

Abstract
Background
The interplay between branched‐chain amino acid (BCAA) metabolism, an important pathway in adiposity and cardiometabolic disease, and visceral adipose depots such as hepatic steatosis (HS) and epicardial adipose tissue is unknown. We leveraged the PROMISE clinical trial with centrally adjudicated coronary computed tomography angiography imaging to determine relationships between adipose depots, BCAA dysregulation, and coronary artery disease (CAD).
Methods and Results
The PROMISE (Prospective Multicenter Imaging Study for Evaluation of Chest Pain) trial randomized 10 003 outpatients with stable chest pain to computed tomography angiography versus standard‐of‐care diagnostics. For this study, we included 1798 participants with available computed tomography angiography data and biospecimens. Linear and logistic regression were used to determine associations between a molar sum of BCAAs measured by nuclear magnetic resonance spectroscopy with body mass index, adipose traits, and obstructive CAD. Mendelian randomization was then used to determine if BCAAs are in the causal pathway for adipose depots or CAD. The study sample had a mean age of 60 years (SD, 8.0), body mass index of 30.6 (SD, 5.9), and epicardial adipose tissue volume of 57.3 (SD, 21.3) cm3/m2; 27% had HS, and 14% had obstructive CAD. BCAAs were associated with body mass index (multivariable beta 0.12 per SD increase in BCAA [95% CI, 0.08–0.17]; P=4×10−8). BCAAs were also associated with HS (multivariable odds ratio [OR], 1.46 per SD increase in BCAAs [95% CI, 1.28–1.67]; P=2×10−8), but BCAAs were associated only with epicardial adipose tissue volume (odds ratio, 1.18 [95% CI, 1.07–1.32]; P=0.002) and obstructive CAD (OR, 1.18 [95% CI, 1.04–1.34]; P=0.009) in univariable models. Two‐sample Mendelian randomization did not support the role of BCAAs as within the causal pathways for HS or CAD.
Conclusions
BCAAs have been implicated in the pathogenesis of cardiometabolic diseases, and adipose depots have been associated with the risk of CAD. Leveraging a large clinical trial, we further establish the role of dysregulated BCAA catabolism in HS and CAD, although BCAAs did not appear to be in the causal pathway of either disease. This suggests that BCAAs may serve as an independent circulating biomarker of HS and CAD but that their association with these cardiometabolic diseases is mediated through other pathways.
Keywords: branched‐chain amino acids, coronary artery disease, coronary computed tomography angiography, epicardial adipose tissue, hepatic steatosis, Mendelian randomization analysis
Subject Categories: Biomarkers
Nonstandard Abbreviations and Acronyms
- BCAA
branched‐chain amino acid
- EAT
epicardial adipose tissue
- HRP
high‐risk plaque
- HS
hepatic steatosis
- MR
Mendelian randomization
- NAFLD
nonalcoholic fatty liver disease
- oCAD
obstructive coronary artery disease
- PROMISE
Prospective Multicenter Imaging Study for Evaluation of Chest Pain
Research Perspective.
What New Question Does This Study Raise?
Our study supports that higher circulating branched‐chain amino acid levels are associated with hepatic steatosis but that branched‐chain amino acids are not in the causal pathway of this disease.
These data highlight the knowledge gap for how branched‐chain amino acids relate to hepatic steatosis.
What Question Should Be Addressed Next?
The association between branched‐chain amino acids with epicardial adipose tissue and coronary artery disease is mediated by other factors; how branched‐chain keto acids relate to these pathways deserves further study.
Obesity is an established risk factor for cardiovascular disease (CVD). However, body mass index (BMI) does not capture all of the risk related to obesity. As such, there has been increased interest in tissue‐specific adipose depots. These organ‐specific adipose depots may provide a better, or more granular, snapshot of cardiometabolic risk related to their possible endocrine and paracrine effects on local tissues (ie, that these depots report more finely on cardiometabolic risk than traditional measurements of obesity). 1 For example, visceral adipose tissue (adipose tissue in internal organs) has been shown to be associated with higher mortality compared with traditional risk factors such as BMI. 2 Hepatic steatosis (HS) has been shown to be a strong predictor of diabetes and CVD. 3 Epicardial adipose tissue (EAT) have been shown to be increased in heart failure, 4 atrial fibrillation, diabetes, and myocardial injury. 5
The biological underpinnings of the epidemiologic link between tissue‐specific adipose depots such as HS with coronary artery disease (CAD) and CVD have also begun to be investigated. For example, the role of branched‐chain amino acid (BCAA) metabolism has been implicated in HS and its pathophysiology. 6 One study showed that plasma levels of immediate substrates of the branched‐chain alpha‐keto acid dehydrogenase kinase are associated with HS in individuals with obesity. 7 Studies have also established that higher circulating levels of BCAAs are associated with heart failure, metabolic health, and diabetes, 8 , 9 and BCAA levels have been shown to be associated with CAD even when adjusted for diabetes and insulin resistance. 10
In the background of this potential biology of BCAA catabolism in HS, there remains a need for a better understanding of the interrelationships between BCAAs, organ‐specific adipose depots, and CAD. We report here a biomarker substudy of the PROMISE (Prospective Multicenter Imaging Study for Evaluation of Chest Pain) clinical trial evaluating the use of a computed tomography angiography (CTA)‐guided anatomic strategy for patients with stable outpatient chest pain. We sought to evaluate the relationships between circulating BCAAs, adipose tissue depots (HS and EAT phenotyped from CTA), CAD, and high‐risk plaque (HRP). Given prior literature suggesting the liver as a source of BCAAs, we hypothesized that (1) BCAA levels are associated with HS but not EAT, (2) BCAA levels are associated with CAD, and (3) BCAAs are in the causal pathway of CAD development but not for adipose depot development.
Methods
Study Population
PROMISE was a pragmatic comparative effectiveness randomized trial that enrolled 10 003 patients without known CAD who presented with stable chest pain and were clinically determined to require noninvasive cardiovascular testing. 11 Study participants were randomized to standard of care (ie, stress testing) or a CTA‐guided anatomic arm. The study design and results have been previously described. 12 This study is a biomarker substudy of PROMISE participants who consented and provided biospecimens. A total of 1798 participants included in this study had both diagnostic CTA imaging and nuclear magnetic resonance–based measurement of BCAAs.
