

Abstract
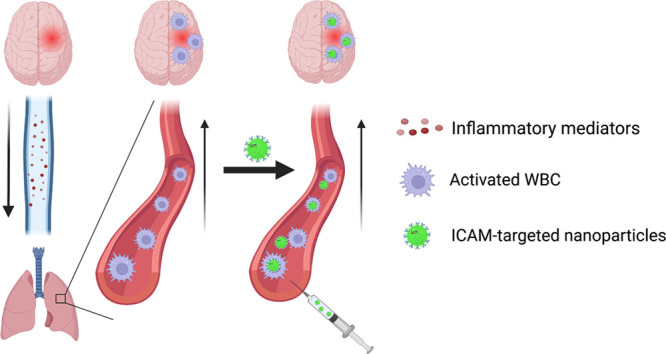
Ex vivo-loaded white blood cells (WBC) can transfer cargo to pathological foci in the central nervous system (CNS). Here we tested affinity ligand driven in vivo loading of WBC in order to bypass the need for ex vivo WBC manipulation. We used a mouse model of acute brain inflammation caused by local injection of tumor necrosis factor alpha (TNF-α). We intravenously injected nanoparticles targeted to intercellular adhesion molecule 1 (anti-ICAM/NP). We found that (A) at 2 h, >20% of anti-ICAM/NP were localized to the lungs; (B) of the anti-ICAM/NP in the lungs >90% were associated with leukocytes; (C) at 6 and 22 h, anti-ICAM/NP pulmonary uptake decreased; (D) anti-ICAM/NP uptake in brain increased up to 5-fold in this time interval, concomitantly with migration of WBCs into the injured brain. Intravital microscopy confirmed transport of anti-ICAM/NP beyond the blood–brain barrier and flow cytometry demonstrated complete association of NP with WBC in the brain (98%). Dexamethasone-loaded anti-ICAM/liposomes abrogated brain edema in this model and promoted anti-inflammatory M2 polarization of macrophages in the brain. In vivo targeted loading of WBC in the intravascular pool may provide advantages of coopting WBC predisposed to natural rapid mobilization from the lungs to the brain, connected directly via conduit vessels.
Keywords: brain, drug delivery, inflammation, nanoparticles, pharmacokinetics, white blood cells
Introduction
Targeted drug delivery to the brain promises breakthroughs in treatment of debilitating and lethal pathologies, including stroke, traumatic brain injury, glioblastoma and other brain tumors, meningitis, and neurodegenerative diseases.1,2 Various carriers with distinct chemistry, geometry, mechanical flexibility, and affinity have been devised to achieve this elusive goal.3−7 One approach to enhancing delivery employs targeting to and across the cerebral vasculature using antibodies, peptides, and other ligands of molecules that are stably expressed on the luminal surface of brain vessels. However, targeting these molecules, including receptors for transferrin, insulin, and growth factors, does not provide selectivity for sites of injury and inflammation.8,9 In order to achieve enhanced specificity for injured regions of the brain, targeting inducible cell adhesion molecules (CAMs) expressed on endothelial cells, such as vascular CAM (VCAM),9−11 has been tested and has shown improved delivery and pharmacologic effects. Despite these inroads, direct, specific delivery to the parenchyma of the injured region of the brain remains an elusive goal.
To achieve the formidable goal of delivering drugs to the brain parenchyma within injured areas of the brain, it is tempting to utilize the natural homing of leukocytes to the pathologically altered region of the brain.12−15 Innate immune cells, namely, neutrophils and monocytes, are the “first responders” in acute inflammatory conditions and, as such, have the potential to be used to overcome physiological barriers to achieving high levels of specific drug delivery to the injured brain. This natural tropism is mediated by several including (A) activation of endothelial cells and white blood cells (WBC) by inflammatory mediators, (B) increased exposure of adhesion molecules on both endothelial cells and WBC, (C) widening of endothelial tight junctions,16,17 (D) attraction of circulating leukocytes via a chemokine gradient emanating from the site of injury,18 and (E) abnormal vasoreactivity due to nitric oxide quenching and pathological hydrodynamics of blood flow. In concert, these mechanisms of WBC activation, binding, adherence, and extravasation provide a strong, redundant, and relatively common tropism of WBC into the sites of CNS injury.
The initial findings and encouraging characteristics of WBC-mediated drug delivery to CNS have been achieved using the extracorporeal (ex vivo) isolation, loading, and re-administration of modified WBC in animals,19−23 just as for CAR-T immunotherapies.24,25 Indeed, ex vivo loading of WBC is attractive, and several groups have reported therapeutic benefits of injecting drug-loaded WBC in animal models of neurological disorders.19,21,23,26−31
There are several approaches for WBC loading, which may differently use the mechanisms described above for local tropism. An alternative strategy to ex vivo WBC loading would be to specifically load those WBC that are predisposed to localize to the injured brain with drugs or drug carriers in vivo, bypassing the need for any ex vivo manipulation. This would bypass the technically complicated procedures required to extract, modify, and load WBC ex vivo, which could result in pro-inflammatory, prothrombotic complications that, while they are often acceptable risks in the treatment of cancer, may not be tolerated in critically ill patients, such as in sepsis and stroke. Additionally, in rapidly evolving conditions such as stroke, the need to treat patients as quickly as possible would likely preclude the use of any strategy involving ex vivo manipulation of cells. Direct, in vivo targeting of WBC has been reported to promote nanoparticle delivery32 and penetration33 into tumors, among other organs.34,35 Additionally, targeting of pulmonary intravascular leukocytes has been reported as a therapeutic strategy in treatment of mouse models of acute respiratory distress syndrome by our group and others.36−39 This approach represents a significant step forward in WBC-based drug delivery. It has been reported that intercellular adhesion molecule 1 (ICAM) is expressed on the surface of many activated WBC, including monocytes and neutrophils.40 Following inflammatory stimuli, the surface expression of ICAM on immune cells is significantly upregulated,41−44 providing selectivity for delivery to activated WBC by targeting to ICAM.
The intravascular leukocyte pool consists of those that are freely flowing in the bloodstream and those that are marginated within vascular beds. The pulmonary circulation hosts the largest and most dynamic pool of intravascular WBC that are poised to quickly respond to local and remote signals from damaged tissues.45,46 The marginated pool of WBC consists of peripheral WBC that transiently dwell in the pulmonary circulation associated with the endothelial surface, constantly exchanging with the pool that is freely flowing in circulation.37 In fact, following acute inflammatory insults, WBC margination in the pulmonary capillaries is significantly enhanced, creating a “carpet” of WBC coating the endothelial cells lining the vessel wall.47−49 Targeting to this pool of leukocytes has been shown to have therapeutic benefits in the treatment of acute lung injury;36,38,39 however, it has not been explored to date for drug delivery for nonpulmonary conditions.
We postulate that the ideal cells for achieving WBC-mediated brain delivery are the intravascular leukocytes that are marginated within the pulmonary vasculature. These cells are predominantly localized to the capillaries50−52 where the low blood flow rate, low shear stress, small diameter, and vessel tortuosity are expected to provide favorable conditions for nanoparticle binding.53,54 WBC-targeted agents will encounter this major pool of marginated leukocytes within seconds as the first vascular bed encountered following intravenous injection is the pulmonary circulation. There are no intervening microvascular beds between the directly interconnected cerebral and pulmonary vasculatures. Hence, the pulmonary marginated leukocyte pool is the first major extracerebral set of cells to receive danger signals emanating from brain injuries (e.g., cytokines, exosomes, damage-associated molecular patterns). In fact, distal injuries often induce a secondary pulmonary pathology,55−58 which results in an increase in and hyperactivation of the pulmonary WBC pool. There are several reports that host defense cells responding to chronic neurological disorders mature in the lungs prior to trafficking to the injured brain.59−61 These considerations imply that the dynamic pool of pulmonary WBCs is ideally positioned to shuttle drugs directly from the lungs to sites of brain pathology. However, strategies for controllable, specific, effective, and safe loading of drugs into intravascular WBCs have not been reported.
In the current study, we characterized the dynamic localization of leukocytes and ICAM-targeted pharmacological agents in the blood, lungs, and brain in a murine model of acute neurovascular inflammation induced by direct injection of tumor necrosis factor α (TNF-α) into the brain parenchyma. This injection induces inflammation in the brain, including cerebral edema, similar to the inflammatory phase that occurs following stroke or aseptic meningitis.62 Our data presented below indicated that following TNF-α injection into the brain there is a rapid increase in the number of intravascular WBC and ICAM-expressing WBC in the lungs, resulting in elevated delivery of ICAM-targeted agents to the lungs, largely through uptake by intravascular WBC. In the brain, in contrast, these cells gradually accumulated and increased up to 5-fold 24 h after TNF-α injury. Dexamethasone-loaded, ICAM-targeted liposomes were able to harness this mechanism to completely prevent cerebral edema and increase anti-inflammatory M2 macrophage polarization in the TNF-α brain injury model. Overall, direct, in vivo targeting of the intravascular, marginated WBC pool shortly after brain injury provides a mechanism to harness this dynamic pool of cells for selective drug delivery to the brain.
Results
Systemic Response to TNF-α-Induced Acute Neurovascular Inflammation
Local, brain injection of TNF-α has been shown to reproducibly induce brain inflammation;8,9,63 however, it is well-known that localized injury/inflammation can also result in a systemic inflammatory response. Dynamics of immune markers (cytokines, chemokines, and white blood cells) were studied in the systemic circulation in order to probe the immune response. Multiplex analysis of cytokines revealed trends toward increasing concentrations of pro-inflammatory cytokines in the plasma after brain injury, at both 2 h (monocyte chemoattractant protein-1) and 24 h (interleukin-27, interferon-γ, interleukin-12p70), and a decrease in anti-inflammatory cytokines at 2 h (interleukin-10) and 24 h (interferon-β) (Supplemental Figure 1). Additionally, complete blood counts revealed that following TNF-α injury, there was a reduction in circulating lymphocytes and a marked elevation in neutrophils and monocytes (Supplemental Figure 2). Taken together, these results demonstrated that there was a response to local TNF-α injury in the brain that could be detected systemically.
As the lungs are the central station of the cardiovascular system and represent a key barrier to the environment, there is often a pulmonary response to distal inflammation to preempt any assault on the indispensable respiratory system. However, the secondary pulmonary response often is overzealous, resulting in severe complications such as acute lung injury/acute respiratory distress syndrome.64−67 Experiments were performed to follow up on the pro-inflammatory response detected in the circulation by assessing the uptake of isotope-labeled mAb targeted to a pan-leukocyte marker (CD45) and to a marker of activated leukocytes and endothelial cells (ICAM). Uptake of mAb in the lungs at various time points after injury was used as a marker of changes in the intravascular leukocyte population in response to brain inflammation. Direct measurements of isotope-labeled mAbs 30 min after IV injection showed (A) no significant differences in blood concentrations with time (Figure 1a,b, Supplemental Table 1), (B) peak lung uptake of both αCD45 and αICAM mAbs 2 h after brain injury (Figure 1d,e), (C) a return to baseline (naïve) levels of lung uptake for αICAM by 6 h after injury (Figure 1e), (D) a progressive increase in αCD45 and αICAM uptake in the brain up to 24 h after injury (Figure 1i,j). Taken together, the biodistribution data suggested that there was a transient increase in the activated, pulmonary marginated leukocyte pool following TNF-α injection into the brain as IV-injected isotope control mAb did not have significant lung uptake at any time point (Supplemental Table 1). On the other hand, the targeting dynamics in the brain suggests that there was likely both de novo synthesis of ICAM by activated endothelial cells along with an influx of activated leukocytes into the injured region of the brain.
