

Abstract
Cancer-associated fibroblasts (CAFs), a significant component of the tumor microenvironment (TME), contribute to cancer progression through the secretion of extracellular matrix (ECM), growth factors, and metabolites. It is now well recognized that CAFs are a heterogenous population with ablation experiments leading to reduced tumor growth and single-cell RNA sequencing demonstrating CAF subgroups. CAFs lack genetic mutations yet substantially differ from their normal stromal precursors. Here, we review epigenetic changes in CAF maturation, focusing on DNA methylation and histone modifications. DNA methylation changes in CAFs have been demonstrated globally, while roles of methylation at specific genes affect tumor growth. Further, loss of CAF histone methylation and gain of histone acetylation has been shown to promote CAF activation and tumor promotion. Many CAF activating factors, such as transforming growth factor β (TGFβ), lead to these epigenetic changes. MicroRNAs (miRNAs) serve as targets and orchestrators of epigenetic modifications that influence gene expression. Bromodomain and extra-terminal domain (BET), an epigenetic reader, recognizes histone acetylation and activates the transcription of genes leading to the pro-tumor phenotype of CAFs.
1. Heterogeneity of CAFs: an emerging concept
Cancer-associated fibroblasts (CAFs), a major component of the tumor stroma, play roles in all hallmarks of cancer. One of their well-known functions is the synthesis, deposition, and remodeling of extracellular matrix (ECM) proteins, such as collagens, laminins, and glycoproteins, which promote tumor progression and metastasis1. In addition, CAFs can also promote tumor growth through the secretion of metabolites, extracellular vesicles containing microRNAs (miRNAs), and growth factors. These growth factors include transforming growth factor β (TGFβ), hepatocyte growth factor (HGF), leukemia inhibitory factor (LIF), fibroblast growth factor 5 (FGF5), epidermal growth factor (EGF), and connective tissue growth factor (CTGF), which have been linked to proliferation, invasion, and therapy resistance in cancer cells1–3. Further, CAFs modulate other cells in the tumor microenvironment (TME), such as endothelial cells and immune cells, leading to angiogenesis, inflammation, and evasion of the antitumor immune response1. These immune-modulating roles have been shown to contribute to resistance to immunotherapy4–6.
However, recent studies have challenged the traditional tumor-promoting role of CAFs. In mouse models of pancreatic cancer (PC), depleting alpha-smooth muscle actin (αSMA+) CAFs or targeting stromal signaling led to the development of aggressive, poorly differentiated tumors and immunosuppression with decreased survival7–9. Further studies in breast and colon cancers have also shown the tumor-restraining roles of CAFs10. These dichotomist functions of CAFs have led to the concept of their heterogeneous nature with both tumor-promoting and tumor-restraining roles, which is supported by single-cell RNA sequencing (RNA-seq) in both murine and human tumors. This transcriptional heterogeneity of CAFs allows for their division into different subgroups. While there is yet to be a consensus nomenclature for CAF subgroups, various studies have categorized CAFs based on their transcriptional profiles into different categories. Some common CAF subtypes include myofibroblast CAFs (myCAFs), inflammatory CAFs (iCAFs), antigen-presenting CAFs (apCAFs), matrix CAFs (mCAF), and vascular CAFs (vCAFs), as outlined in Table 111–13. The mechanistic contribution of these CAF subtypes to cancer pathobiology is an active area of investigation.
Table 1:
Characteristic features of the unique cancer-associated fibroblasts subtypes.
| CAF subtype | CAF subtype markers | CAF subtype functions | References |
|---|---|---|---|
| LRRC15+ CAF | LRRC15 | Poor response to anti-PD-L1 cancer immunotherapy increases with tumor progression | Dominguez et al.149 |
| PDPN+ CAFs | PDPN | Worse prognosis | Hu et al. 150, Friedman et al.151 |
| FAP+ CAF | FAP | Block CD8+ antitumor T cells | Zhang et al.152, Feig et al.153, Grout et al. 154 |
| Steady state-like CAF | PI16, DPP4 | Similar to NF | Foster et al.155 |
| Mechanorespo nsive CAF | αSMA, POSTN, FSP1, PDGFRα | Express mechanosensitive signaling mediators and ECM components | Foster et al.155 |
| Immunomodul atory CAF | Il1r1, Myd88, Il6st, Cxcl1 | Modulates inflammatory TME | Foster et al.155 |
| CD10+GPR77 + CAF | CD10, GPR77 | Sustain cancer stemness, promote cancer formation and chemoresistance | Su et al. 156 |
| ECM CAF | MMP14, LOXL2, POSTN | ECM remodeling, fatty acid metabolism, peroxisome, invasion, shorter overall survival | Li et al.157, Valdés-Mora et al.158 |
| PDGFRα+ SAA1+ CAF | PDGFRα, SAA1 | Stimulate tumor growth in mice | Djurec et al.159 |
| Cancer-restraining CAF | Meflin | Favorable patient outcome, tumor vessel perfusion, regulate collagen structure | Mizutani et al.160 |
| Activated metabolic state CAF | PLA2G2A, CRABP2 | Highly active glycolysis, increased metastasis, and poor prognosis, found in patients with low desmoplasia | Wang et al.161 |
| Igfbp5+ CAF | IGFBP5, FN1, LY6C1 | Found in early PanIN lesions | Schlesinger et al.162 |
| Complement-secreting CAF | C3, C7, CFD | Complement system, regulates immune and inflammation response | Chen et al.163 |
| Classical CAF | COL1A1, FAP | ECM deposition | Chen et al.163 |
| FAP+ αSMA+ CAF | αSMA, FAP | Dense ECM deposition, decreases T-cell infiltration, present in early-stage tumors | Grout et al.154 |
| MYH11+ αSMA+ CAF | αSMA, MYH11 | Dense ECM deposition, which decreases T-cell infiltration, appears in more advanced tumors | Grout et al.154 |
| S100A4+ CAF | FSP1 | ECM remodeling, antigen presentation | Friedman et al.151 |
| CD53high CAF | CD53 | Small proportion of CAFs, cytoplasmic intermediate filament, matrix glycoproteins, integrins, MMP inhibitors | Sebastian et al.164 |
| Crabp1high CAF | CRABP1 | Small proportion of CAFs, ECM proteins, collagens, laminins, MMPs | Sebastian et al.164 |
| Mesothelial CAF | UPK1B, MSLN, KRT19 | Portal fibroblast and mesothelial markers | Affo et al.165 |
| Portal CAF | PRELP, PDGFRα, MMP23B | Minority CAF population, widely interacts with other TME cells, decreases angiogenesis, tumor restraining function | Chiavarina et al. 166 |
| Vascular smooth muscle CAF | CNN1, MYH11 | Unknown | Chiavarina et al. 166 |
| Entoderm-related CAF | COL1A1, POSTN, CTHRC1 | Secrete growth factors, negatively associated with the abundance of M1 macrophages | Zhao et al.167 |
| Adhesion-related CAF | RGS5, NDUFA4L2, ADIRF | Adherens junctions, decreases patient survival | Zhao et al.167 |
| Vascular-related CAF | IGLC7, SPINK4, TFF1 | Vasculature development | Zhao et al.167 |
| Mesenchyme-related CAF | CXCL14, TMEM176B, F3 | Immune checkpoint interactions with other CAF subtypes | Zhao et al.167 |
| Endoplasmic reticulum-related CAF | SERPINE1, IGF1, S100A10 | IL-6 signaling, negatively associated with the abundance of M2 macrophages | Zhao et al.167 |
| Cell cycle-related CAF | HIST1H4C, TK1, BIRC5 | Increases patient survival, interacts with other CAF subtypes via the TIMP1-CD63 signaling pathway | Zhao et al.167 |
| Divergent CAF | PAX3, NRP2, EDNRB | Mesenchymal/neural crest development and amoeboidal cell movement | Costea et al.168 |
| Interferon-regulated CAF | SLC14A1 | Induced by IFN signaling, confers stemness to cancer cells via the WNT5A paracrine pathway, unfavorable clinical outcomes | Ma et al.169 |
| STAR+ CAF | STAR, TSPAN8, ALDH1A1 | Enriched after chemotherapy | Loret et al.170 |
CAF heterogeneity is derived partly from the various sources of precursor cells that contribute to CAFs during cancer progression. One obvious source of CAFs is tissue resident normal fibroblasts (NF). These cells have been shown to be activated in vitro, developing a CAF-like phenotype. Additional sources of CAF precursor cells have been recognized, including bone marrow-derived mesenchymal stem cells (BM-MSCs), adipose tissue-derived MSCs (AD-MSCs), adipocytes, pericytes, endothelial cells, fibrocytes, and mesothelial cells14. Whether and to what percentage these precursor cells contribute to the CAF population varies with cancer type. In E0771 murine breast tumors, around half of the myCAFs were derived from local adipose tissues such as adipocytes and AD-MSCs15. A recent study in PC demonstrated that local pancreatic stellate cells (PSCs) contribute to only 10% to 15% of the CAF population in a KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx1-Cre (KPC) orthotopic cell model, demonstrating the likely contribution by distantly recruited precursor cells16. BM-MSCs contributed to approximately 40% of myCAFs in PC and 20% in gastric and ovarian cancers15,17,18.
In addition to variation from cancer type, it has been suggested that the proportion of CAF precursors varies within the cancer type15. Supporting this, Helms et al. defined a marker combination based on RNA-seq analysis to define PSC-derived CAFs, which was used to examine a patient population. The study revealed that some patients harbored relatively high levels of putative PSC-derived CAFs while others had very low levels16. Recent studies have also suggested that some of the heterogeneity arises from the tumor genotype demonstrated by differences in CAFs between p53-mutant and p53-null PC19. This heterogeneity was shown to be due to the differential contribution of PSCs to the CAF population with loss of p53 reducing the PSC-derived CAF frequencies16. To contribute to the CAF population, the distant precursor cells must localize to the tumor. We have recently shown that mucin 5AC, secreted by PC cells, localizes to adipose tissue and promotes CD44/CD29 clustering, leading to the migration of AD-MSCs20. Once recruited to the tumor, cancer, and immune cell-secreted factors educate these precursor cells to mature into their activated form21. Some of these factors include TGF-β, interleukin-6 (IL-6), platelet-derived growth factor (PDGF), extracellular vesicles containing miRNAs, hypoxia, oxidative stress, and matrix stiffness1–3. Activation of CAFs begins early in neoplastic lesions, demonstrated by the presence of a proinflammatory gene signature in CAFs from early lesions21. The ability of cancer cells to activate CAFs is demonstrated by their activation in NF or BM-MSC co-implantation models, leading to larger tumors compared to cancer cells alone22,23.
CAFs lack genetic mutations yet substantially differ in functions and characteristics from their precursor cells24–28. This has been demonstrated by the transcriptional differences between CAF precursor cells, such as NFs and MSCs, compared to tumor-isolated CAFs29–32. Further, marked functional differences between NFs and CAFs are observed during co-implantation experiments, where the presence of CAFs significantly promotes tumor growth compared to NFs33. Therefore, recent studies focus on the epigenetic regulation of CAFs, including changes in DNA methylation and histone modifications during CAF maturation that may contribute to this functional difference and transcriptional heterogeneity. Furthermore, epigenetic modifications in myofibroblasts in the context of fibrosis are well characterized34–37. Additionally, normal differentiation of the common CAF precursor, MSCs, is epigenetically mediated38.
Here we review the role of two main epigenetic processes: DNA methylation and histone modifications during CAF maturation. These epigenetic modifications regulate the expression of genes and are heritable, allowing for the continued expression of genes associated with the CAF phenotype.
2. Epigenetic modifications during CAF maturation
2.1. DNA methylation
DNA methylation is a covalent modification occurring on approximately 70% of cytosines in CpG sequences in normal cells39. These CpGs are often found in clusters called CpG islands. Hypermethylation can occur at CpG islands located in gene promoters, causing a closed chromatin state and gene repression. DNA methylation is regulated by DNA methyltransferases (DNMTs). DNMT1 is responsible for maintenance methylation, while DNMT3a and DNMT3b are involved in de novo methylation40. The ten-eleven translocation (TETs) methylcytosine dioxygenases oxidize 5-methylcytosines (5mC), leading to the removal of DNA methylation marks. DNA demethylation can also occur passively when methylation patterns are not maintained after cell division leading to replication-dependent dilution of 5mCs41.
