

Abstract
Background
Obstructive sleep apnea (OSA) is an independent risk factor for the development of hypertension. We have demonstrated that OSA induces gut dysbiosis, and this dysbiotic microbiota contributes to hypertension. However, the mechanisms linking gut dysbiosis to blood pressure regulation remain unclear. Recent studies demonstrate that gut dysbiosis can induce a proinflammatory response of the host resulting in peripheral and neuroinflammation, key factors in the development of hypertension. We hypothesized that OSA induces inflammation in the gut that contributes to neuroinflammation and hypertension.
Methods and Results
OSA was induced in 8‐week‐old male rats. After 2 weeks of apneas, lymphocytes were isolated from aorta, brain, cecum, ileum, mesenteric lymph node, and spleen for flow cytometry. To examine the role of interleukin‐17a, a monoclonal antibody was administered to neutralize interleukin‐17a. Lymphocytes originating from the gut were tracked by labeling with carboxyfluorescein succinimidyl ester dye. OSA led to a significant decrease in T regulatory cells along with an increase in T helper (TH) 17 cells in the ileum, cecum, and brain. Interleukin‐17a neutralization significantly reduced blood pressure, increased T regulatory cells, and decreased TH1 cells in the ileum, cecum, and brain of OSA rats. TH1, TH2, and TH17 cells from the gut were found to migrate to the mesenteric lymph node, spleen, and brain with increased frequency in rats with OSA.
Conclusions
OSA induces a proinflammatory response in the gut and brain that involves interleukin‐17a signaling. Gut dysbiosis may serve as the trigger for gut and neuroinflammation, and treatments to prevent or reverse gut dysbiosis may prove useful in reducing neuroinflammation and hypertension.
Keywords: dysbiosis, gut microbiota, inflammation, microbiota‐gut‐brain axis, neuroinflammation
Subject Categories: Hypertension, High Blood Pressure, Animal Models of Human Disease, Basic Science Research, Inflammation
Nonstandard Abbreviations and Acronyms
- CFSE
carboxyfluorescein succinimidyl ester
- MLN
mesenteric lymph node
- nIL‐17a
neutralizing antibody targeted to interleukin‐17a
- OSA
obstructive sleep apnea
- SBP
systolic blood pressure
- SHR
spontaneously hypertensive rat
- TH
T helper cell
- TNF‐α
tumor necrosis factor alpha
- Treg
T regulatory cell
Research Perspective.
What Is New?
The development of obstructive sleep apnea‐induced hypertension involves gut dysbiosis, gut and neuroinflammation, and tracking of proinflammatory immune cells from gut to brain.
Neutralization of interleukin‐17a prevents obstructive sleep apnea‐induced hypertension.
What Question Should be Addressed Next?
What are the signals originating from the dysbiotic microbiota that initiate the proinflammatory response, and ultimately hypertension, in the host?
Obstructive sleep apnea (OSA) is characterized by repeated closure of the upper airway during sleep. These apneic events result in intermittent hypoxia, hypercapnia, and increased sympathetic activity. OSA is a significant risk factor for numerous cardiovascular diseases, including myocardial infarction, stroke, and hypertension. 1 , 2 , 3 The prevalence of OSA in patients with primary hypertension is ≈35%, and as high as ≈80% in drug‐resistant hypertension. 4 Recent evidence supports the idea that OSA‐induced inflammation underlies many of the associated cardiovascular consequences. 5 , 6 , 7 Specifically, neuroinflammation is a hallmark of OSA‐induced hypertension. 8 , 9 , 10 However, the mechanisms by which OSA induces neuroinflammation are not fully understood.
In recent years, the gut microbiota has been shown to play a major role in modulating host immunity. 11 Microbes and microbial products influence the host immune response, with some microbial signals being proinflammatory and others anti‐inflammatory. 12 Furthermore, microbial influence on host immune response is not restricted to the gut wall. Microbial signals entering the circulation can affect immune cells at distant sites. 13 , 14 Additionally, alterations to the gut microbiota (ie, gut dysbiosis) can induce inflammation in the gut, and these immune cells originating from the gut can migrate to distant tissues, including the brain. 15
We previously developed a rat model of OSA that incorporates true apneic events. 16 By implanting a balloon in the trachea, we are able to induce intermittent apneas in unanesthetized rats. We've demonstrated that this model results in arousals, intermittent hypoxia and hypercapnia, and exaggerated negative intrathoracic pressures. 16 In rats fed a normal chow diet, apneas did not significantly affect blood pressure (BP). However, in combination with a high‐fat diet, OSA resulted in significant elevations in BP within 2 weeks. 17 , 18 In addition, we and others have now demonstrated that various models of OSA (eg, true apneas and intermittent hypoxia) significantly alter the makeup of the gut microbiota. 17 , 18 , 19 , 20 , 21 , 22 , 23 To examine if OSA‐induced gut dysbiosis contributes to elevations in BP, we transplanted the microbiota from hypertensive rats with OSA to normotensive recipients, which significantly elevated the recipient BP. 17 Given the relationship between gut dysbiosis and host inflammation, and the importance of neuroinflammation in the pathogenesis of OSA‐induced hypertension, we hypothesized that OSA induces inflammation in the gut that contributes to neuroinflammation and hypertension.
The studies herein demonstrate that OSA‐induced hypertension is associated with a significant loss of anti‐inflammatory T regulatory cells (Tregs) and increased proinflammatory T helper (TH) 1 and TH17 cells in the ileum, cecum, and brain. Neuroinflammation is significant since it is an important component of OSA‐induced hypertension. Furthermore, systemic neutralization of the proinflammatory cytokine interleukin‐17a prevented OSA‐induced hypertension. Finally, through cell tracking studies, we demonstrate that OSA results in increased trafficking of TH1, TH2, and TH17 cells from Peyer's patches in the small intestine to the mesenteric lymph nodes (MLNs), spleen, and brain. Collectively, these results demonstrate that OSA induces a proinflammatory response in the gut and brain that contribute to the development of OSA‐induced hypertension.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
All animal protocols were approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine (Houston, TX) and conformed to the Guide for the Care and Use of Laboratory Animals, 8th edition, published by the National Institutes of Health. Rats were subjected to a 12 hours light (6 am–6 pm): 12 hours dark (6 pm–6 am) cycle.
OSA Rat Model
We previously developed and characterized a model of OSA involving true apneas in unanesthetized rats. 16 , 24 We have demonstrated that male rats fed a high‐fat diet and exposed to 2 weeks of apneas develop gut dysbiosis that contributes to the development of hypertension in this model. 17 After 2 weeks of high‐fat diet rats were implanted with an endotracheal obstruction device. Following 1 week of recovery, rats with OSA were exposed to 60 apneas, 10 s each, per hour for 8 hours during the sleep phase (9 am–5 pm). Sham rats underwent identical surgical procedures and device implantation, but endotracheal obstruction devices were never inflated.
Noninvasive Blood Pressure Measurements
The CODA Volume Pressure Relationship tail‐cuff system (Kent Scientific Corporation) was used to measure systolic blood pressure (SBP) in unanesthetized rats. A minimum of 10 consecutive readings, without movement artifact, were averaged for each measurement. We have demonstrated that SBP values obtained using the tail‐cuff method are comparable to direct arterial measurements made in the same rat.
Gut Microbiota Analysis
On day 14, sham or OSA cecal content was collected and stored at −80 °C. The samples were sent to the Center for Metagenomics and Microbiota Research at the Baylor College of Medicine, where 16S rRNA gene sequence libraries were generated from the V4 primer region using the Illumina MiSeq platform after extracting DNA using MO BIO PowerMag Soil Isolation Kit (MO BIO Laboratories). Reads were denoised and merged into amplicon sequence variants by DADA2 pipeline in R. Taxonomic annotations were also generated against DADA2‐formatted training FASTA files derived from SILVA138 Database. Amplicon sequence variants with identical taxonomic assignment were grouped into taxonomic bins. The results were analyzed and visualized with the Agile Toolkit for Incisive Microbial Analyses version 2 developed by the Center for Metagenomics and Microbiota Research at the Baylor College of Medicine.
Interleukin‐17a Neutralization
Beginning the day before the onset of sham or OSA, rats were administered 1 μg of purified rat monoclonal anti–interleukin‐17a immunoglobulin G (IgG) (clone 50 104, R&D Systems, No. MAB421) or isotype control purified rat monoclonal IgG2A (clone 54 447, R&D Systems, #MAB006) by intraperitoneal injection. Injections were administered every other day during the 2 weeks of sham or OSA.
