

Abstract
Hepatitis C virus (HCV) infection is one of the major factors to trigger a sustained hepatic inflammatory response and hence hepatocellular carcinoma (HCC), but direct-acting-antiviral (DAAs) was not efficient to suppress HCC development. Heat shock protein 90 kDa (HSP90) is highly abundant in different types of cancers, and especially controls protein translation, endoplasmic reticulum stress, and viral replication. In this study we investigated the correlation between the expression levels of HSP90 isoforms and inflammatory response marker NLRP3 in different types of HCC patients as well as the effect of a natural product celastrol in suppression of HCV translation and associated inflammatory response in vivo. We identified that the expression level of HSP90β isoform was correlated with that of NLRP3 in the liver tissues of HCV positive HCC patients (R2 = 0.3867, P < 0.0101), but not in hepatitis B virus-associated HCC or cirrhosis patients. We demonstrated that celastrol (3, 10, 30 μM) dose-dependently suppressed the ATPase activity of both HSP90α and HSP90β, while its anti-HCV activity was dependent on the Ala47 residue in the ATPase pocket of HSP90β. Celastrol (200 nM) halted HCV internal ribosomal entry site (IRES)-mediated translation at the initial step by disrupting the association between HSP90β and 4EBP1. The inhibitory activity of celastrol on HCV RNA-dependent RNA polymerase (RdRp)-triggered inflammatory response also depended on the Ala47 residue of HSP90β. Intravenous injection of adenovirus expressing HCV NS5B (pAde-NS5B) in mice induced severe hepatic inflammatory response characterized by significantly increased infiltration of immune cells and hepatic expression level of Nlrp3, which was dose-dependently ameliorated by pretreatment with celastrol (0.2, 0.5 mg/kg, i.p.). This study reveals a fundamental role of HSP90β in governing HCV IRES-mediated translation as well as hepatic inflammation, and celastrol as a novel inhibitor of HCV translation and associated inflammation by specifically targeting HSP90β, which could be developed as a lead for the treatment of HSP90β positive HCV-associated HCC.
Keywords: hepatitis C virus, celastrol, heat shock protein 90β, inflammatory response, RNA-dependent RNA polymerase
Introduction
Hepatitis C virus (HCV) infected about 1% of the world population, 20%‒30% of which leads to end-stage liver diseases, including liver fibrosis, cirrhosis, and hepatocellular carcinoma (HCC) [1]. Chronic HCV and/or hepatitis B virus (HBV) infection causes approximately 80% of HCC [2]. Direct-acting-antiviral (DAAs) led to a new paradigm for HCV treatment, which dramatically improved sustained viral response (SVR) in all genotypes. Treatment with HCV nonstructural (NS) protein 5B inhibitor Sofosbuvir achieved a 97% SVR in combination with Daclastsvir and Ledipasvir [3]. Sofosbuvir and Velpatasvir improved SVR to as high as 99% to genotype 1, 2, 4, 5, and 6 infections, and 94% to genotype 2 and 3 infections, respectively [4, 5]. Despite recent success in DAAs for HCV treatment, the number of chronic HCV-associated HCC incidences is still on the rise [6]. After a median follow-up of 57 months, a recent study showed that patients with a prior history of HCC and who have been treated with DAAs for HCV infection have a much higher than expected early recurrence rate of their liver cancer much higher than previously predicted, with the rate exceeding 40% in some subgroups [7]. DAA therapy was not as efficient to suppress HCC development and hospitalization in Child–Pugh class C (CP-C) patients as in CP-A or CP-B patients [8]. HCV replication is dependent on the internal ribosomal entry site (IRES) initiated translation, but not the cap-dependent translation utilized by host genes. Thus targeting host factors highly expressed in HCV-associated HCC patients and are actively involved in the HCV replication cycle could be a promising alternative strategy to treatment HCV positive HCC patients.
Heat shock protein 90 kDa (HSP90) are highly abundant in different types of cancers, and especially control protein translation, endoplasmic reticulum stress, and viral replication. For example, HSP90 interacted with HCV NS5A protein through the immunophilin protein FKBP38 [9]. HSP90 also colocalized with the component of the processing GW bodies GW182, which modulated the function of microRNA-122 and promoted the transcription of HCV RNA [10, 11]. Despite the high homology of HSP90 isoforms [12], the distinct isoform-specific function and expression levels of HSP90α and HSP90β are related to off target toxicity and/or enhanced potency of isoform-specific inhibitors [13, 14]. Known inhibitors of HSP90, for example, radicicol, geldanamycin, and analog 17-allylamino-17-demethoxy-geldanamycin (17-AAG) interrupted the ATPase activity of HSP90 without isoform specificity. The distinct isotype expression level in different disease condition and the development of isoform-specific inhibitors are demanding for further translational development targeting HSP90 [15].
Celastrol (Fig. 1a) is a pentacyclic triterpene first extracted from Tripterygium wilfordii Hook F. (“Thunder of God Vine” or雷公藤in Chinese) of the Celastraceae family [16]. Celastrol exhibits diverse activities in different types of cancer and metabolic diseases [17, 18]. Treatment with celastrol suppressed the expression of key pro-inflammatory cytokines including IL-6, IL-17, and IFN-γ in the adjuvant-induced rheumatoid arthritis rats [19], and changed the T helper 17/T regulatory cells ratio in the inflamed joints of arthritis rats [20]. Celastrol induced degradation of HSP90 client proteins at micromolar concentration [21, 22] and has been recognized as a pan-HSP90 inhibitor since then. However, no study has revealed the association of HSP90 inhibition to the anti-inflammation activity of celastrol, especially in HCV infection.
Fig. 1. Correlation of the expression levels of NLRP3, HSP90α, and HSP90β in patients’ samples.

a Chemical structure of celastrol. b Representative IHC result of the expression levels of NLRP3, HSP90α, and HSP90β in the liver tissue from normal individuals, and HCV-HCC, HBV-HCC, and cirrhosis patients. Scale bar = 20 µm. c The expression level of each protein was scored semi-quantitatively by two researchers in blind, showing a strong correlation between NLRP3 and HSP90β in the liver tissues of normal individuals and HCC patients with HCV infection. The correlation analysis results are shown in Table 1.
In the present study, we determined the correlation between the expression levels of HSP90 isoform and inflammatory response marker NLRP3 in different types of HCC patients. We then investigated the potential isoform specific inhibition activity of celastrol in suppression of HCV translation and associated inflammatory response in vivo.
Materials and methods
Materials and reagents
Cell culture media and fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Celastrol was purchased from Cayman Chemical (Ann Arbor, MI, USA), purity ≥98%. Cel-2 and Cel-6 were synthesized in Prof. Yu-qiang Wang’s laboratory (Jinan University, Guangzhou, China) [23]. Stock solutions of all compounds were prepared with dimethylsulfoxide (DMSO) and frozen at −20 °C until further use. Final concentrations of 0.05 to 1 μM were obtained by dilution in culture media so that the final concentration of DMSO did not exceed 0.05%. DMSO was used as vehicle control in all experiments. All other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) except for those noted otherwise.
Biopsy and tissue chip
The LV8013 tissue chip obtained from US Biomax, Inc. (Derwood, MD, USA) contains a replicate of 16 unique cases of liver tissues from HCV positive HCC, and 20 cases of HBV positive HCC, cirrhosis, and normal individuals, respectively. The detailed information of the diagnosis was provided in Supplementary Table S1. The Ethics of using human samples was approved by The University of Macau Research Committee (No. BSERE16-APP009-ICMS).
Animal, adenovirus administration, and treatment condition
Male C57BL/6 J mice (20 ± 2 g body weight, 6–8 weeks of age) were raised in the Animal Facility at The University of Macau. The animals received human care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals [24], and have been reviewed and approved by Panel on Animal Research Ethics of University of Macau (No. UMARE-017-2018). All animals were kept in a standard environment with a 12 h light-dark cycle, and had free access to food and water.
Recombinant adenovirus was intravenously injected into mice via the tail-vein at 1 × 109 p.f.u./mouse. Mice that received intravenous injection of pAde-EGFP were used as the control to exclude the impact of delivery.
Celastrol was first dissolved in DMSO and then diluted with PBS to the final solution in 5% DMSO in PBS. Celastrol was administered through intraperitoneal (i.p.) injection twice a day. Mice in the control group were i.p. injected with 5% DMSO in PBS all through the experiment. Mice were anaesthetized by i.p. injection of pentobarbital sodium or isoflurane inhalation, and sacrificed to death at the end of the treatment to collect blood and liver tissue for further analysis.
