

Abstract
The activity-dependent plasticity of synapses is believed to be the cellular basis of learning. These synaptic changes are mediated through the coordination of local biochemical reactions in synapses and changes in gene transcription in the nucleus to modulate neuronal circuits and behavior. The protein kinase C (PKC) family of isozymes has long been established as critical for synaptic plasticity. However, because of a lack of suitable isozyme-specific tools, the role of the novel subfamily of PKC isozymes is largely unknown. Here, through the development of fluorescence lifetime imaging-fluorescence resonance energy transfer activity sensors, we investigate novel PKC isozymes in synaptic plasticity in CA1 pyramidal neurons of mice of either sex. We find that PKCδ is activated downstream of TrkB and DAG production, and that the spatiotemporal nature of its activation depends on the plasticity stimulation. In response to single-spine plasticity, PKCδ is activated primarily in the stimulated spine and is required for local expression of plasticity. However, in response to multispine stimulation, a long-lasting and spreading activation of PKCδ scales with the number of spines stimulated and, by regulating cAMP response-element binding protein activity, couples spine plasticity to transcription in the nucleus. Thus, PKCδ plays a dual functional role in facilitating synaptic plasticity.
SIGNIFICANCE STATEMENT Synaptic plasticity, or the ability to change the strength of the connections between neurons, underlies learning and memory and is critical for brain health. The protein kinase C (PKC) family is central to this process. However, understanding how these kinases work to mediate plasticity has been limited by a lack of tools to visualize and perturb their activity. Here, we introduce and use new tools to reveal a dual role for PKCδ in facilitating local synaptic plasticity and stabilizing this plasticity through spine-to-nucleus signaling to regulate transcription. This work provides new tools to overcome limitations in studying isozyme-specific PKC function and provides insight into molecular mechanisms of synaptic plasticity.
Keywords: CREB, FRET, PKC, plasticity, sensor, synapse
Introduction
Learning induces structural and functional changes in subsets of active dendritic spines to modulate neuronal circuits and behavior (Hayashi-Takagi et al., 2015; Abdou et al., 2018; Gobbo and Cattaneo, 2020). These changes require early, local plasticity of spines that is stabilized later through gene transcription in the nucleus. Therefore, the complex signaling cascades that mediate these changes must transduce local and transient inputs (μm and ms) into long-lasting changes in cell-wide protein expression (mm and hours). The coordination of early, local events at spine heads and later changes in the nucleus is not well understood, but spine-to-nucleus signaling through kinase cascades, which can extend local, transient signals in both space and time, have been implicated (Smolen et al., 2019).
One of the first kinase families identified as essential for spine plasticity, learning, and memory was the protein kinase C (PKC) family (Farley and Auerbach, 1986; Malenka et al., 1986; Hu et al., 1987; Olds et al., 1989). Gain-of-function studies demonstrated that PKC activation induces plasticity (Malenka et al., 1986; Reymann et al., 1988), and loss-of-function studies disrupted plasticity, including the transition from early to long-lasting, transcription-dependent potentiation (Wang and Feng, 1992; Van der Zee and Douma, 1997). It is now evident that individual isozymes of the PKC kinase family play unique roles in plasticity. Even single isozymes, whose specificity of signaling is defined primarily by their spatiotemporal activation pattern, likely have complex roles depending on the nature of the plasticity-inducing stimulus (Mukherjee et al., 2016). This complexity, combined with few existing tools to measure and distinguish PKC isozyme activity (Kajimoto et al., 2010), has limited understanding of the processes by which plasticity is expressed and stabilized at the molecular level.
The PKC family comprises 12 isozymes grouped into three subfamilies: classic, novel, and atypical. These subfamilies are delineated according to their domain structure and activation mechanisms, largely defining their isozyme-specific differences. Classic PKC isozymes, which integrate calcium and the lipid signaling molecule diacylglycerol (DAG) have been implicated in the induction of plasticity. PKCα activates rapidly (ms) and transiently (s) in dendritic spines undergoing plasticity (Colgan et al., 2018). This rapid and local activation pattern paralleled its upstream activator, calcium, and was consistent with its critical role in the early, spine-specific phase of memory formation (Colgan et al., 2018). On the other hand, atypical isozymes, such as PKMζ, have been proposed to regulate the spine-specific maintenance of long-lasting forms of memory on the timescale of hours to days. Although their requirement and mechanisms in memory maintenance remain unclear (Borodinova et al., 2017), the proposed plasticity-dependent upregulation of a constitutively active PKMζ enzyme through local translation is consistent with a role in long-lasting phases of plasticity maintenance.
The function of the novel subfamily of PKC isozymes (PKCδ, PKCε, PKCη) in synaptic plasticity, however, remains largely unknown. These isozymes, which are recruited by DAG, may be activated by the plasticity-induced increase of DAG in dendritic spines (Colgan et al., 2018). Lacking a binding domain for calcium, however, the activation of novel isozymes may be slower, longer-lasting, and affect a larger spatial area than classic isozymes (Steinberg, 2008; Yasuda, 2017). Thus, novel PKC isozymes may be well positioned to transduce spine-specific inputs (μm and ms) into long-lasting changes in cell-wide protein expression to stabilize memory formation.
Here, through loss-of-function studies and the use of newly developed, isozyme-specific sensors, we explore the functions of novel PKC isozymes in a robust model of synaptic potentiation and learning: structural plasticity (sLTP) of dendritic spines (Matsuzaki et al., 2004; Hayashi-Takagi et al., 2015; Choi et al., 2018; Goto et al., 2021). We find that, among the novel PKC isozymes, PKCδ is uniquely required to induce sLTP of dendritic spines in hippocampal CA1 neurons. Further, we characterize a dual functional role of PKCδ in the local induction of spine plasticity and the stabilization of this plasticity through spine-to-nucleus signaling.
Materials and Methods
DNA constructs
Isozyme-specific TRAnslocation of C Kinase (ITRACK)
Donor plasmids using restriction site independent cloning were constructed into CMV-promotor containing mEGFP C1 vectors such that mEGFP was fused to the N-terminus of PKCδ (mus musculus), PKCε (mus musculus), and PKCη (mus musculus). The acceptor plasmid, mCh-CAAX, consisted of mCherry followed by a 14 aa linker and a K-Ras derived lipid targeting motif (DGKKKKKKSKTKCVIM) driven by a CMV promoter. Negative controls for ITRACK (ITRACK CTL) consisted of the same donor construct and a control acceptor construct named mCh-CAAXneg in which a stop codon was introduced before the lipid targeting domain to make cytosolic mCh.
Isozyme-specific Docking Of C Kinase Substrate (IDOCKS)
Donor plasmids consisted of PKCδ, PKCε, and PKCη, which were tagged on their C termini with mEGFP driven by CMV promoter. The acceptor construct, 2mCh-PS, consisted of two copies of mCherry fluorophores separated by an eight amino acid linker followed by the 18 amino acid pseudosubstrate region from PKCα/β (19RFARKGALRQKNVHEVKN36) driven by the CMV promoter. The negative control IDOCKS consisted of the same acceptor construct with a single point mutation in the pseudosubstrate domain (R27E, 19RFARKGALEQKNVHEVKN36). GFP-PKCδ-AS was made through a single point mutation in the gatekeeper residue of the ATP binding pocket (PKCδ M425A) in the donor construct of GFP tagged PKCδ (Kumar et al., 2015). PKCδ-AS was made by removing GFP from GFP-PKCδ-AS. cAMP response-element binding protein (CREB) and extracellular signal-regulated kinase (ERK) activity (EKAR) sensors consisted of DNA constructs as previously described (Harvey et al., 2008a; Laviv et al., 2020).
HeLa cell maintenance, transfection, and imaging
HeLa cells (ATCC CCL-2) were grown in DMEM supplemented with 10% FBS at 37°C in 5% CO2. Plasmids were transfected into HeLa cells using Lipofectamine 2000 (Invitrogen) at a ratio of donor plasmid to acceptor plasmid of 1:3 for ITRACK and 1:2 for IDOCKS. Imaging was performed 24-48 h following transfection in a HEPES-buffered aCSF solution (20 mm HEPES, pH 7.3, 130 mm NaCl, 2 mm NaHCO3, 25 mm D-glucose, 2.5 mm KCl, 1.25 mm NaH2PO4) with 2 mm CaCl2 and 2 mm MgCl2 by 2pFLIM as described below. When indicated, cells were stimulated with 1 μm phorbol 12,13-dibutyrate (PdBu, Tocris) or DMSO (0.02%) vehicle, or when comparing with classic isozymes 1 μm PdBu (Tocris) followed by 1 μm ionomycin. 1NM-PP1 (Santa Cruz Biotechnology, working dilution [WD] 1 μm) or (2R)-amino-5-phosphonovaleric acid (APV; Sigma, WD 50 μm) was added to the HEPES-buffered aCSF at least 10 min before stimulation when indicated.
Nontransgenic and transgenic animals
All experimental procedures were approved by the Max Planck Florida Institute for Neuroscience Animal Care and Use Committee, following guidelines by the National Institutes of Health. Either nontransgenic or transgenic P3-P8 mouse pups from both sexes were used for organotypic slices for imaging studies as indicated. All experiments consisted of neurons in slices made from at least 4 animals from at least 2 different litters. Nontransgenic animals were used for the initial characterization of the PKCδ sensors in Figure 4. All other figures used transgenic animals and WT littermates as indicated below. P30-P50 mice of both sexes were used for acute slices for electrophysiological studies in Figure 2.
Figure 4.

Characterization of FLIM-FRET sensors for novel PKC isozymes in neurons. A, Representative lifetime images of IDOCKS and IDOCKS CTL for PKCδ in nontransgenic hippocampal CA1 neurons in response to PKC activation by phorbol ester (1 μm PdBu). Warmer colors represent lifetime decrease and sensor activation. B, C, Time course and quantification of the change in the lifetime of ITRACKx (B) or IDOCKSx (C) for PKCδ, PKCε, or PKCη, measured in hippocampal CA1 neurons in response to bath application of PdBu or DMSO vehicle as indicated. Gray shading represents the time over which the average change was quantified (right). One-way ANOVA (F(3,19) = 54.98; p < 0.0001). *p < 0.05; ****p < 0.001; lowest level of significance between each PdBu and vehicle group (Sidak's multiple comparison post-test). n ≥ 5. D, Time course and quantification of mean change in the lifetime of ITRACK, IDOCKS, and CTL sensors for PKCδ in response to PdBu. Gray shading represents the time over which the average change was quantified (right). One-way ANOVA (F(3,21) = 57.85; p < 0.0001). Asterisks indicate significance between indicated comparisons tested by Sidak's multiple comparison post-test. p < 0.001. n ≥ 6 neurons. E, Change in volume of dendritic spines 5 min after stimulation by a glutamate uncaging sLTP protocol in hippocampal CA1 neurons expressing ITRACKδ, IDOCKSδ, or GFP-PKCδ (n (neurons, spines ≥ 4,13, one-way ANOVA; ns, p > 0.16) F, Time course and quantification of mean change in the lifetime of IDOCKSδ expressed in hippocampal CA1 neurons from nontransgenic WT or PKCδ KO animals in response to PdBu (n ≥ 4 neurons, two-way unpaired t test, ns, p = 0.75).
Figure 2.

PKCδ is required for functional plasticity. A, Input–output curve of the fEPSP slopes between PKCδ WT and KO littermate mice. ns indicates results of repeated-measures two-way ANOVA by genotype (F(1,30) = 1.829, p = 0.19). B, C, Quantification (B) and time course (C) of change in fEPSPs' slope in PKCδ WT (n (neurons/animals) = 17/8) and PKCδ KO (n = 16/4) CA1 hippocampal neurons after field stimulation of Schaffer collaterals with a 3× or 5× TBS stimulation protocol. Gray shading represents the time of quantification in B. Insets, Left, Representative traces of fEPSPs before (pre) and after (post) stimulation from WT and KO slices. **p = 0.009 (unpaired t test).
Nontransgenic animals (C57Bl/6N Crl) were received from Charles River Lab. These animals were used in Figure 4.
PKCδ KO mice (B6.129X1-Prkcdtm1Msg/J) were obtained from The Jackson Laboratory (JAX stock #028055) and developed as described previously (Chou et al., 2004). Mice were backcrossed to Swiss Webster mouse line (CRL CFW) to improve breeding efficiency, litter size, and mothering characteristics. Mice were bred het × het to generate WT and KO littermates. These animals were used for Figures 1C, D, 2, 5A–C, 6–9.
Figure 1.