Figure 1.

Local injection of TNF-α into the brain induces a systemic response. Following IV injection of αCD45, (A) blood, (D) lung, and (I) brain targeting was assessed at several time points post-TNF. Similar studies were performed for αICAM biodistribution in (B) blood, (E) lungs, and (J) brain. Data represented as percent of injected dose per gram tissue (%ID/g). Mice were injected with 5 μg mAb at the designated time points following TNF-α injection and mAb was allowed to circulate for 30 min prior to organ harvest. Naïve mice are represented as t = 0 h. Flow cytometry was used to measure the dynamics of ICAM+ leukocytes in the (C) peripheral blood 2 h after TNF-α injection, (G,H) pulmonary intravascular compartment 2 h after TNF-α injection, and (K) brain 24 h after TNF-α injection. (F) Histological analysis of lungs 2 h after TNF-α injection into the brain. Red: Leukocytes (CD45); Green: Autofluorescence (tissue structure); Scale bar = 50 μm. N = 3 mice/group. Comparisons in A, B, D, E, G, and H made via one-way ANOVA with Dunnett’s posthoc test vs naïve and in C, F, and I via unpaired Student’s t test.
To provide further support for this hypothesis, flow cytometry was performed on single cell suspensions derived from the blood, lungs, and brain to evaluate TNF-α-induced changes in the leukocyte populations. Analysis of total CD45+ cells in lungs and in brain revealed a trend toward increased intravascular leukocytes in the lungs 2 h after injury and a significant increase in infiltrating leukocytes in the brain (14-fold) 24 h after injury (Supplemental Figure 3). Cells were then further analyzed to assess the fraction of cells that expressed ICAM at the designated time points. Leukocytes were extracted from whole blood and incubated with radiolabeled αICAM in order to determine the number of ICAM molecules expressed on the surface of mouse leukocytes, and it was determined that there were (1.65 × 104) ± (1.4 × 103) αICAM binding sites/leukocyte (Supplemental Figure 4). Flow cytometry showed no changes in the fraction of leukocytes that were ICAM+ (Figure 1c) in the circulating pool; however, there was a significant increase in the fraction of leukocytes and endothelial cells that were ICAM+ both in the pulmonary intravascular compartment (1.9-fold) (Figure 1g,h, Supplemental Figure 5a) and in the brain (3-fold) (Figure 1k). Subtyping of intravascular leukocytes in the lungs showed that, 2 h after TNF-α injury, 54.2 ± 5.5% of recovered monocyte/macrophage (CD64+CD45+) and 14.8 ± 4.0% of recovered neutrophils (Ly6G+CD45+) expressed ICAM (Supplemental Figure 5b). Histological analysis of lung tissue 2 h after TNF-α injection showed a large number of leukocytes in the tissue (Figure 1f, Supplemental Figure 6). In summary, both mAb targeting data and flow cytometry of tissue single cell suspensions revealed an increase in ICAM-expressing WBC in the pulmonary intravascular pool, which suggests that targeting of activated leukocytes in the marginated pool using αICAM is a viable drug delivery strategy.
ICAM-Targeted Monoclonal Antibodies (mAbs) Migrate to the Brain
Encouraged by the increase in ICAM+ pulmonary intravascular leukocytes following brain injury (Figure 2d,e,f), we performed studies to appraise whether targeting mAbs or nanoparticles to these cells would provide selective drug delivery to the injured brain. First, we injected isotope-labeled mAbs into mice 2 h post-TNF-α injury to evaluate the role of target epitope/cell type on pharmacokinetics and biodistribution (Figure 2b).
Figure 2.

αICAM and αCD45 mAbs rapidly accumulate in the lungs and then migrate to the brain. (A) Schematic of proposed mechanism underlying leukocyte migration. (B) PK study timeline. Lung and brain pharmacokinetics of mAbs directed against (C) PECAM, (D) ICAM, and (E) CD45 following IV injection 2 h post-TNF-α injury. Uptake of mAbs directed against (F) PECAM, (G) ICAM, and (H) CD45 in clearance organs (liver, spleen) following IV injection 2 h post-TNF-α injury. Time points reflect the time post-mAb injection when organs were harvested. Solid bars: 30 min, Striped bars: 4 h, Checkered bars: 22 h. Data represented as mean ± SEM. Comparisons made by one-way ANOVA with Tukey’s posthoc test. N = 3 mice/group. Portions of figure created using www.biorender.com.
αPECAM behaved as expected for ligands of epitopes constitutively and stably expressed on the surface of a nonmigratory cell type (endothelial cells): (A) specific, rapid (vs IgG, see below) uptake in most organs at early time points; (B) decreasing tissue concentrations over time (Figure 2c, Supplemental Table 2); (C) relatively stable uptake in clearance organs (Figure 2f); and (D) more rapid blood clearance vs control IgG (Supplemental Figure 7). In part due to prolonged circulation time, control IgG slowly accumulated in the brain via enhanced vascular permeability, which has been previously reported in this model9 (Supplemental Table 2, Supplemental Figure 8).
The pharmacokinetics and biodistribution of αICAM were more complex and rather unanticipated in some aspects. Over time, lung concentrations of αICAM decreased with a simultaneous increase in the brain uptake of αICAM (Figure 2d, Supplemental Table 2). The distribution pattern of αCD45 was similar to that of αICAM, with specific accumulation in the lungs at early time points, followed by lung clearance and slow delivery to the brain (Figure 2e, Supplemental Table 2). Because CD45 is a pan-leukocyte marker, its accumulation can be attributed to an influx of mAb-tagged leukocytes at the injury in the brain. Distribution patterns of αICAM and αCD45 in typical clearance organs for mAbs were as expected with a transient accumulation of αICAM in clearance organs (Figure 2g) and rapid uptake and clearance of αCD45 in the liver, likely due to rapid targeting of tissue-resident macrophages (Kupffer cells) lining the hepatic sinusoids (Figure 2h). It is not expected that uptake by clearance organs would contribute significantly to brain delivery, as recipient cells in these organs were likely tissue-resident cells that do not migrate in response to distal stimuli. There was a significant correlation between clearance from the lung and changes in brain uptake with time (Supplemental Figure 9). It was hypothesized that this unexpected distribution pattern of αICAM was due to the initial delivery of αICAM to activated leukocytes in the pulmonary vasculature followed by migration of leukocytes to the injured brain (Figure 2a).
ICAM-Directed Loading of Nanoparticles to Lung WBC for Brain Targeting
To apply results obtained with αICAM to demonstrate a strategy for nanoparticle drug delivery to WBC in brain inflammation, we compared three different types of ICAM-targeted nanoparticles: polystyrene nanoparticles, liposomes, and lipid nanoparticles (LNP) (Figure 3a). IgG-coated and ICAM-targeted particles had similar sizes, polydispersity indices (PDI), and zeta potentials, as measured by dynamic light scattering (DLS) (Table 1). Incubation of liposomes in mouse plasma revealed no time-dependent changes in size, suggesting that particles were stable in plasma and did not aggregate (Supplemental Figure 10). ICAM-targeted nanoparticles were largely cleared from the blood within 30 min; however, there was a rebound in blood concentrations over the next several hours for ICAM-targeted nanoparticles, potentially reflecting redistribution of leukocytes carrying nanoparticles into blood (Supplemental Figure 11). Notably, this pattern was not observed for IgG-coated nanoparticles (Supplemental Figure 11). Similar to αICAM mAb, ICAM-targeted nanoparticles were largely taken up in the lungs within 30 min of injection (polystyrene nanoparticles: 147 ± 1%ID/g, liposome: 174 ± 6%ID/g, LNP: 123 ± 9%ID/g), followed by clearance from the lungs over several hours (Figure 3b,c,d, Supplemental Tables 3, 4, 5). Both polystyrene nanoparticles and LNP displayed monotonic increases in brain concentrations with time after injection, while liposomes had a transient increase in brain uptake (Figure 3b,c,d, and Supplemental Table 4). To evaluate the interplay between lung clearance and brain uptake of nanoparticles, lung/brain ratios were calculated at different time points postdose. All three particles displayed a steady increase in this ratio with time, reflecting the opposite trends in tissue targeting kinetics: rising brain uptake and falling lung retention (Figure 3e,f,g, Supplemental Tables 3, 4, 5). Untargeted control IgG nanoparticles did not display significant accumulation in either lungs or brain (concentrations >10-fold lower than ICAM-targeted) (Supplemental Figure 12a,b, Supplemental Tables 3, 4, 5). Nanoparticles were rapidly taken up and cleared by organs of the reticuloendothelial system (liver, spleen), irrespective of targeting to ICAM (Figure 3h,i,j, Supplemental Figure 12c,d), suggesting that similar to mAbs, nanoparticle delivery to static pools of phagocytic cells is not contributing to nanoparticle uptake in the brain.
Figure 3.

ICAM-targeted nanoparticles accumulate in the inflamed brain. (A) Study timeline. Pharmacokinetics of (B) polystyrene nanoparticles, (C) liposomes, and (D) lipid nanoparticles in the lungs and brain following injection. Kinetic changes in the ratio of nanoparticles in brain vs lungs for (E) polystyrene nanoparticles, (F) liposomes, and (G) lipid nanoparticles. Pharmacokinetics in clearance organs of (H) polystyrene nanoparticles, (I) liposomes, and (J) lipid nanoparticles. (K) Transmission electron microscopy of ICAM-targeted polystyrene nanoparticles in lung endothelium and leukocytes 30 min postinjection. (L) Cranial window intravital microscopy of ICAM-targeted polystyrene nanoparticles in the TNF-α injured brain. Data represented as mean ± SEM. Comparisons were made by one-way ANOVA with Tukey’s posthoc test. N ≥ 3 mice/group.
Table 1. Characterization of the Nanoparticles Used in Pharmacokinetic Studiesa.
| Polystyrene
Nanoparticles |
Liposomes |
Lipid Nanoparticles |
||||
|---|---|---|---|---|---|---|
| Coating | IgG | ICAM | IgG | ICAM | IgG | ICAM |
| Diameter (nm) | 278 ± 5 | 241 ± 6 | 144 ± 5 | 139 ± 3 | 93.5 ± 0.4 | 101 ± 1 |
| PDI | 0.0874 ± 0.016 | 0.103 ± 0.043 | 0.145 ± 0.008 | 0.133 ± 0.007 | 0.170 ± 0.020 | 0.150 ± 0.070 |
| Zeta Potential (mV) | –13.4 ± 0.8 | –16.1 ± 0.6 | –6.78 ± 0.18 | –7.00 ± 0.48 | –10.0 ± 0.7 | –8.33 ± 1.05 |
All values represented as mean ± SEM.
