

Abstract
During the first two postnatal weeks, intraneuronal chloride concentrations in rodents gradually decrease, causing a shift from depolarizing to hyperpolarizing GABA responses. The postnatal GABA shift is delayed in rodent models for neurodevelopmental disorders and in human patients, but the impact of a delayed GABA shift on the developing brain remains obscure. Here we examine the direct and indirect consequences of a delayed postnatal GABA shift on network development in organotypic hippocampal cultures made from 6- to 7-d-old mice by treating the cultures for 1 week with VU0463271, a specific inhibitor of the chloride exporter KCC2. We verified that VU treatment delayed the GABA shift and kept GABA signaling depolarizing until DIV9. We found that the structural and functional development of excitatory and inhibitory synapses at DIV9 was not affected after VU treatment. In line with previous studies, we observed that GABA signaling was already inhibitory in control and VU-treated postnatal slices. Surprisingly, 14 d after the VU treatment had ended (DIV21), we observed an increased frequency of spontaneous inhibitory postsynaptic currents in CA1 pyramidal cells, while excitatory currents were not changed. Synapse numbers and release probability were unaffected. We found that dendrite-targeting interneurons in the stratum radiatum had an elevated resting membrane potential, while pyramidal cells were less excitable compared with control slices. Our results show that depolarizing GABA signaling does not promote synapse formation after P7, and suggest that postnatal intracellular chloride levels indirectly affect membrane properties in a cell-specific manner.
SIGNIFICANCE STATEMENT During brain development, the action of neurotransmitter GABA shifts from depolarizing to hyperpolarizing. This shift is a thought to play a critical role in synapse formation. A delayed shift is common in rodent models for neurodevelopmental disorders and in human patients, but its consequences for synaptic development remain obscure. Here, we delayed the GABA shift by 1 week in organotypic hippocampal cultures and carefully examined the consequences for circuit development. We find that delaying the shift has no direct effects on synaptic development, but instead leads to indirect, cell type-specific changes in membrane properties. Our data call for careful assessment of alterations in cellular excitability in neurodevelopmental disorders.
Keywords: brain development, chloride, homeostasis, membrane excitability, neurodevelopmental disorders, synapse formation
Introduction
During postnatal development of rodents, intracellular chloride concentrations in neurons gradually decrease. Since GABAA receptors mainly conduct chloride, the developmental decrease in intracellular chloride causes the reversal potential for GABA currents to gradually drop below resting membrane potential (RMP). As a result, the GABAergic driving force (DF) shifts from depolarizing in immature neurons to hyperpolarizing in mature neurons (Rivera et al., 1999; Hübner et al., 2001; Tyzio et al., 2007; Romo-Parra et al., 2008; Kirmse et al., 2015; Tsukahara et al., 2015; Sulis Sato et al., 2017; Murata and Colonnese, 2020). This GABA shift is a key event during development and is suggested to play a critical role in the formation and maturation of synapses (Leinekugel et al., 1997; Akerman and Cline, 2006; Wang and Kriegstein, 2008, 2011; Chancey et al., 2013; van Rheede et al., 2015; Oh et al., 2016).
The shift in postnatal chloride levels is the direct result of an increased function of the chloride exporter KCC2 (K-Cl cotransporter 2) relative to the chloride importer NKCC1 (Na-K-2Cl cotransporter 1) (Rivera et al., 1999; Gulyás et al., 2001; Yamada et al., 2004; Dzhala et al., 2005; Otsu et al., 2020). In humans, the shift in chloride transporter expression occurs during the first year after birth (Dzhala et al., 2005; Sedmak et al., 2016), but in some patients with neurodevelopmental disorders (NDDs) chloride homeostasis seems impaired and alterations in the expression of both NKCC1 and KCC2 have been widely reported (Talos et al., 2012; Duarte et al., 2013; Merner et al., 2015; Ruffolo et al., 2016; Birey et al., 2022). In rodents, the GABA shift occurs during the first two postnatal weeks (Rivera et al., 1999; Stein et al., 2004; Tyzio et al., 2007; Romo-Parra et al., 2008; Kirmse et al., 2015; Sulis Sato et al., 2017; Murata and Colonnese, 2020). In many animal models for NDDs, a delayed GABA shift and alterations in chloride cotransporter expression have been reported (He et al., 2014; Tyzio et al., 2014; Banerjee et al., 2016; Corradini et al., 2017; Roux et al., 2018; Fernandez et al., 2019; Lozovaya et al., 2019; Bertoni et al., 2021), suggesting that this is a key feature of many NDDs. In NDD mouse models, alterations in early spontaneous network activity are observed (Gonçalves et al., 2013; Cheyne et al., 2019), and coordination between excitatory and inhibitory transmission is impaired in adult mice (Gogolla et al., 2009; Antoine et al., 2019). These observations suggest that a delay in the GABA shift during early postnatal development disturbs synaptic connectivity and network development with consequences for adulthood (Meredith, 2015; Molnár et al., 2020). However, a direct link between the timing of the GABA shift and synaptic connectivity remains elusive.
Depolarizing, excitatory GABA signaling supports spontaneous oscillations in the immature rodent brain (Ben-Ari et al., 1989; Khazipov et al., 2004; Sipila et al., 2006; Rheims et al., 2008; Spoljaric et al., 2019) and can induce the formation and maturation of excitatory synapses until the first week after birth (Leinekugel et al., 1997; Wang and Kriegstein, 2008, 2011; van Rheede et al., 2015; Oh et al., 2016). After the GABA shift, the KCC2 protein takes over the synapse-promoting role, as it directly facilitates spine growth by promoting actin dynamics in spines and supports AMPA receptor insertion and confinement (Li et al., 2007; Gauvain et al., 2011; Fiumelli et al., 2013; Chevy et al., 2015; Puskarjov et al., 2017; Awad et al., 2018; Kesaf et al., 2020). The effect of depolarizing GABA on inhibitory synapses is less clear, as studies have reported both positive and negative effects (Chudotvorova et al., 2005; Akerman and Cline, 2006; Nakanishi et al., 2007; Wang and Kriegstein, 2008, 2011). It is important to note that the shift from depolarizing to hyperpolarizing GABA signaling does not necessarily go hand in hand with a shift from excitatory to inhibitory action. Depolarizing GABA can already have an inhibitory action and limit neuronal activity when GABA-mediated depolarization is subthreshold and opening of GABAA receptors shunts excitatory inputs (Staley and Mody, 1992; Kirmse et al., 2015; Murata and Colonnese, 2020; Salmon et al., 2020). When GABA signaling becomes inhibitory, it restrains excitatory synapse formation (Kang, 2019; Salmon, 2020) and network oscillations disappear (Ben-Ari et al., 1989; Khazipov et al., 2004). Recent studies have suggested that depolarizing GABA is already inhibitory in the brain of newborn rodents after P3-P7 (Kirmse et al., 2015; Valeeva et al., 2016; Murata and Colonnese, 2020). This would imply that the role of depolarizing GABA in promoting synapse formation is limited to the period before and up to the first week after birth.
Many NDD mouse models show a delay in the postnatal GABA shift, although the precise delay is variable. For instance, the GABA shift is delayed with 3 d in the cerebellum of mice exposed in utero to valproate (Roux et al., 2018), the delay is 2-7 d in the hippocampus of Magel2 KO mice (Bertoni et al., 2021), and 6 d in the cortex of fragile X mice (He et al., 2014). Longer delays have also been reported (Banerjee et al., 2016; Corradini et al., 2017; Fernandez et al., 2019; Lozovaya et al., 2019). Here, we carefully assessed the direct and indirect consequences of a delayed postnatal GABA shift on the development of synapses. We used hippocampal organotypic cultures, as in these slices, the anatomy and the development of excitatory and inhibitory synapses are largely preserved compared with the in vivo situation (De Simoni, 2003). We treated the cultures with the KCC2 antagonist VU0463271 (VU) to block chloride extrusion for 1 week (from day in vitro 1 (DIV1) to DIV8) while preserving the structural role of the KCC2 protein in spines (Li et al., 2007; Kesaf et al., 2020). VU treatment delayed the GABA shift without affecting expression levels of the chloride transporters. We found that excitatory and inhibitory synapses were not affected after 1 week of VU treatment. However, 14 d after the VU treatment had ended, we observed an increased frequency of sIPSC, while sEPSCs were unaltered. We found that this was correlated with specific alterations in membrane excitability of pyramidal cells and interneurons. Our data suggest that delaying the GABA shift does not directly affect synaptic development but rather leads to indirect cell type-specific changes in membrane properties.
Materials and Methods
Animals
All animal experiments were performed in compliance with the guidelines for the welfare of experimental animals issued by the Federal Government of The Netherlands and were approved by the Dutch Central Committee Animal experiments, Project AVD1080020173847 and Project AVD1150020184927. In this study, we used male and female transgenic mice: GAD65-GFP mice (López-Bendito et al., 2004) (bred as a heterozygous line in a C57BL/6JRj background), VGAT-Cre mice (JAX stock #028862), and SuperClomeleon (SClm) mice. The SClm mice (Herstel et al., 2022) are SuperClomeleonlox/– mice (Rahmati et al., 2021), a gift from Kevin Staley (Massachusetts General Hospital) crossed with CamKIIαCre/– mice (Tsien et al., 1996; Casanova et al., 2001). SClm mice express the chloride sensor SClm in up to 70% of the pyramidal neurons in the hippocampus (Casanova et al., 2001; Wang et al., 2013). VGAT-Cre mice express Cre recombinase expression in all inhibitory GABAergic neurons (Vong et al., 2011), and GAD65-GFP mice express GFP in ∼20% of GABAergic interneurons in the CA1 region of the hippocampus (Wierenga et al., 2010). As we did not detect any differences between slices from male and female mice, all data were pooled.
Organotypic hippocampal culture preparation and VU treatment
Organotypic hippocampal cultures were made from P6-P7 mice as described previously (Hu et al., 2019; Herstel et al., 2022), based on Stoppini et al. (1991). Mice were decapitated, and their brain was rapidly placed in ice-cold Gray's Balanced Salt Solution containing the following (in mm): 137 NaCl, 5 KCl, 1.5 CaCl2, 1 MgCl2, 0.3 MgSO4, 0.2 KH2PO4, and 0.85 Na2HPO4, with 25 mm glucose, 12.5 mm HEPES, and 1 mm kynurenic acid (pH set at 7.2, osmolarity set at ∼320 mOsm, sterile filtered); 400-μm-thick transverse hippocampal slices were cut with a McIlwain tissue chopper. Slices were placed on Millicell membrane inserts (Millipore) in wells containing 1 ml culture medium (consisting of 48% MEM, 25% HBSS, 25% horse serum, 25 mm glucose, and 12.5 mm HEPES, with an osmolarity of ∼325 mOsm and a pH of 7.3-7.4). Slices were stored in an incubator (35°C with 5% CO2). Culture medium was replaced by culture medium supplemented with 0.1% DMSO (Sigma-Aldrich) or 1 μm VU (Sigma-Aldrich, in 0.1% DMSO) at DIV1. In addition, a small drop of medium supplemented with DMSO or VU was carefully placed on top of the slices. In this way, cultures were treated 3 times per week until DIV8. From DIV8 onwards, cultures received normal culture medium 3 times per week. Experiments were performed at DIV1-DIV3 (DIV2), 7-8 (DIV8), 8-10 (DIV9), or 20-22 (DIV21). Experiments at DIV9 were performed 1-55 h after cessation of the VU treatment, but within groups there was no correlation between measurements and DIV.
Lentivirus with KCC2 short hairpin (SH)
To transiently knock down KCC2 expression, a KCC2 SH lentivirus was produced. HEK293T cells were maintained at a high growth rate in DMEM supplemented with 10% FCS and 1% pen/strep. At 1 d after plating, cells were transfected using PEI (Polysciences) with second-generation LV packaging plasmids (psPAX2 and 2MD2.G) and KCC2-SH-TetR-GFP (cloned from a KCC2-SH plasmid, a gift from Franck Polleux, Zuckerman Institute Columbia University, New York) at a 1:1:1 molar ratio. Six hours after transfection, cells were washed once with PBS, and medium was replaced with DMEM containing 1% pen/strep. At 48 h after transfection, the supernatant was harvested and briefly centrifuged at 700 × g to remove cell debris. The supernatant was concentrated using Amicon Ultra 15 100K MWCO columns (Milipore) and frozen at −80°C until infection. Virus was injected into the CA1 region of slice cultures at DIV1 using a microinjector (Eppendorf, FemtoJet) aided by a stereoscopic microscope (Leica, M80). Medium was supplemented with 1 µg/ml doxycycline (Abcam). After injection, a 15 μl drop of doxycycline-containing medium was added onto the slice. Medium was replaced 4 times with new medium containing doxycycline, and a 15 μl drop of doxycycline-containing medium was added onto the slice until immunohistochemistry or electrophysiology experiments at DIV8-DIV10.
Electrophysiology and analysis
Organotypic cultures were transferred to a recording chamber and continuously perfused with carbonated (95% O2, 5% CO2) ACSF as follows (in mm): 126 NaCl, 3 KCl, 2.5 CaCl2, 1.3 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, 20 glucose, with an osmolarity of ∼310 mOsm) at a rate of ∼1 ml/min. For acute treatments, ACSF containing 0.1% DMSO or 1 μm VU (dissolved in 0.1% DMSO) was bath-applied for 5 min. Bath temperature was maintained at 29°C-32°C. Recordings were acquired using an MultiClamp 700B amplifier (Molecular Devices), filtered with a 3 kHz Bessel filter, and stored using pClamp10 software.
Perforated patch-clamp recordings were performed in CA1 pyramidal neurons. Recording pipettes with resistances of 2-4 mΩ were pulled from borosilicate glass capillaries (World Precision Instruments). The pipette tip was filled with gramicidin-free KCl solution (140 mm KCl and 10 mm HEPES, pH adjusted to 7.2 with KOH, and osmolarity 285 mOsm/l) and then backfilled with solution containing gramicidin (60 µg/ml, Sigma). Neurons were held at −65 mV, and the access resistance of the perforated cells was monitored constantly before and during recordings. An access resistance of 50 mΩ was considered acceptable to start recording. Recordings were excluded if the RMP exceeded −50 mV or if the series resistance after the recording deviated >30% from its original value. GABAergic currents were induced by local application of 50 μm muscimol dissolved in HEPES-ACSF containing the following (in mm): 135 NaCl, 3 KCl, 2.5 CaCl2, 1.3 MgCl2, 1.25 Na2H2PO4, 20 glucose, and 10 HEPES, every 30s using a Picospritzer II. Muscimol responses were recorded in the presence of 1 μm TTX (Abcam) at holding potentials between −100 and −30 mV in 10 mV steps. We plotted response amplitude as a function of holding potential and calculated the chloride reversal potential from the intersection of the linear current–voltage curve with the x axis.
We performed whole-cell patch-clamp recordings from CA1 pyramidal neurons and GAD65-GFP interneurons in the stratum radiatum (sRad) using borosilicate glass pipettes with resistances of 3-6 MΩ. For recordings of sIPSCs, mIPSCs, and evoked IPSCs (eIPSCs), pipettes were filled with a KCl-based internal solution containing the following (in mm): 70 K-gluconate, 70 KCl, 0.5 EGTA, 4 Na2phosphocreatine, 4 MgATP, 0.4 NaGTP, and 10 HEPES, pH adjusted to 7.2 with KOH; and ACSF was supplemented with 20 μm DNQX (Tocris) and 50 μm DL-APV (Tocris). For recordings of mIPSCs, 1 μm TTX (Abcam) was also added to the ACSF. For recordings of sEPSCs, pipettes were filled with a K-gluconate-based internal solution containing the following (in mm): 140 K-gluconate, 4 KCl, 0.5 EGTA, 10 HEPES, 4 MgATP, 0.4 NaGTP, 4 Na2phosphocreatine, and 30 Alexa-568 (Fisher Scientific), pH adjusted to 7.2 with KOH; and ACSF was supplemented with 6 μm gabazine (HelloBio). Membrane potential was clamped at −65 mV. sIPSCs and mIPSCs were recorded for 6 min, sEPSCs for 10 min. For recording of eIPSCs, a glass electrode filled with ACSF and containing a stimulus electrode was positioned either in sRad or stratum pyramidale (sPyr) of CA1 (respectively, ∼ 250-300 μm or ∼50 μm from the recording electrode). Stimulus intensity was set at half-maximum response. eIPSCs were recorded at least 30 times every 30 s with 100 ms interval. Paired-pulse ratios were calculated as the mean amplitude of all second responses divided by the mean amplitude of all first responses (Kim and Alger, 2001). The coefficient of variation was calculated as the SD divided by the mean amplitude of the first responses (n = 15-60). After recording of sEPSCs, we switched to current clamp to assess excitability. Current was injected at RMP (I = 0) in 10 pA increments with a maximum of 200 pA to assess firing properties. The threshold potential was determined as the accompanying membrane potential at the start of the action potential (AP). Cells were discarded if series resistance was >35 mΩ or if the RMP exceeded −50 mV for pyramidal neurons and −40 mV for GAD65-GFP interneurons or if the series resistance after the recording deviated >30% from its original value.
Cell-attached recordings were performed using borosilicate glass pipettes of 3-5 mΩ, filled with a 150 mm NaCl-based solution. Modified ACSF with higher K+ and lower Ca2+ and Mg2+ levels (Maffei et al., 2004) was used to increase the baseline firing frequency containing the following (in mm): 126 NaCl, 3.5 KCl, 1.0 CaCl2, 0.5 MgCl2, 26 NaHCO3, 1.25 Na2H2PO4, and 20 glucose. CA1 pyramidal neurons were sealed (≥1 GΩ) and voltage-clamped at a holding current of 0 pA, to avoid affecting the firing activity of the cell (Perkins, 2006). Firing was recorded for 6 min before and after wash-in of muscimol (50 μm, Tocris).