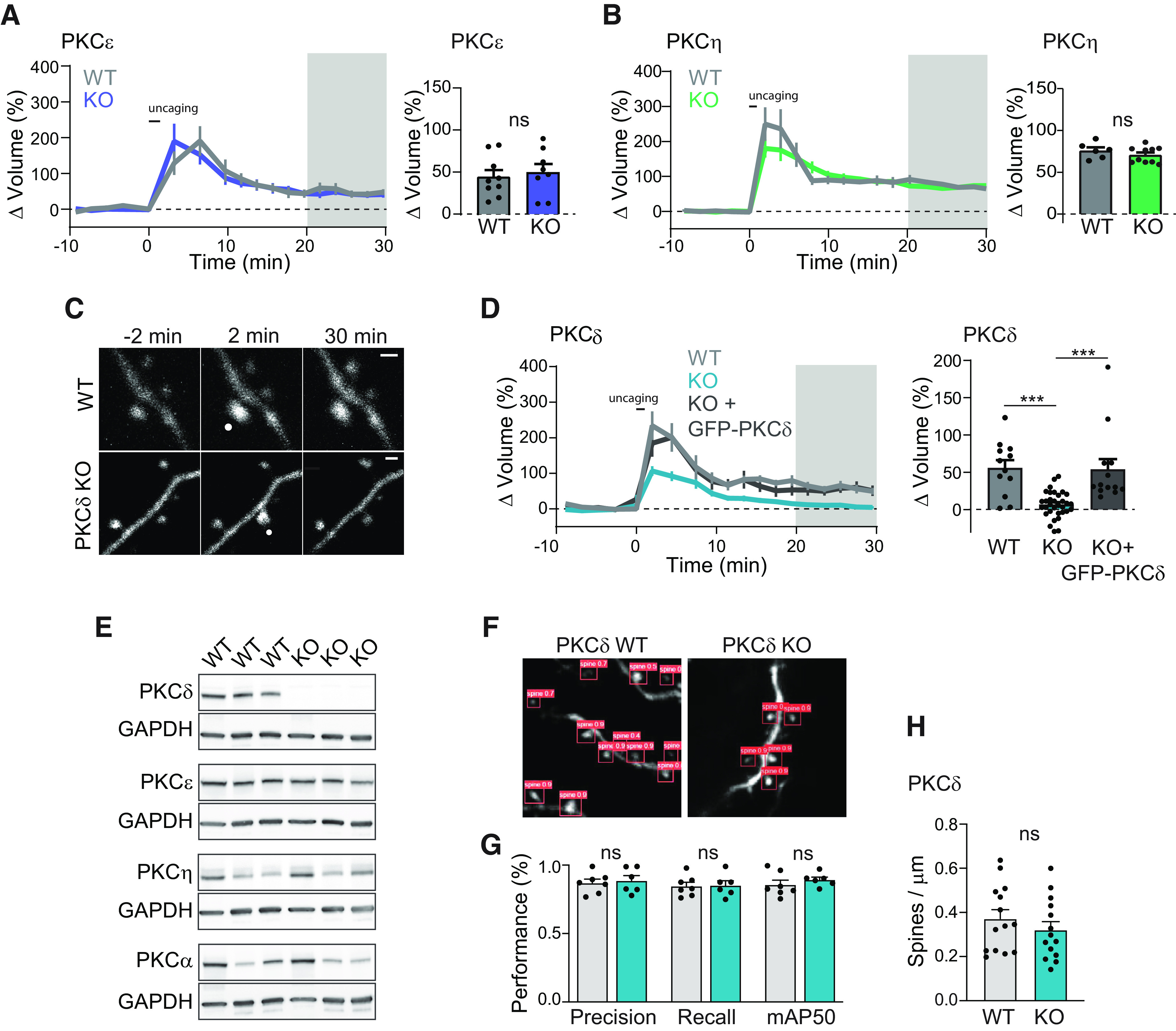
PKCδ, but not PKCε or PKCη, is required for structural plasticity. A, B, Time course and quantification of glutamate uncaging induced change in volume of stimulated dendritic spines in hippocampal CA1 neurons from PKCε WT (n (spines/neurons) = 9/6) or KO (n = 8/6) littermates (A) and PKCη WT (n = 6/5) or KO (n = 10/8) littermates (B). Gray shading represents the time of quantification of mean volume change (right). Two-way, unpaired t test; ns, p > 0.37. C, Representative images of uncaging-induced sLTP in neurons from PKCδ WT or KO littermates. White dot indicates the location of uncaging. Scale bar, 1 μm. D, Time course and quantification of change in volume of stimulated spines from PKCδ WT (n = 12/6), KO (n = 33/15), and KO neurons acutely overexpressing GFP-tagged PKCδ (n = 13/7). p < 0.0001, one-way ANOVA (F(2,55) = 16.85). ***p < 0.001, comparing indicated groups (Sidak's post-test). E, Western blot of hippocampal tissue from 3 PKCδ WT and KO animals probed for PKC isozymes shown as well as GAPDH. F, Example images of automatically detected spines G, Network performance metrics of automated spine segmentation of hippocampal CA1 neurons from PKCδ WT (n (spines/images) = 58/7) and KO (n = 63/6) (Sidak's multiple comparison after two-way ANOVA; ns, p > 0.79) H, Spine density analysis of dendrites from PKCδ WT (n (images/neurons) = 169/14) and KO animals (n = 176/14, two-way, unpaired t test; ns, p = 0.36).
Figure 5.

PKCδ is transiently activated in stimulated spines during plasticity. A, Fluorescence lifetime images of IDOCKSδ in a single spine undergoing sLTP. White dot indicates uncaging location. Scale bar, 1 μm. Warmer colors represent shorter lifetimes and PKCδ activation. B, Mean time course of PKCδ activity in the stimulated spine and underlying dendrite measured by the change in the lifetime of IDOCKS during sLTP. n (spines/neurons) = 26/10. C, D, Time course (C) and quantification (D) of mean lifetime change at the time indicated by gray shading in C and in response to uncaging stimulation in the absence of caged glutamate (No Glu n = 17/9) or the presence of the NMDAR antagonist APV (50 μm, n = 13/6). Two-way ANOVA, by treatment (F(2,105) = 32.96; p < 0.0001). ****p < 0.0001; ***p = 0.0004; Sidak's multiple comparisons. E, Left: Relationship between basal lifetime of sensor and plasticity-induced change in lifetime. Correlation was nonsignificant; ns, p = 0.067. n = 20. Right: Relationship between ratio of donor: acceptor and plasticity-induced change in lifetime. Correlation was ns. p = 0.482. n = 20. F, The mean time course (left) and quantification (right) of PKCδ activity in stimulated spines in neurons in the absence (n = 14/7) or presence of a phospholipase C inhibitor, U73122 (10 μm n = 16/7), two-way, unpaired t-test *p = 0.014. G, The mean time course (left) and quantification (right) of PKCδ activity in stimulated spines in neurons containing an inert analog-sensitive mutation in TrkB-AS in the presence of vehicle (CTL, n = 11/5) or inhibitory ATP analog (1NM-PP1, 1 μm n = 13/5), two-way, unpaired t-test **p = 0.0029.
Figure 6.
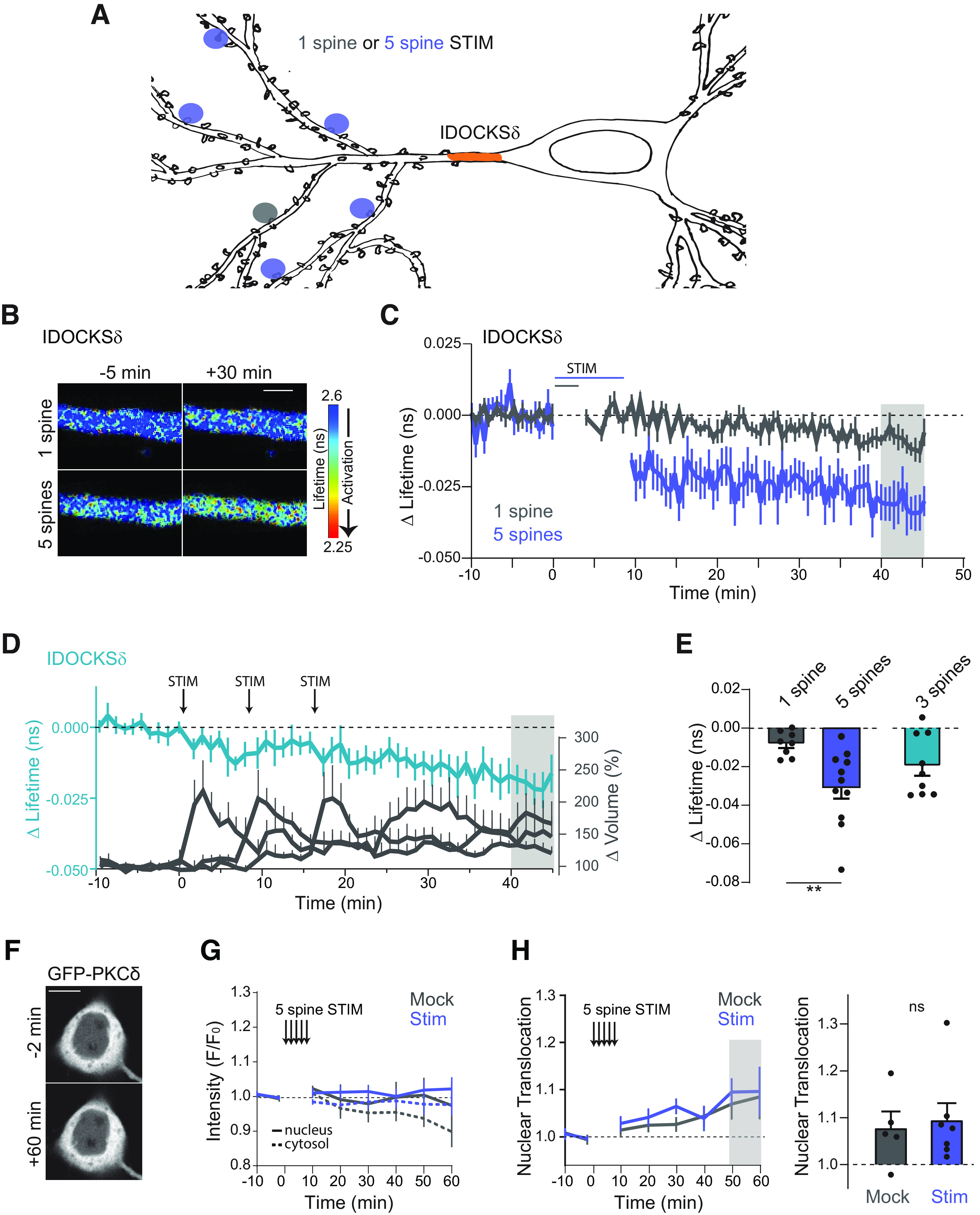
Multispine stimulation enhances long-lasting and spreading activation of PKCδ. A, Schematic of 1 spine or 5 spine stimulation protocol. Orange area represents the region where PKCδ activation was measured (primary dendrite). B, Representative lifetime images of IDOCKSδ in the primary dendrite in response to 1 or 5 spine stimulation. Warmer colors represent decreased lifetime and activation of PKCδ. Scale bar, 2 μm. C, Time course of PKCδ activity measured by the change in the lifetime of IDOCKS in response to induction of single spine sLTP (n (neurons) = 8) or sequential induction of sLTP in 5 spines (n = 13). D, Time course of PKCδ activation in the primary dendrite (left axis) during the spaced induction of 3 spines. Right axis, The change in the volume of stimulated spines. n (neurons) = 9. E, Quantification of mean change in the lifetime at 40-45 min (gray area in B) for 1, 5, or 3 spine stimulation (n = 9). **p = 0.0065 (two-tailed unpaired t test). F, Intensity images of GFP-PKCδ in hippocampal CA1 neurons before and 60 min after 5 spine stimulation. Scale bar, 10 μm. G, Time course of somatic cytosol and nuclear intensity (F/Fo) of GFP-PKCδ before and after 5 spine stimulation (Stim) or stimulation in the absence of caged Glu (Mock). H, Time course and quantification of nuclear translocation (Nuc intensity/Cytosol intensity). Gray shading represents the time of quantification. ns, p = 0.75 (unpaired two-tailed t test).
Figure 9.
