

Abstract
Molecules with bioactivity towards G protein-coupled receptors represent a subset of the vast space of small drug-like molecules. Here, we compare machine learning models, including dilated graph convolutional networks, that conduct binary classification to quickly identify molecules with activity towards G protein-coupled receptors. The models are trained and validated using a large set of over 600,000 active, inactive, and decoy compounds. The best performing machine learning model, dubbed GPCRLigNet, was a surprisingly simple feedforward dense neural network mapping from Morgan fingerprints to activity. Incorporation of GPCRLigNet into a high-throughput virtual screening workflow is demonstrated with molecular docking towards a particular G protein-coupled receptor, the pituitary adenylate cyclase-activating polypeptide receptor type 1. Through rigorous comparison of docking scores for molecules selected with and without using GPCRLigNet, we demonstrate an enrichment of potentially potent molecules using GPCRLigNet. This work provides a proof of principle that GPCRLigNet can effectively hone the chemical search space towards ligands with G protein-coupled receptor activity.
Introduction.
Numerous small-molecule drugs target a superfamily of membrane-bound proteins called G protein-coupled receptors (GPCRs).1 The prevalence of GPCRs as therapeutic targets is a direct result of their ubiquity in a variety of biology processes. Upon agonist binding, GPCRs are capable of transducing signals across a biological membrane thereby acting as hubs for communicating information between and within cells. Many structural features are conserved among GPCRs including the presence of seven membrane (7TM) spanning alpha helices each with approximately 30 amino acids (Figure 1A). The conserved 7TM fold has inspired 3D receptor pharmacophore models that span entire GPCR families2,3. These methods rely at least partially on the structural resemblance of GPCR orthosteric binding pockets which results from the 7TM fold. From the perspective that GPCR homology gives rise to chemical similarity among GPCR active molecules (i.e., molecules who can agonize, antagonize, or otherwise affect signaling at a GPCR), it is reasonable to posit the existence of a subset in chemical space containing molecules active towards GPCRs (Figure 1B). Identification of this subset would expedite the drug development pipeline when targeting GPCRs4 by honing down the search space. Towards this goal, here we develop a machine learning model to classify GPCR active molecules from known chemical databases (Figure 1C) and demonstrate its utility in high-throughput virtual screening (HTVS) methodologies.
Figure 1.
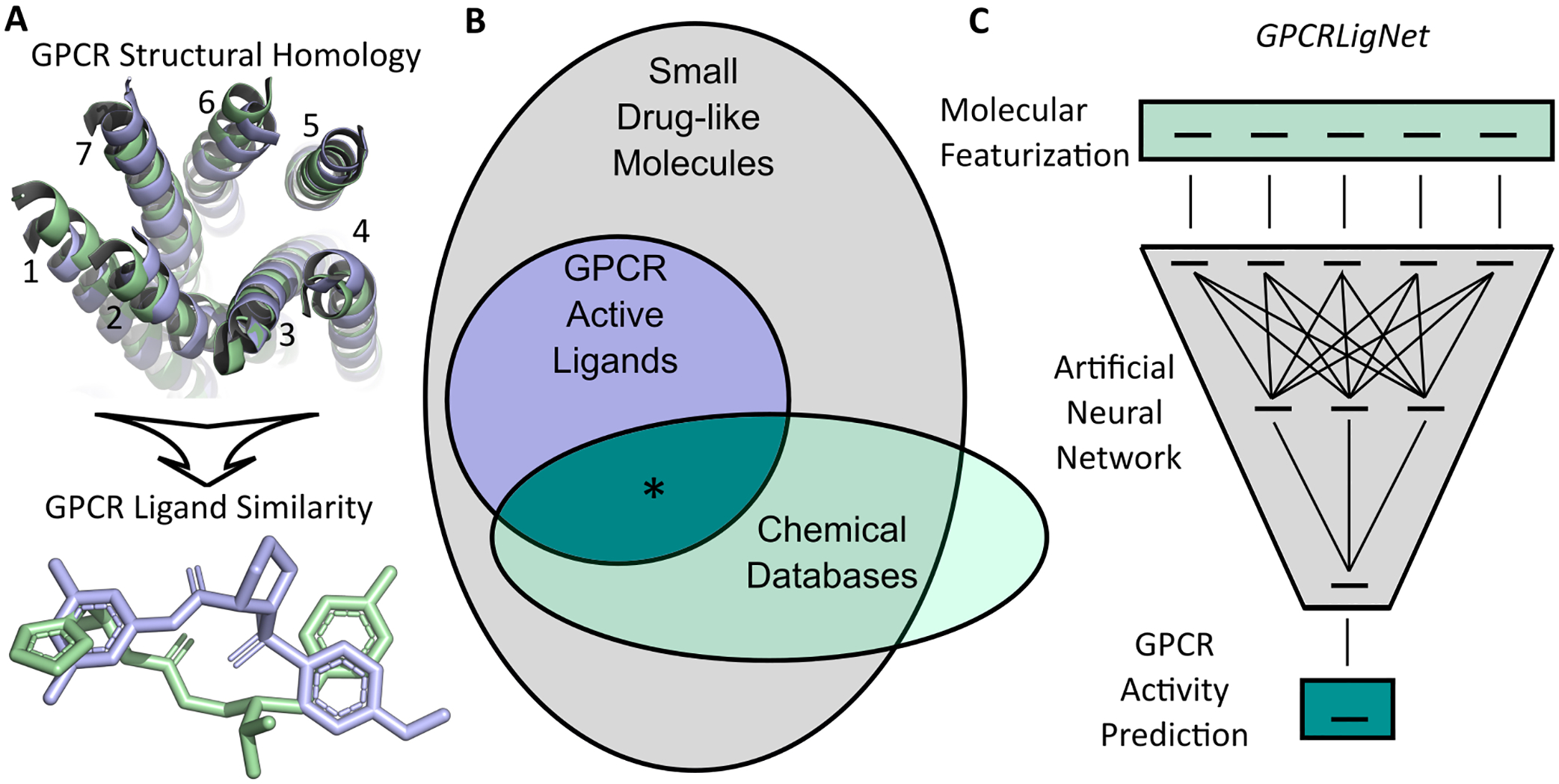
Example of conserved seven transmembrane helices of GPCRs (numbered) with active ligands (PDBIDs: 7SBF and 4UHR green and blue) in A. Diagram of GPCR active molecules as a subset of possible small drug-like molecules B. Identification of GPCR active molecules from known chemical databases (region denoted with *) will aid GPCR drug discovery pipelines. GPCRLigNet uses machine learning to classify GPCR active molecules from their molecular featurization C.
Determining a molecule’s potential activity towards a GPCR based on molecular properties is a challenging but promising task. Accordingly, the use of neural networks for this binary classification problem dates back to at least 20025–12 while modern machine learning approaches have been reviewed more recently.13 These reports generally show the ability to achieve GPCR drug classification with an accuracy of approximately 90%. However, these previous models vary in dataset size and degrees of validation, often stopping short of demonstrating their use at later stages of a drug-development pipeline. Here, we develop an additional machine learning tool, GPCRLigNet, for this task, and trained it using a large dataset of GPCR active and inactive molecules obtained from the GPCR-Ligand Association (GLASS) database14 and for the first time supplemented with decoy molecules from the Database of Useful Decoys: Enhanced (DUD-E).15 GPCRLigNet is designed to guide GPCR drug discovery projects through chemical space towards molecules with activity towards GPCRs. While GPCRLigNet lacks GPCR specificity and mode of activity, we demonstrate its ability to filter out chemicals that have a low chance of modulating any GPCR and thus reduce the load on more time and cost-consuming methodologies that enable GPCR specificity. Finally, we include GPCRLigNet in a HTVS workflow for a novel GPCR target of stress and pain, the human pituitary adenylate cyclase-activating polypeptide receptor type 1 (PAC1R). Our work revealed a robust enhancement of molecules with strong predicted binding affinity.