Imaging Outcomes
Imaging outcomes were measured centrally by a core laboratory and included obstructive CAD (oCAD), HRP, HS, and EAT volume and were measured from CTA in participants randomized to the CTA arm of PROMISE. As previously described, oCAD was defined as ≥50% stenosis in any major epicardial coronary vessel, while HRP was defined as the presence of positive remodeling (remodeling index >1.1), low computed tomography (CT) attenuation (<30 Hounsfield Units, indicating a lipid‐rich necrotic core), or napkin‐ring sign (peripheral higher attenuation of the noncalcified portion of plaque). 13 To separate the possible influence of oCAD on the relationship between BCAAs and HRP, a sensitivity HRP phenotype where participants with oCAD were excluded from analyses was also derived. EAT was defined by CTA‐measured epicardial adipose volume indexed to body surface area in cm3/m2. Specifically, deep learning was used to segment epicardial adipose volume with two U‐nets to identify the pericardial sac and an attenuation‐based mark to identify EAT. 14 For analyses presented here, EAT was modeled as both a continuous and binary (ie, dichotomized as above the median versus at or below the median value) trait. The presence of HS was defined as the presence of at least 1 of the following CT criteria: hepatic CT attenuation minus splenic CT attenuation of <1 Hounsfield Unit, the mean CT number ratio of liver‐to‐spleen parenchyma of <1, or absolute hepatic CT attenuation of <40 Hounsfield Units. 15
Laboratory Methods
Levels of individual BCAAs (valine, leucine, and isoleucine) in μmol/L were measured by Labcorp, Inc. (formerly LipoScience, Inc.) in frozen plasma samples stored in the PROMISE biorepository. Nuclear magnetic resonance spectra were used as per the nuclear magnetic resonance LipoProfile test with annotation through the nuclear magnetic resonance MetaboloProfile analysis using the LP4 lipoprotein profile deconvolution algorithm as previously described. 16 , 17 Total BCAAs were calculated as the molar sum of the individual BCAAs (which are all generally of similar concentrations in plasma).
Statistical Analysis
To evaluate the association between BCAAs and imaging outcomes, and the role of BCAAs as intermediaries of the potential relationship between them, CAD, and HRP, the following analyses were conducted: (1) association testing between BCAAs with BMI and adipose depots (EAT and HS); (2) association testing between BCAAs with oCAD, HRP, and HRP excluding oCAD; (3) mediation analysis between HS and oCAD (where BCAAs are the mediator); and (4) Mendelian randomization (MR) analyses to determine if BCAAs are in the causal pathway of adipose depots or CAD.
For analyses 1 and 2, individual and total BCAA levels were tested for association with imaging outcomes of interest using univariable and multivariable linear (continuous EAT volume and BMI) and logistic regressions (adjusted for diabetes, age, race, sex, low‐density lipoprotein and high‐density lipoprotein cholesterol, metabolic syndrome, and BMI [for all outcomes except BMI]). For univariable associations that were no longer significant in multivariable regressions, we attempted to identify the covariable(s) that most attenuated the relationship between BCAAs and the outcome by running individual regressions incorporating the BCAAs and each covariable, 1 regression per covariable, and determining which brought the BCAAs estimate/odds ratio (OR) closest to 0/1. In exploratory analyses, these same models were constructed stratified by sex. Variance inflation factors for all multivariable analyses were done to assess for possible collinearity between covariables.
For analysis 3, we tested for a mediation effect of total BCAAs upon the relationship between HS and CAD by first establishing the relationship between HS and CAD, then running a separate regression where BCAAs were included as a covariable to determine whether there was any attenuation. To accomplish this and to be consistent with previous work examining the association between HS and CAD, 15 we analyzed CAD as a 4‐level ordinal outcome (“no,” “mild,” “moderate,” and “severe” CAD) and ran ordinal logistic regression models.
For analysis 4, we performed 2‐sample MR using the TwoSampleMR R package. 18 , 19 Genetic instruments previously shown to be associated with individual BCAAs were identified from a prior genome‐wide association study of BCAAs. 20 Specifically, we input 4 single nucleotide polymorphisms (SNPs) for isoleucine (rs7678928, rs75950518, rs58101275, and rs1420601), and 1 SNP (rs1440581) each for leucine and valine as our instrumental variables. As per the prior study, we did not include the lead SNP for isoleucine (rs1260326, GCKR locus) in analyses due to the loci's established pleiotropic effects. A benefit of using the TwoSampleMR package is its built‐in integration with the IEU genome‐wide association study Database (https://gwas.mrcieu.ac.uk/), which we used for the estimates of the association between our instrumental variables and outcomes. Outcomes of interest were defined as follows: for HS, we used ID “finn‐b‐NAFLD” to select results from a FinnGen (https://www.finngen.fi/en) genome‐wide association study of nonalcoholic fatty liver disease (NAFLD), and for oCAD, we used the ID “ebi‐a‐GCST005195” to select results from a CARDIoGRAMplusC4D and UK Biobank genome‐wide association study of CAD. 21 For the CAD analyses, rs1420601 (isoleucine) was unavailable and a proxy SNP was used in its place (rs1420598). Default parameters were used for all TwoSampleMR functions. For isoleucine, which used multiple SNPs, the inverse‐variance weighted method was treated as the primary test, with the other estimates serving as sensitivity estimates.
For all analyses (excluding MR), individual and total BCAA levels were transformed to Z scores, and all estimates thus represent 1 SD change in individual or total BCAAs. Similarly, continuous EAT volume and BMI were also standardized to a Z score. Nominal, unadjusted P values <0.05 were considered significant. All analyses were performed in R (R Foundation for Statistical Computing, Vienna, Austria). 22
Participants in this substudy gave consent to the parent clinical trial (PROMISE) and for use of biospecimens, and the study was approved by the Duke University Institutional Review Board. The data that support the findings of this study are available from the corresponding author upon reasonable request and review by the PROMISE committees. PROMISE data sets are available at https://biolincc.nhlbi.nih.gov/studies/promise. There are no commercial use data restrictions and no data restrictions based on area of research.
Results
A total of 1798 PROMISE participants were included in this study. The baseline characteristics of the study sample and the parent PROMISE trial are presented in Table 1. The mean age for the substudy was 60.2 (SD, 8.0); 951 (52.9%) participants were women; and of self‐reported racial or ethnic groups, 1568 (87.6%) participants were self‐reported White, 154 (8.6%) were self‐reported Black, 30 (1.7%) were self‐reported Asian, 13 (0.7%) were self‐reported American Indian or Alaska Native, and 148 (8.2%) reported Hispanic or Latino ethnicity. The mean BMI for the study population was 30.6 (SD, 5.9), 250 (13.9%) had oCAD, and 277 (15.4%) had HRP. Of the participants with CT imaging available to characterize HS (n=1521) and EAT volume (n=1505), 411 (27.0%) had HS, and their mean EAT volume was 57.3 cm3/m2 (SD, 21.3).