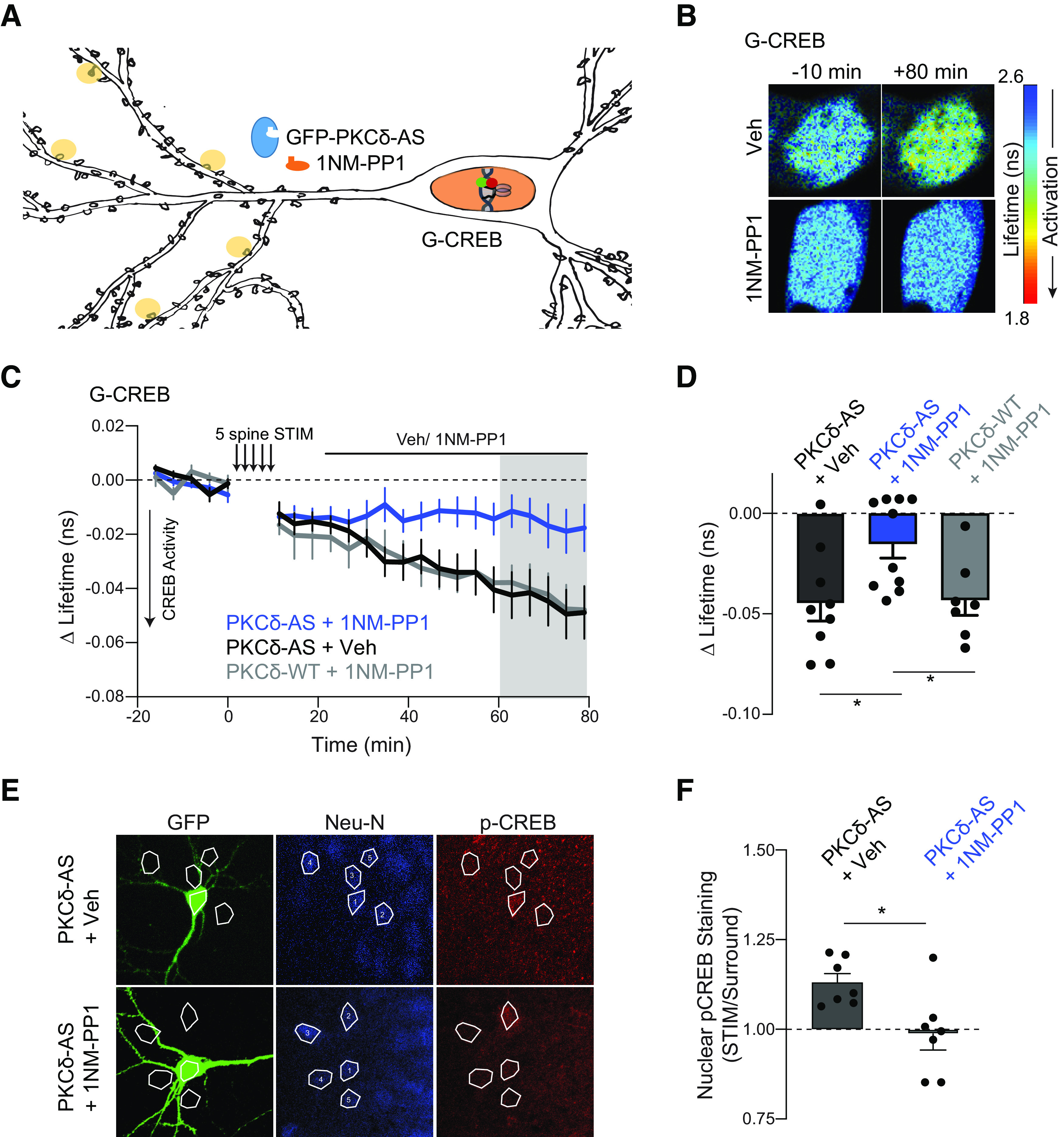
Long-lasting PKCδ activity regulates CREB activity. A, Schematic of five spine stimulation of CA1 neurons in which PKCδ was replaced by PKCδ-AS. CREB activity was monitored in the nucleus using a CREB sensor. B-D, Representative lifetime images (B), time course (C), and quantification (D) of mean lifetime change of CREB sensor in the nucleus before and after 5 spine stimulation and vehicle (n = 9 neurons) or 1NM-PP1 (1 μm, n = 10 neurons) application 10 min after stimulation to inhibit long-lasting PKCδ activity. Gray shading represents the time of quantification in D. *p = 0.016 (two-tailed unpaired t test). E, Representative images and ROIs of p-CREB signal in nuclei of neurons expressing GFP-PKCδ-AS (GFP) and surrounding neurons. Neurons were simulated with 5 spine stimulation, after which vehicle (n = 7 neurons) or 1NM-PP1 (1 μm, n = 7 neurons) was applied. Neurons were fixed 60 min after stimulation and processed for immunostaining. F, Quantification of p-CREB fluorescence signal in nuclei of stimulated neurons (ROI 1) normalized to signal in nuclei of surrounding neurons (average ROI 2-5, one sample t test of normalized p-CREB signal compared with 1, Veh: p = 0.002, NMPP1 ns p = 0.78). *p = 0.016 (between-group comparison of unpaired t test).
PKCε KO mice (B6.129S4-Prkcetm1Msg/J) were obtained from The Jackson Laboratory (JAX stock #004189) and were developed as described previously (Khasar et al., 1999). Mice were bred het × het to generate WT and KO littermates. These animals were used in Figure 1A.
PKC η KO mice (B6.Cg-Prkchtm1.2Gasc/J) were obtained from The Jackson Laboratory (JAX stock #018988) and were developed as previously described (Fu et al., 2011). Mice were bred het × het to generate WT and KO littermates. These animals were used in Figure 1B.
Double transgenic mice (PKCδKO/TrkB-AS) were generated by crossing PKCδKO mice with TrkB-AS mutant mice (TrkBF616A C57bl/6). TrkBF616A C57bl/6 mutant mice were developed and provided by David Ginty as previously described (Chen et al., 2005). Breeding pairs were KO on one gene and Het on the other gene. These animals were used in Figure 5G.
Organotypic hippocampal slice cultures and transfection
Organotypic hippocampal slices were prepared from nontransgenic or transgenic postnatal 3- to 8-d-old pups of both sexes as previously described (Stoppini et al., 1991). In brief, pups were deeply anesthetized by isoflurane and then killed by decapitation. After removing the brain, hippocampi were dissected and cut into 350-µm-thick transverse slices using a McIlwain tissue chopper (Ted Pella). Slices were plated on hydrophilic PTFE membranes (Millicell, Millipore) and maintained at 37°C and a 5% CO2 environment in culture medium (MEM medium, Invitrogen), 20% horse serum, 1 mm L-glutamine, 1 mm CaCl2, 2 mm MgSO4, 12.9 mm D-glucose, 5.2 mm NaHCO3, 30 mm HEPES, 0.075% ascorbic acid, 1 µg/ml insulin) placed beneath the membranes. Seventy-five percent of the culture medium was exchanged every other day. Organotypic slices were transfected after 7-10 DIV with biolistic gene transfer (O'Brien and Lummis, 2006) using 1.6 µm gold beads (8 mg) coated with plasmids containing DNA of interest in the following amounts. mEGFP: 15 µg, PKCδ_mEGFP: 30 µg, mEGFP- PKCδAS: 30 µg, ITRACK or ITRACKneg: 15 µg donor (mEGFP- PKCδ, mEGFP-PKCε, or mEGFP-PKCη) and 40 µg acceptor (mCh-CAAX or mCh-CAAX_neg), IDOCKS or IDOCKSneg: 10 µg donor (PKCδ-mEGFP, PKCε-mEGFP, or PKCη-mEGFP) and 20 µg Acceptor (2mCh-PS or 2mCh-PSmut), PKCδ-AS and EKAR sensor: 20 μg each, GFP-PKCδ-AS or GFP-PKCδ and CREB sensor: GFP-PKCδ-AS or GFP-PKCδ 15 μg and 15 μg donor (mEGFP-CREB) and 30 μg acceptor (mCherry-KIX-mCherry). Secondary or tertiary apical dendrites in the stratum radiatum of transfected CA1 pyramidal neurons were imaged 2-7 d after transfection by 2-photon microscopy or 2pFLIM as described below.
2-photon microscopy and 2pFLIM
PKC isozyme activity was measured using 2pFLIM. For quantification of spine volume change, we monitored the fluorescence intensity change of mEGFP in the spines using regular two-photon microscopy that was simultaneous with lifetime measurements. Intensity measurements and 2pFLIM imaging in HeLa cells and slices were performed using a custom 2p microscope. mEGFP and mCherry were excited with a Ti: sapphire laser (Chameleon, Coherent) at a wavelength of 920 nm and a power of 1.4-1.6 mW measured below the objective. The fluorescence was collected with an objective (60×, 1.0 NA, Olympus), divided with a dichroic mirror (565 nm), and detected with two separated photoelectron multiplier tubes placed after wavelength filters (Chroma, 510/70-2p for green and 620/90-2p for red). Photoelectron multiplier tubes with low transfer time spread (H7422-40p; Hamamatsu) were used for both red and green channels. The system was controlled via PCIe 6323, and data are acquired by TimeHarp 260 pico dual (Picoquant). Software for data acquisition and analysis is FLIMage (Fluorescence Lifetime Imaging). FLIM images were taken at 64 × 64 or 128 × 128 resolution with 2, 6, or 12 frames averaged. Intensity images for analysis of sLTP volume change were collected by 128 × 128 pixels as a z stack of three slices with 1 µm separation and averaging 6 frames/slice. Spine volume change was calculated as the background-subtracted F – F0 where F0 was the average fluorescence intensity before stimulation. In some cases of sLTP volume imaging of PKC KO neurons and WT littermates, parallel automated imaging of 2-3 spines per neuron was done using a custom-built interface in MATLAB by using algorithms for autofocusing and drift correction to maintain position and focus (Smirnov et al., 2017). For these experiments, a 2 min stagger of uncaging events was incorporated to avoid data loss during uncaging events. Nuclear translocation was calculated from intensity measurements of the nucleus (nuc) and surrounding cytoplasmic fluorescence (cyt) in the central cross-section of the nucleus and calculated as the background-subtracted [Fnuc/Fonuc]/[Fcyt/Focyt], where Fo was the average fluorescence intensity before stimulation.
Two-photon glutamate uncaging
Structural plasticity of dendritic spines was stimulated through uncaging of 4-methoxy-7-nitroindolinyl-caged-L-glutamate (MNI-caged glutamate, Tocris) using a Ti: Sapphire laser tuned at a wavelength of 720 nm. The uncaging laser was focused to a small region ∼0.5 µm from the spine and 2.7-2.9 mW of laser power (measured at the objective) and was pulsed 30 times in a 0.5 Hz train with a 6 ms pulse width. Spines deeper than 50 µm were not selected for uncaging experiments. Experiments were performed in Mg2+ fee aCSF (127 mm NaCl, 2.5 mm KCl, 4 mm CaCl2, 25 mm NaHCO3, 1.25 mm NaH2PO4, and 25 mm glucose) containing 1 μm TTX and 4 mm MNI-caged L-glutamate aerated with 95% O2 and 5% CO2. Experiments were performed at room temperature (RT, 24°C-26°C). For multispine stimulation experiments, 5 spines were stimulated sequentially on 3 or 4 secondary or tertiary dendrites. Spines stimulated on the same dendritic branches were spaced by >25 μm.
Western blot
Isolated hippocampi were lysed in ice-cold T-PER Tissue Protein Extraction Reagent (Fisher Scientific) supplemented with cOmplete ULTRA EDTA-free Protease Inhibitor Cocktail tablets (Millipore Sigma). This lysis buffer mixture was added at the ratio of 10 μl per 1 mg of tissue, and the tissue was homogenized using hand-held motor pestle mixer on ice followed by five passages through a 31G needle. Following 10 min incubation on ice, the lysates were centrifuged for 10 min at 10,000 × g in 4°C.
Protein concentration was determined using BCA Protein Assay, and the samples for Western blot were prepared using 4× Laemmli sample buffer with the addition of β-mercaptoethanol. The samples were not boiled but incubated at RT for at least 30 min; 30 μg of total protein for each sample was loaded into each well of Any kDa Mini-PROTEAN TGX Precast Protein gel (Bio-Rad). Following the electrophoresis, the protein was transferred into the Nitrocellulose membrane using Trans-Blot Turbo Transfer system (Bio-Rad).