Methods.
Curation of the Dataset for GPCR Active and Inactive Compounds.
Molecules active and inactive towards GPCRs were first selected from the GLASS database.14 The full dataset was obtained and the InChI keys and activities of molecules were selected based on having IC50, Ki, or EC50 values with nM units. InChI keys were converted into smiles strings using cactus web server.16 RDKit was used to convert the SMILES strings into a molecular data structure with hydrogens added.17 These molecules were then filtered based on the following criteria: (1) chemical formula containing H, C, N, O, P, S, Cl, F, Br, Na, or K only, (2) fewer than 80 total atoms, and (3) formal charge of −1, 0, or 1. The scalar activity values for each molecule were then converted to active and inactive using a 2D one-hot encoding where the 1st digit is 1 when the activity is better than 1 μM (and vice-versa for the 2nd digit). This resulted in 423,166 active molecules, and 120,309 inactive molecules. Decoy molecules towards GPCRs were obtained from the DUD-E database to increase the number of inactive molecules by 79,262. These molecules were put through the same selection criteria as the compounds from GLASS and the activity encoding was set to inactive for all molecules. This increased the final number of inactive molecules to 199,571 for a total dataset size of 622,737. Our 1 μM activity cut-off was chosen because it represented a practical milestone for HTVS. A histogram of GLASS compound activities (Figure S3) showed how changes in the activity cut-off affect the relative numbers of active and inactive molecules.
Each of the molecules in the dataset was converted into molecular fingerprints using RDKit and a molecular graph representation (MGR) using custom python scripts and the NumPy library. The fingerprints and bit vector sizes in parenthesis used in this study were the default RDKFingerprints (2048), MACCs keys (167),18 circular fingerprints denoted circular-l for lengths l = 4, 6, or 8 (each of size 1024),19 and circular fingerprints with additional features (using the flag useFeatures=True set in RDKit to include hydrogen bond donors, acceptors, aromatic, halogen, basic, and acidic features) of length 4, 6, or 8 (each of size 1024). The MGR included the molecule’s rescaled adjacency matrix, A, defined in Eq. 1.
| Eq. 1 |
In Eq. 1, is the matrix with entry i, j = 1 if atoms i and j are bonded and is the diagonal matrix with the number of atoms bonded to atom i on the diagonal. The 17 atomic features, , used in the MGR were one-hot encodings of atomic element and formal charge along with an aromaticity flag. While bond information was not directly included, the use of explicit hydrogens and formal charges provided the network with enough information (based on Valance Bond Theory) to derive relevant molecular features.
Architecture of the Machine Learning Approaches.
Machine learning models to map from either traditional fingerprints or MGR to GPCR activity were built using the TensorFlow library20 and are outlined in Figure 2. For the traditional fingerprints, the input fingerprints were passed directly into a densely connected feedforward neural network with two hidden layers. The first layer with width equal to the floor of one half the fingerprint size and the second layer of size two. The activations used for all layers were exponential linear units. When comparing the ability of different fingerprinting methods to capture GPCR activity, models were also built using the same number of learnable parameters by first applying a random, non-trainable, linear map to 2048-dimensional space (the largest fingerprint size). The final layer of width two was passed into a SoftMax layer and the first output channel interpreted as the probability of a molecule being active.
Figure 2.
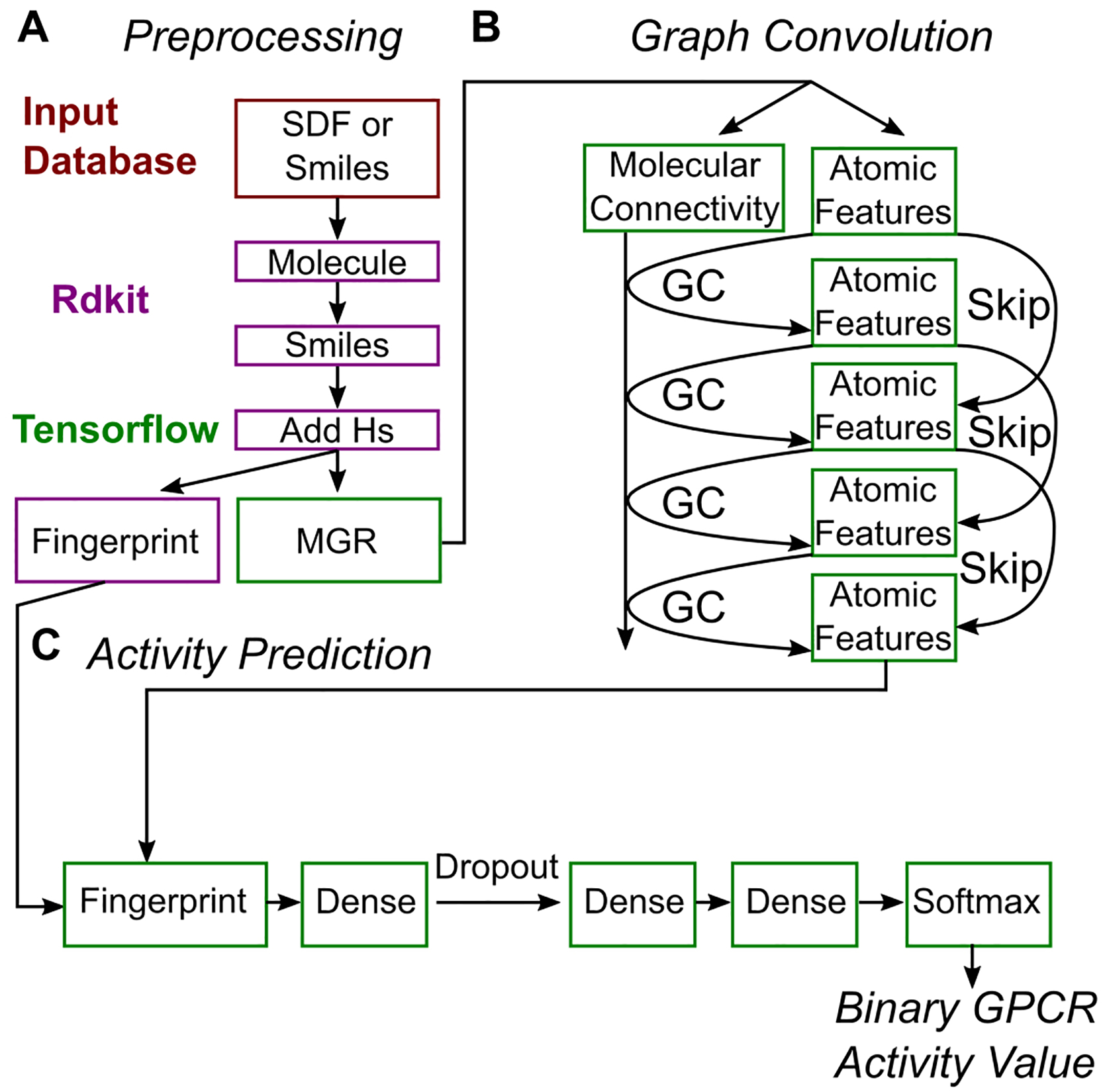
Overview of the molecule preprocessing A, graph convolutional networks B, and activity prediction networks C. Steps that use the input database (red), RDKit (purple), or TensorFlow (green) are highlighted.
