

Abstract
Background and Objectives
Corticobasal syndrome (CBS) is a clinical phenotype characterized by asymmetric parkinsonism, rigidity, myoclonus, and apraxia. Originally believed secondary to corticobasal degeneration (CBD), mounting clinicopathologic studies have revealed heterogenous neuropathologies. The objectives of this study were to determine the pathologic heterogeneity of CBS, the clinicoradiologic findings associated with different underlying pathologies causing CBS, and the positive predictive value (PPV) of current diagnostic criteria for CBD among patients with a CBS.
Methods
Clinical data, brain MRI, and neuropathologic data of patients followed at Mayo Clinic and diagnosed with CBS antemortem were reviewed according to neuropathology category at autopsy.
Results
The cohort consisted of 113 patients with CBS, 61 (54%) female patients. Mean ± SD disease duration was 7 ± 3.7 years; mean ± SD age at death was 70.5 ± 9.1 years. The primary neuropathologic diagnoses were 43 (38%) CBD, 27 (24%) progressive supranuclear palsy (PSP), 17 (15%) Alzheimer disease (AD), 10 (9%) frontotemporal lobar degeneration (FTLD) with TAR DNA-binding protein 43 (TDP) inclusions, 7 (6%) diffuse Lewy body disease (DLBD)/AD, and 9 (8%) with other diagnoses. Patients with CBS-AD or CBS-DLBD/AD were youngest at death (median [interquartile range]: 64 [13], 64 [11] years) while CBS-PSP were oldest (77 [12.5] years, p = 0.024). Patients with CBS-DLBD/AD had the longest disease duration (9 [6] years), while CBS-other had the shortest (3 [4.25] years, p = 0.04). Posterior cortical signs and myoclonus were more characteristic of patients with CBS-AD and patients with CBS-DLBD/AD. Patients with CBS-DLBD/AD displayed more features of Lewy body dementia. Voxel-based morphometry revealed widespread cortical gray matter loss characteristic of CBS-AD, while CBS-CBD and CBS-PSP predominantly involved premotor regions with greater amount of white matter loss. Patients with CBS-DLBD/AD showed atrophy in a focal parieto-occipital region, and patients with CBS-FTLD-TDP had predominant prefrontal cortical loss. Patients with CBS-PSP had the lowest midbrain/pons ratio (p = 0.012). Of 67 cases meeting clinical criteria for possible CBD at presentation, 27 were pathology-proven CBD, yielding a PPV of 40%.
Discussion
A variety of neurodegenerative disorders can be identified in patients with CBS, but clinical and regional imaging differences aid in predicting underlying neuropathology. PPV analysis of the current CBD diagnostic criteria revealed suboptimal performance. Biomarkers adequately sensitive and specific for CBD are needed.
Corticobasal syndrome (CBS) affects movement, language, cognition, and behavior. Diagnosis may be challenging because of the wide variety of signs and symptoms that may be present in addition to the classic asymmetric akinetic rigid apraxic motor syndrome. CBS was originally believed to be secondary to corticobasal degeneration (CBD), but mounting clinicopathologic studies have revealed heterogenous underlying pathologies, including Alzheimer disease (AD), progressive supranuclear palsy (PSP), Creutzfeldt-Jacob disease (CJD), TAR DNA-binding protein 43 (TDP), and Pick disease.1-4
Increasingly, potentially disease-modifying agents are being studied for their potential to target specific pathologies underlying CBS.5 A barrier to successful treatment trials is the lack of diagnostic biomarkers across the different possible pathologies causing CBS, which is needed to select patients based on predicted pathology.1 Therefore, improving the understanding of the clinical and imaging biomarkers associated with different underlying pathologies in CBS is necessary. It has been suggested that identification of select clinical clues can aid in diagnostic accuracy. For example, earlier age at onset or myoclonus in the early stages of CBS are both more common in patients with Alzheimer pathology.6
CBD, a progressive neurodegenerative disorder characterized by circumscribed cortical atrophy with spongiosis, ballooned neurons and tau inclusion in neurons and glia,7 is diagnosed postmortem in only 23%–50% of patients with CBS.1,2,4,7-9 In the absence of definitive biomarkers for CBD, in 2013, an international consortium of subspecialists proposed an updated set of criteria to better the diagnosis of CBD.10 In 2 studies,11,12 small cohorts of patients with neuropathologically confirmed CBD were used to investigate the sensitivity and specificity of these clinical criteria. As reported in both studies, the criteria lacked specificity, and no clinical feature could reliably distinguish CBD pathology from non-CBD pathology, but larger studies were needed to understand the utility of the criteria.