Interleukin‐17a ELISA
Following 2 weeks of sham or OSA, plasma was isolated and stored at −80°C. Plasma was diluted 1:10 in sterile PBS. Plasma was incubated overnight at 4°C with anti–interleukin‐17a rat monoclonal IgG (clone 50 104, R&D Systems, No. MAB421) that was immobilized to wells in a clear round‐bottom 96‐well plate. Biotinylated anti‐rat interleukin‐17a (Rockland, No. 212‐406‐b32) was added and incubated for 2 hours at room temperature. Streptavidin‐conjugated HRP was added and incubated for 20 minutes at room temperature, and 3,3′,5,5′‐tetramethylbenzidine substrate (100 μL; Biolegend 421 501) was added, followed by stop solution. Absorbance was measured at 450 nm.
Interleukin‐10 ELISA
Following 2 weeks of sham or OSA, plasma was isolated and stored at −80 °C. Plasma was diluted 1:2 in sample diluent Z buffer (Invitrogen Rat interleukin‐10 Uncoated ELISA kit, Catalog No. 88‐50 629‐88). Pretitrated, purified anti‐rat interleukin‐10 monoclonal antibody was immobilized to wells in a clear round‐bottom 96‐well plate with overnight incubation. The following day, wells were blocked with blocking buffer provided in the kit with overnight incubation at 4 °C. Standards (recombinant rat interleukin‐10) and diluted plasma samples were added to the wells along with pretitrated, biotin‐conjugated anti‐rat interleukin‐10 and incubated for 2 hours at room temperature. After washing, streptavidin‐conjugated HRP was added and incubated for 1 hour at room temperature. 3,3′,5,5′‐tetramethylbenzidine substrate provided in the kit was added, followed by incubation at room temperature for 15 minutes. Finally, stop solution (Biolegend, No. 77316) was added to the wells. Absorbance was measured at 450 nm.
Carboxyfluorescein Succinimidyl Ester Cell Labeling
Rats were initially exposed to 2 days of sham or OSA. Following the initial 2 days of sham or OSA, rats were anesthetized and the small intestine exteriorized to visualize Peyer's patches. Cells in Peyer's patches were fluorescently labeled by microinjection with 2 μL of 25 μM carboxyfluorescein succinimidyl ester (CFSE; Biolegend, No. 423 801) per Peyer's patch. Subsequently, rats recovered from CFSE microinjections for 24 hours before undergoing an additional 2 days of sham or OSA. On day 5 of the experiment, rats were euthanized and tissues harvested for analyses of T cells positive for CFSE by flow cytometry.
Isolation of Lymphocytes and Flow Cytometry
Rats were transcardially perfused with ice‐cold PBS solution, and the brain was removed. The right hemisphere was homogenized and incubated in RPMI‐1640 (Corning) containing collagenase type 1A (1 mg/mL, Sigma, C2674) and DNase I (10 mg/mL, Roche, 10 104 159 001) for 45 minutes at 37 °C. The cell suspension was passed through a 70‐μM cell strainer and washed, and a gradient centrifugation was performed using 30% and 70% percoll (Cytiva 17 089 101) at 500g for 30 minutes. The interphase layer containing the lymphocytes was collected.
Chopped aorta was incubated in dissociation buffer containing collagenase type I‐S (1.5 mg/mL, Millipore, SCR103), collagenase type XI (115 μg/mL, Sigma, C7667), hyaluronidase type 1‐s (72.5 μg/mL, Sigma, H3506), and DNase I (10 mg/mL) for 60 minutes at 37 °C. The cell suspension was passed through a 70‐μM cell strainer and washed, and the lymphocytes were collected for downstream analysis.
For cecum and ileum, the tissues were cut into small pieces, and the epithelial layer was dissociated with EDTA buffer (2 mM) and removed using a 100‐μM cell strainer. The tissue pieces were retrieved and further digested with collagenase type IV (1 mg/mL; Gibco, 17 104‐019) and DNase I (10 mg/mL) for 45 minutes at 37 °C. Cells were filtered through a 100‐μM cell strainer, followed by 70‐ and 40‐μM cell strainers, and washed, and lymphocytes were collected for downstream analysis.
MLN and spleen were removed and processed by mechanical disruption on a 70‐μM mesh filter. Red blood cell lysis was achieved using the red blood cell lysis buffer (BioLegend, 420 302). Lymphocytes from Peyer's patches were isolated by incubating the tissue with collagenase type IV (1 mg/mL; Gibco, 17 104‐019) for 15 minutes at 37 °C and mechanical disruption through a 70‐μM filter.
Cells from all tissues were washed, stained using live/dead Fixable Near‐IR Dead Cell Stain Kit (Invitrogen, L34975) followed by blocking with anti‐rat Fc block (mouse anti‐rat CD32, clone D34‐485; BD Pharmingen, 550 271) before staining for extracellular markers. Cells were stained with anti‐CD 45 (Alexa Fluor 700, clone OX‐1; BioLegend, 202 218), anti‐CD3 (FITC, clone eBioG4.18, eBioscience, 11‐0030‐85), anti‐CD4 (V450, clone: OX‐35; BD Horizon, 561 579), anti‐CD25 (APC, clone OX39; eBioscience, 17‐0390‐82), and anti–major histocompatibility complex class II (APC, clone HS19; eBioscience, 17‐0920‐82).
For intracellular staining, T cells (1×106 cells/mL) were stimulated for 6 hours with 2 μl Cell Activation Cocktail (BioLegend, 423 303), followed by fixation and permeabilization with either True‐Nuclear Transcription Factor Buffer (Biolegend, 424 401) for Forkhead box P3 staining or Cyto‐Fast Perm/Wash Buffer (Biolegend, 750 000 135) for cytokines. Cells were stained intracellularly for Foxp3 (PE‐Cyanine 7, clone FJK16S, eBioscience, 25‐57 773‐82), interferon‐γ (Alexa Fluor 647, clone DB‐1; BioLegend, 507 810), interleukin‐17a (PE‐Cyanine 7, clone eBio1787; eBioscience, 25‐7177‐82), tumor necrosis factor‐α (TNF‐α) (PE, clone TN3‐19.12; BioLegend, 506 104), CD68 (FITC; Biorad, MCA341F), and interleukin‐4 (PE, clone OX081; BioLegend, 511 906).
Fluorescence‐minus‐one controls were used to distinguish positively stained cells for each antibody. The cells were analyzed using Attune NxT Flow Cytometer. The data were analyzed using FlowJo software. Data are presented as percentage or CD4+ or CD45+ cells.
Statistical Analysis
Sample sizes were estimated a priori using SDs of SBP data and powered to provide detection of a 15 mm Hg change in SBP using P<0.05 and a power of 0.8. Rats were randomized to control and treatment groups. Whenever possible, cohorts included all treatment groups to minimize potential confounders. Sample IDs were nondescriptive such that individuals performing data analysis were blinded to treatment groups.
SBP was analyzed by 2‐way repeated measures ANOVA with Tukey's multiple comparison test. Two‐way ANOVA with Tukey's multiple comparison test was used for 4‐group flow cytometry, interleukin‐17a, and interleukin‐10 measurements. Two‐tailed unpaired t‐test was used to compare flow cytometry data between 2 groups. Data for taxa abundance were analyzed using the Mann–Whitney U test with a false rate of discovery adjusted for multiple comparisons. Differences were considered statistically significant for multiple comparisons or false rate of discovery if P≤0.05. β diversities were measured using unweighted and weighted UniFrac, visualized by principal coordinate analysis, and statistically analyzed using permutational multivariate ANOVA.
Results
OSA‐Induced Hypertension and Gut Inflammation
Exposing rats to 60 apneas/hour for 8 hours during the sleep phase led to significant elevations in blood pressure after 7 (137±5 versus 165±5 mm Hg; P=0.0044) and 14 (137±4 versus 167±4 mm Hg; P=0.0012) days (Figure 1A). We have previously demonstrated that OSA‐induced gut dysbiosis plays a causal role in the development of hypertension. 17 16S rRNA sequencing was used to assess the effects of OSA on the cecal microbiota. Measures of α diversity showed no significant differences between sham and OSA (Figure S1). However, unweighted (Figure 1B; P=0.004) and weighted (Figure 1C; P=0.005) UniFrac distance analysis revealed significant differences between sham and OSA cecal bacterial community composition. Linear discriminant analysis effect size analysis identified 7 genera representative of sham and 5 genera representative of OSA rats (Figure 1D). The relative abundance of genera identified by linear discriminant analysis effect size, with a mean abundance >1%, are shown in Figure 1E. The relative abundance of the genus Lachnospiraceae_UCG_004 was significantly increased in the cecum of OSA rats (11.7 versus 3.5%; false rate of discovery adjusted P=0.028), while Coprococcus was reduced following OSA (0.02 versus 2.1%, false rate of discovery adjusted P=0.055).
