

Abstract
Objectives
Cag A+ Helicobacter pylori chronic infection cause malignant transformation of the human gastric mucosa. N6-methyladenosine (m6A) modifications are the most common and abundant mRNA modifications and one of the pathways affecting tumorigenicity and tumor progression. However, the role of m6A modification in the process of chronic H. pylori infection leading to malignant transformation of gastric mucosa is unclear.
Methods
In this study, we used Cag A− and Cag A+H. pylori chronic infection to establish cellular models in GES-1 cells and analyzed the cellular morphology, proliferation, apoptosis, invasiveness and tumorigenicity of gastric mucosal epithelial cells. The m6A expression levels of GES-1 cells after chronic infection with Cag A− and Cag A+H. pylori were examined, and modifying effect of FTO (the fat mass and obesity-associated protein) on CD44 was verified by MeRIP–qPCR. Finally, the FTO expression changes and m6A expression levels were further validated in clinical gastric cancer tissues.
Results
Chronic Cag A+H. pylori-infected GES-1 cells exhibit altered cell morphology, apoptosis inhibition, abnormal proliferation, enhanced migration, colony formation, and increased stem cell-like properties. Meanwhile, FTO and CD44 expression was enhanced, and FTO may induce malignant transformation of gastric mucosa by regulating CD44 mRNA m6A methylation modifications.
Conclusions
We verified the effect of chronic stimulation of Cag A+H. pylori on malignant transformation of gastric mucosal epithelium. revealing the possibility of FTO in promoting malignant transformation of gastric mucosa by modifying CD44 mRNA methylation, suggesting that FTO expression is a potential molecule for malignant transformation of gastric mucosal epithelial cells.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00432-023-04684-4.
Keywords: Helicobacter pylori, Cag A, FTO, m6A, CD44, Malignant transformation
Introduction
Helicobacter pylori (H. pylori) is one of the most contagious pathogens, infecting more than 50% of the world's population (Maixner et al. 2019). However, more than 80% of infected people have no obvious symptoms, and this highly contagious and insidious bacterium can cause life-long infection and continue to infect more people (Wang et al. 2014; Chojnacki et al. 2019; Baj et al. 2020). H. pylori is significantly associated with active gastritis, peptic ulcer, atrophic gastritis, intestinal metaplasia, Mail's lymphoma, and gastric cancer (Kotilea et al. 2019; Naumann and Crabtree 2004). From chronic non-atrophic gastritis, atrophic gastritis, intestinal metaplasia, dysplasia to gastric carcinogenesis, this process is called Correa's cascade (Sukri et al. 2020), usually triggered by H. pylori infection and influenced by synergistic genetic and environmental influences (Ansari 2018; Lahner et al. 2018). H. pylori is a class I carcinogen that has a significant role in malignant transformation of normal gastric mucosa into gastric carcinogenesis (Keilberg and Ottemann 2016; Maroney and Ciurli 2021). H. pylori can produce cytotoxin-associated gene A (Cag A), vacuolating cytotoxin A (Vac A), outer membrane proteins, urease and other virulence factors that affect host cell signaling pathways and contribute to progressive abnormal changes in the gastric epithelium (Chmiela et al. 2018). The cag pathogenicity island (cag PAI), which encodes the type IV secretion system (T4SS), injects the Cag A into host cells (Müller 2012; Merino et al. 2017), resulting in altered epithelial cell polarity, inducing the production of cells with a hummingbird phenotype, increased cell migration and cell invasion, and formation of tumor-like spheres, with a higher propensity for invasiveness and malignancy (Bessède et al. 2014; Mueller et al. 2012). It is recognized that H. pylori expressing cag PAI is more carcinogenic, and under chronic infection with Cag A+H. pylori the long-lasting inflammatory response and accumulation of "blows" stimulate the dysfunction and structure of gastric epithelial cells, eventually leading to the development of gastric cancer.
Gastric cancer (GC) is the fifth most common cancer and the fourth most lethal cancer in the world (Sung et al. 2021), particularly prevalent in East Asia (Patrad et al. 2022). Patients are usually first diagnosed with advanced gastric cancer with poor treatment and prognosis. Therefore, finding biomarkers and therapeutic targets for malignant transformation of the gastric mucosa is beneficial to prevent gastric cancer.
N6-methyladenosine (m6A) is the most abundant modification ubiquitously occurring in eukaryotic mRNAs and it plays crucial roles in tumorigenicity and tumor progression (Cai et al. 2022; Chen et al. 2019). It has been demonstrated that m6A methylation affects stem cell differentiation and the growth and proliferation of tumor cells. "writers" (methyltransferases, including WTAP, KIAA1429, RBM15/15B, ZC3H13 and METTL3/14/16), "readers" (YTHDF1/2/3, YTHDC1/2, IGF2BP, HNRNPC and HNRNPA2B1) and "erasers" (demethylases, including ALKBH5 and FTO) are involved in the dynamic regulation process of m6A (Lan et al. 2019). The fat mass- and obesity-associated protein (FTO) is the first identified demethylation enzyme, involved in a variety of regulatory biological processes (Zhang et al. 2022). There is growing evidence that FTO promotes the development of a variety of cancers including acute myeloid leukemia (AML), melanoma, breast cancer and lung cancer (Li et al. 2017a, b; Castillo et al. 2012; Kaklamani et al. 2011; You et al. 2022). In addition, FTO is associated with self-renewal and immune evasion of cancer stem cells, and plays an essential role in the progression and metastasis of gastric cancer (Zhang et al. 2021). FTO is also involved in the proliferation, invasion and metastasis of gastric cancer cells, and has excellent prognostic value (Guan et al. 2020).m6A modifications are involved in the regulation of tumor stem cells (CSC) and tumor immune microenvironment leading to malignant phenotypes (Kumari et al. 2022). For example, in pancreatic cancer, FTO regulates the expression of CSC markers involved in pancreatic carcinogenesis (Garg et al. 2022).CD44, a gastric cancer stem cell marker, is not only involved in tumor growth and maintenance of cancer cell stemness, but also highly correlated with tumorigenicity, invasiveness, lymphatic metastasis and drug resistance of gastric cancer cells, which greatly affects the treatment and prognosis of gastric cancer (Wang et al. 2011; Qi et al. 2022).Interestingly, it was found that CD44 expression gradually increased during the process of Correa's cascade, and the abnormal alteration of CD44 was earlier than CD133, which is also a biomarker of gastric cancer stem cells, so CD44 is considered as a biomarker and key molecule of malignant transformation (Zavros 2017; Wang et al. 2011). However, the mechanism of CD44 involvement in malignant transformation of gastric mucosa is still unclear.
