

Abstract
The underdiagnosis of alpha-1 antitrypsin (AAT) deficiency (AATD) has been recognized for many years, yet little progress has been made in treatment of the disease. In this review, we summarize the AATD disease process as well as its diagnosis and treatment by AAT augmentation therapy. AATD is a rare autosomal disease that primarily affects the lungs and liver. AATD is associated with an increased susceptibility to developing pulmonary emphysema. The specific pharmacological treatment for AATD is intravenous administration of exogenous AAT. Augmentation therapy with AAT increases serum and pulmonary epithelial AAT levels, restores anti-elastase capacity, and decreases inflammatory mediators in the lung. Augmentation therapy reduces the loss of lung density over time, thus slowing progression of the disease. The effects of augmentation therapy on outcomes, such as frequency/duration of flare-ups, quality of life, lung function decline and mortality, are assessed. Wider testing for AATD, potentially through primary care physicians, could result in earlier treatment and better outcomes for individuals with AATD-induced lung respiratory disease.
Keywords: alpha-1 antitrypsin, alpha-1 antitrypsin deficiency, augmentation therapy, computed tomography densitometry, forced expiration volume in 1 s, lung density
Introduction
Alpha-1 antitrypsin (AAT) deficiency (AATD) is a genetic disorder that was first recognized 60 years ago. The underdiagnosed nature of AATD has been extensively described in the literature.1–4 Despite the recent advances in identification of the genetic variants underlying AATD,5–8 the lack of widespread testing persists. In this review, the incidence, pathogenesis, diagnosis and treatment of AATD and the opportunity for primary care physicians to improve the diagnosis and treatment of AATD are addressed.
For this narrative review, the databases MEDLINE and EMBASE were used for literature research. Search terms included combinations of the following keywords: alpha-1 antitrypsin, antitrypsin deficiency, chronic obstructive pulmonary disease, liver disease, lung disease, lung density, forced expiration volume in 1 s, computed tomography densitometry diagnosis, treatment or augmentation therapy. Databases were searched for peer-review articles published in different languages without any date restriction.
Review
Incidence and pathogenesis of AATD
AATD is a rare autosomal codominant disease that affects up to 20 people per 100,000 individuals.9 AAT (Figure 1), the most abundant protease inhibitor in human serum, is synthetized by hepatocytes and transported to the lungs, where it is the primary anti-protease. It also has significant anti-inflammatory functions in a wide range of cell types.10
Figure 1.
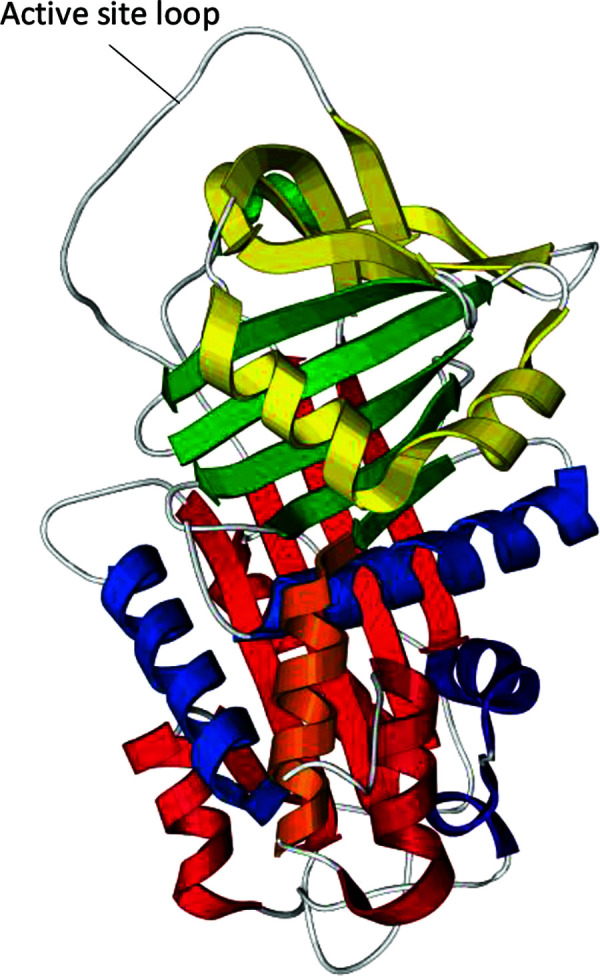
3D structure of the alpha-1 antitrypsin molecule. The polypeptide chain is composed of three β-sheets (yellow, green and red strips) and nine α-helices (in yellow, blue, orange and red coils). The active site loop mediates inhibitory specificity.
Adapted from a generic image of the free-use protein.