Figure 1. OSA induces hypertension and gut dysbiosis.
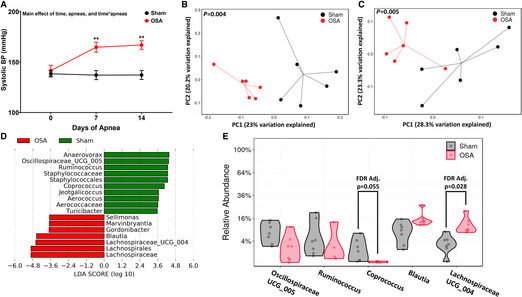
Systolic blood pressure in sham and OSA rats (A). Measures of cecal community β‐diversity in sham and OSA, unweighted UniFrac (B), and weighed UniFrac (C). LefSe analysis was used to identify taxa representative of sham vs OSA (D). Taxa identified by LefSe with a relative abundance >1% are plotted in (E). n=6–7 per group, **P<0.01 using 2‐way repeated‐measures ANOVA and multiple comparisons with Tukey's multiple comparison test (A), permutational multivariate ANOVA (B and C), linear discriminate analysis (LDA; D), and false rate of discovery adjusted 2‐tailed Student's t‐test (E). Error bars represent ±SEM (A). BP indicates blood pressure; LefSe, linear discriminant analysis effect size; and OSA, obstructive sleep apnea.
Given the strong relationship between the gut microbiota and host immune response, we assessed the inflammatory status of ileum and cecum by flow cytometry following 2 weeks of sham or OSA. Figure 2A illustrates the gating strategy used to distinguish T regulatory (Treg; CD4+ Forkhead box P3+), TH1 (CD4+ interferon‐γ+), and TH17 (CD4+interleukin‐17+) cells. Following 2 weeks of OSA, anti‐inflammatory Tregs were significantly reduced and proinflammatory TH1 and TH17 were significantly increased in the ileum and cecum (Figure 2B and 2C). This resulted in a significant increase in the TH17/Treg ratio, which has also been observed as an indicator of inflammation in other models of hypertension (Figure 2B and 2C). 25 , 26 , 27 Plasma levels of TNF‐α have previously been shown to be elevated in OSA patients and were proposed as a diagnostic biomarker for OSA. 5 , 7 We found that CD4+ cells expressing TNF‐α were significantly increased in the ileum and cecum following OSA (Figure 2B and 2C). No significant difference was observed in ileum or cecum macrophages (CD45+CD68+MHCII+) following OSA.
Figure 2. Gut inflammation following 14 days of OSA.
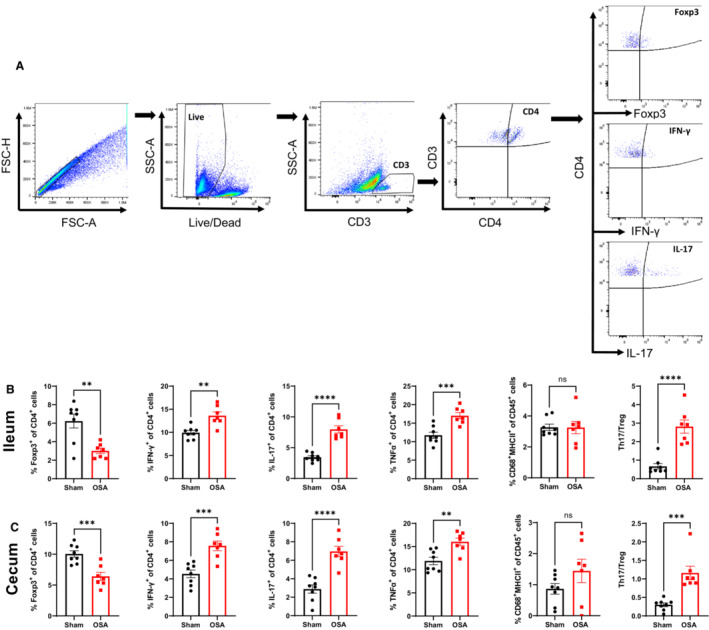
Flow cytometry gating strategy to identify immune cell subtypes (A). Effects of OSA on Tregs, TH1, TH17, TNF‐α+, macrophages, TH17/Treg ratio in ileum (B) and cecum (C). n=8 and 7 in sham and OSA, respectively; **P<0.005, ***P<0.001, ****P<0.0001 using 2‐tailed Student's t‐test. Error bars represent ±SEM (B and C). Foxp3 indicates Forkhead box P3; FSC‐H, forward scatter, IFN, interferon; IL, interleukin; OSA, obstructive sleep apnea; SSC‐A, side scatter; TH, T helper cell; TNF‐α, tumor necrosis factor‐α; and Tregs, T regulatory cells.
OSA‐Induced Inflammation Extends Beyond the Gut
We have previously shown that OSA is associated with gut barrier disruption. 18 Following barrier disruption, bacteria and bacterial antigens can cross the epithelium, leading to activation and migration of antigen‐presenting cells to the MLN, T‐cell activation, and peripheral inflammation. Therefore, we assessed inflammation in the MLN, spleen, and aorta using the same gating strategy presented in Figure 2A. We found significant increases in TH1, macrophages, and TNF‐α+ cells in spleen, and increased TH1 and TNF‐α+ cells in aorta (Figure S2).
OSA‐Induced Neuroinflammation
Neuroinflammation has been shown to play a key role in the pathogenesis of hypertension in patients and animal models. 28 , 29 , 30 , 31 In addition, alterations to the gut microbiota have been shown to influence brain homeostasis and neuroinflammation. 32 Two weeks of apneas led to a significant decrease in Tregs, as well as increased TH17, macrophage/microglia, and CD4+ TNF‐α+ cells, in the brains of OSA rats (Figure 3A through 3E). The ratio of TH17/Treg cells was significantly increased in the brain of OSA versus sham rats (Figure 3F).
Figure 3. Neuroinflammation following 14 days of OSA.
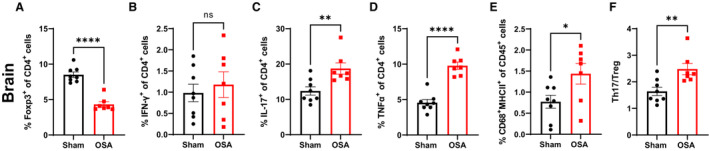
Effects of OSA on Tregs (A), TH1 (B), TH17 (C), TNF‐α+ (D), macrophages (E), and TH17/Treg ratio (F) in brain. n=8 and 7 in sham and OSA, respectively; **P<0.005, ***P<0.001, ****P<0.0001 using 2‐tailed Student's t‐test. Error bars represent ±SEM. OSA indicates obstructive sleep apnea; TH, T helper cell; TNF‐α, tumor necrosis factor‐α; and Tregs, T regulatory cells.
Involvement of Interleukin‐17 in the Systemic Inflammation and Hypertension of OSA
Given the elevations in TH17 cells in the gut and brain following OSA, we chose to examine the involvement of the cytokine interleukin‐17a. Elevated TH17 and interleukin‐17a have been previously implicated in the spontaneously hypertensive rat (SHR) model. 33 , 34 To examine the role of interleukin‐17a in OSA‐induced hypertension, rats were treated with a neutralizing antibody targeted to interleukin‐17a (nIL‐17a) or a nonimmune IgG during the 2 weeks of sham or OSA. First, we observed that circulating interleukin‐17a was significantly elevated in OSA versus sham rats receiving the nonimmune IgG. Neutralization of interleukin‐17a successfully reduced the circulating interleukin‐17a in OSA rats to a similar level measured in sham rats (Figure 4A). Additionally, OSA significantly reduced plasma interleukin‐10 by 50%, and neutralizing interleukin17a significantly increased circulating interleukin‐10 in OSA rats (Figure 4B). As compared with OSA rats receiving the nonimmune IgG, OSA rats receiving nIL‐17a had significantly lower SBP after 2 weeks (143±5 versus 169±5 mm Hg; P=0.022) of OSA (Figure 4C). The SBP of OSA rats receiving nIL‐17a was not significantly different from sham rats, demonstrating that neutralization of interleukin‐17a prevented hypertension.
Figure 4. Role of interleukin‐17a in OSA‐induced hypertension.