Microscopy studies visualized the ICAM-targeted nanoparticle delivery mechanisms in the lungs and brain. Transmission electron microscopy (TEM) demonstrated ICAM-targeted polystyrene nanoparticle localization to both endothelial cells and leukocytes in the lungs 30 min after IV injection (Figure 3k). Cranial window intravital microscopy (Figure 3l) showed that (A) ICAM-targeted polystyrene nanoparticles were associated with the walls of inflamed brain–blood vessels immediately following IV injection; (B) consistent with radiotracing experiments, the number of nanoparticles in the cranial window increased over time after injection; (C) 4 h after injection, nanoparticles appeared in clusters and some beads were detected in the parenchyma; and (D) 22 h after injection, nanoparticle fluorescence was no longer confined to large vessel walls and had spread into the parenchyma, suggesting that ICAM-targeted nanoparticles access a mechanism to cross the blood–brain barrier. Similar data were obtained for ICAM-targeted liposomes using cranial window intravital microscopy, with liposome fluorescence lining the vessel walls immediately postinjection and gradually accumulating in the brain parenchyma over 22 h (Supplemental Figure 13). The fluorescent signal for liposomes was more diffuse than that for polystyrene nanoparticles, possibly reflecting displacement of fluorophore-tagged lipids from liposomes with time after internalization in WBC.
ICAM-Targeted Nanoparticles Are Predominantly Delivered to Leukocytes
From the observations that (1) there was an increase in ICAM+ leukocytes in the pulmonary vasculature and in the brain following TNF-α injury and (2) intravenously injected ICAM-targeted agents (mAb, nanoparticles) had blood and tissue PK consistent with leukocyte targeting and migration, experiments were performed to test the hypothesis that leukocytes were the primary target cell for ICAM-targeted nanoparticles. Single cell suspensions were prepared from lungs 30 min after injection of ICAM-targeted nanoparticles (2 h post TNF-α injury) (Figure 4a). Flow cytometry showed similar affinity for endothelial cells (64.5 ± 2.1% nanoparticle-positive) and leukocytes (69.0 ± 4.0% nanoparticle-positive) in the lungs 30 min after nanoparticle injection (Figure 4b). On the other hand, there was a strong preference for leukocytes (29.4 ± 1.3% nanoparticle-positive) over endothelial cells (4.83 ± 0.4% nanoparticle-positive) 22 h after nanoparticle injection (Figure 4b), suggesting that the gradual uptake of nanoparticles into the brain was mediated by interactions with migrating leukocytes. Further analysis showed that nearly all nanoparticle-positive cells in the lungs were leukocytes (CD45+) (93.4 ± 1.4% of recovered cells), with the remaining nanoparticle-positive cells identified as endothelial cells (CD31+) (Figure 4c). This was a significant shift from naïve mice, where ICAM-targeted nanoparticles were nearly evenly distributed between leukocytes and endothelial cells in the lungs (Supplemental Figure 14). Due to the size of the administered particles (Table 1), it was unlikely that nanoparticles would be able to reach tissue-resident macrophages present in the interstitial space and airways. The significant disconnect between affinity (fraction of cells that are nanoparticle-positive) and targeting selectivity (fraction of nanoparticle-positive cells) could be explained by an accessibility limitation. Pulmonary intravascular leukocytes form a “carpet” over endothelial cells and therefore have the first opportunity to interact with intravenously administered nanoparticles. As these cells dynamically exchange with the circulating leukocyte pool, it was hypothesized that these cells then carry ICAM-targeted nanoparticles to the inflamed brain.
Figure 4.

Cellular specificity of ICAM-targeted polystyrene nanoparticles. (A) Flow cytometry was performed on single cell suspensions obtained from lungs and brain at the designated times post-nanoparticle injection. (B) Relative affinity of nanoparticles for endothelial cells and leukocytes was assessed by considering the fraction of cells that took up nanoparticles. The fraction of nanoparticles recovered in (C) lungs and (D) brain that were associated with specific cell types. Leukocytes: CD45+, Endothelium: CD31+CD45–. Histology of brain tissue sections collected 22 h postinjection of polystyrene nanoparticles in TNF-α challenged mice. Nanoparticle association with macrophages (CD68+) and endothelial cells (VCAM+) was measured for (E,F) ICAM-targeted and (G,H) IgG nanoparticles. Scale bar: 50 μm. Data represented as mean ± SEM. N = 3/group.
We tested this hypothesis in the context of leukocyte migration to the brain in acute neurovascular inflammation induced by a local injection of TNF-α. In neuron-depleted, single cell suspensions prepared from the brain, essentially all nanoparticle-positive cells were leukocytes (98.7 ± 0.2% of recovered cells) (Figure 4d). This suggests that among non-neuronal cells, leukocytes were the preferential target for ICAM-targeted nanoparticle delivery. These data are consistent with a general increase in ICAM+CD45+ cells in the disaggregated brain (∼3-fold increase over naïve) following the TNF-α injury (Figure 1i). Flow cytometry showed negligible polystyrene nanoparticle uptake by cells in the brain for bare and nonspecific IgG-coated polystyrene nanoparticles, consistent with biodistribution data (Supplemental Figure 15). A subtyping of cells in the brain revealed that most nanoparticle-positive leukocytes in the brain were monocytes/macrophages (73.0 ± 9.7%), with the bulk of the remainder being neutrophils (24.5 ± 9.9%) (Supplemental Figures 16 and 17). A large fraction of monocytes/macrophages were nanoparticle-positive (40.5 ± 3.6%). Among other leukocytes, 27.4 ± 6.6% of neutrophils and 25.2 ± 1.2% of other myeloid cells were nanoparticle-positive. We observed minimal ICAM-targeted nanoparticle uptake in CD45mid and T-cells (Supplemental Figure 17b).
Brain histology confirmed the association of nanoparticles with monocytes/macrophages (CD68-stained) (Figure 4e, Supplemental Figure 18a) and endothelial cells (VCAM-stained) (Figure 4f, Supplemental Figure 18b). Histology confirmed greater uptake of ICAM-targeted nanoparticles vs IgG-coated nanoparticles, both in the vasculature and in the brain parenchyma (Figure 4g,h, Supplemental Figure 18a, b). Parenchymal nanoparticle fluorescence was largely colocalized with macrophages, consistent with flow cytometry results.
Dexamethasone Loaded, ICAM-Targeted Liposomes Reduce Brain Edema
Brain injection of TNF-α leads to reproducible brain edema, as assessed by measuring extravasation of radiolabeled albumin.9 Liposomes were loaded with dexamethasone-21-phosphate (henceforth termed dexamethasone) (Supplemental Figure 19) and loading efficiency was determined to be 13.3 ± 0.4% (2.66 mg/mL) via HPLC. Free dexamethasone, dexamethasone-loaded IgG liposomes, and dexamethasone-loaded ICAM-targeted liposomes were assessed for effects on brain edema (Figure 5a, Supplemental Figure 20). No significant effects were detected for IV injection of 0.5 mg/kg free dexamethasone (−0.531 ± 26.3% protection) or dexamethasone-loaded IgG liposomes with equivalent drug dose (24.3 ± 18.9% protection). Dexamethasone-loaded ICAM-targeted liposomes provided near complete protection from edema (88.5 ± 14.6% protection; Figure 5b). Precluding the effects of the liposomes themselves, neither empty IgG liposomes (40.7 ± 23.5% protection) nor empty ICAM-targeted liposomes (−4.14 ± 29.81% protection) provided significant protection against edema (Figure 5b).
Figure 5.

ICAM-targeted dexamethasone (Dex) liposomes protect mice from TNF-induced brain edema. (A) Experimental timeline. (B) Protective effects of ICAM-targeted dexamethasone liposomes (0.5 mg/kg dexamethasone). As controls for Dex-loaded liposomes, equivalent doses of empty IgG or ICAM-targeted liposomes were tested. % protection was calculated assuming 100% protection as equivalent to edema induced by sham injury and 0% protection as equivalent to edema induced by TNF injury without treatment (Supplemental Figure 19). (C) Impact of treatment strategies on leukocyte abundance and phenotype in the brain as measured by flow cytometry. Data displayed as mean ± SEM. Comparisons made by one-way ANOVA with Dunnett’s posthoc test vs untreated (solid line, 0% protection) in B and Tukey’s posthoc test in C. N ≥ 3/group.
Complete blood counts were performed to assess the impact of dexamethasone-loaded ICAM-targeted liposomes and other formulations on circulating blood cells (Supplemental Figure 21). ICAM-targeted dexamethasone liposomes led to a reduction in lymphocytes, consistent with the known mechanism of action of the drug, but no other blood cell parameters were affected by treatment, indicating that the therapeutic effect of ICAM-targeted liposomal dexamethasone represents localized action in the brain rather than a systemic effect. Shortly after injection of dexamethasone loaded, ICAM-targeted liposomes (30 min), minimal effects on complete blood counts were seen (Supplemental Figure 22). The largest observed change was a significant increase in circulating neutrophils (2.2-fold increase vs untreated mice), which is consistent with a model where ICAM-targeted particles are delivered to pulmonary marginated leukocytes, which then dissociate from the vascular wall and migrate to the brain in response to emanating signals.
Additional studies were performed to elucidate the impact of ICAM-targeted liposomal dexamethasone on leukocyte populations in the brain. Flow cytometry of brains harvested at the same time point as measurements of brain edema were made revealed that no tested formulation (free dexamethasone, empty ICAM-targeted liposome, dexamethasone-loaded, ICAM-targeted liposome) affected the abundance of leukocytes (CD45+) or of CD11b+ leukocytes (monocyte/macrophage) (Figure 5c). This was in contrast to studies with rigid polystyrene nanoparticles, which significantly reduced the infiltration of monocytes, in particular (∼50% reduction) into the brain (Supplemental Figure 23), suggesting that material properties of ICAM-targeted nanoparticles determine their impact on leukocyte migration. However, more detailed analysis of the monocyte/macrophage population revealed that only dexamethasone-loaded, ICAM-targeted liposomes increased the relative abundance of anti-inflammatory M2-polarized macrophages (inducible nitric oxide synthase (iNOS), Arginase (Arg); iNOS–Arg+) (∼2-fold over untreated; Figure 5c). These results point to impacts on the phenotype of immune cells in the brain as a key mechanism underlying the efficacy of the ICAM-targeted liposomal dexamethasone formulation.
Discussion
Development of effective therapies for neurological disorders presents formidable challenges including limited success in targeted drug delivery to the brain and especially into the required components of the parenchyma, neurons, glia, etc. The pressing need for effective targeted therapies is especially aggravated in patients suffering from acute brain injuries, including stroke, traumatic brain injury, neuroinflammation, and intracranial hemorrhage.
In general, direct targeting of the blood–brain barrier via transferrin receptor results in low levels of tissue uptake (≤1%ID/g) and no specificity for sites of injury.68,69 In the present work and in our recent publications, we describe the use of CAM targeting to provide specific delivery to the injured brain. We achieved high levels (∼4%ID/g) of delivery to the brain by targeting to PECAM in the brain; however, similar to targets such as transferrin receptor, PECAM does not provide specificity for injured regions.10 We have recently reported studies of targeting to the brain in the same injury model described here (TNF-α), but with injection of targeting ligands occurring the day after injury (rather than 2 h postinjury in this study). Targeting the inducible marker VCAM provided a 27-fold increase in specificity for the brain vs transferrin receptor when injected in mice following establishment of a local inflammatory state in the brain.9 We have also demonstrated specificity of delivery to ICAM in the same model at levels exceeding that of transferrin receptor.8 The approach described here provided a similar peak uptake in the brain as direct targeting to ICAM the day after injury (∼2–3%ID/g); however, concentrations in the brain steadily increased over time due to the mechanism of targeting (leukocyte-based) and time of injection (shortly after injury). Delivery to leukocytes likely provides additional selectivity for sites of injury vs direct targeting due to the complex and specific homing process of leukocytes.