Genome-wide hypomethylation and promoter hypermethylation are hallmarks of many cancers and are associated with tumorigenesis. These methylation changes occur early in tumorigenesis and often increase with tumor progression42. The genome-wide hypomethylation can increase genomic instability, and hypermethylation of the CpG islands in gene promoters can silence tumor suppressor genes affecting key cellular processes such as cell cycle, apoptosis, DNA repair, and angiogenesis43. DNMTs are frequently overexpressed in cancers and correlate with poor prognosis in patients44. On the other hand, decreased expression and inhibition of TET enzymes occur in cancer. The miRNAs (miR-22) overexpressed in cancers have been reported to directly target TET proteins. Likewise, mutations in the metabolic genes ldh1/2, Sdh, and Fh are found in a wide variety of solid tumors and can inhibit TETs, leading to a profound impact on the DNA methylation patterns41.
2.1.1. DNA hypomethylation in CAFs
DNA methylation is a well-studied epigenetic alteration in CAFs, perhaps due to recent advancements in genome-wide DNA methylation profiling techniques. Cancer cells present global DNA hypomethylation combined with local DNA hypermethylation. Several studies have suggested a similar pattern in CAFs. As early as 1990, hypomethylation of stromal genes was demonstrated in colon cancer45,46. More recently, this hypomethylation of colon CAFs was shown to be more global47. Likewise, CAF DNA hypomethylation with focal gains of methylation was also observed in breast, gastric, and lung cancers48–51. In lung CAFs, these DNA methylation changes affected pathways associated with ECM/focal adhesions and the FC-γ receptor49. Genes that were hypomethylated include runt-related transcription factor 1 (Runx1), C22orf9m, mi-R1249, and neurotrimin (Ntm)49. Dietary folic acid is necessary for DNA methylation52. In a murine model, folic acid supplementation prevented this loss of global DNA methylation in both dysplastic gastric epithelial cells and gastric CAFs53, and a dietary folate deficiency has been associated with an increased risk of colon cancer54. Additionally, a population of CAFs has the inflammatory senescence-associated secretory phenotype (SASP)55,56. Senescent fibroblasts were also shown to have DNA hypomethylation with focal gains of methylation57.
2.1.2. Aberrant DNA Methylation
While the pattern of global DNA hypomethylation holds in CAFs from several cancers, recent studies demonstrate that CAF DNA methylation depends on the cancer type, with some CAFs showing aberrant, not just decreased methylation (Figure 1). Whole-genome bisulfite sequencing of prostate cancer patients in matched NFs and CAFs showed differential methylation in CAFs with 7,534 distinct differentially hypomethylated or hypermethylated regions both in gene bodies and promoter regions. These differentially methylated regions (DMRs) are enriched for regulatory elements, including strong enhancers and active promoters that are associated with functional changes in genes related to developmental processes and binding of transcription factors such as the T-box (TBX), forkhead box (FOX), and homeobox (HOX) gene families. Additional changes were seen in ligand-activated cell signaling, including the TGFβ pathway and estrogen receptor (ERα) signaling 58. Hypermethylated genes in CAFs included EBF transcription factor 1, EPH receptor B6, and homeobox D8, while syndecan 2, ATP binding cassette subfamily B member 4, estrogen receptor 1, and thrombospondin 2 were hypomethylated58. In another study using CAFs from 18 prostate cancer patients, the methylation profile changed consistently in 80% of DMRs, while the remaining DMRs varied according to the severity of the disease. Consistently changed DMRs were in genes relating to cell adhesion and ligand-activated cell signaling, including TGFβ, insulin, and PDGF signaling pathways. Notably, GATA binding protein 6 was hypermethylated and hypomethylated genes were paired like homeodomain 2 and A-kinase anchoring protein 2. DMRs in patients with more aggressive tumors showed hypomethylation at the promoter region of the ectodysplasin-A receptor-associated adapter protein, which was associated with poor clinical features such as stage and increased lymph node involvement and patient outcomes59.
Figure 1. DNA methylation changes during CAF maturation.
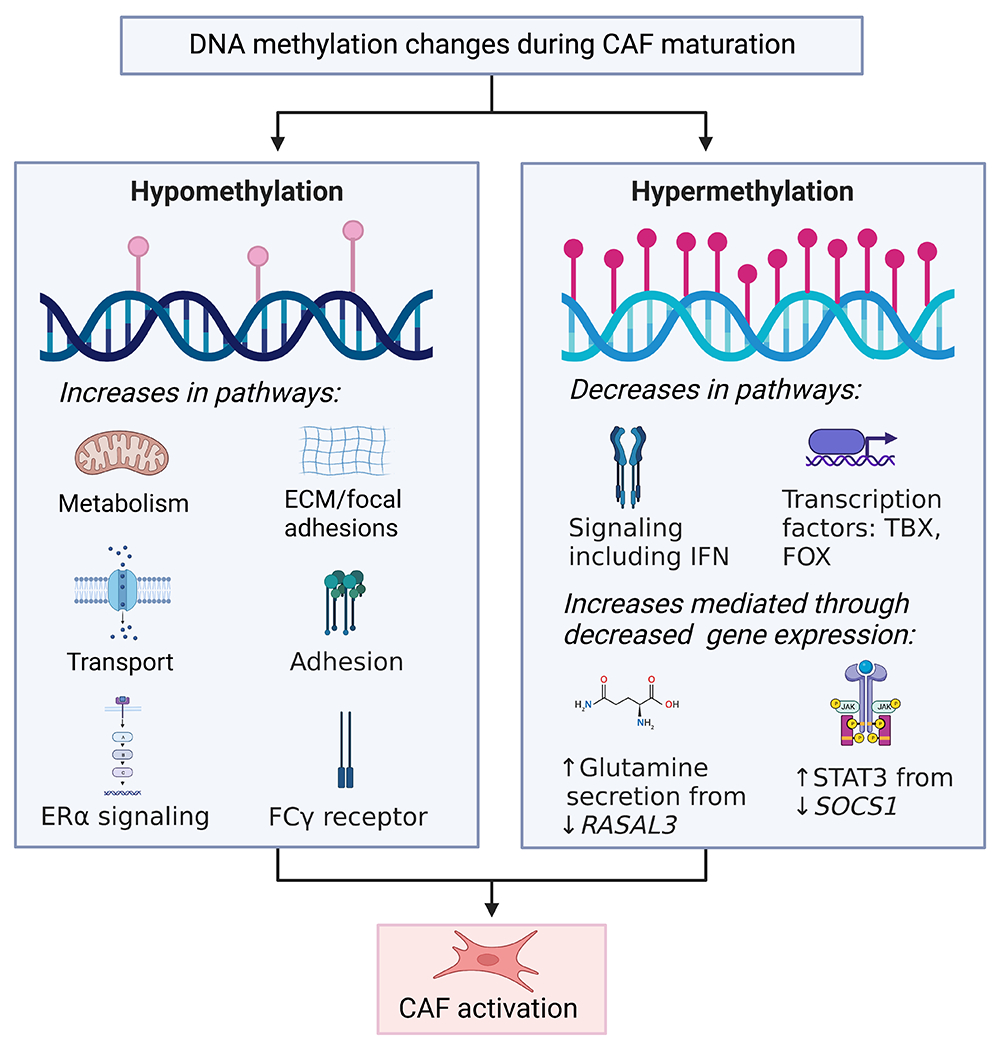
CAFs undergo both DNA hypermethylation and hypomethylation, leading to increases and decrease in signaling pathways causing CAF activation.
Similarly, in colon cancer, differential methylation of 1,772 cytosine residues at CpG dinucleotides was identified between NFs and CAFs. Of these, 60% were hypomethylated, and 40% were hypermethylated in CAFs compared to NFs. This aberrant methylation pattern leads to the upregulated expression of genes involved in metabolism/transport, including albumin, ankyrin 1, and argininosuccinate lyase and adhesion/signaling, including CD83, cholinergic receptor nicotinic alpha-1-subunit, and collagen (Col4a6) due to promoter hypomethylation. Decreased expression of genes involved in signaling and transcription factors, including AT-hook transcription factor, Foxa2, Tbx1, paired box (Pax)-3, and Pax8, was associated with promoter hypermethylation60. On similar lines, 12,364 genome-wide DMRs were found in podoplanin (PDPN+) mouse breast CAFs compared to NF, consisting of approximately 80% hypomethylation and 20% hypermethylation regions. Specifically, hypomethylation and gene upregulation corresponding to tumor necrosis factor(TNF)-α and TGFβ signaling, transcription factors such as Runx1, inflammatory responses, and hypoxia were seen in CAFs. Hypermethylation was enriched in adipogenesis and myogenesis-related genes61. In primary culture from matched CAFs and NFs from 26 lung cancer patients, there were close to 15,000 differentially methylated CpG sites, out of which 60% were hypomethylated, including genes sulfatase 1, Fosl2, homeodomain interacting protein kinase 3, and Fgf14, while 40% were hypermethylated involving genes, BCL2 related protein A1, and tenascin XB in CAFs compared to NF. As smoking is a significant risk factor for cancer development, a study comparing the global methylation differences between NFs and CAFs showed differential methylation of close to 4,000 CpGs in CAF and NF with greater methylation changes in ever-smokers compared to nonsmokers62. Likewise, exposing lung NFs to cigarette smoke condensate increased methylation at their Smad3 promoter63. While not explicitly shown in CAFs, alcohol, another cancer risk factor and CAF inducer, can also alter methylation64,65. Additionally, aging modifies the methylation patterns in CAF precursor cells66,67.
In addition to protein-coding genes, methylation changes have also been seen in interspersed repeat sequences such as long interspersed nuclear element-1 (LINE-1). LINE-1 transcription produces antisense RNAs, which bind to pre-mRNAs targeting them for argonaute-mediated RNA degradation. Hence, the genes containing LINE-1 elements are downregulated. The coculture of breast cancer cells with NFs leads to increased methylation of LINE-1 in the NFs68. This LINE-1 methylation can abrogate the RNA degradation process increasing the expression of genes having global effects on fibroblast biology, as the human genome contains more than 500,000 copies of the LINE-1 transposone69.
Several studies have examined the methylation status of CAF-expressing genes important in carcinogenesis70–75. In prostate cancer, methylation of glutathione S-transferase P1 (Gstp1) was observed in the unique sub-microenvironments of the stromal regions76. Similarly, methylation-mediated silencing of Rasal3 in CAFs leads to macropinocytosis-mediated glutamine synthesis and secretion, which then drives glutamine metabolism in the epithelia. Inhibiting this process with either the macropinocytosis inhibitor 5-(N-ethyl-N-isopropyl)amiloride (EIPA) or the glutamine uptake inhibitor gamma-L-glutamyl-p-nitroanilide (GPNA) in an orthotopic xenograft model reduced the prostate cancer growth77. In PC, methylation of Socs1 increased the expression of pro-cancerous growth factors such as insulin-like growth factor 1 (IGF-1) and activation of STAT378. Additionally, in lung CAFs, Smad3 is silenced by hypermethylation, allowing these CAFs to sensitize to TGFβ signaling, increasing the deposition of ECM in the stroma63. In an interesting monozygous twin study, Brca1 was methylated in one twin, and the skin fibroblasts from the affected twin showed a CAF-like phenotype, overexpressing ECM-associated genes, pro-tumorigenic cytokines, and CAF markers, such as αSMA, fibroblast activation protein, and C-X-C motif chemokine ligand 12. Moreover, these Brca1 methylated fibroblasts exhibited accelerated proliferation and migration and ultimately enhanced the proliferation of lung adenocarcinoma cells, A54979. Importantly, methylation of specific genes in CAFs is associated with organ-specific metastasis in PC. When cocultured with MSCs, cell lines established from the primary tumors of mice with liver metastasis, but not those with lung metastasis, induced the methylation of metabolism genes involved in the glucose metabolic pathway and oxidative phosphorylation, including NAD(P)H quinone dehydrogenase 1 and aldehyde dehydrogenase 1 family member A380.
DNMT1 has been shown to promote the activation of fibroblasts. In the context of liver fibrosis, DNMT1-mediated methylation leads to the conversion of hepatic stellate cells (HSCs) into hepatic myofibroblasts81,82. Moreover, in breast cancer, DNMT1 ectopic expression activated breast NFs and promoted their pro-carcinogenic effects, both in vitro and in orthotopic tumor xenografts. DNMT1 upregulation in CAFs was shown to be mediated by an RNA binding protein, HuR, leading to decreased DNMT1 mRNA decay83.
Targeting DNA methylation with the DNA demethylating drug 5-aza-2’-deoxycytidine (5-AZA-dC) in a PC autochthonous murine model significantly inhibited tumor progression. Furthermore, the role of 5-AZA-dC on the stromal compartment was demonstrated by pretreating CAFs with 5-AZA-dC and co-injecting them with tumor cells in mice leading to an antitumor effect84. In another study, DNMT1 inhibition induced the expression of 42 genes, such as deleted in azoospermia like, gametocyte specific factor1, and Il18 in PC patient derived CAFs85. Further studies have looked at genes induced after 5-AZA-dC treatment and found induction of IFN pathway genes in bladder CAFs86. The 5-AZA-dC as well as a natural DNMT inhibitor, eugenol, suppressed pro-carcinogenic effects of breast CAFs both in vitro and in orthotopic tumor xenografts with MDA-MB-231 cell and CAF coimplantation87. Targeting DNA methylation is a promising clinical direction as aberrant DNA methylation is found both in CAFs and cancer cells.
