 118) |
| 75 (PE) | 3.3–3.7 Gb | 44–50 M (PE)* | 13 h | ||||
| 150 (PE)* | 6.6–7.5 Gb* | 24 h* | |||||
| Illumina MiSeq v2 | 36 (SE) | 540–610 Mb | 12–15 M (SE) | 4 h | 0.1%, substitution‡ | $99,000‡ | ~$1,000 |
| 25 (PE) | 750–850 Mb | 24–30 M (PE)* | 5.5 h | $996 | |||
| 150 (PE) | 4.5–5.1 Gb | 24 h | $212 | ||||
| 250 (PE)* | 7.5–8.5 Gb* | 39 h* | $142‡ | ||||
| Illumina MiSeq v3 | 75 (PE) | 3.3–3.8 Gb | 44–50 M (PE)* | 21–56 h* | 0.1%, substitution‡ | $99,000‡ | $250 |
| 300 (PE)* | 13.2–15 Gb* | $110‡ | |||||
| Illumina NextSeq 500/550 Mid output | 75 (PE) | 16–20 Gb | Up to 260 M (PE)* | 15 h | <1%, substitution‡ | $250‡ | $42 |
| 150 (PE)* | 32–40 Gb* | 26 h* | $40‡ | ||||
| Illumina NextSeq 500/550 High output | 75 (SE) | 25–30 Gb | 400 M (SE)* | 11 h | <1%, substitution‡ | $250‡ | $43 |
| 75 (PE) | 50–60 Gb | 800 M (PE)* | 18 h | $41 | |||
| 150 (PE)* | 100–120 Gb* | 29 h* | $33‡ | ||||
| Illumina HiSeq2500 v2 Rapid run | 36 (SE) | 9–11Gb | 300 M (SE)* | 7 h | 0.1%, substitution‡ | $690‡ | $230 |
| 50 (PE) | 25–30 Gb | 600 M (PE)* | 16 h | $90 | |||
| 100 (PE) | 50–60 Gb | 27 h | $52 | ||||
| 150 (PE) | 75–90 Gb | 40 h | $45 | ||||
| 250 (PE)* | 125–150 Gb* | 60 h* | $40‡ | ||||
| Illumina HiSeq2500 v3 | 36 (SE) | 47–52 Gb | 1.5 B (SE) | 2 d | 0.1%, substitution‡ | $690‡ | $180 |
| 50 (PE) | 135–150 Gb | 3 B (PE)* | 5.5 d | $78 | |||
| 100 (PE)* | 270–300 Gb | 11 d* | $45‡ | ||||
| Illumina HiSeq2500 v4 | 36 (SE) | 64–72 Gb | 2 B (SE) | 29 h | 0.1%, substitution‡ | $690‡ | $150 |
| 50 (PE) | 180–200 Gb | 4 B (PE)* | 2.5 d | $58 | |||
| 100 (PE) | 360–400 Gb | 5 d | $45 | ||||
| 125 (PE)* | 450–500 Gb* | 6 d* | $30‡ | ||||
| Illumina HiSeq3000/4000 | 50 (SE) | 105–125 Gb | 2.5 B (SE)* | 1–3.5 d* | 0.1%, substitution‡ | $740/$900 (REF. 156) | $50 |
| 75 (PE) | 325–375 Gb | $31 | |||||
| 150 (PE)* | 650–750 Gb* | $22 (REF. 157) | |||||
| Illumina HiSeq X | 150 (PE)* | 800–900 Gb per flow cell* | 2.6–3 B (PE)* | <3 d* | 0.1%, substitution‡ | $1,000‡,¶ | $7.0‡ |
| Qiagen GeneReader | NA|| | 12 genes; 1,250 mutations22 | NA|| | Several days22 | Similar to other SBS systems22 | NA|| | $400–$600 per panel22 |
| Sequencing by synthesis: SNA | |||||||
| 454 GS Junior | Up to 600; 400 average (SE, PE)* | 35 Mb* | ~0.1 M* | 10 h* | 1%, indel‡ | NA§ | $40,000‡ |
| 454 GS Junior+ | Up to 1,000; 700 average (SE, PE)* | 70 Mb* | ~0.1 M* | 18 h* | 1%, indel‡ | $108,000‡ | $19,500‡ |
| 454 GS FLX Titanium XLR70 | Up to 600; 450 mode (SE, PE)* | 450 Mb* | ~1 M* | 10 h* | 1%, indel‡ | NA§ | $15,500‡ |
| 454 GS FLX Titanium XL+ | Up to 1,000; 700 mode (SE, PE)* | 700 Mb* | ~1 M* | 23 h* | 1%, indel‡ | $450,000‡ | $9,500‡ |
| Ion PGM 314 | 200 (SE) | 30–50 | 400,000–550,000* | 23 h | 1%, indel‡ | $49‡ | $25–3,500‡ |
| 400 (SE) | 60–100 Mb* | 3.7 h* | |||||
| Ion PGM 316 | 200 (SE) | 300–500 Mb | 2–3 M* | 3 h | 1%, indel‡ | $49‡ | $700–1,000‡ |
| 400 (SE)* | 600 Mb–1 Gb* | 4.9 h* | |||||
| Ion PGM 318 | 200 (SE) | 600 Mb–1 Gb | 4–5.5 M* | 4 h | 1%, indel‡ | $49‡ | $450–800‡ |
| 400 (SE)* | 1–2 Gb* | 7.3 h* | |||||
| Ion Proton | Up to 200 (SE) | Up to 10 Gb* | 60–80 M* | 2–4 h* | 1%, indel‡ | $224‡ | $80‡ |
| Ion S5 520 | 200 (SE) | 600 Mb–1 Gb | 3–5 M* | 2.5 h | 1%, indel‡ | $65 (REF. 158) | $2,400* |
| 400 (SE)* | 1.2–2 Gb* | 4 h* | $1,200* | ||||
| Ion S5 530 | 200 (SE) | 3–4 Gb | 15–20 M* | 2.5 h | 1%, indel‡ | $65 (REF. 158) | $950* |
| 400 (SE)* | 6–8 Gb* | 4 h* | $475* | ||||
| Ion S5 540 | 200 (SE)* | 10–15 Gb* | 60–80 M* | 2.5 h* | 1%, indel‡ | $65 (REF. 158) | $300* |
| Single-molecule real-time long reads | |||||||
| Pacific BioSciences RS II | ~20 Kb | 500 Mb–1 Gb* | ~55,000* | 4 h* | 13% single pass, ≤1% circular consensus read, indel‡ | $695‡ | $1,000‡ |
| Pacific Biosciences Sequel | 8–12 Kb69 | 3.5–7 Gb* | ~350,000* | 0.5–6 h* | NA|| | $350 (REF. 69) | NA|| |
| Oxford Nanopore MK 1 MinION | Up to 200 Kb159 | Up to 1.5 Gb159 | >100,000 (REF. 159) | Up to 48 h160 | ~12%, indel159 | $1,000* | $750* |
| Oxford Nanopore PromethION | NA|| | Up to 4 Tb* | NA|| | NA|| | NA|| | $75* | NA|| |
| Synthetic long reads | |||||||
| Illumina Synthetic Long-Read | ~100 Kb synthetic length* | See HiSeq 2500 | See HiSeq 2500 | See HiSeq 2500 | See HiSeq 2500 (possible barcoding and partitioning errors) | No additional instrument required | ~$1,000* |
| 10X Genomics | Up to 100 Kb synthetic length* | See HiSeq 2500 | See HiSeq 2500 | See HiSeq 2500 | See HiSeq 2500 (possible barcoding and partitioning errors) | $75 (REFS 72,161) | See HiSeq 2500+$500 per sample161 |
Approx., approximate; AT, adenine and thymine; B, billion; bp, base pairs; d, days; Gb, gigabase pairs; h, hours; indel, insertions and deletions; Kb, kilobase pairs; M, million; Mb, megabase pairs; NA, not available; PE, paired-end sequencing; SBS, sequencing by synthesis; SE, single-end sequencing; Tb, terabase pairs.
Manufacturer’s data.
Rounded from Field Guide to next-generation DNA sequencers160 and 2014 update.
Not available as this instrument will be discontinued or only available as an upgraded version.
As this product has been developed only recently, this information is not available.
Not available as a single instrument.
The SBL technique used by both the SOLiD and Complete Genomics systems affords these technologies a very high accuracy (~99.99%)7,14, as each base is probed multiple times. Although accurate, both platforms also show evidence of a trade-off between sensitivity and specificity, such that true variants are missed while few false variants are called29–31. There is also evidence that the platforms share some under-representation of AT-rich regions26,32, and the SOLiD platform displays some substitution errors and some GC-rich under-representation32. Perhaps the feature most limiting to the widespread adoption of these technologies is the very short read lengths. Although both platforms can generate single-end and paired-end sequencing reads, the maximum read length is just 75 bp for SOLiD and 28–100 bp for Complete Genomics33, limiting their use for genome assembly and structural variant detection applications. Unfortunately, owing to these limitations, along with runtimes on the order of several days, the SOLiD system has been relegated to a small niche within the industry. Furthermore, although the cPAL-based Revolocity system was intended to compete with the Illumina HiSeq in terms of cost and throughout, its launch was suspended in 2016 and it is now only available as a service platform for human WGS33,34, whereas the cPAS-based BGISEQ-500 platform is limited to mainland China.