The first cases of AATD were described more than five decades ago,11 and many studies have been published since. Additionally, over 200 AAT mutations have been described in the literature. Some variants are associated with marked deficiency of serum AAT levels (<11 μM or 0.5 g/L) and increased susceptibility to developing emphysema, even in non-smokers.6 Individuals who have inherited at least one abnormal AAT gene are referred to as heterozygotes for PiMZ or PiMS gene variants or ‘carriers’ (Figure 2). The homozygous Pi*ZZ genotype is associated with the most prevalent severe deficiency and Pi*ZZ predisposes individuals to pulmonary emphysema, bronchiectasis, liver cirrhosis, hepatocellular cancer, vasculitis and panniculitis.12 As the AAT protein is synthesized in the liver, most of the AAT molecules in individuals with Pi*ZZ genotype are retained in the endoplasmic reticulum of hepatocytes rather than being secreted. Consequently, there is an accumulation of abnormally folded AAT, which leads to toxicity of the protein and ultimately liver inflammation, cirrhosis and hepatocellular carcinoma. Overall, the odds of developing advanced fibrosis in Pi*ZZ carriers are 9–20 times higher than in non-carriers.13,14
Figure 2.

Individuals who have inherited at least one abnormal alpha-1 antitrypsin gene (S, Z) are referred to as heterozygotes for PiMZ or PiMS gene variants or carriers. Most prevalent severe deficiency is associated with individuals who are homozygous for Pi*ZZ.
In addition to the homozygous Pi*ZZ genotype, many other variants may result in low AAT levels (Pi*M heerlen, Pi*M procida, Pi*M lowell, etc.). Moreover, Pi*Null variants are characterized by the absence of circulating AAT and are also linked to the development of liver and lung diseases. Overall, the severity of AATD is inversely related to serum AAT levels. The lowest serum AAT levels have been detected in patients with Pi*Null/Null genotype followed by Pi*ZZ, Pi*SZ and Pi*MZ genotypes.2,15
The estimated mean prevalence of Pi*ZZ in America is 1:26,002, whereas in Europe it ranges between 1:491 (Latvia) and 1:86,127 (Russia).16
Diagnosis of AATD
AATD remains as an underdiagnosed genetic disease4 despite awareness regarding its diagnosis and management through the development of national and international guidelines. The first guideline was published by the American Thoracic Society/European Respiratory Society in 2003.12 Since then, several studies have developed more accurate and less expensive methods to diagnose AATD.17 Different strategies have been identified to increase the detection rate of AATD. These include targeted detection of chronic obstructive pulmonary disease (COPD) and liver disease, testing of family members of affected individuals, large-scale screening programmes, newborn screening programmes, identifying rare diseases in electronic medical records and the use of commercial diagnostic tests directly by customers.18 However, it is crucial that the patient has access to therapy after a diagnosis to prevent rapid loss of pulmonary function. Regrettably, much improvement is needed in this area.
Patients with AATD must be adequately identified prior to receiving any treatment. Primary care physicians play a key role in identifying possible patients with AATD because they are the first point of contact in the healthcare system. AATD diagnosis and management can be conducted in primary care centres together with pulmonary specialists. By improving disease awareness of primary care providers and by testing all patients with COPD for AATD, early diagnosis may be faciliated.19
The benefits of an early AATD diagnosis include the implementation of non-pharmacological and pharmacological interventions for disease management at an initial stage. One of the most important non-pharmacological measures in patients with diagnosed AATD is lifestyle modification via smoking cessation. As pulmonary emphysema in patients with AATD is strongly associated with smoking, the early detection of AATD and smoking cessation is key to reducing the risk of developing emphysema. A study demonstrated that smokers with PI*ZZ who knew about their severe AATD deficiency had increased their quit attempts at 3 months after being diagnosed with AATD and were more motivated toward smoking cessation.20 Likewise, cigarette smoke is also a risk factor for airflow obstruction and COPD in patients with AATD heterozygous for Pi*MZ.21 Remarkably, COPD with emphysema can occur even without smoking. Therefore, an early AATD diagnosis may improve outcomes and allow the implementation of interventions that prevent or ameliorate the disease.10
Treatment
General therapies
Patients with emphysema and AATD should be treated similarly to those diagnosed with COPD, including continuous pharmacological treatment with bronchodilators, inhaled steroids for patients with exacerbations, antibiotics and oral corticosteroids during flare-ups, influenza vaccination, antipneumococcal vaccination, pulmonary rehabilitation and oxygen therapy. Surgical interventions, such as lung volume reduction surgery and lung transplantation, are limited to the most severe cases.4,9
Treatment should always include smoking cessation — it is essential at any stage of COPD, whether it is related to AATD or not. Exposure to respiratory irritants, such as tobacco smoke, dust and environmental pollution, should also be avoided. Every effort should be made to prevent smoking initiation in patients with AATD because it is a risk factor for the development of emphysema. Additionally, smoking oxidizes AAT and reduces its anti-elastase activity.22
Augmentation therapy for AATD
Augmentation therapy with intravenous purified AAT is the only specific treatment for AATD. This therapy is aimed at increasing patient survival, symptom control and preventing the progression of AATD-related pulmonary emphysema.23,24 AAT is also indicated for patients with necrotizing panniculitis.3 The efficacy and safety of augmentation therapy have been evidenced by numerous observational and randomized clinical trials. In rare diseases, the difficulty in recruiting patients for clinical trials can be a limiting factor to conducting robust studies. Augmentation therapy with AAT is recommended to restore AAT to physiological levels and ensure its protective action.3,4 According to the recommendations issued by international societies,3,4,12 intravenous AAT treatment is indicated for non-smokers or ex-smokers >18 years of age, with a genetic variant consistent with severe AAT deficiency, a low serum AAT level (<11 μM) and evidence of airflow limitation on pulmonary function tests.