The biological significance of m6A in the malignant transformation of gastric epithelium due to chronic H. pylori infection and its potential regulatory mechanisms remain unclear. In this study, we investigated the role of FTO-mediated m6A modification in the development of malignant transformation of human normal gastric mucosal epithelial cells from chronic infection with Cag A+H. pylori.
Methods
Cell culture
Immortalized human gastric mucosal epithelial cells GES-1 (from the Cell Resource Center, Beijing, China) were used, and the cell culture media were supplemented with 10% fetal bovine serum (FBS, BI) and 1% penicillin–streptomycin (100 U/mL) with RPMI 1640 medium (Gibco), and the cells were cultured at 37 °C in a 5% CO2 thermostatic incubator.
Helicobacter pylori culture
We used 2 types of previously collected H. pylori strains (Li et al. 2017a, b), one of which expresses Cag A protein and the other does not. All strains were cultured on Columbia blood agar plates containing sheep blood and H. pylori screening agent, and after 3–5 days of incubation at 37 °C in a microaerobic constant temperature environment, Cag A−H. pylori strains grew as tiny pinpoint-like dots (Supplementary Fig. S1A) and Cag A+H. pylori strains grew as thicker columnar colonies (Supplementary Fig. S1A). Colonies were scraped using an inoculation loop, DNA was extracted and identified by agarose electrophoresis (Supplementary Fig. S1B). After the bacterial growth was stabilized, the colonies were scraped into RPMI 1640 medium containing 10% FBS and used immediately.
Co-culture of GES-1 with H. pylori
In the H. pylori chronic infection model (Yu et al. 2011; Liu et al. 2021; Lin et al. 2019), GES-1 cells were cultured in antibiotic-free cell culture medium and inoculated in 6-well plates (Corning) at a density of 3 × 105 cells/well. At approximately 80% cell confluency, freshly collected H. pylori were suspended in antibiotic-free cell culture medium and added to the cells for co-culture. H. pylori and GES-1 were co-cultured at MOI (multiplicity of infection) = 1:1, 10:1, 100:1, respectively, and the original medium was replaced every 24 h with freshly collected H. pylori. Co-culture was continued for approximately 6 months in the chronic infection model. In the model of acute infection, H. pylori was co-cultured with GES-1 cells under the same conditions for 48 h. The bacterial concentration was calculated based on the optical density (OD) of 2 × 108 colony forming units (CFU) at 660 nm. Cells were used for experimental analysis during the intervention period.
Cell morphology observation
Co-cultured GES-1 was observed under an inverted phase contrast microscope, and morphological changes were observed in the normal, Cag A−H. pylori and Cag A+H. pylori chronically infected cell.
CCK-8 assay for cell viability and proliferation
Cell viability and proliferation were assessed using the CCK-8 kit (Biosharp). Cells were inoculated into 96-well plates (100 µl/well) at a density of 3 × 104 cells/ml and incubated at 0 h, 12 h, 48 h and 72 h with 10µl of CCK-8 solution in an incubator containing 5% CO2 at 37 °C for 2 h. The absorbance was measured at 450 nm using a multifunctional enzyme marker. OD values were positively correlated with the number of cell proliferation and subsequently plotted cell growth curve.
Cell colony formation assay
Cells were turned into individual cells using trypsin (Biosharp), counted and then inoculated at 300–500 cells/well in 6-well plates, grown for 15 days by adding complete medium, fixed in 4% paraformaldehyde (Biosharp) for at least 30 min, stained with crystal violet (GCLONE) and then counted for analysis.
Transwell assay
Cell migration was determined by transwell assay. 100 µl of cell suspension was added to each well of the apical chamber. At the same time, 500 µl of conditioned medium containing 30% FBS was added to the basolateral chamber. After incubation at 37 °C for 48 h, transwell plates were fixed with 4% paraformaldehyde for 30 min, followed by staining the cells with crystal violet for 30 min at room temperature. The cells were wiped off the surface with a cotton ball. Finally, cells were microscopically observed and counted using at least four randomly selected fields of view.
EdU cell proliferation assay
Cell proliferation capacity was assayed according to the steps in the instructions (Meilunbio). An equal volume of EdU reagent (2X, 50 µM) in complete medium configuration was added to the cell culture medium, incubated for 2 h at 37 °C and then fixed using paraformaldehyde, 0.5% Triton-100 permeabilized cells and stained using 488-Azide with Hoechst 33,342.
Live and dead cell double staining
The cells were washed thoroughly with 1X Assay, and the staining solution was prepared according to the Calcein AM/PI Double Stain Kit (MKBio), added to the cell suspension and incubated at 37 °C for 15 min. The cells were double stained and labeled with Calcein-AM and Propidium Iodide (PI) for live and dead cells, respectively, for analysis at the live and dead cell levels.
Western blot
Proteins were obtained using RIPA lysis buffer with 1 mM PMSF and protein concentration was determined by BCA protein quantification method. Proteins were separated by SDS–PAGE electrophoresis and transferred to PVDF membranes. The membranes were incubated with 5% skim milk for 1 h at 4 °C and overnight with the following primary antibody. Then, the membranes were incubated with horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG for 1 h. Protein bands are detected using the ECL kit (Biosharp).
The primary antibodies and secondary antibodies used were as follows: FTO (ZENBIO), CD44 (ZENBIO) and horseradish peroxidase-conjugated anti-IgG secondary antibodies (Cell Signalling Technology).