All data were blinded before analysis. Network discharges were marked and removed from sEPSC recordings before event analysis. Events were selected using a template in Clampfit10 software. Further analysis was performed using custom-written MATLAB scripts. Rise time of sIPSCs was determined as the time between 10% and 90% of the peak value. The distribution of the rise times recorded in control conditions (generated from 100 randomly selected IPSCs per cell) was fitted with two Gaussians and their crossing point determined the separation between fast and slow IPSCs (Ruiter et al., 2020). We verified that the double Gaussian fit for the rise time distribution in VU conditions gave a similar separation value (control: 0.85 ms; VU: 0.95 ms) and our conclusions did not change by taking the VU separation value. The decay tau was fitted with a single exponential function. Only events with a goodness of fit R2 ≥ 0.75 were included.
Two-photon SClm imaging and analysis
We performed two-photon chloride imaging in CA1 pyramidal cells using the SClm sensor (Grimley et al., 2013) in cultured slices from SClm mice (described above). To target GABAergic interneurons, we used an adeno-associated virus (AAV) approach in slices from VGAT-Cre mice. An AAV2/5 for Cre-dependent SClm expression was prepared from a SCLM-DIO construct (kind gift from Thomas Kuner) (Boffi et al., 2018) using packaging plasmid pAAV2/5 (addgene_104964) and helper plasmid pAdDeltaF6 (addgene_112867). The virus was added on top of the CA1 region of organotypic hippocampal cultures from VGAT-Cre mice at DIV1 using a microinjector (Eppendorf, FemtoJet) aided by a stereoscopic microscope (Leica, M80). This resulted in widespread, but sparse, SClm expression in GABAergic neurons.
At the recording day, slices were transferred to a recording chamber and continuously perfused with carbonated (95% O2, 5% CO2) ACSF supplemented with 1 mm Trolox, at a rate of ∼1 ml/min. Bath temperature was monitored and maintained at 30°C-32°C. Two-photon imaging of CA1 pyramidal neurons or VGAT-positive interneurons in the CA1 area of the hippocampus was performed using a customized two-photon laser scanning microscope (Femto3D-RC, Femtonics) with a Ti-Sapphire femtosecond pulsed laser (MaiTai HP, Spectra-Physics) and a 60× water immersion objective (Nikon NIR Apochromat; NA 1.0) The CFP donor was excited at 840 nm. The emission light was split using a dichroic beam splitter at 505 nm and detected using two GaAsP photomultiplier tubes. We collected fluorescence emission of Cerulean/CFP (485 ± 15 nm) and Topaz/YFP (535 ± 15 nm) in parallel. Of each slice from SClm mice, 2-5 image stacks were acquired in different FOVs. Of each VGAT-Cre slice, 2-5 image stacks were acquired in different FOVs in both sPyr and sRad. The resolution was 8.1 pixels/μm (1024 × 1024 pixels, 126 × 126 μm) with 1 μm z steps.
Image analysis was performed using ImageJ software, as described previously (Herstel et al., 2022). We manually determined ROIs around individual neuron somata. To select a representative cell population in SClm slices, in each image z stack, we selected four z planes at comparable depths in which three pyramidal cells were identified that varied in CFP brightness (bright, middle, and dark). In VGAT-Cre slices with viral SClm expression, all visible neurons were analyzed (range: 1-9). We subtracted the mean fluorescence intensity of the background in the same image plane before calculating the fluorescence ratio of CFP and YFP. We limited our analysis to cells that were located within 450 pixels from the center of the image, as fluorescence resonance energy transfer (FRET) ratios showed slight aberrations at the edge of our images. We excluded cells with a FRET ratio <0.5 or >1.6 to avoid unhealthy cells. FRET-colored images (as shown in Figs. 2D and 9A) were made in ImageJ. We first subtracted the average background and Gaussian filtered the CFP and YFP image z stacks separately. Next, the acceptor (YFP) image was divided by the donor (CFP) image to get the ratiometric image. A mask was created by manually drawing ROIs for each soma in the image. An average projection of the ratiometric image was made in a specific z range and multiplied by the mask. Finally, the masked ratiometric image was combined with the grayscale image. These images were made for illustration purposes only; analysis was done on the raw data.
Figure 2.
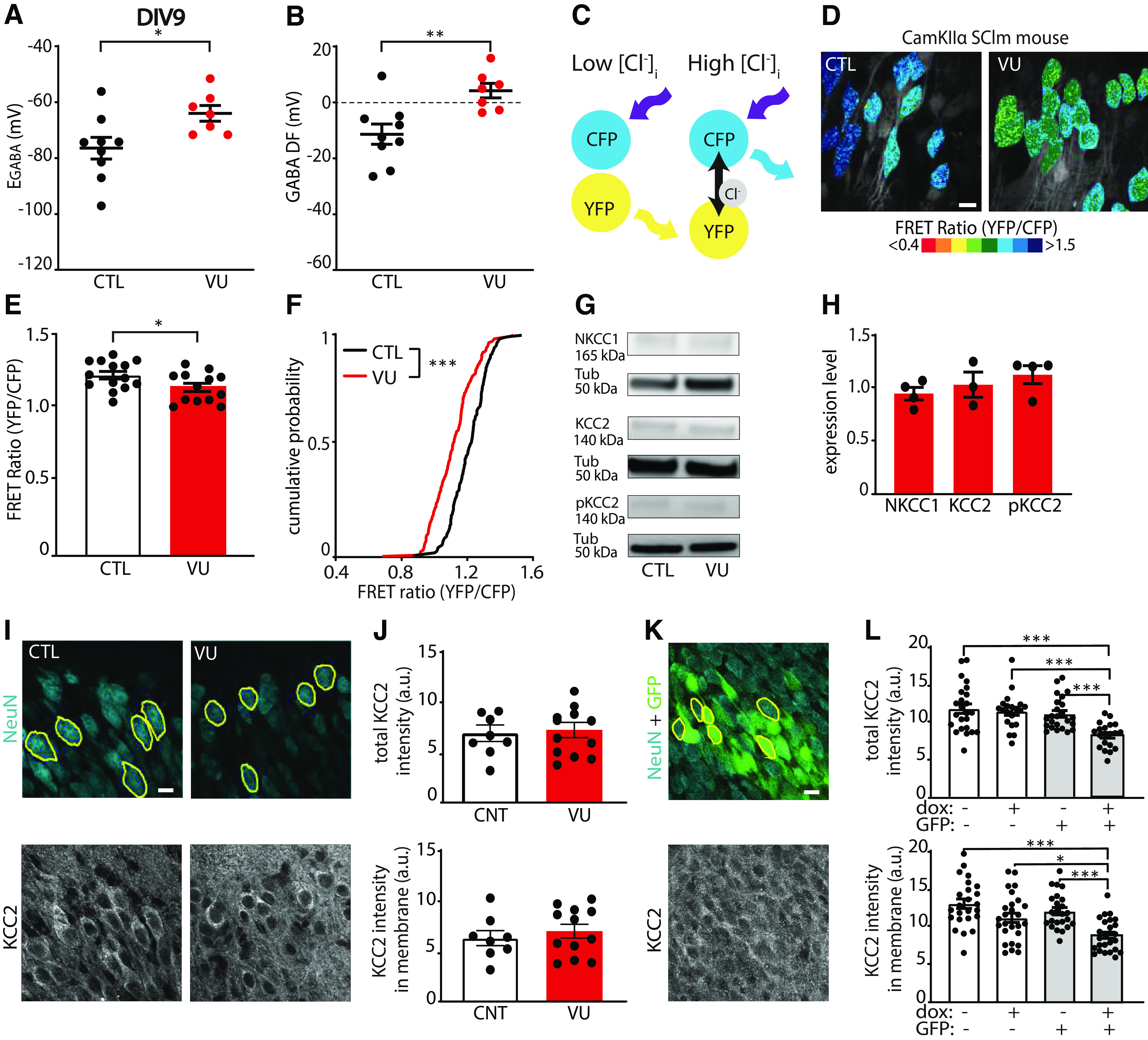
VU depolarizes GABA DF and increases intracellular chloride levels at DIV9. A, B, GABA reversal potential (EGABA) (MW, p = 0.016) and GABA DF (MW, p = 0.005) recorded in CA1 pyramidal cells at DIV9 in control (CTL) and VU-treated slices. Data are from 7-9 cells, 6-8 slices, and 3-5 animals per group. C, Illustration of FRET from CFP donor to YFP acceptor of the SClm sensor. FRET ratios (YFP/CFP fluorescence) decrease with increasing chloride concentrations. D, Two-photon images of SClm FRET ratios in CA1 pyramidal neurons in CTL and VU-treated cultures at DIV9. Individual cells are color-coded to their FRET ratios. Scale bar, 10 μm. E, Average SClm FRET ratios in CTL and VU-treated cultures (UT, p = 0.041). Data are from 16-24 cells per slice, 13 or 14 slices, and 6-8 mice per group. F, Cumulative distribution of FRET ratios in individual cells in CTL and VU-treated cultures (KS, p < 0.0001). Data are from 15 cells per slice, 13 or 14 slices, and 6-8 mice per group. G, Western blot of NKCC1, KCC2, and S940-pKCC2 (pKCC2) expression in CTL and VU-treated cultures. Tubulin (Tub) was used as loading control. H, Summary of data for NKCC1, KCC2, and for s940-pKCC2 (pKCC2) expression levels at DIV9. Values in VU-treated cultures were normalized to the protein level in CTL cultures (NKCC1: MW, p > 0.99; KCC2: MW, p = 0.70; pKCC2: MW, p = 0.31). I, Confocal images of NeuN and KCC2 staining in CTL and VU-treated cultures with ROIs in yellow. Scale bar, 10 μm. J, Total KCC2 levels (UT, p = 0.78) and KCC2 levels in membrane (UT, p = 0.51) in CTL and VU-treated cultures at DIV9. Each data point represents the mean KCC2 intensity of the 5-15 ROIs in one image. Data are from 8-12 images, 4-6 slices, and 3 mice per group. K, Confocal images of NeuN and KCC2 staining in slices were infected with KCC2-SH-TetR-eGFP lentivirus. Expression of shKCC2 was induced in GFP+ cells by doxycycline (dox) treatment. L, Top, Total KCC2 intensity in KCC2-SH-TetR-GFP-positive and -negative CA1 pyramidal neurons in slice cultures with or without dox treatment at DIV9 (KW, p < 0.0001; DMC: GFP– dox– vs GFP– dox+, p > 0.99; GFP– dox– vs GFP+ dox–, p > 0.99; GFP– dox– vs GFP+ dox+, p = 0.0001; GFP– dox+ vs GFP+ dox–, p > 0.99; GFP– dox+ vs GFP+ dox+, p = 0.0003; GFP+ dox– vs GFP+ dox+, p = 0.009). Bottom, KCC2 intensity in membrane in KCC2-SH-TetR-GFP-positive and -negative CA1 pyramidal neurons in slice cultures with or without dox treatment at DIV9 (KW, p < 0.0001; DMC: GFP– dox– vs GFP- dox+, p = 0.20; GFP– dox– vs GFP+ dox–, p > 0.99; GFP– dox– vs GFP+ dox+, p < 0.0001; GFP– dox+ vs GFP+ dox–, p > 0.99; GFP– dox+ vs GFP+ dox+, p = 0.032; GFP+ dox– vs GFP+ dox+, p = 0.0006). Data are from 23 or 24 cells, 3 or 4 slices, and 3 mice per group. *p < 0.05. **p ≤ 0.01. ***p ≤ 0.001.
Figure 9.
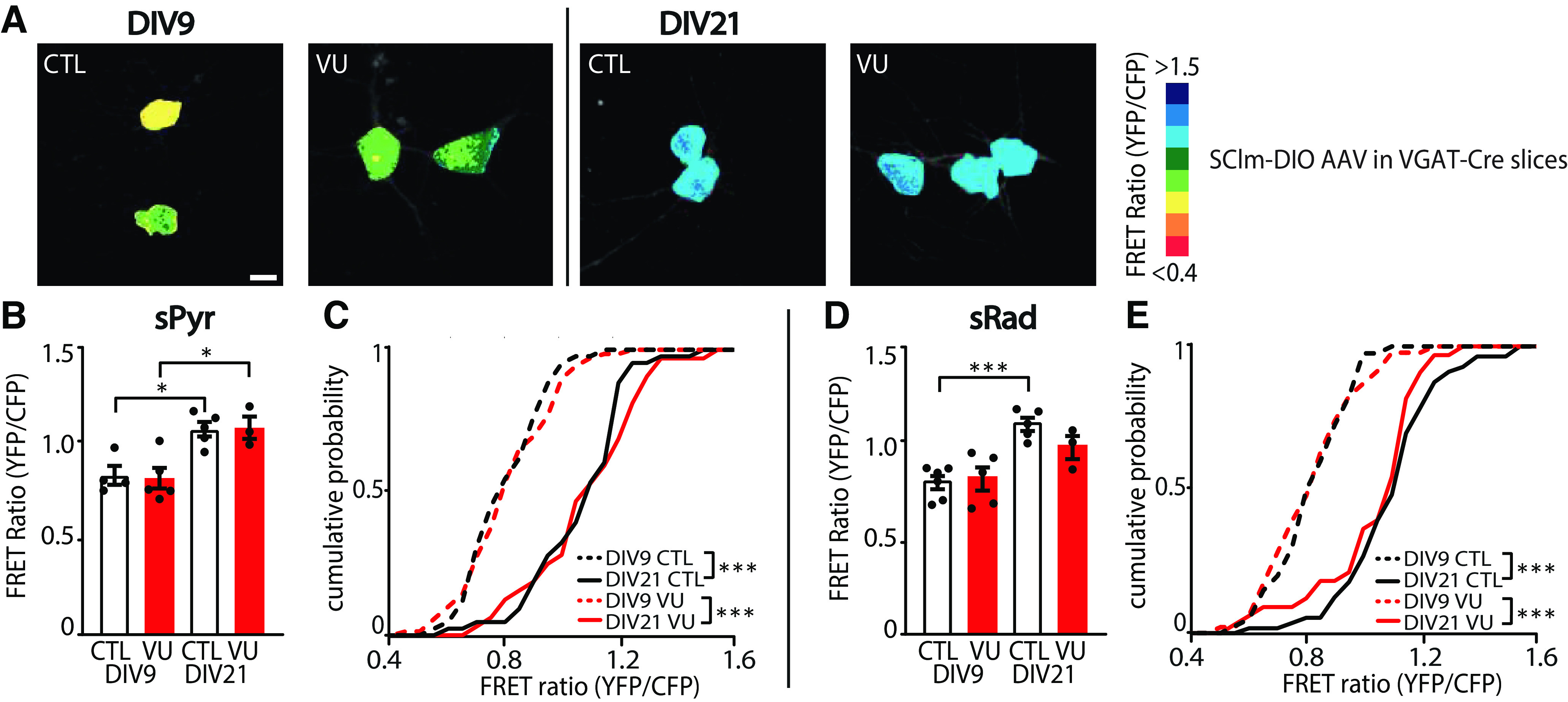
VU treatment does not change chloride concentrations in VGAT-positive interneurons. A, Images of SClm FRET ratios in CA1 sRad GABAergic interneurons in control (CTL) and VU-treated slices from VGAT-Cre mice at DIV9 and DIV21. Scale bar, 10 μm. B, Average SClm FRET ratio in interneurons in the CA1 sPyr in CTL and VU-treated cultures at DIV9 and DIV21 (one-way ANOVA, p = 0.002; SMC: DIV9 DMSO vs DIV9 VU, p = 0.99; DIV9 DMSO vs DIV21 DMSO, p = 0.015; DIV9 VU vs DIV21 VU, p = 0.015; DIV21 DMSO vs DIV21 VU, p = 0.97). C, Cumulative distribution of FRET ratios in individual sPyr interneurons in CTL and VU-treated cultures (KS, p < 0.0001). B, C, Data are from 32-69 cells, 3-5 slices, and 3 mice per group. D, Average SClm FRET ratio in interneurons in the CA1 sRad in CTL and VU-treated cultures at DIV9 and DIV21 (one-way ANOVA, p = 0.02; SMC: DIV9 DMSO vs DIV9 VU, p = 0.77; DIV9 DMSO vs DIV21 DMSO, p = 0.0007; DIV9 VU vs DIV21 VU, p = 0.12; DIV21 DMSO vs DIV21 VU, p = 0.23). An additional one-way ANOVA was performed to compare FRET ratios of individual cells in DIV9 VU vs DIV21 VU cultures and DIV21 DMSO vs DIV21 VU cultures (one-way ANOVA, p < 0.0001; SMC: DIV9 VU vs DIV21 VU, p < 0.0001; DIV21 DMSO vs DIV21 VU, p = 0.37). E, Cumulative distribution of FRET ratios in individual interneurons the CA1 sRad VU-treated (KS, p < 0.0001) and VU-treated cultures (KS, p < 0.0001). An additional KS was performed to compare the distribution of FRET ratios in CTL and VU-treated cultures at DIV21 (KS, p = 0.63). D, E, Data are from 33-56 cells, 3-6 slices, and 2 or 3 mice per group. *p < 0.05. ***p ≤ 0.001.