Cell lines and growth condition
HCV genotype 1b Con1 subgenomic replicon cell line Huh-luc/neo-ET containing a luciferase reporter, Huh 9–13, and HCV genotype 1b plasmid pFK-I389/NS3-3 were provided by Dr. Ralf Bartenschlager from The University of Heidelberg [25]. AD293 cells were purchased from ATCC (Manassas, VA, USA). The Huh-luc/neo-ET cells and AD293 were cultured in high glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) FBS, 1 mM non-essential amino acid, and 250 μg/mL G418 (Life Technologies). All cells were used within the first 10 passages after recovery. The cells were maintained at 37 °C in 95% air and 5% CO2.
Construction of recombinant adenovirus (pAde) expression of HCV NS5B and Hsp90β A47S
ORFs of EGFP, HCV NS5B, and Hsp90β A47S were amplified using PCR and inserted into the CMV-driven pShuttle-IRES-hrGFP-1 vector from Agilent Technologies. The pShuttle plasmids were then recombined with pAdEasy-1 plasmid to generate the adenovirus packed expression system. All adenoviruses amplified in AD293 cells were purified by using two sequential rounds of cesium chloride gradient centrifugation and desalted with Econo-Pac 10 DG disposable chromatography column (Bio-Rad) before use. The titer of each viral stock was determined by using plaque assay on AD293 cells.
Molecular cloning
To construct overexpression plasmids of HSP90α and HSP90β, open reading frames were amplified with primers shown in Supplementary Table S2 by using Phusion High-Fidelity DNA Polymerase (New England Biolabs, Ipswich, MA, USA), and inserted into pcDNA5/TO plasmid (Invitrogen, Carlsbad, CA, USA).
HSP90β A47S and A47D mutants were generated by using QuickChange II Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA). The C-terminal His-tagged NBD domain of HSP90α (C-terminal His-tagged NBD HSP90α) and HSP90β (C-terminal His-tagged NBD HSP90β) were amplified with primers in Supplementary Table S2 and inserted into the pET26-UbCHis vector (a gift from Prof. Craig E. Cameron at Pennsylvania State University, PA, USA) [26].
Transfection of plasmids
All plasmids were cleared of endotoxins using the EndoGO kit (MirusBio, Madison, WI, USA) before transfection. Transfection was performed with either TransIT-LT1 transfection reagent (MirusBio) according to the manufacturers’ instructions or polyethylenimine (PEI) reagent.
Docking and molecular dynamics simulation
The crystal structure of a human HSP90β was obtained from the RCSB Protein Data Bank (PDB code: 3NMQ) [27]. The initial three-dimensional structures of Celastrol were computed and optimized using the B3LYP/6-31 G* method by Gaussian 09 program [12]. Glide module with extra precision in the Schrödinger program was employed to generate the initial receptor-ligand complexes [28]. All the parameters were set to the default values. Top-3 conformations were sampled for a short time molecular dynamic simulation to test the stability of the model.
AMBER 12 was employed for Hsp90β-Celastrol complex MD simulations [29]. Force field ff99SB was applied for protein and the Generalized Amber Force Field (GAFF) for ligand [30]. The ligands’ partial atomic charges were obtained from the restrained electrostatic potential (RESP) charges based on HF/6-31 G* calculations with Gaussian 09 program. Each system was solvated in a truncated cubic box of TIP3P water molecules with a margin distance of 10 Å and then neutralized [31]. Three steps of energy minimization were first performed to gradually remove the unreliable clashes between atoms before the heating step. Then, the system was heated from 0 to 300 K in 50 picoseconds (ps) using Langevin dynamics at constant volume and then equilibrated for 500 ps at a constant pressure of 1 atm. A production simulation run for 10 ns was performed using the NVT ensemble under a target temperature of 300 K. All bonds involving hydrogen atoms were constrained by the SHAKE algorithm and a time step of 2.0 fs was applied for all of the simulations [32].
Expression and purification of recombinant C-terminal His-tagged NBD of HSP90α and HSP90β
The CHis NBD HSP90α and C His NBD HSP90β plasmids were transformed into BL21 cells and grown overnight in 25 mL NZCYM supplemented with 25 μg/mL kanamycin, 20 μg/mL chloramphenicol, and 0.1% dextrose at 37 °C. The overnight culture was used to inoculate 1 L, with 25 μg/mL kanamycin, 20 μg/mL chloramphenicol, and 0.1% dextrose. The cells were grown at 37 °C to an OD600 of 0.8 to 1.0 before IPTG was added to a final concentration of 0.5 mM. The cells were grown for an additional 4 h at 20 °C and then harvested by centrifugation at 4 °C. Recombinant NBD of HSP90α and HSP90β was purified through the Ni-NTA-agarose column (GE Healthcare, Piscataway, NJ, USA) and purified by using the AKTA pure M1 protein purification system (GE Healthcare, Chicago, IL, USA). The fractions containing recombinant protein were pooled and dialyzed against 50 mM HEPES, pH 7.5 and 1 mM DTT overnight. Glycerol was added to a final concentration of 20% after dialysis. The sample was aliquoted and stored at −80 °C until further use.
DARTS assay
DARTS assay was performed based on previous reports [33]. Briefly, AD293 cells transfected with WT HSP90α or HSP90β were treated with either Celastrol, 17-AAG, or geldanamycin for 4 h. Cells were lysed in lysis buffer containing 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 2 mM EDTA, 1 mM ATP, and 0.5% Igepal CA-630 in the presence of complete protease inhibitor cocktail. Cleared lysate after centrifugation was digested with pronase, and analyzed by using immunoblotting.
Isothermal titration calorimetry (ITC) assay
ITC measurements were carried out on MicroCal-ITC microcalorimeter (Malvern Panalytical, Malvern, UK). Recombinant proteins of WT HSP90α and HSP90β were dialyzed in PBS containing 10% glycerol. Celastrol was dissolved in DMSO. All measurements were performed in dialysis buffer containing 5% DMSO at 25 °C. The ITC experiment was done with 20 µM protein in the cell and 400 µM Celastrol in the syringe. The thermogram was analyzed by using GraphPad Prism 9.3 software.
ATPase activity of HSP90
ATPase activity of HSP90 was determined by using the method reported previously [34]. Recombinant HSP90 was incubated with Celastrol or 17-AAG in the reaction buffer (33 mM HEPES, pH 7.4, 30 mM NaCl, 5 mM MgCl2, 1 µM ATP, 1 mM DTT) and incubated at 37 °C for 1 h. The quantity of ATP was measured by using the CellTiter-Glo Assay kit with luminescence spectrometry.
Measurement of ER stress, acidification of lysosome, and mitochondrial transmembrane potential
Huh-luc/neo-EF cells were labeled with ER-Tracker Red, LysoTracker Red, and MitoTracker red for 30 min at 37 °C before Celastrol treatment. The fluorescent intensities of ER-Tracker, Lyso-Tracker red, and mitochondrial potential were measured by using FACS analysis.
Analysis of anti-HCV activity
The anti-HCV activity was determined based on the previously reported method [35]. The EC50 was detected from the dose-effect curve by nonlinear regression analysis using Graphpad (version 9.0).
Analysis of cytotoxicity
Cell viability was determined by using Methylene blue methods following the previous report [36].
Drug combination study
Huh-luc/neo-ET cells were seeded at a density of 5 × 103 cells per well in a 96-well plate. On the following day, a mixture of Celastrol with anti-HCV drugs was applied in a serial dilution and hence kept at a constant ratio in duplicates. Seventy-two hours after incubation with different drug combinations, cells were harvested for luciferase activity assay and the median effect equation was used for dose-effect analysis. Synergistic, antagonistic, or additive effects of drug combination were determined by using an isobolar graph as described earlier [35].
RNA isolation and real-time PCR
Total RNA was extracted by Trizol. Five hundred ng of RNA was reverse transcribed into complementary DNA using random primer with Superscript III Reverse Transcriptase (Invitrogen). Real-time quantitative PCR was performed in triplicates with SYBR supermix (BioRad). Expression levels of each gene were normalized to that of 18 S rRNA, and analyzed following the MIQE guidelines [37]. The primers used are listed in Supplementary Table S3.
Polysome profile
Polysome profile analysis was performed as described previously [38]. In brief, cytoplasmic extracts from control or drug-treated cells were overlayed on the 10%–30% OptiPrepTM gradients and centrifuged at 39,000 rpm for 80 min at 4 °C using the SW 41Ti Beckman Coulter rotor (Beckman Coulter, Brea, CA, USA). Fractions were collected with ISCO continuous autodensi-flow density gradient fractionator (Lincoln, NE). Total RNA from each fraction was isolated by TriRIzol. The amount of specific mRNAs in each fraction was analyzed by real-time PCR.