Table 1.
Baseline Characteristics of PROMISE Clinical Trial Participants and for Biomarker Substudy
| Characteristic | Overall PROMISE | Biomarker and CTA sub‐study |
|---|---|---|
| (N=10 003) | (N=1798) | |
| Age, y, mean (SD) | 60.8 (8.3) | 60.2 (8.0) |
| Female Sex, n (%) | 5270 (52.7) | 951 (52.9) |
| Race, n (%) | ||
| White | 8371 (84.4) | 1568 (87.6) |
| Black or African American | 1096 (11.1) | 154 (8.6) |
| Asian | 253 (2.6) | 30 (1.7) |
| American Indian or Alaska Native | 71 (0.7) | 13 (0.7) |
| Native Hawaiian or Other Pacific Islander | 30 (0.3) | 4 (0.2) |
| Multiracial | 95 (1.0) | 21 (1.2) |
| Hispanic or Latino ethnicity, n (%) | 767 (7.7) | 148 (8.2) |
| BMI, kg/m2, mean (SD) | 30.5 (6.1) | 30.6 (5.9) |
| Diabetes, n (%) | 2144 (21.4) | 365 (20.3) |
| Metabolic Syndrome, n (%) | 3772 (37.7) | 672 (37.4) |
| HDL‐C, mg/dL, mean (SD) | 53.5 (13.8) | 53.5 (13.6) |
| LDL‐C, mg/dL, mean (SD) | 120.9 (33.9) | 121 (34.8) |
| oCAD, n/total n* (%) | 615/4403 (14.0) | 250/1798 (13.9) |
| HRP, n/total n* (%) | 676/4403 (15.4) | 277/1798 (15.4) |
| EAT, cm3/m2, mean (SD)* | 57.5 (22.0) | 57.3 (21.3) |
| HS, n/total n* (%) | 959/3756 (25.5) | 411/1521 (27.0) |
| Total BCAAs, μmol/L, mean (SD) | 468.7 (111.4) | |
| Leucine, μmol/L, mean (SD) | 171.5 (43.3) | |
| Isoleucine, μmol/L, mean (SD) | 52.0 (24.4) | |
| Valine, μmol/L, mean (SD) | 245.2 (53.5) | |
BCAAs indicates branched‐chain amino acids; BMI, body mass index; CTA, computed tomography angiography; EAT, epicardial adipose tissue (volume); HDL‐C, high‐density lipoprotein cholesterol; HRP, high‐risk plaque; HS, hepatic steatosis; LDL‐C, low‐density lipoprotein cholesterol; oCAD, obstructive coronary artery disease; and PROMISE, Prospective Multicenter Imaging Study for Evaluation of Chest Pain.
Available only in individuals randomized to CTA.
BCAA Associations With BMI, HS, and EAT
Total and individual BCAA levels were associated with BMI in univariable (total BCAAs beta, 0.22 [95% CI, 0.18–0.27]; P=3×10−21) and multivariable models (total BCAAs beta, 0.12 [95% CI, 0.08–0.17]; P=4×10−8; Table 2). These associations remained significant in both sexes in sex‐stratified analyses (Table S1), suggesting that these findings are relevant to both women and men. Total and individual BCAAs were also associated with HS in univariable models (total BCAAs OR, 1.70 [95% CI, 1.50–1.92]; P=1×10−17) and remained significant in multivariable models (total BCAAs OR, 1.46 [95% CI, 1.28–1.67]; P=2×10−8; Figure 1, Table 3). Total and individual BCAA levels remained significantly associated with HS in both sexes in sex‐stratified analyses (Table S1).
Table 2.
Associations Between Total and Individual BCAAs and Body Mass Index
| BCAA | Univariable model | Multivariable model | ||
|---|---|---|---|---|
| Beta (95% CI) | P value | Beta (95% CI) | P value | |
| Total | 0.22 (0.18–0.27) | 3×10−21 | 0.12 (0.08–0.17) | 4×10−8 |
| Valine | 0.24 (0.19–0.28) | 3×10−24 | 0.14 (0.10–0.19) | 4×10−10 |
| Leucine | 0.18 (0.14–0.23) | 1×10−14 | 0.09 (0.04–0.13) | 9×10−5 |
| Isoleucine | 0.17 (0.12–0.22) | 6×10−13 | 0.09 (0.05–0.14) | 2×10−5 |
Effect size is per 1 SD change in the BCAA. Multivariable model adjusted for diabetes, age, race, sex, low‐density lipoprotein cholesterol and high‐density lipoprotein cholesterol, and metabolic syndrome. BCAA indicates branched‐chain amino acid.
Figure 1. Associations of total and individual BCAAs with hepatic steatosis in univariable and multivariable models.
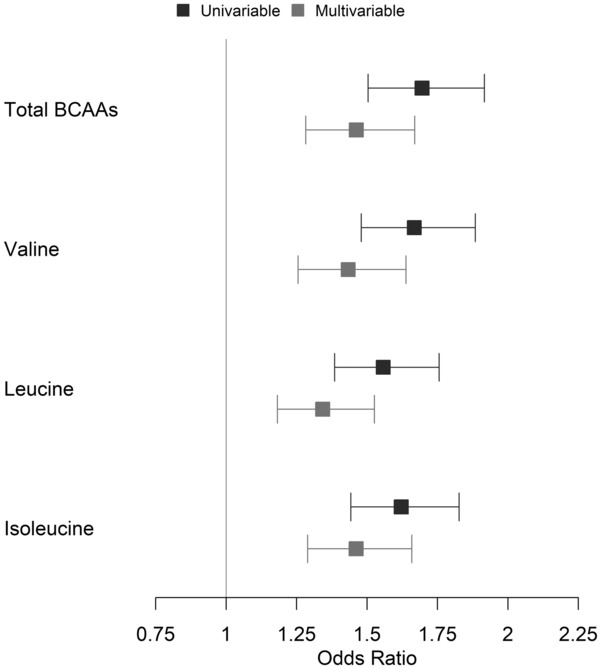
Odds ratio is per 1 SD change in the BCAA. BCAAs indicates branched‐chain amino acids.
Table 3.
Associations Between Total and Individual BCAAs and Hepatic Steatosis
| BCAA | Univariable model | Multivariable model | ||
|---|---|---|---|---|
| OR (95% CI) | P value | OR (95% CI) | P value | |
| Total | 1.70 (1.50–1.92) | 1×10−17 | 1.46 (1.28–1.67) | 2×10−8 |
| Valine | 1.67 (1.48–1.88) | 1×10−16 | 1.43 (1.26–1.64) | 1×10−7 |
| Leucine | 1.56 (1.38–1.76) | 2×10−13 | 1.34 (1.18–1.53) | 6×10−6 |
| Isoleucine | 1.62 (1.44–1.83) | 1×10−15 | 1.46 (1.29–1.66) | 3×10−9 |
Effect size is 1 SD change in the BCAA. Multivariable model adjusted for diabetes, age, race, sex, low‐density lipoprotein cholesterol and high‐density lipoprotein cholesterol, metabolic syndrome, and body mass index. BCAA indicates branched‐chain amino acid; and OR, odds ratio.