The membranes were blocked in 5% nonfat milk solution for 30 min at RT, and subsequently incubated with appropriate primary antibody in 5% nonfat milk overnight at 4°C. Antibodies used in this experiment were rabbit anti-PKCδ (#2058, Cell Signaling, used at 1:500 dilution), rabbit anti-PKCε (#2683, Cell Signaling, used at 1:1000 dilution), rabbit anti-PKC η (#ab179524, Abcam, used at 1:500 dilution), and mouse anti-PKCα (SC8393, Santa Cruz Biotechnology, used at 1:200 dilution).
Following the incubation, the blots were washed 3 times 5 min each with 1× TBST buffer, and goat anti-rabbit IgG (H + L) HRP conjugate (or, in the case of mouse anti-PKCα primary antibody, goat anti-mouse IgG (H + L) HRP conjugate) was added to the membrane at 1:3000 dilution in 5% nonfat milk and incubated for 30 min at RT. Finally, the membranes were washed 3 times 5 min each with 1× TBST buffer, and the resulting bands were visualized using ECL Western Blotting Substrate (Pierce).
Spine density analysis
Spine density was calculated in a blinded and automated fashion using a trained AI network. A dataset consisting of 2456 labeled images was used to train a neural network to segment dendritic spines. The labeled images are a combination of data from an open-source dendritic spine detection dataset (Smirnov et al., 2018) and manually labeled images from our own microscopy. Each image was resized to 416 × 416 pixels. In addition, each training image was augmented with random vertical flip, zoom, and rotation, giving three distinct images for each individual labeled training image. With a train/valid/test split of 70-20-10 in the original dataset, this resulted in a total of 5134 training images, 498 validation images, and 248 test images.
Network architecture and training
Our network uses a pretrained YOLOv5-L model (Jocher et al., 2022) as a backbone. The network was trained with an input dimension of 416 × 416 and a batch size of 32. Otherwise, default parameters and loss functions were used. Spine density was calculated as the total number of spines detected divided by the arc length in pixels, then converted to spines over micrometer using the entered micrometer-to-pixel scale for the images. The code is available at https://github.com/ryoheiyasuda/SpineDetector.
Metrics of performance used included precision: true positives/(true positives + false positives); recall (R): true positives/(true positives + false negatives), and mean average precision (mAP): area under the precision-recall curve as follows:
2pFLIM analysis
To measure the change in fluorescence lifetime, we fit a fluorescence lifetime curve summing all pixels over a whole image with a double exponential function convolved with the Gaussian pulse response function as follows:
where τAD is the fluorescence lifetime of the donor bound with the acceptor, PD and PAD are the fraction of free donor and the donor undergoing FRET with the acceptor, respectively, and H(t) is a fluorescence lifetime curve with a single exponential function convolved with the Gaussian pulse response function as follows:
in which τD is the fluorescence lifetime of the free donor, τG is the width of the Gaussian pulse response function, F0 is the peak fluorescence before convolution and t0 is the time offset, and erfc is the complementary error function.
We fixed τD to 2.65 ns and τAD to 1.33 ns based on previously published work of classic PKC isozyme sensors to obtain stable fitting (Colgan et al., 2018). To generate the fluorescence lifetime image, we calculated the mean photon arrival time, <t>, in each pixel as follows:
Then the mean fluorescence lifetime, <τ>, is calculated as the mean photon arrival time minus an offset arrival time, to, which is obtained by fitting the whole image as follows:
Change in lifetime is calculated as mean fluorescence lifetime, <τ>, in ROI subtracted by the average lifetime in the ROI before stimulation.
The source code of the software is available online: https://github.com/ryoheiyasuda/FLIMage_public.
Pharmacology
1NM-PP1 (Santa Cruz Biotechnology, WD 1 μm), PdBu (Tocris, WD 1 μm), MNI Caged Glu (Tocris, 4 mm), APV (Sigma, WD 50 μm), U73122 (Tocris, WD 10 μm), ionomycin (Sigma, 1 μm), and U0126 (Tocris, WD 0.1 μm) were stored as recommended by supplier and diluted into aCSF to the working dilutions (WD) listed.
Electrophysiology
Acute slice preparation
PKCδ WT or KO littermate mice (P30-P50) were sedated by isoflurane inhalation, and perfused intracardially with a chilled choline chloride solution. Brain was removed and placed in the same choline chloride solution composed of 124 mm choline chloride, 2.5 mm KCl, 26 mm NaHCO3, 3.3 mm MgCl2, 1.2 mm NaH2PO4, 10 mm glucose, and 0.5 mm CaCl2, pH 7.4, equilibrated with 95%O2/5%CO2. Coronal slices (300 µm) containing the hippocampus were cut using a vibratome (Leica) and maintained in a submerged chamber at 32°C for 1 h and then at RT in oxygenated aCSF.
Extracellular recordings and LTP protocol
Slices were perfused with oxygenated aCSF containing 2 mm CaCl2, 2 mm MgCl2, and 100 μm picrotoxin. One glass electrode (resistance ∼4 mΩ) containing the same aCSF solution was placed in the dendritic layer of CA1 area (∼100-200 µm away from the soma) while stimulating Schaffer collateral fibers with current square pulses (0.1 ms) using a concentric bipolar stimulation electrode (FHC). The initial slope of the fEPSP was monitored with custom software (MATLAB). The stimulation strength was set to ∼50% saturation. A 20 min stable baseline was first recorded before induction of LTP. LTP was induced by applying 3-5 trains of TBS stimulation. fEPSP responses were recorded for 1 h after the stimulation protocol. All data were analyzed with an in-house program written with MATLAB.
In vitro kinase activity assay
HEK293FT cell maintenance, transfection, protein purification
HEK293FT cells were grown in 175 cm2 vented cell culture flasks with DMEM supplemented with 10% FBS at 37°C in 5% CO2. Plasmids containing HIS tagged PKCδ- or PKCδ-AS were transfected into HEK293FT cells using Lipofectamine 3000 (Invitrogen). At 24 h following transfection, the maintenance temperature was reduced to 35°C to increase expression levels. The cells were harvested on day 3 and lysed in Mammalian Protein Extraction Reagent (Thermo Scientific) containing EDTA-free phosphatase and protease inhibitor cocktail tablets (Roche) for 20 min and centrifuged at 14,000 rps for 10 min. The supernatant was purified in His GraviTrap Columns (Cytiva) and desalted in PD-10 Columns (Cytiva) in kinase storage buffer (1000 ml ddH2O 300 mm NaCl, 30 mm HEPES, 0.1 mm TCEP with 20% glycerol). The concentration of harvested proteins was calculated using a BCA Protein Assay Kit (Fisher Scientific).
ADP-Glo kinase assay
ADP-Glo Kinase Assay (Promega) was used to determine the titration of PKCδ- or PKCδ-AS and to compare the efficacy of 1NM-PP1 on PKCδ- or PKCδ-AS proteins. All kinase reactions contained Kinase Assay Buffer III (200 mm Tris-HCl, pH 7.4, 100 mm MgCl2, and 0.5 mg/ml BSA), PKC Lipid Activator (Signal Chem), 0.01 m DTT, 0.2 μg/μl CREBtide, and 5 μm ATP. Equally active PKCδ- and PKCδ-AS concentrations were used to determine the IC50 values of 1NM-PP1.The dose–response was created in two replicates for each group with 8 serial 10-fold dilution of 1NM-PP1 (2 m to 0.2 nm). Both groups were tested at the same time and in the same microplate at different wells to minimize the variability. To stop the reaction, ADP-Glo Reagent (Promega) was added to each well. Forty minutes following this step, Kinase Detection Reagent was added to detect the amount of ADP generated in each well. The luminescence was recorded using Glomax Multidetection microplate reader (Promega), and IC50 values were determined by creating a nonlinear inhibitor response curve of the normalized enzyme activity for each group.
Phospho-CREB immunohistochemistry
Hippocampal organotypic slices were fixed with 4% PFA in 0.1 m PB, pH 7.4, for 30 min at RT. The slices were then removed and rinsed with PBS 3 times and stored in PBS at 4°C. For immunohistochemistry, sections were permeabilized with 0.3% Triton X-100 in PBS (PBST) for 20 min, then blocked with 10% NGS in 0.1% PBST for 3.5 h in RT, and then incubated at 4°C for 18-20 h with the following primary antibodies: rabbit anti-pCreb (1:200, MA5-11192; Fisher Scientific), mouse anti-NeuN (1:300, MAB377; Millipore). After 2 h incubation with AlexaFluor-conjugated secondary antibodies (Invitrogen or Jackson ImmunoResearch Laboratories) in 5% NGS in 0.1% PBST, the slices were rinsed with PBST for 20 min and PBS for 1 h and finally mounted in Fluoromount-G (Southern Biotechnology). The stained slices were imaged using a confocal laser-scanning microscope (LSM880 with Airyscan, Zeiss). The acquired images were processed using the Zen (Zeiss) or Adobe Photoshop (Adobe Systems) and analyzed with the ImageJ (http://rsbweb.nih.gov/ij/).
Analysis of phospho-CREB immunostaining
Analysis was done in a blinded fashion. An ROI was drawn outlining the nucleus of the GFP-expressing neuron in the GFP channel. Then the channel was switched to the Neu-N channel, the nuclear NueoN ROI in the GFP-expressing neurons was refined (ROI 1) and four additional ROIs were drawn of nuclei surrounding the GFP-expressing neuron (ROIs 2-5). After ROIs were drawn, the channel was switched to p-CREB and the signal was quantified in these ROIs. The intensity of the p-CREB signal in the GFP-expressing neuron (ROI 1) was normalized to the average of the p-CREB signal in the four surrounding ROIs (average ROI2-5).
Experimental design and statistical analysis
All values are presented as mean ± SEM unless otherwise noted. The number of independent measurements (n) is indicated in the figure legends. For in vitro studies, neurons were assigned to different groups to randomly interleave control and experimental groups from slices prepared from the same animals. Experiments from control and experimental groups were randomly interleaved during experimentation, with the exception that each block of experiments began with one neuron from the control group to ensure the technical success of experiments. Data distribution was assumed to be normal, but this was not formally tested. Unpaired two-tailed Student's t test was used for comparing two independent samples. One-way ANOVA followed by multiple comparison tests was used for comparing more than two independent samples. Two-way ANOVA followed by multiple comparison tests were used to compare grouped datasets. All t tests and one-way ANOVAs included formal testing for differences in variance (e.g., F test or Bartlett's test to compare variances), and appropriate corrections were made and are indicated in instances where this was relevant. Statistical tests and p values are noted in each figure legend and were computed using GraphPad Prism for Windows (GraphPad Software, www.graphpad.com). For in vitro experiments, data collection and analysis were not performed blind to the conditions of the experiments except where noted. Neurons in which there were obvious signs of poor health that developed during the experiment, such as beading of processes, were excluded before analysis. FLIM data in which there was >0.06 ns variation in the baseline (because of low photon number) were excluded from further analysis.
Data and reagent availability
Source data are available at https://www.synapse.org/#!Synapse:syn51174506/wiki/621328.
Results
PKCδ, but not PKCε or PKCη activity, is required for excitatory synaptic potentiation
To assess the potential role of novel PKC isozymes in synaptic plasticity, we induced sLTP through optical uncaging of glutamate (30 pulses at 0.5 Hz) in the presence of TTX in an Mg+2-free aCSF. This approach allows for the precise and robust induction and quantification of sLTP at single dendritic spines of CA1 pyramidal neurons in the stratum radiatum of organotypic hippocampal brain slice. Plasticity was quantified as the uncaging-induced change in the volume of the stimulated spine. Consistent with previous literature, glutamate uncaging induced rapid growth in stimulated spines of WT male and female mice (Matsuzaki et al., 2004; Harvey et al., 2008b). The growth decayed slightly over minutes but remained elevated compared with prestimulus spine size. These changes were both spine-specific and long-lasting. Spine plasticity was not impaired in CA1 neurons in slices prepared from PKCε or PKCη KO mice (Fig. 1A,B) (Khasar et al., 1999; Fu et al., 2011). However, PKCδ KO mice showed impaired induction and expression of spine plasticity (Fig. 1C,D). Spine growth was reduced in the early phases of sLTP; and by 11 min after stimulation, the volume of the stimulated spine was not significantly larger than its prestimulus size (Dunnett's multiple comparisons after mixed model two-way ANOVA). Furthermore, the plasticity deficit seen in PKCδ KO neurons could be fully rescued by sparse, acute (∼24-48 h) overexpression of PKCδ-GFP (Fig. 1D).