Western blot analysis
The cell pellet was extracted in RIPA lysis buffer. The protein concentration was determined by using BCA Protein Assay kit (Thermo Fisher Scientific). Total protein was separated with SDS-PAGE gel and then transferred to PVDF membrane. The following antibodies were used: anti-HSF1 (#cst-9144, Cell Signaling Technology, Danvers, MA, USA), anti-HSC70 (#sc-7298, Santa Cruz, Dallas, TX, USA; #SPA-815, Stressgen, San Diego, CA, USA), anti-HSP90 (#cst-4874, Cell Signaling Technology), anti-HSP90α (#ab79849, Abcam, Cambridge, UK), anti-HSP90β (#ab53497, Abcam), anti-HA (#cst-3724, Cell Signaling Technology), anti-β-actin (#A5316, Sigma Aldrich), and anti-GAPDH (#sc-25778, Santa Cruz). All antibodies were used at optimized dilution for Western blot analysis.
Immunoprecipitation (IP)
Briefly, Huh-luc/neo-EF cells were transiently transfected with HA-HSP90β for 48 h and then treated with 200 nM Celastrol. The cells with different treatments were lysed by freeze and thaw in NT-2 buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM MgCl2, 0.05% NP40 containing proteinase inhibitor cocktail). The protein lysate was cleared by centrifugation, and an equal amount of protein was subjected to IP with anti-HA magnetic beads at 4 °C overnight. The IP complex was then washed with NT-2 buffer, and subjected to Western blot analysis.
Construction of bicistronic reporters containing different IRES
The pLuc-HCV 1b IRES-EGFP plasmid was constructed by first amplifying the firefly luciferase gene from pGL4.2 vector (Promega), and HCV genotype 1b IRES from pFK-I389/N3-3’/LucUbiNeo-ET plasmid (a generous gift from Prof. Ralf Bartenschlager at the University of Heidelberg) using primers shown in Supplementary Table S2. The luciferase and HCV 1b IRES fragments were ligated with the pIRES2-EGFP vector (Clontech Laboratories, Mountain View, CA, USA) using the Gibson Assembly cloning kit (New England Biolabs, Ipswich, MA, USA). pLuc-HCV 2a IRES-EGFP and pLuc-MSCV IRES-EGFP plasmid were constructed by replacing HCV 1b IRES sequence with HCV 2a IRES or MSCV IRES sequence amplified from JFH plasmid or MSCV plasmid (Addgene # 52107) [39], respectively. The WT CrPV-IRES was synthesized [40] and cloned in the pLuc-HCV 1b IRES-EGFP vector between firefly luciferase and EGFP sequences.
Determination of the efficiency of IRES-dependent translation
The bicistronic vectors were transfected into AD293 cells alone or together with WT, A47D, and A47S mutant HSP90β, WT HSP90α. Forty-eight hours after transfection, cells were treated with Celastrol, Cel-2, Cel-6, 17-AAG, and CHX as indicated in Fig. 5. The fluorescent intensity of EGFP was determined by using BD AccuriTM C6 Plus flow cytometer. Cells were then lysed in luciferase lysis buffer, and firefly luciferase activity was determined by using a luciferase assay system (Promega, Madison, WI, USA).
Fig. 5. Treatment with celastrol inhibited the translation of HCV.

a Expression and phosphorylation levels of key enzymes regulating protein translation with the treatment of celastrol, C (CHX 100 μg/mL), LY (LY294002, 1 nM), Ra (Rapamycin, 200 nM), Cel-2, or Cel-6 were examined by using Western blot analysis. b Phosphorylation of Akt and expression levels of transcription enzymes with the treatment of celastrol, C (CHX 100 μg/mL), LY (LY294002, 1 nM), Ra (Rapamycin, 200 nM), or Cel-6 were examined by using Western blot analysis. c Polysome profiling of celastrol- or CHX-treated Huh-luc/neo-ET cells. d Partition of HCV NS3, NS5A, and NS5B, HSP90β, Alix, calnexin, and RPS15 from the fractions obtained in (c) was analyzed by using Western blot. e The relative HCV and GAPDH RNA content in the fractions obtained in (c) was analyzed by using real-time PCR. f Huh-luc/neo-EF cells were transiently transfected with HA-HSP90β for 48 h, and then treated with 200 nM celastrol for different period. Proteins associated with HA-HSP90β were IP with HA magnetic beads and resolved by Western blotting. Results are representative of three independent experiments.
Immunohistochemistry analysis
Formalin-fixed, paraffin-embedded liver tissues were dewaxed with xylene, and gradually hydrated. Antigen retrieval was achieved by 10 mM Sodium citrate pH 6.0 with 0.02% Tween-20 under steaming for 30 min. The primary antibodies to CD68 (#ab31630, Abcam, Cambridge, UK), HSP90α (#ab79849, Abcam), HSP90β (#AB53497, Abcam), and NLRP3 (#AG-20B-0014-C100, Adipogen, San Diego, CA, USA) were diluted using Tris-HCl buffer containing 1% BSA, 2% goat serum, 1% gelatin, and 0.5% Tween-20, and were incubated at 4 °C overnight. After incubation with a secondary antibody at room temperature for 1 h, the signal was detected by DAB peroxidase substrate kit (Vector Laboratories, Burlingame, CA, USA). As a negative control, a set of slides was processed without a primary antibody. The slides were counterstained with hematoxylin and mounted.
Hematoxylin & Eosin (H&E) and Masson’s trichrome staining kits were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China), and performed following the manufacturer’s recommendation.
Serum cytokine and chemokine level
Serum cytokine and chemokine levels were analyzed by using LEGENDplexTM Mouse Inflammation Panel (#740150, BioLegend) following the protocol provided by the manufacturer. Changes in the fluorescent intensity were measured by using BD AccuriTM C6 flow cytometer.
Serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) level
Serum AST and ALT levels were measured by using assay kits from Nanjing Jiancheng Bioengineering Institute following the recommendation from the manufacturer.
Statistical analysis
All statistical analysis was performed according to the guideline on experimental design and analysis in pharmacology [41]. No animal was excluded from the analysis. All results were presented as mean ± SD or mean ± SEM. Differences between groups were analyzed by using one-way or two-way analysis of variance (ANOVA) or Student’s t-test using GraphPad 9.0. P value less than 0.05 was considered statistically significant. All Western blot analysis with representative images was repeated at least three times.
Results
HSP90β expression correlated with hepatic inflammation level in HCV positive HCC patients
HCV infection and hepatic inflammation levels are major carcinogenesis factors that trigger HCC. The expression level of HSP90β significantly correlated with that of NLRP3 in the liver tissues of HCC patients with HCV infection and normal individuals, but not in the liver tissues of HBV-infected HCC or cirrhosis patients (Fig. 1; Table 1). On the contrary, the expression level of HSP90α did not show a similar correlation with that of NLRP3 in these four types of patients (Fig. 1; Table 1). The expression level of NLRP3 were highly elevated in HCC patients with HCV infection, but not in the normal individuals or HBV-infected HCC and cirrhosis patients (Fig. 1; Table 1).
Table 1.
Correlation of the expression levels of NLRP3 with HSP90α and HSP90β in the liver tissue from normal individuals, and patients with HCV-HCC, HBV-HCC, and cirrhosis.
| Normal tissue | HCC with HCV infection | HCC with HBV infection | Cirrhosis with HBV infection | ||
|---|---|---|---|---|---|
| n | 20 | 16 | 20 | 20 | |
| NLRP3 & HSP90α | R2 | 0.1955 | 0.02059 | 0.02841 | 0.1111 |
| P value | 0.0509 | 0.5960 | 0.4775 | 0.1510 | |
| NLRP3 & HSP90β | R2 | 0.3728 | 0.3867 | 0.02139 | 0.1111 |
| P value | 0.0042 | 0.0101 | 0.5384 | 0.1510 |
The R2 and P values were calculated by using Graphpad software 9.0.
Celastrol interacted with HSP90 without inducing heat shock response
Different isoforms of HSP90 share more than 70% similarity in sequence. We predicted the binding between celastrol and HSP90 by using the ligand-based virtual screening with publicly accessible databases. The initial analysis revealed that Alanine (Ala) 47 residue of HSP90β was critical for the differential binding mode between celastrol to HSP90α or HSP90β (Fig. 2a). Results of the drug affinity responsive target stability (DARTS) assay revealed that celastrol protected pronase-mediated degradation of HSP90α and HSP90β in a similar potency as pan HSP90 inhibitor geldanamycin (Geld), but was higher than that of 17-AAG (Fig. 2b). Isothermal titration calorimetry (ITC) results confirmed that celastrol bound to HSP90α or HSP90β in similar affinity (Fig. 2c, d; Table 2). Additionally, celastrol inhibited ATPase activity of both HSP90α and HSP90β in a dose-dependent manner (Fig. 2e).