Illumina dominates the short-read sequencing industry owing, in part, to its maturity as a technology, a high level of cross-platform compatibility and its wide range of platforms. The suite of instruments available ranges from the low-throughput MiniSeq to the ultra-high-throughput HiSeq X, which is capable of sequencing ~1,800 human genomes to 30× coverage per year. Further diversification is derived from the many options available for runtime, read structure and read length (up to 300 bp). As the Illumina platform relies on a CRT approach, it is much less susceptible to the homopolymer errors observed in SNA platforms. Although it has an overall accuracy rate of >99.5%35, the platform does display some under-representation in AT-rich32,36 and GC-rich regions32,37, as well as a tendency towards substitution errors38. In 2008, Bentley et al.35 reported a very high concordance rate between human single-nucleotide polymorphisms (SNPs) identified with Illumina and SNPs identified from genotyping microarrays35. However, this high sensitivity came with a false-positive rate of around 2.5%, leading this and other groups to consider using Sanger sequencing to resequence the called SNPs in order to distinguish between true SNPs and false positives35,39,40. With all of the possible options available, the Illumina suite allows for a wide range of applications: genome sequencing through WGS or exome sequencing; epigenomics applications, such as ChIP–seq (chromatin immunoprecipitation followed by sequencing)41, ATAC–seq (assay for transposase-accessible chromatin using sequencing)42 or DNA methylation sequencing (methyl-seq)43; and transcriptomics applications through RNA sequencing (RNA-seq)44, to name a few. The two-colour labelling system used by the NextSeq and MiniSeq platforms increases speed and reduces costs by reducing scanning to two colour channels and reducing fluorophore usage. However, the two-channel system results in a slightly higher error profile and underperformance for low-diversity samples owing to more ambiguous base discrimination45. HiSeq X is currently the highest-throughput instrument available; however, as a consequence of its optimization, it is limited to just a few applications, such as WGS and whole-genome bisulfite sequencing. HiSeq X is further limited as an all-purpose instrument owing to a required initial purchase of five or ten instruments (additional single instruments can be purchased after the initial commitment), placing this system out of reach of most facilities.
The Qiagen GeneReader is intended to be a clinical device with an explicit focus on cancer gene panels46; although this severely limits its possible applications, it is well optimized within its niche. With a reported several-day runtime, and the use of validated gene panels, it fulfils a similar role to the Illumina MiSeq46. Although no user data are available at this point, it is likely that the Qiagen GeneReader will share many of the same advantages and disadvantages as the Illumina MiSeq platform at a potentially lower cost per gigabase sequenced.
Both the 454 and the Ion Torrent systems offer superior read lengths compared to other short-read sequencers with reads up to an average of 700 bp and 400 bp, respectively, providing some advantages for applications that focus on repetitive or complex DNA. However, as both of these platforms rely on SNA, they share many of the same drawbacks. Insertion and deletion (indel) errors dominate, although the overall error rate is on par with other NGS platforms in non-homopolymer regions. Homopolymer regions are problematic for these platforms, which lack single-base accuracy in measuring homopolymers larger than 6–8 bp47,48. Unfortunately, whereas the Ion Torrent platform has kept pace with the rapidly evolving NGS field, the 454 platform has been unable to complete with other platforms in terms of yield or cost. This limitation has led Roche to discontinue the platform in 2016 (REF. 49).
The Ion Torrent platform offers several different types of chips and instruments to tune sequencer performance to the needs of the researcher. The throughput of these chips ranges from ~50 Mb to 15 Gb, with runtimes between 2 and 7 hours, making it faster than most other current platforms. This makes the device well suited for gene-panel sequencing and for point-of-care clinical applications50, including transcriptome profiling51 and splice site identification (although not to the level of long-read sequencers)51. Ion Torrent is attempting to capitalize on the growing interest in clinical sequencing with the release of its dedicated diagnostic instruments: the Ion Personal Genome Machine (PGM) Dx and the Ion S5 series. When paired with the Ion Chef library preparation and chip loading device, the S5 series in particular aims to be one of the simplest platforms to operate, eliminating the need for the argon required by other Ion Torrent instruments and establishing plug-and-play protocols. An important disadvantage, however, is that although the Ion PGM Dx sequencer can support paired-end sequencing52, the higher-throughput Ion Proton and S5 devices lack the ability to perform paired-end sequencing, thus limiting their utility for elucidating long-range genomic or transcriptomic structure53.
Long-read sequencing
Overview.
It has become apparent that genomes are highly complex with many long repetitive elements, copy number alterations and structural variations that are relevant to evolution, adaptation and disease54–56. However, many of these complex elements are so long that short-read paired-end technologies are insufficient to resolve them. Long-read sequencing delivers reads in excess of several kilobases, allowing for the resolution of these large structural features. Such long reads can span complex or repetitive regions with a single continuous read, thus eliminating ambiguity in the positions or size of genomic elements. Long reads can also be useful for transcriptomic research, as they are capable of spanning entire mRNA transcripts, allowing researchers to identify the precise connectivity of exons and discern gene isoforms.
Currently, there are two main types of long-read technologies: single-molecule real-time sequencing approaches and synthetic approaches that rely on existing short-read technologies to construct long reads in silico. The single-molecule approaches differ from short-read approaches in that they do not rely on a clonal population of amplified DNA fragments to generate detectable signal, nor do they require chemical cycling for each dNTP added. Alternatively, the synthetic approaches do not generate actual long-reads; rather, they are an approach to library preparation that leverages barcodes to allow computational assembly of a larger fragment.
Single-molecule long-read sequencing (PacBio and ONT).
Currently, the most widely used long-read platform is the single-molecule real-time (SMRT) sequencing approach used by Pacific Biosciences (PacBio)57 (FIG. 5a). The instrument uses a specialized flow cell with many thousands of individual picolitre wells with transparent bottoms — zero-mode waveguides (ZMW)58. Whereas short-read SBS technologies bind the DNA and allow the polymerase to travel along the DNA template, PacBio fixes the polymerase to the bottom of the well and allows the DNA strand to progress through the ZMW. By having a constant location of incorporation owing to the stationary enzyme, the system can focus on a single molecule. dNTP incorporation on each single-molecule template per well is continuously visualized with a laser and camera system that records the colour and duration of emitted light as the labelled nucleotide momentarily pauses during incorporation at the bottom of the ZMW. The polymerase cleaves the dNTP-bound fluorophore during incorporation, allowing it to diffuse away from the sensor area before the next labelled dNTP is incorporated. The SMRT platform also uses a unique circular template that allows each template to be sequenced multiple times as the polymerase repeatedly traverses the circular molecule. Although it is difficult for DNA templates longer than ~3 kb to be sequenced multiple times, shorter DNA templates can be sequenced many times as a function of template length57,59. These multiple passes are used to generate a consensus read of insert, known as a circular consensus sequence (CCS).
Figure 5 |. Real-time and synthetic long-read sequencing approaches.
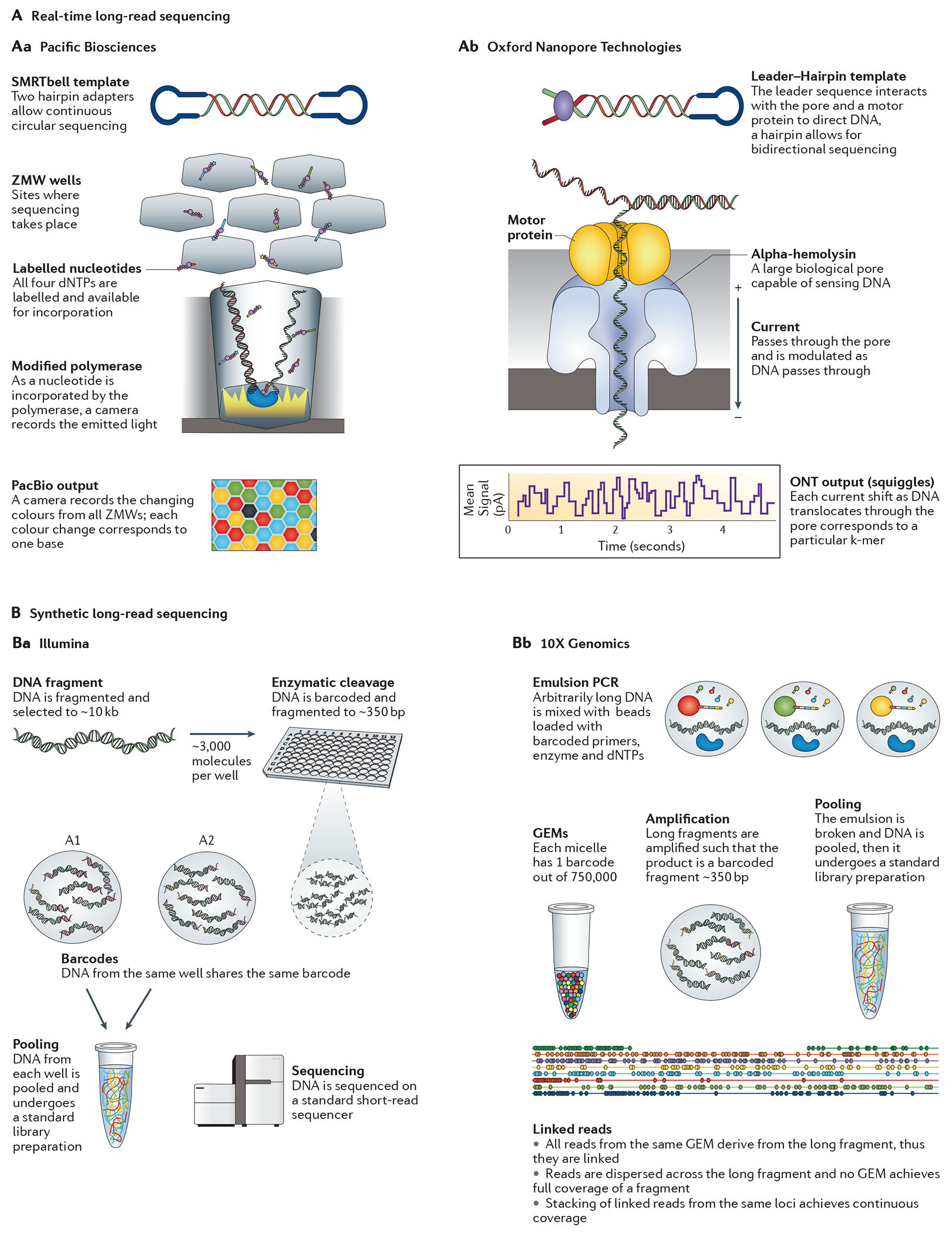
