

Significance
Oxidative damage is one of the earliest events in AD pathogenesis. Nuclear factor erythroid 2-related factor 2 (Nrf2) is activated in response to oxidative stress, which protects the brain from oxidative damage and AD pathogenesis. However, as oxidative stress increases in AD, Nrf2-mediated neuroprotection declines. The mechanistic basis for this dysfunction has been a mystery. Here, we show that the actin-associated protein Slingshot homolog-1 (SSH1) mediates the loss of neuroprotection in AD. Specifically, both Nrf2 and SSH1 are activated by oxidative stress; however, activated SSH1 sequesters Nrf2 to block its natural neuroprotection in disease. This offers insight into the brain’s progressive vulnerability to oxidative stress as AD advances and may provide a unique neuroprotective strategy for AD and related dementias.
Keywords: Nrf2, slingshot, tauopathy, oxidative stress, alzheimer
Abstract
Oxidative damage in the brain is one of the earliest drivers of pathology in Alzheimer’s disease (AD) and related dementias, both preceding and exacerbating clinical symptoms. In response to oxidative stress, nuclear factor erythroid 2-related factor 2 (Nrf2) is normally activated to protect the brain from oxidative damage. However, Nrf2-mediated defense against oxidative stress declines in AD, rendering the brain increasingly vulnerable to oxidative damage. Although this phenomenon has long been recognized, its mechanistic basis has been a mystery. Here, we demonstrate through in vitro and in vivo models, as well as human AD brain tissue, that Slingshot homolog-1 (SSH1) drives this effect by acting as a counterweight to neuroprotective Nrf2 in response to oxidative stress and disease. Specifically, oxidative stress-activated SSH1 suppresses nuclear Nrf2 signaling by sequestering Nrf2 complexes on actin filaments and augmenting Kelch-like ECH-associated protein 1 (Keap1)–Nrf2 interaction, independently of SSH1 phosphatase activity. We also show that Ssh1 elimination in AD models increases Nrf2 activation, which mitigates tau and amyloid-β accumulation and protects against oxidative injury, neuroinflammation, and neurodegeneration. Furthermore, loss of Ssh1 preserves normal synaptic function and transcriptomic patterns in tauP301S mice. Importantly, we also show that human AD brains exhibit highly elevated interactions of Nrf2 with both SSH1 and Keap1. Thus, we demonstrate here a unique mode of Nrf2 blockade that occurs through SSH1, which drives oxidative damage and ensuing pathogenesis in AD. Strategies to inhibit SSH1-mediated Nrf2 suppression while preserving normal SSH1 catalytic function may provide new neuroprotective therapies for AD and related dementias.
Oxidative damage is one of the earliest events in Alzheimer’s disease (AD) pathogenesis (1, 2) and precedes the onset of clinical symptoms (3, 4). Nuclear factor erythroid 2-related factor 2 (Nrf2) is a redox-signaling transcription factor that mediates the expression of genes involved in the antioxidant response, xenobiotic detoxification, antiinflammation, and proteostasis (5–8), which together orchestrate a neuroprotective effect. Notably, AD is associated with nuclear, but not cytoplasmic, depletion of neuronal Nrf2 (9, 10). Loss of Nrf2 replicates transcriptomic changes seen in AD and exacerbates amyloid-β (Aβ) and tau deposition, neuroinflammation, and cognitive impairment in preclinical models of AD (6, 11–13). Importantly, the induction of Nrf2 signaling protects AD mice from these phenotypes (14–17). Thus, Nrf2 is a crucial component of natural neuroprotection in the brain, which unfortunately becomes dysfunctional as AD progresses due to depleted nuclear Nrf2. However, the underlying mechanism for this decrease in nuclear Nrf2 has not previously been determined.
A well-established control point for the Nrf2-mediated oxidative stress response occurs through Kelch-like ECH-associated protein 1 (Keap1) (18, 19). In the absence of oxidative stress, the Keap1 homodimer binds Nrf2 and recruits the E3 ligase complex Cul3–RBX1, which targets Nrf2 for ubiquitination and proteosomal degradation. Under oxidative stress, however, reactive oxygen species (ROS) modify cysteines in Keap1, which suppresses its ability to bind Nrf2. The liberated Nrf2 then translocates into the nucleus where it heterodimerizes with small Maf (sMaf) proteins to bind antioxidant response elements (AREs) of target genes (18). An additional pathway to activate Nrf2 is through the autophagy cargo receptor SQSTM1/p62, which competitively binds Keap1 to also liberate Nrf2 (20, 21). SQSTM1/p62 and multiple autophagy-related genes are also targets of Nrf2/ARE-mediated gene expression (6), providing a feed-forward neuroprotective link between Nrf2 signaling and SQSTM1/p62-mediated autophagy.
Slingshot homolog-1 (SSH1) is an actin filament (F-actin)–associated protein phosphatase that normally resides in an inactivated state in a reserve pool bound to 14-3-3 protein. In response to oxidative stress, however, SSH1 is liberated from 14-3-3 and then bundles F-actin (22, 23), where it subsequently activates the F-actin severing protein cofilin (24–28). Notably, SSH1 and cofilin are activated in the brains of APP/PS1 mice (29) (a model of AD), as well as in human AD brains (30). SSH1 also contains a modular and separable activity on SQSTM1/p62-mediated autophagy (31). Specifically, the binding of SQSTM1/p62 to ubiquitinated cargo depends on its phosphorylation at Ser403 (32–36). SSH1 dephosphorylates SQSTM1/p62 at pSer403, thereby preventing cargo binding and impeding SQSTM1/p62-mediated autophagy flux (31, 37, 38).
Based on the feed-forward neuroprotective link between Nrf2 signaling and SQSTM1/p62-mediated autophagy, we investigated the intersection between SSH1 and Nrf2 in cellular and animal models of oxidative stress and AD. As described below, this revealed that SSH1 acts as a counterweight to Nrf2 in response to oxidative stress and disease in the brain, which occurs independently of its phosphatase activity. Indeed, under conditions of oxidative stress, SSH1 inhibits Nrf2-mediated neuroprotection in AD. This is the first mechanistic explanation for how Nrf2-protective signaling is suppressed in AD brains and offers a possible new neuroprotective strategy for AD and related dementias.
Results
SSH1 Inhibits Nrf2/ARE Target Gene Expression Independently of Its Phosphatase Activity.
SQSTM1/p62 is a multifunctional protein with vital roles in autophagy and Nrf2 activation (20, 21, 32–36). Hereafter, we refer to SQSTM1/p62 simply as p62. Because SSH1 binds p62 and negatively regulates p62-mediated autophagy flux (31), we tested whether SSH1 impacts Nrf2 signaling. We first used an Nrf2 reporter construct in which 8x Nrf2-responsive AREs within a minimal promoter drive GFP expression, while TagBFP expression is constitutively driven by the SV40 promoter (Fig. 1A1) (39). Hence, GFP is normalized to TagBFP as a reporter for Nrf2/ARE-mediated gene expression. As expected, Nrf2 overexpression in mouse hippocampus-derived HT22 neuroblastoma cells increased GFP normalized to TagBFP (~4.4-fold). By contrast, coexpression of SSH1 (Fig. 1A2) with Nrf2 suppressed Nrf2 reporter activity by ~40% (Fig. 1 B and C). Hydrogen peroxide (H2O2) treatment, which activates both Nrf2 (40) and SSH1 (27), increased endogenous Nrf2/ARE-driven reporter activity by ~35%, while SSH1 overexpression drastically suppressed this reporter by ~70% (Fig. 1 D and E). Notably, SSH1–CS lacking phosphatase activity (Fig. 1A2) also blocked Nrf2/ARE reporter activity to a similar extent as wild-type (WT) SSH1 (Fig. 1 F and G), indicating that the phosphatase activity of SSH1 is not required for its Nrf2 inhibitory activity. Conversely, H2O2 treatment increased Nrf2/ARE reporter activity by ~45% and knockdown of SSH1 increased Nrf2/ARE reporter activity by ~60% at steady state, which increased further by >40% with SSH1 siRNA and H2O2 treatment (Fig. 1 H and I). Likewise, increasing concentrations of other Nrf2 activators sodium arsenite (NaAsO2) (41), 15d-PGJ2 (42), or bardoxolone methyl (CDDO-Me) (43, 44) also enhanced Nrf2/ARE reporter activity in a dose-dependent manner, and SSH1 knockdown further augmented their Nrf2/ARE reporter activities (SI Appendix, Fig. S1 A–F). Together, these results indicate that endogenous SSH1 suppresses Nrf2/ARE-mediated gene expression under diverse conditions.
Fig. 1.
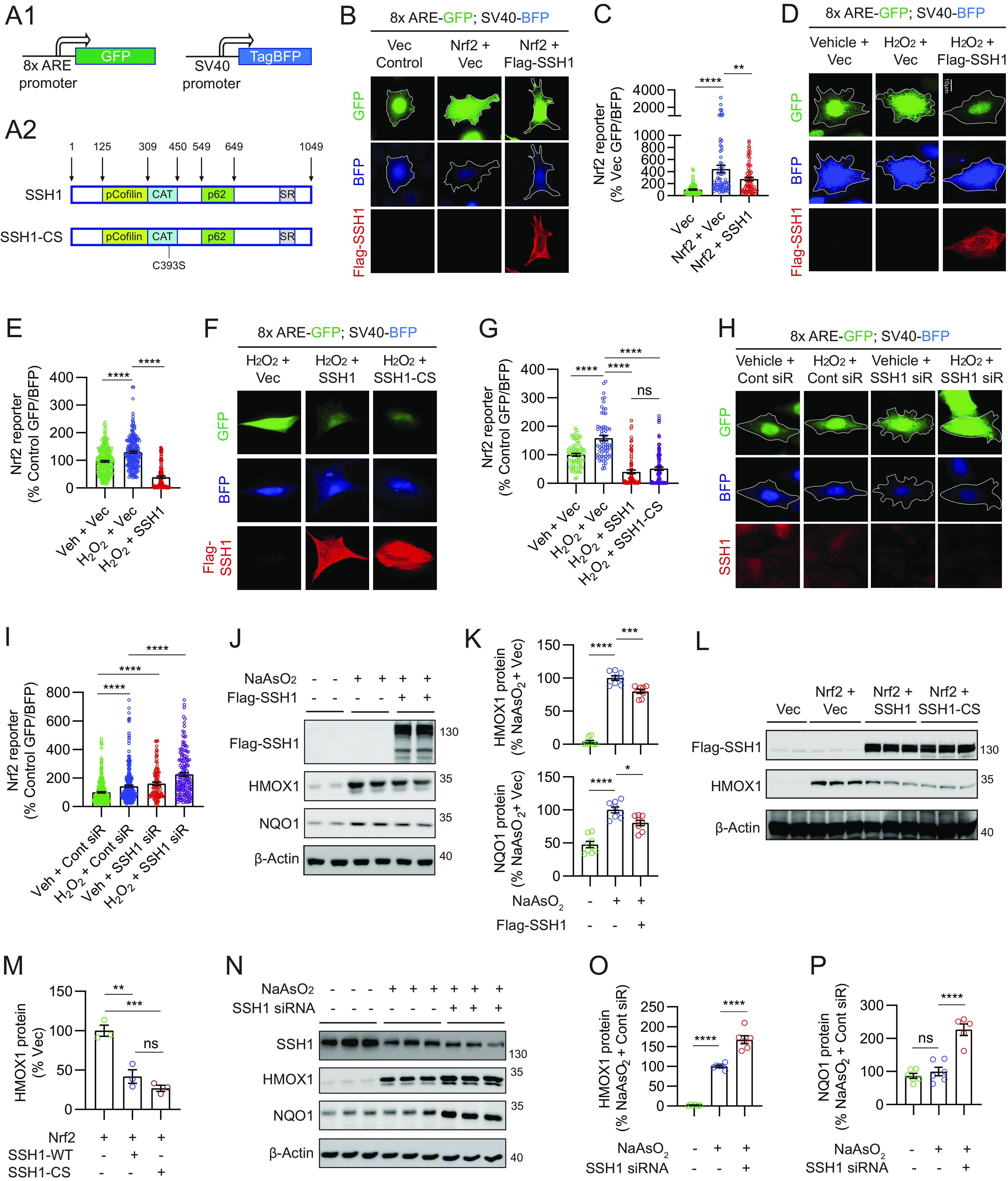
SSH1 inhibits Nrf2/ARE target gene expression independent of SSH1 phosphatase activity. (A1) Schematic of the Nrf2 reporter construct pREP-8xARE-GFP-SV40-BFP. (A2) Schematic of SSH1 and SSH1-CS proteins showing the catalytic domain (CAT) and binding sites for cofilin and p62. (B) Representative images of HT22 cells coexpressing the Nrf2 reporter (green and blue), with vector control, myc-Nrf2, and/or Flag-SSH1 (red). (C) Quantification of Nrf2 reporter [one-way ANOVA, F (2, 218) = 18.62, P < 0.0001, post hoc Dunnett, ****P < 0.0001, **P = 0.0076, n = 15 to 20 images/condition/experiment from three experiments]. (D) Representative images of HT22 cells coexpressing the Nrf2 reporter (green and blue) and vector or Flag-SSH1 (red), ± 200 μM H2O2 (2 h). (E) Quantification of Nrf2 reporter [one-way ANOVA, F (2, 519) = 124.7, P < 0.0001, post hoc Dunnett, ****P < 0.0001, n = 15 to 20 images/condition/experiment from four experiments]. (F) Representative images of HT22 cells coexpressing the Nrf2 reporter (green and blue), vector, Flag-SSH1, or Flag-SSH1CS (red), ± 250 μM H2O2 (3 h). (G) Quantification of Nrf2 reporter [one-way ANOVA, F (3, 308) = 61.03, P < 0.0001, post hoc Dunnett, ****P < 0.0001, ns = not significant, n = 8 to 12 images/condition/experiment from four experiments]. (H) Representative images of HT22 cells cotransfected with the Nrf2 reporter (green and blue) and control siRNA or SSH1 siRNA, stained for SSH1 (red), ± 250 μM H2O2 (3 h). (I) Quantification of Nrf2 reporter [one-way ANOVA, F (3, 855) = 41.31, P < 0.0001, post hoc Dunnett, ****P < 0.0001, n = 15 to 20 images/condition/experiment from four experiments]. (J) Representative immunoblots from lysates of HEK293T cells expressing vector or Flag-SSH1 ± 15 µM NaAsO2 (18 h). (K) Quantification of HMOX1 and NQO1 proteins [one-way ANOVA; HMOX1: F (2, 21) = 310.2, P < 0.0001; NQO1: F (2, 21) = 34.82, P < 0.0001; post hoc Dunnett, ****P < 0.0001, ***P < 0.001, *P < 0.05. n = 8 samples/condition]. (L) Representative immunoblots from lysates of HEK293T cells coexpressing vector or myc-Nrf2 plus vector, Flag-SSH1, or Flag-SSH1-CS. (M) Quantification of HMOX1 protein [one-way ANOVA, F (2, 6) = 32.61, P = 0.0006, post hoc Dunnett, ***P = 0.006, **P = 0.0022, ns = not significant, n = 3 samples/condition]. (N) Representative immunoblots from lysates of HEK293T cells transfected with control or SSH1 siRNA ± 15 µM NaAsO2 (18 h). (O) Quantification of HMOX1 protein [one-way ANOVA, F (2, 15) = 185.4, P < 0.0001, post hoc Dunnett, ****P < 0.0001, n = 6 samples/condition]. (P) Quantification of NQO1 protein [one-way ANOVA, F (2, 14) = 34.49, P < 0.0001, post hoc Dunnett, ****P < 0.0001, ns = not significant, n = 6 samples/condition].