2.2. Histone Modifications
Histone post-translational modifications (PTMs) cooperate with DNA methylation to decide the chromatin state88. The two most well-studied histone PTMs are methylation and acetylation. Unlike DNA methylation, histone PTMs are regulated by a myriad of enzymes, with over 120 different enzymes adding or removing PTMs on histones. Histone methylation leads to both gene activation and repression and is regulated by histone methyltransferases (HMTs) and histone demethylases (HDMs). These enzymes add or remove one to three methyl groups on histone lysine and arginine residues. Histone acetylation increases the accessibility to the gene promoters and enhancers of target genes, allowing for transcription factor binding and promoting gene expression. This process is controlled by histone acetyltransferases (HATs) and histone deacetylases (HDACs)89.
2.2.1. Histone Methylation
Several recent studies have highlighted the importance of histone methylation on CAF function. Maeda et al. examined H3K27me3 modification in 12 surgically resected gastric CAFs and matched NFs. While there was intertumoral heterogeneity, CAFs had distinct H3K27me3 patterns compared with matched NFs. Notably, loss of H3K27me3 in CAFs was enriched in genes involved in stem cell niche, tissue development, and stromal–epithelial interactions, such as Wnt family member 5A, noggin, insulin-like growth factor 2, and gremlim1, leading to their increased expression90. Of these genes, Wnt5a most frequently loses H3K27me3 marks and has been reported to promote gastric cancer cell migration and invasion90,91. Interestingly, CAFs had much higher expression of WNT5A than gastric cancer cell lines, and inhibiting WNT5A-mediated signaling in cancer cells with an antagonist, Box 5, reduced the protumorigenic effects of CAF-conditioned media90. Loss of H3K27me3 was also shown in breast CAFs92. Additionally, in breast cancer, overexpression of the H3K36 demethylase, KDM2A, induced p53-dependent senescence in hTERT immortalized normal human breast fibroblasts and promoted an iCAF phenotype with enhanced secretion of cytokines, including IL-6, IL-8, and CXCL1. The knockdown of KDM2A decreased CAF-promoted growth of MDA-MB-231 cells in a co-implantation study93. Furthermore, in ovarian cancer, nicotinamide N-methyltransferase (NNMT) expression in CAFs promotes cytokine secretion and deposition of ECM due to the depletion of S-adenosyl methionine-mediated histone hypomethylation. The NNMT knockdown significantly increased H3K4 and H3K27 trimethylation. In a syngeneic co-implantation model, the knockdown of stromal NNMT reduced the tumor burden94. Taken together, these studies demonstrate pro-tumor effects from loss of CAF histone methylation and suggest targeting histone demethylases like KDM2A as a promising therapeutic option (Figure 2).
Figure 2. Epigenetic writers, erasers, and readers that promote or inhibit CAF maturation.
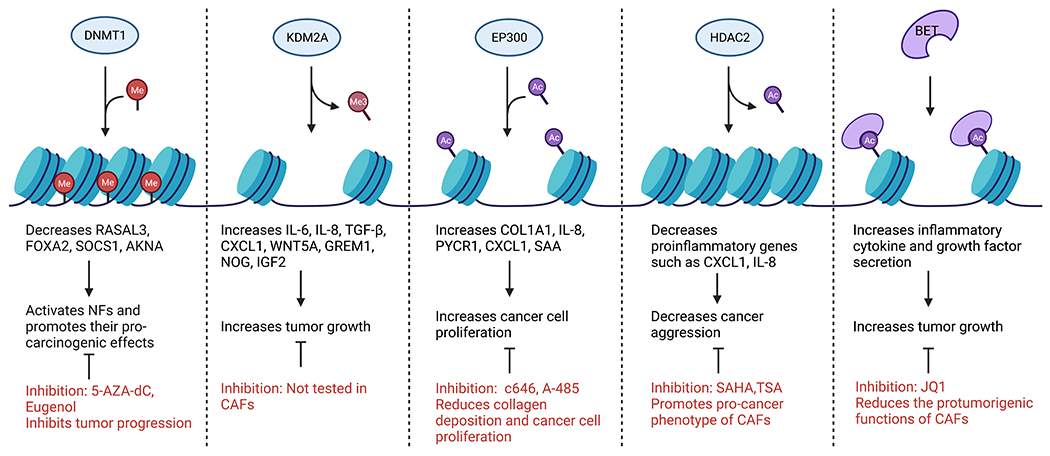
Epigenetic enzymes add, remove, or interact with histone PTMs and DNA methylation. Inhibiting these enzymes can have positive or negative effects on CAF function.
2.2.2. Histone Acetylation
A common histone mark of active transcription, H3K27ac, has been shown to be important for CAF activation in several studies. In PC stroma, the presence of H3K27ac was shown with immunohistochemical analysis95. In gastric cancer, ChIP-qPCR analysis shows increased H3K27ac in enhancer regions and the promoter region of one of the highly upregulated genes in gastric CAFs, serum amyloid 1, from patient-matched CAFs compared to NFs96. Likewise, breast CAFs were shown to have increased H3K27ac compared to matched NFs in the promoter regions of genes, such as Col1a1 and pyrroline-5-carboxylate reductase 1, which are important for collagen biosynthesis. Mechanistically, this acetylation was mediated by the HAT, EP300, and inhibition or knocking down of EP300 decreased collagen deposition and reduced cancer cell proliferation under coculture conditions97.
Cancer cell-derived exosomal HSPC111 promotes CXCL5 secretion by HSCs due to increased H3K27ac marks at the Cxcl5 promoter. An HDAC inhibitor, Trichostatin A (TSA), significantly enhanced HSPC111-induced CXCL5 expression, and TSA alone increased H3K27ac in the Cxcl5 promoter. Importantly, CXCL5 further promoted HSPC111 exosomal secretion from cancer cells leading to colon cancer liver metastases98. Additionally, in a 3D lung cancer model addition of CAFs increased the IC50 of three different HDAC inhibitors99. Likewise, fibroblasts treated with HDAC inhibitors, sodium butyrate or suberanilohydroxamic acid (SAHA) promoted tumor growth of preneoplastic cells and an invasive phenotype by an associated increase in the expression of osteopontin in vivo100.
Further evidence also supports that HDAC inhibitors promote the pro-cancer phenotype of CAFs in PC. The HDAC inhibitor, SAHA (Vorinostat), enhanced the aggressiveness of cancer cells via promoting CAF secretion of tumor-supportive, proinflammatory cytokines CXCL1 and IL-8. This was demonstrated both in vitro and in subcutaneously implanted MIA PaCa-2 cells. Moreover, HDAC2 was shown to bind the regulatory regions for a large group of proinflammatory genes in CAFs, especially Ap1101. Therefore, blocking this deacetylation would promote the expression of these tumor-supportive genes. However, another study using PSCs found that HDAC inhibition decreased cell proliferation, αSMA expression, and collagen synthesis102. These dichotomist results could possibly be explained by the small proportion of PSCs that contribute to the PC CAF population16. Overall, histone acetylation has been shown to promote CAF activation and tumor-promoting function. Therefore, the use of HDAC inhibitors like TSA and SAHA needs caution, while HAT inhibitors, c646, and A-485, could be promising therapeutic drugs.
3. Mediators of epigenetic regulation in CAFs
3.1. Transforming Growth Factor β (TGFβ)
One of the well-recognized soluble factors leading to CAF activation is TGFβ. Notably, TGFβ is well characterized to induce epigenetic changes, both DNA methylation and histone modifications103–106. In the context of CAFs, TGFβ has been shown to regulate DNA methylation. Lung NF treated with TGFβ exhibited reduced DNA methylation, recapitulating changes seen between CAFs and NFs49. Furthermore, breast NFs treated with TGFβ enhanced DNMT3B expression leading to methylation at the promoter region of miRNA200s, setting up an autoregulatory loop of TGF-β1/miR-200s/miR-221/DNMT3B leading to sustained activation of CAFs even in the absence of cancer cells107. Using a different approach of knocking out the TGFβ receptor (TGFβR) or TGFβ antagonist treatment of prostate CAFs increased DNMT1 expression and activity by decreasing proteasome-mediated DNMT1 degradation leading to increased DNA methylation at promoter regions108. The genes involved in DNA damage repair and metabolizing reactive oxygen species (ROS) carry higher promoter hypermethylation with TGFβR knockout, leading to a biological consequence of DNA damage indicated by γ-H2AX and Rad52 expression108. This hypermethylation of ROS metabolizing genes can lead to increased ROS levels inducing further epigenetic changes and differentiation of NF to myofibroblasts109.
As DNA methylation and histone modifications cooperate to cause changes in chromatin structure, it is not surprising that TGFβ also leads to histone modifications. In fact, an HDAC 1/3/8 inhibitor, Scriptaid, prevented the TGFβ-induced maturation of endothelial cells into myCAFs. Functionally, this selective HDAC inhibitor decreased TGFβ induced expression of ECM components and reduced CAF contractility, stiffness, and the invasion of CAF/ D4M tumor cell spheroid cocultures. Moreover, Scriptaid decreased tumor growth in murine melanoma B16F10 models using both Scriptaid intraperitoneal injections and pretreatment of CAFs with Scriptaid110. In addition to histone acetylation, TGFβ also regulates histone methylation during CAF maturation. In immortalized mouse embryonic fibroblasts, TGFβ increased protein arginine methyltransferase 1 and 4 (PRMT1 and PRMT4) expression in a Snail1-mediated manner leading to H3 and H4 methylation at the fibronectin promoter. Importantly, this methylation was required for fibroblast activation111. Furthermore, TGFBR2 knockout in human prostate CAFs elevated H3 trimethylation lysine 9 (H3K9me3) levels while it decreased H3K9Ac108. Overall, these studies show that TGFβ plays a significant role in promoting epigenetic changes during CAF maturation, which in turn modulates the growth of cancer cells.
3.2. Other factors
In addition to the well-known CAF activator, TGFβ, several other CAF activating factors have been shown to lead to epigenetic changes during CAF maturation (Figure 3). The proinflammatory cytokine LIF induces CAF maturation leading to ECM remodeling, cancer cell invasion, and poor clinical outcome112. Albrengues et al. showed that LIF initiates epigenetic modifications in human primary dermal fibroblasts, leading to the constitutive activation of JAK1/STAT3 signaling through the upregulation of DNMT3b113. Importantly, JAK/STAT3 signaling leads to CAF differentiation114. Further studies have examined the role of other proinflammatory cytokine-driven epigenetic reprogramming in CAFs. Specifically, IL-1α, IL-1β, and tumor necrosis factor α (TNFα) downregulate the expression of histone methyltransferase, EZH2, leading to demethylation of H3K27me3 and enhanced peritoneal tumor formation of gastric cancer through JAK/STAT3 signaling in a mouse model115. Furthermore, in breast cancer, IL-6 activates NFs through the JAK2/STAT3 pathway, which is mediated by DNMT183,116. In addition to cytokines, cancer cell-secreted metabolites have been shown to mediate epigenetic changes during CAF maturation. For example, in PC, the neoplastic cell produced lactate increased α-ketoglutarate (αKG) production in MSCs, leading to TET activation117.
Figure 3. Common CAF subtypes are derived from various CAF precursors after exposure to CAF activating factors.