Total BCAAs were also positively associated with EAT in univariable models (beta [continuous EAT], 0.09 [95% CI, 0.03–0.14]; P=0.001; OR [binary EAT], 1.18 [95% CI, 1.07–1.32]; P=0.002), as were individual valine and leucine levels. However, only leucine remained significant in multivariable models (beta, 0.07 [95% CI, 0.02–0.12]; P=0.008; OR, 1.19 [95% CI, 1.06–1.35]; P=0.003; Table 4). The inclusion of BMI attenuated the relationships between total BCAAs and valine with binary and continuous EAT (Table S2). Furthermore, the addition of metabolic syndrome attenuated the relationship between valine with binary and continuous EAT. In sex‐stratified analyses, the associations between total and individual BCAAs with EAT were significant in both univariable and multivariable in women but not in men (Table S1), suggesting that while BCAAs are not associated with EAT overall, they may be associated with EAT even after adjustment for BMI in women.
Table 4.
Associations Between BCAAs and EAT Volume
| BCAA | Continuous EAT volume | Dichotomous EAT volume | ||||||
|---|---|---|---|---|---|---|---|---|
| Univariable model | Multivariable model | Univariable model | Multivariable model | |||||
| Beta (95% CI) | P value | Beta (95% CI) | P value | OR (95% CI) | P value | OR (95% CI) | P value | |
| Total | 0.09 (0.03 to 0.14) | 0.001 | 0.05 (0.00 to 0.10) | 0.07 | 1.18 (1.07–1.32) | 0.002 | 1.12 (1.00–1.27) | 0.06 |
| Valine | 0.07 (0.02 to 0.12) | 0.008 | 0.03 (−0.02 to 0.08) | 0.3 | 1.13 (1.02–1.26) | 0.02 | 1.06 (0.94–1.19) | 0.3 |
| Leucine | 0.11 (0.06 to 0.16) | 5×10−5 | 0.07 (0.02 to 0.12) | 0.008 | 1.26 (1.13–1.40) | 2×10−5 | 1.19 (1.06–1.35) | 0.003 |
| Isoleucine | 0.05 (−0.01 to 0.10) | 0.08 | … | … | 1.10 (0.99–1.22) | 0.07 | … | … |
Dichotomous is defined as above or at or below the median, continuous EAT is in SD units. Effect size is per 1 SD change in the BCAA. Multivariable model adjusted for diabetes, age, race, sex, low‐density lipoprotein cholesterol and high‐density lipoprotein cholesterol, metabolic syndrome, and body mass index. BCAA indicates branched‐chain amino acid; EAT, epicardial adipose tissue; and OR, odds ratio.
BCAA Associations With Obstructive CAD and HRP
Given these results, we then evaluated whether BCAAs were associated with oCAD and HRP. Total and individual BCAAs were associated with oCAD in univariable models (total BCAAs OR, 1.18 [95% CI, 1.04–1.34]; P=0.009), but the associations were attenuated in multivariable models (Table 5). High‐density lipoprotein cholesterol, sex, and diabetes were the strongest and most common attenuating covariables for the relationship between BCAAs and oCAD (Table S2). In exploratory sex‐stratified analyses, BCAAs were associated with oCAD in both univariable and multivariable models in women but not in men (Table S1).
Table 5.
Associations Between Total and Individual BCAA Levels and Obstructive Coronary Artery Disease and High‐Risk Plaque
| BCAA | Outcome | Univariable model | Multivariable model | ||
|---|---|---|---|---|---|
| OR (95% CI) | P value | OR (95% CI) | P value | ||
| Total | oCAD | 1.18 (1.04–1.34) | 0.009 | 1.06 (0.91–1.23) | 0.4 |
| Valine | 1.14 (1.00–1.30) | 0.05 | 1.01 (0.86–1.17) | 0.9 | |
| Leucine | 1.20 (1.05–1.35) | 0.005 | 1.09 (0.94–1.25) | 0.3 | |
| Isoleucine | 1.17 (1.03–1.33) | 0.01 | 1.10 (0.96–1.27) | 0.2 | |
| Total | High‐risk plaque | 1.07 (0.94–1.21) | 0.3 | … | … |
| Valine | 1.06 (0.93–1.20) | 0.4 | … | … | |
| Leucine | 1.10 (0.97–1.24) | 0.1 | … | … | |
| Isoleucine | 1.00 (0.88–1.13) | 0.99 | … | … | |
| Total | High‐risk plaque (without oCAD) | 1.05 (0.90–1.23) | 0.5 | … | … |
| Valine | 1.07 (0.91–1.25) | 0.4 | … | … | |
| Leucine | 1.10 (0.93–1.28) | 0.3 | … | … | |
| Isoleucine | 0.92 (0.77–1.08) | 0.3 | … | … | |
Effect size is 1 SD change in the BCAA. Multivariable model adjusted for diabetes, age, race, sex, low‐density lipoprotein cholesterol and high‐density lipoprotein cholesterol, metabolic syndrome, and body mass index. BCAA indicates branched‐chain amino acid; oCAD, obstructive coronary artery disease; and OR, odds ratio.
Total and individual BCAAs were not associated with HRP, either allowing for (total BCAAs OR, 1.07 [95% CI, 0.94–1.21]; P=0.3) or excluding oCAD (total BCAAs OR, 1.05 [95% CI, 0.90–1.23]; P=0.5; Table 5), including in sex‐stratified analyses (Table S1).
The variance inflation factors for all multivariable analyses were <5 (max of 1.8), so collinearity is unlikely to have affected our results.
BCAAs Mediate the Association Between HS and oCAD
Given prior results showing that HS is associated with oCAD, 15 and our results showing that HS is associated with BCAA levels, we then evaluated whether BCAAs mediate the association between HS and oCAD. In these analyses, we found that the inclusion of BCAAs attenuated the association between HS and oCAD (univariable HS P=0.01 and after inclusion of BCAAs, HS P=0.08) with an 8% decrease in the OR. However, these associations were not significant in multivariable models (without BCAAs, HS P=0.1 and after inclusion of BCAAs, HS P=0.2).