We next examined Nrf2/ARE-responsive antioxidant target genes, HMOX1 and NQO1 (18). Treatment of HEK293T cells with NaAsO2, which induces oxidative stress through H2O2 production (41), significantly increased HMOX1 and NQO1 levels (Fig. 1 J and K), as previously reported (45). However, SSH1 overexpression suppressed the NaAsO2-induced increase in both proteins (Fig. 1 J and K). Likewise, SSH1 overexpression suppressed HMOX1 and NQO1 protein and mRNA expression in Nrf2-transfected cells (SI Appendix, Fig. S1 G–L). Notably, SSH1–CS overexpression suppressed Nrf2-induced HMOX1 expression to levels similar to WT SSH1 (Fig. 1 L and M). By contrast, siRNA-mediated knockdown of SSH1 increased HMOX1 and NQO1 beyond that elevated by NaAsO2 treatment by ~65% and ~130%, respectively (Fig. 1 N–P). These results collectively indicate that SSH1 suppresses Nrf2/ARE-mediated gene expression independently of SSH1 phosphatase activity.
SSH1 Augments Keap1–Nrf2 Interaction under Oxidative Stress.
The binding of p62 to Keap1 competitively frees Nrf2 from Keap1 inhibitory control, resulting in Nrf2 activation (18, 19). This competitive binding of p62 to Keap1 is enhanced by p62 phosphorylation at Ser349 (pS349-p62) (45). Here, we transfected HT22 cells with vector control or SSH1 (also GFP at 1:5 ratio to visualize transfected cells) for 48 h and treated cells with NaAsO2, which increases pS349-p62 (45). We then performed Duolink® proximity ligation assay (PLA) to detect endogenous p62–Keap1 protein complexes, a method that preserves protein–protein interactions in situ and does not require detergent-based cell lysis. Indeed, NaAsO2 treatment robustly increased endogenous p62–Keap1 complexes by ~75%, which was prevented by SSH1 overexpression (Fig. 2 A and B and SI Appendix, Fig. S2A, negative controls). SSH1-mediated reduction in p62–Keap1 interaction was accompanied by the expected increase in SSH1–p62 binding (Fig. 2 C and D and SI Appendix, Fig. S2B, negative controls), indicating that SSH1 sequesters p62 away from Keap1. We also observed that as p62 competitively liberated Nrf2 from Keap1 inhibitory control, consistent with previous reports (18), NaAsO2 decreased endogenous Keap1–Nrf2 complexes, which was reversed and strongly enhanced by SSH1 overexpression (Fig. 2 E and F and SI Appendix, Fig. S2C, negative controls). This demonstrates that SSH1 augments inhibitory Keap1–Nrf2 interaction under oxidative stress. Similar SSH1-mediated reduction of p62–Keap1 interactions and enhancement of Keap1–Nrf2 interactions were seen with Nrf2 overexpression (SI Appendix, Fig. S2 D–F, G, and H, negative controls). Conversely, knockdown of SSH1 significantly increased p62–Keap1 complexes and decreased Keap1–Nrf2 complexes (Fig. 2 G–I), indicating that endogenous SSH1 enhances the inhibitory Keap1–Nrf2 interaction.
Fig. 2.

SSH1 augments Keap1-mediated Nrf2 inhibitory control under oxidative stress and sequesters Nrf2 on actin filaments. (A) Representative images of HT22 cells transfected with GFP (green) and vector or Flag-SSH1, treated ± 8 µM NaAsO2 (14 h), and subjected to PLA for endogenous Keap1–p62 (red). (B) Quantification of Keap1–p62 PLA puncta area/cell [one-way ANOVA, F (2, 296) = 21.7, P < 0.0001, post hoc Dunnett, ****P < 0.0001, ns = not significant, n = 10 to 15 images/condition/experiment from four experiments]. (C) Representative images of HT22 cells transfected with GFP (green) and vector or Flag-SSH1, treated ± 8 µM NaAsO2 (14 h), and subjected to PLA for SSH1–p62 (red). (D) Quantification of SSH1–p62 PLA puncta area/cell [one-way ANOVA, F (2, 341) = 76.79, P < 0.0001, post hoc Dunnett, ****P < 0.0001, ns=not significant, n = 8 to 10 images/condition/experiment from four experiments]. (E) Representative images of HT22 cells transfected with GFP (green) and vector or Flag-SSH1, treated ± 8 µM NaAsO2 (14 h), and subjected to PLA for endogenous Keap1–Nrf2 (red). (F) Quantification of Keap1–Nrf2 PLA puncta area/cell [one-way ANOVA, F (2, 307) = 59.38, P < 0.0001, post hoc Dunnett, ****P < 0.0001, n = 10 to 15 images/condition/experiment from four experiments]. (G and H) Representative images of HT22 cells transfected with GFP (green) and control or SSH1 siRNA and subjected to PLA for endogenous p62–Keap1 (G, red) or Keap1–Nrf2 (H, red). (I) Quantification of p62–Keap1 and Keap1–Nrf2 PLA puncta area/cell (two-tailed t test; p62–Keap1 PLA: t = 4.0812, df = 204; Keap1–Nrf2 PLA: t = 8.855, df = 208; ****P < 0.0001, n = 8 to 10 images/condition/experiment from four experiments). (J) Representative images of HT22 cells coexpressing GFP-Nrf2 (green) and vector or Flag-SSH1 (red) and treated with ± 300 µM H2O2 (3 h). (K) Quantification of nuclear/cytoplasmic Nrf2 [one-way ANOVA, F (3, 366) = 84.88, P < 0.0001, post hoc Dunnett, ****P < 0.0001, n = 10 to 15 images/condition/experiment from four experiments]. (L) Representative immunoblots from HEK293T coexpressing myc-Nrf2 and vector or Flag-SSH1, subjected to cell fractionation for cytosol, nucleus, and cytoskeleton. (M) Quantification of cytoskeletal/cytosolic and nuclear/cytosolic Nrf2, (two-tailed t test; cytoskel/cyto: t = 3.474, df = 10; nucl/cyto: t = 3.19, df = 10; **P < 0.01, n = 6 samples/condition). (N) Representative images of HT22 cells transfected with GFP or GFP-Nrf2 (green) and vector or Flag-SSH1, subjected to PLA for SSH1–Nrf2 (red) and F-actin phalloidin staining (blue). (O) Quantification of SSH1–Nrf2 PLA puncta intensity/cell [one-way ANOVA, F (3, 255) = 306.3, P < 0.0001, post hoc Dunnett, ****P < 0.0001, n = 10 to 15 images/condition/experiment from four experiments]. (P) Quantification of SSH1–Nrf2 PLA colocalized with F-actin [G: one-way ANOVA, F (3, 372) = 11.70, P < 0.0001, post hoc Dunnett, ***P < 0.001, **P < 0.01, ns = not significant; n = 10 to 15 images/condition/experiment from four experiments].
We next investigated whether endogenous SSH1 would alter Nrf2 ubiquitination in the presence or absence of oxidative stress (NaAsO2). SSH1 knockdown showed a trend for reduced Nrf2 ubiquitination both in the presence and absence of NaAsO2 without reaching statistical significance by ANOVA and post hoc Dunnett test (SI Appendix, Fig. S2 I and J). However, post hoc test for linear trend revealed that the combination of NaAsO2 and SSH1 siRNA caused significantly lower Nrf2 ubiquitination, with the highest Nrf2 ubiquitination in the no NaAsO2 and no SSH1 siRNA condition, intermediate Nrf2 ubiquitination with either NaAsO2 or SSH1 siRNA, and the lowest Nrf2 ubiquitination with both NaAsO2 and SSH1 siRNA (SI Appendix, Fig. S2 I and J). This indicates that SSH1 knockdown, which reduces Keap1–Nrf2 interaction, also modestly reduces Nrf2 ubiquitination. However, the ability of SSH1 to significantly suppress Nrf2 signaling was only moderately affected by loss of endogenous SQSTM1/p62 (SI Appendix, Text and Fig. S2 K–O), suggesting that SSH1-mediated sequestration of p62 to indirectly enhance Keap1–Nrf2 interaction is unlikely to fully account for the activity of SSH1 to suppress Nrf2 signaling.
SSH1 Reduces Nuclear Nrf2 by Sequestering Nrf2 on Actin Filaments.