The results from structural MRI with voxel-based morphometry (VBM) and 18F fluorodeoxyglucose (FDG) PET studies suggest that patterns of atrophy or hypometabolism could be useful in predicting underlying pathology in CBS.3,13-15 Focal asymmetric atrophy in the frontoparietal cortex and in the basal ganglia was found in patients with CBS and autopsy-proven CBD or PSP,3,6 while widespread atrophy pointed toward a pathologic diagnosis of frontotemporal lobar degeneration (FTLD)–TDP or AD, with frontotemporal loss suggestive of FTLD-TDP, and temporoparietal loss suggestive of AD. A structural brainstem biomarker of midbrain/pons ratio has been proven to differentiate PSP from other Parkinsonian disorders,16-18 but whether it can distinguish CBS-PSP from other CBS subtypes remains to be determined. According to Pardini et al.,19 hypometabolism patterns in patients with CBS differ depending on the underlying pathology: Patients with CBS-CBD show a marked basal ganglia involvement, patients with CBS-AD show widespread posterior hypometabolism, and patients with CBS-PSP show an anterior hypometabolic pattern, including the medial frontal and anterior cingulate regions.
The objectives of this study were to study the neuropathologic heterogeneity of CBS, key clinical and neuroimaging measures that can predict neuropathology, whether midbrain/pons ratio is a useful biomarker of pathology in CBS, and the positive predictive value (PPV) of proposed clinical CBD criteria among patients with a probable CBS phenotype.
Methods
Standard Protocol Approvals
Study protocols were approved by the Mayo Clinic Institutional Review Board (approval no. 22-001618).
Study Participants
The Mayo Clinic autopsy database was searched to identify all study participants who had antemortem diagnosis of CBS between 1990 and 2021. Neuropathologic diagnosis was extracted from pathology records of the 113 participants included in the study. These cases were part of a review article reporting the neuropathologic diagnosis of all Florida brain bank cases with a CBS diagnosis by Koga et al.,9 but in this study, we focused on those who were clinically evaluated and treated at Mayo Clinic and included clinical and imaging findings. Demographics and clinical data at presentation and at last visit were abstracted from medical records and included age at onset, age of death, sex, family history (>1 relative with a similar neurodegenerative disease), and symptoms and signs reported by the patient and recorded at the neurologic examination. The following clinical features were recorded as present or absent at presentation and at last visit: dominant involved side, limb rigidity, limb dystonia, myoclonus, apraxia, alien limb phenomenon, right-left disorientation, agraphia, acalculia, finger agnosia, oculomotor apraxia, optic ataxia, simultanagnosia, other visuospatial features (including visual field defect and space/object perception deficit) as well as alexia, dressing apraxia, prosopagnosia, behavioral or personality changes, aphasia, apraxia of speech, postural instability, vertical supranuclear gaze palsy or decreased velocity of vertical saccade, fluctuating cognition, well-formed visual hallucinations, REM sleep behavior disorder (RBD, based on history and not sleep study), parkinsonism, pyramidal signs, gait impairment, episodic memory loss, yes/no speech reversals, and mirror movements of the limbs. Scores on cognitive evaluations (Short Test of Mental Status [STMS]) were recorded.20-22 Disease duration was defined as the difference between the age at onset of the first sign or symptom and the age at death.
Clinical Classification
Accepted clinical diagnostic criteria for CBS and related phenotypes10 were used to verify inclusion in the study. Probable CBS included asymmetric presentation of 2 of the following: (a) limb rigidity or akinesia, (b) limb dystonia, (c) and limb myoclonus plus 2 of (d) orobuccal or limb apraxia, (e) cortical sensory deficit, and (f) alien limb phenomena. Possible CBS included 1 of a–c, 1 of d–f, and permitted symmetry. In 7 participants with overlapping CBS phenotypes (including nonfluent/agrammatic variant of primary progressive aphasia (PPA), PSP syndrome, or frontal behavioral-spatial syndrome), asymmetrical motor core characteristics of CBS were also observed, categorizing them as “possible CBD.”10 Participants with pure PSP or PPA or frontal behavioral syndromes were excluded.