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted and cDNA was obtained according to the reverse transcription instructions (Vazyme). Primer sequences were as follows: GAPDH, 5′-GGTCACCAGGGCTGCTTTA-3′ (Forward); 5′-GGATCTCGCTCCTGGAAGATG-3′(Reverse); FTO,5′-ACTTGGCTCCCTTATCTGACC-3′(Forward); 5′-TGTGCAGTGTGAGAAAGGCTT-3′(Reverse); CD44, 5′-CTGCCGCTTTGCAGGTGTA-3′ (Forward); 5′-CATTGTGGGCAAGGTGCTATT-3′ (Reverse); and the relative expression of target genes at the mRNA level was calculated by the 2-ΔΔCT method, using GAPDH as an endogenous control.
Immunohistochemistry
Specimens of gastric cancer from primary gastric cancer patients who underwent surgery at The Third Xiangya Hospital were collected, and the specimens were taken at the tumor and > 15 cm from the tumor margin, respectively, and the tissues were fixed in paraformaldehyde and paraffin-embedded sections were used for immunohistochemical diagnosis. This study was approved by the ethics committee of The Third Xiangya Hospital to be conducted.
Flow cytometry analysis
Cells were digested using EDTA-free trypsin, washed twice with Phosphate Buffered Saline (PBS), resuspended by adding 500 µl of Binding Buffer, incubated together with 5 µl of Annexin V and 5 µl of PI (KeyGEN BioTECH), for 15 min at room temperature, and analyzed for apoptosis by flow cytometry.
In vivo tumor xenograft study
For the tumor growth analysis, GES-1 cells were subcutaneously injected into BALB/c nude mice (male, 5 weeks), and tumor volumes were monitored every 5 days. Tumor volumes were estimated based on length and width and calculated using the following formula: tumor volume = (length × width2)/2. About 1 month later, the nude mice were sacrificed, and then tumors were excised, pictured, and weighed.
m6A RNA dot blot assay
The mRNA samples are dissolved in nuclease-free water, denatured at 65 °C for 5 min, then, transferred to Nylon Transfer Membrane and crosslinked by UV, incubated with 5% skim milk for 1 h, then incubated overnight at 4 °C with an antibody specific for m6A and finally incubated with HRP-linked anti-rabbit IgG or anti-mouse IgG for 1 h. Staining with approximately 0.2% methylene blue was used as a control.
MeRIP–qPCR
After obtaining high quality total cellular RNA, Methylated RNA Immunoprecipitation Assay (MeRIP) were performed. m6A RNA fragments were enriched and purified using the EpiQuik™ CUT&RUN m6A RNA Enrichment kit (Epigentek), and CD44 expression at the RNA level was detected by PCR.
Statistical analysis
Data were expressed as the mean ± SEM from at least three independent experiments. Student's t-test was used for two-group comparisons. For multiple comparisons, Two-way analysis of variance (ANOVA) was performed using GraphPad Prism® version 8.0 software. p values < 0.05 were considered statistically significant.
Results
Increased apoptosis of GES-1 after acute infection with H. pylori
H. pylori relies on a powerful chemotactic motility system to colonize the host gastric mucosa, releasing a variety of virulence factors that resist host immune clearance, induce inflammatory responses and damage host mucosal tissue. Flow cytometry showed a significant increase in GES-1 apoptosis within 48 h of H. pylori infection, which was more pronounced in Cag A+H. pylori infection (Fig. 1A, B). The CCK-8 assay revealed a greater effect of Cag A+H. pylori on GES-1 cell activity at 48 h and 72 h of H. pylori infection (Fig. 1C). Further Western Blot results showed that the expression of Bax and Cleaved Caspase-3 increased in GES-1 cells with increasing concentrations of Cag A+H. pylori acute infection compared to control and Cag A−H. pylori infected groups (Fig. 1D). These tend to suggest damage to the host gastric mucosa under the acute impact of H. pylori infection.
Fig. 1.

Increased apoptosis in acute H. pylori infected GES-1 cells. A Flow cytometry results of GES-1 cells after 48 h of acute H. pylori infection, B statistical analysis. C CCK-8 assay for GES-1 cell viability. C BAX, c-caspase3 and FTO were determined by Western blotting after acute infection with H. pylori in GES-1 cells. Hp: H. pylori, *p < 0.05, **p < 0.01, ***p < 0.001
Chronic Cag A+H. pylori infection induces malignant transformation of GES-1
Normal GES-1 shows shuttle-shaped or polygonal cells with regular shape and clear edges. GES-1 cells are bipolarly prolonged and cellularly narrowed after chronic H. pylori infection. Morphological changes were more pronounced in GES-1 cells chronically infected with Cag A+H. pylori, with linearly prolonged cell poles and hummingbird-like cells (Fig. 2A and Supplementary Fig. S1C). It has been shown that H. pylori synthesizes Cag A and binds to SHP-2 via T4SS transport into host cells, disrupting tight intercellular junctions, rearranging the cytoskeleton, and altering cell morphology. In addition, the cells with hummingbird morphology tend to have more potential for transformation.
Fig. 2.

Chronic Cag A+H. pylori infection induces malignant transformation of GES-1. A GES-1 cells show a hummingbird phenotype after chronic infection with Cag A+H. pylori. B Reduced apoptosis in GES-1 cells detected by flow cytometry after chronic infection with Cag A+H. pylori. C Statistical analysis. D Detection of BCL-2, BAX and c-caspase3 protein levels in GES-1 cells after chronic infection with H. pylori by Western blot. E, G Enhanced proliferation and migration of GES-1 cells in Cag A+H. pylori chronic infection detected by colony formation and Transwell assay. F, H Statistical analysis. ns: no statistical significance, *p < 0.05, **p < 0.01
Acute Cag A+H. pylori infection affects cell viability and contributes to apoptosis of GES-1 cells. However, after chronic infection with Cag A+H. pylori, flow cytometry revealed reduced GES-1 apoptosis (Fig. 2B, C) and decreased expression of apoptotic proteins (Fig. 2D), suggesting a disruption of the programmed cell death process in response to long-term H. pylori infection stimulation. In addition, sustained Cag A+H. pylori stimulation caused malignant transformation of normal GES-1 cells, such as increased abnormal proliferation, invasiveness and tumorigenicity. GES-1 cells in the Cag A+H. pylori chronic infection group showed stronger cell proliferation and invasive ability in colony formation and transwell assays (Fig. 2E–H). In addition, EdU Staining revealed increased proliferation and enhanced DNA replication activity of GES-1 cells under chronic stimulation of Cag A+H. pylori (Fig. 3B, D). Live and dead cell staining suggested that the number of cell death in the Cag A+H. pylori chronic infection group was less than that in the Cag A−H. pylori chronic infection group and the control group (Fig. 3A, C). In a subcutaneous tumorigenic assay in nude mice, Cag A+H. pylori chronically stimulated GES-1 cells exhibited tumorigenicity similar to tumor cells (Fig. 3E–G).