Protein extraction and Western blot analysis
Organotypic hippocampal cultures were washed in cold PBS and subsequently lysed in cold protein extraction buffer containing the following: 150 mm NaCl, 10 mm EDTA, 10 mm HEPES, 1% Triton X-100, and 1× protease and 1× phosphatase inhibitors cocktails (Complete Mini EDTA-free and phosSTOP, Roche). Lysates were cleared of debris by centrifugation (14,000 rpm, 1 min, 4°C) and measured for protein concentration before storage at −20°C until use. Lysates were denatured by adding loading buffer and heating to 95°C for 5 min. For each sample, an equal mass of proteins was resolved on 4%-15% polyacrylamide gel (Bio-Rad). The proteins were then transferred (300 mA, 3 h) onto ethanol-activated Immobilon-P PVDF membrane (Millipore) before blocking with 3% BSA in TBS-Tween (TBST, 20 mm Tris, 150 mm NaCl, 0.1% Tween-20) for 1 h. Primary antibodies used in this study were as follows: mouse anti-NKCC1 (T4, Developmental Studies Hybridoma Bank, 1:1000), rabbit anti-KCC2 (07-432, Merck, 1:1000) and s940-pKCC2 (p1551-940, LuBioSciences, 1:1000), and mouse anti-tubulin (T-5168, Sigma, 1:10,000). Primary antibodies were diluted in blocking buffer and incubated with the blots overnight at 4°C under gentle rotation. The membrane was washed 3 times 15 min with TBST before a 1 h incubation of HRP-conjugated antibodies (P0447 goat anti-mouse IgG HRP, Dako, 1:2500 or P0399 swine anti-rabbit IgG HRP, Dako, 1:2500), and washed 3 times 15 min in TBST again before chemiluminescence detection. For chemiluminescence detection, blots were incubated with Enhanced luminol-based Chemiluminescence substrate (Promega), and the exposure was captured using the ImageQuant 800 system (GE Healthcare). Images were analyzed in ImageJ, by drawing rectangular boxes around each band and measuring average intensities. Protein levels were normalized to the tubulin loading controls.
Immunohistochemistry, confocal microscopy, and analysis
Organotypic hippocampal cultures were fixed 4% PFA solution in PBS for 30 min at room temperature. After washing slices in PBS, they were permeabilized for 15 min in 0.5% Triton X-100 in PBS, followed by 1 h in a blocking solution consisting of 10% NGS and 0.2% Triton X-100 in PBS. Slices were incubated in primary antibody solution at 4°C overnight. The following primary antibodies were used: rabbit polyclonal anti-KCC2 (07-432, Merck, 1:1000), rabbit anti-VGAT (131003, Synaptic Systems, 1:1000), guinea pig anti-VGLUT (AB5905, Merck, 1:1000), mouse anti-NeuN (MAB377, Millipore, 1:500), and guinea pig anti-NeuN Merck, 1:1000).
Slices were washed in PBS and incubated in secondary antibody solution for 4 h at room temperature. Secondary antibodies used were as follows: goat anti-mouse AlexaFluor-467 (Invitrogen, A21236, 1:500), goat anti-rabbit AlexaFluor-405 (Invitrogen, A31556, 1:500), goat anti-guinea pig AlexaFluor-488 (A11073, Invitrogen, 1:1000), goat anti-guineapig AlexaFluor-568 (A11075, Invitrogen, 1:500), and goat anti-rabbit AlexaFluor-647 (A21245, Invitrogen, 1:500). After another PBS wash, slices were mounted in Vectashield mounting medium (Vector Labs).
Confocal images were taken on a Zeiss LSM-700 confocal laser scanning microscopy system with a Plan-Apochromat 40 × 1.3 NA immersion objective for KCC2 staining and 63 × 1.4 NA oil immersion objective for spines and VGLUT/VGAT staining. VGLUT/VGAT staining was imaged in the CA1 sPyr and sRad of each slice, KCC2 staining was imaged in two fields of view in the CA1 sPyr of each slice and spines were imaged on the second dendritic branch of the apical dendrite of each Alexa-568 filled neuron. Image stacks (1024 × 1024 pixels) were acquired at 6.55 pixels/μm for KCC2 staining, 20.15 pixels/μm for spines and 10.08 pixels/μm for VGLUT/VGAT staining. Step size in z was 0.5 μm for spines and KCC2 staining and 0.4 μm for VGLUT/VGAT staining.
Confocal images were blinded before analysis. The number of dendritic spines on the second dendritic branch of the apical dendrite were counted using the multipoint tool in ImageJ. As we noticed that spine density was low near the base of dendrites, the first 10 μm of the dendrites was excluded from analysis. For analysis of VGLUT and VGAT puncta, a custom-made macro was used (Ruiter et al., 2020). Briefly, three z planes were averaged and background was subtracted with rolling ball radius of 10 pixels. Puncta were then identified using watershed segmentation. For the analysis of KCC2 levels, ROIs were drawn at ∼5 NeuN-positive cell bodies, and this was repeated in 1-3 z planes (depending on the depth of KCC2 signal) at comparable depths, to measure KCC2 fluorescence in 5-15 neurons per image stack. We determined average fluorescent intensities in the ROI as well as within the ROIs after scaling it with 10 pixels (∼1 μm) in width to assess both the total and membrane fraction of KCC2.
Experimental design and statistical analyses
Statistical analysis was performed in Prism (GraphPad). Normality was tested using the D'Agostino & Pearson test. For the comparison of two groups, we used an unpaired Student's t test (UT; parametric), a Mann–Whitney test (MW; nonparametric), a paired Student's t test (PT; parametric), or a Wilcoxon signed-rank test (WSR; nonparametric). For comparison of two cumulative distributions, we performed a Kolmogorov–Smirnov test (KS; nonparametric). For comparison of multiple groups, a one-way ANOVA (parametric) was used, followed by a by a Sidak's Multiple Comparisons post hoc test (SMC) or a Kruskal–Wallis test (KW; nonparametric) was used, followed by a Dunn's Multiple Comparison post hoc test (DMC). For comparison of two variables in multiple groups, a two-way ANOVA (two-way ANOVA; parametric) was performed, followed by an SMC. Data are presented as mean ± SEM.
Results
The GABA shift takes place in organotypic hippocampal cultures
We assessed the development of neuronal chloride levels in organotypic hippocampal cultures made from P6-P7 mouse pups using gramicidin perforated patch recordings of CA1 pyramidal neurons. We recorded GABAergic currents in response to brief applications of the GABAA receptor agonist muscimol and determined the GABA reversal potentials at DIV2, DIV9, and DIV21 (Fig. 1A). The GABA reversal potential (EGABA) and GABAergic DF (GABA DF) decreased gradually over this period (Fig. 1B,C), indicating that intracellular chloride homeostasis was still maturing in our slice cultures (Salmon et al., 2020; Herstel et al., 2022). The RMP remained stable (Fig. 1D).
Figure 1.
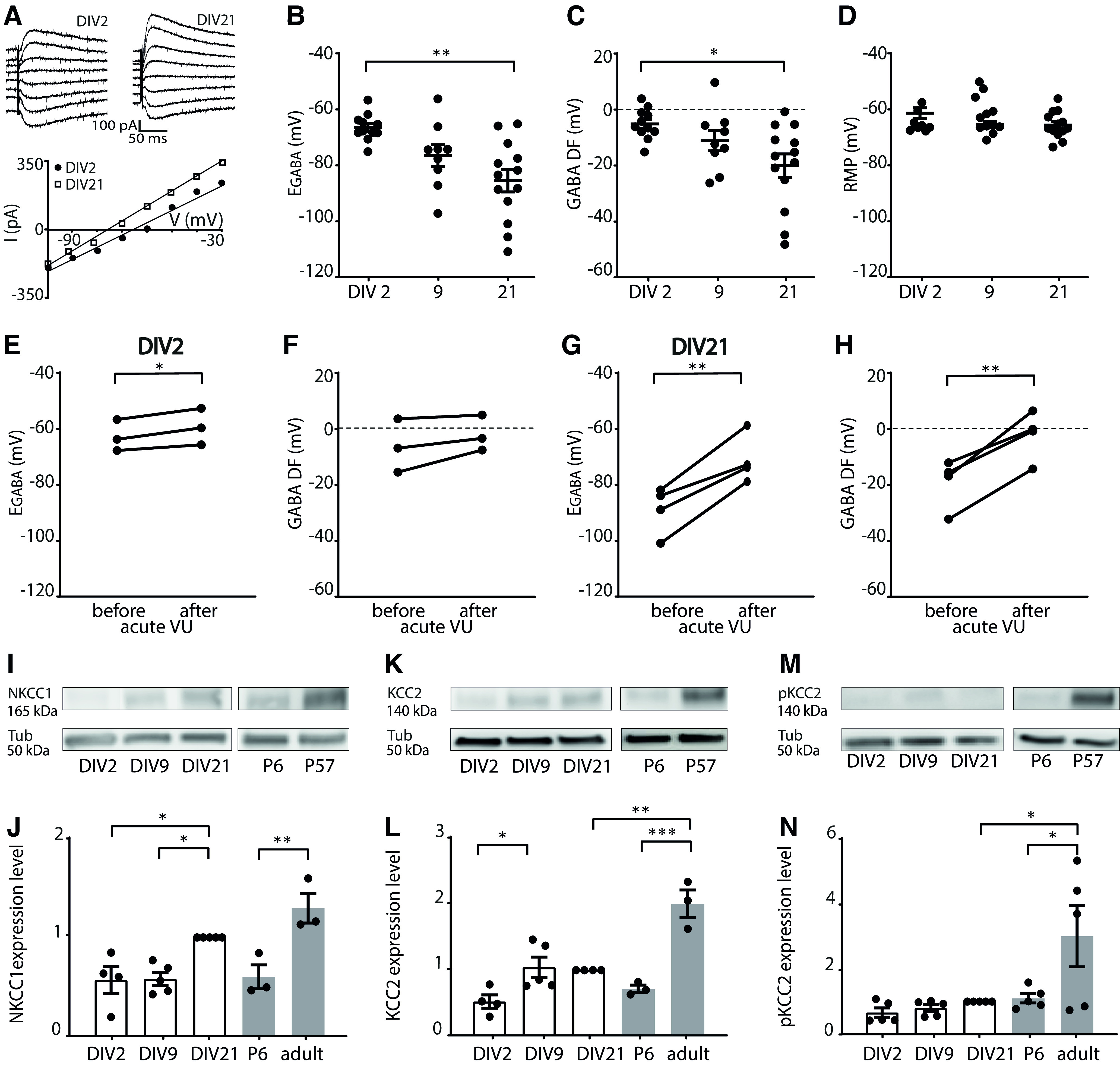
GABA shift is ongoing in organotypic hippocampal slice cultures. A, Perforated patch-clamp recordings in CA1 pyramidal cells in hippocampal slice cultures at DIV2 and DIV21. Responses to muscimol application were recorded at holding potentials from −100 to −30 mV with 10 mV increments, and GABA reversal potential was determined from the intersection of the linear current–voltage curve with the x axis. B, The GABA reversal potential (EGABA) recorded in cultured slices at DIV2, DIV9, and DIV21 (KW, p = 0.019; DMC: DIV2 vs DIV9, p = 0.13; DIV9 vs DIV 21, p = 0.65; DIV2 vs DIV21, p = 0.001). C, GABA DF during in vitro slice development (KW, p = 0.017; DMC: DIV2 vs DIV9, p = 0.35; DIV9 vs DIV21, p = 0.85; DIV2 vs DIV21, p = 0.013). D, The RMP during in vitro slice development (KW, p = 0.23). B-D, Data are from 9-13 cells, 8-11 slices, and 5-10 mice per group. E, F, EGABA (PT, p = 0.038) and GABA DF (PT, p = 0.18) values recorded in CA1 pyramidal cells at DIV2 before and after acute application of VU. Data are from 3 cells, 3 slices, and 3 mice per group. G, H, EGABA (PT, p = 0.009) and GABA DF (PT, p = 0.006) values recorded in CA1 pyramidal cells at DIV21 before and after acute application of VU. Data are from 4 cells, 4 slices, and 4 mice per group. I, Western blot for NKCC1 protein content, measured in hippocampal slices cultures at DIV2, DIV9, and DIV21, and in acute slices from P6 and adult (P57) mice. Tubulin (Tub) was used as loading control. J, Summary of data for NKCC1 expression levels. Protein levels were normalized to DIV21 values (one-way ANOVA, p = 0.0003; SMC: DIV2 vs DIV9, p > 0.99; DIV9 vs DIV21, p = 0.019; DIV2 vs DIV21, p = 0.023; DIV21 vs P57, p = 0.26; P6 vs P57, p = 0.003). K, Western blot for KCC2 protein content, measured in hippocampal slice cultures at DIV2, DIV9, and DIV21, and in acute slices from P6 and adult (P57) mice. Tubulin (Tub) was used as loading control. L, Summary of data for KCC2 expression levels. Protein levels were normalized DIV21 values (one-way ANOVA, p < 0.0001; SMC: DIV2 vs DIV9, p = 0.045; DIV9 vs DIV21, p > 0.99; DIV2 vs DIV21, p = 0.10; DIV21 vs P57, p = 0.0007; P6 vs P57, p = 0.0001). M, Western blot for S940-pKCC2 (pKCC2) protein content, measured in hippocampal slice cultures at DIV2, DIV9, and DIV21, and in acute slices from P6 and adult (P57) mice. Tubulin (Tub) was used as loading control. N, Summary of data for s940-pKCC2 (pKCC2) expression levels. Protein levels were normalized to DIV21 values (one-way ANOVA, p = 0.006; SMC: DIV2 vs DIV9, p > 0.99; DIV9 vs DIV21, p > 0.99; DIV2 vs DIV21, p = 0.99; DIV21 vs P57, p = 0.020; P6 vs P57, p = 0.033). I-N, Data are from 3-5 experiments and 2-5 mice per group. *p < 0.05. **p ≤ 0.01. ***p ≤ 0.001.
Intracellular chloride levels are determined by the relative expression level and activity of the chloride importer NKCC1 and the chloride exporter KCC2 (Rivera et al., 1999; Gulyás et al., 2001; Yamada et al., 2004; Banke and McBain, 2006; Otsu et al., 2020). We therefore determined the contribution of KCC2 to the GABA reversal potential. Application of KCC2 blocker VU in slices at DIV2 resulted only in a small depolarizing shift of EGABA (Fig. 1E), and did not affect GABA DF (Fig. 1F), while in DIV21 slices VU application triggered an acute elevation of EGABA and GABA DF (Fig. 1G,H). This suggests that KCC2 function increased with development in vitro, as expected (Salmon et al., 2020). We assessed the expression levels of NKCC1 and KCC2 in our slice cultures using Western blots. As the function of KCC2 is enhanced during development by phosphorylation at serine 940 (S940) (Lee et al., 2007; Mòdol et al., 2014), we also included a phospho-specific antibody to detect phosphorylated KCC2 (pKCC2) levels. The expression of NKCC1 and KCC2 increased in our organotypic cultures between DIV2 and DIV21 (Fig. 1I–L). For comparison with the developmental trajectory in vivo, we determined NKCC1, KCC2, and pKCC2 levels in hippocampal tissue from P6 and adult mice. NKCC1, KCC2, and pKCC2 expression increased from P6 to adulthood (Fig. 1I–N). We noted that KCC2 and pKCC2 expression in DIV21 slice cultures did not reach adult levels (Fig. 1K–N). Together, these data show that a clear GABA shift occurred in vitro, which is consistent with a previous report (Salmon et al., 2020) and our own previous analysis of chloride maturation in organotypic hippocampal cultures (Herstel et al., 2022).
VU treatment depolarizes GABA DF and elevates intracellular chloride levels at DIV9
Having established that the GABA shift occurs during the first week in vitro, we set out to delay the shift with 1 week to mimic the delayed chloride development in NDD mouse models (He et al., 2014; Tyzio et al., 2014; Deidda et al., 2015b; Banerjee et al., 2016; Corradini et al., 2017; Roux et al., 2018; Fernandez et al., 2019; Lozovaya et al., 2019; Bertoni et al., 2021). We treated slices for 1 week (from DIV1 to DIV8) with VU, a specific blocker of the chloride exporter KCC2 (Delpire et al., 2012), or with DMSO control. One week treatment with VU from resulted in a significant elevation of EGABA in CA1 pyramidal neurons immediately after the VU treatment at DIV9 (Fig. 2A). The DF for GABA signaling (GABA DF) also shifted to positive values (Fig. 2B), which were comparable to control slices at DIV2 (Fig. 1C). This indicates that GABA signaling remained depolarizing in VU-treated slices at DIV9. We assessed intracellular chloride levels in the CA1 pyramidal cell population using the chloride sensor SClm (Grimley et al., 2013; Boffi et al., 2018; Rahmati et al., 2021). The SClm sensor consists of two fluorescent proteins, Cerulean (CFP mutant) and Topaz (YFP mutant), joined by a flexible linker. Depending on the binding of chloride, FRET occurs from the CFP donor to the YFP acceptor (Grimley et al., 2013) (Fig. 2C). We measured SClm FRET ratios (fluorescence intensity of YFP/CFP) using two-photon microscopy. Measured FRET ratios are independent of expression level and tissue depth (Herstel et al., 2022). We observed a prominent decrease in SClm FRET values in CA1 pyramidal neurons (Fig. 2D,E), reflecting an increase in chloride levels after VU treatment. When we analyzed the distribution of individual values, we observed a clear increase in the fraction of cells with low FRET ratios (Fig. 2F). Together, these data show that VU treatment between DIV1 and DIV8 resulted in an elevation of intracellular chloride levels and depolarizing GABA signaling at DIV9.