The complexity of interpreting genetic variants, the screening of patients and the treatment of the disease require experienced medical teams. Therefore, AAT augmentation therapy should be performed in reference centres for rare diseases, with a trained multidisciplinary team properly equipped with immediate relief equipment for any complications.25 Reference centres for rare diseases are structured healthcare centres that provide adequate care for AATD treatment.4
AAT treatment is provided through regulatory approvals in North America (Canada and USA), Europe (Austria, Denmark, France, Germany, Italy, the Netherlands, Spain and Switzerland) and Asia (Japan).26 However, there is still much work to be carried out in developing countries. For instance, in Brazil, treatment with AAT is typically obtained through legal action.26
Optimal therapeutic regimen for AATD
The only approved regimen for patients with AATD is weekly intravenous infusions of AAT (60 mg/kg). This regimen maintains serum trough AAT levels above the protective threshold during the week and long term.23 Serum AAT levels are more consistently above the protective threshold with weekly infusions than with biweekly or monthly therapy, although differences in clinical outcomes have not been formally evaluated. However, weekly visits to the healthcare centre may present inconvenience for patients and may result in a loss of treatment adherence.
High AAT dosage (120 mg/kg) treatment was evaluated in a pilot clinical trial. This treatment restored serum AAT levels above 25 μM.27 Likewise, the SPARTA trial is a multicentre, randomized, double-blind, placebo-controlled, phase III clinical study currently under development, evaluating two dose regimens of AAT versus placebo (60 and 120 mg/kg weekly).28 The rationale for evaluating a higher dose is that AAT levels in patients with AATD treated with the standard dosage (60 mg/kg weekly) are still below the normal range in healthy individuals.29 Similarly, adequate AAT trough concentrations may be achieved with administration of 120 mg/kg in every 2 weeks.30 The dose of 180 mg/kg in every 21 days protected only 67% of the total time, when 60 mg/dL was considered the protective threshold.31 A monthly regimen was assessed in a study of nine patients with AATD (eight Pi*ZZ and one Pi*Z null). AAT 250 mg/kg effectively increased serum AAT levels above protective limits until the next dose and in the long term. However, scientific evidence for biweekly and monthly AAT infusions is limited.3 On the contrary, according to a randomized controlled trial in which patients received AAT infusions (250 mg/kg) at 4-week intervals, mean AAT levels were below the protective threshold at day 28.32
AAT is indicated for patients with Pi*ZZ, Pi*Null or other rare genetic variants, most of whom have serum AAT levels <11 μM. AAT is not recommended for patients with serum AAT level >11 μM, those with emphysema in the absence of documented AATD, or those who are heterozygous for PiMZ or PiMS gene variants as they do not have an increased risk of emphysema in the absence of smoking.33 There is an overlap in serum AAT levels amongst different genotypes. Figure 3 shows the protective and confirmatory testing thresholds in the context of the ranges of serum AAT concentrations for each genetic variant.
Figure 3.
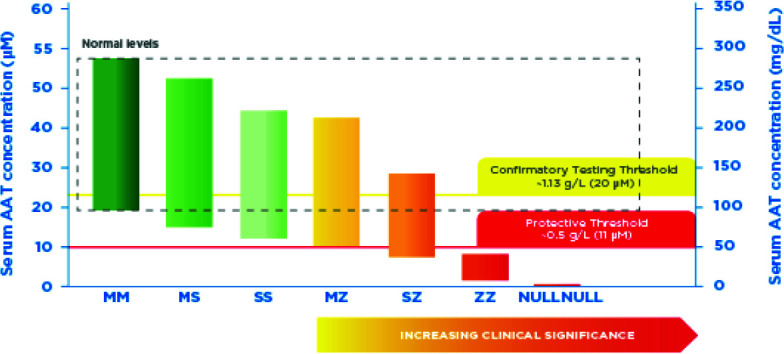
Protective and confirmatory testing thresholds in the context of the ranges of serum alpha-1 antitrypsin (AAT) concentrations for genetic variants.