Loading nanoparticles into leukocytes positioned to respond to distress signals emanating from the injured brain is an attractive strategy for drug delivery to the brain. In this case, delivery to sites of injury would be controlled by the natural homing mechanisms used by leukocytes to reach the injured brain (e.g., emanating chemokine gradients, cell adhesion molecules, etc.). Leukocytes have been used as carriers in chronic neurodegenerative conditions following ex vivo loading of drugs and genes and reinfusion into animals.19,20,70 In these studies, it was suggested that leukocytes (or leukocyte-derived extracellular vesicles) could not only reach the brain but also mediate transfer of their cargo into neurons to elicit a pharmacologic response.22,71
We postulated that targeting the intravascular leukocyte pool in vivo would be a viable strategy to achieve selective drug delivery to injured regions of the brain. Of the intravascular leukocytes, those transiently residing in the pulmonary vasculature (marginated pool) seem to be likely targets following IV administration. This approach would allow for the following: (1) treatment can be initiated immediately after diagnosis, without the need for ex vivo modification of cells, (2) all intravascular leukocytes are potential targets for loading, (3) selection for specific leukocyte phenotypes is possible by targeting to cell surface markers, and (4) leukocytes could be converted into drug depots/biofactories that concentrate drugs in the inflamed region where their activity is required. Multiple mechanisms of leukocyte targeting are possible, including direct targeting to endothelial ICAM and transfer of nanoparticles from the surface of endothelial cells to leukocytes, with the initial anchoring to endothelial cells occurring in an ICAM-dependent fashion, similar to trogocytosis.72−74 Indeed, by targeting leukocytes, a decline in lung concentrations was seen in parallel with delivery to the injured brain.
Following IV administration, affinity ligands directed toward many vascular epitopes have low levels of delivery to organs such as the brain, in part due to first pass binding to the pulmonary endothelium. The data presented above show that targeting to molecules expressed on all (CD45) and on activated (ICAM) leukocytes permits delivery to the brain, despite significant uptake by the lungs, with roughly 3.7-fold higher lung delivery and 5.7-fold higher brain delivery for αICAM vs αCD45. The purported mechanism for this delivery is that ICAM-targeted nanoparticles rapidly bind to pulmonary intravascular leukocytes and remain associated with these leukocytes,75 which then migrate to the brain in response to inflammatory signaling. While not directly studied here, it is anticipated that ICAM-targeted nanoparticles are endocytosed by leukocytes following binding. It has been reported extensively that multivalent, ICAM-targeted agents are efficiently endocytosed by endothelial cells75−77 and neurons78 and that ICAM expression correlates with neutrophil79 and macrophage80 phagocytic function. Alternatively, ICAM-targeted nanoparticles could bind leukocytes circulating in the bloodstream that become part of the dynamic, marginated pool in the pulmonary vasculature. What is abundantly clear is that 30 min postinjection of ICAM-targeted nanoparticles, >20% of the injected dose is recovered in the lungs, mainly in CD45+ cells.
While ICAM is known to be expressed on several cell types, only a few of these are accessible to intravenously administered nanoparticles (activated endothelial cells, leukocytes). The data presented here show that after intravenous administration in acute brain injured mice, ICAM-targeted nanoparticle delivery is biased toward leukocytes in lungs 30 min after injection. These unanticipated results are likely due to the following. The marginated leukocyte pool exists within vascular beds, and cells are transiently associated with the luminal surface of endothelial cells. Under inflammatory conditions, there is a redistribution of the intravascular leukocyte pool with significantly more leukocytes marginating in vascular beds, particularly in the lungs. In addition, the blood velocity in the extensive, torturous pulmonary capillaries is relatively slow, thereby dramatically increasing the chances of interaction and the potential surface area for interaction.
Beyond studies suggesting a mechanism of nanoparticle delivery to the marginated WBC pool, we pursued therapeutic studies to elucidate the therapeutic relevance of this leukocyte-based drug delivery strategy. We selected the small molecule corticosteroid dexamethasone as a therapeutic agent. Notably, dexamethasone has been tested in clinical trials for treatment of acute ischemic stroke but ultimately failed due to off-target effects. Among its pleiotropic effects, dexamethasone downregulates expression of the following: inducible CAMs, inflammatory cytokine expression (IL-1, IL-6, TNF-α), cyclooxygenase-2, collagenase, and NF-κB.81 We hypothesized that ICAM-targeted dexamethasone-loaded liposomes would provide the selective delivery of dexamethasone to the injured brain. The results presented here demonstrate that IV injection of dexamethasone 2 h post-TNF-α injury was only able to prevent brain edema when encapsulated in ICAM-targeted nanoparticles (Figure 5b). These results are likely due to not only changes in local brain concentrations of dexamethasone but also direct effects on leukocytes targeted by this strategy. Direct effects on leukocytes were observed using polystyrene nanoparticles, which reduced infiltration of monocytes/macrophages into the injured brain by 45% (Supplemental Figure 23). On the other hand, liposomes did not impact infiltration of monocytes/macrophages into the inflamed brain regardless of drug loading (Figure 5c), suggesting that material properties of nanoparticles influence trafficking of target leukocytes, consistent with in vitro studies.82 However, treatment with dexamethasone-loaded, ICAM-targeted liposomes led to increased anti-inflammatory macrophage (M2) polarization in the brain (Figure 5c), consistent with the anticipated effects of dexamethasone.83,84 M2 macrophages promote resolution of inflammation in a diverse array of conditions, including in acute neurovascular inflammation.85,86 The data described here suggest that leukocyte-mediated delivery of dexamethasone to the brain via ICAM targeting promotes resolution of inflammation, at least in part, by promoting M2 polarization without affecting leukocyte migration to the site of injury, switching the proinflammatory environment into an anti-inflammatory one.
Conclusions
In summary, we have developed an approach for in vivo loading of activated leukocytes with nanoparticles via targeting to ICAM. We propose the following mechanism for brain delivery whereby the pulmonary intravascular leukocytes (1) respond to signals emanating from the injured brain and change their activation status and local concentration, (2) take up ICAM-targeted nanoparticles, and (3) shuttle the taken up ICAM-targeted nanoparticles from the lungs to the injured region of the brain. Our results show that leukocyte targeting provides a steady accumulation of nanoparticles into the brain parenchyma following induction of acute neurovascular inflammation. Essentially all of the targeted nanoparticles in the brain were associated with leukocytes, namely, monocytes/macrophages and neutrophils. Injection of ICAM-targeted, dexamethasone-loaded liposomes into mice 2 h post-TNF injury was able to completely protect mice from TNF-α-induced brain edema and promote anti-inflammatory M2 macrophage polarization. By harnessing natural leukocyte migration patterns, this strategy provides enhanced selectivity for the injured region of the brain and has the potential for applications in other acute neurovascular inflammatory injuries such as stroke.
Methods and Experimental Section
Reagents
Reagents for the iodination of proteins were obtained from the following sources: Na125I (PerkinElmer, Waltham, MA) and 1,3,4,6-tetrachloro-3α,6α-diphenyl-glycouril (Iodogen) (Pierce, Rockford, IL). Polystyrene beads (190 nm) were purchased from Bangslabs (Fishers, IN). All lipids were obtained from Avanti Polar Lipids (Alabaster, AL). Pooled rat IgG (rIgG) was purchased from Invitrogen (Carlsbad, CA). Antibodies against PECAM (Mec13.3) and CD45 were purchased from BioLegend (San Diego, CA). All other chemicals and reagents were purchased from SigmaAldrich (St. Louis, MO), unless specifically noted.
Animals
All animal studies were carried out in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD), and all animal protocols were approved by the University of Pennsylvania Institutional Animal Care and Use Committee. All animal experiments were carried out using male, 6–8 week old C57BL/6 mice (20–25 g) (The Jackson Laboratory, Bar Harbor, ME).
Protein Production and Purification
Anti-ICAM mAb (YN1) was produced and purified from hybridoma supernatants, as described previously.87 Purification of YN1 was performed using Protein G affinity chromatography.
Radiolabeling
Antibodies (YN1, CD45, Mec13.3, rIgG) were radiolabeled with 125I via the Iodogen method. Briefly, tubes coated with 100 μg of Iodogen reagent were incubated with antibodies (1–2 mg/mL) and Na125I (0.25 μCi/μg protein) for 5 min on ice. Residual free iodine was removed from the bulk solution using a desalting column and thin layer chromatography was used to confirm the efficiency of radiolabeling. As a quality control step, all proteins were confirmed to have <10% free 125I prior to further use.
Polystyrene Nanoparticle Conjugation
Carboxylated, Flash Red (intrinsically fluorescent) polystyrene beads were conjugated to antibodies (rIgG, YN1) via reaction of N-hydroxysulfosuccinimide (sulfo-NHS) (0.275 mg/mL), 1-ethyl-3-(3-dimethylaminopropl)carbodiimide HCl (EDC) (0.1 mg/mL), and 200 antibody molecules/bead. For experiments involving radioisotope tracing, 15% of the total antibody added to the reaction mixture was 125I-labeled rIgG. NC size and polydispersity index (PDI) were confirmed via dynamic light scattering (DLS).
Liposome Formulation
Liposomes were prepared as described previously.87 Briefly, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), cholesterol, and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[azido(polyethylene glycol)-2000 (DSPE-PEG2000-azide) were mixed in a molar ratio of 54:40:6. Liposomes were prepared via the thin film extrusion method. To form drug loaded liposomes, the lipid film was hydrated in a solution containing 20 mg/mL of dexamethasone-21-phosphate in phosphate buffered saline (PBS), at pH 7.4. The resulting vesicles were extruded through 200 nm polycarbonate membranes. For studies requiring fluorescent tracing of liposomes, 0.1 mol % TopFluor PC was spiked in to the liposome formulation.
Conjugation of antibodies to the liposome surface was carried out using strain-promoted alkyne–azide cycloaddition. Antibodies were functionalized by reacting with a 5-fold molar excess of dibenzocyclooctyne-PEG4-NHS ester (DBCO-PEG4-NHS) (Click Chemistry Tools, Scottsdale, AZ) for 30 min at room temperature. Unreacted DBCO-PEG4-NHS was removed via centrifugation through a molecular weight cutoff (MWCO) filter. Liposomes were conjugated with DBCO-functionalized antibodies by reacting for 4 h at 37 °C. For experiments involving radiotracing, 10% of the total antibody added was 125I-labeled rIgG. Unconjugated antibody was removed from the liposomes using gel filtration chromatography. The size, distribution, and concentration of liposomes were determined using DLS and nanoparticle tracking analysis (Malvern Panalytical, Westborough, MA).