Next, we assessed whether the expression levels of the chloride cotransporters NKCC1 and KCC2 were altered after the VU treatment. We did not find any significant difference in the levels of NKCC1, KCC2, or S940-pKCC2 between VU-treated and control slices at DIV9 (Fig. 2G,H). As Western blot only allows for assessment of total KCC2 levels and cannot distinguish between cytosolic KCC2 and KCC2 in the plasma membrane, we also examined KCC2 levels in treated and control slices using immunohistochemistry. We used NeuN to identify individual cell bodies, and we estimated KCC2 levels in these cells. VU treatment did not significantly affect total or membrane KCC2 levels (Fig. 2I,J). To verify that we can detect changes with this method, we performed a positive control experiment. We reduced KCC2 levels in our slices using a lentivirus containing an inducible shKCC2 construct and GFP (KCC2-SH-TetR-GFP). Expression of shKCC2 was induced in GFP-expressing cells by doxycycline treatment, resulting in a cell-specific elevation of GABA DF (data not shown, GABA DF = 12.4 ± 9.3 mV in GFP+ cells and −11.5 ± 2.2 mV in GFP– cells, MW, p = 0.03). We could clearly detect the reduction in KCC2 levels in dox-treated GFP+ cells using immunohistochemistry (Fig. 2K,L) Together, this indicates that blocking KCC2 function between DIV1 and DIV8 delayed the GABA shift without changing the expression or surface levels of KCC2.
Synaptic inputs and firing properties of CA1 pyramidal cells are not affected at DIV9 after VU treatment
Previous experimental studies in which the GABA shift was accelerated showed that depolarizing GABA facilitates excitatory synapse formation (Leinekugel et al., 1997; Akerman and Cline, 2006; Wang and Kriegstein, 2008, 2011; Chancey et al., 2013; van Rheede et al., 2015; Oh et al., 2016). This would predict that delaying the GABA shift (e.g., prolonging the period when GABA signaling is depolarizing) would result in an excess synapse growth.
To assess functional synapses, we recorded sEPSCs and sIPSCs in CA1 pyramidal cells using whole-cell patch-clamp recordings (Fig. 3A,B). We found that sEPSC frequency, amplitudes, and kinetics were not different in CA1 pyramidal cells in VU-treated and control slices at DIV9 (Fig. 3C–F). There were also no changes in sIPSCs (Fig. 3G–J). In addition, we recorded AP firing rates during current injections of increasing amplitude (Fig. 3K). Firing rates, RMP, and AP threshold were comparable in control and VU-treated slices (Fig. 3L–N).
Figure 3.
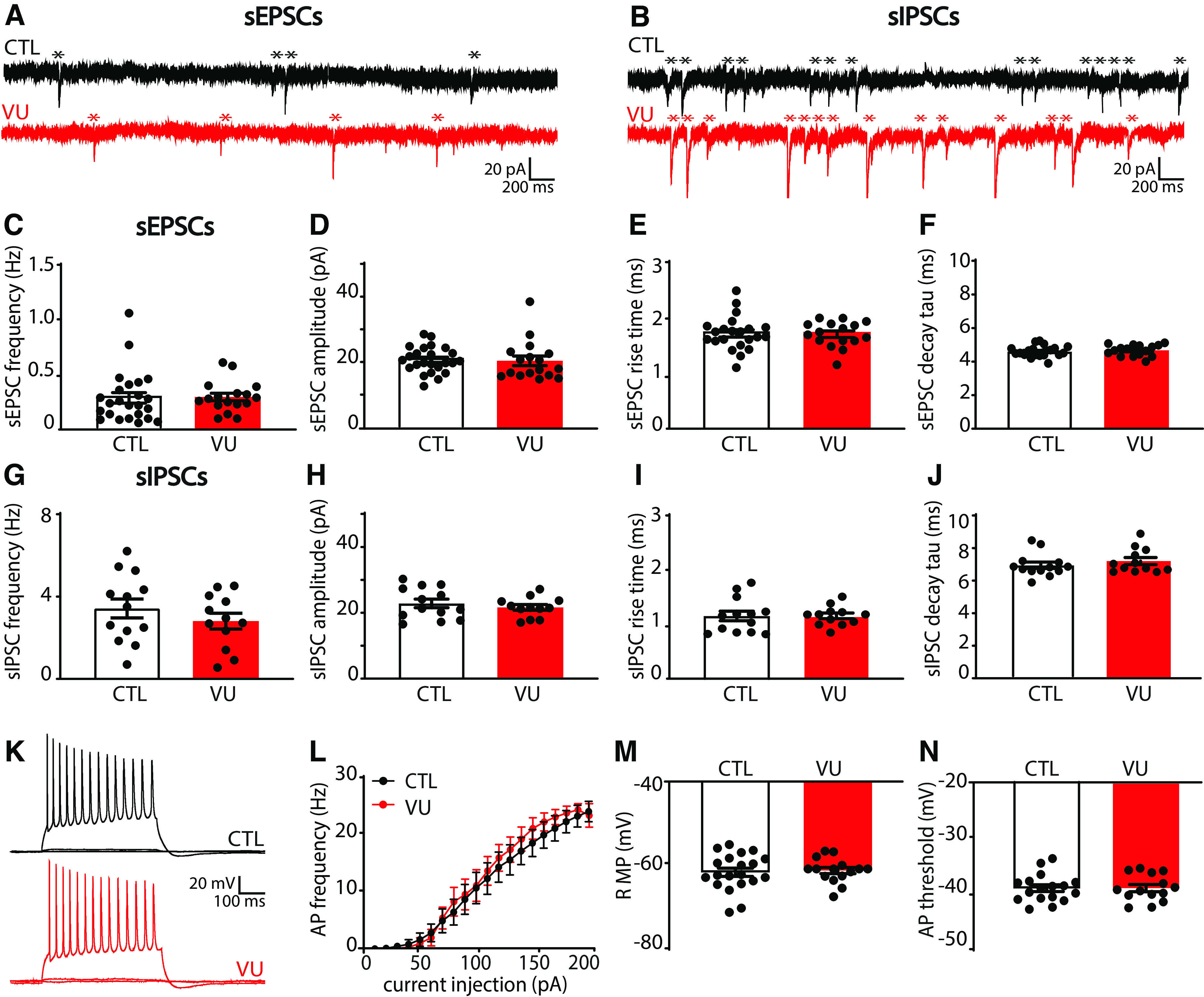
VU treatment does not change excitatory or inhibitory transmission and firing properties at DIV9. A, sEPSC recordings from CA1 pyramidal cells in a control (CTL) and VU-treated culture at DIV9. Asterisks indicate sEPSCs. B, sIPSC recordings in CTL and VU-treated cultures at DIV9. Asterisks indicate sIPSCs. C-F, sEPSC frequency (MW, p = 0.36), amplitude (MW, p = 0.45), rise time (UT, p = 0.87), and decay tau (UT, p = 0.53) in CTL and VU-treated cultures at DIV9. Data are from 16-24 cells, 11-15 slices, and 10-12 mice per group. G-J, sIPSC frequency (UT, p = 0.32), amplitude (UT, p = 0.46), rise time (UT, p = 0.99), and decay tau (UT, p = 0.20) in in CTL and VU-treated cultures at DIV9. Data are from 12-13 cells, 4-7 slices, and 4-6 mice per group. K, Example recordings of APs during current injections in CA1 pyramidal neurons in CTL and VU-treated cultures at DIV9. L, AP firing rates in CTL and VU-treated cultures with increasing current injections at DIV9 (two-way ANOVA, current injection: p < 0.001, treatment: p = 0.24). Data are from 14-17 cells, 10 or 11 slices, and 9 or 10 mice per group. M, N, RMP (UT, p = 0.75) and AP threshold (UT, p = 0.98) in CTL and VU-treated cultures at DIV9. Data are from 14-20 cells, 10 or 11 slices, and 9 or 10 mice per group.
Additionally, we assessed synaptic density by performing immunohistochemistry on VU-treated and control slices. Consistent with our electrophysiological data, the density and size of VGLUT and VGAT puncta in the sPyr and sRad of the CA1 area were similar in VU-treated and control slices at DIV9 (Fig. 4A–J). Furthermore, we filled pyramidal neurons with Alexa-568 via a patch pipette to assess dendritic spines at which excitatory synapses are located (Sheng and Hoogenraad, 2007). Spine density of CA1 pyramidal neurons was similar in control and VU-treated slices (Fig. 4K,L).
Figure 4.
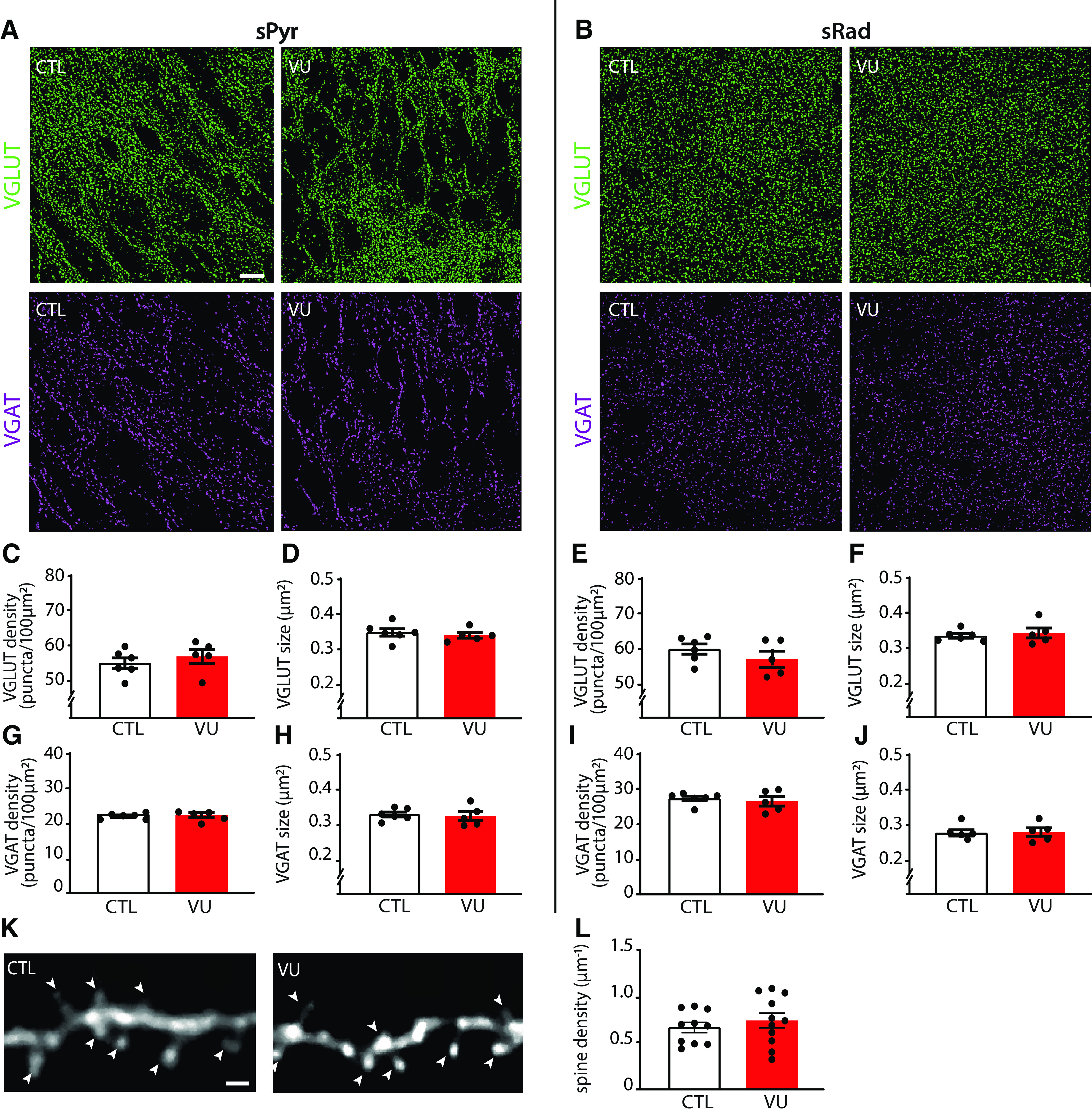
VU treatment does not change excitatory or inhibitory synapses at DIV9. A, B, Thresholded images of VGLUT and VGAT puncta in the CA1 sPyr and sRad of control (CTL) and VU-treated cultures at DIV9. Scale bar, 10 μm. C, D, Density (MW, p = 0.43) and size (MW, p = 0.33) of VGLUT puncta in the sPyr in CTL and VU-treated cultures at DIV9. E, F, Density (MW, p = 0.27) and size (MW, p = 0.83) of VGLUT puncta in the sRad. G, H, Density (MW, p = 0.96) and size (MW, p = 0.54) of VGAT puncta in the sPyr. I, J, Density (MW, p = 0.89) and size (MW, p = 0.84) of VGAT puncta in the sRad. C-J, Data are from 5 or 6 slices, and 4 or 5 mice per group. K, Example images of the apical dendrite of CA1 pyramidal neurons in CTL and VU-treated cultures at DIV9. Arrowheads indicate spines. Scale bar, 1 μm. L, The average density of dendritic spines (UT, p = 0.82) in CTL and VU-treated cultures at DIV9. Data are from 10 or 11 cells, 6-8 slices, and 6 mice per group.
Together, these data show that VU treatment from DIV1 to DIV8 induced a clear depolarizing shift in GABA signaling, but this did not affect excitatory and inhibitory synapses and firing properties in CA1 pyramidal cells at DIV9.
Depolarizing GABA signaling in VU-treated slices acts inhibitory
As eluded to above, it is important to note that depolarizing GABA signaling does not necessarily mean that GABA acts inhibitory. Depolarizing GABA can have an inhibitory action and limit neuronal activity when the GABA-mediated depolarization is subthreshold and shunts excitatory inputs (Staley and Mody, 1992; Kirmse et al., 2015; Murata and Colonnese, 2020; Salmon et al., 2020). We noticed that, during the sEPSC recordings, when GABAA-mediated inhibitory currents were blocked with gabazine, network discharges often occurred. These discharges probably reflect periods of synchronous firing of pyramidal cells that can occur in the absence of fast GABAA-mediated inhibition, when only the slower GABAB-mediated inhibition is left to counteract excitatory synaptic transmission (Scanziani et al., 1994; Menendez De La Prida et al., 2006). The frequency and duration of these network discharges were not different in VU-treated and control slices at DIV9 (Fig. 5A–C). We directly assessed whether depolarizing GABA was already inhibitory in our VU-treated slices. To determine the role of GABA in regulating AP firing, we washed in GABAA receptor agonist muscimol while recording spontaneous firing in cell-attached configuration. As spontaneous activity of hippocampal neurons is very low in standard ACSF, we used a modified ACSF with higher K+ and lower Ca2+ and Mg2+ levels, which elevates spontaneous firing (Maffei et al., 2004). Muscimol inhibited firing in control as well as in VU-treated slices at DIV8 (Fig. 5D–F). This indicates that GABAA receptor-mediated signaling is inhibitory in both control and VU-treated slices at DIV8, despite being hyperpolarizing in control and depolarizing in VU-treated slices (Fig. 2B). This suggests that GABA signaling mostly acts inhibitory in hippocampal slice cultures from P6-P7 mice, which is in line with previous reports in P6-P9 acute hippocampal slices (Khalilov et al., 1997; Valeeva et al., 2016), cultured hippocampal slices at P5 DIV3-5 (Salmon et al., 2020), and in vivo recordings in the hippocampus after P3-P7 (Valeeva et al., 2010; Murata and Colonnese, 2020). Together, our data suggest that depolarizing GABA signaling after the first postnatal week acts mostly inhibitory and does not contribute to synapse formation in the hippocampus.
Figure 5.

GABA is inhibitory in control and VU-treated slice cultures at DIV8 and DIV9. A, Whole-cell voltage-clamp recordings from CA1 pyramidal cells in the presence of gabazine in control (CTL) and VU-treated cultures at DIV9. Asterisks indicate network discharges. B, C, Network discharge (ND) frequency (MW, p = 0.61) and duration (MW, p = 0.20) in CTL and VU-treated cultures at DIV9. Data are from 17-24 cells, 11-15 slices, and 10-12 mice per group. D, Cell-attached recordings from CA1 pyramidal cells showing AP firing in modified ACSF in CTL and VU-treated cultures at DIV8 before (baseline) and after muscimol wash-in. Asterisks indicate APs. E, F, Average AP frequency before and after muscimol wash-in in CTL and VU-treated cultures at DIV8. Muscimol decreases firing rates in CTL (WSR, p = 0.008) and VU-treated (WSR, p = 0.016) cultures at DIV8. Data are from 7 or 8 cells, 7 or 8 slices, and 3 or 4 mice per group. An MW was performed to compare baseline firing rates in CTL and VU-treated cultures (MW, p = 0.79). *p < 0.05. **p ≤ 0.01.
Indirect effects on inhibitory synaptic inputs in VU-treated slices at DIV21
Previous studies demonstrated that transient alterations in GABA function during early postnatal development can have long-lasting consequences for network function and ultimately behavior (He et al., 2019; Bertoni et al., 2021; Matsushima et al., 2022). We therefore assessed possible long-lasting consequences of the VU treatment on the CA1 network at DIV21. After DIV8, the VU treatment was stopped and cultures were maintained in normal medium until DIV21. We verified that, at DIV21, EGABA and GABA DF in CA1 pyramidal neurons in VU-treated slices were back to control levels (Fig. 6A,B), indicating that chloride levels were fully restored.
Figure 6.