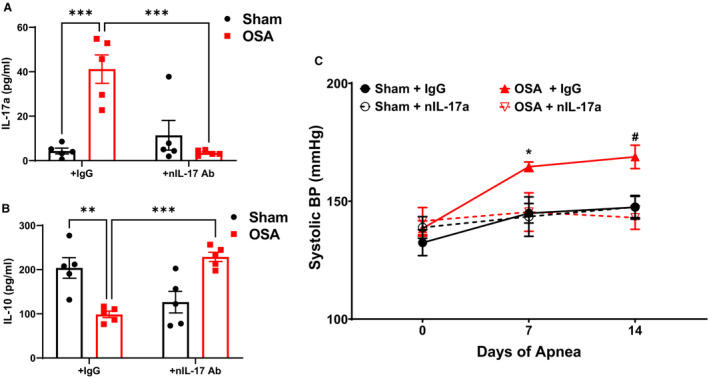
Plasma levels of interleukin‐17a (A) and interleukin‐10 (B). Systolic blood pressure (C) in sham and OSA rats treated with a nonimmune IgG or interleukin‐17a neutralizing antibody. n=5 in (A and B), n=5–6 in (C); *P<0.05 for Sham+IgG vs OSA+IgG, # P<0.05 of OSA+nIL‐17a vs OSA+IgG, ***P<0.001 using two‐way ANOVA (A, B) or 2‐way repeated measures ANOVA and multiple comparisons with Tukey's multiple comparisons test (C). Error bars represent ±SEM. BP indicates blood pressure; IgG, immunoglobulin G; and OSA, obstructive sleep apnea.
We next examined the effects of interleukin‐17a neutralization on the proinflammatory response to OSA in the gut and brain (Figure 5). nIL‐17a treatment did not significantly alter the percentage of TH17 cells in ileum or cecum. OSA rats treated with nonimmune IgG or nIL‐17a both exhibited increased TH17 cells in ileum and cecum as compared with sham rats. However, nIL‐17a prevented the loss of Tregs in ileum and cecum following OSA (Figure 5A and 5B). Additionally, OSA rats receiving nIL‐17a had a significantly lower percentage of TH1 and macrophages in ileum and cecum, as compared with OSA receiving nonimmune IgG (Figure 5A and 5B). Overall, these data indicate that interleukin‐17a neutralization reduced gut inflammation. In brain, nIL‐17a prevented the OSA‐induced increase of TH17 cells (Figure 5C). This was associated with decreased TH1, microglia, and CD4+ TNF‐α+ cells, and increased Tregs in brain following OSA (Figure 5C). Overall, neutralization of interleukin‐17a prevented OSA‐induced inflammation in gut and brain.
Figure 5. Interleukin‐17a neutralization reduces OSA‐induced gut and neuroinflammation.
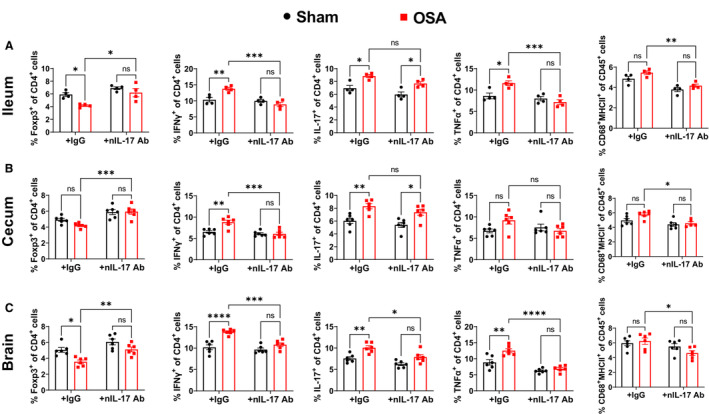
Effects of interleukin‐17a neutralization on Tregs, TH1, TH17, TNFα+, and macrophages in the ileum (A), cecum (B), and brain (C) of sham and OSA rats. n=4 in ileum, n=6 in cecum and brain; *P<0.05, **P<0.005, ***P<0.0005, ****P<0.0001 using 2‐way ANOVA with Tukey's multiple comparisons test. Error bars represent ±SEM. OSA indicates obstructive sleep apnea; TH, T helper cell; and Tregs, T regulatory cells.
No effects of nIL‐17a were observed on the percentages of immune cells in MLN or spleen (Figure S3A and S3B). Neutralization of interleukin‐17a significantly reduced the percentage of CD4+ TNF‐α+ cells found in the aorta following OSA (Figure S3C).
T Cells Migrate From Gut to Brain Following OSA
Previous studies have demonstrated that immune cells originating in gut can migrate to brain. 15 Given that the immune cell response to OSA was similar in gut and brain (Figures 2 and 3), we sought to track the migration of gut‐derived immune cells following OSA. Following 2 days of sham or OSA, cells in Peyer's patches of the small intestine were fluorescently labeled by microinjection with CFSE dye. Following an additional 2 days of sham or OSA, fluorescently labeled T cells were assessed in Peyer's patches, MLNs, spleen, and brain. The gating strategy for identifying CFSE‐labeled TH subsets is shown in Figure 6A. Following OSA, there was a significant reduction in CFSE‐labeled CD4+ cells in the Peyer's patches (Figure 6B). This reduction of TH cells following OSA was observed for TH1, TH2, and TH17 subsets. OSA also led to significant increases in gut‐derived (CFSE+) TH1, TH2, and TH17 cells found in MLNs and spleen (Figure 6C and 6D). Interestingly, we did not observe elevations in TH1 or TH17 in MLN at 14 days of OSA (Figure S2). This suggests that trafficking of proinflammatory immune cells through the MLNs may represent an early response to the onset of OSA. Finally, in sham rats we did not detect any gut‐derived CD4+CFSE+ cells in brain. However, following OSA there were significant increases in gut‐derived CFSE+ TH1, TH2, and TH17 cells found in brain. These findings indicate that OSA induces an efflux of T lymphocytes from Peyer's patches in the small intestine, and these cells migrate to MLNs, spleen, and brain.
Figure 6. Immune cells originating from the gut migrate to the periphery and brain following OSA.
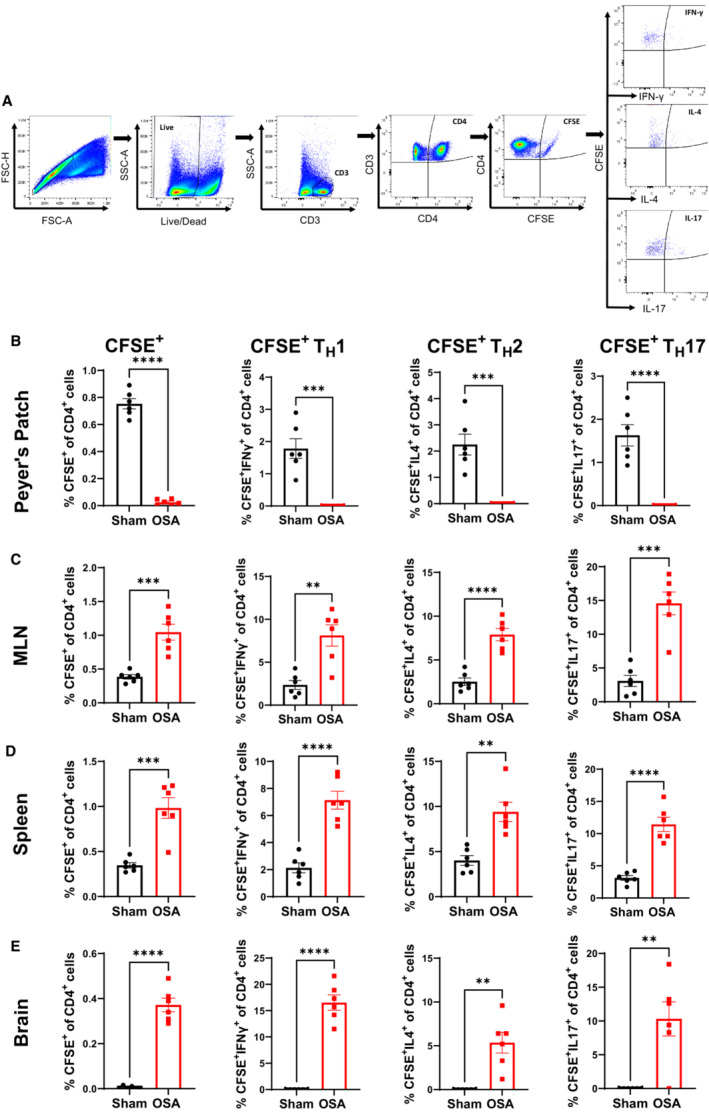
Flow cytometry gating strategy to identify CFSE+ immune cells (A). Percentage of CD4+, TH1, TH2, and TH17 CFSE‐labeled cells in Peyer's patches (B), MLN (C), spleen (D), and brain (E) of sham and OSA rats. n=6 per group; **P<0.005, ***P<0.0005, ****P<0.0001 using 2‐tailed Student's t‐test. Error bars represent ±SEM. CFSE indicates carboxyfluorescein succinimidyl ester; FSC‐H, forward scatter; MLN, mesenteric lymph node; OSA, obstructive sleep apnea; SSC‐A, side scatter; and TH, T helper cell.
