

Abstract
Vinyl bisphosphonates can be readily prepared by condensation of an aromatic aldehyde with the tetraester of a methylenebisphosphonate, and reduction of the resulting olefin is an attractive strategy for the preparation of monoalkyl geminal bisphosphonates. Conjugate reduction through use of variations on the Stryker approach has proven to be an efficient method for that reduction, even in the presence of aromatic substituents that also could be reduced. Furthermore, remote olefins in an isoprenoid chain survive this conjugate reduction unaffected, allowing access to isoprenoid-substituted triazole bisphosphonates of interest as potential inhibitors of terpenoid biosynthesis.
Keywords: Bisphosphonate, Synthesis, Conjugate reduction, Triazole, Isoprenoid biosynthesis
Geminal bisphosphonates can be viewed as metabolically stable analogues of diphosphates, and have become prominent drugs for treatment of bone diseases [1]. The earliest examples that reached clinical use, compounds such as etidronate (1, Fig. 1), were viewed as analogues of diphosphate that could be incorporated metabolically into ATP analogues of greater stability than ATP itself. Subsequent generations, including zoledronate (2) and risedronate (3), were found to serve as inhibitors of isoprenoid biosynthesis resulting in disruption of protein prenylation [2,3]. It is now clear that the primary enzymatic target of these nitrogenous bisphosphonates is farnesyl diphosphate synthase (FDPS) [4,5], with zoledronate reported to display a 3 nM IC50 against this enzyme [6]. Lowered cellular levels of farnesyl diphosphate (FPP) would be expected to impact both protein farnesylation and steroid biosynthesis. However, FPP also is a substrate for the enzyme geranylgeranyl diphosphate synthase (GGDPS), and inhibition of FDPS diminishes cellular levels of the C20 isoprenoid geranylgeranyl diphosphate (GGPP). Thus it is possible that the biological consequences of FDPS inhibition result at least in part from depletion of GGPP [7], especially given the role of key geranylgeranylated proteins (e.g. cdc42, Rac, Rho, Rab7, Rab3D) in osteoclast polarization [8–11].
Fig. 1.
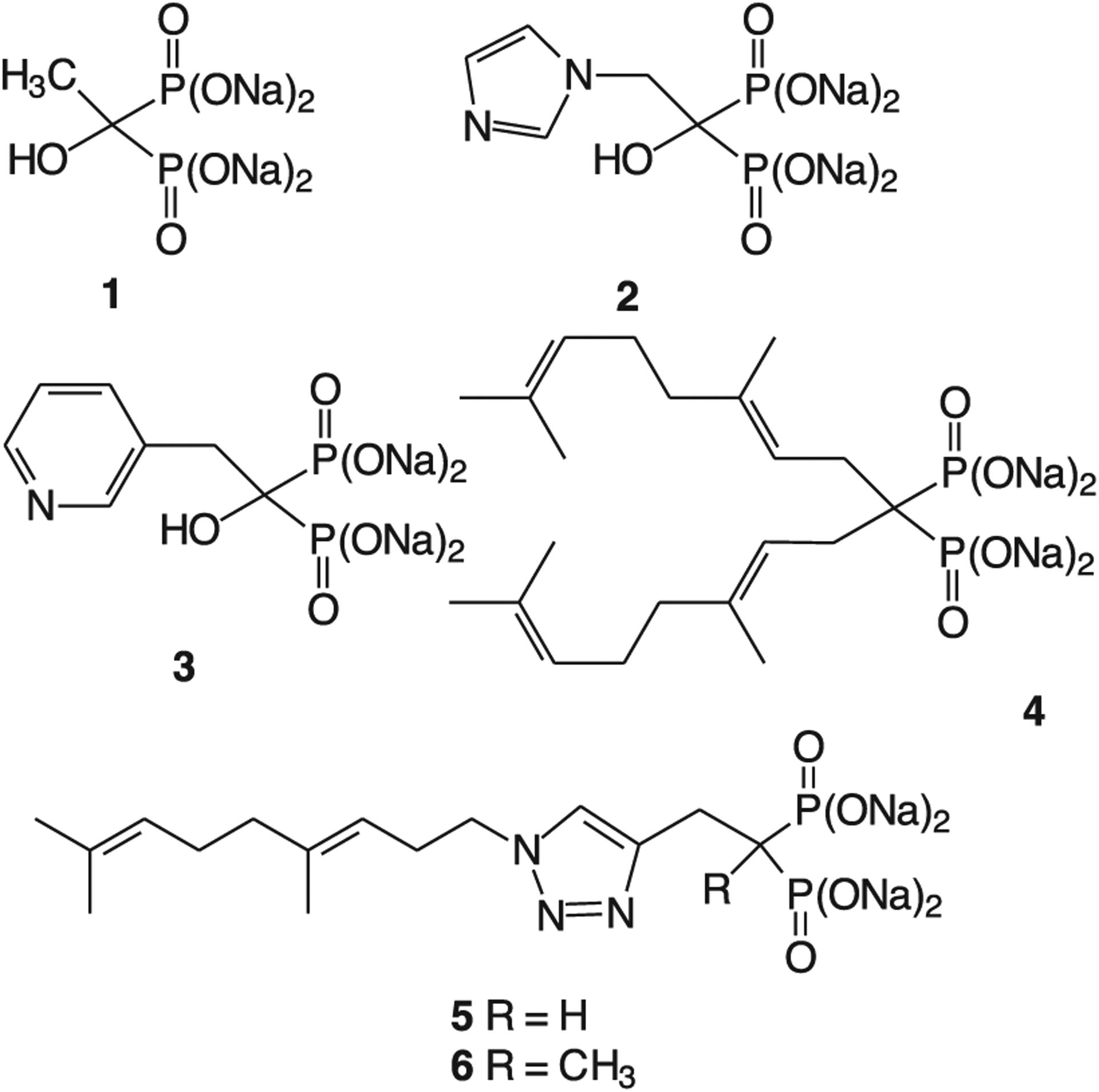
Some geminal bisphosphonates with biological activity.