Fig. 3.

Chronic infection with Cag A+H. pylori induces malignant transformation of GES-1. A Live-dead cell staining reveals reduced GES-1 cell death after chronic infection with Cag A+H. pylori. B EdU Staining finds increased proliferation of GES-1 cells after chronic infection with Cag-A+H. pylori. C, D Statistical analysis. E, F Chronic Cag A+H. pylori infection induces the tumorigenicity of GES-1 cells in vivo, body weight, tumor volume and anatomical tumor images of nude mice were recorded and analyzed. ns: no statistical significance, #: NC and Cag A+H. pylori groups were statistically significant. *p < 0.05, **p < 0.01
FTO is involved in the malignant transformation of GES-1 under persistently stimulation of Cag A+H. pylori
With increasing duration of infection, the expression of FTO was significantly higher in Cag A+H. pylori infection group in comparison with the control and Cag A−H. pylori infection groups, and showed a dose-dependent relationship with the concentration of Cag A+H. pylori infection (Fig. 4A) and higher expression at the RNA level (Fig. 4B). GES-1 was more tumorigenic in vivo after chronic infection with Cag A+H. pylori, and immunohistochemistry assays in mouse tumors suggested higher FTO expression in the Cag A+H. pylori chronic infection group (Fig. 4C). After we down-regulated FTO expression in the Cag A+H. pylori chronic infection group using lentiviral transfection and confirmed FTO low expression by qRT-PCR (Fig. 4D), we found that inhibition of FTO expression rescued the signs of malignant transformation of GES-1 cells caused by Cag A+H. pylori chronic infection with abnormal proliferation, cell migration ability and tumorigenicity in animal assays. Functionally, silencing FTO inhibits cell proliferation and metastasis. Reduced cell proliferation was observed by colony formation assays (Fig. 4E, F), and transwell assays revealed a significant inhibition of cell metastatic ability (Fig. 4G, H), and the effect of FTO on the tumorigenicity of GSE-1 cells chronically infected with Cag A+H. pylori was also verified in in vivo experiments in animals. In tumor xenograft studies, we found that normal GES-1 hardly formed tumors, while GES-1 cells in the Cag A+H. pylori chronic infection group could form significant subcutaneous tumors and were statistically significant with the Cag A−H. pylori chronic infection group (Fig. 4I–K). These results suggest that chronic infection with Cag A+H. pylori induces morphological changes in GES-1 cells and significantly increases the proliferation and invasive capacity of GES-1 cells, which are more tumorigenic in nature, and that FTO is involved in the malignant transformation process of GES-1.
Fig. 4.

FTO is involved in the malignant transformation of GES-1 in response to sustained stimulation by Cag A+H. pylori. A, B Elevated FTO protein and RNA expression after chronic Cag A+H. pylori Infection. C IHC staining of tumor tissues from nude mice verified that the FTO expression was elevated after chronic infection with Cag A+H. pylori. D Efficiency of knockdown of FTO in Cag A+H. pylori chronically infected GES-1 cells by qRT-PCR. E, G Knockdown of FTO inhibited cell proliferation and migration of GES-1 cells measured by colony formation and Transwell assay. F, H Statistical analysis. I, J knockdown of FTO inhibited the tumorigenicity of GES-1 cells in nude mice. ns: no statistical significance, *p < 0.05, **p < 0.01, ***p < 0.001
FTO is involved in CD44 mRNA m6A modification in Cag A+H. pylori chronic infection
GES-1 cells chronically infected by Cag A+H. pylori showed a significant increase in CD44 expression at the RNA and protein levels (Fig. 5A, B). CD44 is a recognized tumor stem cell marker molecule, and its increased expression predicts a malignant transformation change of GES-1 cells under continuous stimulation of Cag A+H. pylori (Ryu et al. 2012). CD44 is not only a marker of gastric cancer CSC, it is functionally participating in the tumorigenicity of gastric cancer cells (Takaishi et al. 2009). A number of studies have found that CD44 is progressively elevated in the Correa's cascade model and plays an active role in the development of gastric cancer (He et al. 2022; Yang et al. 2013; Choi et al. 2019). Daniel Brungs et al. found that CD44 + gastric cancer cells showed enhanced resistance to chemotherapy or radiation-induced cell death and were associated with poor prognosis (Brungs et al. 2016). Meanwhile, we found that CD44 expression was significantly correlated with FTO expression. When we silenced the FTO expression of GES-1 after chronic infection of H. pylori, the expression of CD44 was suppressed, and the expression level of CD44 was significantly increased after exogenous supplementation of FTO (Fig. 5A). qRT-PCR at the RNA level revealed a positive correlation between CD44 expression levels and FTO expression, and silencing of FTO hindered CD44 expression levels in Cag A+H. pylori chronically infected GES-1 cells (Fig. 5B, C).
Fig. 5.