Fig. 2. Celastrol interacted with Hsp90β by binding to the NBD ATPase pocket.

a Complex binding model of celastrol (shown in ball-and-stick mode) in the NBD of Hsp90β. Key residues were shown in stick mode and labeled in green. The corresponding residues in NBD of Hsp90α were labeled in orange (PDB code: 3T0Z). b Huh-luc/neo-ET cells were treated with celastrol (Cel), 17-AAG, or Geldanamycin (Geld) for 2 h. The cell pellet was lysed in the presence of these compounds, and then treated with pronase (75 ng/µg of total protein) at RT. The levels of HSP90α and HSP90β were determined by using Western blot analysis. ITC results of the binding between celastrol with (c) HSP90α or (d) HSP90β. e The ATPase activity of HSP90. (f) Heat map generated from real-time PCR data showing mRNA level of heat shock response genes in Huh-luc/neo-ET cells treated with celastrol, Cel-2, and Cel-6 for up to 72 h. g The expression level of HSP90α, HSP90β, HSC70, and HOP with the treatment of 200 nM celastrol, Cel-2, and Cel-6 for up to 72 h. The blot of β-Actin was used as the internal loading control. The results are representative of three separate experiments. (**P < 0.05).
Table 2.
Thermodynamic parameters obtained from the ITC measurements in Fig. 2c, d.
| Protein | HSP90α | HSP90β |
|---|---|---|
| N (sites) | 2.19 ± 0.111 | 1.34 ± 0.087 |
| KD (µM) | 3.31 ± 0.876 | 2.13 ± 0.934 |
| ΔH (kCal/mol) | −36.4 ± 3.26 | −80 ± 9.78 |
| ΔG (kCal/mol) | −7.48 | −7.74 |
| -TΔS (kCal/mol) | 28.9 | 72.3 |
We next determined heat shock response in Huh-luc/neo-ET cells treated with Celastrol. Celastrol did not induce significant changes in mRNA levels of HSC70, HSP90α, HSP90β, HOP, transcription factors HSF1, HSF2, HSF4, and activator of heat shock 90 kDa protein ATPase homolog (AHSA1 and AHSA2) for up to 72 h (Fig. 2f). Neither did celastrol treatment change the expression of HSP90α, HSP90β, and HSC70, but decreased the expression of HOP within 36 h (Fig. 2g). The changes of HSPs and HSFs in Huh-luc/neo-ET cells treated with structural analogs of celastrol, Cel-2 or Cel-6, were similar to those treated with celastrol for 72 h (Fig. 2g).
Celastrol exhibited potent anti-HCV activity
We next explored the anti-HCV activity of celastrol and its structural analogs, Cel-2, Cel-6. Celastrol emerged as a nanomolar inhibitor of HCV replicons in vitro, with relatively high selectivity for HCV against cytotoxicity (Fig. 3a). Celastrol suppressed the expression levels of HCV RNA, nonstructural (NS) proteins NS3 and NS5B in a time-dependent manner; while Cel-2 and Cel-6 failed to show similar potency against HCV nor similar selectivity (Fig. 3b–e). The combined effect of Celastrol with currently used DAAs, Sofosbuvir, Telaprevir, Simeprevir, and Declastasvir was additive (Fig. 3f–i).
Fig. 3. Celastrol exhibited a potent inhibitory effect against HCV in Huh-luc/neo-ET genotype 1b replicon cells.

a The anti-HCV activity and cytotoxicity of celastrol. b The relative expression level of HCV RNA with the treatment of celastrol, Cel-2, and Cel-6 for up to 72 h. c The expression levels of HCV NS proteins with the treatment of celastrol, Cel-2, and Cel-6 for up to 72 h. The anti-HCV activity and cytotoxicity of (d) Cel-2 and (e) Cel-6. Huh-luc/neo-ET cells were co-incubated with various concentrations of celastrol alone or with (f) Sofosbuvir, (g) Telaprevir, (h) Simeprevir, and (I) Daclatasvir at different potency ratios for 72 h. Ratios of the apparent EC50 of each drug in combination with its EC50 when applied alone were plotted against each other in isobolograms. The hypotenuse represented the linear additive response to the action of two drugs. Isoboles that bow below the hypotenuse indicate synergism, and isoboles that bow above the hypotenuse indicate antagonism. Experimental data points on the isobole represent a combination that inhibits the HCV replication by 50% and is hence isoeffective with the line of additivity. Results are representative of at least three independent experiments and presented as mean ± SD (**P < 0.05).
The Ala47 residue of HSP90β was critical to the anti-HCV activity of celastrol
Treatment with celastrol did not change the expression level of HSP90β in Huh-luc/neo-EF cells (Fig. 2g). The activity of celastrol to suppress expression of HCV RNA and NS3 was abrogated by overexpression of WT HSP90β (Fig. 4a–d) but not HSP90α (Fig. 4e, f). Mutation of Ala47 on HSP90β to Asp (A47D) or Ser (A47S) abrogated the anti-HCV activity of celastrol (Fig. 4c, d).
Fig. 4. The anti-HCV activity of celastrol is dependent on the ATPase activity of HSP90β but not HSP90α.

Huh-luc/neo-ET cells were transiently transfected with HA-HSP90β expression plasmid and treated with celastrol, Cel-2, or Cel-6 for 48 h. The expression levels of (a) HCV RNA and (b) HCV NS proteins were determined by using real-time PCR and Western blot, respectively. Huh-luc/neo-ET cells were transiently transfected with wild-type (WT), A47D, or A47S mutant HA-HSP90β plasmids, and then treated with celastrol for 48 h. The expression levels of (c) HCV RNA and (d) HCV NS3 protein were determined by using real-time PCR and Western blot analysis, respectively. Huh-luc/neo-ET cells were transiently transfected with HA-HSP90α expression plasmid, and treated with different doses of celastrol for 48 h. The expression levels of (e) HCV RNA and (f) HCV NS3 protein were analyzed as in (a) and (b), respectively. Results are representative of three independent experiments. The HCV RNA level and relative band intensity of NS3 protein are presented as mean ± SD (**P < 0.05 compared with untreated control cells or between each other).
Celastrol inhibited HCV RNA translation
Celastrol initially suppressed phosphorylation of eIF2α, eIF4e, and 4EBP1 after 2 to 6 h treatment in Huh-luc/neo-ET cells (Fig. 5a). Phosphorylation of S6 ribosomal protein peaked at 2 h with celastrol treatment and then tapered off (Fig. 5a). The phosphorylation of p70-S6 kinase, Akt, transcription factors IIH subunit 1 L(TF1L) and NELF-E remained unchanged (Fig. 5a, b). Cycloheximide (CHX) only suppressed phosphorylation of 4EBP1, but induced phosphorylation of other proteins examined (Fig. 5a, b). LY294002 and Rapamycin significantly reduced phosphorylation of 4EBP1, S6 ribosomal protein, and p70-S6 kinase (Fig. 5a). Rapamycin elevated the expression level of phosphorylated Akt (Fig. 5b). Cel-2 and Cel-6 induced similar changes as celastrol did under the same conditions (Fig. 5a, b).
We further analyzed the sedimentation shift of HCV proteins and RNA on polysome profiling. Post-nuclear fractions from control, celastrol-, and CHX-treated Huh-luc/neo-ET cells were fractionated on continuous optiprep iodixanol density gradients (Fig. 5c–e). Treatment with celastrol and CHX induced accumulation of the polysome fraction (Fig. 5c). The HCV NS3, NS5A, and NS5B proteins were colocalized with HSP90β, Alix, and Rps15 in the light fractions with celastrol treatment (Fig. 5d). The sedimentation of NS3, NS5A, and NS5B were accumulated in the fractions containing calnexin in the CHX-treated cells (Fig. 5d). Celastrol treatment blocked HCV (grey area) and GAPDH (dot line) RNAs in the monosome but not the polysome fractions (Fig. 5e). CHX treatment increased the distribution of HCV RNA in the polysome fractions (Fig. 5e). The amount of eIF2α bound to HA-HSP90β was decreased by celastrol after 24 h treatment (Fig. 5f). The binding between eIF4E and HSP90β remained unchanged (Fig. 5f). The amount of 4EBP1 bound to HA-HSP90β was increased after 4 h treatment with celastrol, and then diminished after 12 h (Fig. 5f).