The basic element of our graph convolution (GC) network models (Figure 2) were GC layers that pass information between atoms of the molecule according to rules determined by the molecules chemical bonding pattern. Following previous work,21–23 we used GC layers to apply learnable linear combinations (defined by weight matrices Wi) of adjacent atom’s atomic features (computed using the matrix multiplication AFi-1). In the initial GC layer, this result is directly passed into a non-linear activation function, T, (Eq. 2).
| Eq. 2 |
For the remaining GC layers, i, skip (or residual) connections were included that add the output of the previous GC layer before application of the activation function Eq. 3.
| Eq. 3 |
In addition, here we also implemented dilated graph convolutions by expanding the simple adjacency matrix into a dilated one. We define the dilated adjacency matrix with a dilation of d, as the matrix with the (j,k)th entry set to 1 if the shortest path between atoms j and k in the molecular graph is of length d and 0 otherwise. Thus, d=1 is the usual adjacency matrix (where non-zero entries correspond to chemical bonds), while, for d >1, a direct flow of information from distant regions of the molecule is allowed. Examples for a Chemokine receptor type 3 active molecule in the training set is shown in Figure S1. In our GCN model, dilated adjacency matrices with d = 1, 2, 4, and 6 were used in parallel to compute GCs. Four different GC layers (arranged according to Figure 2B) were applied to each of the four dilations resulting in up to 16 total GC layers. The resulting features from the last GC layer of each dilation were then concatenated and averaged over the atoms in the molecule. This later operation helped ensure that the molecular fingerprints generated by the GC layers would be invariant to permutations of the atom indexing in the MGR. Finally, the fingerprints were put through similarly designed feed forward densely connected neural networks as the traditional fingerprints which resulted in the final activity prediction.
Training the Networks.
For each of three replicates, the full dataset was randomly shuffled and split into training (70%) and validation (30%) datasets. Dropout layers (rate 25%) were applied in the dense feed forward networks to reduce overfitting to the dataset. The Adam optimizer was used with a batch size of 100 for all models. For training, a learning rate of 2×10−5 was used for all models with the remaining parameters being the default in TensorFlow 2. Early stopping was used for all training runs by halting training after the loss function, evaluated over the validation set, failed to decrease after 10 epochs (a patience of 10). The cross-entropy loss was used in all training runs where the output was GPCR activity. The hyperparameters were chosen based on a manual optimization on a subset (1/10th of the training set size) of the training data using heuristic considerations. The primary considerations used were model size (hidden widths etc..) should be increased when underfitting was apparent; dropout should be increased when overfitting was observed and decreased when stability of the training loss with respect to epochs was affected; the batch size was tuned to maximize training speed within the constraints of memory usage; and finally, the learning rate was tuned to ensure a smoother decrease in training loss.
Exploring GPCR Activity in Other Chemical Databases.
The Fragment Database (FDB-17) of 10 million small molecules with fewer than 17 atoms of C, N, O, and S was obtained and screened for GPCR activity. For all fragments, the circular-4 fingerprint was computed and passed through the activity prediction networks. The activity scores were then correlated over the different keys in the circular-4 fingerprint. Towards discovering novel PAC1R antagonists, 100,508,729 (referred to hereafter as 100 million) compounds from PubChem were downloaded and screened for GPCR activity using the same circular-4 machine learning model.
Molecular Docking to PAC1R
The receptor grid model was generated using Schrödinger’s Receptor Grid Generation on a homology model of the PAC1R created as previously reported by our group4. All ligands were prepared using Schrödinger ligprep software with the OPLS3e forcefield with all possible stereoisomers and protonation states between pH 5.0 and 9.0 generated by Epik. The center of the docking box was taken as ARG199 with a cutoff of 20 Å in each spatial dimension. Docking was carried out using Schrödinger Virtual Screening Workflow with the 1 μM level of precision.
Results and Discussion.
The ability to predict GPCR activity from the information encoded in traditional molecular fingerprints was tested by comparing machine learning models trained on a dataset of 622,737 molecules (Figure 3). The fingerprints had different dimensionalities, which meant fully connected feedforward neural networks that mapped from the fingerprints to activity would have a different number of learnable parameters. To account for this potential bias, the fingerprints were first mapped to the same size as the largest fingerprint using a random non-learnable matrix so their ability to predict GPCR activity could be compared (see Methods). After training, the final models were evaluated on a validation set which was separated from the training set. Performance of the models on the validation set were judged by receiver operating characteristic (ROC) curves and the area under the ROC curve (AUC). Overall, all fingerprints performed quite well at determining true active compounds in the region of the ROC curve where the false positive rate (FPR) was greater than 0.5 (Figure 3A). The different fingerprints diverged more significantly at the other end of the ROC curve where all the circular fingerprints were able to achieve much higher true positive rates (TPR) than the RDKit topological fingerprints (rdkfs) or the MACCS. This analysis translated into the AUC values with the circular fingerprints outperforming the RDKit topological and MACCS fingerprints which had values of [0.947, 0.959], 0.916, and 0.853, respectively (Figure 3B). Selecting a cut-off between active and inactive molecules based on the slope of the ROC curve reaching unity revealed that the best circular fingerprint of diameter four (circular-4) achieved a false negative rate (FNR = 1 - TPR) of 10.0% and a FPR of 13.0%. Both the high AUC values, and the low FNR and FPRs suggest the circular fingerprints are excellent at distinguishing GPCR active small molecules from both inactive molecules and decoys.
Figure 3.
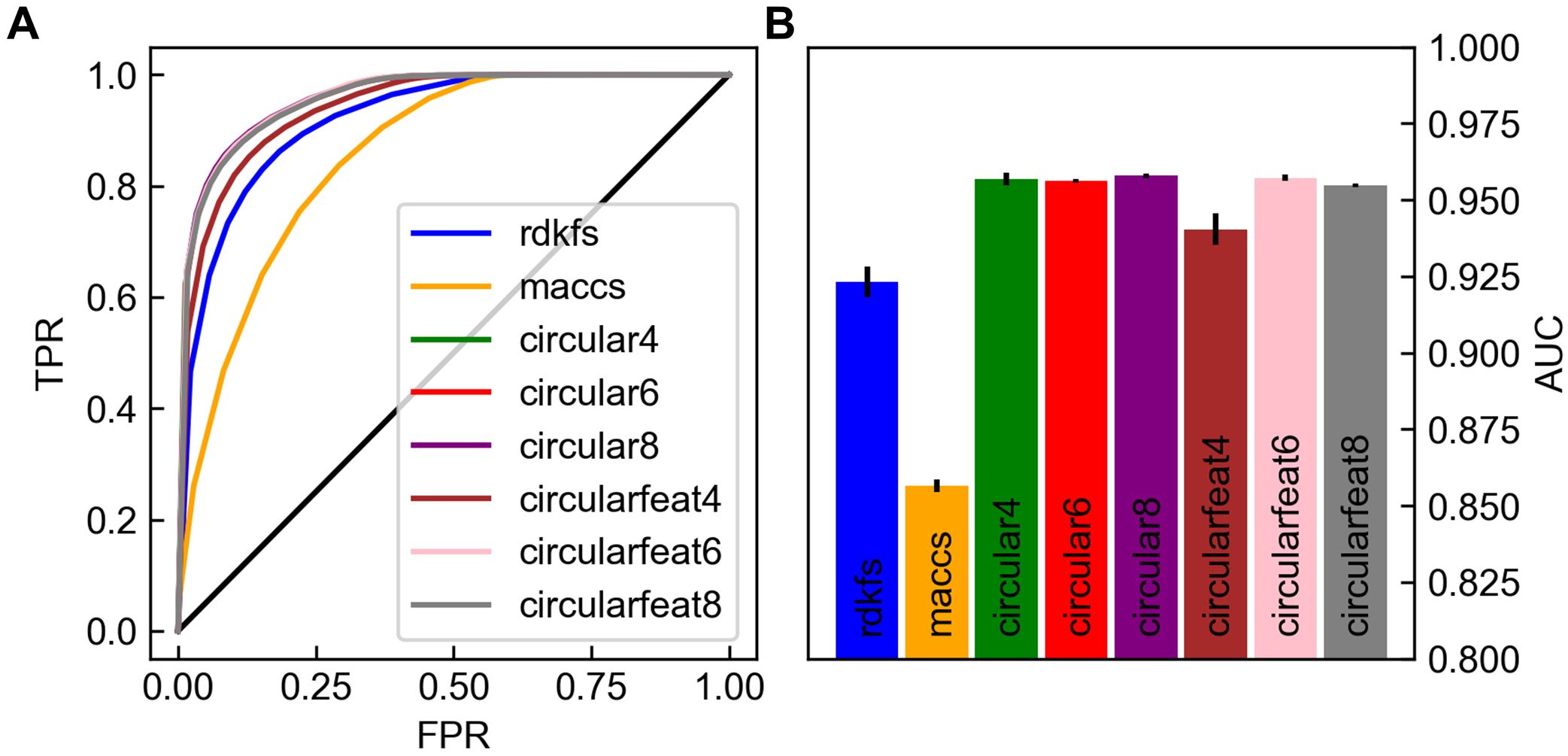
Performance of traditional fingerprints at GPCR activity classification. Receiver operating characteristic curves for different fingerprints A and corresponding area under the curve (AUC) values B were evaluated over the validation data sets. In A the false positive rate (FPR) and true positive rate (TPR) are plotted parametrically for different values of the cut-off used to distinguish active and inactive molecules from the output of the neural networks. Error bars in B are from repeating training with the dataset shuffled three different ways. These panels demonstrate that the circular fingerprints outperform other fingerprinting approaches at the task of predicting GPCR activity.