Two‐Sample MR
Given the association with BCAAs with HS in multivariable and with oCAD in univariable models, we sought to understand whether BCAAs were in the causal pathway for these diseases using 2‐sample MR analyses. We did not find evidence that valine, leucine, or isoleucine were within the causal pathway of NAFLD (valine OR, 1.26 [95% CI, 0.48–3.34]; P=0.6; leucine OR, 1.33 [95% CI, 0.41–4.30]; P=0.6; isoleucine inverse‐variance weighted OR, 0.91 [95% CI, 0.48–1.74]; P=0.8; Table 6). Similarly, we did not observe evidence of BCAAs being within the causal pathway of CAD (valine OR, 1.08 [95% CI, 0.94–1.25]; P=0.3; leucine OR, 1.10 [95% CI [0.93–1.30], P=0.3; isoleucine inverse‐variance weighted OR, 1.08 [95% CI, 0.99–1.19]; P=0.09).
Table 6.
Mendelian Randomization Results for BCAAs With NAFLD
| BCAA exposure | Method | NAFLD | CAD | ||
|---|---|---|---|---|---|
| OR (95% CI) | P value | OR (95% CI) | P value | ||
| Valine | Wald ratio | 1.26 (0.48–3.34) | 0.6 | 1.08 (0.94–1.25) | 0.3 |
| Leucine | Wald ratio | 1.33 (0.41–4.30) | 0.6 | 1.10 (0.93–1.30) | 0.3 |
| Isoleucine | MR egger | 1.49 (0.02–114.96) | 0.9 | 0.84 (0.45–1.58) | 0.6 |
| Weighted median | 1.05 (0.48–2.28) | 0.9 | 1.11 (0.99–1.23) | 0.07 | |
| Inverse variance weighted | 0.91 (0.48–1.74) | 0.8 | 1.08 (0.99–1.19) | 0.09 | |
| Simple mode | 1.13 (0.38–3.33) | 0.8 | 1.12 (0.97–1.28) | 0.2 | |
| Weighted mode | 1.08 (0.40–2.91) | 0.9 | 1.11 (0.97–1.27) | 0.2 | |
BCAA indicates branched‐chain amino acid; CAD, coronary artery disease; MR, Mendelian randomization; NAFLD, nonalcoholic fatty liver disease; and OR, odds ratio.
Discussion
Leveraging a unique large clinical trial of noninvasive imaging in patients with stable chest pain, we sought to test the hypothesis that BCAA biology plays an intermediary role in the relationship between CTA‐determined adiposity traits with oCAD and HRP. We found that total BCAAs are associated with BMI, HS, and EAT and that the association between BCAAs with HS remained significant after adjustment of covariables, notably including BMI. However, we did not find evidence that BCAAs are in the causal pathway of HS. BCAAs were also associated with oCAD but not HRP, but unlike prior studies, this association was attenuated in multivariable models, and BCAAs did not appear to be on the causal pathway for CAD. Our results corroborate prior studies in other cohorts showing an association between BCAAs and BMI and HS, but we extend these findings by analyzing BCAA associations with EAT and conducting MR. Our results support a model whereby HS is associated with circulating BCAA levels even after adjustment for total body adiposity (ie, BMI), and that BCAAs are not causal for HS, 7 but that BCAA associations with EAT are likely primarily related to total body adiposity (and not EAT specifically) given attenuation in models inclusive of BMI. Furthermore, our results illustrate that BCAAs mediate the relationship between HS and CAD but are not directly in the causal pathway for CAD, suggesting that other effects along the pathway either proximal or distal to BCAAs and CAD are responsible for the mediation effect (Figure 2).
Figure 2. Conceptual model.
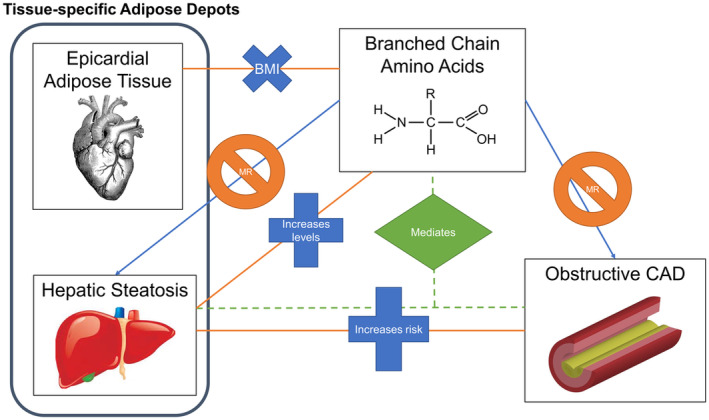
We analyzed the relationship between levels of BCAAs with tissue‐specific adipose depots (EAT and HS) and oCAD. We looked at both associations (orange lines), causal relationships (blue arrows), and mediation analyses (green dotted lines). Our results support a model whereby BCAAs are associated with HS but are not in the causal pathway. This supports current literature implicating BCAA by‐products as causal in development of HS. BCAAs were also found to mediate the association between HS and oCAD; however, BCAAs are not in the causal pathway for oCAD, suggesting that the mediation results we see are due to a proximal (ie, obesity) or distal (ie, branched‐chain keto acids) effect rather than BCAAs themselves. Furthermore, BCAA association with EAT disappeared after adjustment for total body adiposity (body mass index). BCAAs indicates branched‐chain amino acids; EAT, epicardial adipose tissue; HS, hepatic steatosis; MR, Mendelian randomization; and oCAD, obstructive coronary artery disease.
Our results for the association between BCAAs with BMI and HS are overall consistent with prior studies implicating BCAAs in metabolic diseases. For example, we previously identified circulating BCAAs as metabolites most significantly associated with obesity and insulin resistance. 23 Studies have also established circulating BCAAs as a biomarker for metabolic health and weight loss. We have also previously shown that BCAAs are novel biomarkers to differentiate metabolic wellness with BCAAs, discriminating individuals who are metabolically well versus unwell, independent of BMI and other traditional metabolic syndrome features. 8 Organ‐specific adipose depots may serve as more granular markers of CVD risk through their endocrine and paracrine effects. In fact, HS has been shown to be an independent risk factor for CAD and incident CVD events. 15 , 24 Our results specifically implicate BCAA biology in HS. Prior studies have shown that increasing levels of individual BCAAs are associated with worsening degrees of HS (progression from normal, steatosis, to steatohepatitis). 25 , 26 One study of fatty liver hemorrhagic syndrome in hens found that excessive dietary valine leads to adverse metabolic responses that were mediated by amino acid and fatty acid metabolism dysfunction. 27 Another study evaluating individuals with NAFLD without diabetes compared with healthy controls found higher levels of amino acids including BCAAs in individuals with NAFLD, who were also more insulin resistant, suggesting that higher amino acid concentrations in HS may be related to defects in glucose metabolism and increased muscle breakdown during fasting states, 28 also supported by other studies. 29 Data have suggested that BCAAs are involved in many insulin signaling pathways in the liver and may increase hepatic insulin resistance through rapamycin signaling or mitochondrial dysfunction. 23 , 30
Furthermore, our exploratory sex‐stratified analyses highlight that this relationship exists in both sexes. Interestingly, prior research has suggested that BCAA levels increase more across increasing HS disease severity in women as compared with men. 31 While our sex‐stratified analyses were exploratory, our results of higher effect sizes for the association between BCAAs and HS are concordant with this study. We note that these results should be viewed with caution given the exploratory nature and overlap of CIs between men and women.