Celastrol specifically inhibited HCV internal ribosomal entry site (IRES)-initiated translation
The initiation step of HCV RNA translation was driven by IRES on the 5’ untranslated region (UTR), which is distinct in different types of viruses. We constructed a bicistronic vector containing IRES sequence of HCV genotype 1b strain between the open reading frames of firefly luciferase and GFP (HCV genotype 1b IRES-EGFP, Fig. 6a). Celastrol and 17-AAG inhibited HCV genotype 1b IRES-dependent translation; while Cel-2, Cel-6, or CHX did not (Fig. 6b). Celastrol also markedly attenuated translation driven by HCV genotype 2a and MSCV IRES (Fig. 6c, d), but did not impact on translation driven by CrPV IRES (Fig. 6e). The inhibition of celastrol on HCV genotype 1b IRES-initiated translation was abrogated by HSP90β A47S and A47D mutants, but not by WT HSP90α or HSP90β (Fig. 6f).
Fig. 6. Role of HSP90β in celastrol-inhibited IRES-dependent translation in HCV genotype 1b cells.
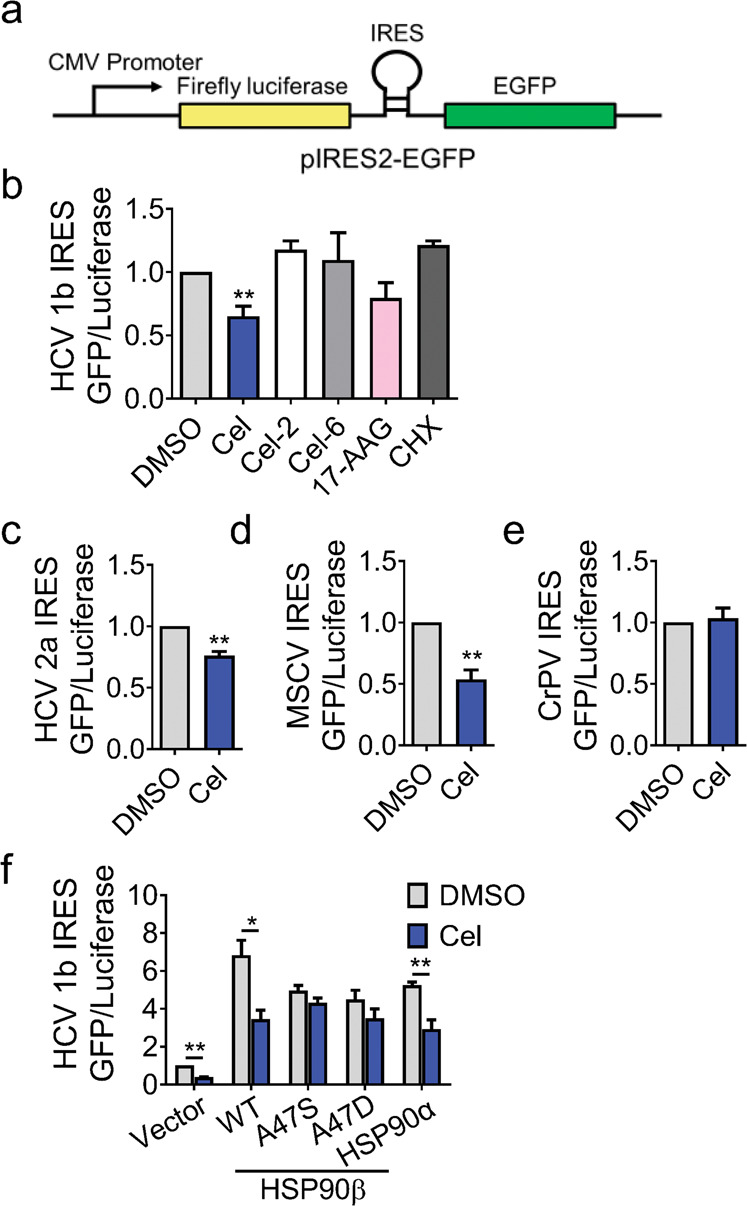
a Diagram of the luciferase pIRES2-EGFP vector. b Impact of celastrol, Cel-2, Cel-6, 17-AAG, and CHX on the translation of HCV genotype 1b IRES-dependent reporters in AD293 stable cell line. Impact of celastrol treatment on (c) HCV genotype 2a, (d) MSCV, and (e) CrPV IRES-dependent translation in AD293 cells harboring the pIRES2-EGFP plasmid was analyzed by the ratio of firefly luciferase and EGFP. f Huh-luc/neo-ET cells were transiently transfected with HA-HSP90β for 48 h and then treated with celastrol as indicated. The HA-HSP90β-associated proteins were pulled down with HA magnetic beads and analyzed by using Western blot analysis. Results are representative of three independent experiments. Data in (b–f) were presented as mean ± SD (**P < 0.05 compared with each other or to DMSO treatment).
Celastrol attenuated HCV RdRp-triggered inflammatory response in vivo depending on HSP90β
HCV translation was dependent on RdRp-triggered RNA replication [42]. HCV RdRp triggered innate immune signaling and inflammatory response is the primary cause of liver injury [43]. Therefore, we triggered hepatic inflammation by using adenovirus expressing HCV NS5B (pAde-NS5B) and used pAde-EGFP as a control. C57BL/6 J mice were pretreated with 0.2 mg/kg or 0.5 mg/kg celastrol through intraperitoneal injection 2 d before intravenous injection of pAde-NS5B or pAde-NS5B. Administration of pAde-NS5B markedly increased immune cell infiltration, hepatic expression of CD68, Nlrp3, and Hsp90β, and serum alanine transaminase (ALT) and aspartate transaminase (AST) levels compared to pAde-EGFP injection (Fig. 7a–d). Results of the Masson’s trichrome staining revealed that no scar tissue formation was detected (Fig. 7a). The short-term in vivo expression of pAde-NS5B significantly increased the serum levels of Ifn-γ, Mcp-1, and Il-1β (Fig. 7e–g). Administration of pAde-NS5B highly elevated the hepatic expression of genes promoting liver fibrogenesis, including Itga1, Gdf15, and Col1a1 (Fig. 7h–j). Celastrol treatment potently decreased these markers of hepatic inflammatory response in a dose-dependent manner (Fig. 7a–j).
Fig. 7. Celastrol suppressed HCV NS5B induced inflammatory response in vivo depending on HSP90β.

C57BL/6 J mice were injected with pAde-EGFP or pAde-NS5B at 1 × 109 p.f.u./mouse through the tail vein (i.v.). Celastrol was intraperitoneally (i.p.) injected 48 h after pAde injection at the dose indicated on the figure twice a day continuously for 5 d. a Mice liver sections were stained with H&E and Masson’s trichrome staining. b Mice liver sections with stained with antibodies against CD68, Nlrp3, Hsp90α, and Hsp90β. Serum levels of (c) AST, (d) ALT, (e) Ifn-γ, (f) Mcp-1, and (g) Il-1β. mRNA expression levels of (h) Itga1, (i) Gdf15, and (j) Col1a1 in the liver. C57BL/6 J mice injected with pAde-NS5B alone or together with pAde-HSP90β A47S were treated with celastrol as in (a). Serum levels of (k) Ifn-γ, (l) Mcp-1, and (m) Il-1β. The results are representative of three independent experiments, data in (c–m) are presented as mean ± SEM (*P < 0.1; **P < 0.05; ***P < 0.01; n.s. not significant). n ≥ 5 mice in each group. Scale bar = 100 µm.
Co-injection of pAde-HSP90β A47S with pAde-NS5B abrogated increased serum levels of Ifn-γ and Mcp-1, but not Il-1β compared to pAde-NS5B alone (Fig. 7k–m). The co-injection of pAde-HSP90β A47S also perturbed the serum levels of Ifn-γ, Mcp-1, and Il-1β in mice that received Celastrol treatment (Fig. 7k–m).