Diagnostic clinical research criteria for probable CBD included insidious onset and gradual progression, minimum duration of 1 year, age at onset 50 years or older, probable CBS, or frontal behavioral-spatial syndrome or nonfluent/agrammatic variant of PPA with at least 1 core CBS feature (a–f), with exclusion of patients with a family history of 2 or more relatives with similar neurologic symptoms or genetic mutation in MAPT.10 Clinical criteria for possible CBD included insidious onset and gradual progression, minimum duration of 1 year, no minimum age at onset, possible CBS or frontal behavioral-spatial syndrome, or nonfluent/agrammatic variant of PPA with at least 1 CBS feature (a–f). Possible CBD permitted family history or genetic mutation.
Neuropathologic Assessment
All neuropathologic examinations were conducted by neuropathologists at Mayo Clinic (R.R.R, D.W.D., A.T.N.), maintaining uniformity. Neuropathologic diagnosis was based on consensus criteria for CBD,23 PSP,24,25 AD,26 FTLD,27 Lewy body dementia (LBD),28,29 argyrophilic grain disease (AGD),30,31 and aging-related tau astrogliopathy (ARTAG).32 Patients who had both DLBD and AD listed as neuropathologic diagnoses or were labeled as “mixed” or “concomitant” were categorized as DLBD/AD. Neuropathologic assessment and diagnosis were completed on either the right or left hemibrains. Hemibrains were weighted and multiplied by 2 to calculate approximate total brain weight.
Radiologic Assessment
Volumetric head MRI was performed on 60 patients. A standardized MRI protocol on either a 3.0 or 1.5 T MRI (GE Healthcare, Waukesha, WI) scanner included a 3-dimensional T1-weighted volumetric sequence (magnetization prepared rapid acquisition gradient echo at 3 T or coronal spoiled gradient-recalled echo sequence at 1.5 T). All scans underwent corrections for intensity inhomogeneity and gradient unwarping before analysis. Patterns of gray matter and white matter atrophy were assessed using automated technique of VBM, with voxel-wise t tests in statistical parametric mapping (SPM) 12 used for statistical comparisons of pathology categories. To account for the asymmetric nature of CBS, the more severely affected (i.e., dominant) hemisphere for each participant was designated by D.S. (contralateral to the most affected limb) using clinical features at time of MRI, without reference to imaging. Each MRI was flipped in the X dimension so that the more severely affected hemisphere was positioned on the left side of the image in all participants, as previously described.33 The control group consisted of 40 healthy participants matched for age and sex distribution at group level who were initially recruited into the Mayo Alzheimer's Disease Patient Registry. Analyses were corrected for multiple comparisons using the family-wise error correction at p < 0.05.
Midbrain and pons area measurements were completed according to published criteria34,35 on T1-weighted MRI images by 1 trained rater (D.S.) using ITK-SNAP software. All measurements were performed blinded to clinical information. Midbrain and pons areas were measured on the first midsagittal slice from the left, where the superior colliculi completely separated from the midbrain, as previously described.17 The ratio was then calculated as midbrain area divided by pons area.35
Available FDG-PET scans of a subset of 28 patients were visually categorized (mean ± SD time from first clinical evaluation to scan: 3.2 ± 10.6 months). For each neuropathologic variant, representative scan was selected. Statistical map shows regions of significant hypometabolism relative to age-matched controls (GE Cortex ID).
Statistical Analyses
All statistical analyses were performed with SPSS statistical software program (version 28.0; IBM Corp., Armonk, NY) with significance established at p < 0.05. Demographic and clinical characteristics were summarized using descriptive statistics. Differences in demographics, neuropathologic findings, cognitive scores, and midbrain/pons ratio were assessed using the Kruskal-Wallis test. Differences in the frequency of diagnostic features across pathology category were evaluated using the Fisher exact test. Voxel-wise t tests in SPM12 were used for statistical comparisons of MRI scans comparing structural differences for each pathology category compared with controls, including age, sex, and field strength as covariates. Deidentified studies using autopsy samples are considered exempt from human subject research by the Mayo Clinic Institutional Review Board.
Data Availability
The data that support the findings of this study are available from the corresponding author, on request.
Results
Patient Characteristics
Patients' demographics and neuropathologic features are summarized in Table 1. The cohort consisted of 113 cases diagnosed with CBS with 61 (54%) female patients and 52 (46%) male patients. Mean ± SD disease duration was 7 ± 3.7 years, and mean ± SD age at death was 70.5 ± 9.1 years. Clinical information at presentation was available for 104 patients; among those, 79 patients were followed by Neurodegenerative Research Group or Alzheimer's Disease Research Center at Mayo Clinic in Rochester, MN, and had longitudinal clinical data available (mean follow-up: 36 months, range 2–99 months).