Discussion
OSA is characterized by chronic low‐grade systemic inflammation, which is believed to contribute to its cardiovascular and cerebrovascular consequences. 8 , 9 , 10 Specifically, neuroinflammation plays an important role in the development of OSA‐induced hypertension. 5 , 6 , 8 Therefore, a better understanding of the mechanisms linking OSA to neuroinflammation is of great importance. We observed that 2 weeks of OSA led to significantly elevated BP, altered gut microbiota, and inflammation in the gut and brain. Additionally, neutralization of interleukin‐17a reduced gut and brain inflammation and prevented OSA‐induced hypertension. Finally, cell‐tracking studies demonstrate that TH cells originating in the gut migrate to the brain in response to OSA. These results support the idea that neuroinflammation plays a key role in the development of OSA‐induced hypertension and suggest that the proinflammatory response is initiated in the gut.
Convincing evidence over the past several decades has demonstrated the importance of inflammation in the development and progression of hypertension. T‐cell infiltration of the kidney, vascular wall, and brain contribute to elevated BP and end‐organ damage. 14 , 30 , 31 , 35 , 36 Recently, several studies have revealed that inflammation in the gut may also be a key component of hypertension. 18 , 37 , 38 , 39 The first significant finding of our studies is that OSA induces gut dysbiosis and a proinflammatory response in the gut. Models of intermittent hypoxia also demonstrate significant alterations to the gut microbiota. 8 , 19 , 20 , 21 , 22 , 23 , 40 This suggests that intermittent hypoxia may be a key component of OSA contributing to gut dysbiosis. We observed trends for decreased abundance of Coprococcus and increased Blautia, along with a significant increase of Lachnospiraceae_UCG_004 in response to OSA. Coprococcus was previously identified as a potentially protective genus in a mouse model of hypoxia‐induced pulmonary hypertension and the SHR model. 41 , 42 , 43 Blautia has been suggested as a potentially harmful genus in the angiotensin II model of hypertension and shown to positively correlate with BP in patients. 44 , 45 Associated with alterations to the microbiota, we found significantly increased TH1 and TH17, along with decreased Tregs, following OSA. A high‐salt diet has also been shown to alter the gut microbiota and induce gut inflammation. 46 , 47 , 48 , 49 High‐salt–induced alterations to the gut microbiota led to increases in immunogenic isolevuglandin adducts in antigen‐presenting cells and subsequent T‐cell activation. 46 Additionally, transfer of the microbiota from high‐salt fed mice to germ‐free mice resulted in gut inflammation and hypertension, demonstrating that gut inflammation was secondary to the effects of high salt on the gut microbiota. 46 We and others have also demonstrated pathophysiological alterations in the gut of the SHR and stroke‐prone SHR models of hypertension, including gut dysbiosis, barrier disruption, and inflammation. 34 , 37 , 41 , 50 , 51 , 52 , 53 , 54 , 55 Intriguingly, signs of gut inflammation and barrier disruption have been reported in prehypertensive SHRs, suggesting gut inflammation may precede and trigger the eventual systemic inflammation contributing to hypertension. 37 Finally, patients with hypertension exhibit elevated circulating TH17 cells expressing markers for gut homing, which was associated with elevated markers of gut barrier disruption. 38 Altogether, these findings support the idea that gut inflammation is a key component in multiple models of hypertension.
The second major finding of our studies is that OSA‐induced hypertension is associated with neuroinflammation, characterized by decreased Tregs and elevated TH17 cells. Neurogenic models of hypertension, including OSA and SHR, exhibit increased sympathetic outflow that is thought to play a key role in the initiation and maintenance of elevated BP. 29 , 56 In the context of OSA, the increased sympathetic outflow has classically been attributed to augmented carotid body chemosensory discharge in response to intermittent hypoxia. 57 However, in recent years, neuroinflammation has also been shown to contribute to elevated sympathetic output and subsequent hypertension. 29 , 58 , 59 Hypertensive animal models have elevated proinflammatory cytokines in cardiovascular‐regulating brain regions. 60 , 61 , 62 Furthermore, central administration of the proinflammatory cytokine interleukin‐1β or TNF‐α lead to elevated sympathetic activity and BP in animal models. 63 , 64 Conversely, central administration of the anti‐inflammatory cytokine interleukin‐10 or minocycline, an anti‐inflammatory agent, reduces neuroinflammation and BP. 61 , 65 These findings support the idea of neuroinflammation as causal, rather than consequential, in these models of hypertension.
Given the observed elevations in TH17 cells in the gut and brain of OSA rats, we examined the role of interleukin‐17a in OSA‐induced hypertension. We show that neutralization of interleukin‐17a reduced gut and brain inflammation as well as prevented the development of hypertension. Interleukin‐17a has been shown to induce a proinflammatory response in other T cells, dendritic cells, endothelial cells, and macrophages/microglia. Recently, interleukin‐17a has been implicated in many inflammatory and autoimmune diseases, including rheumatoid arthritis, multiple sclerosis, and hypertension. 14 , 36 , 66 , 67 In response to angiotensin II infusion, interleukin‐17a−/− mice exhibit significantly blunted BP elevations. 68 Similar to our finding, neutralization of interleukin‐17a lowers BP in the SHR and angiotensin II infusion models. 34 , 68 , 69 The mechanisms by which interleukin‐17a contributes to hypertension have largely focused on the vasculature and kidney. 36 In the vasculature, interleukin‐17a contributes to decreased nitric oxide synthesis, hypertrophic remodeling, and arterial stiffening. 68 , 70 In the kidney, interleukin‐17a has been shown to regulate sodium transporters, leading to sodium and water retention and subsequent elevations in BP. 36 , 69 Our findings demonstrate that interleukin‐17a contributes to OSA‐induced neuroinflammation and hypertension. Similarly, Cao et al. showed that intravenous administration of interleukin‐17a resulted in elevated BP, microglia activation in hypothalamic paraventricular nucleus, and increased renal sympathetic nerve activity. These responses to interleukin‐17a were markedly attenuated by delivery of an interleukin‐17a receptor small interfering RNA to the hypothalamic paraventricular nucleus. 71 Given the increasing evidence supporting neuroinflammation as a key component of hypertension, further understanding of the effects of interleukin‐17a in neurogenic hypertension is needed.
The third major finding of our study is that OSA induces an efflux of TH cells from the gut, some of which traffic to the brain. Historically, the brain has been considered a site of immune privilege. However, recent studies demonstrate that immune cells activated in the periphery can traffic to and invade the brain, thereby contributing to inflammatory neurological diseases. 15 One such site of peripheral immune cell activation is the gut‐associated lymphoid tissue, which includes the Peyer's patches. Immune cells that are activated in the gut and then traffic to brain constitute 1 pathway of the “gut‐brain axis.” We demonstrate that following OSA, TH cells originating in Peyer's patches track to the brain. After 1 week of OSA, we identified TH1, TH2, and TH17 cells that had tracked from gut to brain. Similarly, trafficking of gut‐derived immune cells to the brain has been described in multiple sclerosis. The observation that germ‐free and antibiotic‐treated mouse models of multiple sclerosis failed to develop central nervous system–targeted autoimmunity suggested a role for the gut microbiota. 66 , 72 , 73 Subsequent studies have revealed that segmented filamentous bacteria contribute to activation of TH17 in the gut, which then enter the circulation and migrate to the brain by adhesion of α4β1 integrin to vascular cell adhesion protein 1 expressed on the brain endothelium. 66 , 74 Alternatively, Bacteroides fragilis has been shown to promote activation of Tregs that traffic to the brain and have protective effects in models of demyelinating disease. 73 , 75 , 76 , 77 Trafficking of immune cells to the brain has also been described following stroke. Studies by Benakis et al 78 and Singh et al 79 demonstrated that gut‐derived immune cells traffic to the meninges and peri‐infarct region of the brain following stroke. Our studies demonstrate a role for gut‐derived immune cells in OSA‐induced neuroinflammation and hypertension. Further studies to examine the signals involved in immune cell activation in the gut wall, and trafficking to the brain is needed.