We next tested whether endogenous SSH1 could alter endogenous nuclear Nrf2 levels under various Nrf2 activating conditions (NaAsO2, 15d-PGJ2, or CDDO-Me). Endogenous Nrf2 was detected mainly in the nucleus, and all Nrf2-activating agents significantly increased Nrf2 intensity in the nucleus (SI Appendix, Fig. S3 A–F). Knockdown of SSH1 also significantly augmented nuclear Nrf2 levels in the presence of Nrf2 activating agents (SI Appendix, Fig. S3 A–F), indicating that endogenous SSH1 suppresses nuclear Nrf2 levels under diverse conditions. Furthermore, cells transfected with GFP-Nrf2 alone showed robust nuclear localization of GFP-Nrf2, which increased by ~25% with H2O2 treatment (Fig. 2 J and K). However, GFP-Nrf2 transfection with SSH1 significantly decreased nuclear to cytoplasmic Nrf2 ratio to the same extent regardless of H2O2 treatment (Fig. 2 J and K). Notably, cytoplasmic GFP-Nrf2 was distributed in a discrete and punctate pattern that partially colocalized with SSH1 (SI Appendix, Fig. S3 G and H).
SSH1 contains multiple F-actin binding sites that stabilize and bundle actin filaments (25, 26). Like SSH1, the C-terminal region of Keap1 also binds F-actin, which is required to retain Nrf2 in the cytoplasm and suppress Nrf2-mediated gene expression (46). This, together with observations that SSH1 increases Keap1–Nrf2 interaction, reduces Nrf2 nuclear localization and partially colocalizes with Nrf2 prompted further investigation of the mechanism. Given the physical localization of SSH1 primarily to the F-actin cytoskeleton (25, 26), we hypothesized that SSH1 might alter Nrf2 localization by sequestering Nrf2 on actin filaments. We initially tested this hypothesis by performing subcellular fractionation using the ThermoFisher subcellular protein fraction kit to isolate cytoskeletal, cytosolic, and nuclear fractions in transfected HEK293T cells. The cytoskeletal fraction was enriched with the cytoskeletal marker vimentin, and the nuclear fraction was enriched with the nuclear marker Lamin B1 (Fig. 2L). As expected, Flag-SSH1 was highly enriched in the cytoskeletal fraction, although some Flag-SSH1 was also seen in cytosolic and nuclear fractions (Fig. 2L). Coexpression of myc-Nrf2 with Flag-SSH1 strongly increased myc-Nrf2 in the cytoskeletal fraction with Flag-SSH1 (Fig. 2L). Accordingly, Flag-SSH1 expression increased the ratio of cytoskeletal to cytosolic myc-Nrf2, as well as the ratio of nuclear to cytosolic myc-Nrf2 (Fig. 2M). Likewise, we observed that the catalytic SSH1–CS mutant sequestered myc-Nrf2 in the cytoskeletal fraction as effectively as WT SSH1 (SI Appendix, Fig. S3 I and J).
To validate this finding in a different way and determine whether SSH1 physically associates with Nrf2 in close proximity to F-actin, we performed in situ PLA to detect SSH1–Nrf2 complexes followed by counterstaining with phalloidin to label F-actin. In cells transfected with GFP control alone, we detected a low level of endogenous SSH1–Nrf2 PLA puncta mainly in the nucleus, with a small minority of puncta associated with F-actin in the cytoplasm (Fig. 2 N, Upper and Fig. 2 O and P). Negative PLA controls using only one probe or one primary antibody detected no signal (SI Appendix, Fig. S3K). Upon cotransfection of GFP and SSH1, however, we observed a marked and significant increase in SSH1–Nrf2 PLA puncta concentrated in proximity to phalloidin-labeled actin filaments (Fig. 2N, 2nd row, and Fig. 2 O and P). GFP–Nrf2 transfection alone also increased SSH1–Nrf2 PLA puncta, which localized mainly to the nucleus (Fig. 2N, 3rd row, and Fig. 2 O and P). Cotransfection of GFP–Nrf2 with SSH1 drastically increased SSH1–Nrf2 PLA such that individual dots blended throughout the cell (Fig. 2N, 4th row, and Fig. 2 O and P). Under this condition, the highest intensity of PLA signal was seen at the cell cortex and membrane protrusions. The catalytically inactive SSH1–CS mutation did not disrupt SSH1–Nrf2 interaction (SI Appendix, Fig. S3 L and M), in line with preceding observations. Consistent with the observation that SSH1-mediated Nrf2 inhibition occurs independent of its catalytic activity, this activity of SSH1 was also distinct and separable from its ability to inhibit p62-mediated autophagy and augment cofilin activation (SI Appendix, Text and Fig. S4 A–O).
Having established that SSH1 augments Keap1–Nrf2 interaction and sequesters Nrf2 on F-actin, we next wondered whether Keap1 might also be part of the SSH1 complex associated with F-actin. Indeed, we observed a detectable level of endogenous SSH1–Keap1 PLA puncta, which increased significantly with SSH1 overexpression (SI Appendix, Fig. S4 P and Q). SSH1 overexpression also increased the localization of the SSH1–Keap1 complex on F-actin by ~2.5-fold (SI Appendix, Fig. S4 P and R), indicating that this complex occurs primarily on actin filaments. Surprisingly, transfection of SSH1 with Nrf2 more than doubled SSH1–Keap1 PLA puncta compared to SSH1 transfection alone (SI Appendix, Fig. S4 P and Q), without altering F-actin localization (SI Appendix, Fig. S4 P and R). This suggests that Nrf2 physically facilitates the association of Keap1 with SSH1. Accordingly, coincubation of recombinant GST-SSH1 with recombinant His-Nrf2 demonstrated direct interaction by in vitro SSH1 pulldown assay (SI Appendix, Fig. S4 S and T), whereas recombinant GST-Keap1 did not show specific and direct interaction with recombinant GST-SSH1 in vitro (SI Appendix, Fig. S4 U and V).
Excessive Nrf2 Interactions with SSH1 and Keap1 in AD and FTLD-tau Brains.
Nuclear but not cytoplasmic Nrf2 is depleted in AD (9), despite elevated levels of oxidative stress (47, 48). Furthermore, tauP301S expression (the mutant form of tau associated with some cases of the AD-related condition of frontotemporal lobar dementia (FTLD) also preferentially depletes nuclear versus cytoplasmic Nrf2 in the brain and transfected cells (49). Oxidative stress decreases Keap1 activity (18) but increases SSH1 activity (27). Accordingly, we observed that oxidative stress significantly reduced endogenous Keap1–Nrf2 interaction, which reversed and drastically increased with SSH1 transfection (Fig. 2 E and F). Conversely, oxidative stress increased endogenous SSH1–Nrf2 interaction by ~sixfold and further increased with SSH1 transfection (Fig. 3 A and B). These results indicate that SSH1 controls and titrates Nrf2 activation under oxidative stress, a condition in which Keap1 activity is low. Based on this premise, we performed PLA to detect SSH1–Nrf2 and Keap1–Nrf2 complexes in human frontal cortex tissues of nondementia controls, AD, and FTLD-tau subjects (case information, SI Appendix, Fig. S5A). Compared to nondementia control brains, SSH1–Nrf2 complexes were increased by ~23-fold in AD brains and ~fivefold in FTLD-tau brains (Fig. 3 C, Upper two panels, and Fig. 3 D and E and SI Appendix, Fig. S5B, negative controls). Unlike HT22 cells, SSH1–Nrf2 PLA puncta in AD and FTLD-tau brains were absent from DAPI-positive nuclei and much larger than those in control brains (Fig. 3C). Staining for SSH1 and actin in control brains showed a thin filamentous pattern of both proteins throughout cell bodies and neuropil (SI Appendix, Fig. S5C). By contrast, in AD and FTLD-tau brains, SSH1 and actin filaments were highly disorganized, often with thicker and irregular actin bundles (SI Appendix, Fig. S5 C and D, negative control), indicative of perturbations in SSH1 and F-actin. Furthermore, Keap1–Nrf2 complexes were also elevated by ~four-fold in AD brains and ~17-fold in FTLD-tau brains compared to controls and notably absent from the nucleus (Fig. 3 C, Lower two panels, and Fig. 3 F and G and SI Appendix, Fig. S5E, negative controls)
Fig. 3.
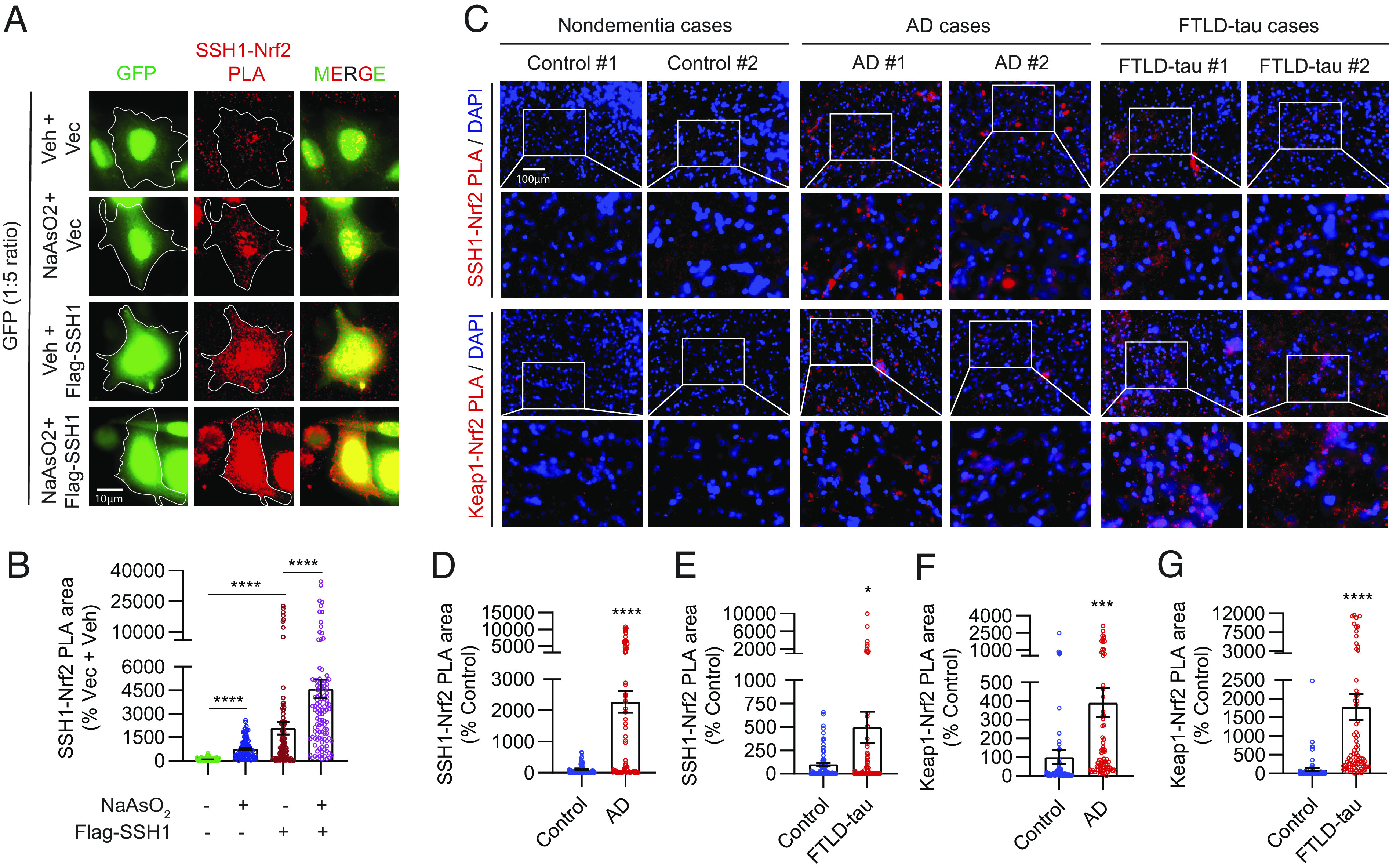
AD and FTLD-tau brains exhibit excessive levels of inhibitory SSH1–Nrf2 and Keap1–Nrf2 interactions. (A) Representative images of HT22 cells transfected with GFP (green) and vector or Flag-SSH1, treated ± 8 µM NaAsO2 (14 h) and subjected to PLA for SSH1–Nrf2 (red). (B) Quantification of SSH1–Nrf2 PLA puncta area/cell [Brown-Forsythe and Welch ANOVA, F (3, 202.7) = 30.48, F (3, 183.2) = 62.13, P < 0.0001, post hoc Dunnett, ****P < 0.0001, n = 10 to 15 images/condition/experiment from four experiments]. (C) Representative images of human frontal gyrus sections from nondementia, AD, and FTLD-tau cases showing DAPI (blue) and PLA for SSH1–Nrf2 (red) and Keap1–Nrf2 (red). (D and E) Quantification of SSH1–Nrf2 PLA area in (D) control vs. AD (two-tailed t test, t = 6.329, df = 156, ****P < 0.0001, n = 8 to 10 images/case from 7 to 8 case/condition) and (E) control vs. FTLD-tau (two-tailed t test, t = 2.353, df = 157, *P = 0.0199, n = 8 to 10 images/case from eight cases/condition). (F and G) Quantification of Keap1–Nrf2 PLA area in (F) control vs. AD (two-tailed t test, t = 3.371, df = 154, ***P = 0.0009; n = 8 to 10 images/case from 7 to 8 cases/condition) and (G) control vs. FTLD-tau (two-tailed t test, t = 4.752, df = 152; ****P < 0.0001; n = 8 to 10 images/case from eight cases/condition).