A | Real-time long-read sequencing platforms. Aa | Single-molecule real-time (SMRT) sequencing from Pacific Biosciences (PacBio). Template fragments are processed and ligated to hairpin adapters at each end, resulting in a circular DNA molecule with constant single-stranded DNA (ssDNA) regions at each end with the double-stranded DNA (dsDNA) template in the middle. The resulting ‘SMRTbell’ template undergoes a size-selection protocol in which fragments that are too large or too small are removed to ensure efficient sequencing. Primers and an efficient φ29 DNA polymerase are attached to the ssDNA regions of the SMRTbell. The prepared library is then added to the zero-mode waveguide (ZMW) SMRT cell, where sequencing can take place. To visualize sequencing, a mixture of labelled nucleotides is added; as the polymerase-bound DNA library sits in one of the wells in the SMRT cell, the polymerase incorporates a fluorophore-labelled nucleotide into an elongating DNA strand. During incorporation, the nucleotide momentarily pauses through the activity of the polymerase at the bottom of the ZMW, which is being monitored by a camera. Ab | Oxford Nanopore Technologies (ONT). DNA is initially fragmented to 8–10 kb. Two different adapters, a leader and a hairpin, are ligated to either end of the fragmented dsDNA. Currently, there is no method to direct the adapters to a particular end of the DNA molecule, so there are three possible library conformations: leader–leader, leader–hairpin and hairpin–hairpin. The leader adapter is a double-stranded adapter containing a sequence required to direct the DNA into the pore and a tether sequence to help direct the DNA to the membrane surface. Without this leader adapter, there is minimal interaction of the DNA with the pore, which prevents any hairpin–hairpin fragments from being sequenced. The ideal library conformation is the leader–hairpin. In this conformation the leader sequence directs the DNA fragment to the pore with current passing through. As the DNA translocates through the pore, a characteristic shift in voltage through the pore is observed. Various parameters, including the magnitude and duration of the shift, are recorded and can be interpreted as a particular k-mer sequence. As the next base passes into the pore, a new k-mer modulates the voltage and is identified. At the hairpin, the DNA continues to be translocated through the pore adapter and onto the complement strand. This allows the forward and reverse strands to be used to create a consensus sequence called a ‘2D’ read. B | Synthetic long-read sequencing platforms. Ba | Illumina. Genomic DNA templates are fragmented to 8–10 kb pieces. They are then partitioned into a microtitre plate such that there are around 3,000 templates in a single well. Within the plate, each fragment is sheared to around 350 bp and barcoded with a single barcode per well. The DNA can then be pooled and sent through standard short-read pipelines. Bb | 10X Genomics’ emulsion-based sequencing. With as little as 1 ng of starting material, the GemCode can partition arbitrarily large DNA fragments, up to ~100 kb, into micelles (also called ‘GEMs’) along with gel beads containing adapter and barcode sequences. The GEMs typically contain ~0.3× copies of the genome and 1 unique barcode out of 750,000. Within each GEM, the gel bead dissolves and smaller fragments of DNA are amplified from the original large fragments, each with a barcode identifying the source GEM. After sequencing, the reads are aligned and linked together to form a series of anchored fragments across a span of ~50 kb. Unlike the Illumina system, this approach does not attempt to get full end-to-end coverage of a single DNA fragment. Instead, the reads from a single GEM are dispersed across the original DNA fragment and the cumulative coverage is derived from multiple GEMs with dispersed — but linked — reads. Part Aa is adapted from REF. 18, Nature Publishing Group. Part Ba is adapted from REF. 62.
In 2014, the first consumer prototype of a nanopore sequencer — the MinION from Oxford Nanopore Technologies (ONT) — became available. Unlike other platforms, nanopore sequencers do not monitor incorporations or hybridizations of nucleotides guided by a template DNA strand. Whereas other platforms use a secondary signal, light, colour or pH, nanopore sequencers directly detect the DNA composition of a native ssDNA molecule. To carry out sequencing, DNA is passed through a protein pore as current is passed through the pore60 (FIG. 5b). As the DNA translocates through the action of a secondary motor protein, a voltage blockade occurs that modulates the current passing through the pore. The temporal tracing of these charges is called squiggle space, and shifts in voltage are characteristic of the particular DNA sequence in the pore, which can then be interpreted as a k-mer. Rather than having 1–4 possible signals, the instrument has more than 1,000 — one for each possible k-mer, especially when modified bases present on native DNA are taken into account. The current MK1 MinION flow cell structure is composed of an application-specific integrated circuit (ASIC) chip with 512 individual channels that are capable of sequencing at ~70 bp per second, with an expected increase to 500 bp per second in 2016. The upcoming PromethION instrument is intended to be an ultra-high-throughput platform reported to include 48 individual flow cells, each with 3,000 pores running at 500 bp per second. This works out to ~2–4 Tb for a 2-day run on a fully loaded device, placing this device in potential competition with Illumina’s HiSeq X. Similar to the circular template used by PacBio, the ONT MinION uses a leader–hairpin library structure. This allows the forward DNA strand to pass through the pore, followed by a hairpin that links the two strands, and finally the reverse strand. This generates 1D and 2D reads in which both ‘1D’ strands can be aligned to create a consensus sequence ‘2D’ read.
Synthetic long-reads.
Unlike true sequencing platforms, synthetic long-read technology relies on a system of barcoding to associate fragments that are sequenced on existing short-read sequencers61. These approaches partition large DNA fragments into either microtitre wells or an emulsion such that very few molecules exist in each partition. Within each partition the template fragments are sheared and barcoded. This approach allows for sequencing on existing short-read instrumentation, after which data are split by barcode and reassembled with the knowledge that fragments sharing barcodes are derived from the same original large fragment62. Similar to an earlier technology, BAC-by-BAC sequencing, synthetic barcoded reads provide an association among small fragments derived from a larger one. By segregating the fragments, repetitive or complicated regions can be isolated, allowing each to be assembled locally. This prevents unresolvable branch points in the assemblies, which lead to breaks (gaps) and shorter assembled contiguous sequences.
There are currently two systems available for generating synthetic long-reads: the Illumina synthetic long-read sequencing platform (FIG. 5c) and the 10X Genomics emulsion-based system (FIG. 5d). The Illumina system (formerly Moleculo) partitions DNA into a microtitre plate and does not require specialized instrumentation. However, the 10X Genomics instruments (GemCode and Chromium) use emulsion to partition DNA and require the use of a microfluidic instrument to perform pre-sequencing reactions. With as little as 1 ng of starting material, the 10X Genomics instruments can partition arbitrarily large DNA fragments, up to ~100 kb, into micelles called ‘GEMs’, which typically contain ≤0.3× copies of the genome and one unique barcode. Within each GEM, a gel bead dissolves and smaller fragments of DNA are amplified from the original large fragments, each with a barcode identifying the source GEM. After sequencing, the reads are aligned and linked together to form a series of anchored fragments across the span of the original fragment. Unlike the Illumina system, this approach does not attempt gapless, end-to-end coverage of a single DNA fragment. Instead it relies on linked reads, in which dispersed, small fragments that are derived from a single long molecule share a communal barcode. Although these fragments leave segments of the original large molecule without any coverage, the gaps are overcome by ensuring that there are many long fragments from the same genomic region in the initial preparation, thus generating a read cloud wherein linked reads from each long fragment can be stacked, combining their individual coverage into an overall map (FIG. 5d).
Comparison of single-molecule and synthetic long-read sequencing.
There is growing interest in the field of long-read sequencing, and each system has its own advantages and drawbacks (TABLE 1) Currently, the most widely used instrument in long-read sequencing is the PacBio RS II instrument. This device is capable of generating single polymerase reads in excess of 50 kb with average read lengths of 10–15 kb for a long-insert library. Such properties are ideal for de novo genome assembly applications63, for revealing complex long-range genomic structures64 and for full-length transcript sequencing. There are, however, several notable limitations. The single-pass error rate for long reads is as high as 15% with indel errors dominating65, raising concerns about the utility of the instrument66. Fortunately, these errors are randomly distributed within each read and hence sufficiently high coverage can overcome the high error rate67. The use of a circular template by PacBio also provides a level of error correction. The more frequently a single molecule is sequenced, the higher the resulting accuracy — up to ~99.999% for insert sequences derived from at least 10 subreads59,68.This high accuracy rivals that of Sanger sequencing, leading researchers to speculate that this technology can be used in a manner analogous to Sanger-based SNP validation65. The runtimes and throughput of this instrument can be tuned by controlling the length of time for which the sensor monitors the ZMW; longer templates require longer times. For example, a 1 kb library that is run for 1 hour will generate around 7,500 bases of sequence per molecule, with an average of 8 passes, whereas a 4-hour run will generate around 30,000 bases per molecule and ~30 passes. Conversely, a 10 kb library requires a 4-hour run to generate ~30,000 bases with ~3 passes. The limited throughput and high costs of PacBio RS II (around $1,000 per Gb), in addition to the need for high coverage, place this instrument out of reach of many small laboratories. However, in an attempt to ameliorate these concerns, PacBio has launched the Sequel System, which reportedly has a throughput 7× that of the RS II, thus halving the cost of sequencing a human genome at 30× coverage69.