To minimize impact on steroid biosynthesis and farnesylated proteins, our focus has been on development of inhibitors of the enzyme GGDPS [12]. One of the most selective early inhibitors of this enzyme was digeranyl bisphosphonate (4), a dialkyl bisphosphonate which is surprisingly effective as an inhibitor of GGDPS over FDPS [13,14]. Preparation of bisphosphonates that carry two identical alkyl groups is straightforward, and simply requires treatment of commercially available tetraalkyl bisphosphonates with an excess of the alkylating agent under basic conditions and subsequent hydrolysis of the phosphonate esters [15]. Preparation of dialkyl bisphosphonates with two different alkyl groups is more challenging because direct alkylation generally affords a mixture of un-, mono- and dialkylated products and the polarity of the bisphosphonate group can make separations challenging [16]. After discovery of more potent inhibitors of GGDPS that carried two different alkyl groups, compounds such as the triazoles 5 [17,18] and 6 [19], it became important to explore other routes for selective synthesis of monoalkyl bisphosphonates. That is the subject of this study.
The simplest monoalkylated geminal bisphosphonates can be prepared through short synthetic sequences based on condensation with a carbonyl compound and subsequent reduction. For example, treatment of tetramethyl methylenebisphosphonate (7, Fig. 2) with base and paraformaldehyde, followed by treatment with acid to induce dehydration, long has been known to provide convenient access to the olefin 8 [20]. The monomethylated compound 9 then easily can be obtained by catalytic hydrogenation [19]. For aromatic aldehydes the corresponding vinylbisphosphonate can be obtained with greater efficiency by treatment of a tetraalkyl methylenebisphosphonate with TiCl4 in the presence of an amine base [21–24]. Once the vinylbisphosphonate is in hand, catalytic hydrogenation again allows facile conversion to, and subsequent purification of, the desired monoalkyl product. Unfortunately, the olefins of isoprenoid substituents and other easily reduced groups also may be susceptible to reduction via this approach, thus limiting the variety of compounds available through this strategy [25,26].