Ssh1 Elimination Increases Nuclear Nrf2, Prevents Oxidative Injury, and Mitigates AD Pathology.
We next utilized a genetic model of Ssh1 deficiency to determine whether loss of Ssh1 would protect from AD-like neurodegeneration. Ssh1−/− mice are healthy, breed well, and exhibit no overt phenotype (50), and the SSH1 transcript is essentially undetectable in Ssh1−/− brains (SI Appendix, Fig. S6A). We first assessed if genetic elimination of Ssh1 would alter nuclear versus cytoplasmic localization of Nrf2 in tauP301S (PS19) mice. We stained 7-mo-old WT, tauP301S, and tauP301S;Ssh1−/− brain sections for Nrf2 followed by DAPI staining and quantified the intensity of Nrf2 in the nucleus versus cytoplasmic cell body. While tauP301S mice showed significantly reduced Nrf2 nuclear/cytoplasmic ratio by ~20% in the cortex compared to WT mice, tauP301S;Ssh1−/− mice showed significantly higher Nrf2 nuclear/cytoplasmic ratio even beyond WT levels (Fig. 4 A and B and SI Appendix, Fig. S6B, negative control). Given the role of Nrf2 in antioxidant signaling, we next tested if Ssh1 elimination could protect against oxidative injury in AD models: tauP301S and APP/PS1 mice (51). Oxidation of nuclear and mitochondrial DNA is most frequently detected as 8-hydroxy-2′-deoxyguanosine (8-OHdG) and is used as a marker of oxidative injury (52). Seven-mo-old tauP301S mice exhibited significantly higher DNA oxidation than littermate WT mice, which was reversed below WT levels in 7-mo-old tauP301S;Ssh1−/− mice (Fig. 4 C–E and SI Appendix, Fig. S6C, negative control). Likewise, 8-mo-old APP/PS1 mice showed significantly higher 8-OHdG staining compared to littermate WT mice, whereas 8-mo-old APP/PS1;Ssh1−/− mice exhibited 8-OHdG intensity comparable to WT mice (Fig. 4 F and G and SI Appendix, Fig. S6D, negative control). These results indicate that loss of Ssh1 prevents oxidative injury in AD models. To directly interrogate neurodegeneration, we employed silver staining of degenerating axons. While tauP301S mice showed markedly elevated axon degeneration in both hippocampus and cortex, tauP301S;Ssh1−/− mice were protected from this feature and indistinguishable from wild-type littermates (Fig. 4 H–J). Thus, elimination of Ssh1 effectively blocks neurodegeneration in the brain of tauP301S mice.
Fig. 4.
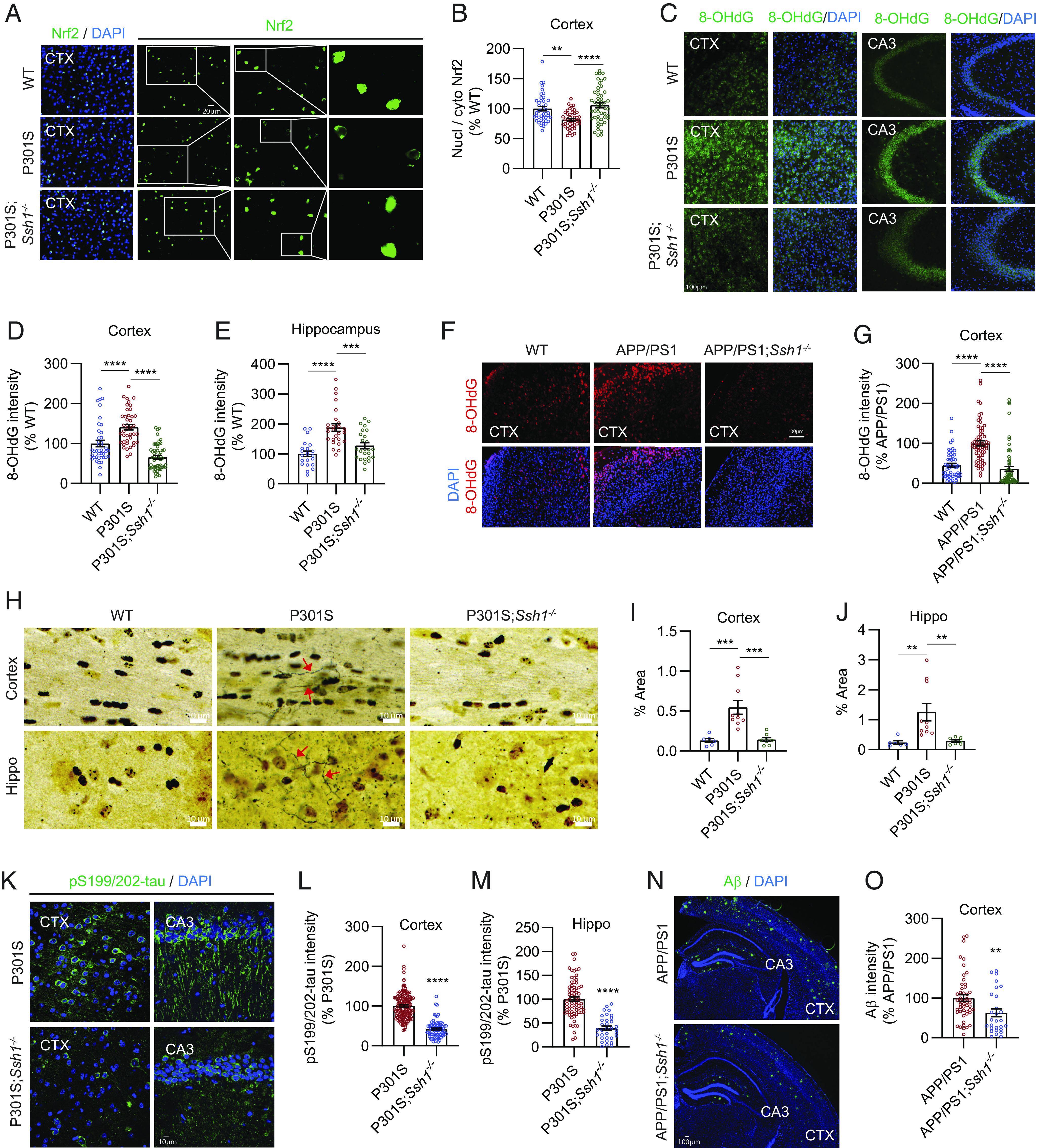
Ssh1 elimination increases nuclear Nrf2, reduces oxidative injury, and alleviates AD pathology. (A) Representative images of the cortex from 7-mo-old WT, P301S, and P301S;Ssh1−/− mice stained for Nrf2 (green) and DAPI (blue). White boxes serially magnified to the right. (B) Quantification of nuclear/cytoplasmic Nrf2 intensity [one-way ANOVA, F (2, 134) = 13.06, P < 0.0001, post hoc Dunnett, ****P < 0.0001, **P = 0.0013, n = 10 to 12 images/mouse from four mice/genotype]. (C) Representative images of the cortex and hippocampus (CA3) stained for 8-OHdG (green) and DAPI (blue) from 7-mo-old WT, P301S, and P301S;Ssh1−/− mice. (D and E) Quantification of 8-OHdG intensity in the (D) cortex [one-way ANOVA, F (2, 134) = 37.79, P < 0.0001, post hoc Dunnett, ****P < 0.0001, n = 10 to 13 images/mouse from four mice/genotype) and (E) hippocampus [one-way ANOVA, F (2, 70) = 17.85, P < 0.0001, post hoc Dunnett, ****P < 0.0001, ***P = 0.0003, n = 5 to 7 images/mouse from four mice/genotype]. (F) Representative images of the cortex from 8-mo-old WT, APP/PS1, and APP/PS1;Ssh1−/− mice stained for 8-OHdG (red) and DAPI (blue). (G) Quantification of 8-OHdG intensity [one-way ANOVA, F (2, 181) = 36.83, P < 0.0001, post hoc Dunnett, ****P < 0.0001, n = 10 to 12 images/mouse from four mice/genotype]. (H) Representative images of silver staining of the cortex and hippocampus from 7-mo-old WT, P301S, and P301S;Ssh1−/− mice. Red arrows indicate silver-positive degenerating axons. (I and J) Quantification of silver-positive axons in the (I) cortex [one-way ANOVA, F (2, 21) = 14.5, P = 0.0001, post hoc Dunnett, ***P < 0.0005, n = 6 to 10 mice/genotype] and (J) hippocampus [one-way ANOVA, F (2, 21) = 7.545, P = 0.0034, post hoc Dunnett, **P < 0.0081, n = 6 to 10 mice/genotype]. (K) Representative images of the cortex and hippocampus (CA3) stained for pS199/202-tau (green) and DAPI (blue) from 7-mo-old P301S and P301S;Ssh1−/− mice. (L and M) Quantification of pS199/202-tau intensity in the (L) cortex (two-tailed t test, t = 13.85, df = 221, ****P < 0.0001, n = 20 to 30 images/mouse from four mice/genotype) and (M) hippocampus (two-tailed t–test, t = 8.147, df = 104, ****P < 0.0001, n = 13 to 18 images/mouse from four mice/genotype). (N) Representative images of the cortex and hippocampus stained for Aβ (green) and DAPI (blue) from 8-mo-old WT, APP/PS1, and APP/PS1;Ssh1−/− mice. (O) Quantification of Aβ intensity in the cortex (two-tailed t test, t = 2.784, df = 76, **P = 0.0068, n = 6 to 8 images/mouse from 4 to 6 mice/genotype).
Loss of Nrf2 (Nfe2l2) exacerbates tau and Aβ pathology in multiple animal models, whereas Nrf2 activation suppresses both pathologies (10–15). Hence, we next investigated whether Ssh1 elimination could protect against pathological tau and Aβ accumulation in tauP301S and APP/PS1 mice, respectively. As expected, 7-mo-old tauP301S mice exhibited intense pS199/202-tau staining in the cortex and hippocampus (Fig. 4 K–M). By contrast, tauP301S;Ssh1−/− showed ~60% reduction in pS199/202-tau pathology compared to tauP301S mice in both brain regions (Fig. 4 K–M), in line with that seen for enhanced Nrf2 activation (12, 14). Eight-mo-old APP/PS1;Ssh1−/− mice also showed significantly decreased Aβ deposition by ~40% compared to age-matched APP/PS1 littermates (Fig. 4 N and O and SI Appendix, Fig. S6E, negative control), consistent with independent results of increased Nrf2 signaling (10, 15, 53).
Loss of Ssh1 Corrects Gliosis and Synaptic Plasticity Deficits in tauP301S Mice.
Next, we assessed gliosis in 7-mo-old WT, tauP301S, and tauP301S;Ssh1−/− mice. Iba1, a marker of microgliosis, was elevated in tauP301S mice compared to WT mice, while Ssh1 elimination in tauP301S mice significantly reversed this measure to WT levels (Fig. 5 A and B). Loss of Ssh1 partially but significantly prevented astrogliosis in tauP301S mice, as assessed by GFAP (Fig. 5 A and C).
Fig. 5.