Lipid Nanoparticle Formulation
LNP were prepared via microfluidic mixing as previously described.88 Briefly, an ethanol phase was prepared by combining ionizable lipid, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), cholesterol, and 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (C14-PEG2000) at molar ratios of 35:16:46.5:2.5, respectively. Separately, an aqueous phase was prepared by resuspending scrambled siRNA sequences in 10 mM citrate buffer to a concentration of. 75 mg/mL. Ethanol and aqueous phases were then mixed in a single channel microfluidic device at a 3:1 ratio using a syringe pump.89 LNP were dialyzed against 1× PBS for 2 h at room temperature, followed by sterile filtration using 22 μm syringe filters.
LNP were functionalized for conjugation with antibodies with DSPE-PEG2000-maleimide using a previously described postinsertion technique.90,91 Micelles were formed with DSPE-PEG2000 and DSPE-PEG-maleimide using a 4:1 molar ratio. Following solvent evaporation, lipids were hydrated in PBS at 65 °C while vortexing. Micelles were then incubated with 1 mg/mL LNP for 3 h at 37 °C. Antibodies were conjugated to maleimide-functionalized LNP using N-succinimidyl S-acetylthioacetate (SATA)-maleimide chemistry. Briefly, antibodies were modified with functionalized with SATA at a 5:1 molar ratio (SATA:antibody) for 30 min at room temperature and were purified using molecular weight cutoff filters. Prior to conjugation to LNP, deprotection was carried out by adding 0.5 M hydroxylamine. SATA-antibodies were then mixed with maleimide-functionalized LNP for 30 min at room temperature. Following conjugation, the reaction was quenched by the addition of cysteine. LNP size, distribution, and concentration were determined as described for liposomes (DLS, nanoparticle tracking analysis).
Liposome Stability in Plasma
IgG-coated liposomes containing a fluorescent lipid were incubated in mouse plasma at 37 °C for designated time points (0–2 h). Liposome size was measured following dilution in deionized water using a nanoparticle tracking analysis (NanoSight NS300).
Dexamethasone Loading and Release
Both the amount of dexamethasone loading into liposomes and the kinetics of release were assessed using reverse phase high performance liquid chromatography (HPLC). The mobile phase consisted of 30% (v/v) acetonitrile, 70% (v/v) water, and 0.1% (v/v) trifluoroacetic acid. Buffer was run at a flow rate of 0.6 mL/min through a C8 column (Eclipse XDB-C8, 3 μm, 3.0 × 100 mm, Phenomenex). Dexamethasone was detected using UV absorbance at 240 nm, and the assay had a linear range of 1.56–100 μg/mL (Supplemental Figure 24). Drug release was measured by dialyzing loaded particles against a large excess of PBS, pH 7.4, at 37 °C and collecting samples at designated time points.
TNF Injury Model
Neurovascular inflammation was induced in mice via a unilateral injection of TNF-α (0.5 μg/mouse, 2.5 μL, BioLegend) into the striatum using a stereotaxic frame at the following coordinates relative to the bregma: 0.5 mm anterior, 2.0 mm lateral, −3 mm ventral.10 At different times relative to TNF-α injection (1–24 h), mice were injected intravenously with a bolus dose of either mAbs (5 μg) or nanoparticles (polystyrene beads, liposomes). Animals were perfused with 20 mL of PBS, pH 7.4, prior to collecting organs for further analysis. For pharmacokinetic and biodistribution studies, the amount of radioactivity in blood and organs was measured using a gamma counter (Wizard2, PerkinElmer, Waltham, MA).
Transmission Electron Microscopy
Visualization of NC uptake in the lungs shortly after injection was performed using TEM, as previously described.92 Briefly, 30 min postinjection, lungs were fixed with 2.5% glutaraldehyde and 4% paraformaldehyde in 0.1 M sodium cacodylate buffer and then processed into 80–90 nm-thin resin-embedded sections for visualization by TEM.
Intravital Microscopy
After the meninges were removed, a cranial window was opened in one parietal bone of mice. This window was sealed with a glass coverslip, and a cannula (PlasticsOne, Roanoke, VA) was placed into the subarachnoid space adjacent to the window (1 mm depth). Animals were allowed to recover for 5 days between opening of the cranial window and injection of TNF-α to prevent any artifacts related to surgery-induced inflammation. In vivo imaging was performed in real time with a Stereo Discovery V20 fluorescence microscope (Carl Zeiss AG, Oberkochen, Germany).
Multiplex Cytokine Analysis
Plasma was collected from naïve mice and from mice following TNF-α injury (2 and 24 h postinjury). Cytokines were measured using the LEGENDplex Mouse Inflammation Panel (BioLegend), according to the manufacturer’s instructions.
Brain Flow Cytometry
Single cell suspensions of brain were produced as described previously.9,93 Briefly, tissues were enzymatically digested with Dispase and collagenase for 1 h at 37 °C, followed by addition of 600 U/mL DNase grade II. Tissue digests were demyelinated in Percoll and ACK buffer (Quality Biological, Gaithersburg, MD) was added to lyse any residual RBCs. Samples were then filtered through (1) 100 μm nylon strainers and (2) 70 μm nylon strainers (ThermoFisher).
Cells were then stained with appropriate antibodies (Supplemental Table 6). Briefly, 2 × 106 cells were labeled per tube in PBS containing 2% v/v fetal bovine serum (FBS). Fc receptors were blocked using TruStain FcX PLUS (antimouse CD16/32, 1:200 dilution) (BioLegend). In pilot experiments to determine localization of NC in leukocytes (CD45+) vs endothelial cells (CD31+CD45–), flow cytometry was performed using an Accuri C6plus (Benton Dickinson, San Jose, CA). Detailed subtyping of white blood cells in the brain was performed using the strategy described by Posel et al. using a BD LSRFortessa (Benton Dickinson, San Jose, CA) flow cytometer. Live/dead staining was performed using LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (1:1000 dilution, ThermoFisher). In this assay uptake by the following cell types was defined: (1) CD45mid, T-cells (CD45hiCD3+), neutrophils (CD45hiLy6G+), and monocytes/macrophages (CD45hiCD3–Ly6G–CD11b+Ly6C+). Analysis of flow cytometry data was performed using the BD Accuri C6 software (Benton Dickinson, San Jose, CA) and FlowJo v10.6.2 (Tree Star). To assess macrophage polarization, a single-cell suspension was fixed with 4% paraformaldehyde and permeabilized by 0.15% Trion X-100 in PBS. Macrophages were identified as CD45+/CD11b+, M1 as iNOS+/Arginase–, and M2 as iNOS–/Arginase+. Anaysis of flow cytometry data was performed using the CytoFLEX S flow cytometry (Beckman, Indianapolis, IN) and FlowJo.
Lung Flow Cytometry
To distinguish intravascular vs extravascular leukocytes, mice were preinjected with PE-labeled CD45 5 min prior to sacrifice. Mice were then perfused with ice-cold PBS at a flow rate of 2 mL/min to remove excessive blood without disturbing the intravascular leukocyte pool. Lung single cell suspensions were prepared as described previously.94 Lungs were digested with collagenase and DNase for 45 min at 37 °C with mixing and then filtered through a 70 μm strainer. RBCs were lysed with ACK buffer and then stained for extravascular leukocytes (PerCP Cy5.5-labeled CD45+), endothelial cells (CD31+CD45–), and ICAM positive cells (Alexa Fluor-647-labeled YN1).
Blood Flow Cytometry and ICAM Binding
Blood was drawn from TNF mice 2 h post injury in an EDTA-coated tube. After centrifugation at 1000 g for 10 min, plasma was removed and the buffy coat containing white blood cells was collected. Excessive red blood cells were removed with ACK buffer. For flow cytometry, ICAM expressing leukocytes was determined by CD45+/YN1+ and analyzed by CytoFLEX S flow cytometry and FlowJo. For the ICAM binding assay, an excessive amount of radiolabeled antibody was mixed with 106 cells at 4 °C for 1 h. Unbounded antibody was washed, and ICAM copies per cell were determined and calculated by a gamma counter.
Histology
For lung histology, Alexa Fluor-647-labeled CD45 antibody was IV injected into TNF mouse 2 h post injury. Five minutes later, the animal was perfused with ice-cold PBS at a flow rate of 2 mL/min, and lungs were freshly frozen. Tissue was sectioned at 10 μm thickness, and sections were counterstained with nuclei dye 4′-6-diamidino-2-phenylindole (DAPI, Southern Biotech). The images were taken by a Leica Stellaris 5 Confocal Microscope.
TNF brains injected with IgG or αICAM conjugated polystyrene nanoparticles were perfused, harvested 24 h postinjected, and fixed in 4% paraformaldehyde. After freezing in tissue freezing medium, the brains were sectioned at a 20 μm thickness. Tissue sections were then permeabilized and blocked in blocking solution (5% normal goat serum and 0.3% Triton X-100 in PBS) for 1 h at room temperature, and then incubated overnight at 4 °C with primary antibodies (Supplemental Table 7) in blocking solution. After washing with PBS, the sections were incubated with secondary antibodies conjugated with Alexa fluorophores (1:200, Invitrogen) in PBS for 1 h at room temperature. After being washed, the sections were counterstained with DAPI. The images were taken by Leica DM6000 Widefield Microscope.
Therapeutic Studies
The effects of dexamethasone on TNF-induced brain edema were assessed as described in our previous publication.9 Briefly, 2 h post-TNF injection, mice were dosed IV with (1) 0.5 mg/kg dexamethasone, (2) empty liposomes (either αICAM or IgG coated), or (3) 0.5 mg/kg liposomal dexamethasone (either αICAM or IgG coated). Twenty hours after TNF injection, mice were injected with 125I-labeled bovine serum albumin (BSA, ∼3 × 106 cpm/mouse), which was then allowed to circulate for 4 h. After BSA circulation, mice were perfused with 20 mL of PBS, pH 7.4, over 5 min and organs were harvested. Edema was determined by measuring the relative concentration of extravasated BSA in the brain compared to the concentration in the bloodstream. For calculations of therapeutic efficacy, 0% protection was defined using PBS-treated, TNF-injured mice, and 100% protection was defined using PBS-treated, sham-injured mice.
Complete Blood Counts
At designated time points, blood was collected from mice into tubes containing EDTA. Blood cells were analyzed using an Abaxis VetScan HM5 Hematology Analyzer, and all values were normalized to the mean value obtained for naïve mice.
Statistics
All statistical tests were performed using GraphPad Prism 8 (GraphPad Software, San Diego, CA). * denotes p < 0.05, ** denotes p < 0.01, *** denotes p < 0.001, **** denotes p < 0.0001.
Acknowledgments
O.A.M.C. received support from the American Heart Association (Grant 19CDA345900001). V.R.M. and J.S.B. received support from the Cardiovascular Institute of the University of Pennsylvania. V.R.M. received funding from the National Institutes of Health (NIH) (R01 HL155106, R01 HL128398, R01 HL143806). J.N. received support from the American Heart Association (Grant 916172). P.M.G. received funding from the National Institutes of Health (R00 HL153696). M.J.M. acknowledges support from a US National Institutes of Health Director’s New Innovator Award (DP2 TR002776), a Burroughs Wellcome Fund Career Award at the Scientific Interface (CASI), a grant from the American Cancer Society (129784-IRG-16-188-38-IRG), and the National Institutes of Health (NCI R01 CA241661, NCI R37 CA244911, and NIDDK R01 DK123049). Some figures created using BioRender.com.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.2c08275.