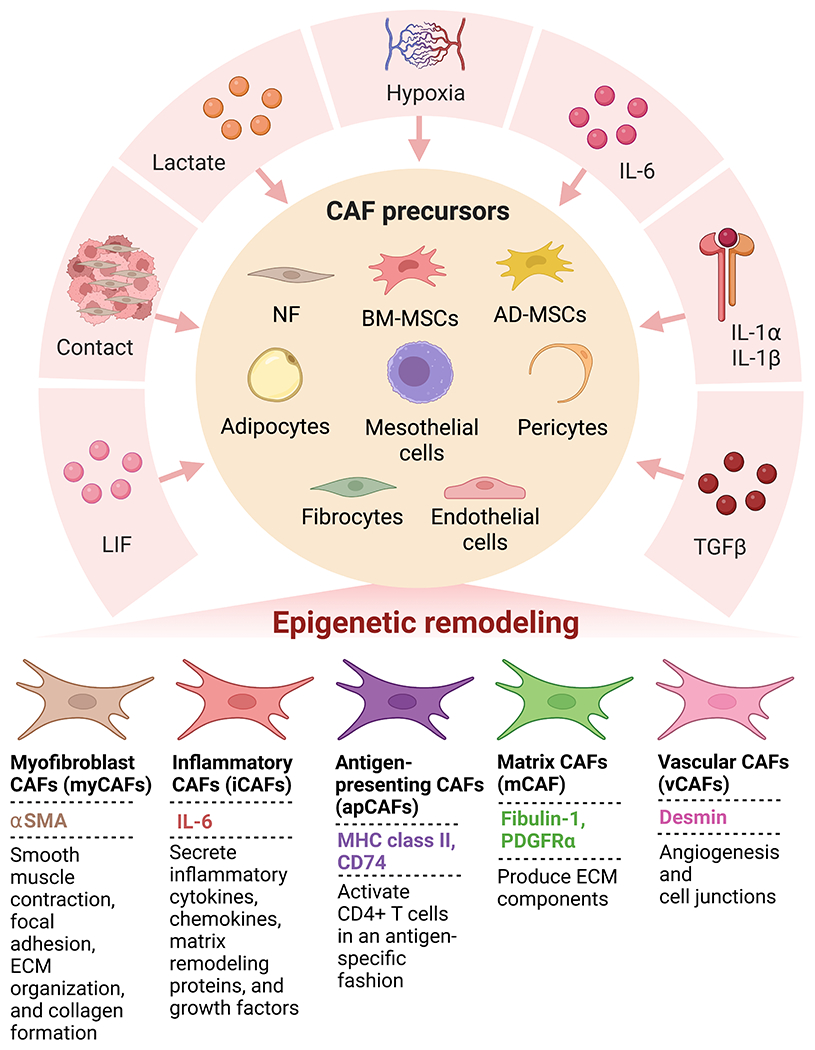
Factors secreted by cancer cells and other TME cells cause CAF precursors to mature into CAFs through epigenetic mechanisms.
Along with these cancer cell-secreted molecules, hypoxia also induced NF epigenetic reprogramming leading to the development of a pro-glycolytic phenotype with a CAF-like transcriptome. In human breast NFs, hypoxia leads to hypoxia-inducible factor 1 (HIF-1α) promoter hypomethylation and elevated HIF-1α levels, which remained high even upon reoxygenation118. HIF-1α directly activated the transcription of metabolic genes encoding glucose transporters and glycolytic enzymes, allowing CAFs to provide metabolic support to proliferating cancer cells119,120. A study in PC showed that direct cell-to-cell contact was necessary for the induction of Socs1 methylation in BM-MSCs78. Furthermore, in dermal NFs, UVA exposure increased Notch2 promoter methylation, leading to the downregulation of Notch2. Loss of downstream Notch signaling in these NF promoted increased tumor cell proliferation mediated through higher levels of diffusible growth factors, inflammatory cytokines, and matrix-remodeling enzymes121.
4. MiRNAs in epigenetic regulation during CAF maturation
MiRNAs are an important constituent of the epigenomic regulatory network as they serve as targets and orchestrators of DNA methylation, RNA modifications, and histone modifications122. This give-and-take of epigenetic pathways and miRNAs creates a feedback loop that has a well-documented influence on gene expression within the tumor cells and fibroblast populations. During CAF maturation, the surrounding TME drives the transition from NFs to CAFs. The miRNAs have been implicated in this transitory process through their ability to both contribute to and respond to the surrounding microenvironment. A study conducted by Li et al. deconstructed the crosstalk between tumor cells, PGE2 signaling, and CAF, IL-6 signaling in gastric cancer. They identified hypermethylation of miR-149 in CAFs, via H. pylon-induced COX2/PGE2 pathway, which regulates proinflammatory IL-6 secretion leading to the induction of epithelial-mesenchymal transition (EMT) and stem-like traits in SGC-7901 cancer cells promoting gastric cancer tumorigenesis. In NFs, miR-149 inhibits fibroblast activation and tumor-promoting functions, as demonstrated by increased SGC-7901 colony formation, invasion, and migration with conditioned media derived from antagiomiR-149 treated NF. Further, these findings were validated in subcutaneously co-implanted SGC-7901 and NF cells. However, hypermethylation-induced silencing allows for CAF activation in a manner that takes advantage of the crosstalk between CAFs and tumor cells during their codependent evolution123. A separate study assessed miRNA expression in primary canine NF after coculturing with C2 mast tumor cells, which induced downregulation of miR-27a and members of the let-7 family in NF. This is interesting in the context of tumor transformation as cyclin G1 (CCNG1), a miR-27a target, is a growth-promoting cell cycle regulator124. Additionally, ROS, which promotes CAF maturation and modifies the proportion of CAF subtypes, is regulated by miRNAs in AD-MSCs125,126. Specifically, miR-29a-3p and miR-30c-5p downregulate DNMT3A, reducing its ability to methylate the upstream regulatory region and suppress the expression of the antioxidant enzyme superoxide dismutase 2 (Sod2), which is prudent in the highly hypoxic TME of many cancers125. The resulting low oxidative stress could suppress CAF maturation and tumor progression and dissemination127.
A notable consideration of the CAF lifecycle is the epigenetic mechanisms governing the sustained activated state of CAFs post-maturation. Hypermethylation is a prominent epigenetic regulatory process, with miRNA promoters commonly being subject to hypermethylation, leading to miRNA silencing128. While most studies are centered around these processes in tumor cells, it is possible to extrapolate the foundation of these findings to CAF epigenomic regulation. In fact, the capacity to form regulatory feedback loops allows CAFs to support their protumorigenic nature independent of tumor-CAF crosstalk. Considerably, most studies fail to address the capacity of CAFs to remain active in the absence of tumor cells. Holes in this field of research are being addressed in some studies; for example, a study conducted in breast cancer has shed light on an epigenetic regulatory loop that maintains pro-tumorigenic CAF activation107. This study dissected the miR-200s/miR-221/DNMT3B signaling axis and demonstrated that DNMT3B methylation reduces the levels of miR200s, which helps establish the activated state of CAFs. Additionally, the miR-200s/miR-221/DNMT3B signaling axis sustained TGFβ1 signaling, which maintained the activated CAF state. In fact, destroying the autocrine TGFβ1/miR200s/miR221/DNMT3B signaling restored the NF phenotype by demethylation of the miR200 promoters107.
5. Epigenetic readers: BETs are promising targets for CAFs
Epigenetic readers are proteins with docking sites that recognize and bind to different modifications laid down by epigenetic modifiers called “writers”129. These readers can be categorized into bromodomains, which recognize acetylated lysine residues on histones, and chromodomains, which recognize methylated lysine residues on histones and are linked to transcriptional repression through the formation of heterochromatin130. Currently, studies on chromodomains in CAFs are limited. Mechanistic studies show that bromodomain and extra-terminal domain (BET) proteins are regulators of multiple genes involved in carcinogenesis, and targeting this family is a promising therapeutic approach. BET inhibitors such as JQ1 and OTX015 can disrupt the interaction between BET proteins and acetylated histones, thus suppressing the transcription of multiple oncogenes. In the context of CAFs, there are three proposed mechanisms by which BET inhibitors could function. First, as previously discussed in this review, TGFβ has a multifaceted role in regulating the differentiation and heterogeneity of CAFs. A previous study in cardiac fibroblasts demonstrates reduced TGFβ levels upon BET inhibition131. An additional study done in PC CAFs adds weight to this theory as it showed that TGFβ increases the occupancy of the BET family member, bromodomain-containing protein 4 (BRD4), at the promoter regions of the profibrogenic and proinflammatory genes Col1a1 and Il6 where it served to recruit transcriptional factors and machinery. The inhibitor, JQ1, prevents BRD4 binding and acts as a suppressor of the TGFβ pathway132. The second proposed mechanism is through inhibiting hedgehog (Hh) signaling, another major activator of CAFs. In mouse embryonic fibroblasts, JQ1 reduces GL1, a major Hh target gene, transcription by preventing BRD4 binding132. While both these studies show BRD4 functions through recruiting transcriptional machinery, BRD4 is also able to recruit chromatin remodeling complexes133. Lastly, BET inhibition could target the non-epigenetic function of BRD4 in which it interacts with the acetylated NFκB p65 subunit leading to suppression of proinflammatory genes and induction of cell cycle arrest in CAFs. In fact, a study done by Wen et al. established the foundation for the aforementioned theories by demonstrating that BET inhibition decreases the protumorigenic functions of CAFs in colorectal cancer134. A study conducted by Kim et al. demonstrated the effectiveness of BET inhibitors in the context of CAF maturation. ATF3 and CSL, which are transcriptional repressors of growth factors, proinflammatory cytokines, and matrix remodeling proteins, work together to negatively regulate CAF activation135. These genes are downregulated in fibroblasts from precancerous actinic keratoses, skin lesions, and skin squamous cell carcinoma135,136. BET inhibition can counteract the effects of ATF3 or CSL loss and suppress CAF tumor-promoting properties in an in vivo model of SCC13 cells co-injected with CAFs135. Likewise, BET inhibition in PC CAFs reduces inflammatory cytokine and growth factor secretion, attenuates desmoplasia, and significantly reduces subcutaneous tumor growth132. These studies support the theory that BET inhibition can be exploited in targeting the tumor promoting functions of CAFs.
6. Perspectives/Conclusions
The combination of dense stroma and inherent drug resistance of the tumors are obstacles that hinder cancer therapeutic interventions. Remarkably, targeting CAFs has the potential to supersede both of these major obstacles. CAFs are major constituents of the impermeable stroma that plagues most therapeutic interventions. Aside from behaving as a physical barrier, CAFs facilitate the crosstalk between tumor cells and the surrounding TME137. The CAF secretome enhances chemoresistance by maintaining cancer stemness, inducing EMT, and promoting cancer cell survival138,139. In addition, many characteristic cancer-driving features of the tumor are ultimately rooted in the genetic instability observed within the tumor subpopulation, resulting in challenges at preclinical and clinical levels. In light of this, the genetic stability demonstrated by CAFs conceptually invites the model of developing therapeutics that target tumor-promoting CAFs.
An additional consideration of CAF regulation is their lifecycle. Similar to the approaches taken with chronic fibrosis, in which a mass of myofibroblasts deposit a surplus of ECM through their recruitment, proliferation, and maturation, it stands to reason that each stage of the CAF lifecycle should also be considered in the context of cancer140,141. Upon further inspection of each CAF phase–priming, maturing, fully mature, and senescent–a few concerns arise regarding targetability. The priming phase likely requires constant stimuli from the TME, which is already a challenge to target with tumor-directed therapies, so the feasibility of this approach is low with our existing knowledge 142. However, increasing understanding of the events and players involved in priming provides an opportunity for their targeting. As for the senescent phase, there is speculation about whether this is an obligatory fate for CAFs140,143. At the stage of full CAF maturation, stromal heterogeneity and the microenvironment are intertwined; therefore, treatment would have to be tailored to targeting interactions between all the TME constituents, which are still poorly understood. Targeting the early maturation phase, though a seemingly irreversible process, holds promise. This conjecture is founded on the premise that the reversible/irreversible regulation of CAF maturation is epigenetic in nature, and targeting the epigenetic modifiers could potentially result in the transdifferentiation to an inactive fibroblast phenotype.
Another, albeit intertwined, field of study, epigenetics, also provides druggable targets. As discussed in-depth throughout this review article, DNA methylation, histone methylation, and acetylation are key regulatory cogs of the epigenetic machinery, and dysregulation in this space is a bona fide hallmark of cancer. Epigenetic regulation is crucial in sustaining CAF heterogeneity as well as their transition from NF to activated fibroblasts141. The dysregulation of epigenetic modifications in cancer provides an opportunity for therapeutic intervention.
While there have been positive strides in epigenetic targeting for hematological malignancies, the same cannot be said for solid tumors144. Perhaps this is due to differential epigenetic regulation in the CAF population of solid tumors. It is important to note that most studies have been done to investigate epigenetic targeting of the tumor cells themselves145. However, as discussed, common epigenetic drugs such as the FDA-approved histone deacetylase inhibitor, SAHA, actuality promote the pro-cancer phenotype of CAFs. Future development of epigenetic drugs for solid tumors must take into careful consideration both the tumor and stromal populations. Literature screens support the notion that epigenetic therapies might be more effective in combination with other cytotoxic agents or in reversing acquired therapeutic resistance146. Examples of epigenetically targeting chemoresistance include targeting both DNA and histone modifications with the combination of 5-AZA-dC and the histone deacetylase inhibitor belinostat for ovarian cancer146. The FDA-approved histone deacetylase inhibitors include SAHA and Romidepsin for cutaneous T-cell lymphoma and Panobinostat against multiple myeloma, while many more are in clinical trials147;148.