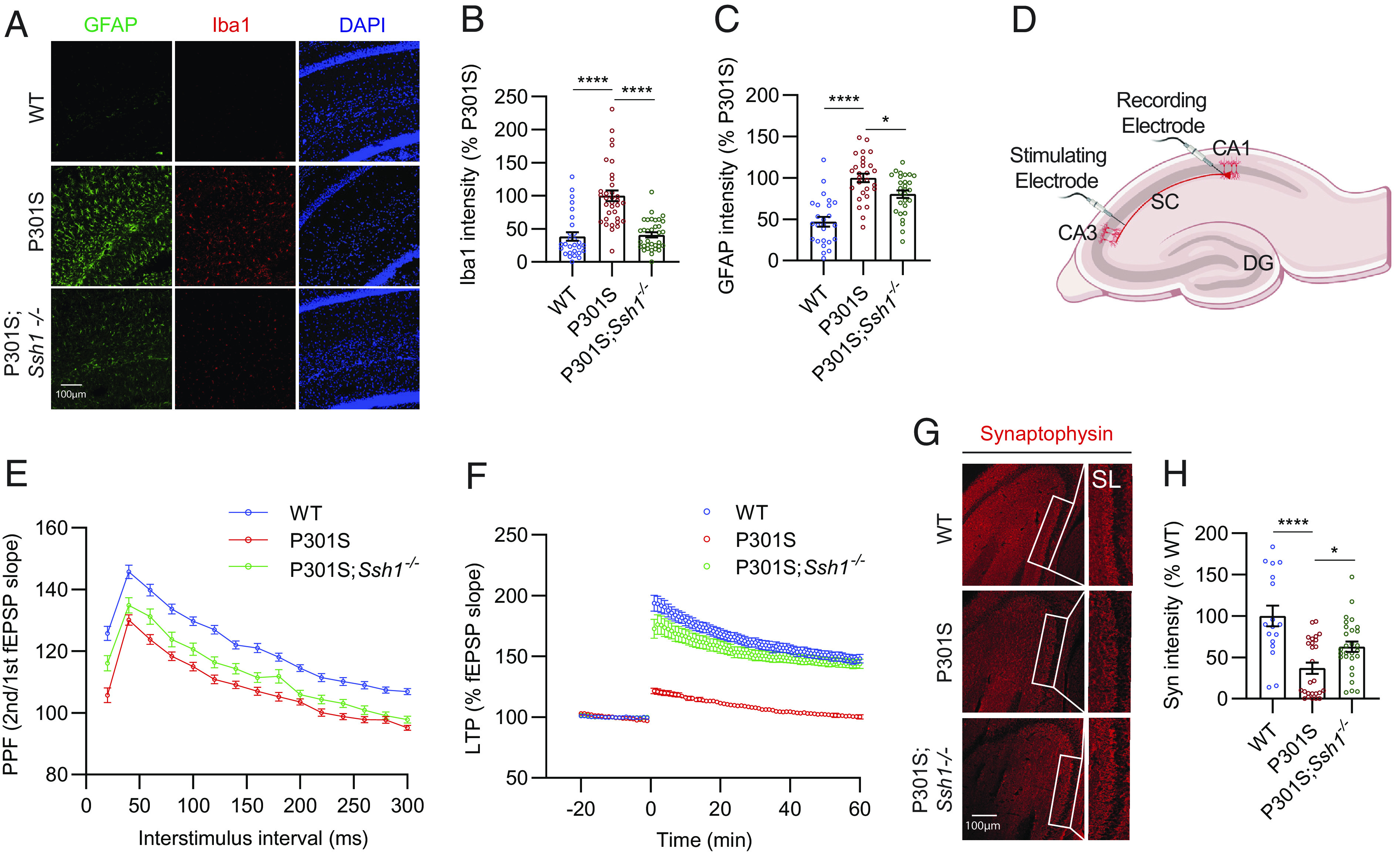
Loss of Ssh1 reverses gliosis and synaptic plasticity deficits in tauP301S mice. (A) Representative images of GFAP (green), Iba1 (red), and DAPI (blue) staining from 7-mo-old WT, P301S, and P301S;Ssh1−/− mice. (B) Quantification of Iba1 intensity [one-way ANOVA, F (2, 93) = 30.78, P < 0.0001, post hoc Dunnett, ****P < 0.0001, n = 7 to 9 images/mouse from four mice/genotype). (C) Quantification of GFAP intensity [one-way ANOVA, F (2, 78) = 26.39, P < 0.0001, post hoc Dunnett, ****P < 0.0001, *P = 0.014, n = 5 to 7 images/mouse from four mice/genotype]. (D) Schematic of electrophysiological recordings from acute brain slices. (E) PPF showing fEPSP slope as a function of increasing interstimulus intervals from 30 to 300 ms from WT, P301S, and P301S;Ssh1−/− brain slices [two-way ANOVA, F (2, 2055) = 324.4; post hoc Tukey, main genotype effects, WT vs. P301S: P < 0.0001; WT vs P301S;Ssh1−/−: P < 0.0001; P301S vs. P301S;Ssh1−/−: P < 0.0001; n = 33 to 62 slices/genotype from 4 to 7 mice/genotype). (F) LTP induced by theta burst stimulation, showing fEPSP slope from WT, P301S, and P301S;Ssh1−/− slices [two-way ANOVA, F (79, 10320) = 5736; post hoc Dunnett, main genotype effects, WT vs. P301S: P < 0.0001; P301S vs. P301S;Ssh1−/−: P < 0.0001; n = 29 to 60 slices from 4 to 7 mice/genotype]. (G) Representative images of the hippocampus stained for synaptophysin (red) from 7-mo-old WT, P301S, and P301S;Ssh1−/− mice. (H) Quantification of synaptophysin intensity [one-way ANOVA, F (2, 68) = 13.66, P < 0.0001, post hoc Dunnett, ****P < 0.0001, *P = 0.0316, n = 5 to 7 images/mouse from four mice/genotype].
We subsequently evaluated mice for any changes in short- and long-term synaptic plasticity using acute brain slices from 4-mo-old WT, tauP301S, and tauP301S;Ssh1−/− mice as we previously showed (29, 54–56). We placed the stimulating electrode at the Schaffer collaterals (SC) of the hippocampus and the recording electrode at the stratum radiatum of CA1 (Fig. 5D) and measured input/output (I/O) curves of the field excitatory postsynaptic potential (fEPSP) by increasing the stimulus voltage stepwise. There were no significant changes in I/O profiles between genotypes (SI Appendix, Fig. S7A), indicating normal basal synaptic efficacy. To measure presynaptic facilitation, we performed paired-pulse facilitation (PPF) experiments by pairing two pulses with increasing interstimulus intervals from 20 ms to 300 ms. The ratio of the second versus the first fEPSP was reduced in tauP301S slices compared to WT slices at all interstimulus intervals, indicating impaired presynaptic facilitation (Fig. 5E). In tauP301S;Ssh1−/− slices, PPF partially but significantly recovered, such that tauP301S;Ssh1−/− slices differed significantly from both WT and tauP301S slices (Fig. 5E). Lastly, to measure long-term potentiation (LTP), we recorded basal fEPSP for 20 min, applied theta-burst stimulation to induce LTP, and recorded slices for 60 min. Consistent with previous reports (54, 57, 58), tauP301S slices exhibited severely impaired LTP compared to WT slices (Fig. 5F). However, tauP301S;Ssh1−/− slices significantly recovered LTP to near WT levels, which was maintained over the entire 60-min recording period (Fig. 5F). Compared to WT mice, 7-mo-old tauP301S mice showed severely reduced synaptophysin intensity in the stratum lucidum (SL), while tauP301S;Ssh1−/− mice of the same age showed significant recovery (Fig. 5 G and H).
Ssh1 Elimination Normalizes Transcriptomic Perturbations in tauP301S Mice and Activates the Nrf2 Pathway.
Nrf2 (Nfe2l2) deficiency replicates transcriptional perturbations seen in AD brains (13). We thus compared the transcriptomic profiles of 7-mo-old tauP301S mice cortex to littermate WT controls by RNA-sequencing. We also determined whether SSh1 elimination altered transcriptomic perturbations in tauP301S mice. From 26,020 individual transcripts read by RNA-seq, 545 were up-regulated and 476 were down-regulated in tauP301S mice compared to WT mice (Fig. 6A and Dataset S1). In tauP301S;Ssh1−/− mice, 740 transcripts were up-regulated and 523 transcripts were down-regulated compared to age-matched tauP301S mice (Fig. 6B and Dataset S2). Of these, 165 transcripts were commonly altered in tauP301S mice relative to WT mice, and in tauP301S;Ssh1−/− mice relative to tauP301S mice (Dataset S3). Remarkably, 160 of the 165 common transcripts were altered in the opposite direction (Fig. 6C and SI Appendix, Fig. S8A, heatmap) with correlation r = −0.932 (Fig. 6D), indicating that Ssh1 elimination normalizes transcriptional perturbations in tauP301S mice.
Fig. 6.
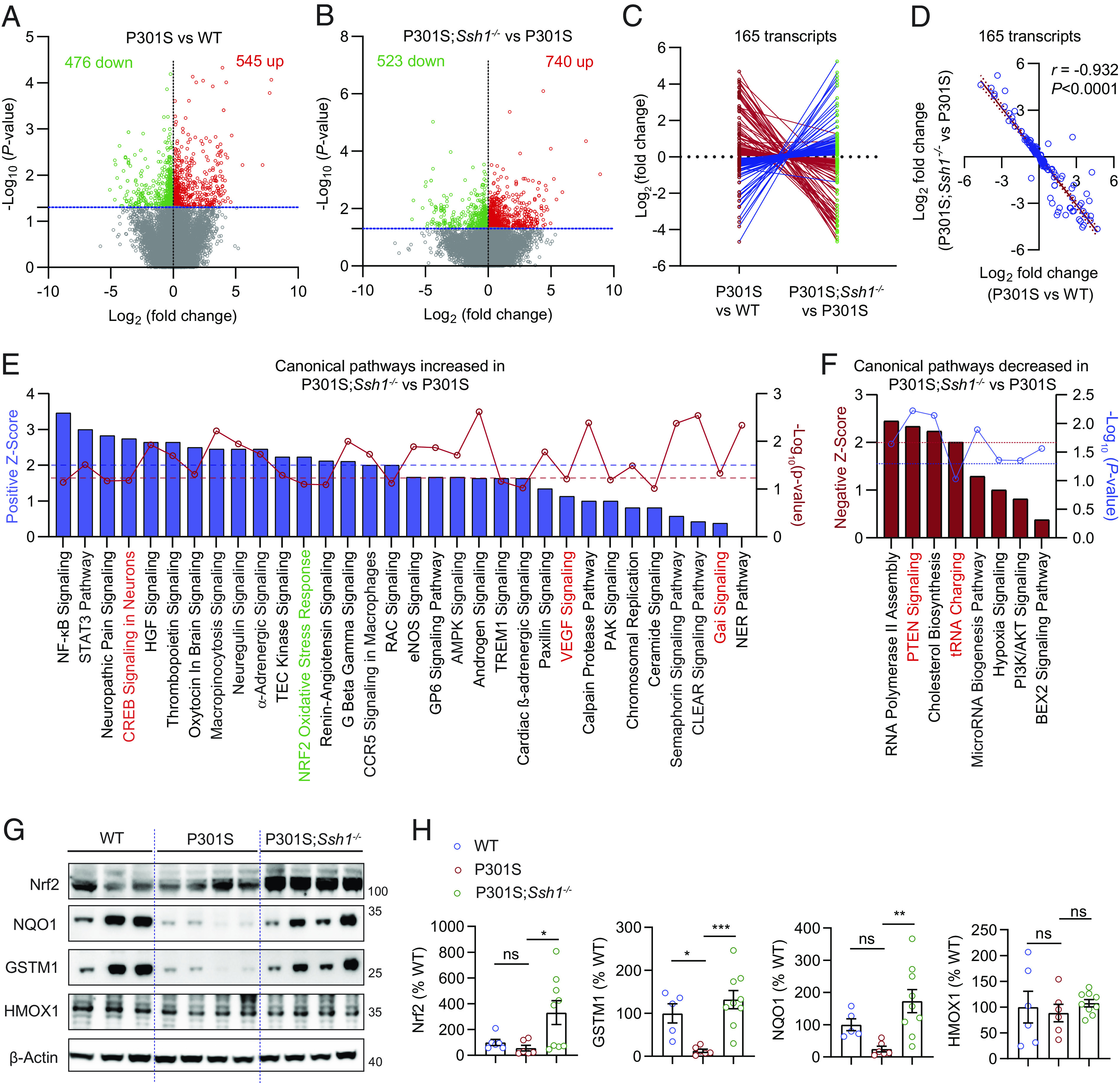
Ssh1 elimination normalizes transcriptomic perturbations in tauP301S mice and activates the Nrf2 pathway. (A and B) Volcano plots of differentially expressed genes (DEGs) based on RNA-seq of transcripts in the cortex of 7-mo-old (A) P301S mice compared to WT mice and (B) P301S;Ssh1−/− mice compared to P301S mice. Log2 (fold change) is plotted against -Log10(P-value) with the dotted blue line representing nominal significance (≥1.3). Each circle represents one transcript, with red circles showing significant upregulation and green circles showing significant downregulation (n = 3 to 4 mice/genotype). (C and D) Of all common transcripts significantly altered in P301S mice relative to WT mice and in P301S;Ssh1−/− mice relative to P301S mice (165 transcripts), (C) butterfly line graph shows 160 transcripts normalize expression in the reverse direction in P301S;Ssh1−/− compared to P301S mice. (D) Spearman correlation analysis of Log2 fold changes of transcripts in P301S relative to WT mice and Log2 fold changes in P301S;Ssh1−/− relative to P301S mice shows a steep negative correlation (r = −0.932, P < 0.0001). (E) Qiagen® Ingenuity® pathway analysis (IPA) showing top canonical pathways increased in the cortex of P301S;Ssh1−/− mice compared to P301S mice, plotted by Z-score (blue bars) and -Log10(P-value) (red circles). Dotted blue line (≥2) = significant pathway activation Z-score. Dotted red line (≥1.3) = significant adjusted -Log10(P-value) for overlap of genes in each pathway. (F) IPA showing top canonical pathways decreased in the cortex of P301S;Ssh1−/− mice compared to P301S mice, plotted by negative Z-score (red bars) and -Log10(P-value) (blue circles). Dotted red line (≥2) = significant pathway activation Z-score. Dotted blue line (≥1.3) = significant adjusted -Log10(P-value) for overlap of genes in each pathway. (G) Representative blots of Nrf2 and Nrf2 targets (NQO1, GSTM1, & HMOX1) from cortical lysates of 7-mo-old WT, P301S, and P301S;Ssh1−/− mice. (H) Quantification of Nrf2, NQO1, GSTM1, and HMOX1 [one-way ANOVA; Nrf2: F (2, 18) = 4.645, P = 0.0236; NQO1: F (2, 17) = 6.763, P = 0.0069; GSTM1: F (2, 17) = 10.35, P = 0.0011; HMOX1: F (2, 18) = 0.2911, P = 0.7509; post hoc Dunnett, ***P = 0.0006, **P = 0.0037, *P < 0.05. n = 5 to 9 mice/genotype].