Created with data taken from ref.31
Commercial preparations of plasma-derived AAT contain IgA. Patients with undetectable serum IgA levels and known antibodies to IgA may develop systemic reactions to human plasma containing IgA.34 Therefore, serum IgA levels should be measured in patients treated with AATD prior to treatment.
Assessment and monitoring of lung function in AATD
After diagnosis, patients should be monitored regularly by a multidisciplinary team comprised by hepatologists, pulmonologists, dermatologists and radiologists during the first year. Adequate monitoring should assess parameters such as forced expiratory volume in 1 second (FEV1), computed tomography densitometry and health-related quality of life.4
FEV1
FEV1 is a clinical parameter that evaluates lung function progression and severity of lung disease. The Global Initiative for Chronic Obstructive Lung Disease (GOLD) document suggests that only patients with severe AATD and a predicted FEV1 between 35% and 65% should receive augmentation therapy.9 The rationale for selecting this FEV1 range is that, in observational studies, individuals with FEV1 35–65% appear to have best response in slowing lung function decline. Pulmonary function is not routinely evaluated to assess the clinical evolution. By contrast, the American Thoracic Society suggests weekly augmentation therapy with human AAT for individuals who have serum AAT levels <11 μM and established airflow obstruction, defined as a predicted FEV1 <80%.12 Routine monitoring of serum AAT levels during therapy is not indicated. However, an initial evaluation with complete lung function testing and an annual follow-up measurement of lung function, including post-bronchodilator FEV1 and gas transfer, are recommended. This monitoring of lung function provides useful data about disease progression according to the patients’ needs.3,4
The clinical efficacy of intravenous AAT administration is demonstrated in several randomized studies that evaluated outcomes such as serum AAT levels, lung function and lung density. Long-term clinical trial data on patient outcomes, such as frequency of flare-ups, quality of life, need for lung transplantation and mortality, are more limited.25 Patients with moderate obstruction (predicted FEV1 30–65%) were more likely to benefit from AAT therapy, whereas there is a lack of evidence for benefit in patients with severe airflow obstruction (e.g. predicted FEV1 <35%) as there is no parenchyma/lung function to be preserved.3
The results of some observational studies of AAT augmentation therapy showed a beneficial effect on survival and lung function and strengthened the evidence for its clinical efficacy.35 In this context, a multicentre, prospective cohort study of 1129 patients found increased survival in patients who received augmentation therapy.36 In patients with predicted FEV1 between 35% and 49%, the rate of decline in FEV1 was also delayed amongst those receiving augmentation therapy with AAT. A systematic review including 5632 patients concluded that augmentation therapy had a slight effect in reducing the decline in lung function since the decline in FEV1 was slower (23%) (13.4 mL/year, 95% CI 1.5–25.3 mL/year) in those patients who received augmentation therapy compared with the placebo group.35 A second study compared the FEV1 decline rate amongst ex-smokers with severe AATD who received weekly infusions of AAT at 60 mg/kg or placebo for an average of 3.2 years. Overall, the rate of decline in FEV1 was significantly lower in patients treated with AAT (−53 mL/year) compared with untreated patients (−75 mL/year; P=0.02).37 A longitudinal study followed 96 patients with severe AATD and analysed the rate of decline in FEV1 before and after starting weekly augmentation therapy. This study showed that the rate of FEV1 decline during augmentation treatment was slower in patients with mild airflow obstruction.38
The effect of AAT therapy in decreasing flare-ups has been described for patients with severe COPD. In an observational study, the rate of flare-ups decreased from 3–5 infections/year before therapy to 0–1 infections/year after initiation of therapy.39 Although randomized trials have not shown an overall effect on the frequency of flare-ups, a post hoc analysis of a clinical trial found a decrease in their severity.34
CT densitometry
Computed tomography (CT) densitometry of the chest is essential for the diagnosis of emphysema caused by AATD.40 In clinical trials, sensitive measurements of lung density by high-resolution CT (HRCT) have shown a decrease in lung density over time in patients with severe AATD. In addition, some patients have evidence of emphysema on HRCT without airflow limitation on spirometry, showing that chest HRCT is more sensitive, at least initially, than functional assessment.