Loss of PKCδ protein in the KO was confirmed through Western blotting. Other PKC isozyme protein levels did not show substantial compensatory changes (Fig. 1E). PKCδ KO neurons also did not show any obvious morphologic changes. To determine whether PKCδ KO deficits in plasticity were associated with differences in spine density, the average spine density of GFP-transfected hippocampal neurons in PKCδWT and PKCδKO littermates was calculated in a blinded and automated manner using a newly developed custom code. This neural network-based code was trained to segment dendritic spines on a publicly available labeled dataset (Smirnov et al., 2018), as well as a small number of manually annotated images from our PKCδWT and PKCδKO dataset. In order to determine whether the network performed at similar levels across our two groups, we calculated the performance metrics of the network across a sample of test images from PKCδWT and PKCδKO datasets. The network correctly identified structures as spines ∼87% of the time and identified ∼85% of all spines (Fig. 1F,G). Importantly, across all metrics, the code performed at comparable levels for images from PKCδWT and PKCδKO neurons. We therefore used this code to compare the spine density of neurons from PKCδWT and PKCδKO animals and found no significant differences (Fig. 1H). Together, these data suggest an acute, postsynaptic, cellular role for PKCδ in structural plasticity and that deficits in PKCδ KO neurons are not because of changes in neuron health, developmental changes, or circuit-level changes in the KO animals.
As sLTP has been shown to correlate strongly with functional plasticity (Matsuzaki et al., 2004), our results suggested that this structural deficit would be concomitant with deficits in functional, electrically induced potentiation. To test this, electrophysiological LTP was induced in acute hippocampal slices made from PKCδ KO or WT littermate animals. Field EPSPs were recorded in the stratum radiatum of the CA1 region before and after stimulation of Schaffer collaterals with a theta-burst stimulation protocol. While no significant differences were seen in basal synaptic transmission (Fig. 2A), neurons lacking PKCδ had significantly impaired potentiation (Fig. 2B,C).
Development of isozyme-specific sensors for novel PKC isoforms
As loss of function experiments demonstrated a critical role of PKCδ in structural and functional plasticity, we sought to investigate the spatial and temporal activity of PKCδ during plasticity. Therefore, we developed and characterized isozyme-specific sensors for PKCδ and the other novel PKC isozymes (PKCε and PKCη). To do this, we extended our previously developed and broadly applicable sensor design for the classic PKC isozymes (Colgan et al., 2018) to each of the three novel isozymes (Fig. 3A schematic). This approach measures two aspects of PKC activation: (1) translocation of the kinase to the plasma membrane and (2) docking of a pseudosubstrate to the active kinase. These two approaches are named ITRACK (Isozyme-specific TRAnslocation of C Kinase, Fig. 3A, left) and IDOCKS (Isozyme-specific Docking Of C Kinase Substrate, Fig. 3A, right), respectively. ITRACK consists of the novel PKC of interest, N-terminally tagged with a FRET donor fluorophore, mEGFP, and expressed with a plasma membrane-targeted FRET acceptor fluorophore mCherry. Upon activation of the PKC isozyme, translocation of the isozyme to membrane leads to increased FRET between the donor and acceptor fluorophores. This increase in FRET is detected as a decrease in the fluorescence lifetime of the mEGFP-tagged PKC isozyme. Similarly, IDOCKS consists of an mEGFP-tagged PKC isozyme expressed with an acceptor construct consisting of a pseudosubstrate sequence (PKCα/β [19-36]) tagged with two mCherry fluorophores. Upon activation, the pseudosubstrate construct binds the active PKC isozyme, increasing FRET between the GFP on the novel PKC isozyme of interest and the mCherry on the pseudosubstrate. Therefore, the activation of novel PKC isozymes is detected as a decrease in the fluorescence lifetime of the donor fluorophore. These designs provide a robust, isozyme-specific measurement of novel PKC activity. It should be noted that ITRACK primarily will measure novel PKC signaling at the plasma membrane, whereas IDOCKS will measure PKC signaling throughout subcellular compartments.
Figure 3.

Characterization of FLIM-FRET sensors for novel PKC isozymes in HeLa cells. A, Schematic of ITRACK and IDOCKS sensors and respective control sensors (ITRACK CTL, IDOCKS CTL). B, Representative lifetime images of IDOCKSδ in HeLa cells before and 15 min after PdBu (1 μm) application. Warmer colors represent a shorter lifetime and activation of PKCδ. Scale bar, 10 μm. C, Time course and quantification of mean lifetime change of PKCδ [n (cells/experiments)= 13/4], PKCε(6/3), or PKCη(12/4) activity measured by ITRACK in HeLa cells in response to PdBu or DMSO (Veh, 15/4) as indicated. p < 0.0001 one way ANOVA (F(3,42) = 22.9). ***p < 0.001 comparing indicated groups to Veh (Dunnett's multiple comparison test). D, The time course of mean change in the lifetime of ITRACKδ (n (cells/experiments) = 13/4) and IDOCKSδ (12/3) and control sensors in HeLa cells in response to bath application of PdBU (1 μm) p < 0.0001 one way ANOVA (F(3,47) = 42.02). ***p < 0.001 comparing indicated groups (Sidak's post test). E, Response of classic PKC isozyme sensors PKCα (37) PKCβ (29), and PKCγ (22) in response to sequential application of PdBu (1 μm) and ionomycin (1 μm) compared with Novel PKC isozymes PKCδ (13), PKCε (19), and PKCη (12) in HeLa cells. F, The time course of mean change in the lifetime of IDOCKSδ in HeLa cells in response to bath application of PdBU (CTL, 1 μm, n experiments/cells = 3/21) or PdBU and vehicle (Veh, n = 3/19), APV (n = 3/17), or 1NM-PP1 (n = 4/19).
ITRACK and IDOCKS for novel PKC isozymes were first tested in cultured HeLa cells (Fig. 3) in response to strong activation of PKC by the pharmacological PKC activator (PdBu, 1 μm). For each novel isozyme, sensors showed significant reduction in fluorescence lifetime after phorbol ester application compared with vehicle application (Fig. 3B–D). Importantly, control sensors (Fig. 3A schematic), in which the acceptor was not targeted to the plasma membrane (ITRACKCTL, mCherry without CAAX domain) or in which the pseudosubstrate contained a single point mutation that disrupts binding to the PKC kinase site (IDOCKSCTL, PKCα/β [19-36] R27E), showed no significant lifetime changes in response to phorbol ester (Fig. 3D). This suggests that the lifetime changes observed by ITRACK and IDOCKS reflect the translocation of novel PKCs to the membrane and binding of the novel PKCs to the pseudosubstrate, respectively. As novel PKC isozymes are calcium-insensitive, sensors for novel PKCs (PKCδ, PKCε, PKCη) were activated by PdBu (DAG mimetic) alone and did not require calcium. On the other hand, classic isozymes (PKCα, PKCβ, PKCγ) only responded robustly when both PdBu and ionomycin (a calcium ionophore) were applied (Fig. 3E) (Colgan et al., 2018). This result demonstrates that the sensors retain their isozyme-specific activation characteristics.
In addition, we tested whether sensor activation and FLIM imaging is robust to nonrelevant pharmacological application. We examined whether applying APV (an NMDAR antagonist) or 1NM-PP1 (an inert ATP analog at low concentrations), neither of which should have a target in HeLa cells, altered IDOCKSδ sensor activation by the phorbol ester PdBu. As seen in Figure 3F, neither pharmaceutical disrupted sensor performance. Because of the isozyme-specific and sensitive nature of these sensors in HeLa cells, we moved forward to characterizing the sensors in organotypic hippocampal slices (Fig. 4).
Sensors were introduced into CA1 hippocampal neurons of organotypic slice cultures after 10 DIV and were imaged between 2 and 8 d after transfection. Similar to what was seen in HeLa cells, the application of PdBu (1 μm) led to robust activation of both ITRACK and IDOCKS sensors for each of the novel isozymes (δ, e, n) (Fig. 4A–C). Importantly, this sensor activation was not seen when control sensors were used (Fig. 4A, D). Moreover, GFP-PKCδ, ITRACKδ, or IDOCKSδ were all able to rescue the PKCδ KO plasticity phenotype (Figs. 1D, 4E), suggesting that the sensors functionally replace the PKC isozyme and do not significantly inhibit downstream signaling. In addition, IDOCKSδ sensor activity in response to pharmacological activation was similar in WT neurons and PKCδ KO neurons (Fig. 4F). Our results suggest that these sensor approaches enable specific and dynamic measurements of novel PKC activity in neurons with minimal perturbation to the system. As the IDOCKS sensor showed higher sensitivity in neurons (Fig. 4D) and measures PKCδ signaling throughout various subcellular compartments, we replaced PKCδ with IDOCKSδ sensor to further investigate the role of PKCδ in plasticity.
PKCδ is activated in stimulated spines during plasticity induction
Using IDOCKS, we measured the spatial and temporal activation pattern of PKCδ during the induction of single spine plasticity. To do this, we introduced IDOCKSδ into hippocampal CA1 neurons of organotypic slices from PKCδ KO animals and monitored sensor activation during uncaging-induced sLTP. During plasticity induction, there was a rapid decrease in the fluorescence lifetime of IDOCKSδ in the stimulated spine and, to a lesser extent, the underlying dendrite (Fig. 5A). This activation of PKCδ peaked during uncaging in the stimulated spine (∼1 min) and in the underlying dendrite with a slightly slower time course. Activation decayed by ∼70% over the next several minutes but did not return completely to baseline within the time imaged (Fig. 5B). Importantly, in the absence of glutamate, or when NMDARs were blocked with APV, IDOCKSδ showed no activation (Fig. 5C,D), suggesting that activation is induced downstream of NMDARs and is not an artifact of laser stimulation. Moreover, the magnitude of sensor activation was not significantly correlated to the sensor's basal lifetime or the relative expression ratio of the donor and acceptor constructs (Fig. 5E). The time course of PKCδ activation was significantly slower than the activation of the classic PKCα isozyme during sLTP, which activated in milliseconds and decayed within ∼1 s in response to each uncaging pulse of the sLTP induction protocol (Colgan et al., 2018). These results demonstrate an isozyme-specific activation of PKCδ in the stimulated spine and suggest an acute and local role of PKCδ in modulating spine volume and synaptic strength.
PKCδ is downstream of TrkB activation
Novel PKC isozymes are primarily activated by the production and binding to the lipid second messenger DAG. Recently, increased DAG production during NMDA stimulation or the induction of sLTP was demonstrated (Codazzi et al., 2006; Colgan et al., 2018). To test whether in spines PKCδ was activated by acute DAG production, we induced structural plasticity in the presence and absence of a phospholipase C inhibitor (U73122) to inhibit DAG production. Blocking DAG production impaired PKCδ activation during spine plasticity, particularly the early activation of PKCδ (Fig. 5F). While there could be multiple sources of stimulation-induced DAG production, one mechanism described to support spine plasticity has been through NMDA-dependent autocrine release of BDNF and subsequent activation of TrkB (Harward et al., 2016). As we already demonstrated the requirement of the NMDAR for PKCδ activation (Fig. 5C,D), we tested whether PKCδ was also downstream of TrkB activation. To do this, we monitored IDOCKSδ in genetically modified neurons that contained an analog-sensitive point mutation in TrkB (TrkB-AS). This mutation is a single-point mutation in the ATP binding pocket of TrkB (Trkb F616A), which does not alter its kinase activity but renders it sensitive to inhibition by a synthetic ATP analog, 1NM-PP1 (Chen et al., 2005). In neurons containing the TrkB-AS mutation that were untreated or treated only with vehicle, PKCδ activation was normal (Fig. 5G, CTL). However, TrkB-AS-containing neurons treated with the inhibitor 1NM-PP1 showed impaired PKCδ activation (Fig. 5G, 1NM-PP1). These results demonstrate that TrkB is upstream of PKCδ activity during plasticity, suggesting that PKCδ can monitor levels of BDNF-mediated activation of TrkB to facilitate plasticity.