Our study has several limitations; first, our study examines the effects of OSA and gut dysbiosis only on young male rats. Sex and age have both been shown to influence the makeup of the microbiota, and these clinically relevant variables will need to be investigated. Second, the signals from the microbiota that influence T‐cell activation in the gut were not investigated. Third, the mechanisms involved in tracking of gut‐derived immune cells to the brain remain unknown. Finally, we acknowledge that the sample sizes in these experiments prohibit the ability to check whether the assumption of normality is reasonable.
Sources of Funding
This work was supported by grants RO1HL134838 (Dr Durgan), R01NS102594 (Dr Bryan), and DK56338 (Texas Medical Center Digestive Diseases Center; Dr Durgan).
Disclosures
None.
Supporting information
Figures S1–S3.
Acknowledgments
The authors appreciate the support of the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the Cancer Prevention and Research Institute of Texas Core Facility Support Award (CPRIT‐RP180672), the National Institutes of Health (CA125123 and RR024574), and the assistance of Joel M. Sederstrom.
This manuscript was sent to Sakima Smith, MD, MPH, Associate Editor, for review by expert referees, editorial decision, and final disposition.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.122.029218
For Sources of Funding and Disclosures, see page 11.
References
- 1. Cowie MR, Linz D, Redline S, Somers VK, Simonds AK. Sleep disordered breathing and cardiovascular disease: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2021;78:608–624. doi: 10.1016/j.jacc.2021.05.048 [DOI] [PubMed] [Google Scholar]
- 2. Somers VK, White DP, Amin R, Abraham WT, Costa F, Culebras A, Daniels S, Floras JS, Hunt CE, Olson LJ, et al. Sleep apnea and cardiovascular disease: an American Heart Association/American College Of Cardiology Foundation Scientific Statement from the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council On Cardiovascular Nursing. In collaboration with the National Heart, Lung, and Blood Institute National Center on Sleep Disorders Research (National Institutes of Health). Circulation. 2008;118:1080–1111. doi: 10.1161/CIRCULATIONAHA.107.189375 [DOI] [PubMed] [Google Scholar]
- 3. Dempsey JA, Veasey SC, Morgan BJ, O'Donnell CP. Pathophysiology of sleep apnea. Physiol Rev. 2010;90:47–112. doi: 10.1152/physrev.00043.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yeghiazarians Y, Jneid H, Tietjens JR, Redline S, Brown DL, El‐Sherif N, Mehra R, Bozkurt B, Ndumele CE, Somers VK. Obstructive sleep apnea and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2021;144:e56–e67. doi: 10.1161/CIR.0000000000000988 [DOI] [PubMed] [Google Scholar]
- 5. Kheirandish‐Gozal L, Gozal D. Obstructive sleep apnea and inflammation: proof of concept based on two illustrative cytokines. Int J Mol Sci. 2019;20:459. doi: 10.3390/ijms20030459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gozal D, Kheirandish‐Gozal L. Cardiovascular morbidity in obstructive sleep apnea: oxidative stress, inflammation, and much more. Am J Respir Crit Care Med. 2008;177:369–375. doi: 10.1164/rccm.200608-1190PP [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wali SO, Al‐Mughales J, Alhejaili F, Manzar MD, Alsallum F, Almojaddidi H, Gozal D. The utility of proinflammatory markers in patients with obstructive sleep apnea. Sleep Breath. 2021;25:545–553. doi: 10.1007/s11325-020-02149-3 [DOI] [PubMed] [Google Scholar]
- 8. Mashaqi S, Gozal D. Obstructive sleep apnea and systemic hypertension: gut dysbiosis as the mediator? J Clin Sleep Med. 2019;15:1517–1527. doi: 10.5664/jcsm.7990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li T, Chen Y, Gua C, Wu B. Elevated oxidative stress and inflammation in hypothalamic paraventricular nucleus are associated with sympathetic excitation and hypertension in rats exposed to chronic intermittent hypoxia. Front Physiol. 2018;9:840. doi: 10.3389/fphys.2018.00840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu X, Gong L, Xie L, Gu W, Wang X, Liu Z, Li S. NLRP3 deficiency protects against intermittent hypoxia‐induced neuroinflammation and mitochondrial ROS by promoting the PINK1‐Parkin pathway of mitophagy in a murine model of sleep apnea. Front Immunol. 2021;12:628168. doi: 10.3389/fimmu.2021.628168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee YK, Mazmanian SK. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science. 2010;330:1768–1773. doi: 10.1126/science.1195568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T‐cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci USA. 2010;107:12204–12209. doi: 10.1073/pnas.0909122107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Main BS, Minter MR. Microbial immuno‐communication in neurodegenerative diseases. Front Neurosci. 2017;11:151. doi: 10.3389/fnins.2017.00151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Madhur MS, Elijovich F, Alexander MR, Pitzer A, Ishimwe J, Van Beusecum JP, Patrick DM, Smart CD, Kleyman TR, Kingery J, et al. Hypertension: do inflammation and immunity hold the key to solving this epidemic? Circ Res. 2021;128:908–933. doi: 10.1161/CIRCRESAHA.121.318052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zundler S, Günther C, Kremer AE, Zaiss MM, Rothhammer V, Neurath MF. Gut immune cell trafficking: inter‐organ communication and immune‐mediated inflammation. Nat Rev Gastroenterol Hepatol. 2023;20:50–64. doi: 10.1038/s41575-022-00663-1 [DOI] [PubMed] [Google Scholar]
- 16. Crossland RF, Durgan DJ, Lloyd EE, Phillips SC, Reddy AK, Marrelli SP, Bryan RM. A new rodent model for obstructive sleep apnea: effects on ATP‐mediated dilations in cerebral arteries. Am J Physiol Regul Integr Comp Physiol. 2013;305:R334–R342. doi: 10.1152/ajpregu.00244.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Durgan DJ, Ganesh BP, Cope JL, Ajami NJ, Phillips SC, Petrosino JF, Hollister EB, Bryan RM. Role of the gut microbiome in obstructive sleep apnea‐induced hypertension. Hypertension. 2016;67:469–474. doi: 10.1161/HYPERTENSIONAHA.115.06672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ganesh BP, Nelson JW, Eskew JR, Ganesan A, Ajami NJ, Petrosino JF, Bryan RM, Durgan DJ. Prebiotics, probiotics, and acetate supplementation prevent hypertension in a model of obstructive sleep apnea. Hypertension. 2018;72:1141–1150. doi: 10.1161/HYPERTENSIONAHA.118.11695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moreno‐Indias I, Torres M, Montserrat JM, Sanchez‐Alcoholado L, Cardona F, Tinahones FJ, Gozal D, Poroyko VA, Navajas D, Queipo‐Ortuño MI, et al. Intermittent hypoxia alters gut microbiota diversity in a mouse model of sleep apnoea. Eur Respir J. 2015;45:1055–1065. doi: 10.1183/09031936.00184314 [DOI] [PubMed] [Google Scholar]
- 20. Poroyko VA, Carreras A, Khalyfa A, Khalyfa AA, Leone V, Peris E, Almendros I, Gileles‐Hillel A, Qiao Z, Hubert N, et al. Chronic sleep disruption alters gut microbiota, induces systemic and adipose tissue inflammation and insulin resistance in mice. Sci Rep. 2016;6:35405. doi: 10.1038/srep35405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xue J, Allaband C, Zhou D, Poulsen O, Martino C, Jiang L, Tripathi A, Elijah E, Dorrestein PC, Knight R, et al. Influence of intermittent hypoxia/hypercapnia on atherosclerosis, gut microbiome, and metabolome. Front Physiol. 2021;12:663950. doi: 10.3389/fphys.2021.663950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tripathi A, Xu ZZ, Xue J, Poulsen O, Gonzalez A, Humphrey G, Meehan MJ, Melnik AV, Ackermann G, Zhou D, et al. Intermittent hypoxia and hypercapnia reproducibly change the gut microbiome and metabolome across rodent model systems. mSystems. 2019;4:4. doi: 10.1128/mSystems.00058-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tripathi A, Melnik AV, Xue J, Poulsen O, Meehan MJ, Humphrey G, Jiang L, Ackermann G, McDonald D, Zhou D, et al. Intermittent hypoxia and hypercapnia, a hallmark of obstructive sleep apnea, alters the gut microbiome and metabolome. mSystems. 2018;3:3. doi: 10.1128/mSystems.00020-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Durgan DJ, Crossland RF, Lloyd EE, Phillips SC, Bryan RM. Increased cerebrovascular sensitivity to endothelin‐1 in a rat model of obstructive sleep apnea: a role for endothelin receptor B. J Cereb Blood Flow Metab. 2015;35:402–411. doi: 10.1038/jcbfm.2014.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Robles‐Vera I, de la Visitación N, Toral M, Sánchez M, Romero M, Gómez‐Guzmán M, Yang T, Izquierdo‐García JL, Guerra‐Hernández E, Ruiz‐Cabello J, et al. Probiotic Bifidobacterium breve prevents DOCA‐salt hypertension. FASEB J. 2020;34:13626–13640. doi: 10.1096/fj.202001532R [DOI] [PubMed] [Google Scholar]