The ONT MinION is a small (~3 cm × 10 cm for the MK1) USB-based device that runs off a personal computer, giving it the smallest footprint of any current sequencing platform. This affords the MinION superior portability, highlighting its utility for rapid clinical responses and hard-to-reach field locations. Although substantial adjunct equipment is still required for library preparation (for instance, a thermocycler), improvements in library preparation and equipment optimization could conceivably reduce the space required for a fully functional sequencing system to the size of a single bag of luggage. Unlike other platforms, the MinION has few constraints on the size of the fragments to be sequenced. In theory, a DNA molecule of any size can be sequenced on the device, but in practice there are some limitations when dealing with ultra-long fragments70. As a consequence of the unique nature of the ONT technology, in which there are more than 1,000 distinct signals, ONT MinION has a large error rate — up to 30% for a 1D read — and is dominated by indel errors. Effective homopolymer sequencing also remains a challenge for ONT MinION. When a homopolymer exceeds the k-mer length, it can be difficult to identify when one k-mer leaves the pore and another k-mer enters. Modified bases also pose a challenge to the device, as a modified base will alter the typical voltage shift for a given k-mer. Fortunately, recent improvements in the chemistry and the base calling algorithms are improving accuracy71.
The Illumina synthetic long-read approaches are a direct response to the costs, error rates and throughput of true long-read sequencers. Relying on the existing Illumina infrastructure affords researchers the ability to simply purchase a kit for long-read sequencing. Accordingly, the throughput and error profile are identical to those of current Illumina devices. However, as a consequence of how the DNA is partitioned, the system requires more coverage than is required for a typical short-read project, thus increasing the costs associated with this technology relative to other Illumina applications62.
Like the Illumina synthetic long-read platform, the 10X Genomics emulsion-based platform relies on an existing short-read infrastructure to provide the sequencing. The microfluidic instrument is a one-time additional equipment cost, and the emulsion approach used allows for as little as 1 ng of starting material, which can be beneficial for situations in which the DNA is precious, such as biopsy samples. Currently, data output from the GemCode instrument is partially limited by the number of barcodes used and the somewhat inefficient DNA partitioning. Inefficient partitioning can lead to a surplus of DNA fragments within a droplet, thus complicating sequence deconvolution, which is further exacerbated by the limited number of barcodes. Both of these conditions lead to ambiguity regarding the positional relationship between reads sharing the same barcode, making analysis more difficult. To rectify this, 10X Genomics plans to release the Chromium System in mid-2016; this will be an upgrade from the GemCode device. Although the chemistry will remain fundamentally the same, the number of possible micelle partitions will increase from 100,000 to 1 million, and the number of barcodes will increase from 750,000 to ~4 million72.
Applications
WGS is becoming one of the most widely used applications in NGS. Through this technology, researchers can obtain the most comprehensive view of genomic information and associated biological implications73. For example, in 2012, Ellis et al.74 published an exploration of the interactions between genes and aromatase inhibitor therapy in patients with breast cancer. They outlined a range of correlations between mutations, outcomes and clinical features, as well as mutational enrichment in genes linked to other cancers, providing additional support for the idea that breast cancer is a highly complex pathology with variable phenotypes that are based on different repertoires of mutations75. However, the recent diversification of NGS platforms has revealed new and more ambitious opportunities that were not possible just a few years ago. In 2010, the 1000 Genomes Project released its initial results from WGS of 179 individuals and targeted sequencing of 697 individuals76. As of 2015, the genomes of 2,504 people from 26 different populations have been reconstructed77,78, providing an unparalleled insight into human variation on a population level, and projects to sequence even larger sets of people are nearing completion or are underway79–81. Population-level sequencing is proving to be an essential tool in understanding human disease, with exciting results. In one example, Sidore et al.82 performed WGS on 2,120 Sardinians and discovered new loci for lipid levels and inflammatory markers, providing greater insight into the mechanisms driving blood cholesterol levels.
Whole-exome and targeted sequencing83 are also proving invaluable to sequencing research. By limiting the size of the genomic material used, more individual samples can be sequenced within a sequencing run, which can increase both the breadth and the depth of a genomic study. Using exome sequencing, Iossifov et al.84 sequenced more than 2,500 simplex families, each with a child who was diagnosed with autism spectrum disorder (ASD), and they discovered de novo missense mutations, de novo gene-disrupting mutations and copy number variants in ~30% of all cases. This and other work regarding the classes of genes mutated is providing a framework for possible causes of ASD85,86. There is also increasing evidence that even high-coverage WGS is inadequate to resolve all variants in complex and/or limited material clinical samples. In 2015, Griffith et al.87 demonstrated the use of an integrated, cross-platform approach (including targeted sequencing) to identify high-confidence SNPs from tumour samples. In this approach, the authors demonstrated that coverage as high as 10,000× can be required to validate rare variants. Although such high coverage is prohibitive for WGS, targeted sequencing approaches are uniquely suited for clinical applications.
NGS is also providing insight into the regulatory mechanisms of the genome. Protein–DNA interactions can be probed by enriching for protein-interacting DNA fragments, often through immunoprecipitation as in the case of ChIP–seq41. Conversely, ATAC–seq uses a hyperactive transposase to generate short-read NGS-compatible DNA fragments from regions unprotected by proteins or nucleosomes42. Analysis of modified bases is also possible. For example, methyl-seq involves the capture and enrichment of methylated DNA88, selective digestion of methylated or unmethylated regions89,90, and/or modification of a methylated base such that it introduces a SNP into the DNA sequence91. Although important discoveries have been made using these approaches, limitations exist, which are primarily derived from the modification or capture process. In 2010, Flusberg et al.92 published a proof-of-concept study of using PacBio to discriminate between methylated and un-methylated bases, as well as between methylated adenine and methylated cytosine. As the polymerase attempts to elongate DNA containing modified bases, it pauses for longer at modified sites compared with unmodified controls, increasing a metric called the interpulse duration and thus indicating the presence of a modified base. Similarly, nanopore platforms also show promise for the direct detection of modified bases, as the characteristic shift in voltage across the pore is modulated by base modifications, allowing for discrimination without the need for chemical manipulations93.
A recent paradigm shift in NGS is the ability to sequence very long stretches of DNA. Repetitive and complex regions have historically been difficult to assemble and resolve using short-read sequencing approaches94–96. Recently, using long-read sequencing technology, Chaisson et al.97 were able to add more than 1 Mb of novel sequence to the human GRCh37 reference genome through gap closure and extension, and they identified >26,000 indels that were ≥50 bp in length, providing one of the most comprehensive reference genomes available. Beyond simply improving reference genomes, long reads are proving to be more effective in identifying clinically relevant structural variation than short-read approaches98. Furthermore, synthetic approaches are enhancing the ability of researches to phase genomes99. In 2014, Kuleshov et al.100 showed how synthetic approaches can reduce the coverage required for genome phasing99 by 10-fold while also phasing up to 99% of all SNPs. This allows researchers to track the history of a mutation across generations — an essential tool for family studies.
Transcriptomic research has also benefited from greater accessibility to NGS. Today, researchers are leveraging the power of NGS to deeply sequence down to single-transcript sensitivity. In 2014, Treutlein et al.101 demonstrated the power of single-cell RNA-seq to characterize different cell populations in developing tissue and to discover new markers for cell subpopulations. Although long-read approaches currently lack the ability to effectively measure transcript abundance levels, long reads provide superior performance for detecting transcriptomic structure51. For example, a recent long-read profile of the human transcriptome showed that >10% of the reads represented novel splice isoforms102.
The newest instrument in the NGS landscape, the nanopore sequencer, is still in the process of finding its niche in the field. Nevertheless, researchers are capitalizing on its rapid library preparation time, real-time generation of data and its small size. Recently, researchers at the Stanley Royd Hospital in the United Kingdom used MinION sequencing to monitor an outbreak of Salmonella enterica. Using phylogenetic placement, the authors were able to unambiguously identify the serovar within 50 minutes after the start of sequencing, indicating that the MinION device is a viable platform for rapid pathogen profiling103. Perhaps one of the most striking applications of MinION sequencing in the field was its use during the 2014 Ebola outbreak, which is outlined in Quick et al.104. Under the auspices of the European Mobile Laboratories in Guinea, the authors were able to monitor the transmission history and evolution of the Ebola virus as the outbreak unfolded.
Closing remarks
We are sitting at the cusp of a new revolution in NGS technologies. Rather than being a novelty, NGS technologies are now a routine part of biological research. The advent of ultra-high-throughput sequencing is propelling research that was considered impossible only a few years ago and is becoming more widespread within the clinical sector. This includes recent precision medicine initiatives105 and plans from Illumina to develop a pan-cancer screening method using circulating tumour DNA106, each with goals of sequencing tens of thousands of genomes. Thus, rapid and low-cost sequencing is providing physicians with the tools needed to translate genomic information into clinically actionable results.
This revolution brings with it a new set of challenges. As NGS aims to be ubiquitous in the clinical setting, time remains a challenge. In cases of serious neonatal disease and aggressive cancer, the weeks it can take for WGS data generation and analysis can be the difference between success and failure. In the case of aggressive infections, that time is reduced to mere days. Although substantial advances have been made in reducing response time, most of the current systems do not yet generate enough data fast enough for a truly rapid response.