In contrast to the entries in traditional fingerprints, where molecular components are predetermined and then tallied over a given molecule, graph convolutional neural network (GCN) based fingerprints have the potential to adapt the recognized molecular features for the target task. GCNs have been proposed as a powerful method of generating molecular fingerprints23. Here, we also implemented a GCN based fingerprint for the classification of GPCR active molecules. We hypothesized that the flexibility of the GCN derived fingerprints would enhance the predictive power of the machine learning models. However, even when using skip connections, the GCNs were only able to reach an AUC of 0.901 (d = 1 in Figure S2). Convolutional neural networks can be improved through the addition of dilations that can effectively increase the receptive field of the network with fewer parameters than simply increasing the kernel size. However, analogous dilations have, to our knowledge, not been proposed for molecular GCN. To further improve our GCN model we derived an implementation of a conceptually similar idea that we term as molecular graph dilations. At the heart of our implementation of molecular graph dilations is the dilated adjacency matrix with non-zero entries when the shortest path along chemical bonds in the molecule is of length d (Figure S1). This definition has the net effect of allowing a single GCN layer to pass atom descriptors from distal regions of the molecule. Using this definition of a dilation, we were able to increase the AUC value of the GCNs to 0.928 by using four dilations concurrently up to a maximum d = 6 (Figure S2). This brought the AUC above the value of the RDKit topological fingerprints (0.916) and MACCS (0.853), but the dilated GCN model still underperformed models using Morgan fingerprints (0.959). Despite the allure of GCN fingerprints, their subpar performance here suggests difficulties in training and implementation, which may hinder their usefulness. Future work to improve GPCRLigNet could take inspiration from message passing or attentive graph neural networks.24,25
Based on the aggregate performance data and the network with the highest AUC, the model trained on the circular-4 fingerprint was best at predicting potential GPCR activity. Thus, we chose this model for further characterization and term it the GPCRLigNet. To reveal chemical groups associated with GPCR activity, we used GPCRLigNet to screen molecules from FDB-17, a large molecular fragment dataset. FDB-17 is a 10 million molecule subset of the expansive database (GDB-17) of 166.4 billion molecules which are composed of a maximum of seventeen atoms of only C, N, O, and S.26 The FDB-17 subset was curated to contain similar chemical diversity as GDB-17. Here, we use post processing of chemical fingerprints from FDB-17 to determine if certain chemical features are present in molecular fragments predicted to be active or inactive. The bits in the average fingerprint of a set of molecules is the prevalence of the bit in the dataset and can therefore be used to compare molecules predicted to be active with those predicted to be inactive. Intriguingly, fingerprint bits diverge in a scatter plot comparing the average fingerprints for the predicted active and inactive molecules (Figure 4). In Figure 4, bits near the line, y = x were not more or less prevalent in molecules predicted to be active or inactive, but bits far from y = x were either more or less prevalent in the active or inactive molecules. For instance, a hydrogen on an aromatic ring (1) was present in over 90% of active molecules and missing in 90% of inactive ones. Similarly, examples of carbon and sulfur containing aromatic groups (2–4) and more complex chemical species (5) were enhanced in the active molecules. On the other hand, quaternary amines were far more prevalent in the inactive compounds (6–9), but this rule was not exclusive as (5) also contains a quaternary amine. We note that while the prevalence of individual fingerprint bits in the FDB-17 dataset does approach 90% and may suggest they could be used directly for GPCR ligand classification, the same plot of the training data revealed no single bit with a strong deviation between inactive and active molecules. Thus, this simple analysis does not capture the complex relationship between fingerprints used by the neural networks, but it does begin to show what chemical features the neural networks determined were relevant for GPCR activity. The non-linear nature of the neural networks enables them to grab onto smaller deviations from y = x than the larger ones seen by eye in Figure 4.
Figure 4.