- 26. Robles‐Vera I, Toral M, de la Visitación N, Sánchez M, Gómez‐Guzmán M, Muñoz R, Algieri F, Vezza T, Jiménez R, Gálvez J, et al. Changes to the gut microbiota induced by losartan contributes to its antihypertensive effects. Br J Pharmacol. 2020;177:2006–2023. doi: 10.1111/bph.14965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Toral M, Romero M, Rodríguez‐Nogales A, Jiménez R, Robles‐Vera I, Algieri F, Chueca‐Porcuna N, Sánchez M, de la Visitación N, Olivares M, et al. Lactobacillus fermentum improves tacrolimus‐induced hypertension by restoring vascular redox state and improving eNOS coupling. Mol Nutr Food Res. 2018;62:e1800033. doi: 10.1002/mnfr.201800033 [DOI] [PubMed] [Google Scholar]
- 28. Toral M, Robles‐Vera I, de la Visitación N, Romero M, Yang T, Sánchez M, Gómez‐Guzmán M, Jiménez R, Raizada MK, Duarte J. Critical role of the interaction gut microbiota‐sympathetic nervous system in the regulation of blood pressure. Front Physiol. 2019;10:231. doi: 10.3389/fphys.2019.00231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zubcevic J, Waki H, Raizada MK, Paton JFR. Autonomic‐immune‐vascular interaction: an emerging concept for neurogenic hypertension. Hypertension. 2011;57:1026–1033. doi: 10.1161/HYPERTENSIONAHA.111.169748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Santisteban MM, Ahmari N, Carvajal JM, Zingler MB, Qi Y, Kim S, Joseph J, Garcia‐Pereira F, Johnson RD, Shenoy V, et al. Involvement of bone marrow cells and neuroinflammation in hypertension. Circ Res. 2015;117:178–191. doi: 10.1161/CIRCRESAHA.117.305853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Richards EM, Li J, Stevens BR, Pepine CJ, Raizada MK. Gut microbiome and neuroinflammation in hypertension. Circ Res. 2022;130:401–417. doi: 10.1161/CIRCRESAHA.121.319816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cryan JF, Mazmanian SK. Microbiota‐brain axis: context and causality. Science. 2022;376:938–939. doi: 10.1126/science.abo4442 [DOI] [PubMed] [Google Scholar]
- 33. Robles‐Vera I, Toral M, de la Visitación N, Sánchez M, Gómez‐Guzmán M, Romero M, Yang T, Izquierdo‐Garcia JL, Jiménez R, Ruiz‐Cabello J, et al. Probiotics prevent dysbiosis and the rise in blood pressure in genetic hypertension: role of short‐chain fatty acids. Mol Nutr Food Res. 2020;64:e1900616. doi: 10.1002/mnfr.201900616 [DOI] [PubMed] [Google Scholar]
- 34. Toral M, Robles‐Vera I, de la Visitación N, Romero M, Sánchez M, Gómez‐Guzmán M, Rodriguez‐Nogales A, Yang T, Jiménez R, Algieri F, et al. Role of the immune system in vascular function and blood pressure control induced by faecal microbiota transplantation in rats. Acta Physiol (Oxf). 2019;227:e13285. doi: 10.1111/apha.13285 [DOI] [PubMed] [Google Scholar]
- 35. Dixon KB, Davies SS, Kirabo A. Dendritic cells and isolevuglandins in immunity, inflammation, and hypertension. Am J Physiol Heart Circ Physiol. 2017;312:H368–H374. doi: 10.1152/ajpheart.00603.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Davis GK, Fehrenbach DJ, Madhur MS. Interleukin 17A: key player in the pathogenesis of hypertension and a potential therapeutic target. Curr Hypertens Rep. 2021;23:13. doi: 10.1007/s11906-021-01128-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Santisteban MM, Qi Y, Zubcevic J, Kim S, Yang T, Shenoy V, Cole‐Jeffrey CT, Lobaton GO, Stewart DC, Rubiano A, et al. Hypertension‐linked pathophysiological alterations in the gut. Circ Res. 2017;120:312–323. doi: 10.1161/CIRCRESAHA.116.309006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim S, Goel R, Kumar A, Qi Y, Lobaton G, Hosaka K, Mohammed M, Handberg EM, Richards EM, Pepine CJ, et al. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin Sci. 2018;132:701–718. doi: 10.1042/CS20180087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Saha P, Mell B, Golonka RM, Bovilla VR, Abokor AA, Mei X, Yeoh BS, Doris PA, Gewirtz AT, Joe B, et al. Selective IgA deficiency in spontaneously hypertensive rats with gut dysbiosis. Hypertension. 2022;79:2239–2249. doi: 10.1161/HYPERTENSIONAHA.122.19307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moreno‐Indias I, Torres M, Sanchez‐Alcoholado L, Cardona F, Almendros I, Gozal D, Montserrat JM, Queipo‐Ortuño MI, Farré R. Normoxic recovery mimicking treatment of sleep apnea does not reverse intermittent hypoxia‐induced bacterial dysbiosis and low‐grade endotoxemia in mice. Sleep. 2016;39:1891–1897. doi: 10.5665/sleep.6176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang T, Santisteban MM, Rodriguez V, Li E, Ahmari N, Carvajal JM, Zadeh M, Gong M, Qi Y, Zubcevic J, et al. Gut dysbiosis is linked to hypertension. Hypertension. 2015;65:1331–1340. doi: 10.1161/HYPERTENSIONAHA.115.05315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Luo L, Chen Q, Yang L, Zhang Z, Xu J, Gou D. MSCs therapy reverse the gut microbiota in hypoxia‐induced pulmonary hypertension mice. Front Physiol. 2021;12:712139. doi: 10.3389/fphys.2021.712139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li H‐B, Yang T, Richards EM, Pepine CJ, Raizada MK. Maternal treatment with captopril persistently alters gut‐brain communication and attenuates hypertension of male offspring. Hypertension. 2020;75:1315–1324. doi: 10.1161/HYPERTENSIONAHA.120.14736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kashtanova DA, Tkacheva ON, Doudinskaya EN, Strazhesko ID, Kotovskaya YV, Popenko AS, Tyakht AV, Alexeev DG. Gut microbiota in patients with different metabolic statuses: Moscow study. Microorganisms. 2018;6:6. doi: 10.3390/microorganisms6040098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sharma RK, Yang T, Oliveira AC, Lobaton GO, Aquino V, Kim S, Richards EM, Pepine CJ, Sumners C, Raizada MK. Microglial cells impact gut microbiota and gut pathology in angiotensin II‐induced hypertension. Circ Res. 2019;124:727–736. doi: 10.1161/CIRCRESAHA.118.313882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ferguson JF, Aden LA, Barbaro NR, Van Beusecum JP, Xiao L, Simmons AJ, Warden C, Pasic L, Himmel LE, Washington MK, et al. High dietary salt‐induced dendritic cell activation underlies microbial dysbiosis‐associated hypertension. JCI Insight. 2019;4:5. doi: 10.1172/jci.insight.126241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Elijovich F, Laffer CL, Sahinoz M, Pitzer A, Ferguson JF, Kirabo A. The gut microbiome, inflammation, and salt‐sensitive hypertension. Curr Hypertens Rep. 2020;22:79. doi: 10.1007/s11906-020-01091-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Avery EG, Bartolomaeus H, Maifeld A, Marko L, Wiig H, Wilck N, Rosshart SP, Forslund SK, Müller DN. The gut microbiome in hypertension: recent advances and future perspectives. Circ Res. 2021;128:934–950. doi: 10.1161/CIRCRESAHA.121.318065 [DOI] [PubMed] [Google Scholar]