Discussion
Celastrol was previously reported to inhibit HSP90 chaperone complex through decreasing its mRNA expression [44, 45]. Recent studies revealed that the molecular target of Celastrol varies in different models. For example, celastrol sensitized the anti-obesity activity of leptin without affecting the ATPase activity of HSP90 [17]. Narayan et al. synthesized Celastrol-biotinamide (bCel) by conjugating the biotin moiety to the carboxyl group on the C29 position of celastrol, and identified that bCel physically bound to HIV Tat protein [46]. Luo et al. also conjugated the biotin moiety on the same position and discovered that celastrol interacted with the ER protein GRP78 [47]. The binding between celastrol and GRP78 reduced the chaperone activity of GRP78 and was essential for its anti-inflammation and anti-obesity activities. The study from Yang’s group revealed that celastrol inhibited the chaperone activity of peroxiredoxin I in the same fashion as natural products triptolide and withaferin A [48]. Yet they did not provide detailed biological activities associated with the binding. Based on the distinct anti-HCV activity of celastrol and its analogs Cel-2 and Cel-6 and the affinity evaluation to HSP90 (Figs. 2–4), our study demonstrated that the carboxyl group of celastrol was critical for its anti-HCV activity and the binding with Ala47 residue of HSP90β. This binding model of celastrol to HSP90β was not reported previously.
Celastrol suppressed the replication and inflammatory response of several different viruses. Treatment with celastrol inhibited HCV replication and protected the degradation of VE-cadherin in dengue virus-infected EA.hy926 cells by upregulating heme oxygenase-1 (HO-1) expression [49, 50]. The immunosuppressive activity of celastrol in HIV-1 Tat infected astrocytes also depends on the expression level of HO-1 [51]. Celastrol treatment decreased the expression levels of proinflammatory cytokines TNF-α and IL-6 in influenza A (H1N1) infected Madin-Barby Canine kidney cells [52]. Celastrol also suppressed HCV replication through stimulating the transcriptional factor activity of HO-1 [49]. However, no mechanistic insights have been reported.
We demonstrated that celastrol bound to both HSP90α and HSP90β with similar affinity (Fig. 2). But the anti-HCV and immunosuppressive activities of celastrol were dependent on the ATPase activity of HSP90β (Figs. 4, 6, 7). Mutagenesis of the Ala47 to Ser or Asp markedly abrogated the inhibitory activity of celastrol on the expression of HCV RNA and NS proteins (Fig. 4) or HCV RdRp-triggered inflammatory cytokines in vivo (Fig. 7).
Translation is of pivotal importance for HCV replication, which is executed by the translational complex containing HCV NS proteins and various host factors [25]. HCV infection triggered phosphorylation of eIF2α to inhibit global protein translation of host proteins at the initiation step. The subsequent binding of the eIF4e and eIF4f to the complex was the rate-limiting step in translation initiation [53]; while phosphorylation of 4EBP1 was critical for the assembly of the HCV translation complex [54].
HSP90 was involved in multi-steps of the HCV life cycle. HSP90 participated in HCV replication through binding to the FKBP8-NS5A complex [9]. Treatment with HSP90 inhibitor 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (17-DMAG) suppressed phosphorylation of NS5B through destabilizing phosphoinositide-dependent kinase-1 [55]. We demonstrated that HSP90 was a cellular factor required for HCV translation. Celastrol interrupted the initiation of translation by diminishing the association of 4EBP1 to the translational complex (Fig. 5). This process was also dependent on the sequence and structure of different viral IRES, and the ATPase activity of HSP90β (Fig. 6). Celastrol treatment did not reduce cell viability, alter cell cycle distribution, nor induce heat shock response at the dose efficient to inhibit HCV replication (Figs. 2, 3). Celastrol did not change phosphorylation of p70S6 kinase or Akt as Rapamycin did (Fig. 5a, b), indicating that celastrol had minimal impact on cap-dependent initiation of translation commonly utilized by host proteins.
The complete cure of all-oral DAAs was markedly decreased in HCV patients with HCC, especially since DAAs treatment failed in HCV patients who bared active HCC tumor at the initial stage of the therapy [56]. Reversely, the progression of HCC tumors was more aggressive in HCV patients who received DAA treatment, which in turn affected the SVR in this group of patients [57]. Chronic HCV infection promoted the sustained progression of liver inflammation and consequently liver fibrosis and HCC [43, 58]. Especially the intrahepatic IL-1β produced through activation of the NLRP3 inflammasome in Kupffer cells, the liver resident macrophages, was considered as one of the key features of liver inflammation [58, 59].
The clinical application of HSP90 inhibitors for the treatment of HCC is under intensive investigation [60]. HSP90 inhibitors also demonstrated in vivo potency in treating hepatic inflammation. Natural products Echinatin and Carnosol effectively inactivated NLRP3 inflammasome through inhibition of the ATPase activity of HSP90, and exhibited therapeutic potential in NLRP3-driven nonalcoholic steatohepatitis [34, 61]. HSP90β directly regulated the turnover rate of hepatocyte nuclear factor 4α [62], the transcription factor that governed the proliferation and development of liver cancer [63].
Our study revealed that celastrol significantly reduced infiltration of immune cells and hepatic expression level of Nlrp3 in mice injected with pAde-NS5B (Fig. 7a, b). Celastrol treatment markedly attenuated serum levels of proinflammatory cytokines triggered by HCV RdRp in vivo, including Ifn-γ, Mcp-1, and Il-1β (Fig. 7e–g). The HCV NS5B-induced liver inflammation was ameliorated with celastrol treatment, including the elevated levels of serum ALT and AST (Fig. 7c, d), and mRNA levels of markers for liver fibrosis and HCV-driven HCC, including Itga1, Gdf15, and Col1a1 (Fig. 7h–j) [64]. Treatment with celastrol also alleviated inflammatory response triggered by autoimmune hepatitis [65], while the inhibitory activity of celastrol to HCV RdRp-triggered elevated serum level of Ifn-γ, Mcp-1, and Il-1β was lost when mice expressed HSP90βA47S (Fig. 7k–m). The expression levels of NLRP3 highly correlated with that of HSP90β but not HSP90α in HCC patients with HCV infection (Fig. 1; Table 1), as well as in HCV RdRp-expressed mice (Fig. 7b). These results suggest that HSP90β is also directly related to the anti-inflammation activity of celastrol, as inhibition of the ATPase activity of HSP90 lead to the dissociation of NLRP3 to HSP90 and cochaperon protein, which subsequently lead to degradation of NLRP3 [34]. The anti-HCV activity of celastrol was additive to other DAAs (Fig. 3f–i). Therefore, celastrol has great potential to be developed as a treatment agent for HCV alone or in combination with DAAs in positive HCC patients with high expression of HSP90β.
The limitation of the study is that the efficacy of celastrol could be further confirmed in humanized mice infected with HCV. Another limitation is that overexpression of WT HSP90β failed to up-regulate NS3 (Fig. 4a, c), which is in line with previous reports that exogenous expression of HSP90 did not impact on the HCV NS protein [9, 66]. This was due to the nature that the HCV replicon systems in the culture were already at the maximal capacity of HCV replication [25]. The detailed binding mode between HSP90β but not HSP90α to the HCV translational machinery, and the effect on HCV RNA and NS protein expression could be further analyzed by using real virus infected systems.
Conclusion
In this study, we discovered celastrol as a potentially novel and potent HSP90 inhibitor, which markedly suppressed IRES-dependent translation of HCV genotype 1b RNA and HCV RdRp-triggered inflammatory response in vivo. The inhibitory activity of celastrol on HCV replication is dependent on the Ala47 residue of HSP90β but not on HSP90α. The significant correlation between the expression levels of HSP90β and NLRP3 also suggests that celastrol could be developed as a therapeutic agent for HCV positive HCC patients, especially those with high expression of HSP90β.
Supplementary information
Acknowledgements
The authors thank Mr. Rui-feng Li and Dr. Zi-feng Yang from The First Affiliated Hospital of Guangzhou Medical University for anti-influenza activity, Dr. Martin Holcik from the University of Ottawa, Dr. Tyson Graber from McGill University, and Dr. Yan-dong Zhang from South University of Science and Technology of China for discussion of polysome profiling. This work was partially supported by Macao Science and Technology Development Fund (grant number 0092/2021/A2), Research Fund of the University of Macau (grant number MYRG 2019-00015-ICMS), and Shenzhen-Hong Kong-Macao Science and Technology Innovation Project (Category C) (grant number SGDX2020110309260100). The authors also appreciate the support from Animal Research Core at the University of Macau.
Author contributions
SRC and ZQL conducted the major experiments. JX, MYD, YMS, YWS, and GXZ took part and assisted in some experiments. YCC performed part of the data analysis. YQW and YW designed the experiments and performed data analysis. YW summarized all the results and gave indispensable guidance to the entire study, wrote and revised the manuscript. All authors read and approved the submitted manuscript.
Competing interests
The authors declare no competing interests.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-023-01067-w.