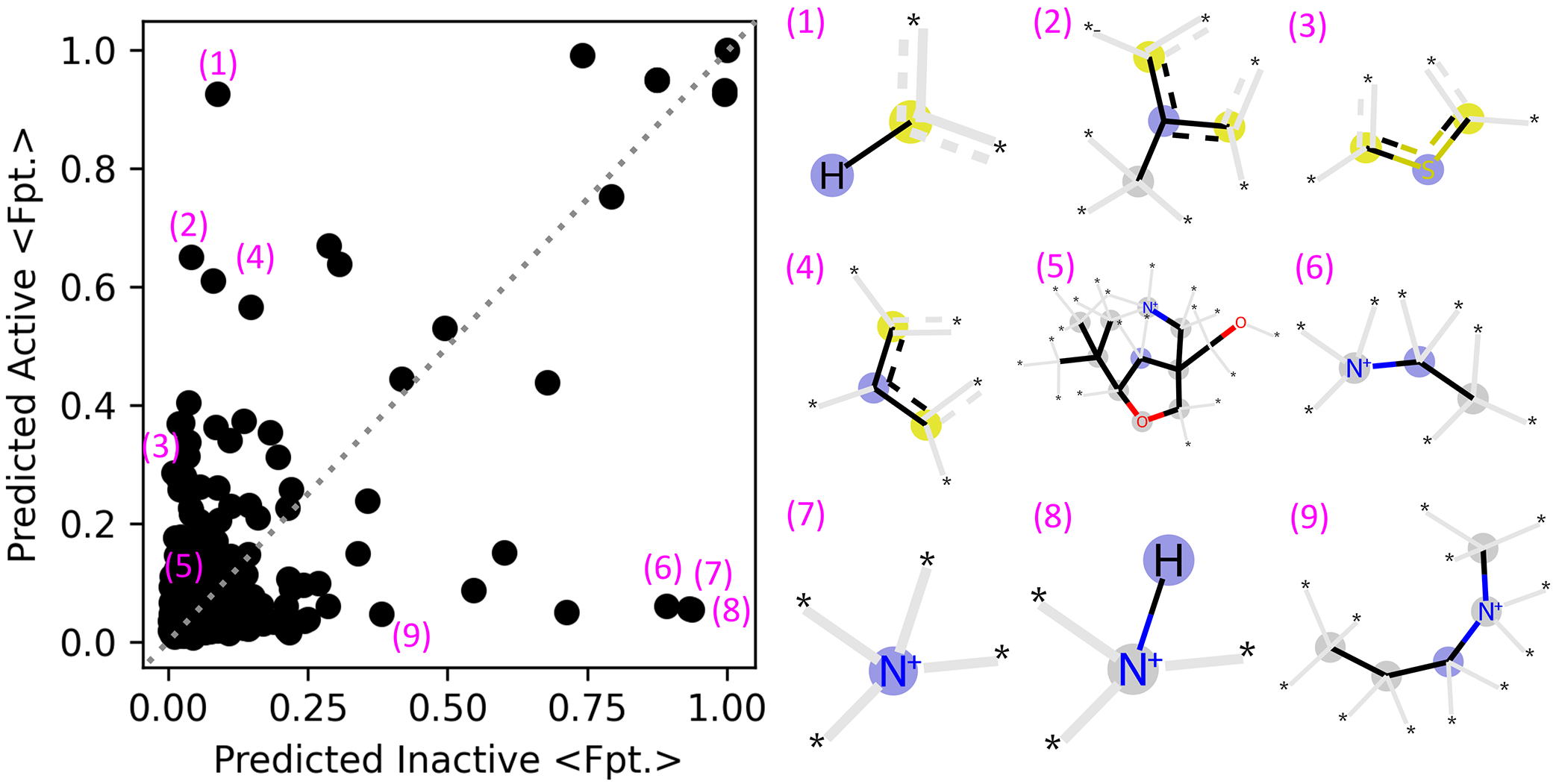
Fingerprint bit prevalence of molecules from FDB-17 whose GPCR activity was screened using the highest scoring machine learning model. The average fingerprint bits (<Fpt.>) for active and inactive molecules is plotted on the left, with select fingerprint bits shown on the right. The <Fpt.> values correspond to the fraction of molecules with the bit present and points away from y = x (grey dashed) were therefore more present in either active or inactive molecules. On the right, the blue circles are the central atoms, yellow circles are aromatic atoms, and wild-card atoms and bonds not included directly in the fingerprints are shown with an asterisk and grey bars respectively.
Additional physical and chemical properties besides activity are critical for drug discovery and here we explore relationships between GPCRLigNet’s predictions and a molecule’s absorption, distribution, metabolism, and excretion (ADME) profile. Many quick screening tools exist for filtering molecules based on desirable ADME profiles including Lipinski’s rule of five (Ro5),27 the Verber method,28 Ghose method,29 and quantitative estimate of drug-likeness(QED).30 Using GPCRLigNet we screened 1 million compounds selected randomly from PubChem to compute their continuous GPCR activity score and compared it to their drug-likeness (Figure 5). Interestingly, we found that GPCR activity anticorrelated with QED, Verber, and Lipinski (Figures 5A, B, and D). For these three measures this suggests that to optimize for oral availability and ADME, GPCRLigNet should be used in concert with the drug-like filters. On the other hand, drug-likeness as judged by Ghose, slightly correlated with GPCR activity (Figure 5C). The key physiochemical feature that differs in the Ghose filter compared with the others is the use of polarizability in the form of the molar refractivity. This finding may be related to the discovery that including polarization in GPCR/ligand binding calculations improves their accuracy.31–33
Figure 5.
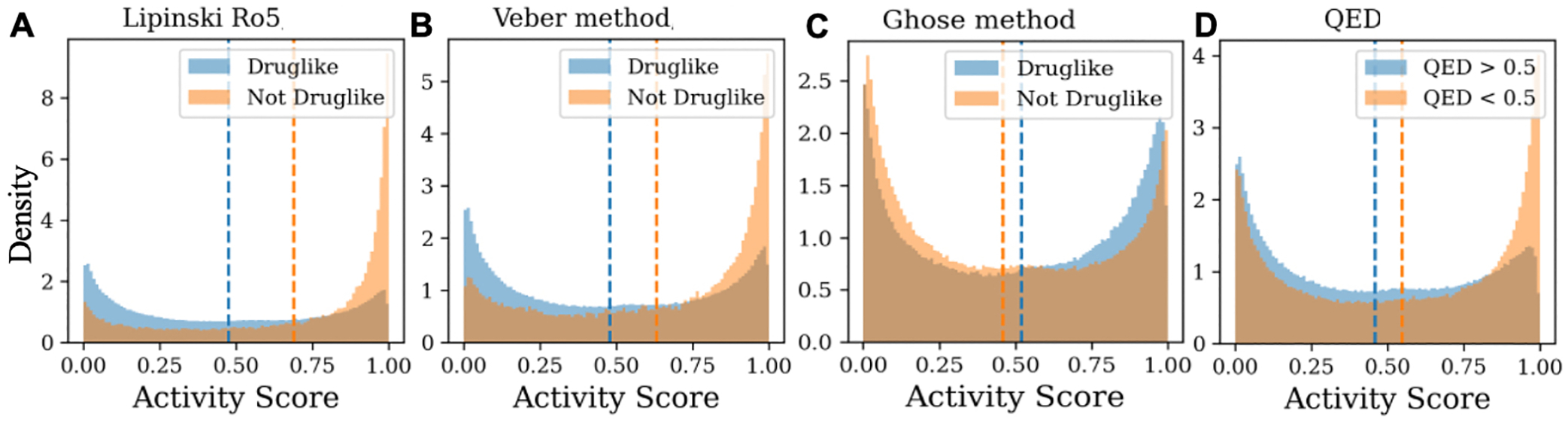
Area normalized histograms of GPCRLigNet activity score for 1 million molecules from PubChem found to be druglike (blue) and not druglike (orange) using Lipinski rule of 5 (Ro5), Verber, Ghose, and quantitative estimate of drug-likeness (QED) in A, B, C, and D respectively. For QED, a cutoff of 0.5 was used to define drug-likeness. Based on the average activity score (dashed lines) and Kolmogorov-Smirnov tests (p-values < 1e-5), QED, Verber, and Lipinski druglikeness increasingly anticorrelate with GPCR activity while Ghose slightly correlates.
To enhance the consideration of druglikeness factors in a HTVS workflow with GPCRLigNet, we propose two approaches. The simplest approach would be to apply the druglikeness filters utilized in the above analysis on post-model output. By adding rigid, computationally inexpensive druglikeness filters to screen model output, we would be ensuring that the ligands that pass these filters are both GPCR-active and druglike according to the metrics applied. The primary problem with this approach is that these quick, simple druglikeness metrics do not capture the entire space of small druglike molecules.34 Therefore, it is likely that some GPCR-active potential leads would be unnecessarily excluded by these filters. The second, more rigorous approach would be to pre-process the dataset of GPCR ligands with these same filters, and then train the model on the dataset. Though this pre-filter step may also suffer from the same unnecessary exclusion problem as the post-filtering method, this cost may be outweighed by the benefit of training the model on GPCR-active, druglike ligands. Training the model in this way would not explicitly enforce simplistic, rigid druglikeness rules, but rather would encourage the model to learn complex patterns that simultaneously optimize both GPCR activity and druglikeness. Though this pre-filtering approach is more expensive than the post-model filtering approach due to retraining, it will likely make GPCRLigNet more effective at classifying druglike, GPCR-active ligands without the need for rigid post-model filters.