Fig. 2.
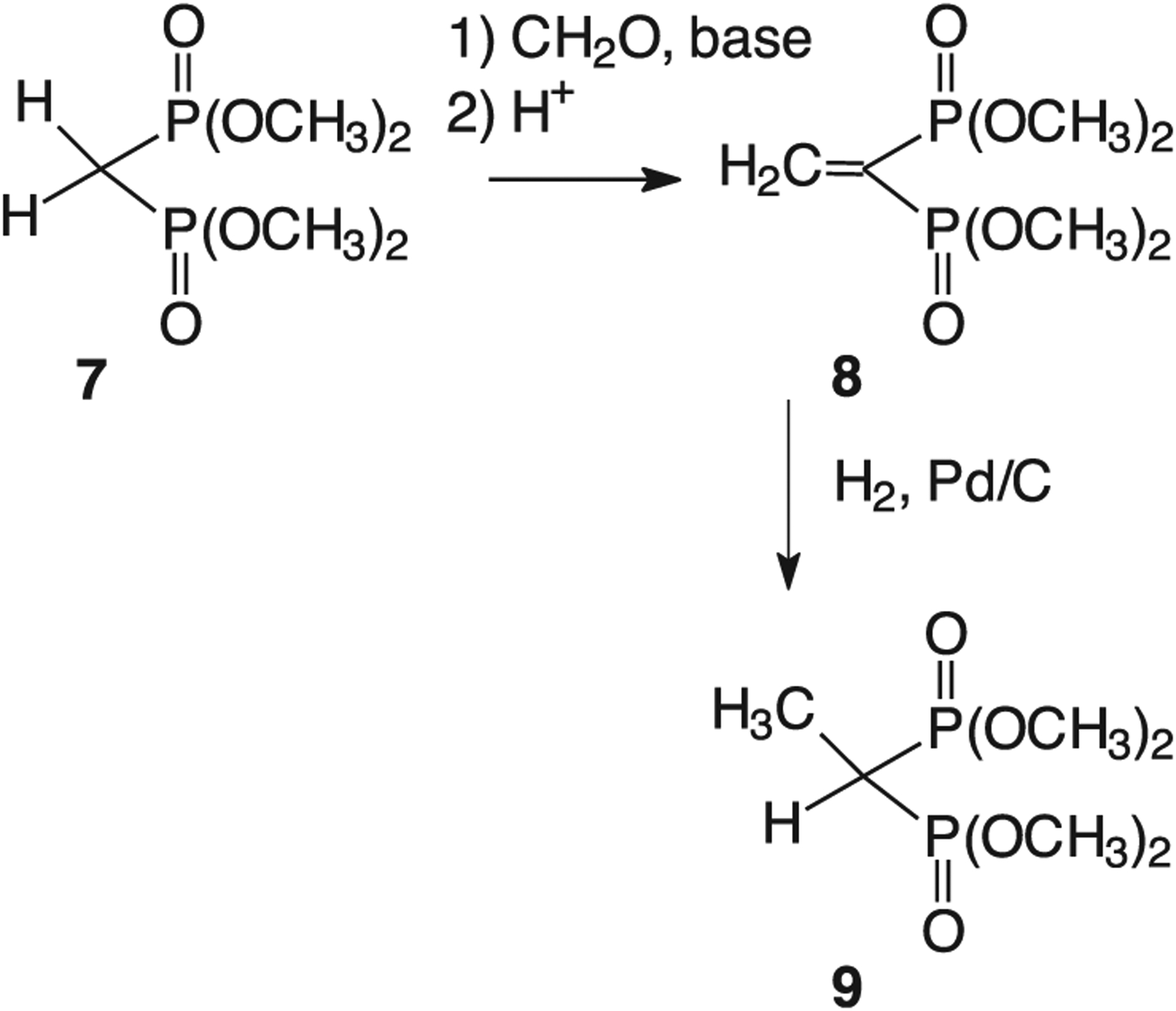
Synthesis of the monomethylated bisphosphonate 9.