Augmentation therapy with AAT should be initiated in adults with clinical evidence of emphysema due to severe AATD. In the absence of more robust data to initiate AAT therapy in the view of CT changes, it is critical to follow these patients closely (e.g. every 6 months) with spirometry to identify early or progressive declines in FEV1. This would help to identify patients with a greater potential to benefit from augmentation therapy.3,4
CT densitometry is a more sensitive measure than spirometry to assess the progression of emphysema, but its role in routine monitoring requires further investigation to evaluate the future role of lung densitometry loss in the initiation of AATD treatment.4 This parameter is important during the follow-up, because patients being treated with AAT have a slower decrease in lung density.
To date, the RAPID and RAPID Open Label Extension (RAPID-OLE)41,42 studies are the largest multicentre randomized placebo-controlled trials of AAT therapy in patients with AATD. In the RAPID trial, patients were randomly assigned to receive AAT intravenously 60 mg/kg/week or placebo for 24 months.41 After 2 years of AAT treatment, a statistically significant reduction in the annual rate of CT lung density loss and the total lung capacity (TLC) was observed, whilst functional residual capacity alone or TLC plus functional residual capacity did not reach statistical significance. In the RAPID-OLE study, patients previously on placebo switched to AAT therapy for 2 years (delayed-onset group) and had a similar rate of lung density loss between months 24 and 48 as those from the early-onset group.42 Between day 1 and month 24, TLC annual decline rate was 33% greater in the delayed-onset group compared with the early-onset group. The efficacy of AAT treatment was maintained over 4 years. Between months 24 and 48, lung density loss was not recovered in the delayed-onset group. These results showed the effectiveness of early intervention with AAT to delay lung density loss. Desmosine and isodesmosine are two amino acid cross-links that are used as biomarkers of elastin degradation.43 A post hoc study of the RAPID/RAPID OLE trials showed that desmosine and isodesmosine were significantly reduced in plasma from patients treated with AAT between baseline and month 48.44
Future perspectives
Clinical practice guidelines from the Medical and Scientific Advisory Committee of the Alpha-1 Foundation recommend that all individuals with COPD, unexplained chronic liver disease, necrotizing panniculitis, granulomatosis with polyangiitis or unexplained bronchiectasis be tested for AATD. First degree relatives of patients known to have AATD should also be tested.3 Several articles that have highlighted the inadequacy of the reach of current testing have also identified primary care physicians as the potential solution to expanding the breadth of AATD testing.19,45,46
In several countries, medical associations and reference centres have reinforced the need for careful diagnosis and access to intravenous augmentation treatment in patients with severe AATD.47 However, there is an urgent need to identify new biomarkers of emphysema progression and response to therapy. It is paramount to unravel the clinically important minimum difference in the rate of lung density loss to help clinicians and researchers to better understand the therapy outcomes. The optimal AAT treatment should be personalized, with individualized selection of the therapeutic regimen, according to patient needs.4,47,48
Conclusion
In summary, patients with severe AATD benefit from augmentation therapy with AAT. A decrease in FEV1 is the main point to evaluate effectiveness. Previous studies have shown benefits with specific AAT treatment, especially when lung density is assessed by HRCT. We recognize difficulties in the diagnosis and treatment of AATD. However, it is necessary that patients are adequately treated with the specific AAT treatment, which will delay the loss of lung parenchyma and an unfavourable clinical evolution. Primary care physicians can play a valuable role in the early detection of AATD and early initiation of treatment.
Acknowledgements
Zvezda Drobnic MD, Eugenio Rosado PhD, Michael James PhD, CMPP, and Jordi Bozzo PhD, CMPP (Grifols) are acknowledged for their expert revision of the text and editorial support.
Footnotes
Contributions: The named author meets the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, takes responsibility for the integrity of the work as a whole, and has given approval for this version to be published.
Disclosure and potential conflicts of interest: The author declares that he has no conflicts of interest relevant to this manuscript. The International Committee of Medical Journal Editors (ICMJE) Potential Conflicts of Interests form for the authors is available for download at: https://www.drugsincontext.com/wp-content/uploads/2023/07/dic.2023-3-1-COI.pdf
Funding declaration: Grifols, a manufacturer of plasma-derived alpha-1 antitrypsin products, granted financial and editorial support to the Sociedade Brasileira de Pneumologia e Tisiologia (Brazilian Society of Pulmonology and Phthisiology) for the preparation of this manuscript.
Correct attribution: Copyright © 2023 Feitosa PH. https://doi.org/10.7573/dic.2023-3-1. Published by Drugs in Context under Creative Commons License Deed CC BY NC ND 4.0.
Provenance: Submitted; externally peer reviewed.