PKCδ activity is not restricted to stimulated spine
During plasticity induction, the activation of PKCδ was highest in the stimulated spine but was present at a lower level throughout the small region of dendrite imaged (Fig. 5B), suggesting that PKCδ activity might be able to spread some distance from the synapse. Consistently, TrkB activation during plasticity has previously been shown to regulate long-distance signaling in neurons, including signals from synapses to the nucleus (Harward et al., 2016; Esvald et al., 2020; Moya-Alvarado and Bronfman, 2023). Therefore, we hypothesized that TrkB activation of PKCδ might be a mechanism to extend local, transient signals in both space and time. To investigate whether PKCδ activity was able to spread over long distances, IDOCKSδ was monitored in the primary apical dendrite before and after inducing plasticity in a single dendritic spine or multiple spines spread across the dendritic tree (Fig. 6A). The induction of plasticity in multiple spines was shown previously to enhance long-distance signaling to the nucleus of another kinase, the ERK (Zhai et al., 2013). In response to plasticity in one spine, there was little activation of PKCδ in the primary dendrite. However, multiple spine stimulation (sequential induction of sLTP in 5 spines on three dendrites) led to a significant increase in PKCδ activity in the primary dendrite that was long-lasting (Fig. 6B,C). To monitor the integration of multiple spine stimulation, we imaged PKCδ activity in the primary dendrite during the sequential stimulation of three spines on different branches with more extended time intervals between spine stimulations (∼8 min). In response to this spaced stimulation of three spines, PKCδ activity integrated the stimuli in the primary dendrite to an intermediate level between the activation of 1 and 5 spines (Fig. 6D,E). This finding suggests that, in addition to a local role in regulating plasticity in the spine, PKCδ may also play a long-distance signaling role in response to multiple spine plasticity.
Previously, PKCδ was found to translocate to the nucleus in response to apoptotic-inducing stimuli (DeVries et al., 2002). To determine whether the long-distance spreading of PKCδ activity was because of the directed translocation of PKCδ into the soma or nucleus, we monitored the somatic localization of GFP-tagged PKCδ (Fig. 6F–H). Multispine stimulation did not induce significant somatic or nuclear translocation of PKCδ compared with mock stimulation, and PKCδ remained largely excluded from the nucleus. Thus, PKCδ does not show activity-dependent translocation to the somatonuclear compartment but, instead, is activated over long distances.
Acute and long-lasting PKCδ signaling have dual functions
To further investigate the potential functional role of this long-distance and long-lasting PKCδ activity, we needed a tool to inhibit the long-lasting phase of PKCδ activity selectively. Because of a lack of isozyme-specific pharmacologic PKC inhibitors, we replaced endogenous PKCδ with a previously characterized PKCδ mutant, PKCδ-AS (Kumar et al., 2015). The PKCδ-AS mutant consists of a point mutation in ATP binding pocket (PKCδ M425A, analogous to TrkB-AS mutation) that leaves its kinase activity unaltered but renders it sensitive to inhibition by a synthetic ATP analog, 1NM-PP1. In vitro analysis of PKCδ kinase activity confirmed that the PKCδ-AS significantly left-shifted the IC50 curve of 1NM-PP1 (Fig. 7A). Importantly, 1NM-PP1 application to WT neurons does not impair structural plasticity (Fig. 7B), suggesting that any drug effects would be because of inhibition of PKCδ kinase activity. We therefore expressed PKCδ-AS in a PKCδ KO background and induced sLTP in the presence of vehicle or 1NM-PP1. In the presence of vehicle, stimulated spines showed normal sLTP (Fig. 7C–E). However, neurons treated with 1NM-PP1 shortly before the induction of sLTP showed impaired sLTP (−10 min, Fig. 7C–E). The return of spine size to prestimulus values at 20-30 min was consistent with the PKCδ KO plasticity phenotype and confirmed that PKCδ kinase activity is required for the spine plasticity. To determine whether sustained PKCδ activity was necessary to maintain early phases of sLTP in the spine, we applied 1NM-PP1 10 min after spine plasticity was induced. Blocking PKCδ kinase activity after plasticity induction, however, did not impair spine structural plasticity (10 min, Fig. 7D,E). Therefore, unlike early activity, long-lasting PKCδ activity is not required to maintain early sLTP of dendritic spines.
Figure 7.
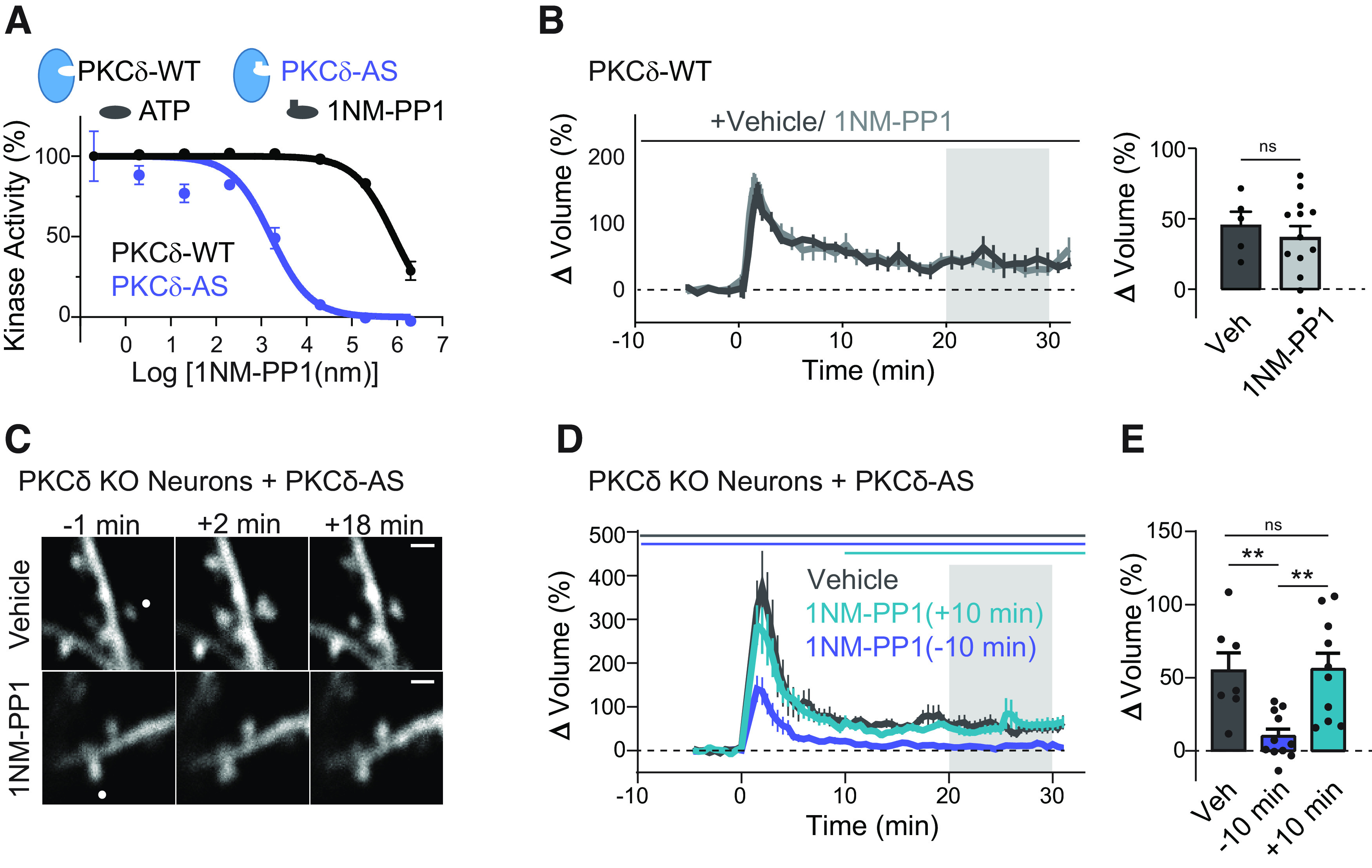
Acute but not long-lasting PKCδ kinase activity is required during plasticity of spines. A, In vitro kinase activity of PKCδ and PKCδ with an inert ATP binding pocket mutation (PKCδ-AS) to varying concentrations of the ATP analog 1NM-PP1. B, Time course and quantification of sLTP in PKCδ WT neurons in the presence of vehicle and 1NM-PP1 (n spines/neurons > 5/5, ns). C, Representative images of sLTP in neurons expressing PKCδ with an inert ATP binding pocket mutation (PKCδ-AS) in place of PKCδ. sLTP was induced in the presence of vehicle or the ATP analog (1NM-PP1, 1 μm) to inhibit PKCδ kinase activity. White dot indicates the location of uncaging. Scale bar, 1 μm. D, Averaged time course of induced volume change of stimulated spines in the presence of vehicle (n = 7) or 1NM-PP1 applied 10 min before (−10 min, n = 11) or 10 min after (+10 min, n = 10) uncaging stimulation. Gray shading represents the time of quantification in E. E, Quantification of mean volume change. One-way ANOVA (F(2,25) = 8.688). **p < 0.0089 (Sidak's multiple comparison post-test), ns, p = 0.999.
Long-lasting PKCδ activity is required for plasticity-transcription coupling
The ability of PKCδ to integrate the plasticity of multiple spines into a long-lasting and spreading signal suggested that it might regulate plasticity-dependent transcriptional programs, which are required for late phases of plasticity and memory formation (Alberini and Kandel, 2014). Multispine stimulation was previously shown to induce long-lasting activation and nuclear translocation of ERK to regulate spine-to-nucleus signaling (Zhai et al., 2013). Interestingly, this signaling was reversed by the application of bisindolylmaleimide I, a broad-spectrum PKC, PDK, and GSK3 inhibitor. Therefore, we tested whether ERK was a downstream target of long-lasting PKCδ activity. To do this, PKCδ-AS was expressed in a PKCδ KO background to render it sensitive to inhibition and expressed together with a previously developed FLIM sensor for ERK activity (EKAR, Fig. 8A) (Harvey et al., 2008a). We directly measured ERK activity in the primary dendrite of neurons during multispine stimulation through the EKAR sensor. Consistent with previous data, long-lasting sensor activity was induced (Fig. 8B,C). This activity was blocked by U0126, an inhibitor of ERK activation by MEK, validating the sensor (Zhai et al., 2013). Next, we inhibited ongoing PKCδ activity by 1NM-PP1 application after uncaging stimulation, so that the manipulation would not inhibit spine plasticity. This manipulation did not affect ERK activity in the primary dendrite, suggesting that ERK is not a target of long-lasting PKCδ.
Figure 8.

Long-lasting PKCδ activity does not regulate ERK. A, Schematic of experimental design B, The time course of ERK activity as measured by the mean change in the lifetime of the EKAR sensor in the primary dendrite after 5 spine stimulation. ERK activity was compared when either vehicle (n (neurons) = 9) or 1NM-PP1 (n = 8) was added 10 min after the end of the stimulation to inhibit long-lasting PKCδ activity or when activation of ERK was blocked by bath application of U0176 (n = 8). Mixed affects analysis **p = 0.009 fixed effect by drug treatment F(2,22) = 5.874. C, Quantification of mean change in lifetime at the time indicated by gray shading in B. p = 0.033 one way ANOVA (F(2,22) = 4.00). *p = 0.034, ns p = 0.98 comparing indicated groups (Sidak's post test).
We therefore tested whether PKCδ might regulate plasticity-induced transcription independent of ERK. One potential downstream target is the activity-dependent transcription factor CREB. CREB is activated by plasticity-inducing protocols and learning and plays a critical role in converting early forms of plasticity into long-lasting forms (Kandel, 2012). Moreover, PKCδ regulation of CREB has been demonstrated in vitro and in other cellular contexts, such as cardiac tissue (Yamamoto et al., 1988; Zhao, 2007; Ozgen et al., 2008; Garg et al., 2013; Morioka et al., 2013). To test whether long-lasting PKCδ activity regulates CREB activity, we expressed PKCδ-AS and a previously developed sensor for CREB in PKCδ KO neurons (Fig. 9A) (Laviv et al., 2020). Following multispine stimulation, CREB was activated robustly in the nucleus (Fig. 9B–D). However, if 1NM-PP1 was applied after multispine stimulation to inhibit the long-lasting phase of PKCδ activity, CREB activation was significantly impaired (Fig. 9B–D). To confirm the regulation of CREB activation by PKCδ, we also measured nuclear p-CREB through immunofluorescence. In a similar experimental design, neurons expressing GFP-PKCδ-AS were stimulated through multispine glutamate uncaging. After stimulation, either vehicle or 1NM-PP1 to inhibit the long-lasting phase of PKCδ was applied. Slices were fixed 1 h after stimulation and stained for p-CREB. Stimulated neurons showed increased p-CREB in the nucleus compared with surrounding nonstimulated neurons (Veh, Fig. 9E,F). However, the application of 1NM-PP1 blocked this enrichment (one-sample t test, p = 0.78 1NM-PP1, Fig. 9E,F). These data suggest that PKCδ serves a dual functional role. In addition to facilitating local spine plasticity, PKCδ integrates multispine plasticity to regulate the magnitude of CREB activity in the nucleus, an efficient mechanism of spine-to-nucleus signaling.