Whereas clinics may be challenged by a paucity of rapid data, other aspects of NGS are suffering from an abundance of data. To date, more than 14,000 genomes have been deposited within the US National Center for Biotechnology Information (NCBI) genome repositories, with new genomes deposited regularly. In 2013, Schatz and Langmead reported that the world can generate ~15 petabytes of sequencing data per year, and the number and throughput of sequencers has only increased since then107. This wealth of data is proving challenging for both analysis and infrastructure, requiring innovative storage and bioinformatic solutions108. Translation of this vast pool of genetic data into biological contexts remains a challenge, requiring both integrative approaches to NGS research and well-validated guidelines for discovery87,109,110. The proliferation of NGS in the clinical arena also raises concerns about the utility and ethics associated with genomic information. With so many genetic tests available, including direct-to-consumer testing, questions exist regarding the consumer response to genetic results and the impact of false positives and false negatives on health care111,112.
Recently, Ulumina’s highly successful suite of instruments have been the juggernaut of NGS, relegating technologies that could not keep pace to niche applications or outright dissolution. The casualties of the NGS arms race have included the Helicos Genetic Analysis System113, the Revolocity system from Complete Genomics and 454 pyrosequencing from Roche. Yet, despite these setbacks, new applications and instruments are being developed at an astounding pace. As Illumina’s market share grows, so too do challenges to its dominance. The BGISEQ-500 and the forthcoming GenoCare114 from Direct Genomics (based on the Helicos technology) seek to gain a foothold in Asia, where NGS research is still developing. Platforms such as the ONT PromethION115 and Illumina HiSeq X are poised to push the limits of cost and yield. With the growing interest in clinical sequencing, established NGS providers are offering rapid solutions, such as the Ion Torrent S5 and the Illumina MiniSeq, and newcomers such as Qiagen’s GeneReader22 are also competing to fill the void. Finally, pre-sequencing approaches such as 10X Genomics aim to fundamentally change how existing sequencing is carried out by providing long-range information on short-read sequencing platforms.
In the next few years, additional players seek to further democratize the field with novel sequencing solutions. GenapSys (in partnership with Sigma-Aldrich), with its electronic ‘lunchbox’-sized sequencer116; Genia (Roche), with a new nanopore sequencing approach117; and the recently announced Firefly (Illumina), with its one-channel CMOS technology118, all claim to deliver superior cost and time savings for clinical applications. Finally, NanoString Technologies with its enzyme-free hybridization method119, GnuBio (Bio-Rad) with a fluorescence resonance energy transfer (FRET)-based approach120, and Electron Optica with an electron microscopy-based system121 all aim to revolutionize sequencing with unique technologies. These existing and forthcoming NGS tools have the potential to allow for revolutionary science, including direct sequencing of RNA or proteins, real-time genomic pathogen monitoring or precision medicine based on personal genome sequencing. These current and future advances make today an astonishing time for the field of NGS.
Glossary
- Read
The sequence of bases from a single molecule of DNA.
- Sanger sequencing
An approach in which dye-labelled normal deoxynucleotides (dNTPs) and dideoxy-modified dNTPs are mixed. A standard PCR reaction is carried out and, as elongation occurs, some strands incorporate a dideoxy-dNTP, thus terminating elongation. The strands are then separated on a gel and the terminal base label of each strand is identified by laser excitation and spectral emission analysis.
- Template
A DNA fragment to be sequenced. The DNA is typically ligated to one or more adapter sequences where DNA sequencing will be initiated.
- Fragmentation
The process of breaking large DNA fragments into smaller fragments. This can be achieved mechanically (by passing the DNA through a narrow passage), by sonication or enzymatically.
- Clusters
Groups of DNA templates in close spatial proximity generated either though bead-based amplification or by solid-phase amplification. Bead-based approaches rely on emulsions to maintain template isolation during amplification. Solid-phase approaches rely on the template-to-bound-adapter ratio to probabilistically bind template molecules at a sufficient distance from each other.
- Flow cells
Disposable parts of a next-generation sequencing routine. Template DNA is immobilized within the flow cell where fluid reagents can be streamed into the cell and flushed away.
- Rolling circle amplification (RCA).
A method of DNA amplification using a circular template. Briefly, DNA polymerase binds to a primed section of a circular DNA template. As the polymerase traverses the template, a new strand is synthesized. When the polymerase completes a full circle and encounters the double-stranded DNA (dsDNA) template, it displaces the template without degradation, thus creating a long ssDNA fragment composed of many copies of the template sequence.
- One-base-encoded probes
Oligonucleotides that contain a single interrogation base in a known position. The base corresponds to a fluorescent label on each probe. The remaining bases are either degenerate (any of the four bases) or universal (unnatural bases with nonspecific hybridization), allowing the probe to interact with many different possible template sequences.
- Two-base-encoded probes
Oligonucleotides that contain two adjacent interrogation bases in a known position. The bases correspond to a fluorescent label on each probe. The remaining bases are either degenerate (any of the four bases) or universal (unnatural bases with nonspecific hybridization] allowing the probe to interact with many different possible template sequences.
- Colour-space
A system exclusively used by SOLiD. When a two-base-encoded probe is used, the bound label corresponds to two bases rather than one. Thus, the signal derived from a SOLiD run is in a series of colours that represent overlapping dinucleotides, rather than each colour being directly correlated to a single base. A reference-based alignment is the most efficient way to translate colour-space into base-space. For example, in the sequence ATGT the first probe will match AT, the second will match TG and the third GT. If the AT is known, then the subsequent colour order is uniquely solved as TG and GT, leading to a readout of ATGT. Final sequence deconvolution of colour-space is achieved with the knowledge of the second base identity in one round and the colour of the subsequent round in which the ligation is offset by one nucleotide, allowing for the identification of the next base.
- Base-space
A system used by most next-generation sequencing platforms. When a one-base-encoded probe or a sequencing-by-synthesis approach is used, each signal is correctly correlated to a base.
- Whole-genome sequencing (WGS).
Sequencing of the entire genome without using methods for sequence selection.
- Two-fluorophore system
A system in which bases are discriminated by labelling Cs and Ts with a red or green fluorophore, respectively. Each A base is labelled with either a red or green fluorophore, but the two populations are mixed. During base discrimination, clusters that are either red or green are called either C or T, whereas clusters with a red and green mixed signal are called A. The G base is unlabelled, thus any cluster without a fluorophore signal is called G.
- Homopolymer
A sequence run of identical bases.
- Charge-coupled device (CCD).
A device composed of an integrated circuit that forms light-sensitive elements: pixels. When a photon interacts with the device, the light generates a charge that can be interpreted by an electronic device.
- Integrated complementary metal-oxide-semiconductor (CMOS).
An integrated circuit design that is printed on a microchip that contains different types of semiconductor transistors to create a circuit that both uses very little power and is resistant to high levels of electronic noise.
- Ion-sensitive field-effect transistor (ISFET).
A type of transistor that is sensitive to changes in ion concentration.
- Single-end and paired-end sequencing
In single-end sequencing, a DNA template is sequenced only in one direction. In paired-end sequencing, a DNA template is sequenced from both sides; the forward and reverse reads may or may not overlap. A deviation in the expected genome alignment between two ends of a paired-end read can indicate astructural variation.
- Structural variant
A variation larger than single-nucleotide polymorphisms (SNPs). This can include the insertion or deletion of blocks of DNA, inversions or translocations of DNA segments, and copy-number differences.
- ChIP–seq
(Chromatin immunoprecipitation followed by sequencing). A method used to analyse protein interactions with DNA by combining Chip with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins.
- ATAC–seq
(Assay for transposase-accessible chromatin with high-throughput sequencing). A method that uses the activity of a hyperactive transposase to cleave exposed DNA and add sequencing adapters. Regions that cannot be sequenced are inferred to be chromatin interacting.
- RNA sequencing (RNA-seq).
A method of sequencing cDNA derived from RNA. This approach can be used to sequence both coding and non-coding RNA.
- Real-time sequencing
A sequencing strategy used in the Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) platforms. In these approaches there is no pause after the detection of a base or series of bases, thus the sequence is derived in real-time.
- Barcodes
A series of known bases added to a template molecule either through ligation or amplification. After sequencing, these barcodes can be used to identify which sample a particular read is derived from.
- Zero-mode waveguides (ZMW).
Nanostructure devices used in the Pacific Biosciences (PacBio) platform. Each ZMW well (also called a waveguide) is several nanometres in diameter and is anchored to a glass substrate. The size of each well does not allow for light propagation, thus the fluorophores bound to bases can only be visualized through the glass substrate in the bottom-most portion of the well, a volume in the zeptolitre range.
- Read of insert
The highest-quality single sequence for an insert, regardless of the number of passes.
- Consensus sequence
In next-generation sequencing (NGS) routines that allow multiple overlapping reads from a single molecule of DNA, all related reads are aligned to each other and the most likely base at each position is determined. This process helps to overcome high, single-pass error rates. A high-quality consensus sequence derived from the circular template from Pacific Biosciences (PacBio) is called a circular consensus sequence (CCS).
- Squiggle space
A system exclusively used by Oxford Nanopore Technologies (ONT). As DNA translocates through the pore, a shift in voltage occurs that is directly correlated to a k-mer within the pore. Thus, the signal derived from a nanopore run is a continuous series of voltage shifts (squiggles) that represent a series of overlapping k-mers.
- K-mer
A substring within a sequence of bases of some (k) length. Currently, k-mer sizes of Oxford Nanopore Technologies (ONT) range from 3 to 6 bases.