- 49. Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, Haase S, Mähler A, Balogh A, Markó L, et al. Salt‐responsive gut commensal modulates TH17 axis and disease. Nature. 2017;551:585–589. doi: 10.1038/nature24628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stewart DC, Rubiano A, Santisteban MM, Shenoy V, Qi Y, Pepine CJ, Raizada MK, Simmons CS. Hypertension‐linked mechanical changes of rat gut. Acta Biomater. 2016;45:296–302. doi: 10.1016/j.actbio.2016.08.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nelson JW, Phillips SC, Ganesh BP, Petrosino JF, Durgan DJ, Bryan RM. The gut microbiome contributes to blood‐brain barrier disruption in spontaneously hypertensive stroke prone rats. FASEB J. 2021;35:e21201. doi: 10.1096/fj.202001117R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Adnan S, Nelson JW, Ajami NJ, Venna VR, Petrosino JF, Bryan RM, Durgan DJ. Alterations in the gut microbiota can elicit hypertension in rats. Physiol Genomics. 2017;49:96–104. doi: 10.1152/physiolgenomics.00081.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shi H, Zhang B, Abo‐Hamzy T, Nelson JW, Ambati CSR, Petrosino JF, Bryan RM, Durgan DJ. Restructuring the gut microbiota by intermittent fasting lowers blood pressure. Circ Res. 2021;128:1240–1254. doi: 10.1161/CIRCRESAHA.120.318155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shi H, Nelson JW, Phillips S, Petrosino JF, Bryan RM, Durgan DJ. Alterations of the gut microbial community structure and function with aging in the spontaneously hypertensive stroke prone rat. Sci Rep. 2022;12:8534. doi: 10.1038/s41598-022-12578-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gómez‐Guzmán M, Toral M, Romero M, Jiménez R, Galindo P, Sánchez M, Zarzuelo MJ, Olivares M, Gálvez J, Duarte J. Antihypertensive effects of probiotics lactobacillus strains in spontaneously hypertensive rats. Mol Nutr Food Res. 2015;59:2326–2336. doi: 10.1002/mnfr.201500290 [DOI] [PubMed] [Google Scholar]
- 56. Javaheri S, Barbe F, Campos‐Rodriguez F, Dempsey JA, Khayat R, Javaheri S, Malhotra A, Martinez‐Garcia MA, Mehra R, Pack AI, et al. Sleep apnea: types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol. 2017;69:841–858. doi: 10.1016/j.jacc.2016.11.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Iturriaga R, Oyarce MP, Dias ACR. Role of carotid body in intermittent hypoxia‐related hypertension. Curr Hypertens Rep. 2017;19:38. doi: 10.1007/s11906-017-0735-0 [DOI] [PubMed] [Google Scholar]
- 58. Mikolajczyk TP, Guzik TJ. Adaptive immunity in hypertension. Curr Hypertens Rep. 2019;21:68. doi: 10.1007/s11906-019-0971-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Oyarce MP, Iturriaga R. Proinflammatory cytokines in the nucleus of the solitary tract of hypertensive rats exposed to chronic intermittent hypoxia. Adv Exp Med Biol. 2018;1071:69–74. doi: 10.1007/978-3-319-91137-3_8 [DOI] [PubMed] [Google Scholar]
- 60. Shi P, Diez‐Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, Sriramula S, Francis J, Sumners C, Raizada MK. Brain microglial cytokines in neurogenic hypertension. Hypertension. 2010;56:297–303. doi: 10.1161/HYPERTENSIONAHA.110.150409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shi P, Raizada MK, Sumners C. Brain cytokines as neuromodulators in cardiovascular control. Clin Exp Pharmacol Physiol. 2010;37:e52–e57. doi: 10.1111/j.1440-1681.2009.05234.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zubcevic J, Jun JY, Kim S, Perez PD, Afzal A, Shan Z, Li W, Santisteban MM, Yuan W, Febo M, et al. Altered inflammatory response is associated with an impaired autonomic input to the bone marrow in the spontaneously hypertensive rat. Hypertension. 2014;63:542–550. doi: 10.1161/HYPERTENSIONAHA.113.02722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Takahashi H, Nishimura M, Sakamoto M, Ikegaki I, Nakanishi T, Yoshimura M. Effects of interleukin‐1 beta on blood pressure, sympathetic nerve activity, and pituitary endocrine functions in anesthetized rats. Am J Hypertens. 1992;5:224–229. doi: 10.1093/ajh/5.4.224 [DOI] [PubMed] [Google Scholar]
- 64. Sriramula S, Haque M, Majid DSA, Francis J. Involvement of tumor necrosis factor‐alpha in angiotensin II‐mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension. 2008;51:1345–1351. doi: 10.1161/HYPERTENSIONAHA.107.102152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Guggilam A, Haque M, Kerut EK, McIlwain E, Lucchesi P, Seghal I, Francis J. TNF‐alpha blockade decreases oxidative stress in the paraventricular nucleus and attenuates sympathoexcitation in heart failure rats. Am J Physiol Heart Circ Physiol. 2007;293:H599–H609. doi: 10.1152/ajpheart.00286.2007 [DOI] [PubMed] [Google Scholar]
- 66. Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T‐cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4615–4622. doi: 10.1073/pnas.1000082107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin II‐induced hypertension and vascular dysfunction. Hypertension. 2010;55:500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Saleh MA, Norlander AE, Madhur MS. Inhibition of interleukin 17‐a but not interleukin‐17F signaling lowers blood pressure and reduces end‐organ inflammation in angiotensin II‐induced hypertension. JACC Basic Transl Sci. 2016;1:606–616. doi: 10.1016/j.jacbts.2016.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin‐17 causes Rho‐kinase‐mediated endothelial dysfunction and hypertension. Cardiovasc Res. 2013;97:696–704. doi: 10.1093/cvr/cvs422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cao Y, Yu Y, Xue B, Wang Y, Chen X, Beltz TG, Johnson AK, Wei S‐G. IL (interleukin)‐17A acts in the brain to drive neuroinflammation, sympathetic activation, and hypertension. Hypertension. 2021;78:1450–1462. doi: 10.1161/HYPERTENSIONAHA.121.18219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, Wekerle H, Krishnamoorthy G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. 2011;479:538–541. doi: 10.1038/nature10554 [DOI] [PubMed] [Google Scholar]
- 73. Ochoa‐Repáraz J, Mielcarz DW, Ditrio LE, Burroughs AR, Foureau DM, Haque‐Begum S, Kasper LH. Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J Immunol. 2009;183:6041–6050. doi: 10.4049/jimmunol.0900747 [DOI] [PubMed] [Google Scholar]
- 74. Kent SJ, Karlik SJ, Rice GP, Horner HC. A monoclonal antibody to alpha 4‐integrin reverses the MR‐detectable signs of experimental allergic encephalomyelitis in the guinea pig. J Magn Reson Imaging. 1995;5:535–540. doi: 10.1002/jmri.1880050510 [DOI] [PubMed] [Google Scholar]
- 75. Erturk‐Hasdemir D, Ochoa‐Repáraz J, Kasper DL, Kasper LH. Exploring the gut‐brain axis for the control of CNS inflammatory demyelination: immunomodulation by Bacteroides fragilis' polysaccharide A. Front Immunol. 2021;12:662807. doi: 10.3389/fimmu.2021.662807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang Y, Begum‐Haque S, Telesford KM, Ochoa‐Repáraz J, Christy M, Kasper EJ, Kasper DL, Robson SC, Kasper LH. A commensal bacterial product elicits and modulates migratory capacity of CD39(+) CD4 T regulatory subsets in the suppression of neuroinflammation. Gut Microbes. 2014;5:552–561. doi: 10.4161/gmic.29797 [DOI] [PubMed] [Google Scholar]
- 77. Ochoa‐Repáraz J, Mielcarz DW, Wang Y, Begum‐Haque S, Dasgupta S, Kasper DL, Kasper LH. A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal Immunol. 2010;3:487–495. doi: 10.1038/mi.2010.29 [DOI] [PubMed] [Google Scholar]
- 78. Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, Sita G, Racchumi G, Ling L, Pamer EG, et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat Med. 2016;22:516–523. doi: 10.1038/nm.4068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Singh V, Roth S, Llovera G, Sadler R, Garzetti D, Stecher B, Dichgans M, Liesz A. Microbiota dysbiosis controls the neuroinflammatory response after stroke. J Neurosci. 2016;36:7428–7440. doi: 10.1523/JNEUROSCI.1114-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S3.