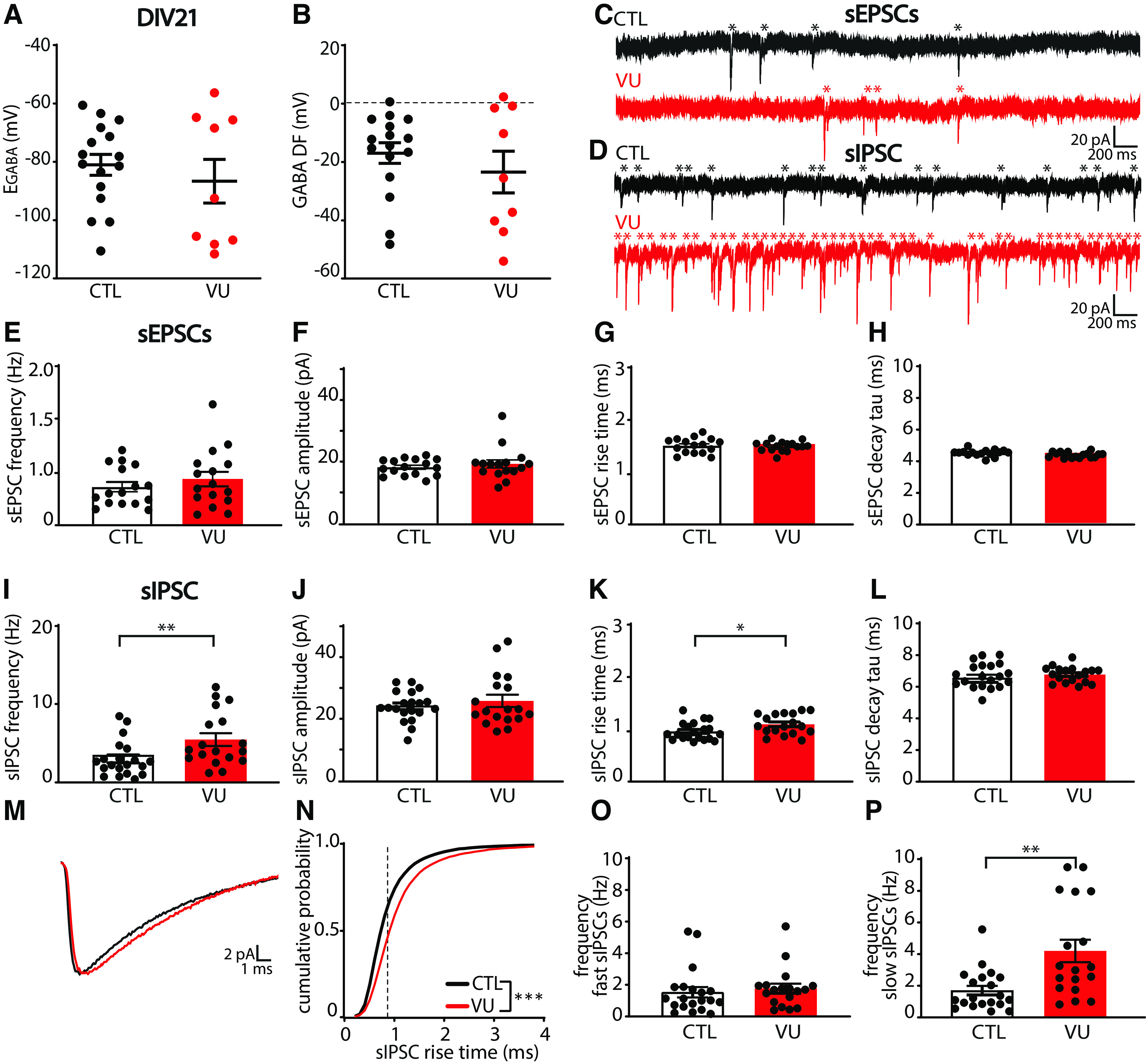
VU treatment does not affect excitatory transmission but increases spontaneous inhibitory transmission at DIV21. A, B, GABA reversal potential (EGABA) (MW, p = 0.56) and GABA DF (MW, p = 0.76) recorded in CA1 pyramidal cells in control (CTL) and VU-treated cultures at DIV21, 2 weeks after cessation of treatment. Data are from 9-16 cells, 3-10 slices, and 3-10 mice per group. C, D, sEPSC and sIPSC recordings from CA1 pyramidal cells in CTL and VU-treated cultures at DIV21. Asterisks indicate sEPSCs and sIPSCs. E-H, sEPSC frequency (UT, p = 0.26), amplitude (MW, p = 0.59), rise time (UT, p = 0.80), and decay tau (UT, p = 0.94) in CTL and VU-treated cultures at DIV21. Data are from 16 cells, 9 slices, and 9 mice per group. I-L, sIPSC frequency (MW, p = 0.006), amplitude (UT, p = 0.52), rise time (UT, p = 0.026), and decay tau (UT, p = 0.37) in CTL and VU-treated cultures at DIV21. M, Average sIPSC recorded in CA1 pyramidal cells in CTL and VU-treated cultures at DIV21. N, Cumulative distribution of sIPSC rise times in control and VU-treated cultures at DIV21 (KS, p < 0.0001). Dotted line indicates value used to split fast from slow rise time sIPSCs. O, Frequency of fast sIPSCs in CTL and VU-treated cultures at DIV21 (MW, p = 0.20). P, Frequency of fast slow sIPSCs in CTL and VU-treated cultures at DIV21 (MW, p = 0.003). I-P, Data are from 18-20 cells, 11 or 12 slices, and 8 or 9 mice per group. *p < 0.05. **p ≤ 0.01.
We performed whole-cell recordings in CA1 pyramidal cells to record sEPSCs and sIPSCs (Fig. 6C,D). Similar to DIV9, we found no differences in sEPSCs between control and VU-treated slices at DIV21 (Fig. 6E–H). Surprisingly, we observed that sIPSC frequency in VU-treated slices was increased compared with control slices at DIV21 (Fig. 6I), whereas sIPSC amplitudes were not different (Fig. 6J). When we analyzed the kinetics of the individual sIPSCs, we noticed that sIPSCs in VU-treated slices had slightly larger rise times compared with control (Fig. 6K), while decay times were not different (Fig. 6L). The IPSC rise time depends on the location of synapse where the current originates from, with somatic synapses generating currents with faster rise times while currents from dendritic synapses will be slower because of dendritic filtering (Rall, 1967; Bekkers and Clements, 1999; Wierenga and Wadman, 1999; Ruiter et al., 2020). When we split slow and fast sIPSCs (Ruiter et al., 2020), we observed that the increase was most prominent in slow sIPSCs (Fig. 6M–P), possibly reflecting an increase in IPSCs originating from dendritic synapses.
To assess whether the increase in inhibitory transmission after VU was activity-dependent, we also measured mIPSCs in control and VU-treated slices at DIV21. We found that mIPSCs were not different in VU-treated and control slices (Fig. 7A–D), suggesting that VU treatment increased activity-dependent GABA release at DIV21. To assess possible changes in synaptic density, we determined the density and size of VGLUT and VGAT puncta in the CA1 area, and we found no differences between control and VU-treated slices, both in sPyr and sRad (Fig. 7E–L). Spine density in CA1 pyramidal neurons was also not different in control and VU-treated slices at DIV21 (Fig. 7M,N). This indicates that excitatory and inhibitory synaptic density remained unaffected 14 d after the VU treatment and suggests that the observed increase in sIPSC frequency is because of an increased activity-dependent release from GABAergic terminals, possibly preferentially at dendritic inhibitory synapses.
Figure 7.
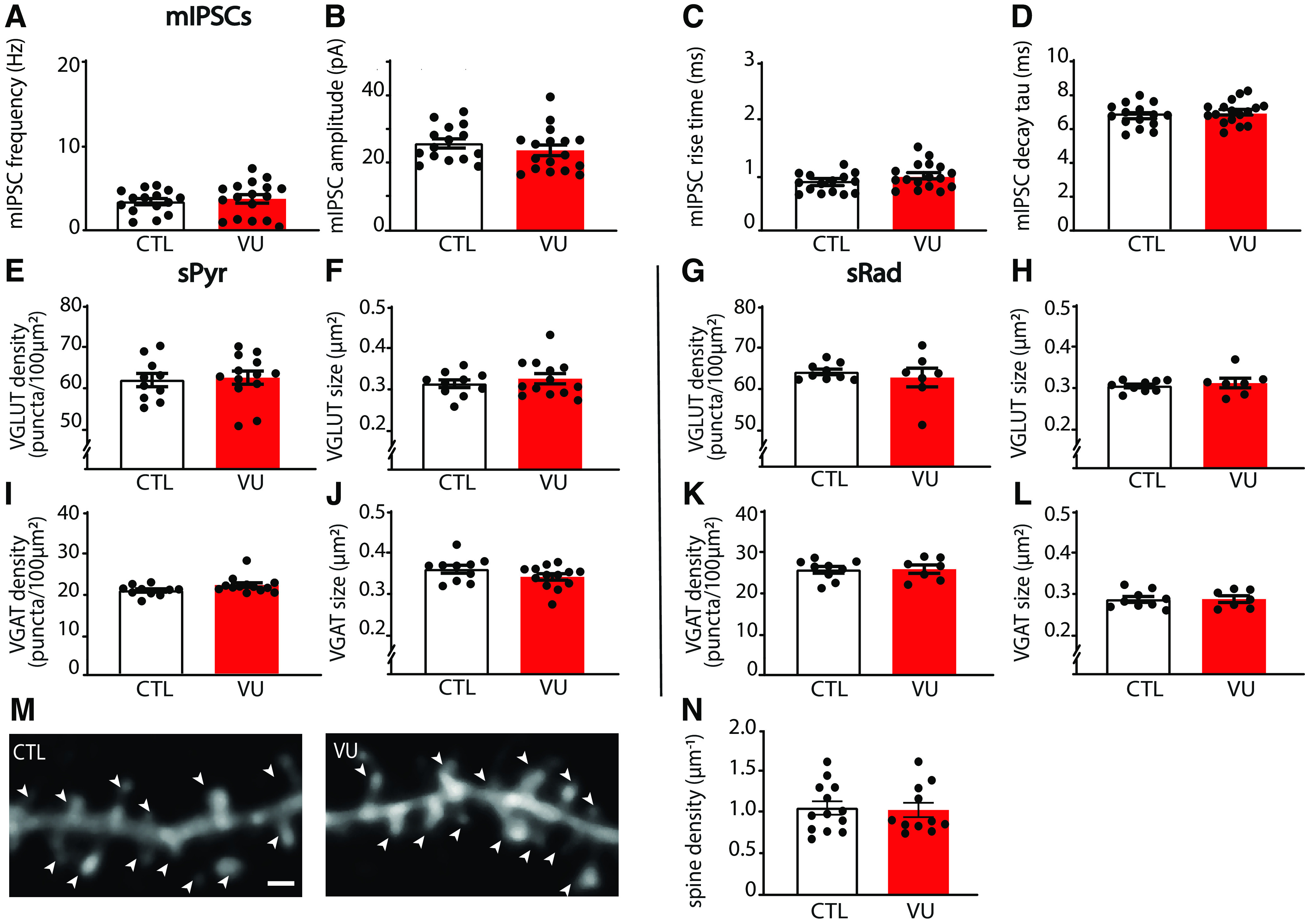
VU treatment does not affect mIPSCs, or excitatory and inhibitory synapses at DIV21. A-D, mIPSC frequency (UT, p = 0.60), amplitude (UT, p = 0.34), rise time (UT, p = 0.077), and decay tau (UT, p = 0.40) recorded from CA1 pyramidal cells in CTL and VU-treated cultures at DIV21. Data are from 15-17 cells, 9-12 slices, and 6-9 mice per group. E, F, Density (UT, p = 0.80) and size (UT, p = 0.61) of VGLUT puncta in the sPyr in CTL and VU-treated slices at DIV21. G, H, Density (MW, p = 0.91) and size (MW, p = 0.76) of VGLUT puncta in the sRad. I, J, Density (MW, p = 0.078) and size (UT, p = 0.14) of VGAT puncta in the sPyr. K, L, Density (MW, p = 0.76) and size (MW, p > 0.99) of VGAT puncta in the sRad. E-L, Data are from 10-13 slices and 6 or 7 mice per group. M, Maximal projection of confocal images of the apical dendrite of DIV21 CA1 pyramidal neurons in CTL and VU-treated cultures. Arrowheads indicate spines. Scale bar, 1 μm. N, Dendritic spine densities in control and VU-treated cultures at DIV21 (MW, p = 0.77). Data are from 10-11 cells, 8 slices, and 7 or 8 mice per group.
Network discharges during sEPSC recordings were rare at DIV21 and not different between control and VU-treated slices (Fig. 8A–C). One week VU treatment did not affect firing properties (Fig. 8D,E) or RMP (Fig. 8F) of CA1 pyramidal neurons at DIV21, but we observed a slight elevation of the AP threshold in VU-treated slices at DIV21 (Fig. 8G). The relative AP threshold (defined as the difference between AP threshold and RMP, Fig. 8H) was not different VU-treated and control pyramidal cells. It is noteworthy that the AP threshold was not different when we recorded with high chloride internal solution, which resulted in a more negative AP threshold (DMSO −41.9 ± 3.3 mV; VU −42.9 ± 3.4 mV; UT, p = 0.45; data not shown). A similar decrease in AP threshold after chloride loading was recently described in CA3 pyramidal neurons (Sørensen et al., 2017), but the mechanism remained unresolved.
Figure 8.
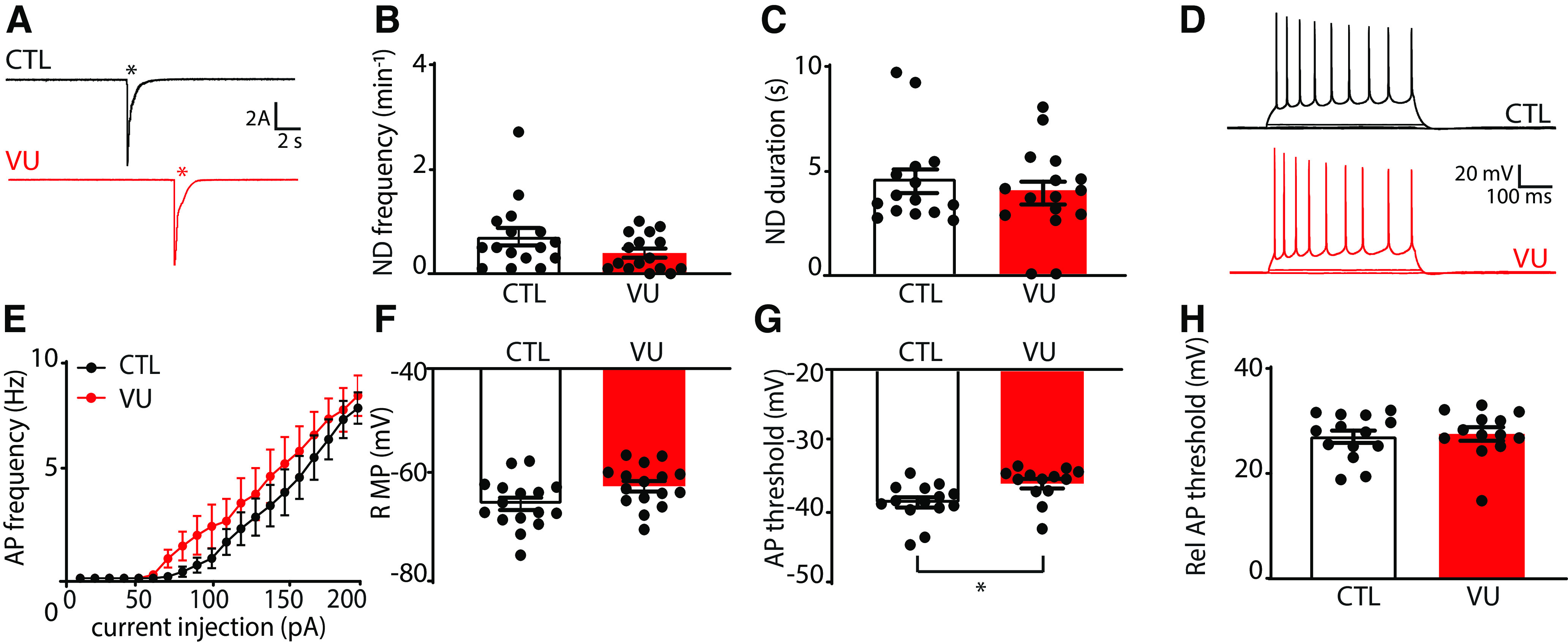
VU treatment increases the firing threshold of pyramidal neurons at DIV21. A, Whole-cell voltage-clamp recordings from CA1 pyramidal cells in control (CTL) and VU-treated cultures at DIV21, in the presence of gabazine. Asterisks indicate network discharges. B, C, Network discharge (ND) frequency (MW, p = 0.14) and duration (MW, p = 0.20) in CTL and VU-treated cultures at DIV21. Data are from 16 cells, 9 slices, and 9 mice per group. D, Whole-cell current-clamp recordings of APs after current injections in CA1 pyramidal neurons in CTL and VU-treated cultures at DIV21. E, AP firing rates in CTL and VU-treated cultures with increasing current injections at DIV21 (two-way ANOVA, current injection: p < 0.001, treatment: p = 0.36). Data are from 13 or 14 cells, 9 slices, and 9 mice per group. F-H, RMP (UT, p = 0.75), AP threshold (MW, p = 0.006), and relative AP (Rel AP) threshold (MW, p = 0.72) in CTL and VU-treated cultures at DIV21. Data are from 13-16 cells, 9 slices, and 9 mice per group. *p < 0.05.