- 1D and 2D reads
Oxford Nanopore Technologies (ONT) sequencing allows for both the full forward and full reverse strand of a double-stranded DNA (dsDNA) molecule to be sequenced and associated. A 1D read is the sequence of DNA bases derived from either the forward or reverse DNA strand. A 2D read is a consensus sequence derived from both the forward and the reverse reads.
- BAC-by-BAC sequencing
A sequencing method where a physical map is generated from overlapping bacterial artificial chromosome (BAC) clones tiled across a chromosome. Each BAC is then fragmented and sequenced. The sequenced fragments are aligned with the knowledge of the originating BAC.
- Linked reads
Reads derived from the 10X Genomics synthetic long-read platform. These are discontinuous reads each sharing the same barcode, thus they are derived from the same original long molecule.
- Read cloud
The means by which the 10X Genomics platform determines a synthetic long read. Discontinuous linked reads from the same genomic region are aligned to each other. No single linked read contains the entire long sequence; however, when they are stacked, full coverage is achieved.
- Polymerase reads
Contiguous sequences of nucleotides incorporated by the DNA polymerase while reading a template. These reads include sequences from adapters and can represent sequences from multiple passes around a circular template.
- Single-pass
The single-molecule real-time (SMRT) sequencing approach from Pacific Biosciences (PacBio) enables a single molecule of DNA to be sequenced multiple times. A single pass is one single iteration through a molecule.
- Subreads
The sequences derived from a single pass as a polymerase traverses a DNA molecule multiple times. A subread is trimmed to exclude any adapter sequence.
- Whole-exome and targeted sequencing
Sequencing of only exons or other selected regions. A system of capture or amplification is used to isolate or enrich for only exons or target regions. This is done by designing probes or primers for the regions of interest.
- Genome phasing
A method to identify which chromosome a DNA sequence is derived from. By examining polymorphisms, the chromosome of origin can be inferred by matching the reads that share the same variation.
- Family studies
A study design in which many members of a family across several generations are sequenced. These studies are used to understand how phenotypes manifest within a particular genotype background.
- Helicos Genetic Analysis System
A sequencing technology based on single nucleotide addition. Each nucleotide contains a ‘virtual terminator’ that prevents the incorporation of multiple nucleotides per cycle.
- Fluorescence resonance energy transfer
(FRET; also known as Förster resonance energy transfer). A system in which energy can be transferred from one light-sensitive molecule to another. When the two molecules are in close proximity (≤30 nm), energy transferred between the two molecules modulates the intensity of a fluorescence signal.
Footnotes
Competing interests statement
The authors declare competing interests: see Web version for details.
References
- 1.Watson JD & Crick FH The structure of DNA. Cold Spring Harb. Symp. Quant. Biol 18, 123–131 (1953). [DOI] [PubMed] [Google Scholar]
- 2.Mardis ER Next-generation sequencing platforms. Annu. Rev. Anal. Chem. (Palo Alto Calif.) 6, 287–303 (2013). [DOI] [PubMed] [Google Scholar]; This article provides a concise description of technological advancements supporting NGS.
- 3.Wetterstrand KA DNA sequencing costs: data from the NHGRI Genome Sequencing Program (GSP). National Human Genome Research Institute; [online], http://www.genome.gov/sequencingcosts (updated 15 Jan 2016). [Google Scholar]
- 4.Kircher M & Kelso J High-throughput DNA sequencing — concepts and limitations. Bioessays 32, 524–536 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Veritas Genetics. Veritas genetics launches $999 whole genome and sets new standard for genetic testing — Press Release. Veritas Genetics; [online], https://www.veritasgenetics.com/documents/VG-launches-999-whole-genome.pdf (updated 4 Mar 2016). [Google Scholar]
- 6.Veritas Genetics. Veritas genetics breaks $1,000 whole genome barrier — Press Release. Veritas Genetics; [online], https://www.veritasgenetics.com/documents/VG-PGP-Announcement-Final.pdf (29 Sep 2015). [Google Scholar]
- 7.Liu L et al. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol 2012, 251364 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dressman D, Yan H, Traverso G, Kinzler KW & Vogelstein B Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc. Natl Acad. Sci. USA 100, 8817–8822 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shendure J. et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309, 1728–1732 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Kim JB et al. Polony multiplex analysis of gene expression (PMAGE) in mouse hypertrophic cardiomyopathy. Science 316, 1481–1484 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Leamon JH et al. A massively parallel PicoTiterPlate based platform for discrete picoliter-scale polymerase chain reactions. Electrophoresis 24, 3769–3777 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Fedurco M, Romieu A, Williams S, Lawrence I & Turcatti G BTA, a novel reagent for DNA attachment on glass and efficient generation of solid-phase amplified DNA colonies. Nucleic Acids Res. 34, e22 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hairis TD et al. Single-molecule DNA sequencing of a viral genome. Science 320, 106–109 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Drmanac R et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327, 78–81 (2010). [DOI] [PubMed] [Google Scholar]; This paper describes the use of DNA nanoballs to achieve clonal amplification and the use of cPAL to achieve human genome sequencing as implemented by Complete Genomics (BGI).
- 15.Tomkinson AE, Vijayakumar S, Pascal JM & Ellenberger T DNA ligases: structure, reaction mechanism, and function. Chem. Rev 106, 687–699 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Landegren U, Kaiser R, Sanders J & Hood L A ligase-mediated gene detection technique. Science 241, 1077–1080 (1988). [DOI] [PubMed] [Google Scholar]
- 17.Valouev A et al. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res. 18, 1051–1063 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the use of cleavable two-base-encoded probes to achieve genome-wide nucleosome mapping in Caenorhabditis elegans. This technology is implemented by Applied Biosystems (Thermo Fisher) for the SOLiD platform.
- 18.Metzker ML Sequencing technologies — the next generation. Nat. Rev. Genet 11, 31–46 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Ju J. et al. Four-color DNA sequencing by synthesis using cleavable fluorescent nucleotide reversible terminators. Proc. Natl Acad. Sci. USA 103, 19635–19640 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo J et al. Four-color DNA sequencing with 3’-O-modified nucleotide reversible terminators and chemically cleavable fluorescent dideoxynucleotides. Proc. Natl Acad. Sci. USA 105, 9145–9150 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Timmerman L DNA sequencing market will exceed $20 billion, says Illumina CEO Jay Flatley. Forbes; [online], http://www.forbes.com/sites/luketimmerman/2015/04/29/qa-with-jay-flatley-ceo-of-illumina-the-genomics-company-pursuing-a-20b-market/#4dbd19943bf5 (29 Apr 2015). [Google Scholar]
- 22.Karow J Qiagen launches GeneReader NGS System at AMP; presents performance evaluation by broad. GenomeWeb; [online], https://www.genomeweb.com/molecular-diagnostics/qiagen-launches-genereaderngs-system-amp-presents-performance-evaluation (4 Nov 2015). [Google Scholar]
- 23.Smith DR & McKernan K Methods of producing and sequencing modified polynucleotides. US Patent 8058030 (2011).
- 24.Margulies M et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the development of the first NGS technology through the use of pyrosequencing. The authors demonstrate this method through sequencing of the Mycoplasma genitalium genome.
- 25.Rothberg JM et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 475, 348–352 (2011). [DOI] [PubMed] [Google Scholar]; This paper describes the first non-optical sequencing technology using a massively parallel semi-conductor device to monitor H+ release during DNA synthesis, as implemented by the Ion Torrent platform (Thermo Fisher). The authors demonstrate this technology by sequencing both bacterial and human DNA.
- 26.Rieber N et al. Coverage bias and sensitivity of variant calling for four whole-genome sequencing technologies. PLoS ONE 8, e66621 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zook JM et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol 32, 246–251 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Nothnagel M et al. Technology-specific error signatures in the 1000 Genomes Project data. Hum. Genet 130, 505–516 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Shen Y Sarin S, Liu Y, Hobert O & Pe’er I Comparing platforms for C. elegans mutant identification using high-throughput whole-genome sequencing. PLoS ONE, e4012 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan M et al. Development of a next-generation sequencing method for BRCA mutation screening: a comparison between a high-throughput and a benchtop platform. J. Mol. Diagnost 14, 602–612 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Wall JD et al. Estimating genotype error rates from high-coverage next-generation sequence data. Genome Res. 24, 1734–1739 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harismendy O et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 10, R32 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.BGI. Revolocity Whole Genome Sequencing Service — Press Release. BGI; [online], http://u70g92ptbyk941g21dd41fc4.wpengine.netdna-cdn.com/wp-content/uploads/2015/10/Global-WGSRevolocity-ENG-10-15.pdf (2015). [Google Scholar]
- 34.Karow J BGI halts revolocity launch, cuts complete genomics staff as part of strategic shift. GenomeWeb; [online], https://www.genomeweb.com/sequencing-technology/bgi-halts-revolocity-launch-cuts-complete-genomics-staff-part-strategic-shift (23 Nov 2015). [Google Scholar]
- 35.Bentley DR et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates the use of reversible dye-terminator chemistry for human genome sequencing. This platform is used by the Illumina suite of platforms.
- 36.Dohm JC, Lottaz C, Borodina T & Himmelbauer H Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 36, e105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakamura K et al. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res. 39, e90 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Minoche AE, Dohm JC & Himmelbauer H Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and genome analyzer systems. Genome Biol. 12, R112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ley TJ et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 456, 66–72 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarin S, Prabhu S, O’Meara MM, Pe’er I & Hobert O Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat. Methods 5, 865–867 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park PJ ChIP–seq: advantages and challenges of a maturing technology. Nat. Rev. Genet 10, 669–680 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This review provides an overview of ChIP–seq methods for detecting chromatin–DNA interactions and their importance to epigenetics research.