References
- 1.Carrat F, Fontaine H, Dorival C, Simony M, Diallo A, Hezode C, et al. Clinical outcomes in patients with chronic hepatitis C after direct-acting antiviral treatment: a prospective cohort study. Lancet. 2019;393:1453–64. doi: 10.1016/S0140-6736(18)32111-1. [DOI] [PubMed] [Google Scholar]
- 2.Arzumanyan A, Reis HM, Feitelson MA. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat Rev Cancer. 2013;13:123–35. doi: 10.1038/nrc3449. [DOI] [PubMed] [Google Scholar]
- 3.Opar A. Excitement grows for potential revolution in hepatitis C virus treatment. Nat Rev Drug Discov. 2010;9:501–3. doi: 10.1038/nrd3214. [DOI] [PubMed] [Google Scholar]
- 4.Feld JJ, Jacobson IM, Hezode C, Asselah T, Ruane PJ, Gruener N, et al. Sofosbuvir and velpatasvir for HCV genotype 1, 2, 4, 5, and 6 infection. N Engl J Med. 2015;373:2599–607. doi: 10.1056/NEJMoa1512610. [DOI] [PubMed] [Google Scholar]
- 5.Foster GR, Afdhal N, Roberts SK, Brau N, Gane EJ, Pianko S, et al. Sofosbuvir and velpatasvir for HCV genotype 2 and 3 infection. N Engl J Med. 2015;373:2608–17. doi: 10.1056/NEJMoa1512612. [DOI] [PubMed] [Google Scholar]
- 6.Felmlee DJ, Coilly A, Chung RT, Samuel D, Baumert TF. New perspectives for preventing hepatitis C virus liver graft infection. Lancet Infect Dis. 2016;16:735–45. doi: 10.1016/S1473-3099(16)00120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reig M, Marino Z, Perello C, Inarrairaegui M, Ribeiro A, Lens S, et al. Unexpected high rate of early tumor recurrence in patients with HCV-related HCC undergoing interferon-free therapy. J Hepatol. 2016;65:719–26. doi: 10.1016/j.jhep.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 8.Tahata Y, Hikita H, Mochida S, Enomoto N, Kawada N, Kurosaki M, et al. Liver-related events after direct-acting antiviral therapy in patients with hepatitis C virus-associated cirrhosis. J Gastroenterol. 2022;57:120–32. doi: 10.1007/s00535-021-01845-5. [DOI] [PubMed] [Google Scholar]
- 9.Okamoto T, Nishimura Y, Ichimura T, Suzuki K, Miyamura T, Suzuki T, et al. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 2006;25:5015–25. doi: 10.1038/sj.emboj.7601367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bukong TN, Hou W, Kodys K, Szabo G. Ethanol facilitates hepatitis C virus replication via up-regulation of GW182 and heat shock protein 90 in human hepatoma cells. Hepatology. 2013;57:70–80. doi: 10.1002/hep.26010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bukong TN, Momen-Heravi F, Kodys K, Bala S, Szabo G. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog. 2014;10:e1004424. doi: 10.1371/journal.ppat.1004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu M, Zhao C, Zhu B, Wang L, Zhou H, Yan D, et al. Discovering high potent Hsp90 inhibitors as antinasopharyngeal carcinoma agents through fragment assembling approach. J Med Chem. 2021;64:2010–23. doi: 10.1021/acs.jmedchem.0c01521. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Wang L, You QD, Xu XL. Heat shock protein 90 inhibitors: an update on achievements, challenges, and future directions. J Med Chem. 2020;63:1798–822. doi: 10.1021/acs.jmedchem.9b00940. [DOI] [PubMed] [Google Scholar]
- 14.Li M, Mulkey F, Jiang C, O’Neil BH, Schneider BP, Shen F, et al. Identification of a genomic region between SLC29A1 and HSP90AB1 associated with risk of bevacizumab-induced hypertension: CALGB 80405 (Alliance) Clin Cancer Res. 2018;24:4734–44. doi: 10.1158/1078-0432.CCR-17-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munch C, Harper JW. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature. 2016;534:710–3. doi: 10.1038/nature18302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corson TW, Crews CM. Molecular understanding and modern application of traditional medicines: triumphs and trials. Cell. 2007;130:769–74. doi: 10.1016/j.cell.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu J, Lee J, Salazar Hernandez MA, Mazitschek R, Ozcan U. Treatment of obesity with celastrol. Cell. 2015;161:999–1011. doi: 10.1016/j.cell.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen SR, Dai Y, Zhao J, Lin L, Wang Y, Wang Y. A mechanistic overview of triptolide and celastrol, natural products from tripterygium wilfordii hook F. Front Pharmacol. 2018;9:104. doi: 10.3389/fphar.2018.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Venkatesha SH, Yu H, Rajaiah R, Tong L, Moudgil KD. Celastrus-derived celastrol suppresses autoimmune arthritis by modulating antigen-induced cellular and humoral effector responses. J Biol Chem. 2011;286:15138–46. doi: 10.1074/jbc.M111.226365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Astry B, Venkatesha SH, Laurence A, Christensen-Quick A, Garzino-Demo A, Frieman MB, et al. Celastrol, a Chinese herbal compound, controls autoimmune inflammation by altering the balance of pathogenic and regulatory T cells in the target organ. Clin Immunol. 2015;157:228–38. doi: 10.1016/j.clim.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang T, Hamza A, Cao X, Wang B, Yu S, Zhan CG, et al. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol Cancer Ther. 2008;7:162–70. doi: 10.1158/1535-7163.MCT-07-0484. [DOI] [PubMed] [Google Scholar]
- 22.Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006;10:321–30. doi: 10.1016/j.ccr.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 23.Sun H, Xu L, Yu P, Jiang J, Zhang G, Wang Y. Synthesis and preliminary evaluation of neuroprotection of celastrol analogues in PC12 cells. Bioorg Med Chem Lett. 2010;20:3844–7. doi: 10.1016/j.bmcl.2010.05.066. [DOI] [PubMed] [Google Scholar]
- 24.Guide for the Care and Use of Laboratory Animals 8th edition. The National Academies Press, 2011. [PubMed]
- 25.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–3. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 26.Huang L, Sineva EV, Hargittai MR, Sharma SD, Suthar M, Raney KD, et al. Purification and characterization of hepatitis C virus non-structural protein 5A expressed in Escherichia coli. Protein Expr Purif. 2004;37:144–53. doi: 10.1016/j.pep.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 27.Yun TJ, Harning EK, Giza K, Rabah D, Li P, Arndt JW, et al. EC144, a synthetic inhibitor of heat shock protein 90, blocks innate and adaptive immune responses in models of inflammation and autoimmunity. J Immunol. 2011;186:563–75. doi: 10.4049/jimmunol.1000222. [DOI] [PubMed] [Google Scholar]
- 28.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–9. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 29.Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr., et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–88. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model. 2006;25:247–60. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 31.Huggins DJ. Correlations in liquid water for the TIP3P-Ewald, TIP4P-2005, TIP5P-Ewald, and SWM4-NDP models. J Chem Phys. 2012;136:064518. doi: 10.1063/1.3683447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eastman P, Pande VS. CCMA: a robust, parallelizable constraint method for molecular simulations. J Chem Theory Comput. 2010;6:434–7. doi: 10.1021/ct900463w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lomenick B, Jung G, Wohlschlegel JA, Huang J. Target identification using drug affinity responsive target stability (DARTS) Curr Protoc Chem Biol. 2011;3:163–80. doi: 10.1002/9780470559277.ch110180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu G, Fu S, Zhan X, Wang Z, Zhang P, Shi W, et al. Echinatin effectively protects against NLRP3 inflammasome-driven diseases by targeting HSP90. JCI Insight. 2021;6:e134601. doi: 10.1172/jci.insight.134601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng Y, Tsou LK, Cai J, Aya T, Dutschman GE, Gullen EA, et al. A novel class of meso-tetrakis-porphyrin derivatives exhibits potent activities against hepatitis C virus genotype 1b replicons in vitro. Antimicrob Agents Chemother. 2010;54:197–206. doi: 10.1128/AAC.01206-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ding MY, Peng Y, Li F, Li ZQ, Wang D, Zhou GC, et al. Andrographolide derivative as antagonist of vitamin D receptor to induce lipidation of microtubule associate protein 1 light chain 3 (LC3) Bioorg Med Chem. 2021;51:116505. doi: 10.1016/j.bmc.2021.116505. [DOI] [PubMed] [Google Scholar]
- 37.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–22. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, Gao W, Svitkin YV, Chen AP, Cheng YC. DCB-3503, a tylophorine analog, inhibits protein synthesis through a novel mechanism. PLoS One. 2010;5:e11607. doi: 10.1371/journal.pone.0011607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holst J, Szymczak-Workman AL, Vignali KM, Burton AR, Workman CJ, Vignali DA. Generation of T-cell receptor retrogenic mice. Nat Protoc. 2006;1:406–17. doi: 10.1038/nprot.2006.61. [DOI] [PubMed] [Google Scholar]
- 40.Fernandez IS, Bai XC, Murshudov G, Scheres SH, Ramakrishnan V. Initiation of translation by cricket paralysis virus IRES requires its translocation in the ribosome. Cell. 2014;157:823–31. doi: 10.1016/j.cell.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA, et al. Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol. 2015;172:3461–71. doi: 10.1111/bph.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu HM, Aizaki H, Machida K, Ou JH, Lai MM. Hepatitis C virus translation preferentially depends on active RNA replication. PLoS One. 2012;7:e43600. doi: 10.1371/journal.pone.0043600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu GY, He G, Li CY, Tang M, Grivennikov S, Tsai WT, et al. Hepatic expression of HCV RNA-dependent RNA polymerase triggers innate immune signaling and cytokine production. Mol Cell. 2012;48:313–21. doi: 10.1016/j.molcel.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peng B, Wang Y, Song YT, Zhang X, Cao FF, Xu LM, et al. Therapeutic effects and related molecular mechanisms of celastrol, a triterpenoid natural compound and novel hsp90 inhibitor extracted from plants of the celastraceae family. in: asea AAA, Kaur P, Editors. Heat Shock Protein 90 in Human Diseases and Disorders. Springer International Publishing, Cham, 2019; p 441–70.