The practical utility of our machine learning model, to identify GPCR active molecules, was further tested by using it to aid the search for antagonists of the PAC1R.35,36 First, the known antagonists of PAC1R37 were screened for GPCR activity and compared using a confusion matrix (Figures 6 and S4). The machine learning model correctly classified 15 known antagonists with sub-μM inhibition constants (Ki) as being active. However,17 false positives (for PAC1R) were identified. This demonstrates how this machine learning tool, when applied to a specific GPCR, may still lead to false positives as the identified molecules could be active towards a different receptor besides PAC1R.
Figure 6.
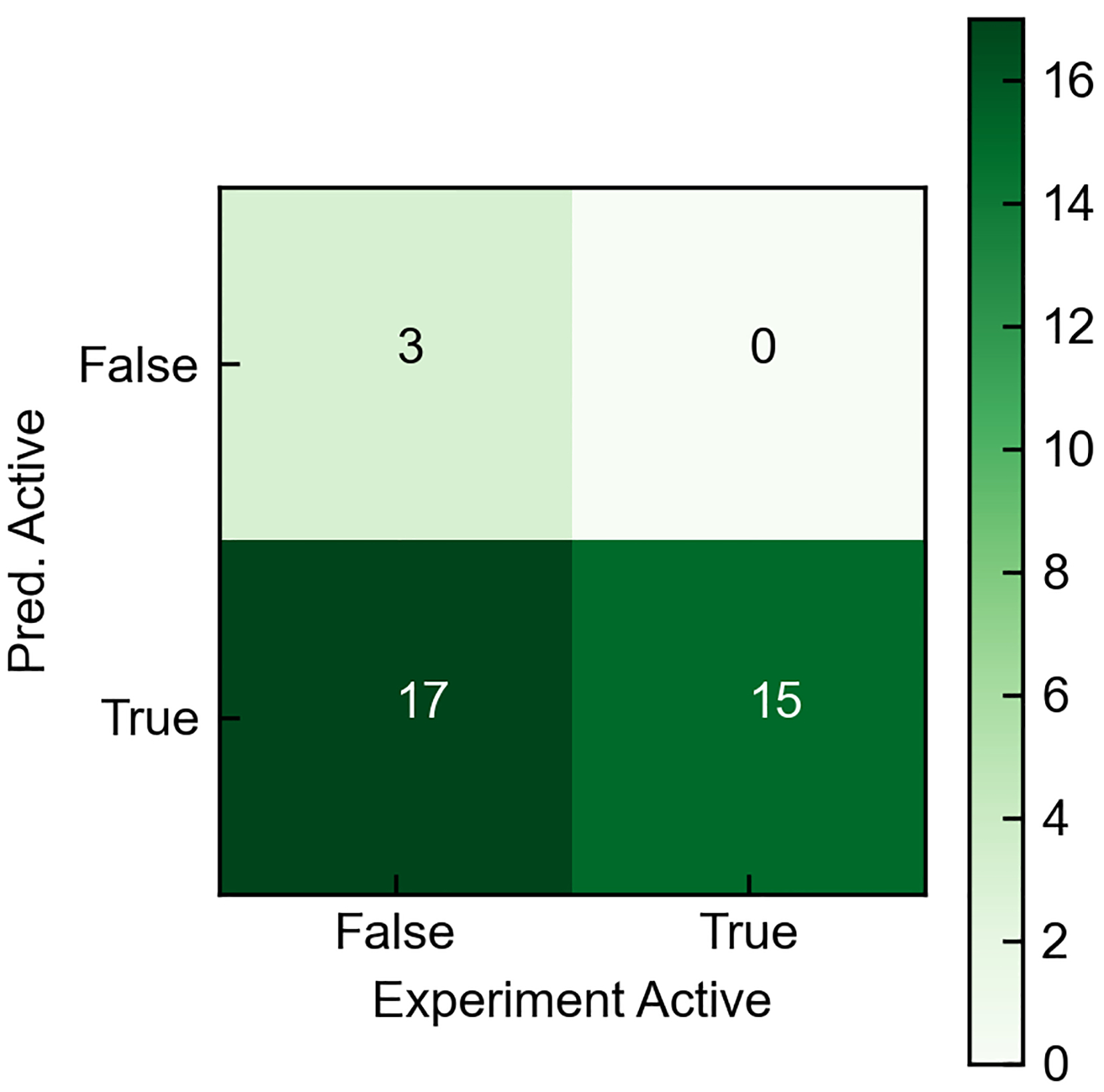
Confusion matrix for GPCR Activity prediction versus experimental values for the known antagonists of PAC1R. The molecules without exact Ki values from Ref. 34 were put in the inactive class.
GPCRLigNet can quickly eliminate GPCR inactive compounds from large chemical databases, so that further receptor specific tools, like molecular docking, can focus on finding which of the GPCR active molecules fit the given receptor. Indeed, GPCRLigNet enriched the number of molecules with strong predicted binding affinity towards PAC1R from a subset of PubChem compounds (Figure 7). For this test, we used the GPCRLigNet to screen 100 million compounds obtained from PubChem and, with an activity cutoff of 0.9, reduced them to ~10 million with predicted GPCR activity. Due to the reduced speed of docking, the docking score of only a 1 million random subset of these molecules was computed using Glide and compared with 1 million molecules selected randomly from the 100 million PubChem compounds. This test enabled a quantitative comparison of how much GPCRLigNet identifies the subset of chemical space corresponding to molecules that are GPCR active (Figure 1). GPCRLigNet achieved this task by shifting the distribution of docking scores from an average of −5.76 to −6.49 kcal/mol (Figure 7). On the low docking score tail (< −8 kcal/mol) we found an enrichment of 2.3 – 3.6 more molecules when prescreening with GPCRLigNet. On the extreme end of the best scoring molecules, GPCRLigNet found 244 more molecules with a docking score less than −11 kcal/mol and the lowest scoring molecule with a score of −13.8 kcal/mol. Notably, GPCRLigNet was incredibly rapid. We achieved a speed of ~6,000 molecules per second from download to activity prediction using a single workstation (16 core AMD Ryzen Threadripper with 2x NVIDIA 2080Ti). For comparison Glide (Schrödinger, Inc.) docking on the same workstation achieves speeds from ~0.1 to 1 molecule per second (although it depends on the number of rotatable bonds and the exhaustiveness of the search). Similar speeds and further parallelization would allow the entire known chemical universe to be screened within a practical period.
Figure 7.