FTO is involved in CD44 mRNA m6A modification in Cag A+H. pylori chronic infection. A Western blot showed that CD44 expression increased with FTO after chronic infection of GES-1 cells by Cag A+H. pylori, and that overexpression or knockdown of FTO promoted or suppressed the protein expression level of CD44. B, C Increased CD44 RNA levels after chronic infection with Cag A+H. pylori and decreased CD44 RNA expression after knockdown of FTO. D, E m6A modification site predictor SRAMP was used to predict the m6A modification site in the CDS region of CD44. F, G Enrichment and validation of m6A-methylated RNA fragments by MeRIP–qPCR in GES-1 cells after chronic infection with H. pylori. The level of CD44 m6A modification decreased when FTO was highly expressed and increased after FTO knock-out, suggesting a role for m6A modification of CD44 by FTO. ns: no statistical significance, *p < 0.05, **p < 0.01, ***p < 0.001
To further explore the role of FTO-mediated m6A methylation in Cag A+H. pylori chronically infected GES-1 cells, we predicted the m6A binding site of CD44 by SRAMP website and found an abundant m6A binding site at the 3' UTR of CD44 (Fig. 5D, E). We enriched m6A-RNA fragments in GES-1 cells by methylated RNA Immunoprecipitation assay (MeRIP), and MeRIP–qPCR revealed that FTO mediated CD44 mRNA m6A modification. The results showed that chronic infection of Cag A+H. pylori induced high expression of FTO, and m6A modification of CD44 was reduced when FTO was up-regulated, but significantly increased after FTO expression was silenced (Fig. 5F, G). The above results suggest the enrichment of m6A RNA in GES-1 after chronic infection with Cag A+H. pylori, and verified the interaction between FTO and CD44 mRNA, suggesting that FTO, as a demethylating enzyme, regulates CD44 mRNA in a m6A-dependent manner to involve in malignant transformation of gastric mucosal epithelial cells.
FTO is enriched in gastric cancer and associated with poor prognosis
The outcome of malignant transformation of normal gastric epithelial cells often points to the development of gastric cancer. Finally, we argue a strong link between FTO and gastric cancer, further suggesting the involvement of FTO in the malignant transformation of normal gastric epithelium and even in the development of tumorigenesis. We used the GEPIA database to analyze and found that FTO expression was significantly elevated in GC tissues (Fig. 6A, B), and then we analyzed the Kaplan–Meier Plotter database to correlate FTO expression levels with overall survival (OS) of GC patients and found that high FTO expression tended to predict poor survival prediction (log-rank p < 0.001) (Fig. 6C). In gastric cancer tissues, Western blot revealed that the expression level of FTO and CD44 was significantly higher than that of adjacent tissues (Fig. 6D), and in immunohistochemistry, FTO expression was significantly elevated in GC patients, with positive staining mainly concentrated in GC cell nuclei (Fig. 6E). Total m6A levels were elevated in gastric cancer tissues and in subcutaneous tumor tissues of mice in Cag A+H. pylori chronic infection group (Fig. 6F, G).
Fig. 6.

FTO expression is up-regulated in GC tissues and its prognostic value in GC. A, B Expression of FTO in gastric cancer tissues and C prognostic value of FTO in gastric cancer tissues were analyzed in the clinical database GEPIA. D Expression of FTO and CD44 protein levels in GC tissues and paracancerous tissues by Western blotting. E Analysis of FTO protein level expression in GC tissues and paraneoplastic tissues by immunohistochemistry. F, G Total m6A levels in gastric cancer tissues and subcutaneous tumor tissues in nude mice were analyzed by m6A RNA Dot Blot Assay, and MB (methylene blue) staining was used as the loading control
Discussion
GC is the fifth most common malignancy in the world, characterized by high mortality, poor differentiation and poor prognosis (Digklia and Wagner 2016; Rugge et al. 2019). Chronic H. pylori infection is the main cause of gastric cancer (Correa 1992; Choi et al. 2018), but the process from H. pylori chronic infection-induced malignant transformation of gastric mucosa, precancerous lesions to gastric carcinogenesis is not clear.
Gastric carcinogenesis and progression are also affected by epigenetic modifications. FTO is an independent prognostic marker for gastric cancer and also the most potent prognostic risk factor among all m6A regulators (Wang et al. 2021; Zhao et al. 2021). GC patients with high expression of FTO were often accompanied by poor TNM staging and prognosis (Su et al. 2019). In addition, FTO can dynamically regulate the structure and function of RNA by reducing m6A modifications, increasing the translational expression of oncogenes or triggering transcriptional changes in oncogenes to induce cellular deterioration and promote the development of malignancy (Zou et al. 2019). Yang Z et al. found that FTO promotes the stability of MYC mRNA by decreasing the m6A methylation of MYC in GC cells, thereby accelerating the proliferation, migration and invasive properties (Yang et al. 2021). FTO was found to promote GC metastasis by regulating m6A levels of integrin β1 (ITGB1) to upregulate ITGB1 expression (Wang et al. 2021). Similarly, our study found that m6A levels were altered in both malignantly transformed gastric epithelial cells and gastric cancer tissues, and CD44 was positively correlated with the expression level of FTO. In addition, combined with the prediction of m6A modification sites, we identified CD44 as a possible demethylation target of FTO. This result supports that FTO plays an important tumor-promoting role before gastric carcinogenesis, which has important implications for the diagnosis and treatment of gastric cancer (Fig. 7).
Fig. 7.