To avoid the potential complications of catalytic hydrogenation, reductions via Stryker’s reagent [27–29] and its variations [30,31] have become popular. There have been a few studies that have examined reagents for conjugate reduction in activated monophosphonate systems [32–34], but the utility of this approach with vinyl bisphosphonates has not been explored. One might assume that a mechanism involving in situ formation of a copper hydride, followed by hydride addition to the β carbon of the vinyl bisphosphonate and protonation at the α-carbon, would result in selective reduction of the conjugated double bond even in the presence of remote olefins.
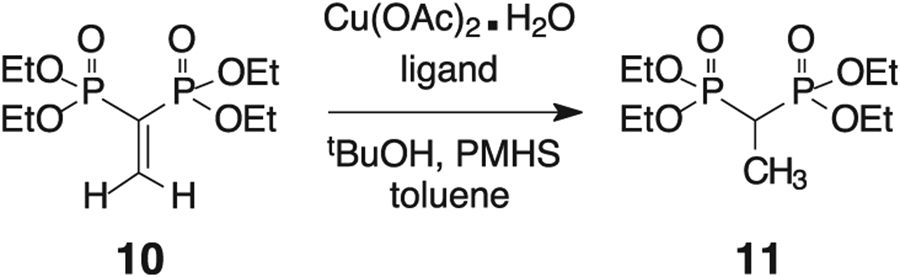
We began these studies with the unsubstituted vinyl bisphosphonate 10 as a model system and conducted a brief survey of potential reaction conditions (Table 1). Slow conversion to the desired product 11 was observed in the absence of any added phosphine ligand (Table 1, Entry 1) when compound 10 was treated with copper (II) acetate and polymethylhydrosiloxane (PMHS) at room temperature, which might be due to the vinyl bisphosphonate serving both as a substrate and as a copper ligand [35,36]. Conjugate reduction took place more quickly and at lower temperature in the presence of a phosphine ligand (Table 1, Entry 2). No change in either the required reaction time or product yield was observed via 31P NMR analysis when the reaction was conducted at elevated temperatures. The reaction proceeded well in the presence of various bidentate phosphine ligands including 1,2-bis (diphenylphosphino)benzene (DPPBZ), bis(diphenylphosphino) methane (DPPM), 1,1′-bis(diphenylphosphino)ferrocene (DPPF), 1,2-bis(diphenylphosphino)ethane (DPPE), BINAP, or cis-vinylenebis(diphenylphosphine (DPPV) (Entries 2–8). With the exception of DPPM. There was no notable loss in yield or requirement for extended reaction times with any of these ligands which suggests that the various bite angles are not an important factor in this reduction. With the DPPM ligand, conversion to the desired product was observed but only under extended reaction times and in modestly reduced yield. The conjugate reduction reaction proceeded well at 10 or even 5 mol % ligand/catalyst loading (Entries 2 and 9). However, as the catalyst and ligand loading were decreased further (Entries 10 and 11), a diminished rate of conversion to the reduced product was observed (Table 1.).
Table 1.
Reaction conditions surveyed for conversion of the vinyl bisphosphonate 10 to the reduced product 11.
| |||||
|---|---|---|---|---|---|
| Entrya | Ligand | Mol % CuOAc2·H2O | T (°C) | Time (h) | Conv (%)b |
| 1 | None | 10 | 70 | 48 | 90 |
| 2 | DPPBZ | 10 | 70 | 1 | >99 |
| 3 | DPPBZ | 10 | 25 | 1 | >99 |
| 4 | DPPM | 10 | 25 | 12 | 95 |
| 5 | DPPF | 10 | 25 | 1 | >99 |
| 6 | DPPE | 10 | 25 | 1 | >99 |
| 7 | BINAP | 10 | 25 | 1 | >99 |
| 8 | DPPV | 10 | 25 | 1 | >99 |
| 9 | DPPBZ | 5 | 25 | 1 | >99 |
| 10 | DPPBZ | 2.5 | 25 | 1 | 50 |
| 11 | DPPBZ | 1 | 25 | 1 | 0 |
Reactions were conducted in toluene, on a 1 mmol scale, with a 1:1 ratio of Cu(OAc)2 to ligand (except for entry #1).