- 42.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brunner AL et al. Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res. 19, 1044–1056 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Z, Gerstein M & Snyder M RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet 10, 57–63 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This review provides an overview of advances and challenges in techniques that are used in transcriptomic research with a specific focus in methods that use NGS technologies.
- 45.Wang X et al. A trimming-and-retrieving alignment scheme for reduced representation bisulfite sequencing. Bioinformatics 31, 2040–2042 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qiagen. Oncology insights enabled by knowledge base-guided panel design and the seamless workflow of the GeneReader NGS system — Press Release. Qiagen; [online], http://www.genereaderngs.com/PROM-9192-001_1100403_WP_GeneReader_NGS_0116_NA.pdf (2016). [Google Scholar]
- 47.Forgetta V et al. Sequencing of the Dutch elm disease fungus genome using the Roche/454 GS-FLX Titanium System in a comparison of multiple genomics core facilities. J. Biomol. Tech 24, 39–49 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loman NJ et al. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol 30, 434–439 (2012). [DOI] [PubMed] [Google Scholar]
- 49.GenomeWeb. Roche shutting down 454 sequencing business. GenomeWeb; [online], https://www.genomeweb.com/sequencing/roche-shutting-down-454-sequencing-business (15 Oct 2015). [Google Scholar]
- 50.Malapelle U et al. Ion Torrent next-generation sequencing for routine identification of clinically relevant mutations in colorectal cancer patients. J. Clin. Pathol 68, 64–68 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Li S et al. Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study. Nat. Biotechnol 32, 915–925 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Life Technologies. Ion semiconductor sequencing uniquely enables both accurate long reads and paired-end sequencing. Life Technologies; [online], https://www3.appliedbiosystems.com/cms/groups/applied_markets_marketing/documents/generaldocuments/cms_098680.pdf (2011) [Google Scholar]
- 53.Campbell PJ et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat. Genet 40, 722–729 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McCarroll SA & Altshuler DM Copy-number variation and association studies of human disease. Nat. Genet 39, S37–S42 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Mirkin SM Expandable DNA repeats and human disease. Nature 447, 932–940 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Stankiewicz P & Lupski JR Structural variation in the human genome and its role in disease. Annu. Rev. Med 61, 437–455 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Eid J et al. Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138 (2009). [DOI] [PubMed] [Google Scholar]; The authors describe the development of a real-time sequencing method using their zero-mode waveguide sensors as implemented by the Pacific Biosciences platform. The authors demonstrate the technique by sequencing synthetic DNA templates.
- 58.Levene MJ et al. Zero-mode waveguides for single-molecule analysis at high concentrations. Science 299, 682–686 (2003). [DOI] [PubMed] [Google Scholar]
- 59.Loomis EW et al. Sequencing the unsequenceable: expanded CGG-repeat alleles of the fragile X gene. Genome Res. 23, 121–128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clarke J et al. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol 4, 265–270 (2009). [DOI] [PubMed] [Google Scholar]; The authors demonstrate the use of a mutant alpha-hemolysin for ordered, continuous detection of free nucleotides in solution. This work provides the basis for the approach used by ONT.
- 61.Voskoboynik A et al. The genome sequence of the colonial chordate, Botryllus schlosseri. eLife 2, e00569 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McCoy RC et al. Illumina TruSeq synthetic long-reads empower de novo assembly and resolve complex, highly-repetitive transposable elements. PLoS ONE 9, e106689 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schatz MC, Delcher AL & Salzberg SL Assembly of large genomes using second-generation sequencing. Genome Res. 20, 1165–1173 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.English AC et al. Assessing structural variation in a personal genome-towards a human reference diploid genome. BMC Genomics 16, 286 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carneiro MO et al. Pacific Biosciences sequencing technology for genotyping and variation discovery in human data. BMC Genomics 13, 375 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Quail MA et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 13, 341 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koren S et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol 30, 693–700 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Larsen PA, Heilman AM & Yoder AD The utility of PacBio circular consensus sequencing for characterizing complex gene families in non-model organisms. BMC Genomics 15, 720 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heger M PacBio launches higher-throughput, lower-cost single-molecule sequencing system. GenomeWeb; [online], https://www.genomeweb.com/business-news/pacbio-launches-higher-throughput-lower-cost-single-molecule-sequencing-system (01 Oct 2015). [Google Scholar]
- 70.Goodwin S et al. Oxford Nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Res. 25, 1750–1756 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jain M et al. Improved data analysis for the MinION nanopore sequencer. Nat. Methods 12, 351–356 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Heger M 10X Genomics, Pacific Biosciences provide business updates at JP Morgan Healthcare Conference. GenomeWeb; [online], https://www.genomeweb.com/sequencing-technology/10x-genomics-pacific-biosciences-provide-business-updates-jp-morgan-healthcare (13 Jan 2016). [Google Scholar]
- 73.Cirulli ET & Goldstein DB Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat. Rev. Genet 11, 415–425 (2010). [DOI] [PubMed] [Google Scholar]; This review provides a comprehensive overview of advances in, and challenges of using, WGS for variant discovery in human disease.
- 74.Ellis MJ et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 486, 353–360 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Prat A & Perou CM Mammary development meets cancer genomics. Nat. Med 15, 842–844 (2009). [DOI] [PubMed] [Google Scholar]
- 76.1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sudmant PH et al. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.UK10K Consortium. The UK10K project identifies rare variants in health and disease. Nature 526, 82–90 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gudbjartsson DF et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet 47, 435–444 (2015). [DOI] [PubMed] [Google Scholar]
- 81.Regalado A U.S. to develop DNA study of one million people. MIT Technology Review; [online], http://www.technologyreview.com/news/534591/us-to-develop-dna-study-of-one-million-people (30 Jan 2015). [Google Scholar]
- 82.Sidore C et al. Genome sequencing elucidates Sardinian genetic architecture and augments association analyses for lipid and blood inflammatory markers. Nat. Genet 47, 1272–1281 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hodges E et al. Genome-wide in situ exon capture for selective resequencing. Nat. Genet 39, 1522–1527 (2015). [DOI] [PubMed] [Google Scholar]; This paper describes the in situ capture and selective enrichment of human exons for downstream NGS. This manuscript provides the methodological basis for whole-exome and targeted sequencing.
- 84.Iossifov I et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.O’Roak BJ et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sanders SJ et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Griffith M et al. Optimizing cancer genome sequencing and analysis. Cell Syst. 1, 210–223 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rauch C et al. Towards an understanding of DNA recognition by the methyl-CpG binding domain 1. J. Biomol. Struct. Dyn 22, 695–706 (2005). [DOI] [PubMed] [Google Scholar]
- 89.Oda M et al. High-resolution genome-wide cytosine methylation profiling with simultaneous copy number analysis and optimization for limited cell numbers. Nucleic Acids Res. 37, 3829–3839 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Irizarry RA et al. Comprehensive high-throughput arrays for relative methylation (CHARM). Genome Res. 18, 780–790 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Meissner A et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 33, 5868–5877 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Flusberg BA et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 7, 461–465 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wescoe ZL, Schreiber J & Akeson M Nanopores discriminate among five C5-cytosine variants in DNA. J. Am. Chem. Soc 136, 16582–16587 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lam ET et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol 30, 771–776 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Eichler EE, Clark RA & She X An assessment of the sequence gaps: unfinished business in a finished human genome. Nat. Rev. Genet 5, 345–354 (2004). [DOI] [PubMed] [Google Scholar]
- 96.Chaisson MJ, Wilson RK & Eichler EE Genetic variation and the de novo assembly of human genomes. Nat. Rev. Genet 16, 627–640 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chaisson MJ et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517, 608–611 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article provides strong support for the utility of long-read sequencing for generating high-quality reference genomes. The authors demonstrate this by closing and/or extending gaps and resolving structural variants in the GRCh37 human reference genome.