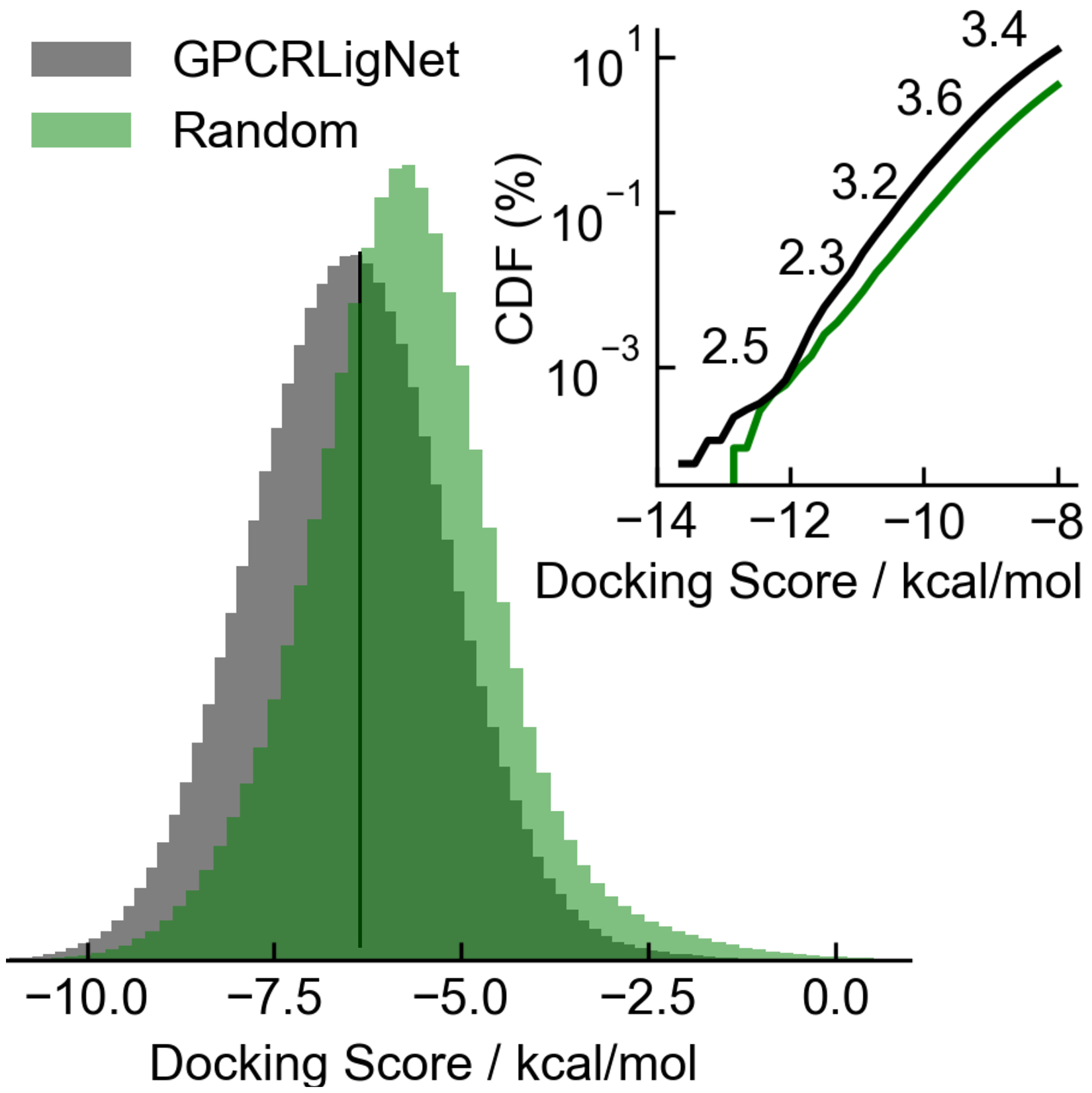
The enrichment of docking scores towards PAC1 is shown by the shift in the probability density functions of 1 million molecules screened using GPCRLigNet (black) and randomly selected (green). The inset shows the low docking score tail of the cumulative density function (CDF) with enrichment factors labeled at different docking scores.
Conclusions.
As an addition to the many computational tools available for drug discovery, we have demonstrated how a machine learning based binary classifier for molecules with GPCR activity, GPCRLigNet, can accelerate the search for potential drug candidates. We explored the use of different fingerprinting techniques and found the circular fingerprints can outperform even more exotic methods such as dilated graph convolutional networks with the model architectures used herein. The best machine learning model was able to exploit the presence and absence of chemical features to achieve very high true positive and low false positive rates. We further applied this method to the search for small molecule antagonists of PAC1R and were able to discover potentially highly potent ligands (docking score < −13 kcal/mol) a random screening approach missed and found a general enhancement of 3.6 times more molecules with scores normally considered acceptable at this stage of drug discovery (score < −10 kcal/mol). The ease of use and prediction speed of the machine learning approach enhanced the receptive field of our overall search through chemical space. As many more GPCRs besides PAC1R are valuable therapeutic targets, this tool is expected to be broadly applicable towards drug discovery applications when searching for new scaffolds or initial hit molecules.
Supplementary Material
Acknowledgements.
The work was mainly supported by an NIH grant R01-GM129431 to J.L. J.B.F. was supported by an NSF award (CHE-1945394 to J.L.). S.T.S. was supported by an NIH R35 award (R35-GM147579).
Data and Software Availability
The Python scripts for generating the training and validation datasets, building, and training machine learning models, and evaluating them are all available on GitHub https://github.com/jrem-chem/GPCRLigNet.git. In addition, the datasets themselves and indexes used for training and validation are provided along with the data and Python scripts to produce the figures in the paper.
References.
- (1).Sriram K; Insel PA G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol Pharmacol 2018, 93 (4), 251. 10.1124/mol.117.111062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kratochwil NA; Malherbe P; Lindemann L; Ebeling M; Hoener MC; Mühlemann A; Porter RHP; Stahl M; Gerber PR An Automated System for the Analysis of G Protein-Coupled Receptor Transmembrane Binding Pockets: ; Alignment, Receptor-Based Pharmacophores, and Their Application. J. Chem. Inf. Model 2005, 45 (5), 1324–1336. 10.1021/ci050221u. [DOI] [PubMed] [Google Scholar]
- (3).H. P. Porter R; Steward L; Kolczewski S; G. Panousis C; Narquizian R; Hertel C; Grether U; Dehmlow H; Winnig M; P. Slack J; A. Kratochwil N; Malherbe P; E. Martin R; Guba W; G. Green L; D. Christ A; Lindemann L; C. Hoener M; Gatti-McArthur S G Protein-Coupled Receptor Transmembrane Binding Pockets and Their Applications in GPCR Research and Drug Discovery: A Survey. Current Topics in Medicinal Chemistry 2011, 11 (15), 1902–1924. 10.2174/156802611796391267. [DOI] [PubMed] [Google Scholar]
- (4).Mckay K; Hamilton NB; Remington JM; Schneebeli ST; Li J Essential Dynamics Ensemble Docking for Structure-Based GPCR Drug Discovery. Front. Mol. Biosci https://doi.org/fmolb.2022.879212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Balakin KV; Tkachenko SE; Lang SA; Okun I; Ivashchenko AA; Savchuk NP Property-Based Design of GPCR-Targeted Library. J. Chem. Inf. Comput. Sci 2002, 42 (6), 1332–1342. 10.1021/ci025538y. [DOI] [PubMed] [Google Scholar]
- (6).Balakin KV; Lang SA; Skorenko AV; Tkachenko SE; Ivashchenko AA; Savchuk NP Structure-Based versus Property-Based Approaches in the Design of G-Protein-Coupled Receptor-Targeted Libraries. J. Chem. Inf. Comput. Sci 2003, 43 (5), 1553–1562. 10.1021/ci034114g. [DOI] [PubMed] [Google Scholar]
- (7).von Korff M; Steger M GPCR-Tailored Pharmacophore Pattern Recognition of Small Molecular Ligands. J. Chem. Inf. Comput. Sci 2004, 44 (3), 1137–1147. 10.1021/ci0303013. [DOI] [PubMed] [Google Scholar]
- (8).Lamb ML; Bradley EK; Beaton G; Bondy SS; Castellino AJ; Gibbons PA; Suto MJ; Grootenhuis PDJ Design of a Gene Family Screening Library Targeting G-Protein Coupled Receptors. Journal of Molecular Graphics and Modelling 2004, 23 (1), 15–21. 10.1016/j.jmgm.2004.03.001. [DOI] [PubMed] [Google Scholar]
- (9).Givehchi A; Schneider G Multi-Space Classification for Predicting GPCR-Ligands. Molecular Diversity 2005, 9 (4), 371–383. 10.1007/s11030-005-6293-4. [DOI] [PubMed] [Google Scholar]
- (10).Kelemen ÁA; Ferenczy GG; Keserű GM A Desirability Function-Based Scoring Scheme for Selecting Fragment-like Class A Aminergic GPCR Ligands. Journal of Computer-Aided Molecular Design 2015, 29 (1), 59–66. 10.1007/s10822-014-9804-5. [DOI] [PubMed] [Google Scholar]
- (11).van der Horst E; Okuno Y; Bender A; IJzerman AP Substructure Mining of GPCR Ligands Reveals Activity-Class Specific Functional Groups in an Unbiased Manner. J. Chem. Inf. Model 2009, 49 (2), 348–360. 10.1021/ci8003896. [DOI] [PubMed] [Google Scholar]
- (12).Seo S; Choi J; Ahn SK; Kim KW; Kim J; Choi J; Kim J; Ahn J Prediction of GPCR-Ligand Binding Using Machine Learning Algorithms. Comput Math Methods Med 2018, 2018, 6565241–6565241. 10.1155/2018/6565241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Raschka S; Kaufman B Machine Learning and AI-Based Approaches for Bioactive Ligand Discovery and GPCR-Ligand Recognition. Methods 2020, 180, 89–110. 10.1016/j.ymeth.2020.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chan WKB; Zhang H; Yang J; Brender JR; Hur J; Özgür A; Zhang Y GLASS: A Comprehensive Database for Experimentally Validated GPCR-Ligand Associations. Bioinformatics 2015, 31 (18), 3035–3042. 10.1093/bioinformatics/btv302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Mysinger MM; Carchia M; Irwin John. J.; Shoichet BK Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem 2012, 55 (14), 6582–6594. 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).NCI/CADD, group. Cactus Web Server, 2021. https://cactus.nci.nih.gov/.