Drugs in Context is published by BioExcel Publishing Ltd. Registered office: 6 Green Lane Business Park, 238 Green Lane, New Eltham, London, SE9 3TL, UK.
BioExcel Publishing Limited is registered in England Number 10038393. VAT GB 252 7720 07.
For all manuscript and submissions enquiries, contact the Editorial office editorial@drugsincontext.com
For all permissions, rights, and reprints, contact David Hughes david.hughes@bioexcelpublishing.com
References
- 1.Stoller JK, Fromer L, Brantly M, Stocks J, Strange C. Primary care diagnosis of alpha-1 antitrypsin deficiency: issues and opportunities. Cleve Clin J Med. 2007;74(12):869–874. doi: 10.3949/ccjm.74.12.869. [DOI] [PubMed] [Google Scholar]
- 2.Stoller JK, Aboussouan LS. A review of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2012;185(3):246–259. doi: 10.1164/rccm.201108-1428CI. [DOI] [PubMed] [Google Scholar]
- 3.Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis. 2016;3(3):668–682. doi: 10.15326/jcopdf.3.3.2015.0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α(1)-antitrypsin deficiency. Eur Respir J. 2017;50(5):1700610. doi: 10.1183/13993003.00610-2017. [DOI] [PubMed] [Google Scholar]
- 5.Renoux C, Odou MF, Tosato G, et al. Description of 22 new alpha-1 antitrypsin genetic variants. Orphanet J Rare Dis. 2018;13(1):161. doi: 10.1186/s13023-018-0897-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seixas S, Marques PI. Known mutations at the cause of alpha-1 antitrypsin deficiency an updated overview of SERPINA1 variation spectrum. Appl Clin Genet. 2021;14:173–194. doi: 10.2147/tacg.S257511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silva D, Oliveira MJ, Guimaraes M, Lima R, Gomes S, Seixas S. Alpha-1-antitrypsin (SERPINA1) mutation spectrum: three novel variants and haplotype characterization of rare deficiency alleles identified in Portugal. Respir Med. 2016;116:8–18. doi: 10.1016/j.rmed.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Presotto MA, Veith M, Trinkmann F, et al. Clinical characterization of a novel alpha1-antitrypsin null variant: PiQ0Heidelberg. Respir Med Case Rep. 2022;35:101570. doi: 10.1016/j.rmcr.2021.101570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orphanet Report Series – Prevalence of rare diseases: bibliographic data – Number 1. Jan, 2022. https://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf .
- 10.Barjaktarevic I, Campos M. Management of lung disease in alpha-1 antitrypsin deficiency: what we do and what we do not know. Ther Adv Chronic Dis. 2021;12(Suppl):20406223211010172. doi: 10.1177/20406223211010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laurell CB, Eriksson S. The electrophoretic α1-globulin pattern of serum in α1-antitrypsin deficiency. 1963. COPD. 2013;10(Suppl 1):3–8. doi: 10.3109/15412555.2013.771956. [DOI] [PubMed] [Google Scholar]
- 12.American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168(7):818–900. doi: 10.1164/rccm.168.7.818. [DOI] [PubMed] [Google Scholar]
- 13.Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med. 1976;294(24):1316–1321. doi: 10.1056/nejm197606102942404. [DOI] [PubMed] [Google Scholar]
- 14.Hamesch K, Mandorfer M, Pereira VM, et al. Liver fibrosis and metabolic alterations in adults with alpha-1-antitrypsin deficiency caused by the Pi*ZZ mutation. Gastroenterology. 2019;157(3):705–719e18. doi: 10.1053/j.gastro.2019.05.013. [DOI] [PubMed] [Google Scholar]
- 15.Fregonese L, Stolk J, Frants RR, Veldhuisen B. Alpha-1 antitrypsin Null mutations and severity of emphysema. Respir Med. 2008;102(6):876–884. doi: 10.1016/j.rmed.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 16.Blanco I, Bueno P, Diego I, et al. Alpha-1 antitrypsin Pi*Z gene frequency and Pi*ZZ genotype numbers worldwide: an update. Int J Chron Obstruct Pulmon Dis. 2017;12:561–569. doi: 10.2147/copd.S125389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jardim JR, Casas-Maldonado F, Fernandes FLA, Castellano M, Torres-Durán M, Miravitlles M. Update on and future perspectives for the diagnosis of alpha-1 antitrypsin deficiency in Brazil. J Bras Pneumol. 2021;47(3):e20200380. doi: 10.36416/1806-3756/e20200380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brantly M, Campos M, Davis AM, et al. Detection of alpha-1 antitrypsin deficiency: the past, present and future. Orphanet J Rare Dis. 2020;15(1):96. doi: 10.1186/s13023-020-01352-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lascano JE, Campos MA. The important role of primary care providers in the detection of alpha-1 antitrypsin deficiency. Postgrad Med. 2017;129(8):889–895. doi: 10.1080/00325481.2017.1381539. [DOI] [PubMed] [Google Scholar]