Epigenetic modifications provide an elegant and holistic explanation of how CAFs function and adapt to accommodate the varying needs and demands of the tumor. Developing a robust understanding of the underlying epigenetic mechanisms driving CAF biology will aid in circumventing CAF-driven resistant phenotypes, which could be key to solving the core issues in the field of aggressive carcinoma today.
Funding and acknowledgments:
The authors/work was partly supported by funding from the National Institutes of Health (P01 CA217798, R01 CA273349, R01 CA263575, R01 CA256973, R01 CA247471, R01 CA206444, R01 CA210637, R01 CA228524, U01 CA210240, U01 CA200466, and R44 CA235991). Figures created with BioRender.com. Affiliation with Center for Heart & Vascular Research.
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Competing interests: S.K.B. is a founder of Sanguine Diagnostics and Therapeutics, Inc. Other authors have no competing interests to declare.
References
- 1.Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, et al. (2020). A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer 20, 174–186. 10.1038/s41568-019-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Santi A, Kugeratski FG, and Zanivan S (2018). Cancer Associated Fibroblasts: The Architects of Stroma Remodeling. Proteomics 18, e1700167. 10.1002/pmic.201700167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Y, McAndrews KM, and Kalluri R (2021). Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat Rev Clin Oncol 18, 792–804. 10.1038/s41571-021-00546-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harper J, and Sainson RC (2014). Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin Cancer Biol 25, 69–77. 10.1016/j.semcancer.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 5.Zheng S, Liang JY, Tang Y, Xie J, Zou Y, Yang A, Shao N, Kuang X, Ji F, Liu X, et al. (2023). Dissecting the role of cancer-associated fibroblast-derived biglycan as a potential therapeutic target in immunotherapy resistance: A tumor bulk and single-cell transcriptomic study. Clin Transl Med 13, e1189. 10.1002/ctm2.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong J, Wang F, Gao X, Zhao H, Zhang J, Wang N, Liu Z, Yan X, Jin J, Ba Y, et al. (2022). Integrated analysis of genome-wide DNA methylation and cancer-associated fibroblasts identified prognostic biomarkers and immune checkpoint blockade in lower grade gliomas. Front Oncol 12, 977251. 10.3389/fonc.2022.977251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. (2014). Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734. 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, et al. (2014). Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25, 735–747. 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JJ, Perera RM, Wang H, Wu DC, Liu XS, Han S, Fitamant J, Jones PD, Ghanta KS, Kawano S, et al. (2014). Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci U S A 111, E3091–3100. 10.1073/pnas.1411679111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, Yang Q, Tan Y, Tang Y, Ye J, Yuan B, and Yu W (2021). Cancer-Associated Fibroblasts Suppress Cancer Development: The Other Side of the Coin. Front Cell Dev Biol 9, 613534. 10.3389/fcell.2021.613534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, Corbo V, Oni TE, Hearn SA, Lee EJ, et al. (2017). Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 214, 579–596. 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, et al. (2019). Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov 9, 1102–1123. 10.1158/2159-8290.Cd-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartoschek M, Oskolkov N, Bocci M, Lövrot J, Larsson C, Sommarin M, Madsen CD, Lindgren D, Pekar G, Karlsson G, et al. (2018). Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat Commun 9, 5150. 10.1038/s41467-018-07582-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biffi G, and Tuveson DA (2021). Diversity and Biology of Cancer-Associated Fibroblasts. Physiol Rev 101, 147–176. 10.1152/physrev.00048.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kidd S, Spaeth E, Watson K, Burks J, Lu H, Klopp A, Andreeff M, and Marini FC (2012). Origins of the tumor microenvironment: quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS One 7, e30563. 10.1371/journal.pone.0030563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Helms EJ, Berry MW, Chaw RC, DuFort CC, Sun D, Onate MK, Oon C, Bhattacharyya S, Sanford-Crane H, Horton W, et al. (2021). Mesenchymal Lineage Heterogeneity Underlies Non-Redundant Functions of Pancreatic Cancer-Associated Fibroblasts. Cancer Discov. 10.1158/2159-8290.Cd-21-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishii G, Sangai T, Oda T, Aoyagi Y, Hasebe T, Kanomata N, Endoh Y, Okumura C, Okuhara Y, Magae J, et al. (2003). Bone-marrow-derived myofibroblasts contribute to the cancer-induced stromal reaction. Biochem Biophys Res Commun 309, 232–240. 10.1016/s0006-291x(03)01544-4. [DOI] [PubMed] [Google Scholar]
- 18.Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, Baik GH, Shibata W, Diprete B, Betz KS, et al. (2011). Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 19, 257–272. 10.1016/j.ccr.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vennin C, Mélénec P, Rouet R, Nobis M, Cazet AS, Murphy KJ, Herrmann D, Reed DA, Lucas MC, Warren SC, et al. (2019). CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat Commun 10, 3637. 10.1038/s41467-019-10968-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ganguly K, Cox JL, Ghersi D, Grandgenett PM, Hollingsworth MA, Jain M, Kumar S, and Batra SK (2022). Mucin 5AC-mediated CD44/ITGB1 clustering mobilizes adipose-derived mesenchymal stem cells to modulate pancreatic cancer stromal heterogeneity. Gastroenterology. 10.1053/j.gastro.2022.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erez N, Truitt M, Olson P, Arron ST, and Hanahan D (2010). Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 17, 135–147. 10.1016/j.ccr.2009.12.041. [DOI] [PubMed] [Google Scholar]
- 22.Wu X, Chen X, Zhou Q, Li P, Yu B, Li J, Qu Y, Yan J, Yu Y, Yan M, et al. (2013). Hepatocyte growth factor activates tumor stromal fibroblasts to promote tumorigenesis in gastric cancer. Cancer Lett 335, 128–135. 10.1016/j.canlet.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 23.Borriello L, Nakata R, Sheard MA, Fernandez GE, Sposto R, Malvar J, Blavier L, Shimada H, Asgharzadeh S, Seeger RC, and DeClerck YA (2017). Cancer-Associated Fibroblasts Share Characteristics and Protumorigenic Activity with Mesenchymal Stromal Cells. Cancer Res 77, 5142–5157. 10.1158/0008-5472.CAN-16-2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, Porter D, Hu M, Chin L, Richardson A, et al. (2004). Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 6, 17–32. 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 25.Hosein AN, Wu M, Arcand SL, Lavallée S, Hébert J, Tonin PN, and Basik M (2010). Breast carcinoma-associated fibroblasts rarely contain p53 mutations or chromosomal aberrations. Cancer Res 70, 5770–5777. 10.1158/0008-5472.Can-10-0673. [DOI] [PubMed] [Google Scholar]
- 26.Bianchi-Frias D, Basom R, Delrow JJ, Coleman IM, Dakhova O, Qu X, Fang M, Franco OE, Ericson NG, Bielas JH, et al. (2016). Cells Comprising the Prostate Cancer Microenvironment Lack Recurrent Clonal Somatic Genomic Aberrations. Mol Cancer Res 14, 374–384. 10.1158/1541-7786.Mcr-15-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walter K, Omura N, Hong SM, Griffith M, and Goggins M (2008). Pancreatic cancer associated fibroblasts display normal allelotypes. Cancer Biol Ther 7, 882–888. 10.4161/cbt.7.6.5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiu W, Hu M, Sridhar A, Opeskin K, Fox S, Shipitsin M, Trivett M, Thompson ER, Ramakrishna M, Gorringe KL, et al. (2008). No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat Genet 40, 650–655. 10.1038/ng.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bauer M, Su G, Casper C, He R, Rehrauer W, and Friedl A (2010). Heterogeneity of gene expression in stromal fibroblasts of human breast carcinomas and normal breast. Oncogene 29, 1732–1740. 10.1038/onc.2009.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campos LT, Brentani H, Roela RA, Katayama ML, Lima L, Rolim CF, Milani C, Folgueira MA, and Brentani MM (2013). Differences in transcriptional effects of 1α,25 dihydroxyvitamin D3 on fibroblasts associated to breast carcinomas and from paired normal breast tissues. J Steroid Biochem Mol Biol 133, 12–24. 10.1016/j.jsbmb.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 31.Pasanen I, Lehtonen S, Sormunen R, Skarp S, Lehtilahti E, Pietilä M, Sequeiros RB, Lehenkari P, and Kuvaja P (2016). Breast cancer carcinoma-associated fibroblasts differ from breast fibroblasts in immunological and extracellular matrix regulating pathways. Exp Cell Res 344, 53–66. 10.1016/j.yexcr.2016.04.016. [DOI] [PubMed] [Google Scholar]
- 32.Vicent S, Sayles LC, Vaka D, Khatri P, Gevaert O, Chen R, Zheng Y, Gillespie AK, Clarke N, Xu Y, et al. (2012). Cross-species functional analysis of cancer-associated fibroblasts identifies a critical role for CLCF1 and IL-6 in non-small cell lung cancer in vivo. Cancer Res 72, 5744–5756. 10.1158/0008-5472.Can-12-1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, and Weinberg RA (2005). Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121, 335–348. 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 34.Tampe B, and Zeisberg M (2014). Contribution of genetics and epigenetics to progression of kidney fibrosis. Nephrol Dial Transplant 29 Suppl 4, iv72–79. 10.1093/ndt/gft025. [DOI] [PubMed] [Google Scholar]
- 35.Duong TE, and Hagood JS (2018). Epigenetic Regulation of Myofibroblast Phenotypes in Fibrosis. Curr Pathobiol Rep 6, 79–96. 10.1007/s40139-018-0155-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeisberg EM, and Zeisberg M (2013). The role of promoter hypermethylation in fibroblast activation and fibrogenesis. J Pathol 229, 264–273. 10.1002/path.4120. [DOI] [PubMed] [Google Scholar]
- 37.Tao H, Yang JJ, Shi KH, Deng ZY, and Li J (2014). DNA methylation in cardiac fibrosis: new advances and perspectives. Toxicology 323, 125–129. 10.1016/j.tox.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 38.Mortada I, and Mortada R (2018). Epigenetic changes in mesenchymal stem cells differentiation. Eur J Med Genet 61, 114–118. 10.1016/j.ejmg.2017.10.015. [DOI] [PubMed] [Google Scholar]
- 39.Moore LD, Le T, and Fan G (2013). DNA methylation and its basic function. Neuropsychopharmacology 38, 23–38. 10.1038/npp.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ambrosi C, Manzo M, and Baubec T (2017). Dynamics and Context-Dependent Roles of DNA Methylation. J Mol Biol 429, 1459–1475. 10.1016/j.jmb.2017.02.008. [DOI] [PubMed] [Google Scholar]
- 41.Rasmussen KD, and Helin K (2016). Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev 30, 733–750. 10.1101/gad.276568.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ehrlich M (2009). DNA hypomethylation in cancer cells. Epigenomics 1, 239–259. 10.2217/epi.09.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang J, Yang C, Wu C, Cui W, and Wang L (2020). DNA Methyltransferases in Cancer: Biology, Paradox, Aberrations, and Targeted Therapy. Cancers (Basel) 12. 10.3390/cancers12082123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang W, and Xu J (2017). DNA methyltransferases and their roles in tumorigenesis. Biomark Res 5, 1. 10.1186/s40364-017-0081-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adany R, and lozzo RV (1990). Altered methylation of versican proteoglycan gene in human colon carcinoma. Biochem Biophys Res Commun 171, 1402–1413. 10.1016/0006-291x(90)90841-a. [DOI] [PubMed] [Google Scholar]