We next performed Qiagen® Ingenuity® Pathway Analysis (IPA) for canonical pathways positively or negatively altered in tauP301S brains compared to WT mouse brains and also in tauP301S;Ssh1−/− brains compared to tauP301S brains. Of the top canonical pathways with attributable activation Z-scores and -Log10(P-value)≥1, PTEN signaling and tRNA charging pathways showed upregulation in tauP301S brains compared to WT brains (SI Appendix, Fig. S8B), which were reversed in tauP301S;Ssh1−/− brains compared to tauP301S brains (Fig. 6E). Conversely, CREB, VEGF, and Gai signaling pathways were decreased in tauP301S brains compared to WT brains (SI Appendix, Fig. S8C), and normalized in tauP301S;Ssh1−/− brains compared to tauP301S brains (Fig. 6E). Of the top canonical pathways, the Nrf2 oxidative stress response pathway showed a significant Z-score = 2.23 in tauP301S;Ssh1−/− brains compared to tauP301S brains, indicating that Ssh1 elimination activates Nrf2 oxidative stress response based on gene expression patterns within the Qiagen® Nrf2 pathway panel (Fig. 6E and SI Appendix, Fig. S8D). However, the degree of overlap with the analysis-ready genes within the Nrf2 pathway panel fell slightly shy of statistical significance, indicating that loss of Ssh1 leads to activation of a subset of Nrf2 target genes, perhaps due to the low level of oxidative injury seen in tauP301S;Ssh1−/− mice. To validate these results, we performed quantitative western blotting for Nrf2 and established Nrf2 targets NQO1, GSTM1, and HMOX1 in WT, tauP301S, and tauP301S;Ssh1−/− mouse cortical tissues. Despite no observed changes in HMOX1 among the genotypes, tauP301S brains showed reduced Nrf2, GSTM1, and NQO1 compared to WT brains, which were all significantly increased in tauP301S;Ssh1−/− compared to tauP301S brains (Fig. 6 G and H). These changes by loss of Ssh1 are in line with observations in Nrf2 knockout mice, in which NQO1 but not HMOX1 (HO-1) levels were significantly reduced (59). We also observed a significant increase in Nrf2 mRNA levels in tauP301S;Ssh1−/− compared to tauP301S brains (SI Appendix, Fig. S8E), consistent with previous observations that the Nrf2 promoter contains ARE-like sequences (60) through which Nrf2 activation increases Nrf2 mRNA levels (60–62). Collectively, these results indicate that Ssh1 elimination positively regulates the Nrf2 oxidative stress response pathway and normalizes transcriptomic perturbations in tauP301S mice.
Discussion
Here, we have uncovered a mechanism by which AD is associated with loss of nuclear Nrf2 and the associated Nrf2-mediated neuroprotective response to oxidative stress. Specifically, we uncovered a new function of SSH1 that inhibits and titrates the Nrf2 oxidative stress response, independent of SSH1 phosphatase activity and distinct from its actions on cofilin-mediated actin dynamics and p62-mediated autophagy (25, 31). We found that this inhibitory activity of SSH1 is achieved primarily in response to oxidative stress by sequestering Nrf2 on actin filaments and by augmenting Keap1-mediated Nrf2 inhibitory control. Indeed, these inhibitory Nrf2 interaction signatures were excessively elevated in the brains of AD and FTLD-tau patients, in agreement with the loss of nuclear but not cytoplasmic Nrf2 (9). Accordingly, genetic elimination of Ssh1 increased nuclear Nrf2, which mitigated tau and Ab deposition and protected against oxidative injury, neuroinflammation, neurodegeneration, and synaptic plasticity deficits in AD mice. Loss of Ssh1 also normalized transcriptomic perturbations in tauP301S mice and activated the Nrf2 oxidative stress response pathway. As oxidative stress activates Nrf2 and SSH1 while deactivating Keap1 (18, 24, 27, 28), the intersection between Nrf2 and SSH1 appears to be a tipping point that impacts the balance between neuroprotection and neurodegeneration under oxidative stress in AD. Our results indicate that SSH1 tips this balance away from Nrf2-mediated neuroprotection in AD and FTLD.
SSH1 appears to act physiologically as a molecular break to prevent Nrf2 overactivation under oxidative stress, which would prevent excessive Nrf2 activation from promoting neoplastic transformation and survival of tumorigenic cells (63). As transient oxidative stress subsides in a physiological setting, SSH1 activity is normally homeostatically suppressed through 14-3-3 binding (22, 23), which then releases Nrf2 from SSH1 (SI Appendix, Fig. S9). By contrast, during AD pathogenesis, elevated oxidative stress strongly activates SSH1 (22, 23, 64, 65) while suppressing Keap1 (18, 19, 66). This allows SSH1 to block and override Nrf2-mediated natural neuroprotection by sequestering the majority of Nrf2 and indirectly augmenting Keap1–Nrf2 interaction (SI Appendix, Fig. S9). Adding further mechanistic support, Aβ42 increases 14-3-3 oxidation in rat brains (67, 68), and SSH1/14-3-3 interaction is reduced in the APP/PS1 AD model (29).
Our results point to the ability of SSH1 to sequester Nrf2 on actin filaments independent of its phosphatase activity as a key mechanism by which SSH1 suppresses Nrf2 signaling (SI Appendix, Fig. S9), which is reminiscent of how SSH1 also binds, stabilizes, and bundles F-actin independently of its phosphatase activity (25, 69). Importantly, we also detected highly elevated SSH1–Nrf2 interaction in the brains of AD and FTLD patients. Notably, pharmacologic disruption of actin filaments increases nuclear Nrf2 translocation and signaling (46), similar to loss of SSH1. The finding that full-length SSH1 but not SSH1 deletion mutants sequesters Nrf2 on F-actin is also consistent with the requirement of both N- and C-terminal regions of SSH1 to stabilize F-actin (69), which is distinct and separable from SSH1 activities that dephosphorylate SQSTM1/pS403-p62 (31, 37, 38) and cofilin (24, 27, 28). Another avenue to suppress Nrf2 signaling occurs through SSH1 sequestering SQSTM1/pS349-p62 away from Keap1, which indirectly increases the association of Keap1 with Nrf2 (SI Appendix, Fig. S9). However, SSH1 only modestly promotes Keap1-mediated Nrf2 ubiquitination under oxidative stress, and loss of SQSTM1/p62 only moderately tempers the ability of SSH1 to suppresses Nrf2 signaling, suggesting that this is a secondary avenue for SSH1-mediated Nrf2 suppression when SQSTM1/p62 drives Nrf2 activation. In human AD and FTLD brains, we indeed observed highly elevated Keap1–Nrf2 interaction. While increased Keap1–Nrf2 interaction is consistent with increased SSH1 activity, the extent to which SSH1 drives this phenotype in the disease setting is unknown. A more precise mechanism by which SSH1 suppresses Nrf2 signaling by enhancing Keap1–Nrf2 interaction in AD and FTLD awaits further investigation. Nonetheless, the activity of SSH1 to sequester Nrf2 on F-actin and enhance Keap1–Nrf2 interaction appears to be most robust under oxidative stress when ROS suppresses Keap1 (19) and activates SSH1 (27).
Based on the above premise, it is plausible that Ssh1 elimination protects from AD in large part through enhanced Nrf2 signaling. Notably, Nrf2 activation or inhibition decreases or increases β-secretase (BACE1) levels, respectively (10), in line with decreased Aβ deposition and increased nuclear Nrf2 seen with Ssh1 elimination. Furthermore, loss of Nrf2 induces neuroinflammation and elevates interferon γ (IFNγ) (11, 13, 15), a cytokine that stimulates Aβ production (70, 71). Hence, enhanced Nrf2 signaling by Ssh1 elimination may also decrease Ab deposition through reduced neuroinflammation. However, the canonical role of SSH1 in cofilin activation (25), which regulates APP endocytosis by F-actin severing (72), cannot be excluded as a potential regulator of Aβ deposition. The Nrf2 oxidative stress response, which is elevated by Ssh1 elimination, not only activates antioxidant and antiinflammatory targets but also multiple autophagy-related genes, including autophagy cargo receptors SQSTM1/p62 (6) and NDP52 (12), both of which are involved in the clearance of pathological tau (12, 31, 73). SQSTM1/p62 also reciprocally activates Nrf2 (18, 19), thereby completing a neuroprotective feed-forward loop. Hence, while the roles for SSH1 in suppressing Nrf2 activation and impeding SQSTM1/p62-mediated autophagy (31) are experimentally separable, Nrf2 and SQSTM1/p62 pathways are intertwined at multiple levels and function as components of a large homeostatic system activated by both oxidative and proteostatic stress. In this sense, activated SSH1 acts as a molecular break in this homeostatic neuroprotective system.
Overall, this study reports a mechanism of AD pathogenesis through SSH1-mediated Nrf2 sequestration, providing a mechanistic basis for selective nuclear depletion of Nrf2 in AD. As most Nrf2-activating drugs are electrophiles that are not well tolerated in patients (8), inhibiting activated SSH1 to unblock and enhance Nrf2 signaling may provide an effective therapeutic strategy for neuroprotection in AD and related disorders. As the catalytic activity of SSH1 does not significantly contribute to Nrf2 inhibition, strategies for therapeutic targeting SSH1-mediated Nrf2 suppression will require innovative approaches beyond inhibiting activity at its catalytic pocket.
Materials and Methods
DNA Constructs and siRNAs.