Conversions were determined by 31P NMR analysis.
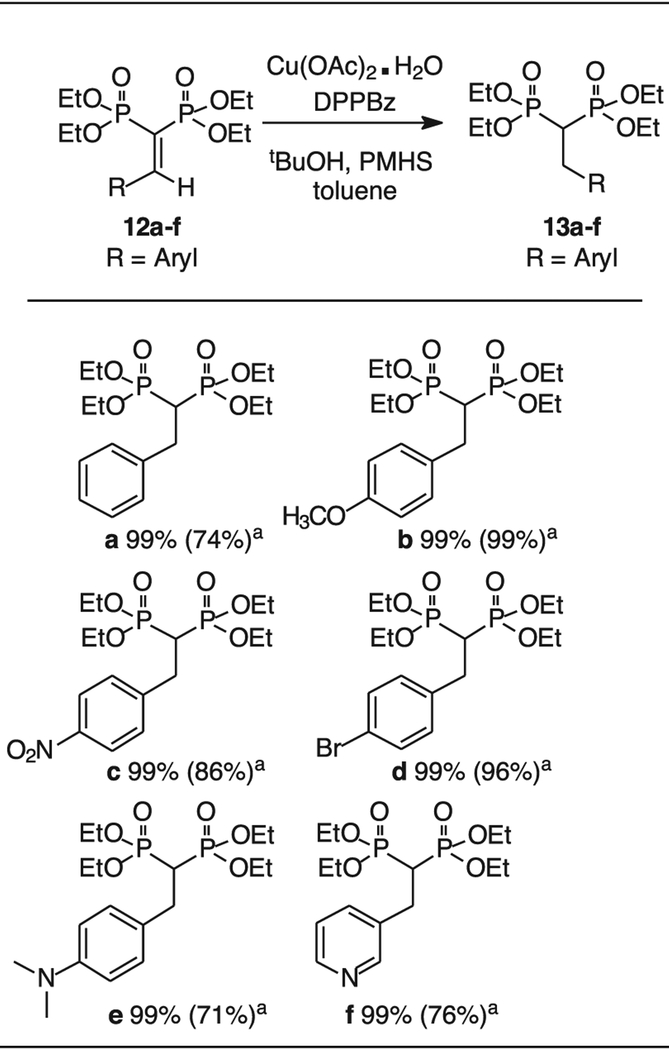
After appropriate reaction conditions were established for the conjugate addition, a small set of vinyl bisphosphonates was prepared by known methods [21,22] to gauge the utility of this process. The parent aromatic system 12a, derived from a Knovenagel condensation of benzaldehyde, undergoes conjugate reduction in quantitative yield by 31P NMR when treated with CuOAc2 H2O, PMHS, and DPPBZ. Reactions conducted on substrates with a phenyl ring bearing a strong electron donating substituent (e.g. 12b and 12e) proceeded equally well, and gave isolated yields >95 % in each case. The p-nitro compound 12c also gave the desired product 13c in high isolated yield, with no evidence of reduction at the nitro group. The p-bromo compound 12d [37] also gave the reduction product in high yield, with no evidence for cleavage of the carbon-bromine bond which would afford compound 13a. Finally, the pyridyl substituted compound 12f also undergoes smooth reduction despite its ability to function as a Lewis base through complexation of the ring nitrogen. In all cases, the conversion to the reduced product was complete based upon 31P NMR analysis (cf SI). Isolated yields were sometimes lower, especially with the compounds that contained a basic nitrogen which may be due to complexation with copper.