- 46.Adany R, Heimer R, Caterson B, Sorrell JM, and Iozzo RV (1990). Altered expression of chondroitin sulfate proteoglycan in the stroma of human colon carcinoma. Hypomethylation of PG-40 gene correlates with increased PG-40 content and mRNA levels. J Biol Chem 265, 11389–11396. [PubMed] [Google Scholar]
- 47.Ling E, Ringel A, Sigal-Batikoff I, Abu-Freha N, Vaknine H, Friah W, Reshef A, Pinsk I, Fich A, and Lamprecht S (2016). Human Colorectal Cancer Stage-dependent Global DNA Hypomethylation of Cancer-associated Fibroblasts. Anticancer Res 36, 4503–4507. 10.21873/anticanres.10996. [DOI] [PubMed] [Google Scholar]
- 48.Jiang L, Gonda TA, Gamble MV, Salas M, Seshan V, Tu S, Twaddell WS, Hegyi P, Lazar G, Steele I, et al. (2008). Global hypomethylation of genomic DNA in cancer-associated myofibroblasts. Cancer Res 68, 9900–9908. 10.1158/0008-5472.Can-08-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vizoso M, Puig M, Carmona FJ, Maqueda M, Velásquez A, Gómez A, Labernadie A, Lugo R, Gabasa M, Rigat-Brugarolas LG, et al. (2015). Aberrant DNA methylation in non-small cell lung cancer-associated fibroblasts. Carcinogenesis 36, 1453–1463. 10.1093/carcin/bgv146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhuang J, Jones A, Lee SH, Ng E, Fiegl H, Zikan M, Cibula D, Sargent A, Salvesen HB, Jacobs IJ, et al. (2012). The dynamics and prognostic potential of DNA methylation changes at stem cell gene loci in women’s cancer. PLoS Genet 8, e1002517. 10.1371/journal.pgen.1002517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu M, Yao J, Cai L, Bachman KE, van den Brûle F, Velculescu V, and Polyak K (2005). Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet 37, 899–905. 10.1038/ng1596. [DOI] [PubMed] [Google Scholar]
- 52.Crider KS, Yang TP, Berry RJ, and Bailey LB (2012). Folate and DNA methylation: a review of molecular mechanisms and the evidence for folate’s role. Adv Nutr 3, 21–38. 10.3945/an.111.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gonda TA, Kim YI, Salas MC, Gamble MV, Shibata W, Muthupalani S, Sohn KJ, Abrams JA, Fox JG, Wang TC, and Tycko B (2012). Folic acid increases global DNA methylation and reduces inflammation to prevent Helicobacter-associated gastric cancer in mice. Gastroenterology 142, 824–833.e827. 10.1053/j.gastro.2011.12.058. [DOI] [PubMed] [Google Scholar]
- 54.Lamprecht SA, and Lipkin M (2003). Chemoprevention of colon cancer by calcium, vitamin D and folate: molecular mechanisms. Nat Rev Cancer 3, 601–614. 10.1038/nrc1144. [DOI] [PubMed] [Google Scholar]
- 55.Yasuda T, Baba H, and Ishimoto T (2021). Cellular senescence in the tumor microenvironment and context-specific cancer treatment strategies. Febs j. 10.1111/febs.16231. [DOI] [PubMed] [Google Scholar]
- 56.Takasugi M, Yoshida Y, and Ohtani N (2022). Cellular senescence and the tumour microenvironment. Mol Oncol. 10.1002/1878-0261.13268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cruickshanks HA, McBryan T, Nelson DM, Vanderkraats ND, Shah PP, van Tuyn J, Singh Rai T, Brock C, Donahue G, Dunican DS, et al. (2013). Senescent cells harbour features of the cancer epigenome. Nat Cell Biol 15, 1495–1506. 10.1038/ncb2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pidsley R, Lawrence MG, Zotenko E, Niranjan B, Statham A, Song J, Chabanon RM, Qu W, Wang H, Richards M, et al. (2018). Enduring epigenetic landmarks define the cancer microenvironment. Genome Res 28, 625–638. 10.1101/gr.229070.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lawrence MG, Pidsley R, Niranjan B, Papargiris M, Pereira BA, Richards M, Teng L, Norden S, Ryan A, Frydenberg M, et al. (2020). Alterations in the methylome of the stromal tumour microenvironment signal the presence and severity of prostate cancer. Clin Epigenetics 12, 48. 10.1186/s13148-020-00836-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mrazek AA, Carmical JR, Wood TG, Hellmich MR, Eltorky M, Bohanon FJ, and Chao C (2014). Colorectal Cancer-Associated Fibroblasts are Genotypically Distinct. Curr Cancer Ther Rev 10, 97–218. 10.2174/157339471002141124123103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Halperin C, Hey J, Weichenhan D, Stein Y, Mayer S, Lutsik P, Plass C, and Scherz-Shouval R (2022). Global DNA Methylation Analysis of Cancer-Associated Fibroblasts Reveals Extensive Epigenetic Rewiring Linked with RUNX1 Upregulation in Breast Cancer Stroma. Cancer Res, Of1–of14. 10.1158/0008-5472.Can-22-0209. [DOI] [PubMed] [Google Scholar]
- 62.Su SF, Ho H, Li JH, Wu MF, Wang HC, Yeh HY, Kuo SW, Chen HW, Ho CC, and Li KC (2021). DNA methylome and transcriptome landscapes of cancer-associated fibroblasts reveal a smoking-associated malignancy index. J Clin Invest 131. 10.1172/jci139552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ikemori R, Gabasa M, Duch P, Vizoso M, Bragado P, Arshakyan M, Luis IC, Marín A, Morán S, Castro M, et al. (2020). Epigenetic SMAD3 Repression in Tumor-Associated Fibroblasts Impairs Fibrosis and Response to the Antifibrotic Drug Nintedanib in Lung Squamous Cell Carcinoma. Cancer Res 80, 276–290. 10.1158/0008-5472.Can-19-0637. [DOI] [PubMed] [Google Scholar]
- 64.Zhou FC, Balaraman Y, Teng M, Liu Y, Singh RP, and Nephew KP (2011). Alcohol alters DNA methylation patterns and inhibits neural stem cell differentiation. Alcohol Clin Exp Res 35, 735–746. 10.1111/j.1530-0277.2010.01391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sanchez-Alvarez R, Martinez-Outschoorn UE, Lin Z, Lamb R, Hulit J, Howell A, Sotgia F, Rubin E, and Lisanti MP (2013). Ethanol exposure induces the cancer-associated fibroblast phenotype and lethal tumor metabolism: implications for breast cancer prevention. Cell Cycle 12, 289–301. 10.4161/cc.23109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cakouros D, and Gronthos S (2020). The changing epigenetic landscape of Mesenchymal Stem/Stromal Cells during aging. Bone 137, 115440. 10.1016/j.bone.2020.115440. [DOI] [PubMed] [Google Scholar]
- 67.Koch CM, Suschek CV, Lin Q, Bork S, Goergens M, Joussen S, Pallua N, Ho AD, Zenke M, and Wagner W (2011). Specific age-associated DNA methylation changes in human dermal fibroblasts. PLoS One 6, e16679. 10.1371/journal.pone.0016679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Puttipanyalears C, Kitkumthorn N, Buranapraditkun S, Keelawat S, and Mutirangura A (2016). Breast cancer upregulating genes in stromal cells by LINE-1 hypermethylation and micrometastatic detection. Epigenomics 8, 475–486. 10.2217/epi-2015-0007. [DOI] [PubMed] [Google Scholar]
- 69.Kitkumthorn N, and Mutirangura A (2011). Long interspersed nuclear element-1 hypomethylation in cancer: biology and clinical applications. Clin Epigenetics 2, 315–330. 10.1007/si3148-011-0032-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hanson JA, Gillespie JW, Grover A, Tangrea MA, Chuaqui RF, Emmert-Buck MR, Tangrea JA, Libutti SK, Linehan WM, and Woodson KG (2006). Gene promoter methylation in prostate tumor-associated stromal cells. J Natl Cancer Inst 98, 255–261. 10.1093/jnci/djj051. [DOI] [PubMed] [Google Scholar]
- 71.Kekeeva TV, Popova OP, Shegaĭ PV, Alekseev B, Adnreeva I, Zaletaev DV, and Nemtsova MV (2007). [Abberant methylation of p16, HIC1, N33 and GSTP1 genes in tumor epitelium and tumor-associated stromal cells of prostate cancer]. Mol Biol (Mosk) 41, 79–85. [PubMed] [Google Scholar]
- 72.Ye F, Zhang SF, Xie X, and Lu WG (2008). OPCML gene promoter methylation and gene expression in tumor and stroma cells of invasive cervical carcinoma. Cancer Invest 26, 569–574. 10.1080/07357900701837044. [DOI] [PubMed] [Google Scholar]
- 73.Wasserkort R, Kalmar A, Valcz G, Spisak S, Krispin M, Toth K, Tulassay Z, Sledziewski AZ, and Molnar B (2013). Aberrant septin 9 DNA methylation in colorectal cancer is restricted to a single CpG island. BMC Cancer 13, 398. 10.1186/1471-2407-13-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fiegl H, Millinger S, Goebel G, Müller-Holzner E, Marth C, Laird PW, and Widschwendter M (2006). Breast cancer DNA methylation profiles in cancer cells and tumor stroma: association with HER-2/neu status in primary breast cancer. Cancer Res 66, 29–33. 10.1158/0008-5472.Can-05-2508. [DOI] [PubMed] [Google Scholar]
- 75.You Y, Ren Y, Liu J, and Qu J (2021). Promising Epigenetic Biomarkers Associated With Cancer-Associated-Fibroblasts for Progression of Kidney Renal Clear Cell Carcinoma. Front Genet 12, 736156. 10.3389/fgene.2021.736156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rodriguez-Canales J, Hanson JC, Tangrea MA, Erickson HS, Albert PS, Wallis BS, Richardson AM, Pinto PA, Linehan WM, Gillespie JW, et al. (2007). Identification of a unique epigenetic sub-microenvironment in prostate cancer. J Pathol 211, 410–419. 10.1002/path.2133. [DOI] [PubMed] [Google Scholar]
- 77.Mishra R, Haldar S, Placencio V, Madhav A, Rohena-Rivera K, Agarwal P, Duong F, Angara B, Tripathi M, Liu Z, et al. (2018). Stromal epigenetic alterations drive metabolic and neuroendocrine prostate cancer reprogramming. J Clin Invest 128, 4472–4484. 10.1172/jci99397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xiao Q, Zhou D, Rucki AA, Williams J, Zhou J, Mo G, Murphy A, Fujiwara K, Kleponis J, Salman B, et al. (2016). Cancer-Associated Fibroblasts in Pancreatic Cancer Are Reprogrammed by Tumor-Induced Alterations in Genomic DNA Methylation. Cancer Res 76, 5395–5404. 10.1158/0008-5472.Can-15-3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Etzold A, Galetzka D, Weis E, Bartsch O, Haaf T, Spix C, Itzel T, Schweiger S, Strand D, Strand S, and Zechner U (2016). CAF-like state in primary skin fibroblasts with constitutional BRCA1 epimutation sheds new light on tumor suppressor deficiency-related changes in healthy tissue. Epigenetics 11, 120–131. 10.1080/15592294.2016.1140295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pan X, Zhou J, Xiao Q, Fujiwara K, Zhang M, Mo G, Gong W, and Zheng L (2021). Cancer-associated fibroblast heterogeneity is associated with organ-specific metastasis in pancreatic ductal adenocarcinoma. J Hematol Oncol 14, 184. 10.1186/s13045-021-01203-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bian EB, Huang C, Wang H, Chen XX, Zhang L, Lv XW, and Li J (2014). Repression of Smad7 mediated by DNMT1 determines hepatic stellate cell activation and liver fibrosis in rats. Toxicol Lett 224, 175–185. 10.1016/j.toxlet.2013.10.038. [DOI] [PubMed] [Google Scholar]
- 82.Bian EB, Huang C, Ma TT, Tao H, Zhang H, Cheng C, Lv XW, and Li J (2012). DNMT1-mediated PTEN hypermethylation confers hepatic stellate cell activation and liver fibrogenesis in rats. Toxicol Appl Pharmacol 264, 13–22. 10.1016/j.taap.2012.06.022. [DOI] [PubMed] [Google Scholar]
- 83.Al-Kharashi LA, Al-Mohanna FH, Tulbah A, and Aboussekhra A (2018). The DNA methyl-transferase protein DNMT1 enhances tumor-promoting properties of breast stromal fibroblasts. Oncotarget 9, 2329–2343. 10.18632/oncotarget.23411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shakya R, Gonda T, Quante M, Salas M, Kim S, Brooks J, Hirsch S, Davies J, Cullo A, Olive K, et al. (2013). Hypomethylating therapy in an aggressive stroma-rich model of pancreatic carcinoma. Cancer Res 73, 885–896. 10.1158/0008-5472.Can-12-1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yu J, Walter K, Omura N, Hong SM, Young A, Li A, Vincent A, and Goggins M (2012). Unlike pancreatic cancer cells pancreatic cancer associated fibroblasts display minimal gene induction after 5-aza-2’-deoxycytidine. PLoS One 7, e43456. 10.1371/journal.pone.0043456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liang G, Gonzales FA, Jones PA, Orntoft TF, and Thykjaer T (2002). Analysis of gene induction in human fibroblasts and bladder cancer cells exposed to the methylation inhibitor 5-aza-2’-deoxycytidine. Cancer Res 62, 961–966. [PubMed] [Google Scholar]