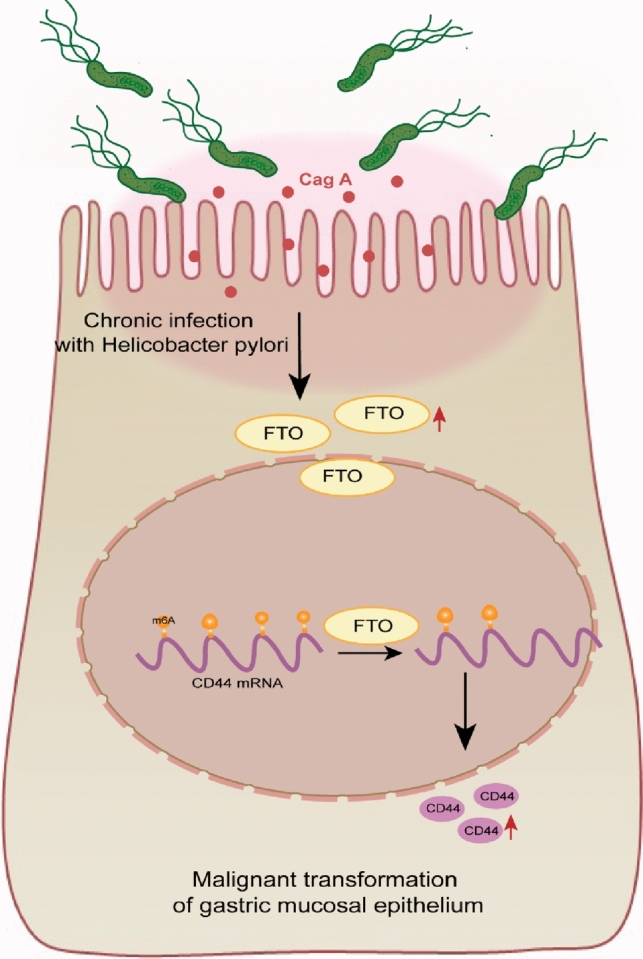
Regulatory axis of Cag A+H. pylori/FTO/CD44 is involved in the malignant transformation of GES-1
In this study, we found that in Cag A+H. pylori chronically stimulated GES-1 cells, upregulated FTO promoted CD44 expression by regulating m6A modification of CD44 RNA, accompanied by abnormal changes in cell morphology and enhanced proliferation invasion, which were associated with malignant transformation of normal gastric epithelium. CD44, a member of the CAM family, is a major receptor for hyaluronic acid (HA) and is normally involved in cell signaling-mediated cell proliferation, invasion and migration (Senbanjo and Chellaiah 2017). CD44 is also a marker molecule for many tumor–stem cells, including gastric cancer. Low differentiation of tumor and poor prognosis of gastric cancer correlate with CD44. CD44 expression rate was found to be as high as 80% in primary gastric cancer resection specimens, and gastric cancer with high CD44 expression was associated with higher level of clinicopathological features and worse prognosis (Brungs et al. 2016). Shigeo Takaishi et al. found that CD44+ gastric cancer cells exhibited self-renewal properties and differentiation of stem cells. In vivo, CD44+ GC cells injected dermally and intragastrically were more tumorigenic in mice, and the same was verified in a xenograft test in mice (Takaishi et al. 2009).
Conclusion
Overall, our findings suggest the involvement of Cag A+H. pylori/FTO/CD44 in the malignant transformation of GES-1 and reveal the possibility of FTO in promoting gastric carcinogenesis by modifying CD44 mRNA methylation, and FTO may be a predictor and therapeutic target for malignant transformation of gastric epithelium, providing a new perspective for the prevention and treatment of gastric cancer. However, the specific mechanisms regulating the process of malignant transformation of the gastric mucosa still need further investigation.
Supplementary Information
Below is the link to the electronic supplementary material.
Author contributions
All authors contributed to the study conception and design. Study supervision was performed by XCX and LXM. Experimental analysis and first draft clutching were performed by CS. Material preparation was performed by LH and CJS, and data collection was performed by ZWF and LJH. In addition, all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the following grants and foundations: National Natural Science Foundation of China (No. 81570509) and Changsha Natural Science Foundation Project (kq2202118).
Data availability
The data sets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
This study was performed in line with the principles of the Declaration of Helsinki. This study was approved by the Ethics Committee of the Department of Laboratory Animals, Central South University (CSU-2022–0598) and the Ethics Committee of the Third Xiangya Hospital, Central South University (No: 22209).
Patient consent for publications
Not applicable.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Ansari S, Gantuya B, Tuan VP, Yamaoka Y. Diffuse gastric cancer: a summary of analogous contributing factors for its molecular pathogenicity. Int J Mol Sci. 2018;19(8):2424. doi: 10.3390/ijms19082424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baj J, Korona-Głowniak I, Forma A, Maani A, Sitarz E, Rahnama-Hezavah M, Radzikowska E, Portincasa P. Mechanisms of the epithelial-mesenchymal transition and tumor microenvironment in helicobacter pylori-induced gastric cancer. Cells. 2020;9(4):1055. doi: 10.3390/cells9041055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessède E, Staedel C, Acuña Amador LA, Nguyen PH, Chambonnier L, Hatakeyama M, Belleannée G, Mégraud F, Varon C. Helicobacter pylori generates cells with cancer stem cell properties via epithelial-mesenchymal transition-like changes. Oncogene. 2014;33:4123–4131. doi: 10.1038/onc.2013.380. [DOI] [PubMed] [Google Scholar]
- Brungs D, Aghmesheh M, Vine KL, Becker TM, Carolan MG, Ranson M. Gastric cancer stem cells: evidence, potential markers, and clinical implications. J Gastroenterol. 2016;51:313–326. doi: 10.1007/s00535-015-1125-5. [DOI] [PubMed] [Google Scholar]
- Cai X, Liang C, Zhang M, Xu Y, Weng Y, Li X, Yu W. N6-methyladenosine modification and metabolic reprogramming of digestive system malignancies. Cancer Lett. 2022;544:215815. doi: 10.1016/j.canlet.2022.215815. [DOI] [PubMed] [Google Scholar]
- Castillo JJ, Mull N, Reagan JL, Nemr S, Mitri J. Increased incidence of non-Hodgkin lymphoma, leukemia, and myeloma in patients with diabetes mellitus type 2: a meta-analysis of observational studies. Blood. 2012;119:4845–4850. doi: 10.1182/blood-2011-06-362830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XY, Zhang J, Zhu JS. The role of m(6)A RNA methylation in human cancer. Mol Cancer. 2019;18:103. doi: 10.1186/s12943-019-1033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmiela M, Walczak N, Rudnicka K. Helicobacter pylori outer membrane vesicles involvement in the infection development and Helicobacter pylori-related diseases. J Biomed Sci. 2018;25:78. doi: 10.1186/s12929-018-0480-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JM, Kim SG, Choi J, Park JY, Oh S, Yang HJ, Lim JH, Im JP, Kim JS, Jung HC. Effects of Helicobacter pylori eradication for metachronous gastric cancer prevention: a randomized controlled trial. Gastrointest Endosc. 2018;88:475–85.e2. doi: 10.1016/j.gie.2018.05.009. [DOI] [PubMed] [Google Scholar]