Supplemental Figures 1–24 and Supplemental Tables 1–7, as described in the text (PDF)
Author Contributions
¶ J.N., P.M.G., and O.A.M.-C. contributed equally.
A previous version of this manuscript was deposited on a preprint server (BioRχiv). Patrick M. Glassman, Jia Nong, Jacob W. Myerson, Viviana Zuluaga-Ramirez, Alba Rodriguez-Garcia, Alvin Mukalel, Serena Omo-Lamai, Landis R. Walsh, Raisa Y. Kiseleva, Carlos H. Villa, Colin F. Greineder, Scott E. Kasner, Drew Weissman, Michael J. Mitchell, Silvia Muro, Yuri Persidsky, Jacob S. Brenner, Vladimir R. Muzykantov, Oscar A. Marcos Contreras. Targeted nanocarriers coopting pulmonary leukocytes for drug delivery to the injured brain. 2022, 479150. BioRχiv. https://www.biorxiv.org/content/10.1101/2022.02.04.479150v1.article-info (Accessed April 28, 2023).
The authors declare no competing financial interest.
Supplementary Material
References
- Patel M. M.; Goyal B. R.; Bhadada S. V.; Bhatt J. S.; Amin A. F. Getting into the brain: approaches to enhance brain drug delivery. CNS Drugs 2009, 23 (1), 35–58. 10.2165/0023210-200923010-00003. [DOI] [PubMed] [Google Scholar]
- Finbloom J. A.; Sousa F.; Stevens M. M.; Desai T. A. Engineering the drug carrier biointerface to overcome biological barriers to drug delivery. Adv. Drug Deliv Rev. 2020, 167, 89–108. 10.1016/j.addr.2020.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen E. M.; Quijano A. R.; Seo Y. E.; Jackson C.; Josowitz A. D.; Noorbakhsh S.; Merlettini A.; Sundaram R. K.; Focarete M. L.; Jiang Z.; et al. Biodegradable PEG-poly(omega-pentadecalactone-co-p-dioxanone) nanoparticles for enhanced and sustained drug delivery to treat brain tumors. Biomaterials 2018, 178, 193–203. 10.1016/j.biomaterials.2018.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song E.; Gaudin A.; King A. R.; Seo Y. E.; Suh H. W.; Deng Y.; Cui J.; Tietjen G. T.; Huttner A.; Saltzman W. M. Surface chemistry governs cellular tropism of nanoparticles in the brain. Nat. Commun. 2017, 8, 15322. 10.1038/ncomms15322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo D.; Magsam A. W.; Kelly A. M.; Stayton P. S.; Kievit F. M.; Convertine A. J. Core-Cross-Linked Nanoparticles Reduce Neuroinflammation and Improve Outcome in a Mouse Model of Traumatic Brain Injury. ACS Nano 2017, 11 (9), 8600–8611. 10.1021/acsnano.7b03426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alghamri M. S.; McClellan B. L.; Hartlage M. S.; Haase S.; Faisal S. M.; Thalla R.; Dabaja A.; Banerjee K.; Carney S. V.; Mujeeb A. A.; et al. Targeting Neuroinflammation in Brain Cancer: Uncovering Mechanisms, Pharmacological Targets, and Neuropharmaceutical Developments. Front Pharmacol 2021, 12, 680021 10.3389/fphar.2021.680021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler D. B.; Kadiyala P.; Nunez F. J.; Nunez F. M.; Carney S.; Alghamri M. S.; Garcia-Fabiani M. B.; Asad A. S.; Nicola Candia A. J.; Candolfi M.; et al. Prospects of biological and synthetic pharmacotherapies for glioblastoma. Expert Opin Biol. Ther 2020, 20 (3), 305–317. 10.1080/14712598.2020.1713085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcos-Contreras O. A.; Brenner J. S.; Kiseleva R. Y.; Zuluaga-Ramirez V.; Greineder C. F.; Villa C. H.; Hood E. D.; Myerson J. W.; Muro S.; Persidsky Y.; et al. Combining vascular targeting and the local first pass provides 100-fold higher uptake of ICAM-1-targeted vs untargeted nanocarriers in the inflamed brain. J. Controlled Release 2019, 301, 54–61. 10.1016/j.jconrel.2019.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcos-Contreras O. A.; Greineder C. F.; Kiseleva R. Y.; Parhiz H.; Walsh L. R.; Zuluaga-Ramirez V.; Myerson J. W.; Hood E. D.; Villa C. H.; Tombacz I.; et al. Selective targeting of nanomedicine to inflamed cerebral vasculature to enhance the blood-brain barrier. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (7), 3405–3414. 10.1073/pnas.1912012117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Esteves S.; Nong J.; Glassman P. M.; Omo-Lamai S.; Ohashi S.; Myerson J. W.; Zamora M. E.; Ma X.; Kasner S. E.; Sansing L.; et al. Targeted drug delivery to the brain endothelium dominates over passive delivery via vascular leak in experimental intracerebral hemorrhage. J. Controlled Release 2023, 356, 185–195. 10.1016/j.jconrel.2023.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glassman P. M.; Walsh L. R.; Villa C. H.; Marcos-Contreras O. A.; Hood E. D.; Muzykantov V. R.; Greineder C. F. Molecularly Engineered Nanobodies for Tunable Pharmacokinetics and Drug Delivery. Bioconjug Chem. 2020, 31 (4), 1144–1155. 10.1021/acs.bioconjchem.0c00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U.; Iadecola C.; Moskowitz M. A. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 1999, 22 (9), 391–397. 10.1016/S0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Abdul-Muneer P. M.; Chandra N.; Haorah J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol. Neurobiol 2015, 51 (3), 966–979. 10.1007/s12035-014-8752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohidpour A.; Morgun A. V.; Boitsova E. B.; Malinovskaya N. A.; Martynova G. P.; Khilazheva E. D.; Kopylevich N. V.; Gertsog G. E.; Salmina A. B. Neuroinflammation and Infection: Molecular Mechanisms Associated with Dysfunction of Neurovascular Unit. Front Cell Infect Microbiol 2017, 7, 276. 10.3389/fcimb.2017.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveed M.; Zhou Q. G.; Han F. Cerebrovascular inflammation: A critical trigger for neurovascular injury?. Neurochem. Int. 2019, 126, 165–177. 10.1016/j.neuint.2019.03.011. [DOI] [PubMed] [Google Scholar]
- Ma Q.; Chen S.; Klebe D.; Zhang J. H.; Tang J. Adhesion molecules in CNS disorders: biomarker and therapeutic targets. CNS Neurol Disord Drug Targets 2013, 12 (3), 392–404. 10.2174/1871527311312030012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frijns C. J.; Kappelle L. J. Inflammatory cell adhesion molecules in ischemic cerebrovascular disease. Stroke 2002, 33 (8), 2115–2122. 10.1161/01.STR.0000021902.33129.69. [DOI] [PubMed] [Google Scholar]
- Ahmad M.; Graham S. H. Inflammation after stroke: mechanisms and therapeutic approaches. Transl Stroke Res. 2010, 1 (2), 74–84. 10.1007/s12975-010-0023-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anselmo A. C.; Gilbert J. B.; Kumar S.; Gupta V.; Cohen R. E.; Rubner M. F.; Mitragotri S. Monocyte-mediated delivery of polymeric backpacks to inflamed tissues: a generalized strategy to deliver drugs to treat inflammation. J. Controlled Release 2015, 199, 29–36. 10.1016/j.jconrel.2014.11.027. [DOI] [PubMed] [Google Scholar]
- Klyachko N. L.; Polak R.; Haney M. J.; Zhao Y.; Gomes Neto R. J.; Hill M. C.; Kabanov A. V.; Cohen R. E.; Rubner M. F.; Batrakova E. V. Macrophages with cellular backpacks for targeted drug delivery to the brain. Biomaterials 2017, 140, 79–87. 10.1016/j.biomaterials.2017.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang L.; Qin J.; Han L.; Zhao W.; Liang J.; Xie Z.; Yang P.; Wang J. Exploiting macrophages as targeted carrier to guide nanoparticles into glioma. Oncotarget 2016, 7 (24), 37081–37091. 10.18632/oncotarget.9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Haney M. J.; Jin Y. S.; Uvarov O.; Vinod N.; Lee Y. Z.; Langworthy B.; Fine J. P.; Rodriguez M.; El-Hage N.; et al. GDNF-expressing macrophages restore motor functions at a severe late-stage, and produce long-term neuroprotective effects at an early-stage of Parkinson’s disease in transgenic Parkin Q311X(A) mice. J. Controlled Release 2019, 315, 139–149. 10.1016/j.jconrel.2019.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Haney M. J.; Mahajan V.; Reiner B. C.; Dunaevsky A.; Mosley R. L.; Kabanov A. V.; Gendelman H. E.; Batrakova E. V. Active Targeted Macrophage-mediated Delivery of Catalase to Affected Brain Regions in Models of Parkinson’s Disease. J. Nanomed. Nanotechnol. 2011, S4, 1. 10.4172/2157-7439.S4-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghassemi S.; Nunez-Cruz S.; O’Connor R. S.; Fraietta J. A.; Patel P. R.; Scholler J.; Barrett D. M.; Lundh S. M.; Davis M. M.; Bedoya F.; et al. Reducing Ex Vivo Culture Improves the Antileukemic Activity of Chimeric Antigen Receptor (CAR) T Cells. Cancer Immunol Res. 2018, 6 (9), 1100–1109. 10.1158/2326-6066.CIR-17-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June C. H.; O’Connor R. S.; Kawalekar O. U.; Ghassemi S.; Milone M. C. CAR T cell immunotherapy for human cancer. Science 2018, 359 (6382), 1361–1365. 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- Batrakova E. V.; Li S.; Reynolds A. D.; Mosley R. L.; Bronich T. K.; Kabanov A. V.; Gendelman H. E. A macrophage-nanozyme delivery system for Parkinson’s disease. Bioconjug Chem. 2007, 18 (5), 1498–1506. 10.1021/bc700184b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H.; Grotepas C. B.; McMillan J. M.; Destache C. J.; Chaubal M.; Werling J.; Kipp J.; Rabinow B.; Gendelman H. E. Macrophage delivery of nanoformulated antiretroviral drug to the brain in a murine model of neuroAIDS. J. Immunol 2009, 183 (1), 661–669. 10.4049/jimmunol.0900274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavas T. E. J.; Su F. Y.; Srinivasan S.; Roy D.; Lee B.; Lovelace-Macon L.; Rerolle G. F.; Limqueco E.; Skerrett S. J.; Ratner D. M.; et al. A macrophage-targeted platform for extending drug dosing with polymer prodrugs for pulmonary infection prophylaxis. J. Controlled Release 2021, 330, 284–292. 10.1016/j.jconrel.2020.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Zang J.; Zhao Z.; Zhang Q.; Chen S. The Advances of Neutrophil-Derived Effective Drug Delivery Systems: A Key Review of Managing Tumors and Inflammation. Int. J. Nanomedicine 2021, 16, 7663–7681. 10.2147/IJN.S328705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che J.; Najer A.; Blakney A. K.; McKay P. F.; Bellahcene M.; Winter C. W.; Sintou A.; Tang J.; Keane T. J.; Schneider M. D.; et al. Neutrophils Enable Local and Non-Invasive Liposome Delivery to Inflamed Skeletal Muscle and Ischemic Heart. Adv. Mater. 2020, 32 (48), e2003598 10.1002/adma.202003598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Qin D.; Zou J.; Li X.; Guo X. D.; Tang Y.; Liu C.; Chen W.; Kong N.; Zhang C. Y. Living Leukocyte-Based Drug Delivery Systems. Adv. Mater. 2023, 35, e2207787 10.1002/adma.202207787. [DOI] [PubMed] [Google Scholar]