Plasmids pcDNA3-EGFP-C4-Nrf2 (Addgene, 21549) (74), pCDNA3-Myc3-Nrf2 (Addgene, 21555) (74), pREP-8xARE-GFP-SV40-BFP (Addgene, 13,4910) (39), and HA-Ubiquitin (Addgene, 18,712) (75) were obtained from Addgene. p3xFlag-SSH1, p3xFlag-SSH1ΔC, p3xFlag-SSH1ΔN, pEGFP-N1-p62-S403E, and pEGFP-N1-p62-S403A were previously generated by the Kang Lab (31, 38). p3xFlag-SSH1-CS was generated by the Kang lab during this study. p62-S349A-pEGFP-N1 (76) was kindly provided by Tanji Konikazu (Hirosaki University, Japan). Signal Silence SQSTM1/p62 siRNA II ® (Cell Signaling Technologies, 6,399) and SSH1 siRNA (Dharmacon, GE Healthcare; 5′- GAGGAGCUGUCCCGAUGAC-3′) were obtained from the indicated sources.
Cell Lines.
Hippocampus-derived mouse neuroblastoma HT22 cells (David Schubert, Salk Institute, La Jolla, CA) and HEK293T cells (ATCC, CRL-3,216) were cultured in Dulbecco’s modified Eagle’s medium (Gibco, 11,965-092) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich, 12,306 C), 1% penicillin/streptomycin (P/S) (Gibco, 15,140–122), and BM-cyclin (Roche, 10799050001). Cells were kept at 37 °C with 5% CO2 levels. For treatment of H2O2, cells were supplemented with 1% FBS during the duration of H2O2 exposure.
DNA and siRNA Transfections and shRNA Transduction.
DNA plasmids or siRNA (100 nM) were transiently transfected in HT22 or HEK293T cells using lipofectamine 2,000 (Invitrogen, 116688500) and Opti-MEM (Gibco, 31,985-070). After 4 to 6 h post-transfection, the medium was replaced with a new complete medium. Cells were grown for 48 h after transfection before experimental assays.
Cell Lysis, Subcellular Protein Fractionation, Western Blotting, and Antibodies.
Cells were lysed with RIPA buffer (50 mM Tris pH 7.4, 0.1% SDS, 2 mM ethylenediaminetetraacetic acid, 150 mM NaCl, 1% Triton X-100) with 1% protease inhibitors (GeneDEPOT, P3100-010) and phosphatase inhibitors (GeneDEPOT, P3200-005). After centrifugation at 16,000 g for 15 min at 4 °C, the supernatant was transferred to new tubes. Protein concentration was measured by bicinchoninic acid (Pierce™ BCA protein assay kit, 23,225). Equal amounts of total protein were subjected to SDS-PAGE. After transferring the proteins to a nitrocellulose membrane (GE Healthcare,10600002), the membrane was blocked with 5% skim milk for 1 h at room temperature (RT). Primary antibodies were applied overnight at 4 °C. The corresponding peroxidase-conjugated secondary antibody was applied for 4 h at RT and detected by ECL western blot reagents (Thermo Scientific, 1863096 and/or A38556). ECL images were obtained with the Fuji LAS-4000 imager (LAS-4000, Pittsburgh, PA, USA) or Amersham ImageQuant 800 (Cytiva) and analyzed with Fiji Image J software (NIH, https://imagej.net/software/fiji/). Quantified bands in Western blots were normalized to the loading control (actin) prior to comparisons between experimental conditions.
Subcellular protein fractionation was performed on HEK293T cells using Subcellular Protein Fractionation Kit for Cultured Cells (Thermo Fisher Scientific, 78,840) following the manufacturer’s protocol. Briefly, the cells were washed with ice-cold PBS, lysed with CEB buffer, and centrifuged at 500 g to isolate cytoplasmic fraction in the supernatants. MEB buffer was then added to pellets which were centrifuged at 3,000 g to isolate membrane fraction. Pellets from this centrifugation were then incubated with NEB buffer and further centrifuged at 5,000 g to isolate nuclear fraction. Resulting pellets were incubated with NEB+CaCl2+micrococcal nuclease and centrifuged at 16,000 g to isolate chromatin fraction in the supernatants. Final pellets were added PEB buffer and centrifuged at 16,000 g to isolate the cytoskeletal fraction in the supernatant. All buffers had protease inhibitors (GeneDEPOT, P3100-010) and phosphatase inhibitors (GeneDEPOT, P3200-005). Protein concentration was measured by bicinchoninic acid (Pierce™ BCA protein assay kit, 23,225).
The following antibodies were used to probe target proteins by immunoblotting: Rabbit polyclonal anti-SSH1 (ECM Biosciences, SP1711); mouse monoclonal anti-6xHis antibody (D1Z9C) (Cell Signaling Technologies, 12,721), rabbit monoclonal anti-SSH1 (E1K3W) (Cell Signaling Technologies, 13,578); rabbit monoclonal anti-Nrf2 (D1Z9C) (Cell Signaling Technologies, 12,721); rabbit monoclonal anti-Keap1 (D6B12) (Cell Signaling Technologies, 8,047); rabbit polyclonal anti-Myc-tag (abcam, ab9106); mouse monoclonal anti-β-Actin (C4) (Santa Cruz Biotech, sc-47778); mouse monoclonal anti-FLAG (M2) (Sigma-Aldrich, F3165); rabbit polyclonal anti HMOX1 (Invitrogen, PA5-27338); mouse monoclonal anti-NQO1 (A180)(Santa Cruz Biotech), sc-32,793); anti-Myc-tag (abcam, ab9106); mouse monoclonal anti-Vimentin (5G3F10) (Cell Signaling Technologies, 3,390); mouse monoclonal anti-Lamin B1 (8D1) (Santa Cruz Biotech, sc-56144); mouse monoclonal anti-GSTM1 antibody (R&D Systems, MAB6894); mouse monoclonal anti-ubiquitin antibody (Cell Signaling Technologies, 3,936); peroxidase-Conjugated AffiniPure goat anti-mouse IgG (Jackson ImmunoResearch, 115-035-033); and peroxidase-conjugated AffiniPure goat anti-rabbit IgG (Jackson ImmunoResearch, 111-035-033).
Immunocytochemistry, PLA, F-actin Staining, and Antibodies.
For ICC, Cells were washed with PBS and fixed with 4% paraformaldehyde (Acros Organics, 41678-5000) for 15 min at RT. Fixed cells were blocked with 3% normal goat serum (Vector Laboratories, S-1000) and permeabilized with 0.2% triton (Fluka, 93426) at RT for 1 h. Indicated primary antibodies were applied overnight at 4 °C, followed by secondary antibodies (Alexa Fluor) and DAPI (Thermo Scientific, 62,248) for 45 min at RT, and mounted with Fluromount-G (Thermo Fisher Scientific, 00-4958-02).
For PLA, primary antibodies were followed by incubation of the Duolink® In Situ PLA® secondary antibody probes (Sigma-Aldrich, DUO92004 and DUO92002) at 37 °C for 1 h. Duolink® In Situ PLA® ligation and amplification reagents (Sigma-Aldrich, DUO92008 or DUO92013) were incubated at 37 °C for 30 min and 100 min, respectively, as previously shown (31) before being mounted with the Duolink® In Situ PLA® Mounting Medium with DAPI (Sigma-Aldrich, DUO82040). Each procedure included samples processed with only one primary or only one probe as negative controls. Slides were saved at −20 °C. In PLA experiments where no fluorescent marker was present, GFP was transfected at a ratio 1:5 as a marker of transfected cells, and only GFP-positive cells were quantified.
F-actin was stained subsequent to secondary antibody for ICC or PLA with F-actin Staining Kit—Blue Fluorescence— Cytopainter (abcam, ab112124), which was applied for 45 min at RT before mounting with Fluromount-G (Thermo Fisher Scientific, 00-4958-02).
The following antibodies were used to probe target proteins: rabbit polyclonal anti-SSH1(ECM Biosciences, SP1711); mouse monoclonal anti-Nrf2 (3G7)(Novus Biologicals, AF4000); rabbit monoclonal anti-Keap1 (D6B12) (Cell Signaling Technologies, 8,047); rabbit monoclonal anti-SQSTM1/p62 (D10E10) (Cell Signaling Technologies, 7,695); mouse monoclonal anti-FLAG (M2) (Sigma-Aldrich, F3165); rabbit monoclonal anti-Nrf2 (D1Z9C) (Cell Signaling Technologies, 12,721); mouse monoclonal anti-Keap1 (1B4) (Abcam, ab119403); mouse monoclonal anti-SSH1 (1A5C8) (Santa Cruz Biotech, sc-517226); Alexa Fluor 594 goat anti-rabbit IgG (Invitrogen, A11037); Alexa Fluor 594 goat anti-mouse IgG (Invitrogen, A11032); and Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen, A11034).
Quantification and Statistical Analysis.
Images and Western blots were analyzed and organized using Fiji Image J (NIH, https://imagej.net/software/fiji/) and Microsoft Excel. Statistical analysis was performed with the Prism 8.0 software (GraphPad Software, San Diego, CA, USA) using two-tailed t tests, one-way ANOVA, or two-way ANOVA. ANOVA (both one-way and two-way) was followed by the indicated post hoc tests. P values ≤ 0.05 were considered significant. All quantitative graphs with error bars are presented as means ± SEM.
Chemicals and Reagents, Recombinant Protein Assays, Ubiquitinated Nrf2 Detection, Real-Time PCR, Silver Staining, RNA Sequencing, Qiagen Ingenuity Pathway Analysis, Mice, Mouse Brain IHC and Western Blotting, Human Brain Tissue, Human Brain IHC/PLA, and Ex Vivo Electrophysiology.
Please see SI Appendix, Methods.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (PDF)
Dataset S02 (PDF)
Dataset S03 (PDF)
Acknowledgments
We thank Dr. Kunikazu Tanji (Hirosaki University, Japan) for providing the GFP-p62-S349A construct. We thank Drs. Allan Levey and Marla Gearing at Emory Alzheimer’s Disease Research Center (P50 AG025688) for providing postmortem brain tissues. We also acknowledge Kyle McGill Percy and Sophia Khan (Case Western Reserve University) for their contributions to experimental protocols. This work was supported by grants from the NIH [R01AG059721 to J.-A.A.W, R01AG067741 to J.-A.A.W. and D.E.K., R01NS122350 to D.E.K, RF1AG053060 to D.E.K]; and Veterans Affairs [BX004680 to D.E.K]. D.E.K is also supported by the Howard T. Karsner Professorship in Pathology, CWRU. A.A.P was supported as the CWRU Rebecca E. Barchas, MD, Professor in Translational Psychiatry and as the University Hospitals Morley-Mather Chair in Neuropsychiatry. A.A.P. also acknowledges the support from The Valour Foundation, the American Heart Association and Paul Allen Foundation Initiative in Brain Health and Cognitive Impairment (19PABH134580006), the Department of Veterans Affairs Merit Award I01BX005976, NIH/NIGMS RM1 GM142002, NIH/NIA RO1AG066707, NIH/NIA 1 U01 AG073323, the Elizabeth Ring Mather & William Gwinn Mather Fund, S. Livingston Samuel Mather Trust, G. R. Lincoln Family Foundation, Wick Foundation, Gordon & Evie Safran, the Leonard Krieger Fund of the Cleveland Foundation, the Maxine and Lester Stoller Parkinson’s Research Fund, and the Louis Stokes Veteran’s Affairs Medical Center resources and facilities.
Author contributions
S.C., J.-A.A.W., T.L., A.A.P., and D.E.K. designed research; S.C., J.-A.A.W., X.W., T.L., S.R., T.R.K., Y.K., E.V.-R., and D.E.K. performed research; S.C., J.-A.A.W., T.L., A.A.P., and D.E.K. contributed new reagents/analytic tools; S.C., J.-A.A.W., X.W., S.R., T.R.K., Y.K., E.V.-R., and D.E.K. analyzed data; and S.C., J.-A.A.W., A.A.P., and D.E.K. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the article and/or supporting information. Raw data are available upon reasonable request to the senior author and in compliance with local ethical regulations.