VU treatment does not affect chloride levels in interneurons at DIV9 or DIV21
Our data suggest that VU treatment specifically affected activity-dependent GABAergic transmission from interneurons to pyramidal neurons. We wondered if VU treatment affected chloride concentrations in inhibitory neurons in the same way as in pyramidal neurons. We therefore expressed the chloride sensor SClm (Grimley et al., 2013) specifically in GABAergic interneurons, using a Cre-dependent adeno associated virus (Boffi et al., 2018) in slices from VGAT-Cre mice. We measured SClm FRET ratios in cell bodies of GABAergic interneurons in sRad and sPyr (Fig. 9A). We observed that FRET values in interneurons were much lower compared with pyramidal cells at DIV9 (Fig. 2E), and clearly increased between DIV9 and DIV21 (Fig. 9B,C), reflecting a decrease of intracellular chloride levels with development. This was remarkable as we previously showed that chloride levels in pyramidal neurons are already mature at DIV9 and remain relatively stable during the this period (Herstel et al., 2022). When we normalized FRET ratios at DIV21 to DIV9 for interneurons and pyramidal neurons, the relative increase was 1.33 ± 0.03% in interneurons compared with 1.02 ± 0.03% in pyramidal neurons (UT, p < 0.0001). This suggests that the GABA shift occurs later in inhibitory neurons compared with excitatory neurons in the CA1 area, which is in line with previous reports (Patenaude et al., 2005; Otsu et al., 2020). Interestingly, and in clear contrast to pyramidal neurons (Fig. 2G,F), VU treatment from DIV1 to DIV8 did not affect SClm FRET values in interneurons at DIV9 (Fig. 9B–E), suggesting that the contribution of KCC2 to chloride levels in interneurons was minimal during the treatment period. FRET ratios increased similarly to DIV21 in VU-treated and control slices (Fig. 9B–E). These results indicate that the 1 week VU treatment had a differential effect on intracellular chloride levels in CA1 pyramidal and interneurons, probably because of cell type-specific timing of chloride maturation.
VU treatment results in elevated membrane potential in sRad interneurons at DIV21
As sIPSC frequency was increased in VU-treated slices at DIV21, while mIPSCs and VGAT puncta were unaffected, we wondered whether release probability at GABAergic synapses was increased. We therefore assessed paired-pulse ratios of evoked IPSCs in CA1 pyramidal cells after stimulation with an extracellular electrode in the sPyr or sRad (Fig. 10A), but paired-pulse ratios of IPSCs were similar in VU-treated and control slices (Fig. 10B,C). We also determined the coefficient of variation of the first IPSCs, another parameter that is dependent on release probability, but we did not find any difference between VU-treated and control slices (Fig. 10D,E). This suggests that IPSC release probability was not affected by VU treatment. We therefore wondered whether the increase in sIPSC frequency in VU-treated slices could be because of an increased activity of GABAergic cells at DIV21. Based on our observation of a specific increase in sIPSC with slow rise times in VU-treated slices (Fig. 6M–P), we focused on dendritically targeting GABAergic cells in the sRad. We performed whole-cell patch-clamp recordings from GFP-labeled interneurons in the sRad in slices from GAD65-GFP mice, which are mostly reelin-positive and preferentially target pyramidal dendrites (Wierenga et al., 2008, 2010). We found that sEPSCs (Fig. 10F–H) and general firing properties (Fig. 10I,J) of sRad interneurons were not different in VU-treated and control slices at DIV21. Intriguingly, we observed that, in VU-treated slices, RMP of the GFP-labeled interneurons was slightly elevated (Fig. 10K). The AP threshold was not significantly altered (Fig. 10L), but the relative AP threshold was lower in VU-treated slices compared with control (Fig. 10M). Input resistance was not different (DMSO 271.1 ± 12.8 mΩ; VU 244.2 ± 13.2 mΩ; UT, p = 0.15). Together, this suggests that sRad interneurons in VU-treated slices receive normal excitatory synaptic input at DIV21, but that less input current is required to reach their firing threshold. We propose that this subtle alteration in membrane excitability contributes to the elevated sIPSC frequency that we observed after VU treatment at DIV21.
Figure 10.
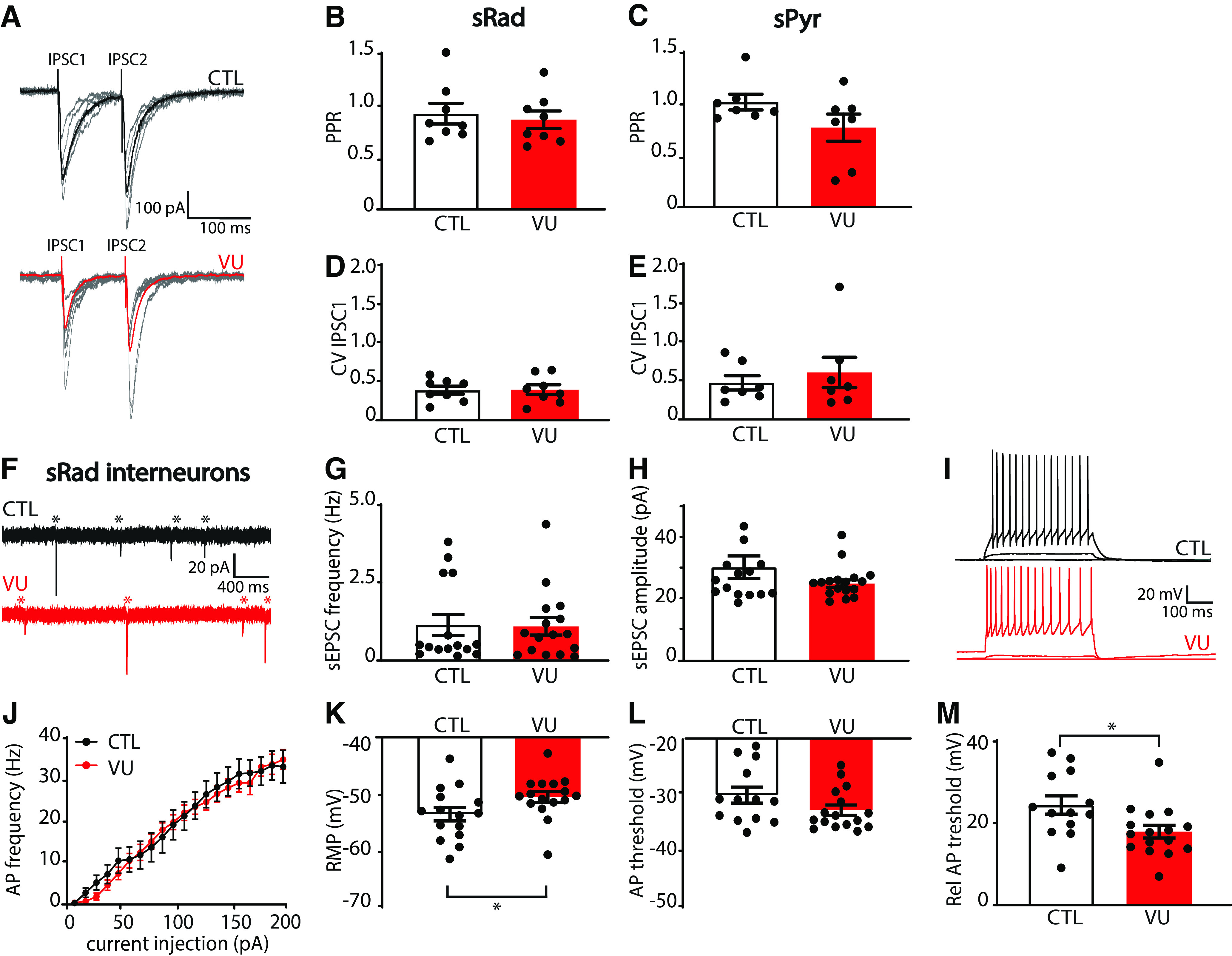
VU treatment results in elevated RMPs in sRad interneurons at DIV21. A, Whole-cell voltage-clamp recordings of evoked IPSCs in control (CTL) and VU-treated cultures at DIV21. Stimulation electrode was placed in the sRad. Bold represents average evoked IPSCs. Gray represents individual traces. B, C, Paired-pulse ratios (IPSC2/IPSC1 amplitudes) evoked in sRad (MW, p = 0.65) and sPyr (MW, p = 0.21) in CTL and VU-treated cultures at DIV21. D, E, Coefficient of variation (CV) of the first IPSCs evoked in sRad (MW, p = 0.96) and sPyr (MW, p = 0.80) in CTL and VU-treated cultures at DIV21. B–E, Data are from 7 or 8 cells, 4-6 slices, and 3-5 mice per group. F, sEPSC recording in GFP-labeled interneurons in sRad from control (CTL) and VU-treated cultures from GAD65-GFP mice at DIV21. Asterisks indicate sEPSC. G, H, sEPSC frequency (MW; p = 0.85) and amplitude (MW; p = 0.38) in these interneurons. Data are from 15 or 16 cells, 11 slices, and 10 mice per group. I, Whole-cell current-clamp recordings of APs after current injections in sRad interneurons in CTL and VU-treated cultures at DIV21. J, AP firing rates in sRad interneurons in CTL and VU-treated cultures at DIV21 (two-way ANOVA, current injection: p < 0.001, treatment: p = 0.42). K–M, RMP (MW, p = 0.023), AP threshold (UT, p = 0.11), and relative AP (Rel AP) threshold (MW, p = 0.012) in sRad interneurons in CTL and VU-treated cultures at DIV21. J-M, Data are from 13-16 cells, 11 slices, and 10 mice per group. *p < 0.05.
Discussion
There is accumulating evidence that the developmental GABA shift is often delayed in NDD patients (Talos et al., 2012; Duarte et al., 2013; Merner et al., 2015; Tang et al., 2016; Ruffolo et al., 2018; T. Wang et al., 2021; Birey et al., 2022) and in NDD animal models (He et al., 2014; Tyzio et al., 2014; Deidda et al., 2015b; Banerjee et al., 2016; Corradini et al., 2017; Roux et al., 2018; Fernandez et al., 2019; Lozovaya et al., 2019; Bertoni et al., 2021). Here, we used organotypic hippocampal cultures to examine the consequences of a delayed GABA shift for the developing CA1 network using the specific KCC2 blocker VU. VU treatment between DIV1 and DIV8 increased intracellular chloride levels without affecting chloride transporter expression levels, thereby effectively delaying the GABA shift in CA1 pyramidal neurons. We found that VU treatment did not have a direct effect on synaptic currents and firing properties of CA1 pyramidal cells at DIV9. However, at DIV21, when chloride levels were fully normalized, we observed a remarkable increase in sIPSC frequency compared with control slices, while synapse numbers remained unaffected. We found that firing thresholds in CA1 pyramidal neurons were slightly elevated while dendrite-targeting interneurons showed an elevated RMP and lower firing threshold at DIV21. Together, this shows that a delay in the postnatal GABA shift does not directly affect synaptic development but rather leads to indirect, cell type-specific changes in membrane properties that may contribute to altered network activity at a later time point.
In this study, we used organotypic hippocampal cultures as a model system to study postnatal development. We demonstrate that the GABA shift occurs in slice cultures around the same time as it does in vivo (Valeeva et al., 2016; Sulis Sato et al., 2017; Murata and Colonnese, 2020; Salmon et al., 2020; Herstel et al., 2022). As previously reported, synapses continue to develop in these cultured slices. Indeed, sEPSC frequency and density of VGLUT puncta and density of spines increased from DIV9 to DIV21 (De Simoni et al., 2003; Berry et al., 2012), while sEPSC amplitude decreased (De Simoni et al., 2003) and the frequency of sIPSCs and density of puncta VGAT remained mostly stable (De Simoni et al., 2003). VU treatment from DIV1 to DIV8 resulted in increased chloride levels in CA1 pyramidal cells at DIV9, that were comparable to untreated slices at DIV2. This indicates that the VU treatment effectively delayed chloride maturation and maintained depolarizing GABA signaling up to DIV9. Chloride maturation in interneurons occurs later compared with pyramidal cells and remained unaffected by the VU treatment. Importantly, VU treatment blocked KCC2 function without altering expression levels of the protein. This suggests that VU treatment altered intracellular chloride levels in pyramidal cells without interfering with the structural role of KCC2 proteins in spines (Li et al., 2007; Gauvain et al., 2011; Fiumelli et al., 2013; Chevy et al., 2015; Puskarjov et al., 2017; Awad et al., 2018; Kesaf et al., 2020), although we can never exclude that blocking chloride transport changes how KCC2 interacts with other proteins (Mahadevan et al., 2017; Smalley et al., 2020). Our experimental design therefore allowed for selectively testing how intracellular chloride concentrations influence the developing CA1 network.
The first key finding from this study is that maintaining depolarizing GABA signaling until DIV9 did not have any direct effects on excitatory and inhibitory synaptic structure or function. Previous groundbreaking research has demonstrated that excitatory GABAergic signaling during prenatal and perinatal development promotes synapse formation and maturation (Leinekugel et al., 1997; Akerman and Cline, 2006; Nakanishi et al., 2007; Wang and Kriegstein, 2008, 2011; van Rheede et al., 2015; Oh et al., 2016). These studies were performed before P8, or when the GABA shift was accelerated. Our slices are made from P6-P7 pups, when GABA signaling is still depolarizing. We did not observe any difference in synapse number between control and VU-treated slices, at DIV9 nor at DIV21. This demonstrates that prolonging the period of depolarizing GABA signaling during the second postnatal week does no longer affect synapse formation. These findings were corroborated by a parallel study, in which inhibitory transmission was found unaltered in slices that were treated with furosemide for 1 week (Peerboom et al., 2023). These observations suggest that the synapse-promoting effect of depolarizing GABA signaling is restricted from embryonic development up to the first postnatal week.
Comparing our current results with previous literature suggests that the effect of chloride manipulation critically depends on whether GABAergic signaling is excitatory or inhibitory for network activity, which is only indirectly related to EGABA. The effect of GABA signaling under physiological conditions does not only depend on the relation between EGABA and the AP threshold, but also on the spatiotemporal distribution of glutamatergic and GABAergic inputs (Staley and Mody, 1992; Gao et al., 1998; Morita et al., 2006; Le Magueresse and Monyer, 2013; Kilb, 2021). With more (excitatory) synaptic activity, shunting inhibition will become more prominent (Woodruff et al., 2011; Branchereau et al., 2016). This means that the precise consequences of alterations in the postnatal GABA shift depend on the effect on local network activity (Wang and Kriegstein, 2011; Seja et al., 2012; Deidda et al., 2015a; Pisella et al., 2019). In our slices, even when GABA signaling was kept depolarizing, GABA signaling already had an inhibitory action on cellular and network activity. Although EGABA in VU-treated slices was depolarized relative to RMP at DIV9 (Fig. 2B), it remained well below AP threshold (Fig. 3N). As a result, GABA signaling was inhibitory in both control and VU-treated slices (Fig. 5). It should be noted that we measured inhibitory GABA signaling after elevating network activity with modified ACSF, which may have skewed GABAergic function toward more inhibitory action via activity-induced chloride changes (Raimondo et al., 2012; Branchereau et al., 2016; Kilb, 2021). However, network discharges were also observed in DMSO and VU-treated slices during sEPSC recordings in normal ACSF (Fig. 5A), indicating a similar inhibitory action. Our results are in line with previous reports which argue that depolarizing GABAergic signaling is inhibitory in hippocampal slices from approximately P6-P9 (Khalilov et al., 1997; Valeeva et al., 2016; Salmon et al., 2020). Also in the hippocampus of living mice, depolarizing GABA was found to inhibit activity in the hippocampus from P7 onwards (Valeeva et al., 2010; Murata and Colonnese, 2020). This suggests that the situation in our slices recapitulates the in vivo situation well. However, it will be important to confirm our findings in vivo in future studies, especially since the precise consequences of altered chloride levels depend on local activity. Together, our results support the notion that depolarizing but inhibitory GABA signaling has limited contribution to postnatal synapse development in the hippocampus.
The second key finding from this study is that subtle, and cell type-specific, alterations in membrane properties were observed 2 weeks after the VU treatment had ended. We found that CA1 pyramidal cells had a slightly increased firing threshold, and interneurons in the sRad showed a slightly elevated membrane potential, which reduced the amount of depolarization required to fire an AP. We speculate that together this will modify spontaneous network activity toward an increased inhibitory tone and contribute to the increased sIPSC frequency that we observed in VU-treated slices (Fig. 6I). It is important to note that our study does not address the possible contribution of other interneurons, for instance oriens-lacunosum moleculare interneurons (Leão et al., 2012). Interestingly, pharmacological inhibition of NKCC1 from P3 to P8 (resulting in decreased intracellular chloride levels) has been reported to transiently decrease inhibitory transmission in the visual cortex several weeks later (at P35) (Deidda et al., 2015a), suggesting that developing GABAergic transmission may be particularly sensitive to intracellular chloride levels.
It remains unclear how blocking KCC2 from DIV1 to DIV8 can alter membrane properties of neurons 2 weeks later. It is important to note that these indirect effects observed in slices cannot easily be translated to the in vivo situation as they will likely be influenced by in vivo activity patterns and neuromodulatory signaling. In addition, although we carefully selected VU for its highest selectivity to KCC2 (Delpire and Weaver, 2016) and lowest off-target effects, we cannot exclude that the latter may have contributed (Sivakumaran et al., 2015), for instance via the Translocator protein (Liu et al., 2017; Shi et al., 2022). However, it is interesting that our study adds to an increasing number of studies that demonstrate that intracellular chloride levels can modify ion channels, and therefore membrane excitability, in often unpredictable ways (Huang et al., 2012; Seja et al., 2012; Goutierre et al., 2019; Sinha et al., 2022). Most notably, it was shown that membrane levels of TASK-3 potassium channels are regulated via KCC2 (Goutierre et al., 2019) and that the chloride-dependent kinase WNK3 regulates inward rectifier potassium channels (Sinha et al., 2022). It will also be important to further examine the role of various chloride channels in membrane excitability (Jentsch, 2016; Jentsch and Pusch, 2018; Akita and Fukuda, 2020). Interestingly, effects seem to strongly depend on cell type (Seja et al., 2012) and timing of the chloride manipulation (Lim et al., 2021; Sinha et al., 2022). The changes in excitability that we observed at DIV21 may therefore reflect a complex sequence of subtle adaptations of ion channel distribution that is likely specific per cell type and per brain region.