- 20.Carpenter MJ, Strange C, Jones Y, et al. Does genetic testing result in behavioral health change? Changes in smoking behavior following testing for alpha-1 antitrypsin deficiency. Ann Behav Med. 2007;33(1):22–28. doi: 10.1207/s15324796abm3301_3. [DOI] [PubMed] [Google Scholar]
- 21.Molloy K, Hersh CP, Morris VB, et al. Clarification of the risk of chronic obstructive pulmonary disease in α1-antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med. 2014;189(4):419–427. doi: 10.1164/rccm.201311-1984OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taggart C, Cervantes-Laurean D, Kim G, et al. Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activity. J Biol Chem. 2000;275(35):27258–27265. doi: 10.1074/jbc.M004850200. [DOI] [PubMed] [Google Scholar]
- 23.Wewers MD, Casolaro MA, Sellers SE, et al. Replacement therapy for alpha 1-antitrypsin deficiency associated with emphysema. N Engl J Med. 1987;316(17):1055–1062. doi: 10.1056/nejm198704233161704. [DOI] [PubMed] [Google Scholar]
- 24.Franciosi AN, Alkhunaizi MA, Woodsmith A, et al. Alpha-1 antitrypsin deficiency and tobacco smoking: exploring risk factors and smoking cessation in a registry population. COPD. 2021;18(1):76–82. doi: 10.1080/15412555.2020.1864725. [DOI] [PubMed] [Google Scholar]
- 25.Miravitlles M, Nuñez A, Torres-Durán M, et al. The importance of reference centers and registries for rare diseases: the example of alpha-1 antitrypsin deficiency. COPD. 2020;17(4):346–354. doi: 10.1080/15412555.2020.1795824. [DOI] [PubMed] [Google Scholar]
- 26.Castellano M, Feitosa PH. How are we in Brazil with the treatment of alpha-1 antitrypsin deficiency? J Bras Pneumol. 2022;48(3):e20210045. doi: 10.36416/1806-3756/e20210045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campos MA, Geraghty P, Holt G, et al. The biological effects of double-dose alpha-1 antitrypsin augmentation therapy. A pilot clinical trial. Am J Respir Crit Care Med. 2019;200(3):318–326. doi: 10.1164/rccm.201901-0010OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sorrells S, Camprubi S, Griffin R, Chen J, Ayguasanosa J. SPARTA clinical trial design: exploring the efficacy and safety of two dose regimens of alpha1-proteinase inhibitor augmentation therapy in alpha1-antitrypsin deficiency. Respir Med. 2015;109(4):490–499. doi: 10.1016/j.rmed.2015.01.022. [DOI] [PubMed] [Google Scholar]
- 29.McElvaney NG, Stoller JK, Buist AS, et al. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of alpha 1-antitrypsin deficiency. Alpha 1-Antitrypsin Deficiency Registry Study Group. Chest. 1997;111(2):394–403. doi: 10.1378/chest.111.2.394. [DOI] [PubMed] [Google Scholar]
- 30.Soy D, de la Roza C, Lara B, Esquinas C, Torres A, Miravitlles M. Alpha-1-antitrypsin deficiency: optimal therapeutic regimen based on population pharmacokinetics. Thorax. 2006;61(12):1059–1064. doi: 10.1136/thx.2005.057943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vidal Pla R, Padullés Zamora N, Sala Piñol F, Jardí Margaleff R, Rodríguez Frías F, Montoro Ronsano JB. Farmacocinética de la alfa-1-antitripsina utilizada en el tratamiento sustitutivo del enfisema congénito grave . [Pharmacokinetics of alpha1-antitrypsin replacement therapy in severe congenital emphysema]. Arch Bronconeumol. 2006;42(10):553–556. doi: 10.1016/S1579-2129(06)60583-1. [DOI] [PubMed] [Google Scholar]
- 32.Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of alpha(1)-antitrypsin augmentation therapy. Am J Respir Crit Care Med. 1999;160(5 Pt 1):1468–1472. doi: 10.1164/ajrccm.160.5.9901055. [DOI] [PubMed] [Google Scholar]