- Sofias A. M.; Toner Y. C.; Meerwaldt A. E.; van Leent M. M. T.; Soultanidis G.; Elschot M.; Gonai H.; Grendstad K.; Flobak A.; Neckmann U.; et al. Tumor Targeting by alphavbeta3-Integrin-Specific Lipid Nanoparticles Occurs via Phagocyte Hitchhiking. ACS Nano 2020, 14 (7), 7832–7846. 10.1021/acsnano.9b08693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z. P.; Nguyen L. N. M.; Ouyang B.; MacMillan P.; Ngai J.; Kingston B. R.; Mladjenovic S. M.; Chan W. C. W. Macrophages Actively Transport Nanoparticles in Tumors After Extravasation. ACS Nano 2022, 16, 6080. 10.1021/acsnano.1c11578. [DOI] [PubMed] [Google Scholar]
- Luo Z.; Lu Y.; Shi Y.; Jiang M.; Shan X.; Li X.; Zhang J.; Qin B.; Liu X.; Guo X.; et al. Neutrophil hitchhiking for drug delivery to the bone marrow. Nat. Nanotechnol 2023, 10.1038/s41565-023-01374-7. [DOI] [PubMed] [Google Scholar]
- Wang F.; Han D.; Qiao Z.; Zuang Y.; Zhang Y.; Jiang Q.; Liu M.; An Q.; Shen D.. Neutrophil-targeted Mn3O4 nanozyme treats myocardial ischemia reperfusion injury by scavenging reactive oxygen species. Research Square 2022, Preprint. 10.21203/rs.3.rs-2288620/v1. [DOI]
- Bachmaier K.; Stuart A.; Singh A.; Mukhopadhyay A.; Chakraborty S.; Hong Z.; Wang L.; Tsukasaki Y.; Maienschein-Cline M.; Ganesh B. B.; et al. Albumin Nanoparticle Endocytosing Subset of Neutrophils for Precision Therapeutic Targeting of Inflammatory Tissue Injury. ACS Nano 2022, 16 (3), 4084–4101. 10.1021/acsnano.1c09762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukasaki Y.; Toth P. T.; Davoodi-Bojd E.; Rehman J.; Malik A. B. Quantitative Pulmonary Neutrophil Dynamics Using Computer-Vision Stabilized Intravital Imaging. Am. J. Respir. Cell Mol. Biol. 2022, 66 (1), 12–22. 10.1165/rcmb.2021-0318MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Li J.; Cho J.; Malik A. B. Prevention of vascular inflammation by nanoparticle targeting of adherent neutrophils. Nat. Nanotechnol 2014, 9 (3), 204–210. 10.1038/nnano.2014.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myerson J. W.; Patel P. N.; Rubey K. M.; Zamora M. E.; Zaleski M. H.; Habibi N.; Walsh L. R.; Lee Y. W.; Luther D. C.; Ferguson L. T.; et al. Supramolecular arrangement of protein in nanoparticle structures predicts nanoparticle tropism for neutrophils in acute lung inflammation. Nat. Nanotechnol 2022, 17 (1), 86–97. 10.1038/s41565-021-00997-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui T. M.; Wiesolek H. L.; Sumagin R. ICAM-1: A master regulator of cellular responses in inflammation, injury resolution, and tumorigenesis. J. Leukoc Biol. 2020, 108 (3), 787–799. 10.1002/JLB.2MR0220-549R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsner J.; Sach M.; Knopf H. P.; Norgauer J.; Kapp A.; Schollmeyer P.; Dobos G. J. Synthesis and surface expression of ICAM-1 in polymorphonuclear neutrophilic leukocytes in normal subjects and during inflammatory disease. Immunobiology 1995, 193 (5), 456–464. 10.1016/S0171-2985(11)80430-4. [DOI] [PubMed] [Google Scholar]
- Hubbard A. K.; Rothlein R. Intercellular adhesion molecule-1 (ICAM-1) expression and cell signaling cascades. Free Radic Biol. Med. 2000, 28 (9), 1379–1386. 10.1016/S0891-5849(00)00223-9. [DOI] [PubMed] [Google Scholar]
- Real E.; Kaiser A.; Raposo G.; Amara A.; Nardin A.; Trautmann A.; Donnadieu E. Immature dendritic cells (DCs) use chemokines and intercellular adhesion molecule (ICAM)-1, but not DC-specific ICAM-3-grabbing nonintegrin, to stimulate CD4+ T cells in the absence of exogenous antigen. J. Immunol 2004, 173 (1), 50–60. 10.4049/jimmunol.173.1.50. [DOI] [PubMed] [Google Scholar]
- Wiesolek H. L.; Bui T. M.; Lee J. J.; Dalal P.; Finkielsztein A.; Batra A.; Thorp E. B.; Sumagin R. Intercellular Adhesion Molecule 1 Functions as an Efferocytosis Receptor in Inflammatory Macrophages. Am. J. Pathol. 2020, 190 (4), 874–885. 10.1016/j.ajpath.2019.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales C.; Demaurex N.; Lowell C. A.; Uribe-Querol E. Neutrophils: Their Role in Innate and Adaptive Immunity. J. Immunol Res. 2016, 2016, 1469780 10.1155/2016/1469780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliseeva S. I.; Knowlden Z. A.; Lester G. M.; Dean D. A.; Georas S. N.; Chapman T. J. Changes in lung immune cell infiltrates after electric field treatment in mice. Sci. Rep 2021, 11 (1), 1453. 10.1038/s41598-021-81174-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupane A. S.; Kubes P. Imaging reveals novel innate immune responses in lung, liver, and beyond. Immunol Rev. 2022, 306 (1), 244–257. 10.1111/imr.13040. [DOI] [PubMed] [Google Scholar]
- Yipp B. G.; Kim J. H.; Lima R.; Zbytnuik L. D.; Petri B.; Swanlund N.; Ho M.; Szeto V. G.; Tak T.; Koenderman L. The Lung is a Host Defense Niche for Immediate Neutrophil-Mediated Vascular Protection. Sci. Immunol 2017, 2 (10), 1. 10.1126/sciimmunol.aam8929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barletta K. E.; Cagnina R. E.; Wallace K. L.; Ramos S. I.; Mehrad B.; Linden J. Leukocyte compartments in the mouse lung: distinguishing between marginated, interstitial, and alveolar cells in response to injury. J. Immunol Methods 2012, 375 (1–2), 100–110. 10.1016/j.jim.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downey G. P.; Worthen G. S.; Henson P. M.; Hyde D. M. Neutrophil sequestration and migration in localized pulmonary inflammation. Capillary localization and migration across the interalveolar septum. Am. Rev. Respir. Dis. 1993, 147 (1), 168–176. 10.1164/ajrccm/147.1.168. [DOI] [PubMed] [Google Scholar]
- Doerschuk C. M.; Allard M. F.; Martin B. A.; MacKenzie A.; Autor A. P.; Hogg J. C. Marginated pool of neutrophils in rabbit lungs. J. Appl. Physiol (1985) 1987, 63 (5), 1806–1815. 10.1152/jappl.1987.63.5.1806. [DOI] [PubMed] [Google Scholar]
- Worthen G. S.; Schwab B. 3rd; Elson E. L.; Downey G. P. Mechanics of stimulated neutrophils: cell stiffening induces retention in capillaries. Science 1989, 245 (4914), 183–186. 10.1126/science.2749255. [DOI] [PubMed] [Google Scholar]
- Sohrabi S.; Yunus D. E.; Xu J.; Yang J.; Liu Y. Characterization of nanoparticle binding dynamics in microcirculation using an adhesion probability function. Microvasc Res. 2016, 108, 41–47. 10.1016/j.mvr.2016.07.005. [DOI] [PubMed] [Google Scholar]
- Papademetriou I.; Vedula E.; Charest J.; Porter T. Effect of flow on targeting and penetration of angiopep-decorated nanoparticles in a microfluidic model blood-brain barrier. PLoS One 2018, 13 (10), e0205158 10.1371/journal.pone.0205158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A.; Zhao J.; Anakk S.; Dash P. K.; Strobel H. W. Brain trauma leads to enhanced lung inflammation and injury: evidence for role of P4504Fs in resolution. J. Cereb Blood Flow Metab 2007, 27 (5), 963–974. 10.1038/sj.jcbfm.9600396. [DOI] [PubMed] [Google Scholar]
- Koutsoukou A.; Katsiari M.; Orfanos S. E.; Kotanidou A.; Daganou M.; Kyriakopoulou M.; Koulouris N. G.; Rovina N. Respiratory mechanics in brain injury: A review. World J. Crit Care Med. 2016, 5 (1), 65–73. 10.5492/wjccm.v5.i1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P. J.; Pittet J. F.; Kerby J. D.; Bosarge P. L.; Wagener B. M. Acute brain trauma, lung injury, and pneumonia: more than just altered mental status and decreased airway protection. Am. J. Physiol Lung Cell Mol. Physiol 2017, 313 (1), L1–L15. 10.1152/ajplung.00485.2016. [DOI] [PubMed] [Google Scholar]
- Samary C. S.; Ramos A. B.; Maia L. A.; Rocha N. N.; Santos C. L.; Magalhaes R. F.; Clevelario A. L.; Pimentel-Coelho P. M.; Mendez-Otero R.; Cruz F. F.; et al. Focal ischemic stroke leads to lung injury and reduces alveolar macrophage phagocytic capability in rats. Crit Care 2018, 22 (1), 249. 10.1186/s13054-018-2164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehl C.; Thati S.; Sullivan B.; Sestak J.; Thompson M.; Siahaan T.; Berkland C. Pulmonary Administration of Soluble Antigen Arrays Is Superior to Antigen in Treatment of Experimental Autoimmune Encephalomyelitis. J. Pharm. Sci. 2017, 106 (11), 3293–3302. 10.1016/j.xphs.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odoardi F.; Sie C.; Streyl K.; Ulaganathan V. K.; Schlager C.; Lodygin D.; Heckelsmiller K.; Nietfeld W.; Ellwart J.; Klinkert W. E.; et al. T cells become licensed in the lung to enter the central nervous system. Nature 2012, 488 (7413), 675–679. 10.1038/nature11337. [DOI] [PubMed] [Google Scholar]