The present work shows that delaying the postnatal GABA shift by 1 week has no direct effects on synaptic development. Instead, we found evidence for indirect, cell type-specific changes in membrane properties, possibly via chloride-dependent regulation of ion channels, which may have long-term consequences for network activity and brain function. Our data call for careful assessment of alterations in cellular excitability in NDDs.
Footnotes
This work was supported by ZonMW TOP Grant 91216021 and Nederlandse Organisatie voor Wetenschappelijk Onderzoek OCENW.KLEIN.150. We thank Prof. Franck Polleux for providing the KCC2 short hairpin construct (Bortone and Polleux, 2009); Prof. Thomas Kuner for providing the Cre-dependent SCLM construct (Boffi et al., 2018); Prof. Kevin Staley for providing the SuperClomeleonlox/– mouse line; Dr. Stefan Berger for the CamKIIαCre/– mice; Dr. Henk Karst for sharing these mice with us; Dunya Selemangel for help with SuperClomeleon data analysis; and René van Dorland for the AAV production and excellent technical support.
The authors declare no competing financial interests.
References
- Akerman CJ, Cline HT (2006) Depolarizing GABAergic conductances regulate the balance of excitation to inhibition in the developing retinotectal circuit in vivo. J Neurosci 26:5117–5130. 10.1523/JNEUROSCI.0319-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akita T, Fukuda A (2020) Intracellular Cl− dysregulation causing and caused by pathogenic neuronal activity. Pflugers Arch Eur Arch 472:977–987. 10.1007/s00424-020-02375-4 [DOI] [PubMed] [Google Scholar]
- Antoine MW, Langberg T, Schnepel P, Feldman DE (2019) Increased excitation-inhibition ratio stabilizes synapse and circuit excitability in four autism mouse models. Neuron: 101:648–661.e5. 10.1016/j.neuron.2018.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad PN, Amegandjin CA, Szczurkowska J, Carriço JN, Fernandes do Nascimento AS, Baho E, Chattopadhyaya B, Cancedda L, Carmant L, Di Cristo G (2018) KCC2 regulates dendritic spine formation in a brain-region specific and BDNF dependent manner. Cereb Cortex: 28:4049–4062. 10.1093/cercor/bhy198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Rikhye RV, Breton-Provencher V, Tang X, Li C, Li K, Runyan CA, Fu Z, Jaenisch R, Sur M (2016) Jointly reduced inhibition and excitation underlies circuit-wide changes in cortical processing in Rett syndrome. Proc Natl Acad Sci USA 113:E7287–E7296. 10.1073/pnas.1615330113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banke TG, McBain CJ (2006) GABAergic input onto CA3 hippocampal interneurons remains shunting throughout development. J Neurosci 26:11720–11725. 10.1523/JNEUROSCI.2887-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Clements JD (1999) Quantal amplitude and quantal variance of strontium-induced asynchronous EPSCs in rat dentate granule neurons. J Physiol 516:227–248. 10.1111/j.1469-7793.1999.227aa.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa J (1989) Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol 416:303–325. 10.1113/jphysiol.1989.sp017762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry CT, Sceniak MP, Zhou L, Sabo SL (2012) Developmental up-regulation of vesicular glutamate transporter-1 promotes neocortical presynaptic terminal development. PLoS One 7:e50911. 10.1371/journal.pone.0050911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoni A, Schaller F, Tyzio R, Gaillard S, Santini F, Xolin M, Diabira D, Vaidyanathan R, Matarazzo V, Medina I, Hammock E, Zhang J, Chini B, Gaiarsa JL, Muscatelli F (2021) Oxytocin administration in neonates shapes the hippocampal circuitry and restores social behavior in a mouse model of autism. Mol Psychiatry 26:7582–7595. 10.1038/s41380-021-01227-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birey F, Li MY, Gordon A, Thete MV, Valencia AM, Revah O, Paşca AM, Geschwind DH, Paşca SP (2022) Dissecting the molecular basis of human interneuron migration in forebrain assembloids from Timothy syndrome. Cell Stem Cell 29:248–264.e7. 10.1016/j.stem.2021.11.011 [DOI] [PubMed] [Google Scholar]
- Boffi JC, Knabbe J, Kaiser M, Kuner T (2018) KCC2-dependent steady-state intracellular chloride concentration and pH in cortical layer 2/3 neurons of anesthetized and awake mice. Front Cell Neurosci 12:1–14. 10.3389/fncel.2018.00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortone D, Polleux F (2009) KCC2 expression promotes the termination of cortical interneuron migration in a voltage-sensitive calcium-dependent manner. Neuron 62:53–71. 10.1016/j.neuron.2009.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branchereau P, Cattaert D, Delpy A, Allain AE, Martin E, Meyrand P (2016) Depolarizing GABA/glycine synaptic events switch from excitation to inhibition during frequency increases. Sci Rep 6:21753. 10.1038/srep21753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova E, Fehsenfeld S, Mantamadiotis T, Lemberger T, Greiner E, Stewart AF, Schtz G (2001) A CamKIIα iCre BAC allows brain-specific gene inactivation. Genesis 31:37–42. 10.1002/gene.1078 [DOI] [PubMed] [Google Scholar]
- Chancey JH, Adlaf EW, Sapp MC, Pugh PC, Wadiche JI, Overstreet-Wadiche LS (2013) GABA depolarization is required for experience-dependent synapse unsilencing in adult-born neurons. J Neurosci 33:6614–6622. 10.1523/JNEUROSCI.0781-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevy Q, Heubl M, Goutierre M, Backer S, Moutkine I, Eugene E, Bloch-Gallego E, Levi S, Poncer JC (2015) KCC2 gates activity-driven AMPA receptor traffic through cofilin phosphorylation. J Neurosci 35:15772–15786. 10.1523/JNEUROSCI.1735-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheyne JE, Zabouri N, Baddeley D, Lohmann C (2019) Spontaneous activity patterns are altered in the developing visual cortex of the Fmr1 knockout mouse. Front Neural Circuits 13:1–8. 10.3389/fncir.2019.00057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudotvorova I, Ivanov A, Rama S, Hübner CA, Pellegrino C, Ben-Ari Y, Medina I (2005) Early expression of KCC2 in rat hippocampal cultures augments expression of functional GABA synapses. J Physiol 566:671–679. 10.1113/jphysiol.2005.089821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradini I, Focchi E, Rasile M, Morini R, Desiato G, Tomasoni R, Lizier M, Ghirardini E, Fesce R, Morone D, Barajon I, Antonucci F, Pozzi D, Matteoli M (2017) Maternal immune activation delays excitatory-to-inhibitory gamma-aminobutyric acid switch in offspring. Biol Psychiatry 83:1–12. [DOI] [PubMed] [Google Scholar]
- De Simoni A, Griesinger CB, Edwards FA (2003) Development of rat CA1 neurones in acute versus organotypic slices: role of experience in synaptic morphology and activity. J Physiol 550:135–147. 10.1113/jphysiol.2003.039099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deidda G, Allegra M, Cerri C, Naskar S, Bony G, Zunino G, Bozzi Y, Caleo M, Cancedda L (2015a) Early depolarizing GABA controls critical-period plasticity in the rat visual cortex. Nat Neurosci 18:87–96. 10.1038/nn.3890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deidda G, Parrini M, Naskar S, Bozarth IF, Contestabile A, Cancedda L (2015b) Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat Med 21:318–326. 10.1038/nm.3827 [DOI] [PubMed] [Google Scholar]
- Delpire E, Weaver CD (2016) Challenges of finding novel drugs targeting the K-Cl cotransporter. ACS Chem Neurosci 7:1624–1627. 10.1021/acschemneuro.6b00366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpire E, Baranczak A, Waterson AG, Kim K, Kett N, Morrison RD, Daniels JS, Weaver CD, Lindsley CW (2012) Further optimization of the K-Cl cotransporter KCC2 antagonist ML077: development of a highly selective and more potent in vitro probe. Bioorg Med Chem Lett 22:4532–4535. 10.1016/j.bmcl.2012.05.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte ST, Armstrong J, Roche A, Ortez C, Pérez A, del Mar O'Callaghan M, Pereira A, Sanmartí F, Ormazábal A, Artuch R, Pineda M, García-Cazorla A (2013) Abnormal expression of cerebrospinal fluid cation chloride cotransporters in patients with Rett syndrome. PLoS One 8:e68851. 10.1371/journal.pone.0068851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, Delpire E, Jensen FE, Staley KJ (2005) NKCC1 transporter facilitates seizures in the developing brain. Nat Med 11:1205–1213. 10.1038/nm1301 [DOI] [PubMed] [Google Scholar]
- Fernandez A, Dumon C, Guimond D, Fernandez A, Burnashev N, Tyzio R, Lozovaya N, Ferrari DC, Bonifazi P, Ben-Ari Y (2019) The GABA developmental shift is abolished by maternal immune activation already at birth. Cereb Cortex 29:3982–3992. 10.1093/cercor/bhy279 [DOI] [PubMed] [Google Scholar]
- Fiumelli H, Briner A, Puskarjov M, Blaesse P, Belem B, Dayer A, Kaila K, Martin J, Vutskits L (2013) An ion transport-independent role for the cation-chloride cotransporter KCC2 in dendritic spinogenesis in vivo. Cereb Cortex 23:378–388. 10.1093/cercor/bhs027 [DOI] [PubMed] [Google Scholar]
- Gao XB, Chen G, van den Pol AN (1998) GABA-dependent firing of glutamate-evoked action potentials at AMPA/kainate receptors in developing hypothalamic neurons. J Neurophysiol 79:716–726. 10.1152/jn.1998.79.2.716 [DOI] [PubMed] [Google Scholar]
- Gauvain G, Chamma I, Chevy Q, Cabezas C, Irinopoulou T, Bodrug N, Carnaud M, Levi S, Poncer JC (2011) The neuronal K-Cl cotransporter KCC2 influences postsynaptic AMPA receptor content and lateral diffusion in dendritic spines. Proc Natl Acad Sci USA 108:15474–15479. 10.1073/pnas.1107893108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogolla N, LeBlanc JJ, Quast KB, Südhof TC, Fagiolini M, Hensch TK (2009) Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J Neurodev Disord 1:172–181. 10.1007/s11689-009-9023-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves JT, Anstey JE, Golshani P, Portera-Cailliau C (2013) Circuit level defects in the developing neocortex of Fragile X mice. Nat Neurosci 16:903–909. 10.1038/nn.3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutierre M, Al Awabdh S, Donneger F, François E, Gomez-Dominguez D, Irinopoulou T, Menendez de la Prida L, Poncer JC (2019) KCC2 regulates neuronal excitability and hippocampal activity via interaction with Task-3 channels. Cell Rep 28:91–103.e7. 10.1016/j.celrep.2019.06.001 [DOI] [PubMed] [Google Scholar]
- Grimley JS, Li L, Wang W, Wen L, Beese LS, Hellinga HW, Augustine GJ (2013) Visualization of synaptic inhibition with an optogenetic sensor developed by cell-free protein engineering automation. J Neurosci 33:16297–16309. 10.1523/JNEUROSCI.4616-11.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyás AI, Sík A, Payne JA, Kaila K, Freund TF (2001) The KCl cotransporter, KCC2, is highly expressed in the vicinity of excitatory synapses in the rat hippocampus. Eur J Neurosci 13:2205–2217. 10.1046/j.0953-816x.2001.01600.x [DOI] [PubMed] [Google Scholar]
- He Q, Nomura T, Xu J, Contractor A (2014) The developmental switch in GABA polarity is delayed in Fragile X mice. J Neurosci 34:446–450. 10.1523/JNEUROSCI.4447-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Arroyo ED, Smukowski SN, Xu J, Piochon C, Savas JN, Portera-Cailliau C, Contractor A (2019) Critical period inhibition of NKCC1 rectifies synapse plasticity in the somatosensory cortex and restores adult tactile response maps in fragile X mice. Mol Psychiatry 24:1732–1747. 10.1038/s41380-018-0048-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herstel LJ, Peerboom C, Uijtewaal S, Selemangel D, Karst H, Wierenga CJ (2022) Using SuperClomeleon to measure changes in intracellular chloride during development and after early life stress. eNeuro 9:ENEURO.0416-22.2022. 10.1523/ENEURO.0416-22.2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HY, Kruijssen DL, Frias CP, Rózsa B, Hoogenraad CC, Wierenga CJ (2019) Endocannabinoid signaling mediates local dendritic coordination between excitatory and inhibitory synapses. Cell Rep 27:666–675.e5. 10.1016/j.celrep.2019.03.078 [DOI] [PubMed] [Google Scholar]
- Huang WC, Xiao S, Huang F, Harfe BD, Jan YN, Jan LY (2012) Calcium-activated chloride channels (CaCCs) regulate action potential and synaptic response in hippocampal neurons. Neuron 74:179–192. 10.1016/j.neuron.2012.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ (2001) Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 30:515–524. 10.1016/s0896-6273(01)00297-5 [DOI] [PubMed] [Google Scholar]
- Jentsch TJ (2016) VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat Rev Mol Cell Biol 17:293–307. 10.1038/nrm.2016.29 [DOI] [PubMed] [Google Scholar]
- Jentsch TJ, Pusch M (2018) CLC chloride channels and transporters: structure, function, physiology, and disease. Physiol Rev 98:1493–1590. 10.1152/physrev.00047.2017 [DOI] [PubMed] [Google Scholar]
- Kesaf S, Khirug S, Dinh E, Saez Garcia M, Soni S, Orav E, Delpire E, Taira T, Lauri SE, Rivera C (2020) The kainate receptor subunit GluK2 interacts with KCC2 to promote maturation of dendritic spines. Front Cell Neurosci 14:1–14. 10.3389/fncel.2020.00252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalilov I, Khazipov R, Esclapez M, Ben-Ari Y (1997) Bicuculline induces ictal seizures in the intact hippocampus recorded in vitro. Eur J Pharmacol 319:5–6. [DOI] [PubMed] [Google Scholar]
- Khazipov R, Khalilov I, Tyzio R, Morozova E, Ben-Ari Y, Holmes GL (2004) Developmental changes in GABAergic actions and seizure susceptibility in the rat hippocampus. Eur J Neurosci 19:590–600. 10.1111/j.0953-816x.2003.03152.x [DOI] [PubMed] [Google Scholar]
- Kilb W (2021) When are depolarizing GABAergic responses excitatory? Front Mol Neurosci 14:747835. 10.3389/fnmol.2021.747835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Alger BE (2001) Random response fluctuations lead to spurious paired-pulse facilitation. J Neurosci 21:9608–9618. 10.1523/JNEUROSCI.21-24-09608.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirmse K, Kummer M, Kovalchuk Y, Witte OW, Garaschuk O, Holthoff K (2015) GABA depolarizes immature neurons and inhibits network activity in the neonatal neocortex in vivo. Nat Commun 6:13. 10.1038/ncomms8750 [DOI] [PubMed] [Google Scholar]
- Le Magueresse C, Monyer H (2013) GABAergic interneurons shape the functional maturation of the cortex. Neuron 77:388–405. 10.1016/j.neuron.2013.01.011 [DOI] [PubMed] [Google Scholar]
- Leão RN, Mikulovic S, Leão KE, Munguba H, Gezelius H, Enjin A, Patra K, Eriksson A, Loew LM, Tort AB, Kullander K (2012) OLM interneurons differentially modulate CA3 and entorhinal inputs to hippocampal CA1 neurons. Nat Neurosci 15:1524–1530. 10.1038/nn.3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Walker JA, Williams JR, Goodier RJ, Payne JA, Moss SJ (2007) Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J Biol Chem 282:29777–29784. 10.1074/jbc.M705053200 [DOI] [PubMed] [Google Scholar]
- Leinekugel X, Medina I, Khalilov I, Ben Ari Y, Khazipov R (1997) Ca2+ oscillations mediated by the synergistic excitatory actions of GABA. Neuron 18:243–255. 10.1016/s0896-6273(00)80265-2 [DOI] [PubMed] [Google Scholar]
- Li H, Khirug S, Cai C, Ludwig A, Blaesse P, Kolikova J, Afzalov R, Coleman SK, Lauri S, Airaksinen MS, Keinänen K, Khiroug L, Saarma M, Kaila K, Rivera C (2007) KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron 56:1019–1033. 10.1016/j.neuron.2007.10.039 [DOI] [PubMed] [Google Scholar]
- Lim WM, Chin EW, Tang BL, Chen T, Goh EL (2021) WNK3 maintains the GABAergic inhibitory tone, synaptic excitation and neuronal excitability via regulation of KCC2 cotransporter in mature neurons. Front Mol Neurosci 14:1–17. 10.3389/fnmol.2021.762142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GJ, Middleton RJ, Kam WW, Chin DY, Hatty CR, Chan RH, Banati RB (2017) Functional gains in energy and cell metabolism after TSPO gene insertion. Cell Cycle 16:436–447. 10.1080/15384101.2017.1281477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Bendito G, Sturgess K, Erdélyi F, Szabó G, Molnár Z, Paulsen O (2004) Preferential origin and layer destination of GAD65-GFP cortical interneurons. Cereb Cortex 14:1122–1133. 10.1093/cercor/bhh072 [DOI] [PubMed] [Google Scholar]
- Lozovaya N, Nardou R, Tyzio R, Chiesa M, Pons-Bennaceur A, Eftekhari S, Bui TT, Billon-Grand M, Rasero J, Bonifazi P, Guimond D, Gaiarsa JL, Ferrari DC, Ben-Ari Y (2019) Early alterations in a mouse model of Rett syndrome: the GABA developmental shift is abolished at birth. Sci Rep 9:16. 10.1038/s41598-019-45635-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A, Nelson SB, Turrigiano GG (2004) Selective reconfiguration of layer 4 visual cortical circuitry by visual deprivation. Nat Neurosci 7:1353–1359. 10.1038/nn1351 [DOI] [PubMed] [Google Scholar]