The range of substrates displayed in Table 2 encouraged examination of the vinyl triazole bisphosphonate 14 [38], to access compounds of interest for our studies of GGDPS inhibitors (Scheme 1). While the triazole system theoretically has the potential to impact the desired transformation in various ways, including through complexation with the catalyst, reductive cleavage, or even loss of nitrogen to form an aziridine, in practice the desired reaction proceeded smoothly. Only the desired reduction product was observed by NMR, and the product 15 was isolated in 76 % yield.
Table 2.
Conjugate reduction of selected vinylbisphosphonates.
|
Conversion by 31P NMR (isolated yield).
Scheme 1.
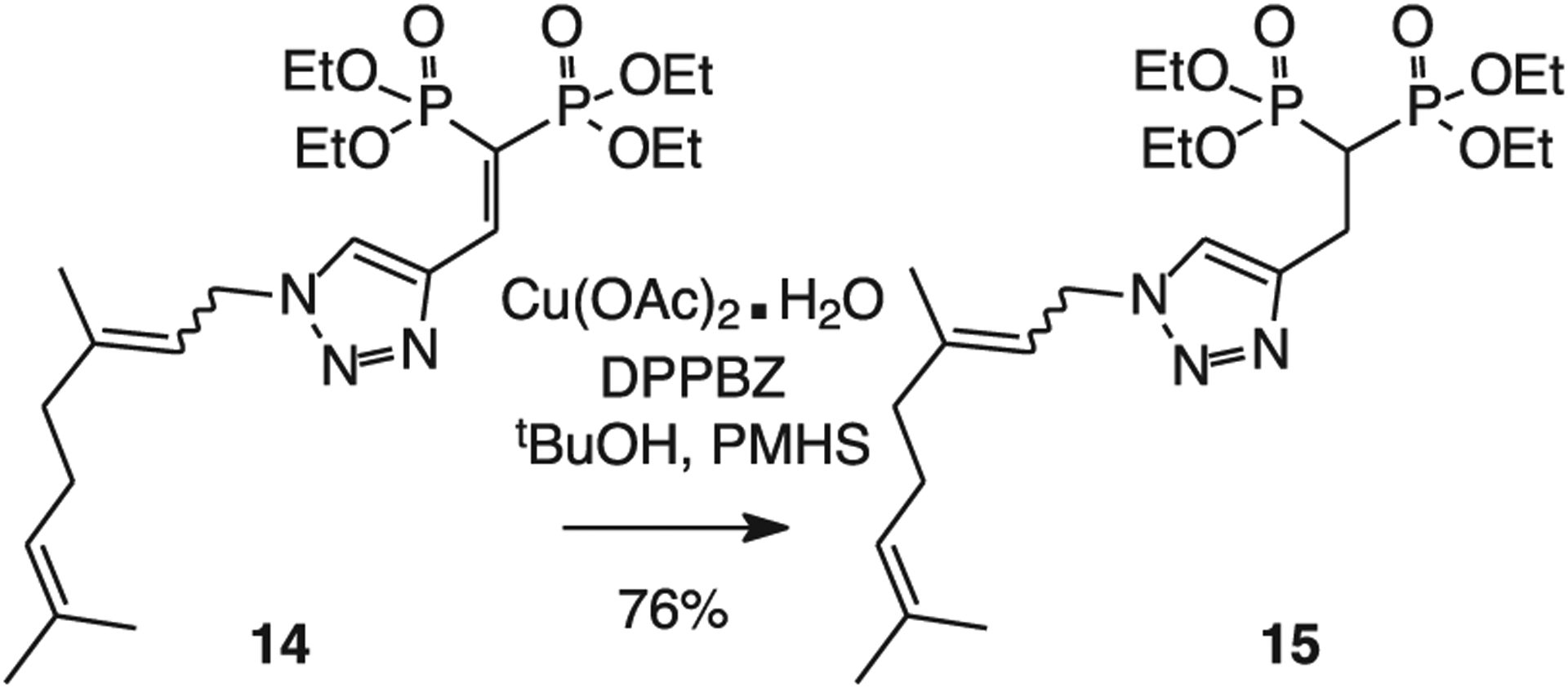
Conjugate reduction of a triazole bisphosphonate.