- Choi SI, Yoon C, Park MR, Lee D, Kook MC, Lin JX, Kang JH, Ashktorab H, Smoot DT, Yoon SS, Cho SJ. CDX1 Expression induced by CagA-expressing Helicobacter pylori promotes gastric tumorigenesis. Mol Cancer Res. 2019;17:2169–2183. doi: 10.1158/1541-7786.MCR-19-0181. [DOI] [PubMed] [Google Scholar]
- Chojnacki C, Popławski T, Błońska A, Błasiak J, Romanowski M, Chojnacki J. Expression of tryptophan hydroxylase in gastric mucosa in symptomatic and asymptomatic Helicobacter pylori infection. Arch Med Sci. 2019;15:416–423. doi: 10.5114/aoms.2018.76928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa P. Human gastric carcinogenesis: a multistep and multifactorial process–first American Cancer Society award lecture on cancer epidemiology and prevention. Cancer Res. 1992;52:6735–6740. [PubMed] [Google Scholar]
- Digklia A, Wagner AD. Advanced gastric cancer: current treatment landscape and future perspectives. World J Gastroenterol. 2016;22:2403–2414. doi: 10.3748/wjg.v22.i8.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg R, Melstrom L, Chen J, He C, Goel A. Targeting FTO suppresses pancreatic carcinogenesis via regulating stem cell maintenance and EMT pathway. Cancers (basel) 2022;14(3):5919. doi: 10.3390/cancers14235919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan K, Liu X, Li J, Ding Y, Li J, Cui G, Cui X, Sun R. Expression status and prognostic value of M6A-associated genes in gastric cancer. J Cancer. 2020;11:3027–3040. doi: 10.7150/jca.40866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Hu W, Ouyang Q, Zhang S, He L, Chen W, Li X, Hu C. Helicobacter pylori infection induces stem cell-like properties in Correa cascade of gastric cancer. Cancer Lett. 2022;542:215764. doi: 10.1016/j.canlet.2022.215764. [DOI] [PubMed] [Google Scholar]
- Kaklamani V, Yi N, Sadim M, Siziopikou K, Zhang K, Xu Y, Tofilon S, Agarwal S, Pasche B, Mantzoros C. The role of the fat mass and obesity associated gene (FTO) in breast cancer risk. BMC Med Genet. 2011;12:52. doi: 10.1186/1471-2350-12-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilberg D, Ottemann KM. How Helicobacter pylori senses, targets and interacts with the gastric epithelium. Environ Microbiol. 2016;18:791–806. doi: 10.1111/1462-2920.13222. [DOI] [PubMed] [Google Scholar]
- Kotilea K, Bontems P, Touati E. Epidemiology, diagnosis and risk factors of Helicobacter pylori infection. Adv Exp Med Biol. 2019;1149:17–33. doi: 10.1007/5584_2019_357. [DOI] [PubMed] [Google Scholar]
- Kumari S, Kumar S, Muthuswamy S. RNA N6-methyladenosine modification in regulating cancer stem cells and tumor immune microenvironment and its implication for cancer therapy. J Cancer Res Clin Oncol. 2022 doi: 10.1007/s00432-022-04158-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahner E, Carabotti M, Annibale B. Treatment of Helicobacter pylori infection in atrophic gastritis. World J Gastroenterol. 2018;24:2373–2380. doi: 10.3748/wjg.v24.i22.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Q, Liu PY, Haase J, Bell JL, Hüttelmaier S, Liu T. The critical role of RNA m(6)A methylation in cancer. Cancer Res. 2019;79:1285–1292. doi: 10.1158/0008-5472.CAN-18-2965. [DOI] [PubMed] [Google Scholar]
- Li R, Jiang XX, Zhang LF, Liu XM, Hu TZ, Xia XJ, Li M, Xu CX. Group 2 innate lymphoid cells are involved in skewed type 2 immunity of gastric diseases induced by Helicobacter pylori infection. Mediators Inflamm. 2017;2017:4927964. doi: 10.1155/2017/4927964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, Huang H, Nachtergaele S, Dong L, Hu C, Qin X, Tang L, Wang Y, Hong GM, Huang H, Wang X, Chen P, Gurbuxani S, Arnovitz S, Li Y, Li S, Strong J, Neilly MB, Larson RA, Jiang X, Zhang P, Jin J, He C, Chen J. FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-Methyladenosine RNA demethylase. Cancer Cell. 2017;31:127–141. doi: 10.1016/j.ccell.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Wei H, Yi J, Xie B, Chen J, Zhou C, Wang L, Yang Y. Chronic CagA-positive Helicobacter pylori infection with MNNG stimulation synergistically induces mesenchymal and cancer stem cell-like properties in gastric mucosal epithelial cells. J Cell Biochem. 2019;120:17635–17649. doi: 10.1002/jcb.29031. [DOI] [PubMed] [Google Scholar]
- Liu JF, Guo D, Kang EM, Wang YS, Gao XZ, Cong HY, Liu P, Zhang NQ, Wang MY. Acute and chronic infection of H pylori caused the difference in apoptosis of gastric epithelial cells. Microb Pathog. 2021;150:104717. doi: 10.1016/j.micpath.2020.104717. [DOI] [PubMed] [Google Scholar]
- Maixner F, Thorell K, Granehäll L, Linz B, Moodley Y, Rattei T, Engstrand L, Zink A. Helicobacter pylori in ancient human remains. World J Gastroenterol. 2019;25:6289–6298. doi: 10.3748/wjg.v25.i42.6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroney MJ, Ciurli S. 'Nickel as a virulence factor in the Class I bacterial carcinogen, Helicobacter Pylori. Semin Cancer Biol. 2021;76:143–155. doi: 10.1016/j.semcancer.2021.04.009. [DOI] [PubMed] [Google Scholar]
- Merino E, Flores-Encarnación M, Aguilar-Gutiérrez GR. Functional interaction and structural characteristics of unique components of Helicobacter pylori T4SS. Febs j. 2017;284:3540–3549. doi: 10.1111/febs.14092. [DOI] [PubMed] [Google Scholar]