- 87.Al-Kharashi LA, Bakheet T, AlHarbi WA, Al-Moghrabi N, and Aboussekhra A (2021). Eugenol modulates genomic methylation and inactivates breast cancer-associated fibroblasts through E2F1-dependent downregulation of DNMT1/DNMT3A. Mol Carcinog 60, 784–795. 10.1002/mc.23344. [DOI] [PubMed] [Google Scholar]
- 88.Hughes AL, Kelley JR, and Klose RJ (2020). Understanding the interplay between CpG island-associated gene promoters and H3K4 methylation. Biochim Biophys Acta Gene Regul Mech 1863, 194567. 10.1016/jbbagrm.2020.194567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Audia JE, and Campbell RM (2016). Histone Modifications and Cancer. Cold Spring Harb Perspect Biol 8, a019521. 10.1101/cshperspect.a019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maeda M, Takeshima H, lida N, Hattori N, Yamashita S, Moro H, Yasukawa Y, Nishiyama K, Hashimoto T, Sekine S, et al. (2020). Cancer cell niche factors secreted from cancer-associated fibroblast by loss of H3K27me3. Gut 69, 243–251. 10.1136/gutjnl-2018-317645. [DOI] [PubMed] [Google Scholar]
- 91.Kurayoshi M, Oue N, Yamamoto H, Kishida M, Inoue A, Asahara T, Yasui W, and Kikuchi A (2006). Expression of Wnt-5a is correlated with aggressiveness of gastric cancer by stimulating cell migration and invasion. Cancer Res 66, 10439–10448. 10.1158/0008-5472.Can-06-2359. [DOI] [PubMed] [Google Scholar]
- 92.Tyan SW, Hsu CH, Peng KL, Chen CC, Kuo WH, Lee EY, Shew JY, Chang KJ, Juan LJ, and Lee WH (2012). Breast cancer cells induce stromal fibroblasts to secrete ADAMTS1 for cancer invasion through an epigenetic change. PLoS One 7, e35128. 10.1371/journal.pone.0035128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen JY, Li CF, Lai YS, and Hung WC (2021). Lysine demethylase 2A expression in cancer-associated fibroblasts promotes breast tumour growth. Br J Cancer 124, 484–493. 10.1038/S41416-020-01112-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Eckert MA, Coscia F, Chryplewicz A, Chang JW, Hernandez KM, Pan S, Tienda SM, Nahotko DA, Li G, Blaženović I, et al. (2019). Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature 569, 723–728. 10.1038/s41586-019-1173-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hosein AN, Huang H, Wang Z, Parmar K, Du W, Huang J, Maitra A, Olson E, Verma U, and Brekken RA (2019). Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 5. 10.1172/jci.insight.129212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yasukawa Y, Hattori N, lida N, Takeshima H, Maeda M, Kiyono T, Sekine S, Seto Y, and Ushijima T (2021). SAA1 is upregulated in gastric cancer-associated fibroblasts possibly by its enhancer activation. Carcinogenesis 42, 180–189. 10.1093/carcin/bgaa131. [DOI] [PubMed] [Google Scholar]
- 97.Kay EJ, Paterson K, Domingo CR, Sumpton D, Daebritz H, Tardito S, Boldrini C, Hernandez-Fernaud JR, Athineos D, Dhayade S, et al. (2021). PYCR1-dependent proline synthesis in cancer-associated fibroblasts is required for the deposition of protumorigenic extracellular matrix. bioRxiv, 2020.2005.2030.125237. 10.1101/2020.05.30.125237. [DOI] [Google Scholar]
- 98.Zhang C, Wang XY, Zhang P, He TC, Han JH, Zhang R, Lin J, Fan J, Lu L, Zhu WW, et al. (2022). Cancer-derived exosomal HSPC111 promotes colorectal cancer liver metastasis by reprogramming lipid metabolism in cancer-associated fibroblasts. Cell Death Dis 13, 57. 10.1038/s41419-022-04506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Onion D, Argent RH, Reece-Smith AM, Craze ML, Pineda RG, Clarke PA, Ratan HL, Parsons SL, Lobo DN, Duffy JP, et al. (2016). 3-Dimensional Patient-Derived Lung Cancer Assays Reveal Resistance to Standards-of-Care Promoted by Stromal Cells but Sensitivity to Histone Deacetylase Inhibitors. Mol Cancer Ther 15, 753–763. 10.1158/1535-7163.Met-15-0598. [DOI] [PubMed] [Google Scholar]
- 100.Pazolli E, Alspach E, Milczarek A, Prior J, Piwnica-Worms D, and Stewart SA (2012). Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res 72, 2251–2261. 10.1158/0008-5472.Can-11-3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nguyen AH, Elliott IA, Wu N, Matsumura C, Vogelauer M, Attar N, Dann A, Ghukasyan R, Toste PA, Patel SG, et al. (2017). Histone deacetylase inhibitors provoke a tumor supportive phenotype in pancreatic cancer associated fibroblasts. Oncotarget 8, 19074–19088. 10.18632/oncotarget.13572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bülow R, Fitzner B, Sparmann G, Emmrich J, Liebe S, and Jaster R (2007). Antifibrogenic effects of histone deacetylase inhibitors on pancreatic stellate cells. Biochem Pharmacol 74, 1747–1757. 10.1016/j.bcp.2007.08.023. [DOI] [PubMed] [Google Scholar]
- 103.Thillainadesan G, Chitilian JM, Isovic M, Ablack JN, Mymryk JS, Tini M, and Torchia J (2012). TGF-β-dependent active demethylation and expression of the p15ink4b tumor suppressor are impaired by the ZNF217/CoREST complex. Mol Cell 46, 636–649. 10.1016/j.molcel.2012.03.027. [DOI] [PubMed] [Google Scholar]
- 104.Martin M, Ancey PB, Cros MP, Durand G, Le Calvez-Kelm F, Hernandez-Vargas H, and Herceg Z (2014). Dynamic imbalance between cancer cell subpopulations induced by transforming growth factor beta (TGF-β) is associated with a DNA methylome switch. BMC Genomics 15, 435. 10.1186/1471-2164-15-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cardenas H, Vieth E, Lee J, Segar M, Liu Y, Nephew KP, and Matei D (2014). TGF-β induces global changes in DNA methylation during the epithelial-to-mesenchymal transition in ovarian cancer cells. Epigenetics 9, 1461–1472. 10.4161/15592294.2014.971608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jjingo D, Conley AB, Yi SV, Lunyak VV, and Jordan IK (2012). On the presence and role of human gene-body DNA methylation. Oncotarget 3, 462–474. 10.18632/oncotarget.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tang X, Tu G, Yang G, Wang X, Kang L, Yang L, Zeng H, Wan X, Qiao Y, Cui X, et al. (2019). Autocrine TGF-β1/miR-200s/miR-221/DNMT3B regulatory loop maintains CAF status to fuel breast cancer cell proliferation. Cancer Lett 452, 79–89. 10.1016/j.canlet.2019.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Banerjee J, Mishra R, Li X, Jackson RS 2nd, Sharma A, and Bhowmick NA (2014). A reciprocal role of prostate cancer on stromal DNA damage. Oncogene 33, 4924–4931. 10.1038/onc.2013.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shrishrimal S, Kosmacek EA, and Oberley-Deegan RE (2019). Reactive Oxygen Species Drive Epigenetic Changes in Radiation-Induced Fibrosis. Oxid Med Cell Longev 2019, 4278658. 10.1155/2019/4278658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kim DJ, Dunleavey JM, Xiao L, Ollila DW, Troester MA, Otey CA, Li W, Barker TH, and Dudley AC (2018). Suppression of TGFβ-mediated conversion of endothelial cells and fibroblasts into cancer associated (myo)fibroblasts via HDAC inhibition. Br J Cancer 118, 1359–1368. 10.1038/s41416-018-0072-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sala L, Franco-Valls H, Stanisavljevic J, Curto J, Vergés J, Peña R, Duch P, Alcaraz J, García de Herreros A, and Baulida J (2019). Abrogation of myofibroblast activities in metastasis and fibrosis by methyltransferase inhibition. Int J Cancer 145, 3064–3077. 10.1002/ijc.32376. [DOI] [PubMed] [Google Scholar]
- 112.Albrengues J, Bourget I, Pons C, Butet V, Hofman P, Tartare-Decked S, Feral CC, Meneguzzi G, and Gaggioli C (2014). LIF mediates proinvasive activation of stromal fibroblasts in cancer. Cell Rep 7, 1664–1678. 10.1016/j.celrep.2014.04.036. [DOI] [PubMed] [Google Scholar]
- 113.Albrengues J, Bertero T, Grasset E, Bonan S, Maiel M, Bourget I, Philippe C, Herraiz Serrano C, Benamar S, Croce O, et al. (2015). Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat Commun 6, 10204. 10.1038/ncomms10204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wu F, Yang J, Liu J, Wang Y, Mu J, Zeng Q, Deng S, and Zhou H (2021). Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct Target Ther 6, 218. 10.1038/s41392-021-00641-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yasuda T, Koiwa M, Yonemura A, Miyake K, Kariya R, Kubota S, Yokomizo-Nakano T, Yasuda-Yoshihara N, Uchihara T, Itoyama R, et al. (2021). Inflammation-driven senescence-associated secretory phenotype in cancer-associated fibroblasts enhances peritoneal dissemination. Cell Rep 34, 108779. 10.1016/j.celrep.2021.108779. [DOI] [PubMed] [Google Scholar]
- 116.Hendrayani SF, Al-Khalaf HH, and Aboussekhra A (2014). The cytokine IL-6 reactivates breast stromal fibroblasts through transcription factor STAT3-dependent upregulation of the RNA-binding protein AUF1. J Biol Chem 289, 30962–30976. 10.1074/jbc.M114.594044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bhagat TD, Von Ahrens D, Dawlaty M, Zou Y, Baddour J, Achreja A, Zhao H, Yang L, Patel B, Kwak C, et al. (2019). Lactate-mediated epigenetic reprogramming regulates formation of human pancreatic cancer-associated fibroblasts. Elife 8. 10.7554/eLife.50663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Becker LM, O’Connell JT, Vo AP, Cain MP, Tampe D, Bizarro L, Sugimoto H, McGow AK, Asara JM, Lovisa S, et al. (2020). Epigenetic Reprogramming of Cancer-Associated Fibroblasts Deregulates Glucose Metabolism and Facilitates Progression of Breast Cancer. Cell Rep 31, 107701. 10.1016/j.celrep.2020.107701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Semenza GL (2010). HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev 20, 51–56. 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li Z, Sun C, and Qin Z (2021). Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics 11, 8322–8336. 10.7150/thno.62378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hu B, Castillo E, Harewood L, Ostano P, Reymond A, Dummer R, Raffoul W, Hoetzenecker W, Hofbauer GF, and Dotto GP (2012). Multifocal epithelial tumors and field cancerization from loss of mesenchymal CSL signaling. Cell 149, 1207–1220. 10.1016/j.cell.2012.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Arif KMT, Elliott EK, Haupt LM, and Griffiths LR (2020). Regulatory Mechanisms of Epigenetic miRNA Relationships in Human Cancer and Potential as Therapeutic Targets. Cancers (Basel) 12. 10.3390/cancers12102922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li P, Shan JX, Chen XH, Zhang D, Su LP, Huang XY, Yu BQ, Zhi QM, Li CL, Wang YQ, et al. (2015). Epigenetic silencing of microRNA-149 in cancer-associated fibroblasts mediates prostaglandin E2/interleukin-6 signaling in the tumor microenvironment. Cell Res 25, 588–603. 10.1038/cr.2015.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Aguilera-Rojas M, Sharbati S, Stein T, and Einspanier R (2020). Deregulation of miR-27a may contribute to canine fibroblast activation after coculture with a mast cell tumour cell line. FEBS Open Bio 10, 802–816. 10.1002/2211-5463.12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jung YD, Park SK, Kang D, Hwang S, Kang MH, Hong SW, Moon JH, Shin JS, Jin DH, You D, et al. (2020). Epigenetic regulation of miR-29a/miR-30c/DNMT3A axis controls SOD2 and mitochondrial oxidative stress in human mesenchymal stem cells. Redox Biol 37, 101716. 10.1016/j.redox.2020.101716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Costa A, Scholer-Dahirel A, and Mechta-Grigoriou F (2014). The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin Cancer Biol 25, 23–32. 10.1016/j.semcancer.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 127.Arcucci A, Ruocco MR, Granato G, Sacco AM, and Montagnani S (2016). Cancer: An Oxidative Crosstalk between Solid Tumor Cells and Cancer Associated Fibroblasts. Biomed Res Int 2016, 4502846. 10.1155/2016/4502846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang S, Wu W, and Claret FX (2017). Mutual regulation of microRNAs and DNA methylation in human cancers. Epigenetics 12, 187–197. 10.1080/15592294.2016.1273308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Damiani E, Duran MN, Mohan N, Rajendran P, and Dashwood RH (2020). Targeting Epigenetic ‘Readers’ with Natural Compounds for Cancer Interception. J Cancer Prev 25, 189–203. 10.15430/jcp.2020.25.4.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bottomley MJ (2004). Structures of protein domains that create or recognize histone modifications. EMBO Rep 5, 464–469. 10.1038/sj.embor.7400146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Antolic A, Wakimoto H, Jiao Z, Gorham JM, DePalma SR, Lemieux ME, Conner DA, Lee DY, Qi J, Seidman JG, et al. (2020). BET bromodomain proteins regulate transcriptional reprogramming in genetic dilated cardiomyopathy. JCI Insight 5. 10.1172/jci.insight.138687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yamamoto K, Tateishi K, Kudo Y, Hoshikawa M, Tanaka M, Nakatsuka T, Fujiwara H, Miyabayashi K, Takahashi R, Tanaka Y, et al. (2016). Stromal remodeling by the BET bromodomain inhibitor JQ1 suppresses the progression of human pancreatic cancer. Oncotarget 7, 61469–61484. 10.18632/oncotarget.11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Josling GA, Selvarajah SA, Petter M, and Duffy MF (2012). The role of bromodomain proteins in regulating gene expression. Genes (Basel) 3, 320–343. 10.3390/genes3020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wen D, Wang Y, Zhu Z, Huang Z, Cui L, Wu T, and Liu CY (2020). Bromodomain and Extraterminal (BET) protein inhibition suppresses tumor progression and inhibits HGF-MET signaling through targeting cancer-associated fibroblasts in colorectal cancer. Biochim Biophys Acta Mol Basis Dis 1866, 165923. 10.1016/j.bbadis.2020.165923. [DOI] [PubMed] [Google Scholar]