In conclusion, these studies have determined that conjugate reduction of vinyl bisphosphonates is a viable route to the monoalkylated triazole bisphosphonates of interest as inhibitors of the enzyme GGDPS. With this sequence now established, it will be possible to explore modifications at the alpha position that might further enhance potency or afford more favorable biodistribution.
Supplementary Material
Acknowledgments
Financial support from the National Institutes of Health (R01 CA258621) is gratefully acknowledged. The Q-Exactive mass spectrometer used in this research was acquired through the National Science Foundation Major Research Instrumentation and the Chemical Instrumentation Programs (CHE-1919422), while the Neo-400 NMR was acquired through NSF Instrumentation grant CHE-2017828.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
The experimental procedures and the 1H and 31P NMR spectra are available free of charge via the Internet. Supplementary data to this article can be found online at https://doi.org/10.1016/j.tetlet.2022.154078.
Data availability
Data will be made available on request.
References
- [1].Ebetino FH, Sun S, Cherian P, Roshandel S, Neighbors JD, Hu E, Dunford JE, Sedghizadeh PP, McKenna CE, Srinivasan V, Boeckman RK, Russell RGG, Bone 156 (2022) 116289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Luckman SP, Coxon FP, Ebetino FH, Russell RGG, Rogers MJ, J. Bone Miner. Res 13 (1998) 1668. [DOI] [PubMed] [Google Scholar]
- [3].Luckman SP, Hughes DE, Coxon FP, Russell RGG, Rogers MJ, J. Bone Miner. Res 13 (1998) 581. [DOI] [PubMed] [Google Scholar]
- [4].van Beek EP, Cohen L, Lowik C, Papapoulos S, Biochem. Biophys. Res. Commun 264 (1999) 108. [DOI] [PubMed] [Google Scholar]
- [5].Bergstrom JD, Bostedor RG, Masarachia PJ, Reszka AA, Rodan G, Arch. Biochem. Biophys 373 (2000) 231. [DOI] [PubMed] [Google Scholar]
- [6].Dunford JE, Thompson K, Coxon FP, Luckman SP, Hahn FM, Poulter CD, Ebetino FH, Rogers MJ, J. Pharmacol. Exp. Ther 296 (2001) 235. [PubMed] [Google Scholar]
- [7].Lee HF, Lacbay CM, Boutin R, Matralis AN, Park J, Waller DD, Guan TL, Sebag M, Tsantrizos YS, J. Med. Chem 65 (2022) 2471. [DOI] [PubMed] [Google Scholar]
- [8].Ory S, Brazier H, Pawlak G, Blangy A, Eur. J. Cell Biol 87 (2008) 469. [DOI] [PubMed] [Google Scholar]
- [9].Zhao HB, Laitala-Leinonen T, Parikka V, Vaananen HK, J. Biol. Chem 276 (2001) 39295. [DOI] [PubMed] [Google Scholar]
- [10].Pavlos NJ, Xu JK, Riedel D, Yeoh JSG, Teitelbaum SL, Papadimitriou JM, Jahn R, Ross FP, Zheng MH, Mol. Cell. Biol 25 (2005) 5253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Itzstein C, Coxon FP, Rogers MJ, Small GTPases 2 (2011) 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Haney SL, Wills VS, Wiemer DF, Holstein SA, Molecules 22 (2017) 886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Shull LW, Wiemer AJ, Hohl RJ, Wiemer DF, Bioorg. Med. Chem 14 (2006) 4130. [DOI] [PubMed] [Google Scholar]