- Mueller D, Tegtmeyer N, Brandt S, Yamaoka Y, De Poire E, Sgouras D, Wessler S, Torres J, Smolka A, Backert S. c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J Clin Invest. 2012;122:1553–1566. doi: 10.1172/JCI61143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller A. Multistep activation of the Helicobacter pylori effector CagA. J Clin Invest. 2012;122:1192–1195. doi: 10.1172/JCI61578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumann M, Crabtree JE. Helicobacter pylori-induced epithelial cell signalling in gastric carcinogenesis. Trends Microbiol. 2004;12:29–36. doi: 10.1016/j.tim.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Patrad E, Khalighfard S, Amiriani T, Khori V, Alizadeh AM. Molecular mechanisms underlying the action of carcinogens in gastric cancer with a glimpse into targeted therapy. Cell Oncol (dordr) 2022;45(6):1073–1117. doi: 10.1007/s13402-022-00715-3. [DOI] [PubMed] [Google Scholar]
- Qi Z, Wang J, Li Y, Xu Y. MZF1 transcriptionally activated MicroRNA-328-3p suppresses the malignancy of stomach adenocarcinoma via inhibiting CD44. J Immunol Res. 2022;2022:5819295. doi: 10.1155/2022/5819295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugge M, Meggio A, Pravadelli C, Barbareschi M, Fassan M, Gentilini M, Zorzi M, Pretis G, Graham DY, Genta RM. Gastritis staging in the endoscopic follow-up for the secondary prevention of gastric cancer: a 5-year prospective study of 1755 patients. Gut. 2019;68:11–17. doi: 10.1136/gutjnl-2017-314600. [DOI] [PubMed] [Google Scholar]
- Ryu HS, Park DJ, Kim HH, Kim WH, Lee HS. Combination of epithelial-mesenchymal transition and cancer stem cell-like phenotypes has independent prognostic value in gastric cancer. Hum Pathol. 2012;43:520–528. doi: 10.1016/j.humpath.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Senbanjo LT, Chellaiah MA. CD44: a multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front Cell Dev Biol. 2017;5:18. doi: 10.3389/fcell.2017.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Huang J, Hu J. m(6)A RNA methylation regulators contribute to malignant progression and have clinical prognostic impact in gastric cancer. Front Oncol. 2019;9:1038. doi: 10.3389/fonc.2019.01038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukri A, Hanafiah A, Mohamad Zin N, Kosai NR. Epidemiology and role of Helicobacter pylori virulence factors in gastric cancer carcinogenesis. APMIS. 2020;128:150–161. doi: 10.1111/apm.13034. [DOI] [PubMed] [Google Scholar]
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- Takaishi S, Okumura T, Shuiping Tu, Wang SSW, Shibata W, Vigneshwaran R, Gordon SAK, Shimada Y, Wang TC. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells. 2009;27:1006–1020. doi: 10.1002/stem.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Ong CW, Shi J, Srivastava S, Yan B, Cheng CL, Yong WP, Chan SL, Yeoh KG, Iacopetta B, Salto-Tellez M. Sequential expression of putative stem cell markers in gastric carcinogenesis. Br J Cancer. 2011;105:658–665. doi: 10.1038/bjc.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Meng W, Wang B, Qiao L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014;345:196–202. doi: 10.1016/j.canlet.2013.08.016. [DOI] [PubMed] [Google Scholar]
- Wang D, Qu X, Lu W, Wang Y, Jin Y, Hou K, Yang B, Li C, Qi J, Xiao J, Che X, Liu Y. N(6)-Methyladenosine RNA demethylase FTO promotes gastric cancer metastasis by down-regulating the m6A methylation of ITGB1. Front Oncol. 2021;11:681280. doi: 10.3389/fonc.2021.681280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Levi E, Zhu S, Du J, Majumdar AP. Cancer stem cells biomarkers in gastric carcinogenesis. J Gastrointest Cancer. 2013;44:428–435. doi: 10.1007/s12029-013-9534-2. [DOI] [PubMed] [Google Scholar]
- Yang Z, Jiang X, Zhang Z, Zhao Z, Xing W, Liu Y, Jiang X, Zhao H. HDAC3-dependent transcriptional repression of FOXA2 regulates FTO/m6A/MYC signaling to contribute to the development of gastric cancer. Cancer Gene Ther. 2021;28:141–155. doi: 10.1038/s41417-020-0193-8. [DOI] [PubMed] [Google Scholar]
- You Y, Fu Y, Huang M, Shen D, Zhao B, Liu H, Zheng Y, Huang L. Recent advances of m6A demethylases inhibitors and their biological functions in human diseases. Int J Mol Sci. 2022;23(10):5815. doi: 10.3390/ijms23105815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu XW, Xu Y, Gong YH, Qian X, Yuan Y. Helicobacter pylori induces malignant transformation of gastric epithelial cells in vitro. APMIS. 2011;119:187–197. doi: 10.1111/j.1600-0463.2010.02709.x. [DOI] [PubMed] [Google Scholar]
- Zavros Y. Initiation and maintenance of gastric cancer: a focus on CD44 variant isoforms and cancer stem cells. Cell Mol Gastroenterol Hepatol. 2017;4:55–63. doi: 10.1016/j.jcmgh.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Zuo Y, Peng Y, Zuo L. Function of N6-Methyladenosine modification in tumors. J Oncol. 2021;2021:6461552. doi: 10.1155/2021/6461552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Wornow S, Peehl DM, Rankin EB, Brooks JD. The controversial role and therapeutic development of the m6A demethylase FTO in renal cell carcinoma. Transl Oncol. 2022;25:101518. doi: 10.1016/j.tranon.2022.101518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Yan X, Wang Y, Zhou J, Yu Y. N6-methyladenosine regulators promote malignant progression of gastric adenocarcinoma. Front Oncol. 2021;11:726018. doi: 10.3389/fonc.2021.726018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou D, Dong L, Li C, Yin Z, Rao S, Zhou Q. The m(6)A eraser FTO facilitates proliferation and migration of human cervical cancer cells. Cancer Cell Int. 2019;19:321. doi: 10.1186/s12935-019-1045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.