Interestingly, our MR results did not support a causal role of BCAAs in HS, suggesting that BCAAs are a by‐product of hepatic fat accumulation or related to common metabolic risk factors such as obesity. Recent work in preclinical models suggests that branched‐chain keto acids, a by‐product of BCAA catabolism in the setting of downregulation of branched‐chain alpha‐keto acid dehydrogenase complex (the first irreversible step of BCAA mitochondrial metabolism) may be causal for HS. 32 This is supported by our recent work showing higher expression of the branched‐chain alpha‐keto acid dehydrogenase complex inhibitory kinase and lower expression of the branched‐chain alpha‐keto acid dehydrogenase complex stimulating phosphatase PPM1K. 7
With regard to other organ‐specific adipose depots, we found that total BCAAs were associated with EAT in univariable models, but only the individual BCAA leucine was associated with EAT in multivariable models. BMI was the strongest attenuator of the relationship between BCAAs and EAT, suggesting that the association between BCAAs and EAT was confounded by total body adiposity and not because of a direct relationship between BCAAs and EAT. EAT has also been shown to be an independent risk factor for oCAD 14 and thus appears to be an important cardiometabolic trait, although it is related to total body adiposity. Our sex‐stratified results suggest that BCAAs are associated with EAT even after adjustment for BMI in women but not men; these results should be viewed with caution, given the exploratory nature of the analyses, but warrant further investigation in other studies. To our knowledge, no prior study has evaluated the association between BCAAs and EAT. These results support and extend the role of BCAA catabolism in obesity and HS but suggest that BCAA biology does not underlie the relationship between EAT and cardiovascular disease.
Our results did not support a significant association between BCAAs with oCAD after adjustment for covariables. While BCAAs were significantly associated with oCAD in univariable models, the association was most consistently and strongly attenuated by the inclusion of sex and high‐density lipoprotein cholesterol covariables. Interestingly, in sex‐stratified analyses the association between BCAAs and oCAD remained significant even after adjustment for BMI in multivariable models in women but not men. Again, these results should be viewed with caution, given the exploratory nature, but suggest sex‐dependent differences in the relationship between BCAAs and oCAD that could be pursued in future studies. Regardless, using 2‐sample MR, we find that BCAAs are not in the causal pathway of CAD.
Prior studies have demonstrated an association between higher BCAA levels with risk of oCAD, 10 even after adjusting for metabolic health and BMI. 33 Multiple subsequent mechanistic studies have established the underlying molecular pathways for the association between BCAAs, metabolic traits, and CAD. 23 , 34 The association was shown in patients diagnosed with CAD via coronary angiography in a Beijing hospital from 2008 to 2011. 35 In patients with ST‐segment–elevation myocardial infarction and heart failure, BCAAs were an independent predictor for adverse cardiovascular events over a 3‐year follow‐up. 36 In a murine model, induction of myocardial infarction resulted in an increase in myocardial BCAA levels and caused adverse effects on cardiac function and structure via remodeling. 37 Our discordant results may be related to the fact that the PROMISE study population is an outpatient cohort without known cardiovascular disease at the time of enrollment and therefore is a healthier population than prior studies. For example, only 21% of PROMISE study participants have diabetes compared with Bhattacharya and colleagues' 34%. 10
While BCAAs were not found to be in the causal pathway of CAD, we did demonstrate that they mediated the association between HS and CAD. This indicates that BCAAs may be related to an effect that is the true mediation between HS and CAD, that is, one that is proximal or distal to the direct relationship of BCAAs to CAD. For example, obesity is a known risk factor for CAD, HS, and higher BCAA levels 8 and could account for this mediation effect. Alternatively, branched‐chain keto acids, a by‐product of mitochondrial BCAA catabolism that have been suggested to be causal to HS, 7 may account for a downstream effect to CAD development. This idea is further supported by the results that showed the association between BCAAs and HS disappeared in the multivariable model. As such, BCAAs can mediate HS and CAD while not being in the causal pathway to CAD.
While this is the largest study of the interrelationships between BCAAs with HS, EAT, and oCAD, important limitations should be noted. Our study population is one of outpatients with stable chest pain and, as such, our results are not generalizable to other populations, for example, those with prevalent CVD or presenting with unstable angina. While CT is a well‐established noninvasive imaging method for evaluating HS, the CTA protocol used only the imaged part of the liver, and the clinical gold standard for HS diagnosis is liver biopsy with histologic confirmation. Further, it is not possible within the PROMISE cohort to differentiate between HS as a result of alcoholic fatty liver disease or hepatitis C. However, given the relatively low percentage of alcoholic fatty liver disease and hepatitis C compared with NAFLD in the general population, 38 , 39 it can be presumed that few participants in the PROMISE study had these as a cause of their HS. Furthermore, our study could not evaluate how waist circumference or waist‐to‐hip ratio played a role in these relationships as these data were not collected on PROMISE participants. However, future studies could examine how these other easily obtained markers of adiposity affect these relationships.
In conclusion, we demonstrate the heterogeneous role of BCAAs depending on the organ specificity of the adipose tissue. Specifically, we confirm the role of BCAAs in the biology of HS by demonstrating that circulating BCAA levels are associated with HS after adjustment for BMI and other factors but show that BCAAs do not appear to play a role in the association between EAT with CVD.