- Mahadevan V, Khademullah CS, Dargaei Z, Chevrier J, Uvarov P, Kwan J, Bagshaw RD, Pawson T, Emili A, De Koninck Y, Anggono V, Airaksinen M, Woodin MA (2017) Native KCC2 interactome reveals PACSIN1 as a critical regulator of synaptic inhibition. Elife 6:1–34. 10.7554/eLife.28270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima T, Miura M, Patzke N, Toji N, Wada K, Ogura Y, Homma KJ, Sgadò P, Vallortigara G (2022) Impaired epigenesis of imprinting predispositions causes autism-like behavioral phenotypes in domestic chicks. bioRxiv 492744. 10.1101/2022.05.19.492744. [DOI] [Google Scholar]
- Menendez De La Prida L, Huberfeld G, Cohen I, Miles R (2006) Threshold behavior in the initiation of hippocampal population bursts. Neuron 49:131–142. 10.1016/j.neuron.2005.10.034 [DOI] [PubMed] [Google Scholar]
- Meredith RM (2015) Sensitive and critical periods during neurotypical and aberrant neurodevelopment: a framework for neurodevelopmental disorders. Neurosci Biobehav Rev 50:180–188. 10.1016/j.neubiorev.2014.12.001 [DOI] [PubMed] [Google Scholar]
- Merner ND, Chandler MR, Bourassa C, Liang B, Khanna AR, Dion P, Rouleau GA, Kahle KT (2015) Regulatory domain or CPG site variation in SLC12A5, encoding the chloride transporter KCC2, in human autism and schizophrenia. Front Cell Neurosci 9:1–10. 10.3389/fncel.2015.00386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mòdol L, Casas C, Llidó A, Navarro X, Pallarès M, Darbra S (2014) Neonatal allopregnanolone or finasteride administration modifies hippocampal K+ Cl− co-transporter expression during early development in male rats. J Steroid Biochem Mol Biol 143:343–347. 10.1016/j.jsbmb.2014.05.002 [DOI] [PubMed] [Google Scholar]
- Molnár Z, Luhmann HJ, Kanold PO (2020) Transient cortical circuits match spontaneous and sensory-driven activity during development. Science 370:eabb2153. 10.1126/science.abb2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K, Tsumoto K, Aihara K (2006) Bidirectional modulation of neuronal responses by depolarizing GABAergic inputs. Biophys J 90:1925–1938. 10.1529/biophysj.105.063164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata Y, Colonnese MT (2020) GABAergic interneurons excite neonatal hippocampus in vivo. Sci Adv 6:1–10. 10.1126/sciadv.aba1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K, Yamada J, Takayama C, Oohira A, Fukuda A (2007) NKCC1 activity modulates formation of functional inhibitory synapses in cultured neocortical neurons. Synapse 61:138–149. 10.1002/syn.20352 [DOI] [PubMed] [Google Scholar]
- Oh WC, Lutzu S, Castillo PE, Kwon HB (2016) De novo synaptogenesis induced by GABA in the developing mouse cortex: supplemental. Science 353:1037–1040. 10.1126/science.aaf5206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu Y, Donneger F, Schwartz EJ, Poncer JC (2020) Cation–chloride cotransporters and the polarity of GABA signalling in mouse hippocampal parvalbumin interneurons. J Physiol 598:1865–1880. 10.1113/JP279221 [DOI] [PubMed] [Google Scholar]
- Patenaude C, Massicotte G, Lacaille JC (2005) Cell type-specific GABA synaptic transmission and activity-dependent plasticity in rat hippocampal stratum radiatum interneurons. Eur J Neurosci 22:179–188. 10.1111/j.1460-9568.2005.04207.x [DOI] [PubMed] [Google Scholar]
- Perkins KL (2006) Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices. J Neurosci Methods 154:1–18. 10.1016/j.jneumeth.2006.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peerboom CNE, Wijne TB, Wierenga CJ (2023) Treatment with furosemide indirectly increases inhibitory transmission in the developing hippocampus. bioRxiv 548438. 10.1101/2023.07.11.548438. [DOI] [Google Scholar]
- Pisella LI, Gaiarsa JL, Diabira D, Zhang J, Khalilov I, Duan J, Kahle KT, Medina I (2019) Impaired regulation of KCC2 phosphorylation leads to neuronal network dysfunction and neurodevelopmental pathology. Sci Signal 12:eaay0300. 10.1126/scisignal.aay0300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puskarjov M, Fiumelli H, Briner A, Bodogan T, Demeter K, Lacoh C, Mavrovic M, Blaesse P, Kaila K, Vutskits L (2017) K-Cl cotransporter 2–mediated Cl− extrusion determines developmental stage–dependent impact of propofol anesthesia on dendritic spine. Anesthesiology 126:855–867. 10.1097/ALN.0000000000001587 [DOI] [PubMed] [Google Scholar]
- Rahmati N, Normoyle KP, Glykys J, Dzhala VI, Lillis KP, Kahle KT, Raiyyani R, Jacob T, Staley KJ (2021) Unique actions of GABA arising from cytoplasmic chloride microdomains. J Neurosci 41:4957–4975. 10.1523/JNEUROSCI.3175-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondo JV, Markram H, Akerman CJ (2012) Short-term ionic plasticity at GABAergic synapses. Front Synaptic Neurosci 4:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rall W (1967) Distinguishing theoretical synaptic potentials computed for different soma-dendritic distributions of synaptic. J Neurophysiol 30:1138–1168. 10.1152/jn.1967.30.5.1138 [DOI] [PubMed] [Google Scholar]
- Rheims S, Minlebaev M, Ivanov A, Represa A, Khazipov R, Holmes GL, Ben-Ari Y, Zilberter Y (2008) Excitatory GABA in rodent developing neocortex in vitro. J Neurophysiol 100:609–619. 10.1152/jn.90402.2008 [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K (1999) The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397:251–255. 10.1038/16697 [DOI] [PubMed] [Google Scholar]
- Romo-Parra H, Treviño M, Heinemann U, Gutiérrez R (2008) GABA actions in hippocampal area CA3 during postnatal development: differential shift from depolarizing to hyperpolarizing in somatic and dendritic compartments. J Neurophysiol 99:1523–1534. 10.1152/jn.01074.2007 [DOI] [PubMed] [Google Scholar]
- Roux S, Lohof A, Ben-Ari Y, Poulain B, Bossu JL (2018) Maturation of GABAergic transmission in cerebellar Purkinje cells is sex dependent and altered in the valproate model of autism. Front Cell Neurosci 12:1–14. 10.3389/fncel.2018.00232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffolo G, Iyer A, Cifelli P, Roseti C, Mühlebner A, van Scheppingen J, Scholl T, Hainfellner JA, Feucht M, Krsek P, Zamecnik J, Jansen FE, Spliet WG, Limatola C, Aronica E, Palma E (2016) Functional aspects of early brain development are preserved in tuberous sclerosis complex (TSC) epileptogenic lesions. Neurobiol Dis 95:93–101. 10.1016/j.nbd.2016.07.014 [DOI] [PubMed] [Google Scholar]
- Ruffolo G, Cifelli P, Roseti C, Thom M, van Vliet EA, Limatola C, Aronica E, Palma E (2018) A novel GABAergic dysfunction in human Dravet syndrome. Epilepsia 59:2106–2117. 10.1111/epi.14574 [DOI] [PubMed] [Google Scholar]
- Ruiter M, Herstel LJ, Wierenga CJ, Ma T (2020) Reduction of dendritic inhibition in CA1 pyramidal neurons in amyloidosis models of early Alzheimer's disease. J Alzheimers Dis 78:951–964. 10.3233/JAD-200527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon CK, Pribiag H, Gizowski C, Farmer WT, Cameron S, Jones EV, Mahadevan V, Bourque CW, Stellwagen D, Woodin MA, Murai KK (2020) Depolarizing GABA transmission restrains activity-dependent glutamatergic synapse formation in the developing hippocampal circuit. Front Cell Neurosci 14:36–16. 10.3389/fncel.2020.00036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M, Debanne D, Müller M, Gähwiler BH, Thompson SM (1994) Role of excitatory amino acid and GABAB receptors in the generation of epileptiform activity in disinhibited hippocampal slice cultures. Neuroscience 61:823–832. 10.1016/0306-4522(94)90405-7 [DOI] [PubMed] [Google Scholar]
- Sedmak G, Jovanov-Milošević N, Puskarjov M, Ulamec M, Krušlin B, Kaila K, Judaš M (2016) Developmental expression patterns of KCC2 and functionally associated molecules in the human brain. Cereb Cortex 26:4574–4589. 10.1093/cercor/bhv218 [DOI] [PubMed] [Google Scholar]
- Seja P, Schonewille M, Spitzmaul G, Badura A, Klein I, Rudhard Y, Wisden W, Hübner CA, De Zeeuw CI, Jentsch TJ (2012) Raising cytosolic Cl– in cerebellar granule cells affects their excitability and vestibulo-ocular learning. EMBO J 31:1217–1230. 10.1038/emboj.2011.488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC (2007) The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem 76:823–847. 10.1146/annurev.biochem.76.060805.160029 [DOI] [PubMed] [Google Scholar]
- Shi Y, Cui M, Ochs K, Brendel M, Strübing FL, Briel N, Eckenweber F, Zou C, Banati RB, Liu GJ, Middleton RJ, Rupprecht R, Rudolph U, Zeilhofer HU, Rammes G, Herms J, Dorostkar MM (2022) Long-term diazepam treatment enhances microglial spine engulfment and impairs cognitive performance via the mitochondrial 18 kDa translocator protein (TSPO). Nat Neurosci 25:317–329. 10.1038/s41593-022-01013-9 [DOI] [PubMed] [Google Scholar]
- Sinha AS, Wang T, Watanabe M, Hosoi Y, Sohara E, Akita T, Uchida S, Fukuda A (2022) WNK3 kinase maintains neuronal excitability by reducing inwardly rectifying K+ conductance in layer V pyramidal neurons of mouse medial prefrontal cortex. Front Mol Neurosci 15:856262. 10.3389/fnmol.2022.856262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipila ST, Schuchmann S, Voipio J, Yamada J, Kaila K (2006) The cation-chloride cotransporter NKCC1 promotes sharp waves in the neonatal rat hippocampus. 573:765–773. 10.1113/jphysiol.2006.107086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivakumaran S, Cardarelli RA, Maguire J, Kelley MR, Silayeva L, Morrow DH, Mukherjee J, Moore YE, Mather RJ, Duggan ME, Brandon NJ, Dunlop J, Zicha S, Moss SJ, Deeb TZ (2015) Selective inhibition of KCC2 leads to hyperexcitability and epileptiform discharges in hippocampal slices and in vivo. J Neurosci 35:8291–8296. 10.1523/JNEUROSCI.5205-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalley JL, Kontou G, Choi C, Ren Q, Albrecht D, Abiraman K, Santos MA, Bope CE, Deeb TZ, Davies PA, Brandon NJ, Moss SJ (2020) Isolation and characterization of multi-protein complexes enriched in the K-Cl co-transporter 2 from brain plasma membranes. Front Mol Neurosci 13:1–16. 10.3389/fnmol.2020.563091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen AT, Ledri M, Melis M, Ledri LN, Andersson M, Kokaia M (2017) Altered chloride homeostasis decreases the action potential threshold and increases hyperexcitability in hippocampal neurons. eNeuro 4:ENEURO.0172-17.2017. 10.1523/ENEURO.0172-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spoljaric I, Spoljaric A, Mavrovic M, Seja P, Puskarjov M, Kaila K (2019) KCC2-mediated Cl− extrusion modulates spontaneous hippocampal network events in perinatal rats and mice. Cell Rep 26:1073–1081.e3. 10.1016/j.celrep.2019.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Mody I (1992) Shunting of excitatory input to dentate gyrus granule cells by a depolarizing GABA(A) receptor-mediated postsynaptic conductance. J Neurophysiol 68:197–212. 10.1152/jn.1992.68.1.197 [DOI] [PubMed] [Google Scholar]
- Stein V, Hermans-Borgmeyer I, Jentsch TJ, Hübner CA (2004) Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol 468:57–64. 10.1002/cne.10983 [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D (1991) A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37:173–182. 10.1016/0165-0270(91)90128-m [DOI] [PubMed] [Google Scholar]
- Sulis Sato S, Artoni P, Landi S, Cozzolino O, Parra R, Pracucci E, Trovato F, Szczurkowska J, Luin S, Arosio D, Beltram F, Cancedda L, Kaila K, Ratto GM (2017) Simultaneous two-photon imaging of intracellular chloride concentration and pH in mouse pyramidal neurons in vivo. Proc Natl Acad Sci USA 114:E8770–E8779. 10.1073/pnas.1702861114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talos DM, Sun H, Kosaras B, Joseph A, Folkerth RD, Poduri A, Madsen JR, Black PM, Jensen FE (2012) Altered inhibition in tuberous sclerosis and type IIb cortical dysplasia. Ann Neurol 71:539–551. 10.1002/ana.22696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Kim J, Zhou L, Wengert E, Zhang L, Wu Z, Carromeu C, Muotri AR, Marchetto MC, Gage FH, Chen G (2016) KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc Natl Acad Sci USA 113:751–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S (1996) Subregion- and cell type-restricted gene knockout in mouse brain. Cell 87:1317–1326. 10.1016/s0092-8674(00)81826-7 [DOI] [PubMed] [Google Scholar]
- Tsukahara T, Masuhara M, Iwai H, Sonomura T, Sato T (2015) Repeated stress-induced expression pattern alterations of the hippocampal chloride transporters KCC2 and NKCC1 associated with behavioral abnormalities in female mice. Biochem Biophys Res Commun 465:145–151. 10.1016/j.bbrc.2015.07.153 [DOI] [PubMed] [Google Scholar]
- Tyzio R, Holmes GL, Ben-Ari Y, Khazipov R (2007) Timing of the developmental switch in GABAA mediated signaling from excitation to inhibition in CA3 rat hippocampus using gramicidin perforated patch and extracellular recordings. Epilepsia 48:96–105. 10.1111/j.1528-1167.2007.01295.x [DOI] [PubMed] [Google Scholar]
- Tyzio R, Nardou R, Ferrari D, Tsintsadze T, Shahrokhi A, Eftekhari S, Khalilov I, Tsintsadze V, Brouchoud C, Chazal G, Lemonnier E, Lozovaya N, Burnashev N (2014) Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 343:675–679. 10.1126/science.1247190 [DOI] [PubMed] [Google Scholar]
- Valeeva G, Abdullin A, Tyzio R, Skorinkin A, Nikolski E, Ben-Ari Y, Khazipov R (2010) Temporal coding at the immature depolarizing GABAergic synapse. Front Cell Neurosci 4:1–12. 10.3389/fncel.2010.00017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valeeva G, Tressard T, Mukhtarov M, Baude A, Khazipov R (2016) An optogenetic approach for investigation of excitatory and inhibitory network GABA actions in mice expressing channelrhodopsin-2 in GABAergic neurons. J Neurosci 36:5961–5973. 10.1523/JNEUROSCI.3482-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rheede JJ, Richards BA, Akerman CJ (2015) Sensory-evoked spiking behavior emerges via an experience-dependent plasticity mechanism. Neuron 87:1050–1062. 10.1016/j.neuron.2015.08.021 [DOI] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S, Lowell BB (2011) Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71:142–154. 10.1016/j.neuron.2011.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang BS, Feng L, Liu M, Liu X, Cang J (2013) Environmental enrichment rescues binocular matching of orientation preference in mice that have a precocious critical period. Neuron 80:198–209. 10.1016/j.neuron.2013.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DD, Kriegstein AR (2008) GABA regulates excitatory synapse formation in the neocortex via NMDA receptor activation. J Neurosci 28:5547–5558. 10.1523/JNEUROSCI.5599-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DD, Kriegstein AR (2011) Blocking early GABA depolarization with bumetanide results in permanent alterations in cortical circuits and sensorimotor gating deficits. Cereb Cortex 21:574–587. 10.1093/cercor/bhq124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Shan L, Miao C, Xu Z, Jia F (2021) Treatment effect of bumetanide in children with autism spectrum disorder: a systematic review and meta-analysis. Front Psychiatry 12:1–11. 10.3389/fpsyt.2021.751575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga CJ, Wadman WJ (1999) Miniature inhibitory postsynaptic currents in CA1 pyramidal neurons after kindling epileptogenesis. J Neurophysiol 82:1352–1362. 10.1152/jn.1999.82.3.1352 [DOI] [PubMed] [Google Scholar]
- Wierenga CJ, Becker N, Bonhoeffer T (2008) GABAergic synapses are formed without the involvement of dendritic protrusions. Nat Neurosci 11:1044–1052. 10.1038/nn.2180 [DOI] [PubMed] [Google Scholar]
- Wierenga CJ, Müllner FE, Rinke I, Keck T, Stein V, Bonhoeffer T (2010) Molecular and electrophysiological characterization of GFP-expressing CA1 interneurons in GAD65-GFP mice. PLoS One 5:e15915. 10.1371/journal.pone.0015915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff AR, McGarry LM, Vogels TP, Inan M, Anderson SA, Yuste R (2011) State-dependent function of neocortical chandelier cells. J Neurosci 31:17872–17886. 10.1523/JNEUROSCI.3894-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann H, Fukuda A (2004) Cl− uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J Physiol 557:829–841. 10.1113/jphysiol.2004.062471 [DOI] [PMC free article] [PubMed] [Google Scholar]