Table 1.
Demographics, Cognitive, and Neuropathologic Features of Patients With CBS According to Pathology Category

Neuropathology Findings
Neuropathologic diagnoses were distributed as follows: 43 (38%) CBD, 27 (24%) PSP, 17 (15%) AD, 10 (9%) FTLD-TDP, 7 (6%) DLBD/AD, and 9 (8%) with other diagnoses (CBS-other, including 1 case with diffuse AGD, 1 case with fused in sarcoma (FUS)–negative neurofilament inclusion body disease (NIBD), 1 case with FUS-positive FTLD, 1 case with brainstem predominant Lewy body disease, 1 case with multifocal necrotizing leukoencephalopathy, 1 case Pick disease, and 3 cases with Creutzfeldt-Jakob disease). Fourteen (33%) patients with CBS-CBD pathology had secondary findings of AGD, and 13 (30%) had coexisting ARTAG. Copathology in patients with CBS-PSP included 7 (27%) with AGD, 5 (19%) with ARTAG, and 10 (31%) with neocortical amyloid plaques.
Differences in Disease Characteristics and Cognition Across Cliniconeuropathologic Category
Cases categorized as CBS-AD or CBS-DLBD/AD were the youngest at death (median [interquartile range (IQR)]: 64 [13] and 64 [11] years), while cases categorized as CBS-PSP were the oldest (77 [12.5], p = 0.024) (Table 1). CBS-DLBD/AD cases had the longest disease duration (median [IQR]: 9 [6] years), while CBS-other subgroup had the shortest disease duration (3 [4.25] years, p = 0.04). The 3 CBS cases with pathology-proven CJD had shortest time between symptom onset and first neurologic evaluation (mean ± SD: 0.9 ± 0.4 years compared with 2.8 ± 1.8 years, p = 0.048) compared with other CBS cases. The duration of the disease was not different at the first visit, but at the last visit, CBS-AD, CBS-DLBD, and CBS-FTLD-TDP had the longest follow-up times (p = 0.019). STMS scores (global cognition) at presentation did not differ among the groups; at last visit, cognitive scores were lowest for patients with CBS-AD (19 [16.25] points) and patients with CBS-DLBD/AD (14 [12] points) and highest for patients with CBS-CBD (27 [11]) or CBS-FTLD-TDP (31.5 [—], p = 0.027).
Clinical Features at Presentation and at Last Visit
Clinical assessment of symptoms and signs by neuropathologic category are summarized in Figure 1 and eTables 1, 2 (links.lww.com/WNL/C848). Considering that the inclusion criteria for the study required a clinical diagnosis of CBS, all but 4 patients had asymmetrical motor signs at presentation, with no preference for dominant side (p = 0.99). The frequency of CBS core clinical features and motor signs at presentation (Figure 1, panel A) differed between the groups: Myoclonus and apraxia were more frequent in CBS-AD and CBS-DLBD/AD (p = 0.003 and p = 0.017, respectively), while alien limb phenomenon was more frequent in CBS-CBD, CBS-PSP, and CBS-AD subgroups (p = 0.036). Throughout the disease course, myoclonus remained more frequent in CBS-AD (p = 0.006). Episodic memory loss (p < 0.001) and posterior cortical signs were more frequent in patients with AD and DLBD/AD pathology, particularly agraphia (p = 0.002), acalculia (p = 0.003), simultanagnosia (p = 0.005), and other visuospatial features (p < 0.001) at presentation, as well as optic ataxia (p = 0.004) throughout the disease course (Figure 1, panels B, E). Core features of LBD were significantly more frequent in patients with CBS-DLBD/AD (p = 0.01 for visual hallucinations, p = 0.032 for RBD, eTable 2). At last visit, typical PSP features (i.e., features of Richardson syndrome) were more frequent in CBS-PSP (supranuclear gaze palsy or decreased velocity of vertical saccades in 64%, postural instability in 82%), but this trend did not reach statistical significance (p = 0.071, p = 0.085, respectively, eTable 2). Speech/language deficits consistent with apraxia of speech were most frequent throughout the disease course in patients with CBS-CBD (15 [58%]), but this trend did not reach significance (p = 0.101).