Discussion
In this study, we have found that PKCδ, among the novel isozymes, is uniquely required to induce sLTP. Moreover, by developing highly sensitive isozyme-specific biosensors, we have identified a dual functional role for PKCδ that is defined by the spatiotemporal nature of its activation. During the plasticity of a single spine, NMDA-dependent TrkB activation leads to local PKCδ activity in the stimulated spine within 1 min that returns close to basal levels over ∼5 min. This early and local activation is essential for inducing structural and functional spine plasticity. However, the induction of plasticity at multiple spines across the dendritic tree leads to long-lasting (>40 min) PKCδ activation that spreads throughout the neuron. This long-lasting activity scales with the number of stimulated spines to couple plasticity and transcription through the regulation of CREB in the nucleus. This dual functional role of PKCδ in plasticity is an efficient mechanism of coordinating the induction of plasticity in spines with protein synthesis in the nucleus required for long-term plasticity stabilization.
The development of isozyme-specific biosensors of the novel PKC isozymes has revealed a unique spatiotemporal activation profile of PKCδ during stimulation. The slower and more widespread activation, compared with classic isozymes during plasticity (Colgan et al., 2018), confirms that different PKC isozymes have specific roles, even in response to the same stimulation. During plasticity, PKCδ extends the local and transient glutamate input (μm and ms) into kinase activation that lasts minutes and spreads over tens of microns. This early PKCδ activity is required for the initial phases of structural and functional plasticity.
PKCδ likely has diverse targets, and further study is required to determine the downstream targets of PKCδ in the spine. However, potential targets likely include upstream actin regulating factors, such as Rho GTPases, many of which are regulated by both TrkB and phosphorylation during plasticity (Hedrick et al., 2016). Other potential sites of PKCδ regulation of actin could be through regulation of the Arp2/3 complex, which, when knocked out, shows a similar deficit in sLTP to the PKCδ KO (Kim et al., 2013). In other cell types, PKCδ phosphorylation of coronin modulates the actin nucleator complex Arp2/3 and actin stabilizer cofilin to regulate actin dynamics and structure (Cai et al., 2008; Usatyuk et al., 2013). However, whether this regulation occurs during plasticity is unknown. Finally, PKCδ has been found to bind to F-actin directly through its C2-like domain (López-Lluch et al., 2001; Smallwood et al., 2005). Further study is necessary to determine whether PKCδ binds to actin in neuronal spines. Consistent with a role in actin regulation, spine volume growth follows PKCδ activation closely. PKC has also been shown to regulate activity-dependent exocytosis presynaptically and in other cell types, as well as phosphorylation of AMPAR subtypes. Therefore, PKCδ may play a role in regulating AMPAR exocytosis and incorporation during plasticity (Summers et al., 2019).
In response to multiple spine stimulation the temporal and spatial scale of PKCδ activity is stretched even longer to a cell-wide activation that lasts > 40 min, effectively coupling early plasticity of spines to gene transcription. The spatial spread of PKCδ activity requires a large amplification of signaling as diffusion alone would negate activation because of the orders of magnitude difference in volume (∼10,000×) between the spine and the entire neuron. A positive feedback loop is likely playing a role in the amplification of PKCδ activation. Although during cerebellar LTD a PKC and ERK positive feedback loop has been described (Tanaka and Augustine, 2008), our results suggest that ERK is not involved. Amplification may occur at the level of PKCδ directly, at upstream activators, such as TrkB, phospholipase C, or DAG, or at multiple levels. The spatiotemporal nature of TrkB activation in response to multiple spine stimulation may provide some insight.
BDNF/TrkB signaling has been previously implicated in the induction, expression, and consolidation of plasticity as well as learning (Korte et al., 1995; Figurov et al., 1996; Kang et al., 1997; Linnarsson et al., 1997; Minichiello et al., 1999; Mu et al., 1999). One mechanism of this regulation is activating CREB-mediated transcription (Finkbeiner et al., 1997; Ying et al., 2002). However, this mechanism has often been hypothesized to occur through activation of Ras/MAPK/ERK signaling (Impey et al., 1998; Ying et al., 2002; Zhai et al., 2013). Our study, however, implicates PKCδ in TrkB signaling to CREB and suggests that PKCδ regulation of CREB during plasticity induction is independent of ERK activity (Fig. 8). This finding is consistent with previous work that generated phospho-site-specific mutations in TrkB to study the role of specific downstream signaling cascades in plasticity (Minichiello et al., 2002). Animals with a mutation in the phospho-site that binds phospholipase C to produce DAG and PKC signaling showed impaired expression and consolidation of LTP and reduced learning (Minichiello et al., 2002; Gruart et al., 2007). Moreover, these neurons showed impaired activation of CREB in response to BDNF application. These deficits were seen despite ERK signaling remaining intact. Together, the results implicate PKCδ as a necessary mediator of BDNF-TrkB signaling to CREB during CA1 plasticity and hippocampal learning. These results also highlight the need for further understanding of plasticity-dependent transcriptional regulation in response to various stimuli. Given the recent implication of TrkB modulation of the CREB pathway in the mode of action of diverse antidepressants (Rantamäki et al., 2007; Casarotto et al., 2021), the role of PKCδ in antidepressant effects and the detailed signaling mechanisms through which PKCδ regulates CREB activity is warranted.
The development and characterization of isozyme-specific sensors for novel PKC activity will further the study of the novel class of PKCs. Particularly, isozyme-specific activity sensors will clarify the multiple roles of PKC isozymes specified by the spatiotemporal nature of their activation. The closely related structure of PKC isozymes and the challenge to develop specific pharmacology to activate or inhibit novel PKC isozymes have limited our understanding of PKC function in dendritic spines and plasticity. Studies using combinations of activators and inhibitors with different affinities for various PKC isozymes have demonstrated that novel PKC isozymes play an important role in spine morphology and function (Sun et al., 2014; Ly et al., 2020; Ge et al., 2023). However, the complex pharmacological properties against different potential targets complicate the interpretation of pharmacological studies (Singh et al., 2021). Nonetheless, these studies suggest that different novel PKC isozymes are involved in unique aspects of cell morphology regulation. For example, pharmacological studies suggest that PKCepsilon may be involved in regulating spinogenesis during development and aging (Hongpaisan et al., 2013; Schaffer et al., 2018). The continued development of new approaches for temporally controlled, isozyme-specific inhibition for PKC isozymes, including chemogenetic/optogenetic approaches will complement sensor work to aid in distentangling isozyme-specific roles. As the novel subfamily of PKC isoforms has been implicated in numerous disorders, including metabolic disease, cardiovascular disease, autoimmune disorders, and neurodegenerative disorders, these tools will be of great use for dissecting signaling pathways leading to cellular phenotypes in these diseases.
Footnotes
This work was supported by National Institutes of Health Grants R35-NS-116804 and R01-MH-080047 to R.Y. and Grant F32MH101954 to L.A.C. We thank David Kloetzer for laboratory management; Long Yan for microscopy development and maintenance; Yuki Hayano and Kathy Liu for technical assistance; Mary Philips for suggestions in figure design; and the MPFI ARC, including Minida Dowdy, Elizabeth Garcia, and Amanda Coldwell, for animal care and maintenance.
R.Y. is the owner of Florida Lifetime Imaging LLC, a company that sells integrated solutions for performing fluorescence lifetime imaging and fluorescence resonance energy transfer imaging. The remaining authors declare no competing financial interests.
References
- Abdou K, Shehata M, Choko K, Nishizono H, Matsuo M, Muramatsu SI, Inokuchi K (2018) Synapse-specific representation of the identity of overlapping memory engrams. Science 360:1227–1231. 10.1126/science.aat3810 [DOI] [PubMed] [Google Scholar]
- Alberini CM, Kandel ER (2014) The regulation of transcription in memory consolidation. Cold Spring Harb Perspect Biol 7:a021741. 10.1101/cshperspect.a021741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodinova AA, Zuzina AB, Balaban PM (2017) Role of atypical protein kinases in maintenance of long-term memory and synaptic plasticity. Biochemistry (Mosc) 82:243–256. 10.1134/S0006297917030026 [DOI] [PubMed] [Google Scholar]
- Cai L, Makhov AM, Schafer DA, Bear JE (2008) Coronin 1B antagonizes cortactin and remodels Arp2/3-containing actin branches in lamellipodia. Cell 134:828–842. 10.1016/j.cell.2008.06.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casarotto PC, et al. (2021) Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 184:1299–1313.e19. 10.1016/j.cell.2021.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ye H, Kuruvilla R, Ramanan N, Scangos KW, Zhang C, Johnson NM, England PM, Shokat KM, Ginty DD (2005) A chemical-genetic approach to studying neurotrophin signaling. Neuron 46:13–21. 10.1016/j.neuron.2005.03.009 [DOI] [PubMed] [Google Scholar]
- Choi JH, Sim SE, Kim J, Choi DI, Oh J, Ye S, Lee J, Kim T, Ko HG, Lim CS, Kaang BK (2018) Interregional synaptic maps among engram cells underlie memory formation. Science 360:430–435. 10.1126/science.aas9204 [DOI] [PubMed] [Google Scholar]
- Chou WH, Choi DS, Zhang H, Mu D, McMahon T, Kharazia VN, Lowell CA, Ferriero DM, Messing RO (2004) Neutrophil protein kinase Cdelta as a mediator of stroke-reperfusion injury. J Clin Invest 114:49–56. 10.1172/JCI21655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codazzi F, Di Cesare A, Chiulli N, Albanese A, Meyer T, Zacchetti D, Grohovaz F (2006) Synergistic control of protein kinase C activity by ionotropic and metabotropic glutamate receptor inputs in hippocampal neurons. J Neurosci 26:3404–3411. 10.1523/JNEUROSCI.0478-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan LA, Hu M, Misler JA, Parra-Bueno P, Moran CM, Leitges M, Yasuda R (2018) PKCα integrates spatiotemporally distinct Ca2+ and autocrine BDNF signaling to facilitate synaptic plasticity. Nat Neurosci 21:1027–1037. 10.1038/s41593-018-0184-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries TA, Neville MC, Reyland ME (2002) Nuclear import of PKCdelta is required for apoptosis: identification of a novel nuclear import sequence. EMBO J 21:6050–6060. 10.1093/emboj/cdf606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esvald EE, Tuvikene J, Sirp A, Patil S, Bramham CR, Timmusk T (2020) CREB family transcription factors are major mediators of BDNF transcriptional autoregulation in cortical neurons. J Neurosci 40:1405–1426. 10.1523/JNEUROSCI.0367-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley J, Auerbach S (1986) Protein kinase C activation induces conductance changes in Hermissenda photoreceptors like those seen in associative learning. Nature 319:220–223. 10.1038/319220a0 [DOI] [PubMed] [Google Scholar]
- Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B (1996) Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 381:706–709. 10.1038/381706a0 [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME (1997) CREB: a major mediator of neuronal neurotrophin responses. Neuron 19:1031–1047. 10.1016/S0896-6273(00)80395-5 [DOI] [PubMed] [Google Scholar]
- Fu G, Hu J, Niederberger-Magnenat N, Rybakin V, Casas J, Yachi PP, Feldstein S, Ma B, Hoerter JA, Ampudia J, Rigaud S, Lambolez F, Gavin AL, Sauer K, Cheroutre H, Gascoigne NRJ (2011) Protein kinase C η is required for T cell activation and homeostatic proliferation. Sci Signal 4:ra84. 10.1126/scisignal.2002058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg R, Caino MC, Kazanietz MG (2013) Regulation of transcriptional networks by PKC isozymes: identification of c-Rel as a key transcription factor for PKC-regulated genes. PLoS One 8:e67319. 10.1371/journal.pone.0067319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge C, Wang X, Wang Y, Lei L, Song G, Qian M, Wang S (2023) PKCε inhibition prevents ischemia-induced dendritic spine impairment in cultured primary neurons. Exp Ther Med 25:152. 10.3892/etm.2023.11851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobbo F, Cattaneo A (2020) Neuronal activity at synapse resolution: reporters and effectors for synaptic neuroscience. Front Mol Neurosci 13:572312. 10.3389/fnmol.2020.572312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto A, Bota A, Miya K, Wang J, Tsukamoto S, Jiang X, Hirai D, Murayama M, Matsuda T, McHugh TJ, Nagai T, Hayashi Y (2021) Stepwise synaptic plasticity events drive the early phase of memory consolidation. Science 374:857–863. 10.1126/science.abj9195 [DOI] [PubMed] [Google Scholar]