Sources of Funding
The project described was supported by grant numbers 1R01HL098237, 1R01HL098236, 1R01HL098305, and 1R01HL098235 from the National Heart, Lung, and Blood Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosures
Dr Pagidipati: research support from Amgen, Boehringer Ingelheim, Eggland's Best, Eli Lilly, Novartis, Novo Nordisk, Verily Life Sciences; consultation/advisory panels for Boehringer Ingelheim, CRISPR Therapeutics, Eli Lilly, Esperion, AstraZeneca, Merck, Novartis, and Novo Nordisk; executive committee member for trials sponsored by Novo Nordisk and by Amgen. Dr Ferencik: consulting Siemens Healthineers, Elucid, HeartFlow. Dr Shah: sponsored research funding from Verily Inc., AstraZeneca, and Lilly; unlicensed patent related to the findings held by Duke University. Dr Voora: research grant funding from Abbott Diagnostics. Dr Foldyna: research support from AstraZeneca, MedImmune, and MedTrace. Dr Hoffmann: employment and stock options for Cleerly Inc.; consultant fees to Innovative Imaging Consulting: Stanford University, Clinical Cardiovascular Sciences, Rapid AI, and Medtrace Inc. The authors report no other pertinent disclosures.
Supporting information
Tables S1–S2
This article was sent to Erik B. Schelbert, Associate Editor, for review by expert referees, editorial decision, and final disposition.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.122.028410
For Sources of Funding and Disclosures, see page 9.
References
- 1. Bermudez V, Duran P, Rojas E, Diaz MP, Rivas J, Nava M, Chacin M, Cabrera de Bravo M, Carrasquero R, Ponce CC, et al. The sick adipose tissue: new insights into defective signaling and crosstalk with the myocardium. Front Endocrinol (Lausanne). 2021;12:735070. doi: 10.3389/fendo.2021.735070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shah RV, Yeri AS, Murthy VL, Massaro JM, D'Agostino RB Sr, Freedman JE, Long MT, Fox CS, Das S, Benjamin EJ, et al. Association of multiorgan computed tomographic phenomap with adverse cardiovascular health outcomes: the Framingham heart study. JAMA Cardiol. 2017;2:1236–1246. doi: 10.1001/jamacardio.2017.3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Davis TME. Diabetes and metabolic dysfunction‐associated fatty liver disease. Metabolism. 2021;123:154868. doi: 10.1016/j.metabol.2021.154868 [DOI] [PubMed] [Google Scholar]
- 4. Obokata M, Borlaug BA. Response by Obokata and Borlaug to letters regarding article, "evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction." Circulation. 2018;137:416–417. doi: 10.1161/CIRCULATIONAHA.117.031394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van Woerden G, Gorter TM, Westenbrink BD, Willems TP, van Veldhuisen DJ, Rienstra M. Epicardial fat in heart failure patients with mid‐range and preserved ejection fraction. Eur J Heart Fail. 2018;20:1559–1566. doi: 10.1002/ejhf.1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Trico D, Biancalana E, Solini A. Protein and amino acids in nonalcoholic fatty liver disease. Curr Opin Clin Nutr Metab Care. 2021;24:96–101. doi: 10.1097/MCO.0000000000000706 [DOI] [PubMed] [Google Scholar]
- 7. Grenier‐Larouche T, Coulter Kwee L, Deleye Y, Leon‐Mimila P, Walejko JM, McGarrah RW, Marceau S, Trahan S, Racine C, Carpentier AC, et al. Altered branched‐chain alpha‐keto acid metabolism is a feature of NAFLD in individuals with severe obesity. JCI Insight. 2022;7:7. doi: 10.1172/jci.insight.159204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Batch BC, Shah SH, Newgard CB, Turer CB, Haynes C, Bain JR, Muehlbauer M, Patel MJ, Stevens RD, Appel LJ, et al. Branched chain amino acids are novel biomarkers for discrimination of metabolic wellness. Metabolism. 2013;62:961–969. doi: 10.1016/j.metabol.2013.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. White PJ, McGarrah RW, Herman MA, Bain JR, Shah SH, Newgard CB. Insulin action, type 2 diabetes, and branched‐chain amino acids: a two‐way street. Mol Metab. 2021;52:101261. doi: 10.1016/j.molmet.2021.101261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bhattacharya S, Granger CB, Craig D, Haynes C, Bain J, Stevens RD, Hauser ER, Newgard CB, Kraus WE, Newby LK, et al. Validation of the association between a branched chain amino acid metabolite profile and extremes of coronary artery disease in patients referred for cardiac catheterization. Atherosclerosis. 2014;232:191–196. doi: 10.1016/j.atherosclerosis.2013.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Douglas PS, Hoffmann U, Lee KL, Mark DB, Al‐Khalidi HR, Anstrom K, Dolor RJ, Kosinski A, Krucoff MW, Mudrick DW, et al. PROspective multicenter imaging study for evaluation of chest pain: rationale and design of the PROMISE trial. Am Heart J. 2014;167:796–803.e1. doi: 10.1016/j.ahj.2014.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Douglas PS, Hoffmann U, Patel MR, Mark DB, Al‐Khalidi HR, Cavanaugh B, Cole J, Dolor RJ, Fordyce CB, Huang M, et al. Outcomes of anatomical versus functional testing for coronary artery disease. N Engl J Med. 2015;372:1291–1300. doi: 10.1056/NEJMoa1415516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ferencik M, Mayrhofer T, Bittner DO, Emami H, Puchner SB, Lu MT, Meyersohn NM, Ivanov AV, Adami EC, Patel MR, et al. Use of high‐risk coronary atherosclerotic plaque detection for risk stratification of patients with stable chest pain: a secondary analysis of the PROMISE randomized clinical trial. JAMA Cardiol. 2018;3:144–152. doi: 10.1001/jamacardio.2017.4973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Foldyna B, Zeleznik R, Eslami P, Mayrhofer T, Ferencik M, Bittner DO, Meyersohn NM, Puchner SB, Emami H, Aerts H, et al. Epicardial adipose tissue in patients with stable chest pain: insights from the PROMISE trial. JACC Cardiovasc Imaging. 2020;13:2273–2275. doi: 10.1016/j.jcmg.2020.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Meyersohn NM, Mayrhofer T, Corey KE, Bittner DO, Staziaki PV, Szilveszter B, Hallett T, Lu MT, Puchner SB, Simon TG, et al. Association of hepatic steatosis with major adverse cardiovascular events, independent of coronary artery disease. Clin Gastroenterol Hepatol. 2021;19:1480–1488.e14. doi: 10.1016/j.cgh.2020.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jeyarajah EJ, Cromwell WC, Otvos JD. Lipoprotein particle analysis by nuclear magnetic resonance spectroscopy. Clin Lab Med. 2006;26:847–870. doi: 10.1016/j.cll.2006.07.006 [DOI] [PubMed] [Google Scholar]