- 33.McElvaney GN, Sandhaus RA, Miravitlles M, et al. Clinical considerations in individuals with α(1)-antitrypsin PI*SZ genotype. Eur Respir J. 2020;55(6):1902410. doi: 10.1183/13993003.02410-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barros-Tizón JC, Torres ML, Blanco I, Martínez MT. Reduction of severe exacerbations and hospitalization-derived costs in alpha-1-antitrypsin-deficient patients treated with alpha-1-antitrypsin augmentation therapy. Ther Adv Respir Dis. 2012;6(2):67–78. doi: 10.1177/1753465812438387. [DOI] [PubMed] [Google Scholar]
- 35.Edgar RG, Patel M, Bayliss S, Crossley D, Sapey E, Turner AM. Treatment of lung disease in alpha-1 antitrypsin deficiency: a systematic review. Int J Chron Obstruct Pulmon Dis. 2017;12:1295–1308. doi: 10.2147/copd.S130440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.The Alpha 1-Antitrypsin Deficiency Registry Study Group. A registry of patients with severe deficiency of alpha 1-antitrypsin. Design and methods. Chest. 1994;106(4):1223–1232. doi: 10.1378/chest.106.4.1223. [DOI] [PubMed] [Google Scholar]
- 37.Seersholm N, Wencker M, Banik N, et al. Does alpha1-antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary alpha1-antitrypsin deficiency? Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WATL) alpha1-AT study group. Eur Respir J. 1997;10(10):2260–2263. doi: 10.1183/09031936.97.10102260. [DOI] [PubMed] [Google Scholar]
- 38.Ficker J, Chapman KR, Turner A, et al. Alpha-1 antitrypsin (A1-PI) treatment slows emphysema progression independent of baseline FEV1. Eur Respir J. 2017;50(Suppl 61):OA3416. doi: 10.1183/1393003.congress-2017.OA3416. [DOI] [Google Scholar]
- 39.Lieberman J. Augmentation therapy reduces frequency of lung infections in antitrypsin deficiency: a new hypothesis with supporting data. Chest. 2000;118(5):1480–1485. doi: 10.1378/chest.118.5.1480. [DOI] [PubMed] [Google Scholar]
- 40.Campos MA, Diaz AA. The role of computed tomography for the evaluation of lung disease in alpha-1 antitrypsin deficiency. Chest. 2018;153(5):1240–1248. doi: 10.1016/j.chest.2017.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chapman KR, Burdon JG, Piitulainen E, et al. Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386(9991):360–368. doi: 10.1016/s0140-6736(15)60860-1. [DOI] [PubMed] [Google Scholar]
- 42.McElvaney NG, Burdon J, Holmes M, et al. Long-term efficacy and safety of α1 proteinase inhibitor treatment for emphysema caused by severe α1 antitrypsin deficiency: an open-label extension trial (RAPID-OLE) Lancet Respir Med. 2017;5(1):51–60. doi: 10.1016/s2213-2600(16)30430-1. [DOI] [PubMed] [Google Scholar]
- 43.Ma S, Lin YY, He J, Rouhani FN, Brantly M, Turino GM. Alpha-1 antitrypsin augmentation therapy and biomarkers of elastin degradation. COPD. 2013;10(4):473–481. doi: 10.3109/15412555.2013.771163. [DOI] [PubMed] [Google Scholar]
- 44.Ma S, Lin YY, Cantor JO, et al. The effect of alpha-1 proteinase inhibitor on biomarkers of elastin degradation in alpha-1 antitrypsin deficiency: an analysis of the RAPID/RAPID extension trials. Chronic Obstr Pulm Dis. 2016;4(1):34–44. doi: 10.15326/jcopdf.4.1.2016.0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barrecheguren M, Monteagudo M, Llor C, et al. Diagnosis of alpha 1 antitrypsin deficiency in primary care. Eur Respir J. 2015;46:PA3866. doi: 10.1183/13993003.congress-2015.PA3866. [DOI] [Google Scholar]
- 46.Molina J, Flor X, García R, Timiraos R, Tirado-Conde G, Miravitlles M. The IDDEA project: a strategy for the detection of alpha-1 antitrypsin deficiency in COPD patients in the primary care setting. Ther Adv Respir Dis. 2011;5(4):237–243. doi: 10.1177/1753465811404919. [DOI] [PubMed] [Google Scholar]
- 47.Brantly ML, Lascano JE, Shahmohammadi A. Intravenous alpha-1 antitrypsin therapy for alpha-1 antitrypsin deficiency: the current state of the evidence. Chronic Obstr Pulm Dis. 2018;6(1):100–114. doi: 10.15326/jcopdf.6.1.2017.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stockley RA. Alpha-1 antitrypsin deficiency: the learning goes on. Am J Respir Crit Care Med. 2020;202(1):6–7. doi: 10.1164/rccm.202004-0922ED. [DOI] [PMC free article] [PubMed] [Google Scholar]