- (17).RDKit: Open-Source Cheminformatics. http://www.rdkit.org.
- (18).Durant JL; Leland BA; Henry DR; Nourse JG Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci 2002, 42 (6), 1273–1280. 10.1021/ci010132r. [DOI] [PubMed] [Google Scholar]
- (19).Rogers D; Hahn M Extended-Connectivity Fingerprints. J. Chem. Inf. Model 2010, 50 (5), 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- (20).Abadi M; Barham P; Chen J; Chen Z; Davis A; Dean J; Devin M; Ghemawat S; Irving G; Isard M; Kudlur M; Levenberg J; Monga R; Moore S; Murray D; Steiner B; Tucker P; Vasudevan V; Warden P; Zhang X TensorFlow: A System for Large-Scale Machine Learning. 2016. [Google Scholar]
- (21).Kipf TN; Welling M Semi-Supervised Classification with Graph Convolutional Networks. CoRR 2016, abs/1609.02907. [Google Scholar]
- (22).Duvenaud D; Maclaurin D; Aguilera-Iparraguirre J; G\’mez-Bombarelli R; Hirzel T; Aspuru-Guzik A. ‘n; Adams RP Convolutional Networks on Graphs for Learning Molecular Fingerprints. Nips’ 15 2015, 2224–2232. [Google Scholar]
- (23).Kearnes S; McCloskey K; Berndl M; Pande V; Riley P Molecular Graph Convolutions: Moving beyond Fingerprints. Journal of computer-aided molecular design 2016, 30 (8), 595–608. 10.1007/s10822-016-9938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Xiong Z; Wang D; Liu X; Zhong F; Wan X; Li X; Li Z; Luo X; Chen K; Jiang H; Zheng M Pushing the Boundaries of Molecular Representation for Drug Discovery with the Graph Attention Mechanism. J. Med. Chem 2020, 63 (16), 8749–8760. 10.1021/acs.jmedchem.9b00959. [DOI] [PubMed] [Google Scholar]
- (25).Yang K; Swanson K; Jin W; Coley C; Eiden P; Gao H; Guzman-Perez A; Hopper T; Kelley B; Mathea M; Palmer A; Settels V; Jaakkola T; Jensen K; Barzilay R Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model 2019, 59 (8), 3370–3388. 10.1021/acs.jcim.9b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Visini R; Awale M; Reymond J-L Fragment Database FDB-17. J. Chem. Inf. Model 2017, 57 (4), 700–709. 10.1021/acs.jcim.7b00020. [DOI] [PubMed] [Google Scholar]
- (27).Lipinski CA; Lombardo F; Dominy BW; Feeney PJ Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv Drug Deliv Rev 2001, 46 (1–3), 3–26. 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- (28).Veber DF; Johnson SR; Cheng H-Y; Smith BR; Ward KW; Kopple KD Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J Med Chem 2002, 45 (12), 2615–2623. 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- (29).Ghose AK; Viswanadhan VN; Wendoloski JJ A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J Comb Chem 1999, 1 (1), 55–68. 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- (30).Bickerton GR; Paolini GV; Besnard J; Muresan S; Hopkins AL Quantifying the Chemical Beauty of Drugs. Nature Chemistry 2012, 4 (2), 90–98. 10.1038/nchem.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Heifetz A; Chudyk EI; Gleave L; Aldeghi M; Cherezov V; Fedorov DG; Biggin PC; Bodkin MJ The Fragment Molecular Orbital Method Reveals New Insight into the Chemical Nature of GPCR–Ligand Interactions. J. Chem. Inf. Model 2016, 56 (1), 159–172. 10.1021/acs.jcim.5b00644. [DOI] [PubMed] [Google Scholar]
- (32).Morao I; Fedorov DG; Robinson R; Southey M; Townsend-Nicholson A; Bodkin MJ; Heifetz A Rapid and Accurate Assessment of GPCR–Ligand Interactions Using the Fragment Molecular Orbital-Based Density-Functional Tight-Binding Method. Journal of Computational Chemistry 2017, 38 (23), 1987–1990. 10.1002/jcc.24850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Chudyk EI; Sarrat L; Aldeghi M; Fedorov DG; Bodkin MJ; James T; Southey M; Robinson R; Morao I; Heifetz A Exploring GPCR-Ligand Interactions with the Fragment Molecular Orbital (FMO) Method. In Computational Methods for GPCR Drug Discovery; Heifetz A, Ed.; Springer New York: New York, NY, 2018; pp 179–195. 10.1007/978-1-4939-7465-8_8. [DOI] [PubMed] [Google Scholar]
- (34).Vistoli G; Pedretti A; Testa B Assessing Drug-Likeness--What Are We Missing? Drug Discov Today 2008, 13 (7–8), 285–294. 10.1016/j.drudis.2007.11.007. [DOI] [PubMed] [Google Scholar]
- (35).Liao C; de Molliens MP; Schneebeli ST; Brewer M; Song G; Chatenet D; Braas KM; May V; Li J Targeting the PAC1 Receptor for Neurological and Metabolic Disorders. Curr Top Med Chem 2019, 19 (16), 1399–1417. 10.2174/1568026619666190709092647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Liao C; Remington JM; May V; Li J Molecular Basis of Class B GPCR Selectivity for the Neuropeptides PACAP and VIP. Frontiers in Molecular Biosciences 2021, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Beebe X; Darczak D; Davis-Taber RA; Uchic ME; Scott VE; Jarvis MF; Stewart AO Discovery and SAR of Hydrazide Antagonists of the Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) Receptor Type 1 (PAC1-R). Bioorganic & Medicinal Chemistry Letters 2008, 18 (6), 2162–2166. 10.1016/j.bmcl.2008.01.052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Python scripts for generating the training and validation datasets, building, and training machine learning models, and evaluating them are all available on GitHub https://github.com/jrem-chem/GPCRLigNet.git. In addition, the datasets themselves and indexes used for training and validation are provided along with the data and Python scripts to produce the figures in the paper.