Supporting Information
References
- 1.Nunomura A., et al. , Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Nunomura A., et al. , Neuronal oxidative stress precedes amyloid-beta deposition in down syndrome. J. Neuropathol. Exp. Neurol. 59, 1011–1017 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Smith M. A., et al. , Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J. Alzheimers Dis. 19, 363–372 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keller J. N., et al. , Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 64, 1152–1156 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Matsumaru D., Motohashi H., The KEAP1-NRF2 system in healthy aging and longevity. Antioxidants (Basel) 10, 1929 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pajares M., et al. , Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 12, 1902–1916 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pajares M., et al. , Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 14, 1310–1322 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gazaryan I. G., Thomas B., The status of Nrf2-based therapeutics: current perspectives and future prospects. Neural Regen. Res. 11, 1708–1711 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramsey C. P., et al. , Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 66, 75–85 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bahn G., et al. , NRF2/ARE pathway negatively regulates BACE1 expression and ameliorates cognitive deficits in mouse Alzheimer’s models. Proc. Natl. Acad. Sci. U.S.A. 116, 12516–12523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Branca C., et al. , Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 26, 4823–4835 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jo C., et al. , Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 5, 3496 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rojo A. I., et al. , NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox. Biol. 13, 444–451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiwaji Z., et al. , Reactive astrocytes acquire neuroprotective as well as deleterious signatures in response to Tau and Ass pathology. Nat. Commun. 13, 135 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uruno A., et al. , Nrf2 suppresses oxidative stress and inflammation in app knock-in Alzheimer’s Disease model mice. Mol. Cell Biol. 40, e00467-19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tapias V., et al. , Benfotiamine treatment activates the Nrf2/ARE pathway and is neuroprotective in a transgenic mouse model of tauopathy. Hum. Mol. Genet. 27, 2874–2892 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stack C., et al. , Methylene blue upregulates Nrf2/ARE genes and prevents tau-related neurotoxicity. Hum. Mol. Genet. 23, 3716–3732 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Itoh K., Mimura J., Yamamoto M., Discovery of the negative regulator of Nrf2, keap1: A historical overview. Antioxid. Redox Signal. 13, 1665–1678 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Itoh K., et al. , Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes. Dev. 13, 76–86 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y., et al. , A genomic screen for activators of the antioxidant response element. Proc. Natl. Acad. Sci. U.S.A. 104, 5205–5210 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Komatsu M., et al. , The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of keap1. Nat. Cell Biol. 12, 213–223 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Soosairajah J., et al. , Interplay between components of a novel LIM kinase-slingshot phosphatase complex regulates cofilin. EMBO J. 24, 473–486 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagata-Ohashi K., et al. , A pathway of neuregulin-induced activation of cofilin-phosphatase slingshot and cofilin in lamellipodia. J. Cell Biol. 165, 465–471 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bernstein B. W., Bamburg J. R., ADF/cofilin: A functional node in cell biology. Trends Cell Biol. 20, 187–195 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niwa R., Nagata-Ohashi K., Takeichi M., Mizuno K., Uemura T., Control of actin reorganization by slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 108, 233–246 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Kurita S., Watanabe Y., Gunji E., Ohashi K., Mizuno K., Molecular dissection of the mechanisms of substrate recognition and F-actin-mediated activation of cofilin-phosphatase slingshot-1. J. Biol. Chem. 283, 32542–32552 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Kim J. S., Huang T. Y., Bokoch G. M., Reactive oxygen species regulate a slingshot-cofilin activation pathway. Mol. Biol. Cell 20, 2650–2660 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eiseler T., et al. , Protein kinase D1 regulates cofilin-mediated F-actin reorganization and cell motility through slingshot. Nat. Cell Biol. 11, 545–556 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woo J. A., et al. , Slingshot-cofilin activation mediates mitochondrial and synaptic dysfunction via abeta ligation to beta1-integrin conformers. Cell Death Differ. 22, 921–934 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim T., et al. , Human LilrB2 is a beta-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science 341, 1399–1404 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fang C., et al. , SSH1 impedes SQSTM1/p62 flux and MAPT/Tau clearance independent of CFL (cofilin) activation. Autophagy 17, 2144–2165 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katsuragi Y., Ichimura Y., Komatsu M., p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 282, 4672–4678 (2015), 10.1111/febs.13540. [DOI] [PubMed] [Google Scholar]
- 33.Matsumoto G., Shimogori T., Hattori N., Nukina N., TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 24, 4429–4442 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Sanchez-Martin P., Komatsu M., p62/SQSTM1—Steering the cell through health and disease. J. Cell Sci. 131, jcs222836 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Pilli M., et al. , TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37, 223–234 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lim J., et al. , Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 11, e1004987 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cazzaro S., et al. , Slingshot homolog-1 mediates the secretion of small extracellular vesicles containing misfolded proteins by regulating autophagy cargo receptors and actin dynamics. Front. Aging Neurosci. 14, 933979 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cazzaro S., Zhao X., Zhao V. K., Kim Y. K., Woo J. A., Slingshot homolog-1 amplifies mitochondrial abnormalities by distinctly impairing health and clearance of mitochondria. Hum. Mol. Genet. 32, 1660–1672 (2023), 10.1093/hmg/ddad006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wyler E., et al. , Single-cell RNA-sequencing of herpes simplex virus 1-infected cells connects NRF2 activation to an antiviral program. Nat. Commun. 10, 4878 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kobayashi M., et al. , The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol. Cell Biol. 29, 493–502 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pi J., Qu W., Reece J. M., Kumagai Y., Waalkes M. P., Transcription factor Nrf2 activation by inorganic arsenic in cultured keratinocytes: Involvement of hydrogen peroxide. Exp. Cell Res. 290, 234–245 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Kansanen E., Kivela A. M., Levonen A. L., Regulation of Nrf2-dependent gene expression by 15-deoxy-Delta 12,14-prostaglandin J2. Free Radic. Biol. Med. 47, 1310–1317 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Sussan T. E., et al. , Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc. Natl. Acad. Sci. U.S.A. 106, 250–255 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y. Y., Yang Y. X., Zhe H., He Z. X., Zhou S. F., Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Devel. Ther. 8, 2075–2088 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ichimura Y., et al. , Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 51, 618–631 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Kang M. I., Kobayashi A., Wakabayashi N., Kim S. G., Yamamoto M., Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc. Natl. Acad. Sci. U.S.A. 101, 2046–2051 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Plascencia-Villa G., Perry G., Preventive and therapeutic strategies in alzheimer’s disease: Focus on oxidative stress, redox metals, and ferroptosis. Antioxid. Redox Signal. 34, 591–610 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martinez A., Portero-Otin M., Pamplona R., Ferrer I., Protein targets of oxidative damage in human neurodegenerative diseases with abnormal protein aggregates. Brain Pathol. 20, 281–297 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xie J. Z., et al. , P301S-hTau acetylates KEAP1 to trigger synaptic toxicity via inhibiting NRF2/ARE pathway: A novel mechanism underlying hTau-induced synaptic toxicities. Clin. Transl. Med. 12, e1003 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Williams H. C., et al. , The cofilin phosphatase slingshot homolog 1 restrains angiotensin II-induced vascular hypertrophy and fibrosis in vivo. Lab. Invest. 99, 399–410 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jankowsky J. L., et al. , Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: Evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 13, 159–170 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Chiorcea-Paquim A. M., 8-oxoguanine and 8-oxodeoxyguanosine biomarkers of oxidative DNA damage: A review on HPLC-ECD determination. Molecules 27, 1620 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tian Y., et al. , Activation of Nrf2/ARE pathway alleviates the cognitive deficits in PS1V97L-Tg mouse model of Alzheimer’s disease through modulation of oxidative stress. J. Neurosci. Res. 97, 492–505 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Yan Y., et al. , X-linked ubiquitin-specific peptidase 11 increases tauopathy vulnerability in women. Cell 185, 3913–3930.e19 (2022), 10.1016/j.cell.2022.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu T., et al. , Modulation of synaptic plasticity, motor unit physiology, and TDP-43 pathology by CHCHD10. Acta Neuropathol. Commun. 10, 95 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Woo J. A., et al. , beta-Arrestin2 oligomers impair the clearance of pathological tau and increase tau aggregates. Proc. Natl. Acad. Sci. U.S.A. 117, 5006–5015 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Woo J. A., et al. , Activated cofilin exacerbates tau pathology by impairing tau-mediated microtubule dynamics. Commun. Biol. 2, 112 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoshiyama Y., et al. , Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Crilly M. J., Tryon L. D., Erlich A. T., Hood D. A., The role of Nrf2 in skeletal muscle contractile and mitochondrial function. J. Appl. Physiol. 1985, 730–740 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kwak M. K., Itoh K., Yamamoto M., Kensler T. W., Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: Role of antioxidant response element-like sequences in the nrf2 promoter. Mol. Cell. Biol. 22, 2883–2892 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramos-Gomez M., et al. , Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 98, 3410–3415 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kwak M. K., Itoh K., Yamamoto M., Sutter T. R., Kensler T. W., Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H–1, 2-dimethiole-3-thione. Mol. Med. 7, 135–145 (2001). [PMC free article] [PubMed] [Google Scholar]
- 63.Panieri E., et al. , NRF2 and mitochondrial function in cancer and cancer stem cells. Cells 11, 2401 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhao J. W., et al. , Regulation of cofilin activity by CaMKII and calcineurin. Am. J. Med. Sci. 344, 462–472 (2012). [DOI] [PubMed] [Google Scholar]
- 65.Wang Y., Shibasaki F., Mizuno K., Calcium signal-induced cofilin dephosphorylation is mediated by slingshot via calcineurin. J. Biol. Chem. 280, 12683–12689 (2005). [DOI] [PubMed] [Google Scholar]
- 66.Oguro A., Sugitani A., Kobayashi Y., Sakuma R., Imaoka S., Bisphenol A stabilizes Nrf2 via Ca(2+) influx by direct activation of the IP(3) receptor. J. Toxicol. Sci. 46, 1–10 (2021). [DOI] [PubMed] [Google Scholar]
- 67.Boyd-Kimball D., et al. , Proteomic identification of proteins specifically oxidized by intracerebral injection of amyloid beta-peptide (1–42) into rat brain: Implications for Alzheimer’s disease. Neuroscience 132, 313–324 (2005). [DOI] [PubMed] [Google Scholar]
- 68.Sultana R., et al. , Protective effect of D609 against amyloid-beta1-42-induced oxidative modification of neuronal proteins: Redox proteomics study. J. Neurosci. Res. 84, 409–417 (2006). [DOI] [PubMed] [Google Scholar]
- 69.Kurita S., Gunji E., Ohashi K., Mizuno K., Actin filaments-stabilizing and -bundling activities of cofilin-phosphatase slingshot-1. Genes. Cells 12, 663–676 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Blasko I., et al. , Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Abeta1-40 and Abeta1-42 by human astrocytes. Neurobiol. Dis. 7, 682–689 (2000). [DOI] [PubMed] [Google Scholar]
- 71.Blasko I., Marx F., Steiner E., Hartmann T., Grubeck-Loebenstein B., TNFalpha plus IFNgamma induce the production of Alzheimer beta-amyloid peptides and decrease the secretion of APPs. FASEB J. 13, 63–68 (1999). [DOI] [PubMed] [Google Scholar]
- 72.Liu T., et al. , Dual role of cofilin in APP trafficking and amyloid-beta clearance. FASEB J. 33, 14234–14247 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu Y., Zhang S., Zheng H., The cargo receptor SQSTM1 ameliorates neurofibrillary tangle pathology and spreading through selective targeting of pathological MAPT (microtubule associated protein tau). Autophagy 15, 583–598 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Furukawa M., Xiong Y., BTB protein keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the cullin 3-Roc1 ligase. Mol. Cell Biol. 25, 162–171 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kamitani T., Kito K., Nguyen H. P., Yeh E. T., Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J. Biol. Chem. 272, 28557–28562 (1997). [DOI] [PubMed] [Google Scholar]
- 76.Tanji K., et al. , Phosphorylation of serine 349 of p62 in Alzheimer’s disease brain. Acta Neuropathol. Commun. 2, 50 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (PDF)
Dataset S02 (PDF)
Dataset S03 (PDF)
Data Availability Statement
All study data are included in the article and/or supporting information. Raw data are available upon reasonable request to the senior author and in compliance with local ethical regulations.