- 98.Ritz A et al. Characterization of structural variants with single molecule and hybrid sequencing approaches. Bioinformatics 30, 3458–3466 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Snyder MW, Adey A, Kitzman JO & Shendure J Haplotype-resolved genome sequencing: experimental methods and applications. Nat. Rev. Genet 16, 344–358 (2015). [DOI] [PubMed] [Google Scholar]
- 100.Kuleshov V et al. Whole-genome haplotyping using long reads and statistical methods. Nat. Biotechnol 32, 261–266 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Treutlein B et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 509, 371–375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sharon D, Tilgner H, Grubert F & Snyder M A single-molecule long-read survey of the human transcriptome. Nat. Biotechnol 31, 1009–1014 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Quick J et al. Rapid draft sequencing and real-time nanopore sequencing in a hospital outbreak of Salmonella. Genome Biol. 16, 114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Quick J et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 530, 228–232 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.GenomeWeb. White House announces efforts to accelerate precision medicine initiative. GenomeWeb; [online], https://www.genomeweb.com/molecular-diagnostics/white-house-announces-efforts-accelerate-precision-medicine-initiative (25 Feb 2016). [Google Scholar]
- 106.Illumina. Illumina forms new company to enable early cancer detection via blood-based screening — Press Release. Illumina; [online], http://www.illumina.com/company/news-center/press-releases/press-release-details.html?newsid=2127903 (10 Jan 2016). [Google Scholar]
- 107.Schatz MC & Langmead B The DNA data deluge: fast, efficient genome sequencing machines are spewing out more data than geneticists can analyze. IEEE Spectr. 50, 26–33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pop M & Salzberg SL Bioinformatics challenges of new sequencing technology. Trends Genet. 24, 142–149 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sunyaev SR Inferring causality and functional significance of human coding DNA variants. Hum. Mol. Genet 21, R10–R17 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gargis AS et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat. Biotechnol 30, 1033–1036 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chrystoja CC & Diamandis EP Whole genome sequencing as a diagnostic test: challenges and opportunities. Clin. Chem 60, 724–733 (2014). [DOI] [PubMed] [Google Scholar]
- 112.McGuire AL et al. Point-counterpoint. Ethics and genomic incidental findings. Science 340, 1047–1048 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bowers J et al. Virtual terminator nucleotides for next-generation DNA sequencing. Nat. Methods 6, 593–595 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Heger M China’s Direct Genomics unveils new targeted NGS system based on Helicos Tech for clinical use. GenomeWeb; [online], https://www.genomeweb.com/business-news/chinas-direct-genomics-unveils-new-targeted-ngs-system-based-helicos-tech-clinical-use (27 Oct 2015). [Google Scholar]
- 115.Karow J Oxford Nanopore presents details on new high-throughput sequencer, improvements to Minion. GenomeWeb; [online], https://www.genomeweb.com/sequencing/oxford-nanopore-presents-details-new-high-throughput-sequencer-improvements-mini (16 Sep 2014). [Google Scholar]
- 116.Karow J Sigma-Aldrich enters co-marketing agreement with GenapSys for Genius sequencer. GenomeWeb; [online], https://www.genomeweb.com/sequencing-technology/sigma-aldrich-enters-co-marketing-agreement-genapsys-genius-sequencer (1 Jul 2015). [Google Scholar]
- 117.Roche. Roche acquires Genia Technologies to strengthen next generation sequencing pipeline — Press Release. Roche; [online], http://www.roche.com/media/store/releases/med-cor-2014-06-02.htm (2 Jun 2014). [Google Scholar]
- 118.Heger M Illumina unveils mini targeted sequencer, semiconductor sequencing project at JP Morgan Conference. GenomeWeb; [online], https://www.genomeweb.com/sequencing-technology/illumina-unveils-mini-targeted-sequencer-semiconductor-sequencing-project-jp (1 Jan 2016). [Google Scholar]
- 119.NanoString. NanoString Technologies presents proof-of-concept data for novel massively parallel single molecule sequencing chemistry at AGBT meeting — Press Release. NanoString; [online], http://investors.nanostring.com/releasedetail.cfm?ReleaseID=954517 (11 Feb 2016). [Google Scholar]
- 120.Raz T & Pascaline M Nucleic acid target detection using a detector, a probe and an inhibitor. US Patent 20130344485 (2013).
- 121.Mankos M et al. A novel low energy electron microscope for DNA sequencing and surface analysis. Ultramicroscopy 145, 36–49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Augenlicht LH & Kobrin D Cloning and screening of sequences expressed in a mouse colon tumor. Cancer Res. 42, 1088–1093 (1982). [PubMed] [Google Scholar]
- 123.Dandy DS, Wu P & Grainger DW Array feature size influences nucleic acid surface capture in DNA microarrays. Proc. Natl Acad. Sci. USA 104, 8223–8228 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Keating BJ et al. Concept, design and implementation of a cardiovascular gene-centric 50 k SNP array for large-scale genomic association studies. PLoS ONE, e3583 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.DeRisi J et al. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat. Genet 14, 457–460 (1996). [DOI] [PubMed] [Google Scholar]
- 126.Alizadeh AA & Staudt LM Genomic-scale gene expression profiling of normal and malignant immune cells. Curr. Opin. Immunol 12, 219–225 (2000). [DOI] [PubMed] [Google Scholar]
- 127.Rhodes DR et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc. Natl Acad. Sci. USA 101, 9309–9314 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Vora GJ, Meador CH, Stenger DA & Andreadis JD Nucleic acid amplification strategies for DNA microarray-based pathogen detection. Appl. Environ. Microbiol 70, 3047–3054 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wilson WJ et al. Sequence-specific identification of 18 pathogenic microorganisms using microarray technology. Mol. Cell Probes 16, 119–127 (2002). [DOI] [PubMed] [Google Scholar]
- 130.Imai K, Kricka LJ & Fortina P Concordance study of 3 direct-to-consumer genetic-testing services. Clin. Chem 57, 518–521 (2011). [DOI] [PubMed] [Google Scholar]
- 131.Dolgin E. Personalized investigation. Nat. Med 16, 953–955 (2010). [DOI] [PubMed] [Google Scholar]
- 132.Jia P, Wang L, Meltzer HY & Zhao Z Common variants conferring risk of schizophrenia: a pathway analysis of GWAS data. Schizophr. Res 122, 38–42 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Welter D et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 42, D1001–D1006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Carter NP Methods and strategies for analyzing copy number variation using DNA microarrays. Nat. Genet 39, S16–S21 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Vrijenhoek T. et al. Recurrent CNVs disrupt three candidate genes in schizophrenia patients. Am. J. Hum. Genet 83, 504–510 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Marshall CR et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet 82, 477–488 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Buck MJ & Lieb JD ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics 83, 349–360 (2004). [DOI] [PubMed] [Google Scholar]
- 138.Liang L et al. A cross-platform analysis of 14,177 expression quantitative trait loci derived from lymphoblastoid cell lines. Genome Res. 23, 716–726 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhao S, Fung-Leung WP, Bittner A, Ngo K & Liu X Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 9, e78644 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Holland PM, Abramson RD, Watson R & Gelfand DH Detection of specific polymerase chain reaction product by utilizing the 5′→3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc. Natl Acad. Sci. USA 88, 7276–7280 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Morin PA & McCarthy M Highly accurate SNP genotyping from historical and low-quality samples. Mol. Ecol. Notes 7, 937–946 (2007). [Google Scholar]
- 142.VanGuilder HD, Vrana KE & Freeman WM Twenty-five years of quantitative PCR for gene expression analysis. Biotechniques 44, 619–626 (2008). [DOI] [PubMed] [Google Scholar]
- 143.Weaver S et al. Taking qPCR to a higher level: Analysis of CNV reveals the power of high throughput qPCR to enhance quantitative resolution. Methods 50, 271–276 (2010). [DOI] [PubMed] [Google Scholar]
- 144.Sedlak RH, Cook L, Cheng A, Magaret A & Jerome KR Clinical utility of droplet digital PCR for human cytomegalovirus. J. Clin. Microbiol 52, 2844–2848(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kulkarni MM in Current Protocols in Molecular Biology Ch. 25 (eds Ausubel FM et al. ) (Wiley, 2011). [Google Scholar]
- 146.Nielsen T et al. Analytical validation of the PAM50-based Prosigna Breast Cancer Prognostic Gene Signature Assay and nCounter Analysis System using formalin-fixed paraffin-embedded breast tumor specimens. BMC Cancer 14, 177 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ku BM et al. High-throughput profiling identifies clinically actionable mutations in salivary duct carcinoma. J. Transl. Med 12, 299 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sailani MR et al. The complex SNP and CNV genetic architecture of the increased risk of congenital heart defects in Down syndrome. Genome Res. 23, 1410–1421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lira ME et al. Multiplexed gene expression and fusion transcript analysis to detect ALK fusions in lung cancer. J. Mol. Diagn 15, 51–61 (2013). [DOI] [PubMed] [Google Scholar]
- 150.Schwartz DC et al. Ordered restriction maps of Saccharomyces cerevisiae chromosomes constructed by optical mapping. Science 262, 110–114(1993). [DOI] [PubMed] [Google Scholar]
- 151.Hastie AR et al. Rapid genome mapping in nanochannel arrays for highly complete and accurate de novo sequence assembly of the complex Aegilops tauschii genome. PLoS ONE 8, e55864 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cao H et al. Rapid detection of structural variation in a human genome using nanochannel-based genome mapping technology. Gigascience 3, 34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pendleton M et al. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat. Methods 12, 780–786 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Life Technologies. 5500 W series genetic analyzers. Life Technologies; [online], https://tools.thermofisher.com/content/sfs/brochures/5500-w-series-spec-sheet.pdf (2012). [Google Scholar]
- 155.Yuzuki D BGISEQ-500 debuts at the International Congress of Genomics 10. Next Generation Technologist; [online], http://www.yuzuki.org/bgiseq-500-debut-at-the-international-congress-of-genomics-10 (24 Oct 2015). [Google Scholar]
- 156.Winnick E Illumina launches four new systems; provides financial, Dx update at JP Morgan. GenomeWeb; [online], https://www.genomeweb.com/business-news/illumina-launches-four-new-systems-provides-financial-dx-update-jp-morgan (12 Jan 2015). [Google Scholar]
- 157.[No authors listed.] Illumina HiSeq 3000 Service Fees. Oregon State University; [online], http://cgrb.oregonstate.edu/core/illumina-hiseq-3000/illumina-hiseq-3000-service-fees (updated 1 Jan 2016) [Google Scholar]
- 158.Heger M Thermo Fisher launches new systems to focus on plug and play targeted sequencing. GenomeWeb; [online], https://www.genomeweb.com/sequencing-technology/thermo-fisher-launches-new-systems-focus-plug-and-play-targeted-sequencing 1 Sep 2015). [Google Scholar]
- 159.Ip CL et al. MinION analysis and reference consortium: Phase 1 data release and analysis. F1000Research 4, 1075 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Glenn TC Field guide to next-generation DNA sequencers. Mol. Ecol. Resour 11, 759–769 (2011). [DOI] [PubMed] [Google Scholar]
- 161.Karow J At AGBT, 10X Genomics launches GemCode platform; shipments slated for Q2 as firm battles IP lawsuits. GenomeWeb; [online], https://www.genomeweb.com/sample-prep/agbt-10x-genomics-launches-gemcode-platform-shipments-slated-q2-firm-battles-ip-lawsuits (2 Mar 2015). [Google Scholar]