- Saito E.; Gurczynski S. J.; Kramer K. R.; Wilke C. A.; Miller S. D.; Moore B. B.; Shea L. D. Modulating lung immune cells by pulmonary delivery of antigen-specific nanoparticles to treat autoimmune disease. Sci. Adv. 2020, 6 (42), 1. 10.1126/sciadv.abc9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rom S.; Zuluaga-Ramirez V.; Dykstra H.; Reichenbach N. L.; Ramirez S. H.; Persidsky Y. Poly(ADP-ribose) polymerase-1 inhibition in brain endothelium protects the blood-brain barrier under physiologic and neuroinflammatory conditions. J. Cereb Blood Flow Metab 2015, 35 (1), 28–36. 10.1038/jcbfm.2014.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne A.; Gauberti M.; Macrez R.; Jullienne A.; Briens A.; Raynaud J. S.; Louin G.; Buisson A.; Haelewyn B.; Docagne F.; et al. Ultra-sensitive molecular MRI of cerebrovascular cell activation enables early detection of chronic central nervous system disorders. Neuroimage 2012, 63 (2), 760–770. 10.1016/j.neuroimage.2012.07.018. [DOI] [PubMed] [Google Scholar]
- Alge J.; Dolan K.; Angelo J.; Thadani S.; Virk M.; Akcan Arikan A. Two to Tango: Kidney-Lung Interaction in Acute Kidney Injury and Acute Respiratory Distress Syndrome. Front Pediatr 2021, 9, 744110 10.3389/fped.2021.744110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn J. M.; Caldwell E. C.; Deem S.; Newell D. W.; Heckbert S. R.; Rubenfeld G. D. Acute lung injury in patients with subarachnoid hemorrhage: incidence, risk factors, and outcome. Crit Care Med. 2006, 34 (1), 196–202. 10.1097/01.CCM.0000194540.44020.8E. [DOI] [PubMed] [Google Scholar]
- Austin V.; Ku J. M.; Miller A. A.; Vlahos R. Ischaemic stroke in mice induces lung inflammation but not acute lung injury. Sci. Rep 2019, 9 (1), 3622. 10.1038/s41598-019-40392-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacon-Aponte A. A.; Duran-Vargas E. A.; Arevalo-Carrillo J. A.; Lozada-Martinez I. D.; Bolano-Romero M. P.; Moscote-Salazar L. R.; Grille P.; Janjua T. Brain-lung interaction: a vicious cycle in traumatic brain injury. Acute Crit Care 2022, 37 (1), 35–44. 10.4266/acc.2021.01193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge W. M. Blood-brain barrier drug delivery of IgG fusion proteins with a transferrin receptor monoclonal antibody. Expert Opin Drug Deliv 2015, 12 (2), 207–222. 10.1517/17425247.2014.952627. [DOI] [PubMed] [Google Scholar]
- Wiley D. T.; Webster P.; Gale A.; Davis M. E. Transcytosis and brain uptake of transferrin-containing nanoparticles by tuning avidity to transferrin receptor. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (21), 8662–8667. 10.1073/pnas.1307152110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Ling C. L.; Pang L.; Wang Q.; Liu J. X.; Wang B. S.; Liang J. M.; Guo Y. Z.; Qin J.; Wang J. X. Direct Macromolecular Drug Delivery to Cerebral Ischemia Area using Neutrophil-Mediated Nanoparticles. Theranostics 2017, 7 (13), 3260–3275. 10.7150/thno.19979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haney M. J.; Zhao Y.; Harrison E. B.; Mahajan V.; Ahmed S.; He Z.; Suresh P.; Hingtgen S. D.; Klyachko N. L.; Mosley R. L.; et al. Specific transfection of inflamed brain by macrophages: a new therapeutic strategy for neurodegenerative diseases. PLoS One 2013, 8 (4), e61852 10.1371/journal.pone.0061852. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Joly E.; Hudrisier D. What is trogocytosis and what is its purpose?. Nat. Immunol 2003, 4 (9), 815. 10.1038/ni0903-815. [DOI] [PubMed] [Google Scholar]
- Miyake K.; Karasuyama H. The Role of Trogocytosis in the Modulation of Immune Cell Functions. Cells 2021, 10 (5), 1255. 10.3390/cells10051255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa S.; Lee Y.; Jacob A.; Aziz M.; Wang P. Neutrophil trogocytosis during their trans-endothelial migration: role of extracellular CIRP. Mol. Med. 2022, 28 (1), 91. 10.1186/s10020-022-00515-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muro S.; Wiewrodt R.; Thomas A.; Koniaris L.; Albelda S. M.; Muzykantov V. R.; Koval M. A novel endocytic pathway induced by clustering endothelial ICAM-1 or PECAM-1. J. Cell Sci. 2003, 116 (8), 1599–1609. 10.1242/jcs.00367. [DOI] [PubMed] [Google Scholar]
- Serrano D.; Manthe R. L.; Paul E.; Chadha R.; Muro S. How Carrier Size and Valency Modulate Receptor-Mediated Signaling: Understanding the Link between Binding and Endocytosis of ICAM-1-Targeted Carriers. Biomacromolecules 2016, 17 (10), 3127–3137. 10.1021/acs.biomac.6b00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papademetriou J.; Garnacho C.; Serrano D.; Bhowmick T.; Schuchman E. H.; Muro S. Comparative binding, endocytosis, and biodistribution of antibodies and antibody-coated carriers for targeted delivery of lysosomal enzymes to ICAM-1 versus transferrin receptor. J. Inherit Metab Dis 2013, 36 (3), 467–477. 10.1007/s10545-012-9534-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu J.; Hoenicka J.; Muro S. Targeting, endocytosis, and lysosomal delivery of active enzymes to model human neurons by ICAM-1-targeted nanocarriers. Pharm. Res. 2015, 32 (4), 1264–1278. 10.1007/s11095-014-1531-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfin A.; Beyrau M.; Voisin M. B.; Ma B.; Whiteford J. R.; Hordijk P. L.; Hogg N.; Nourshargh S. ICAM-1-expressing neutrophils exhibit enhanced effector functions in murine models of endotoxemia. Blood 2016, 127 (7), 898–907. 10.1182/blood-2015-08-664995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H.; Lin H.; Pang Q.; Zhuang J.; Liu X.; Li X.; Liu J.; Tang J. Macrophage ICAM-1 functions as a regulator of phagocytosis in LPS induced endotoxemia. Inflamm Res. 2021, 70 (2), 193–203. 10.1007/s00011-021-01437-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmer B. P.; Funder J. W.. Adrenocorticotropic Hormone, Adrenal Steroids, and the Adrenal Cortex. In Goodman & Gilman’s: The Pharmacological Basis of Therapeutics, 13e, 2017. [Google Scholar]
- Habibi N.; Brown T. D.; Adu-Berchie K.; Christau S.; Raymond J. E.; Mooney D. J.; Mitragotri S.; Lahann J. Nanoparticle Properties Influence Transendothelial Migration of Monocytes. Langmuir 2022, 38 (18), 5603–5616. 10.1021/acs.langmuir.2c00200. [DOI] [PubMed] [Google Scholar]
- Jiang K.; Weaver J. D.; Li Y.; Chen X.; Liang J.; Stabler C. L. Local release of dexamethasone from macroporous scaffolds accelerates islet transplant engraftment by promotion of anti-inflammatory M2 macrophages. Biomaterials 2017, 114, 71–81. 10.1016/j.biomaterials.2016.11.004. [DOI] [PubMed] [Google Scholar]
- Tedesco S.; Bolego C.; Toniolo A.; Nassi A.; Fadini G. P.; Locati M.; Cignarella A. Phenotypic activation and pharmacological outcomes of spontaneously differentiated human monocyte-derived macrophages. Immunobiology 2015, 220 (5), 545–554. 10.1016/j.imbio.2014.12.008. [DOI] [PubMed] [Google Scholar]
- Kanazawa M.; Ninomiya I.; Hatakeyama M.; Takahashi T.; Shimohata T. Microglia and Monocytes/Macrophages Polarization Reveal Novel Therapeutic Mechanism against Stroke. Int. J. Mol. Sci. 2017, 18 (10), 2135. 10.3390/ijms18102135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; Leak R. K.; Shi Y.; Suenaga J.; Gao Y.; Zheng P.; Chen J. Microglial and macrophage polarization-new prospects for brain repair. Nat. Rev. Neurol 2015, 11 (1), 56–64. 10.1038/nrneurol.2014.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood E. D.; Greineder C. F.; Shuvaeva T.; Walsh L.; Villa C. H.; Muzykantov V. R. Vascular Targeting of Radiolabeled Liposomes with Bio-Orthogonally Conjugated Ligands: Single Chain Fragments Provide Higher Specificity than Antibodies. Bioconjug Chem. 2018, 29 (11), 3626–3637. 10.1021/acs.bioconjchem.8b00564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingsley M. M.; Singh N.; Ravikumar P.; Zhang R.; June C. H.; Mitchell M. J. Ionizable Lipid Nanoparticle-Mediated mRNA Delivery for Human CAR T Cell Engineering. Nano Lett. 2020, 20 (3), 1578–1589. 10.1021/acs.nanolett.9b04246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd S. J.; Warzecha C. C.; Yadavali S.; El-Mayta R.; Alameh M. G.; Wang L.; Weissman D.; Wilson J. M.; Issadore D.; Mitchell M. J. Scalable mRNA and siRNA Lipid Nanoparticle Production Using a Parallelized Microfluidic Device. Nano Lett. 2021, 21 (13), 5671–5680. 10.1021/acs.nanolett.1c01353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parhiz H.; Shuvaev V. V.; Pardi N.; Khoshnejad M.; Kiseleva R. Y.; Brenner J. S.; Uhler T.; Tuyishime S.; Mui B. L.; Tam Y. K.; et al. PECAM-1 directed re-targeting of exogenous mRNA providing two orders of magnitude enhancement of vascular delivery and expression in lungs independent of apolipoprotein E-mediated uptake. J. Controlled Release 2018, 291, 106–115. 10.1016/j.jconrel.2018.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parhiz H.; Brenner J. S.; Patel P. N.; Papp T. E.; Shahnawaz H.; Li Q.; Shi R.; Zamora M. E.; Yadegari A.; Marcos-Contreras O. A.; et al. Added to pre-existing inflammation, mRNA-lipid nanoparticles induce inflammation exacerbation (IE). J. Controlled Release 2022, 344, 50–61. 10.1016/j.jconrel.2021.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnacho C.; Dhami R.; Solomon M.; Schuchman E. H.; Muro S. Enhanced Delivery and Effects of Acid Sphingomyelinase by ICAM-1-Targeted Nanocarriers in Type B Niemann-Pick Disease Mice. Mol. Ther 2017, 25 (7), 1686–1696. 10.1016/j.ymthe.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posel C.; Moller K.; Boltze J.; Wagner D.-C.; Weise G. Isolation and Flow Cytometric Analysis of Immune Cells from the Ischemic Mouse Brain. J. Vis Exp 2016, 108, 53658. 10.3791/53658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson L. T.; Hood E. D.; Shuvaeva T.; Shuvaev V. V.; Basil M. C.; Wang Z.; Nong J.; Ma X.; Wu J.; Myerson J. W.; et al. Dual Affinity to RBCs and Target Cells (DART) Enhances Both Organ- and Cell Type-Targeting of Intravascular Nanocarriers. ACS Nano 2022, 16 (3), 4666–4683. 10.1021/acsnano.1c11374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.