- 45.Kannaiyan R, Shanmugam MK, Sethi G. Molecular targets of celastrol derived from thunder of god vine: potential role in the treatment of inflammatory disorders and cancer. Cancer Lett. 2011;303:9–20. doi: 10.1016/j.canlet.2010.10.025. [DOI] [PubMed] [Google Scholar]
- 46.Narayan V, Ravindra KC, Chiaro C, Cary D, Aggarwal BB, Henderson AJ, et al. Celastrol inhibits Tat-mediated human immunodeficiency virus (HIV) transcription and replication. J Mol Biol. 2011;410:972–83. doi: 10.1016/j.jmb.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo D, Fan N, Zhang X, Ngo FY, Zhao J, Zhao W, et al. Covalent inhibition of endoplasmic reticulum chaperone GRP78 disconnects the transduction of ER stress signals to inflammation and lipid accumulation in diet-induced obese mice. Elife. 2022;11:e72182. doi: 10.7554/eLife.72182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao Q, Ding Y, Deng Z, Lee OY, Gao P, Chen P, et al. Natural products triptolide, celastrol, and withaferin A inhibit the chaperone activity of peroxiredoxin I. Chem Sci. 2015;6:4124–30. doi: 10.1039/C5SC00633C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tseng CK, Hsu SP, Lin CK, Wu YH, Lee JC, Young KC. Celastrol inhibits hepatitis C virus replication by upregulating heme oxygenase-1 via the JNK MAPK/Nrf2 pathway in human hepatoma cells. Antivir Res. 2017;146:191–200. doi: 10.1016/j.antiviral.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu YH, Chen WC, Tseng CK, Chen YH, Lin CK, Lee JC. Heme oxygenase-1 inhibits DENV-induced endothelial hyperpermeability and serves as a potential target against dengue hemorrhagic fever. FASEB J. 2022;36:e22110. doi: 10.1096/fj.202100688RRR. [DOI] [PubMed] [Google Scholar]
- 51.Youn GS, Kwon DJ, Ju SM, Rhim H, Bae YS, Choi SY, et al. Celastrol ameliorates HIV-1 Tat-induced inflammatory responses via NF-kappaB and AP-1 inhibition and heme oxygenase-1 induction in astrocytes. Toxicol Appl Pharmacol. 2014;280:42–52. doi: 10.1016/j.taap.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 52.Khalili N, Karimi A, Moradi MT, Shirzad H. In vitro immunomodulatory activity of celastrol against influenza A virus infection. Immunopharmacol Immunotoxicol. 2018;40:250–5. doi: 10.1080/08923973.2018.1440591. [DOI] [PubMed] [Google Scholar]
- 53.Panda S, Vedagiri D, Viveka TS, Harshan KH. A unique phosphorylation-dependent eIF4E assembly on 40S ribosomes co-ordinated by hepatitis C virus protein NS5A that activates internal ribosome entry site translation. Biochem J. 2014;462:291–302. doi: 10.1042/BJ20131530. [DOI] [PubMed] [Google Scholar]
- 54.Shrivastava S, Bhanja Chowdhury J, Steele R, Ray R, Ray RB. Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J Virol. 2012;86:8705–12. doi: 10.1128/JVI.00616-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim MG, Moon JS, Kim EJ, Lee SH, Oh JW. Destabilization of PDK1 by Hsp90 inactivation suppresses hepatitis C virus replication through inhibition of PRK2-mediated viral RNA polymerase phosphorylation. Biochem Biophys Res Commun. 2012;421:112–8. doi: 10.1016/j.bbrc.2012.03.126. [DOI] [PubMed] [Google Scholar]
- 56.Prenner SB, VanWagner LB, Flamm SL, Salem R, Lewandowski RJ, Kulik L. Hepatocellular carcinoma decreases the chance of successful hepatitis C virus therapy with direct-acting antivirals. J Hepatol. 2017;66:1173–81. doi: 10.1016/j.jhep.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abdelaziz AO, Nabil MM, Abdelmaksoud AH, Shousha HI, Hashem MB, Hassan EM, et al. Tumor behavior of hepatocellular carcinoma after hepatitis C treatment by direct-acting antivirals: comparative analysis with non-direct-acting antivirals-treated patients. Eur J Gastroenterol Hepatol. 2019;31:75–9. doi: 10.1097/MEG.0000000000001264. [DOI] [PubMed] [Google Scholar]
- 58.Negash AA, Ramos HJ, Crochet N, Lau DT, Doehle B, Papic N, et al. IL-1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013;9:e1003330. doi: 10.1371/journal.ppat.1003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lebeaupin C, Vallee D, Rousseau D, Patouraux S, Bonnafous S, Adam G, et al. Bax inhibitor-1 protects from nonalcoholic steatohepatitis by limiting inositol-requiring enzyme 1 alpha signaling in mice. Hepatology. 2018;68:515–32. doi: 10.1002/hep.29847. [DOI] [PubMed] [Google Scholar]
- 60.Kawazoe A, Itahashi K, Yamamoto N, Kotani D, Kuboki Y, Taniguchi H, et al. TAS-116 (Pimitespib), an oral HSP90 Inhibitor, in combination with nivolumab in patients with colorectal cancer and other solid tumors: an open-label, dose-finding, and expansion phase Ib trial (EPOC1704) Clin Cancer Res. 2021;27:6709–15. doi: 10.1158/1078-0432.CCR-21-1929. [DOI] [PubMed] [Google Scholar]
- 61.Shi W, Xu G, Zhan X, Gao Y, Wang Z, Fu S, et al. Carnosol inhibits inflammasome activation by directly targeting HSP90 to treat inflammasome-mediated diseases. Cell Death Dis. 2020;11:252. doi: 10.1038/s41419-020-2460-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jing R, Duncan CB, Duncan SA. A small-molecule screen reveals that HSP90beta promotes the conversion of induced pluripotent stem cell-derived endoderm to a hepatic fate and regulates HNF4A turnover. Development. 2017;144:1764–74. doi: 10.1242/dev.146845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bellido Molias F, Sim A, Leong KW, An O, Song Y, Ng VHE, et al. Antisense RNAs influence promoter usage of their counterpart sense genes in cancer. Cancer Res. 2021;81:5849–61. doi: 10.1158/0008-5472.CAN-21-1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee ES, Kim SH, Kim HJ, Kim KH, Lee BS, Ku BJ. growth differentiation factor 15 predicts chronic liver disease severity. Gut Liver. 2017;11:276–82. doi: 10.5009/gnl16049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang S, Huang Z, Lei Y, Han X, Tian D, Gong J, et al. Celastrol alleviates autoimmune hepatitis through the PI3K/AKT signaling pathway based on network pharmacology and experiments. Front Pharmacol. 2022;13:816350. doi: 10.3389/fphar.2022.816350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakagawa S, Umehara T, Matsuda C, Kuge S, Sudoh M, Kohara M. Hsp90 inhibitors suppress HCV replication in replicon cells and humanized liver mice. Biochem Biophys Res Commun. 2007;353:882–8. doi: 10.1016/j.bbrc.2006.12.117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.