- 135.Kim DE, Procopio MG, Ghosh S, Jo SH, Goruppi S, Magliozzi F, Bordignon P, Neel V, Angelino P, and Dotto GP (2017). Convergent roles of ATF3 and CSL in chromatin control of cancer-associated fibroblast activation. J Exp Med 214, 2349–2368. 10.1084/jem.20170724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Procopio MG, Laszlo C, Al Labban D, Kim DE, Bordignon P, Jo SH, Goruppi S, Menietti E, Ostano P, Ala U, et al. (2015). Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat Cell Biol 17, 1193–1204. 10.1038/ncb3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bhatia R, Bhyravbhatla N, Kisling A, Li X, Batra SK, and Kumar S (2022). Cytokines chattering in pancreatic ductal adenocarcinoma tumor microenvironment. Semin Cancer Biol 86, 499–510. 10.1016/j.semcancer.2022.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang L, Zhang F, Cui JY, Chen L, Chen YT, and Liu BW (2018). CAFs enhance paclitaxel resistance by inducing EMT through the IL- 6/JAK2/STAT3 pathway. Oncol Rep 39, 2081–2090. 10.3892/or.2018.6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Feng B, Wu J, Shen B, Jiang F, and Feng J (2022). Cancer-associated fibroblasts and resistance to anticancer therapies: status, mechanisms, and countermeasures. Cancer Cell Int 22, 166. 10.1186/s12935-022-02599-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.D’Arcangelo E, Wu NC, Cadavid JL, and McGuigan AP (2020). The life cycle of cancer-associated fibroblasts within the tumour stroma and its importance in disease outcome. Br J Cancer 122, 931–942. 10.1038/s41416-019-0705-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kalluri R. (2016). The biology and function of fibroblasts in cancer. Nat Rev Cancer 16, 582–598. 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 142.Kakarla S, Song XT, and Gottschalk S (2012). Cancer-associated fibroblasts as targets for immunotherapy. Immunotherapy 4, 1129–1138. 10.2217/imt.12.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Schosserer M, Grillari J, and Breitenbach M (2017). The Dual Role of Cellular Senescence in Developing Tumors and Their Response to Cancer Therapy. Front Oncol 7, 278. 10.3389/fonc.2017.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Morel D, Jeffery D, Aspeslagh S, Almouzni G, and Postel-Vinay S (2020). Combining epigenetic drugs with other therapies for solid tumours - past lessons and future promise. Nat Rev Clin Oncol 17, 91–107. 10.1038/s41571-019-0267-4. [DOI] [PubMed] [Google Scholar]
- 145.Lee YT, Tan YJ, Falasca M, and Oon CE (2020). Cancer-Associated Fibroblasts: Epigenetic Regulation and Therapeutic Intervention in Breast Cancer. Cancers (Basel) 12. 10.3390/cancers12102949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Steele N, Finn P, Brown R, and Plumb JA (2009). Combined inhibition of DNA methylation and histone acetylation enhances gene re-expression and drug sensitivity in vivo. Br J Cancer 100, 758–763. 10.1038/sj.bjc.6604932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Halsall JA, and Turner BM (2016). Histone deacetylase inhibitors for cancer therapy: An evolutionarily ancient resistance response may explain their limited success. Bioessays 38, 1102–1110. 10.1002/bies.201600070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nepali K, and Liou JP (2021). Recent developments in epigenetic cancer therapeutics: clinical advancement and emerging trends. J Biomed Sci 28, 27. 10.1186/s12929-021-00721-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, Breart B, Foreman O, Bainbridge TW, Castiglioni A, et al. (2020). Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov 10, 232–253. 10.1158/2159-8290.Cd-19-0644. [DOI] [PubMed] [Google Scholar]
- 150.Hu G, Wang S, Xu F, Ding Q, Chen W, Zhong K, Huang L, and Xu Q (2018). Tumor-Infiltrating Podoplanin+ Fibroblasts Predict Worse Outcome in Solid Tumors. Cell Physiol Biochem 51, 1041–1050. 10.1159/000495484. [DOI] [PubMed] [Google Scholar]
- 151.Friedman G, Levi-Galibov O, David E, Bornstein C, Giladi A, Dadiani M, Mayo A, Halperin C, Pevsner-Fischer M, Lavon H, et al. (2020). Cancer-associated fibroblast compositions change with breast cancer progression linking the ratio of S100A4(+) and PDPN(+) CAFs to clinical outcome. Nat Cancer 1, 692–708. 10.1038/s43018-020-0082-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhang Y, and Ertl HC (2016). Depletion of FAP+ cells reduces immunosuppressive cells and improves metabolism and functions CD8+T cells within tumors. Oncotarget 7, 23282–23299. 10.18632/oncotarget.7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al. (2013). Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A 110, 20212–20217. 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Grout JA, Sirven P, Leader AM, Maskey S, Hector E, Puisieux I, Steffan F, Cheng E, Tung N, Maurin M, et al. (2022). Spatial positioning and matrix programs of cancer-associated fibroblasts promote T cell exclusion in human lung tumors. Cancer Discov. 10.1158/2159-8290.Cd-21-1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Foster DS, Januszyk M, Delitto D, Yost KE, Griffin M, Guo J, Guardino N, Delitto AE, Chinta M, Burcham AR, et al. (2022). Multiomic analysis reveals conservation of cancer-associated fibroblast phenotypes across species and tissue of origin. Cancer Cell. 10.1016/j.ccell.2022.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Su S, Chen J, Yao H, Liu J, Yu S, Lao L, Wang M, Luo M, Xing Y, Chen F, et al. (2018). CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 172, 841–856.e816. 10.1016/j.cell.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 157.Li X, Sun Z, Peng G, Xiao Y, Guo J, Wu B, Li X, Zhou W, Li J, Li Z, et al. (2022). Single-cell RNA sequencing reveals a pro-invasive cancer-associated fibroblast subgroup associated with poor clinical outcomes in patients with gastric cancer. Theranostics 12, 620–638. 10.7150/thno.60540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Valdés-Mora F, Salomon R, Gloss BS, Law AMK, Venhuizen J, Castillo L, Murphy KJ, Magenau A, Papanicolaou M, Rodriguez de la Fuente L, et al. (2021). Single-cell transcriptomics reveals involution mimicry during the specification of the basal breast cancer subtype. Cell Rep 35, 108945. 10.1016/j.celrep.2021.108945. [DOI] [PubMed] [Google Scholar]
- 159.Djurec M, Graña O, Lee A, Troulé K, Espinet E, Cabras L, Navas C, Blasco MT, Martín-Díaz L, Burdiel M, et al. (2018). Saa3 is a key mediator of the protumorigenic properties of cancer-associated fibroblasts in pancreatic tumors. Proc Natl Acad Sci U S A 115, E1147–e1156. 10.1073/pnas.1717802115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mizutani Y, Kobayashi H, lida T, Asai N, Masamune A, Hara A, Esaki N, Ushida K, Mii S, Shiraki Y, et al. (2019). Meflin-Positive Cancer-Associated Fibroblasts Inhibit Pancreatic Carcinogenesis. Cancer Res 79, 5367–5381. 10.1158/0008-5472.Can-19-0454. [DOI] [PubMed] [Google Scholar]
- 161.Wang Y, Liang Y, Xu H, Zhang X, Mao T, Cui J, Yao J, Wang Y, Jiao F, Xiao X, et al. (2021). Single-cell analysis of pancreatic ductal adenocarcinoma identifies a novel fibroblast subtype associated with poor prognosis but better immunotherapy response. Cell Discov 7, 36. 10.1038/s41421-021-00271-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Schlesinger Y, Yosefov-Levi O, Kolodkin-Gal D, Granit RZ, Peters L, Kalifa R, Xia L, Nasereddin A, Shiff I, Amran O, et al. (2020). Single-cell transcriptomes of pancreatic preinvasive lesions and cancer reveal acinar metaplastic cells’ heterogeneity. Nat Commun 11, 4516. 10.1038/s41467-020-18207-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chen K, Wang Q, Li M, Guo H, Liu W, Wang F, Tian X, and Yang Y (2021). Single-cell RNA-seq reveals dynamic change in tumor microenvironment during pancreatic ductal adenocarcinoma malignant progression. EBioMedicine 66, 103315. 10.1016/j.ebiom.2021.103315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sebastian A, Hum NR, Martin KA, Gilmore SF, Peran I, Byers SW, Wheeler EK, Coleman MA, and Loots GG (2020). Single-Cell Transcriptomic Analysis of Tumor-Derived Fibroblasts and Normal Tissue-Resident Fibroblasts Reveals Fibroblast Heterogeneity in Breast Cancer. Cancers (Basel) 12. 10.3390/cancers12051307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Affo S, Nair A, Brundu F, Ravichandra A, Bhattacharjee S, Matsuda M, Chin L, Filliol A, Wen W, Song X, et al. (2021). Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell 39, 866–882.e811. 10.1016/j.ccell.2021.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chiavarina B, Ronca R, Otaka Y, Sutton RB, Rezzola S, Yokobori T, Chiodelli P, Souche R, Pourquier D, Maraver A, et al. (2022). Fibroblast-derived prolargin is a tumor suppressor in hepatocellular carcinoma. Oncogene 41, 1410–1420. 10.1038/s41388-021-02171-z. [DOI] [PubMed] [Google Scholar]
- 167.Zhao Z, Li W, Zhu L, Xu B, Jiang Y, Ma N, Liu L, Qiu J, and Zhang M (2022). Construction and Verification of a Fibroblast-Related Prognostic Signature Model for Colon Cancer. Front Genet 13, 908957. 10.3389/fgene.2022.908957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Costea DE, Hills A, Osman AH, Thurlow J, Kalna G, Huang X, Pena Murillo C, Parajuli H, Suliman S, Kulasekara KK, et al. (2013). Identification of two distinct carcinoma-associated fibroblast subtypes with differential tumor-promoting abilities in oral squamous cell carcinoma. Cancer Res 73, 3888–3901. 10.1158/0008-5472.Can-12-4150. [DOI] [PubMed] [Google Scholar]
- 169.Ma Z, Li X, Mao Y, Wei C, Huang Z, Li G, Yin J, Liang X, and Liu Z (2022). Interferon-dependent SLC14A1(+) cancer-associated fibroblasts promote cancer stemness via WNT5A in bladder cancer. Cancer Cell 40, 1550–1565.e1557. 10.1016/j.ccell.2022.11.005. [DOI] [PubMed] [Google Scholar]
- 170.Loret N, Vandamme N, De Coninck J, Taminau J, De Clercq K, Blancke G, Jonckheere S, Goossens S, Lemeire K, De Prijck S, et al. (2022). Distinct Transcriptional Programs in Ascitic and Solid Cancer Cells Induce Different Responses to Chemotherapy in High-Grade Serous Ovarian Cancer. Mol Cancer Res 20, 1532–1547. 10.1158/1541-7786.Mcr-21-0565. [DOI] [PubMed] [Google Scholar]