- Gruart A, Sciarretta C, Valenzuela-Harrington M, Delgado-García JM, Minichiello L (2007) Mutation at the TrkB PLCγ-docking site affects hippocampal LTP and associative learning in conscious mice. Learn Mem 14:54–62. 10.1101/lm.428307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CD, Ehrhardt AG, Cellurale C, Zhong H, Yasuda R, Davis RJ, Svoboda K (2008a) A genetically encoded fluorescent sensor of ERK activity. Proc Natl Acad Sci USA 105:19264–19269. 10.1073/pnas.0804598105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CD, Yasuda R, Zhong H, Svoboda K (2008b) The spread of Ras activity triggered by activation of a single dendritic spine. Science 321:136–140. 10.1126/science.1159675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harward SC, Hedrick NG, Hall CE, Parra-Bueno P, Milner TA, Pan E, Laviv T, Hempstead BL, Yasuda R, McNamara JO (2016) Autocrine BDNF–TrkB signalling within a single dendritic spine. Nature 538:99–103. 10.1038/nature19766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi-Takagi A, Yagishita S, Nakamura M, Shirai F, Wu YI, Loshbaugh AL, Kuhlman B, Hahn KM, Kasai H (2015) Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature 525:333–338. 10.1038/nature15257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick NG, Harward SC, Hall CE, Murakoshi H, McNamara JO, Yasuda R (2016) Rho GTPase complementation underlies BDNF-dependent homo- and heterosynaptic plasticity. Nature 538:104–108. 10.1038/nature19784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongpaisan J, Xu C, Sen A, Nelson TJ, Alkon D (2013) PKC activation during training restores mushroom spine synapses and memory in the aged rat. Neurobiol Dis 55:44–62. 10.1016/j.nbd.2013.03.012 [DOI] [PubMed] [Google Scholar]
- Hu GY, Hvalby Ø, Walaas SI, Albert KA, Skjeflo P, Andersen P, Greengard P (1987) Protein kinase C injection into hippocampal pyramidal cells elicits features of long term potentiation. Nature 328:426–429. 10.1038/328426a0 [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR (1998) Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron 21:869–883. 10.1016/s0896-6273(00)80602-9 [DOI] [PubMed] [Google Scholar]
- Jocher G, Chaurasia A, Stoken A, Borovec J, Kwon Y, Michael K, Fang J (2022) ultralytics/yolov5: v7.0-YOLOv5 SOTA Realtime Instance Segmentation. 10.5281/zenodo.7347926 [DOI] [Google Scholar]
- Kajimoto T, Sawamura S, Tohyama Y, Mori Y, Newton AC (2010) Protein kinase C δ-specific activity reporter reveals agonist-evoked nuclear activity controlled by Src family of kinases. J Biol Chem 285:41896–41910. 10.1074/jbc.M110.184028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER (2012) The molecular biology of memory: CAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain 5:14. 10.1186/1756-6606-5-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Welcher AA, Shelton D, Schuman EM (1997) Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron 19:653–664. 10.1016/s0896-6273(00)80378-5 [DOI] [PubMed] [Google Scholar]
- Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO (1999) A novel nociceptor signaling pathway revealed in protein kinase C ε mutant mice. Neuron 24:253–260. 10.1016/s0896-6273(00)80837-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IH, Racz B, Wang H, Burianek L, Weinberg R, Yasuda R, Wetsel WC, Soderling SH (2013) Disruption of Arp2/3 results in asymmetric structural plasticity of dendritic spines and progressive synaptic and behavioral abnormalities. J Neurosci 33:6081–6092. 10.1523/JNEUROSCI.0035-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T (1995) Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA 92:8856–8860. 10.1073/pnas.92.19.8856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Weng YC, Geldenhuys WJ, Wang D, Han X, Messing RO, Chou WH (2015) Generation and characterization of ATP analog-specific protein kinase Cδ. J Biol Chem 290:1936–1951. 10.1074/jbc.M114.598698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviv T, Scholl B, Parra-Bueno P, Foote B, Zhang C, Yan L, Hayano Y, Chu J, Yasuda R (2020) In vivo imaging of the coupling between neuronal and CREB activity in the mouse brain. Neuron 105:799–812.e5. 10.1016/j.neuron.2019.11.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnarsson S, Björklund A, Ernfors P (1997) Learning deficit in BDNF mutant mice. Eur J Neurosci 9:2581–2587. 10.1111/j.1460-9568.1997.tb01687.x [DOI] [PubMed] [Google Scholar]
- López-Lluch G, Bird MM, Canas B, Godovac-Zimmerman J, Ridley A, Segal AW, Dekker LV (2001) Protein kinase C-delta C2-like domain is a binding site for actin and enables actin redistribution in neutrophils. Biochem J 357:39–47. 10.1042/bj3570039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly C, Shimizu AJ, Vargas MV, Duim WC, Wender PA, Olson DE (2020) Bryostatin 1 promotes synaptogenesis and reduces dendritic spine density in cortical cultures through a PKC-dependent mechanism. ACS Chem Neurosci 11:1545–1554. 10.1021/acschemneuro.0c00175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Madison DV, Nicoll RA (1986) Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature 321:175–177. 10.1038/321175a0 [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429:761–766. 10.1038/nature02617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R (1999) Essential role for TrkB receptors in hippocampus-mediated learning. Neuron 24:401–414. 10.1016/S0896-6273(00)80853-3 [DOI] [PubMed] [Google Scholar]
- Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M (2002) Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron 36:121–137. 10.1016/S0896-6273(02)00942-X [DOI] [PubMed] [Google Scholar]
- Morioka N, Yoshida Y, Nakamura Y, Hidaka N, Hisaoka-Nakashima K, Nakata Y (2013) The regulation of exon-specific brain-derived neurotrophic factor mRNA expression by protein kinase C in rat cultured dorsal root ganglion neurons. Brain Res 1509:20–31. 10.1016/j.brainres.2013.03.015 [DOI] [PubMed] [Google Scholar]
- Moya-Alvarado G, Bronfman FC (2023) BDNF/TrkB mediates long-distance dendritic growth by activating CREB/PI3K-mTOR-dependent translation in neuronal cell bodies. eLife 12:e77455. 10.1101/2020.08.22.26292336826992 [DOI] [Google Scholar]
- Mu JS, Li WP, Yao ZB, Zhou XF (1999) Deprivation of endogenous brain-derived neurotrophic factor results in impairment of spatial learning and memory in adult rats. Brain Res 835:259–265. 10.1016/s0006-8993(99)01592-9 [DOI] [PubMed] [Google Scholar]
- Mukherjee A, Roy S, Saha B, Mukherjee D (2016) Spatio-temporal regulation of PKC isoforms imparts signaling specificity. Front Immunol 7:45. 10.3389/fimmu.2016.00045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien JA, Lummis SC (2006) Biolistic transfection of neuronal cultures using a hand-held gene gun. Nat Protoc 1:977–981. 10.1038/nprot.2006.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olds JL, Anderson ML, McPhie DL, Staten LD, Alkon DL (1989) Imaging of memory-specific changes in the distribution of protein kinase C in the hippocampus. Science 245:866–869. 10.1126/science.2772638 [DOI] [PubMed] [Google Scholar]
- Ozgen N, Obreztchikova M, Guo J, Elouardighi H, Dorn GW, Wilson BA, Steinberg SF (2008) Protein kinase D links Gq-coupled receptors to cAMP response element-binding protein (CREB)-Ser133 phosphorylation in the heart. J Biol Chem 283:17009–17019. 10.1074/jbc.M709851200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymann KG, Schulzeck K, Kase H, Matthies H (1988) Phorbol ester-induced hippocampal long-term potentiation is counteracted by inhibitors of protein kinase C. Exp Brain Res 71:227–230. 10.1007/BF00247540 [DOI] [PubMed] [Google Scholar]
- Rantamäki T, Hendolin P, Kankaanpää A, Mijatovic J, Piepponen P, Domenici E, Chao MV, Männistö PT, Castrén E (2007) Pharmacologically diverse antidepressants rapidly activate brain-derived neurotrophic factor receptor TrkB and induce phospholipase-Cγ signaling pathways in mouse brain. Neuropsychopharmacology 32:2152–2162. 10.1038/sj.npp.1301345 [DOI] [PubMed] [Google Scholar]
- Schaffer TB, Smith JE, Cook EK, Phan T, Margolis SS (2018) PKCε inhibits neuronal dendritic spine development through dual phosphorylation of Ephexin5. Cell Rep 25:2470–2483.e8. 10.1016/j.celrep.2018.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, Kumar S, Tomar MS, Verma PK, Kumar A, Kumar S, Kumar N, Singh JP, Acharya A (2021) Putative role of natural products as Protein Kinase C modulator in different disease conditions. Daru 29:397–414. 10.1007/s40199-021-00401-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood ND, Hausman BS, Wang X, Liedtke CM (2005) Involvement of NH2 terminus of PKC-delta in binding to F-actin during activation of Calu-3 airway epithelial NKCC1. Am J Physiol Cell Physiol 288:C906–C912. 10.1152/ajpcell.00484.2004 [DOI] [PubMed] [Google Scholar]
- Smirnov MS, Evans PR, Garrett TR, Yan L, Yasuda R (2017) Automated remote focusing, drift correction, and photostimulation to evaluate structural plasticity in dendritic spines. PLoS One 12:e0170586. 10.1371/journal.pone.0170586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnov MS, Garrett TR, Yasuda R (2018) An open-source tool for analysis and automatic identification of dendritic spines using machine learning. PLoS One 13:e0199589. 10.1371/journal.pone.0199589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolen P, Baxter DA, Byrne JH (2019) How can memories last for days, years, or a lifetime? Proposed mechanisms for maintaining synaptic potentiation and memory. Learn Mem 26:133–150. 10.1101/lm.049395.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg SF (2008) Structural basis of protein kinase C isoform function. Physiol Rev 88:1341–1378. 10.1152/physrev.00034.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D (1991) A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37:173–182. 10.1016/0165-0270(91)90128-m [DOI] [PubMed] [Google Scholar]
- Summers KC, Bogard AS, Tavalin SJ (2019) Preferential generation of Ca2+-permeable AMPA receptors by AKAP79-anchored protein kinase C proceeds via GluA1 subunit phosphorylation at Ser-831. J Biol Chem 294:5521–5535. 10.1074/jbc.RA118.004340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun MK, Hongpaisan J, Lim CS, Alkon DL (2014) Bryostatin-1 restores hippocampal synapses and spatial learning and memory in adult fragile x mice. J Pharmacol Exp Ther 349:393–401. 10.1124/jpet.114.214098 [DOI] [PubMed] [Google Scholar]
- Tanaka K, Augustine GJ (2008) A positive feedback signal transduction loop determines timing of cerebellar long-term depression. Neuron 59:608–620. 10.1016/j.neuron.2008.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usatyuk PV, Burns M, Mohan V, Pendyala S, He D, Ebenezer DL, Harijith A, Fu P, Huang LS, Bear JE, Garcia JG, Natarajan V (2013) Coronin 1B regulates S1P-induced human lung endothelial cell chemotaxis: role of PLD2, protein kinase C and Rac1 signal transduction. PLoS One 8:e63007. 10.1371/journal.pone.0063007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Zee EA, Douma BR (1997) Historical review of research on protein kinase C in learning and memory. Prog Neuropsychopharmacol Biol Psychiatry 21:379–406. [DOI] [PubMed] [Google Scholar]
- Wang JH, Feng DP (1992) Postsynaptic protein kinase C essential to induction and maintenance of long-term potentiation in the hippocampal CA1 region. Proc Natl Acad Sci USA 89: 2576–2580. 10.1073/pnas.89.7.2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto KK, Gonzalez GA, Biggs WH, Montminy MR (1988) Phosphorylation-induced binding and transcriptional efficacy of nuclear factor CREB. Nature 334:494–498. 10.1038/334494a0 [DOI] [PubMed] [Google Scholar]
- Yasuda R (2017) Biophysics of biochemical signaling in dendritic spines: implications in synaptic plasticity. Biophys J 113:2152–2159. 10.1016/j.bpj.2017.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV, Bramham CR (2002) Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of arc synthesis. J Neurosci 22:1532–1540. 10.1523/JNEUROSCI.22-05-01532.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai S, Ark ED, Parra-Bueno P, Yasuda R (2013) Long-distance integration of nuclear ERK signaling triggered by activation of a few dendritic spines. Science 342:1107–1111. 10.1126/science.1245622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D (2007) Protein kinase Cdelta-mediated CREB activation regulates ghrelin-induced cyclooxygenase-2 expression and prostaglandin E2 production in human colonic epithelial cells. J Cell Biochem 102:1245–1255. 10.1002/jcb.21355 [DOI] [PubMed] [Google Scholar]