- 17. Matyus SP, Braun PJ, Wolak‐Dinsmore J, Jeyarajah EJ, Shalaurova I, Xu Y, Warner SM, Clement TS, Connelly MA, Fischer TJ. NMR measurement of LDL particle number using the Vantera clinical analyzer. Clin Biochem. 2014;47:203–210. doi: 10.1016/j.clinbiochem.2014.07.015 [DOI] [PubMed] [Google Scholar]
- 18. Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R, et al. The MR‐base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408. doi: 10.7554/eLife.34408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hemani G, Tilling K, Davey Smith G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 2017;13:e1007081. doi: 10.1371/journal.pgen.1007081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lotta LA, Scott RA, Sharp SJ, Burgess S, Luan J, Tillin T, Schmidt AF, Imamura F, Stewart ID, Perry JR, et al. Genetic predisposition to an impaired metabolism of the branched‐chain amino acids and risk of type 2 diabetes: a mendelian randomisation analysis. PLoS Med. 2016;13:e1002179. doi: 10.1371/journal.pmed.1002179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van der Harst P, Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res. 2018;122:433–443. doi: 10.1161/CIRCRESAHA.117.312086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. R Core Team . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2021. https://www.R‐project.org/ [Google Scholar]
- 23. Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, et al. A branched‐chain amino acid‐related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9:311–326. doi: 10.1016/j.cmet.2009.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mellinger JL, Pencina KM, Massaro JM, Hoffmann U, Seshadri S, Fox CS, O'Donnell CJ, Speliotes EK. Hepatic steatosis and cardiovascular disease outcomes: an analysis of the Framingham heart study. J Hepatol. 2015;63:470–476. doi: 10.1016/j.jhep.2015.02.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lake AD, Novak P, Shipkova P, Aranibar N, Robertson DG, Reily MD, Lehman‐McKeeman LD, Vaillancourt RR, Cherrington NJ. Branched chain amino acid metabolism profiles in progressive human nonalcoholic fatty liver disease. Amino Acids. 2015;47:603–615. doi: 10.1007/s00726-014-1894-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kalhan SC, Guo L, Edmison J, Dasarathy S, McCullough AJ, Hanson RW, Milburn M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism. 2011;60:404–413. doi: 10.1016/j.metabol.2010.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jian H, Xu Q, Wang X, Liu Y, Miao S, Li Y, Mou T, Dong X, Zou X. Amino acid and fatty acid metabolism disorders trigger oxidative stress and inflammatory response in excessive dietary valine‐induced NAFLD of laying hens. Front Nutr. 2022;9:849767. doi: 10.3389/fnut.2022.849767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gaggini M, Carli F, Rosso C, Buzzigoli E, Marietti M, Della Latta V, Ciociaro D, Abate ML, Gambino R, Cassader M, et al. Altered amino acid concentrations in NAFLD: impact of obesity and insulin resistance. Hepatology. 2018;67:145–158. doi: 10.1002/hep.29465 [DOI] [PubMed] [Google Scholar]
- 29. Pozefsky T, Tancredi RG, Moxley RT, Dupre J, Tobin JD. Effects of brief starvation on muscle amino acid metabolism in nonobese man. J Clin Invest. 1976;57:444–449. doi: 10.1172/JCI108295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sunny NE, Kalavalapalli S, Bril F, Garrett TJ, Nautiyal M, Mathew JT, Williams CM, Cusi K. Cross‐talk between branched‐chain amino acids and hepatic mitochondria is compromised in nonalcoholic fatty liver disease. Am J Physiol Endocrinol Metab. 2015;309:E311–E319. doi: 10.1152/ajpendo.00161.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Grzych G, Vonghia L, Bout MA, Weyler J, Verrijken A, Dirinck E, Chevalier Curt MJ, Van Gaal L, Paumelle R, Francque S, et al. Plasma BCAA changes in patients with NAFLD are sex dependent. J Clin Endocrinol Metab. 2020;105:2311–2321. doi: 10.1210/clinem/dgaa175 [DOI] [PubMed] [Google Scholar]
- 32. White PJ, McGarrah RW, Grimsrud PA, Tso SC, Yang WH, Haldeman JM, Grenier‐Larouche T, An J, Lapworth AL, Astapova I, et al. The BCKDH kinase and phosphatase integrate BCAA and lipid metabolism via regulation of ATP‐citrate lyase. Cell Metab. 2018;27:1281–1293.e7. doi: 10.1016/j.cmet.2018.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ellison S, Abdulrahim JW, Kwee LC, Bihlmeyer NA, Pagidipati N, McGarrah R, Bain JR, Kraus WE, Shah SH. Novel plasma biomarkers improve discrimination of metabolic health independent of weight. Sci Rep. 2020;10:21365. doi: 10.1038/s41598-020-78478-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gander J, Carrard J, Gallart‐Ayala H, Borreggine R, Teav T, Infanger D, College F, Streese L, Wagner J, Klenk C, et al. Metabolic impairment in coronary artery disease: elevated serum acylcarnitines under the spotlights. Front Cardiovasc Med. 2021;8:792350. doi: 10.3389/fcvm.2021.792350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang RY, Wang SM, Sun L, Liu JM, Li HX, Sui XF, Wang M, Xiu HL, Wang S, He Q, et al. Association of branched‐chain amino acids with coronary artery disease: a matched‐pair case‐control study. Nutr Metab Cardiovasc Dis. 2015;25:937–942. doi: 10.1016/j.numecd.2015.06.003 [DOI] [PubMed] [Google Scholar]
- 36. Du X, Li Y, Wang Y, You H, Hui P, Zheng Y, Du J. Increased branched‐chain amino acid levels are associated with long‐term adverse cardiovascular events in patients with STEMI and acute heart failure. Life Sci. 2018;209:167–172. doi: 10.1016/j.lfs.2018.08.011 [DOI] [PubMed] [Google Scholar]
- 37. Wang W, Zhang F, Xia Y, Zhao S, Yan W, Wang H, Lee Y, Li C, Zhang L, Lian K, et al. Defective branched chain amino acid catabolism contributes to cardiac dysfunction and remodeling following myocardial infarction. Am J Physiol Heart Circ Physiol. 2016;311:H1160–H1169. doi: 10.1152/ajpheart.00114.2016 [DOI] [PubMed] [Google Scholar]
- 38. Mann RE, Smart RG, Govoni R. The epidemiology of alcoholic liver disease. Alcohol Res Health. 2003;27:209–219. [PMC free article] [PubMed] [Google Scholar]
- 39. Rosenberg ES, Rosenthal EM, Hall EW, Barker L, Hofmeister MG, Sullivan PS, Dietz P, Mermin J, Ryerson AB. Prevalence of hepatitis C virus infection in US states and the District of Columbia, 2013 to 2016. JAMA Netw Open. 2018;1:e186371. doi: 10.1001/jamanetworkopen.2018.6371 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S2