- [14].Wiemer AJ, Tong H, Swanson KM, Hohl RJ, Biochem. Biophys. Res. Commun 353 (2007) 921. [DOI] [PubMed] [Google Scholar]
- [15].McKenna CE, Higa MT, Cheung NH, McKenna MC, Tetrahedron Lett. (1977) 155. [Google Scholar]
- [16].Wiemer AJ, Yu JS, Lamb KM, Hohl RJ, Wiemer DF, Bioorg. Med. Chem 16 (2008) 390. [DOI] [PubMed] [Google Scholar]
- [17].Matthiesen RA, Wills VS, Metzger JI, Holstein SA, Wiemer DF, J. Org. Chem 81 (2016) 9438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wills VS, Allen C, Holstein SA, Wiemer DF, ACS Med. Chem. Lett 6 (2015) 1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Matthiesen RA, Varney ML, Xu PC, Rier AS, Wiemer DF, Holstein SA, Bioorg. Med. Chem 26 (2018) 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Degenhardt CR, Burdsall DC, J. Org. Chem 51 (1986) 3488. [Google Scholar]
- [21].Lehnert W, Tetrahedron 30 (1974) 301. [Google Scholar]
- [22].Chiminazzo A, Sperni L, Scarso A, Strukul G, Catalysts 7 (2017) 90. [Google Scholar]
- [23].Bortolamiol E, Chiminazzo A, Sperni L, Borsato G, Fabris F, Scarso A, New J. Chem 43 (2019) 12641. [Google Scholar]
- [24].Chiminazzo A, Borsato G, Favero A, Fabbro C, McKenna CE, Dalle Carbonare LG, Valenti MT, Fabris F, Scarso A, Chem. Eur. J 25 (2019) 3617. [DOI] [PubMed] [Google Scholar]
- [25].Shilpa ML, Gayathri V, Trans. Met. Chem 41 (2016) 393. [Google Scholar]
- [26].Meemken F, Baiker A, Chem. Rev 117 (2017) 11522. [DOI] [PubMed] [Google Scholar]
- [27].Chen JX, Daeuble JF, Brestensky DM, Stryker JFM, Tetrahedron 56 (2000) 2153. [Google Scholar]
- [28].Lipshutz BH, Wiley-VCH Verlag GmbH (2002) 167.
- [29].Lee D-W, Yun J, Tetrahedron Lett. 46 (2005) 2037. [Google Scholar]
- [30].Roy SR, Sau SC, Mandal SK, J. Org. Chem 79 (2014) 9150. [DOI] [PubMed] [Google Scholar]
- [31].Trose M, Lazreg F, Chang T, Nahra F, Cordes DB, Slawin AMZ, Cazin CSJ, ACS Catal. 7 (2017) 238. [Google Scholar]
- [32].Duan ZC, Hu XP, Wang DY, Yu SB, Zheng Z, Tetrahedron Lett. 50 (2009) 6720. [Google Scholar]
- [33].Zhang L, Fang Y, Jin X, Guo T, Li R, Li Y, Li X, Yang Y, Yuan M, Tian Z, Tetrahedron Lett. 58 (2017) 4538. [Google Scholar]
- [34].Janicki I, Kielbasinski P, Szelag J, Glebski A, Szczesna-Antczak M, Bioorg. Chem 96 (2020) 103548. [DOI] [PubMed] [Google Scholar]
- [35].Kowalik-Jankowska T, Pietruszka M, Jezierska J, Matczak-Jon E, Kafarski P, Polyhedron 30 (2011) 1274. [Google Scholar]
- [36].Bretti C, De Stefano C, Cardiano P, Cataldo S, Pettignano A, Arena G, Sgarlata C, Grasso GI, Lando G, Sammartano S, J. Mol. Liq 343 (2021) 117699. [Google Scholar]
- [37].Xue Z-Y, Li Q-H, Tao H-Y, Wang C-J, J. Am. Chem. Soc 133 (2011) 11757. [DOI] [PubMed] [Google Scholar]
- [38].Fairweather AER, Goetz DB, Schroeder CM, Bhuiyan NH, Varney ML, Wiemer DF, Holstein SA, Biorg. Med. Chem 44 (2021) 116307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.