Figure 1. Radar Charts of Clinical Features by Neuropathology Diagnosis.

(A–C) At presentation. (D–F) At last visit. Axes present percentage. Panel A: At presentation, myoclonus and apraxia were more frequent in CBS-AD (green, p = 0.003) and CBS-DLBD/AD (light blue, p = 0.017); alien limb phenomenon was more frequent in CBS-CBD (dark blue), CBS-PSP (red), and CBS-AD (green, p = 0.036). Panel B: At presentation, posterior cortical signs were more frequent in CBS-AD (green) and CBS-DLBD/AD (light blue, including agraphia [p = 0.002], acalculia [p = 0.003], simultanagnosia [p = 0.005], and other visuospatial features [p < 0.001]). Panel C: Visual hallucinations were more frequent in CBS-DLBD/AD (p = 0.003). Panel D: At last visit, myoclonus remained more frequent in CBS-AD (green) and CBS-DLBD/AD (blue, p = 0.006). Panel E: At last visit, the above mentioned posterior cortical signs remained more frequent among CBS-AD (green) and CBS-DLBD/AD (light blue), in addition to optic ataxia (p = 0.004). Panel F: At last visit, core features of LBD were more frequent in patients with CBS-DLBD/AD (including visual hallucinations [p = 0.01], RBD [p = 0.032]); episodic memory loss was more frequent among CBS-AD (green) and CBS-DLBD/AD (p < 0.001); typical PSP features were more frequent in CBS-PSP (red), and apraxia of speech was more frequent in CBS-CBD (dark blue), but these trends did not reach statistical significance. For Panels B and E: Other visuospatial features at presentation include visual field defect, space/object perception deficit, alexia, dressing apraxia, and prosopagnosia. For Panels A–C: CBD (n = 38), PSP (n = 26), AD (n = 17), FTLD-TDP43 (n = 8), DLBD/AD (n = 7). For Panels D–F: CBD (n = 26), PSP (n = 22), AD (n = 15), FTLD-TDP43 (n = 7), DLBD/AD (n = 5). AD = Alzheimer disease; CBD = corticobasal degeneration; CBS = corticobasal syndrome; DLBD = diffuse Lewy body dementia; LBD = Lewy body dementia; PSP = progressive supranuclear palsy; RBD = REM sleep behavior disorder; TDP = TAR DNA-binding protein 43.
Imaging Findings
Voxel-based morphometry showed different patterns of volume loss according to underlying pathology. Patients with CBS-AD showed widespread gray matter loss compared with controls (Figure 2), involving lateral and medial aspects of parietal, temporal, and occipital lobes. The greatest loss was observed in the most severely affected (i.e., dominant) hemisphere. White matter loss was observed in the splenium of the corpus callosum and in periventricular region of the occipital horn of the lateral ventricle. Patients with CBS-CBD showed gray matter loss predominantly in the superior and medial premotor cortices, spreading into the lateral prefrontal cortex and insula in the dominant hemisphere, compared with controls (Figure 2). White matter loss was observed in the body of the corpus callosum, cingulum, and in the white matter of the premotor and motor cortices. Volume loss was greatest in the dominant hemisphere. Compared with controls, patients with CBS-DLBD/AD had atrophy in a focal parieto-occipital distribution, with no regions of white matter loss. Patients with CBS-FTLD-TDP had predominant lateral and medial prefrontal involvement, including the anterior cingulate cortex and a small area in the posterior temporal lobe. CBS-FTLD-TDP had only a few areas of white matter loss in the frontal lobe of the dominant hemisphere, with findings also observed in the periventricular region. The CBS-PSP group had a few areas of gray matter loss in the posterior frontal lobe of the dominant hemisphere, as well as white matter loss in the body of the corpus callosum, midbrain, thalamus, and the premotor cortex, with greater loss observed in the dominant hemisphere. Scattered findings in the frontal lobe were observed in the CBS-other group.
Figure 2. Regions of Gray Matter Loss in Patients With CBS According to Pathology Category.

All results are FWE-corrected for multiple comparisons, p < 0.05, extent threshold = 20. Legends show t scores with brighter colors representing higher score. N = 103 (24 CBD, 13 AD, 11 PSP, 7 TDP43, 4 DLBD/AD, 4 other, 40 controls). AD = Alzheimer disease; CBD = corticobasal degeneration; CBS = corticobasal syndrome; D = dominant; DLBD = diffuse Lewy body dementia; FWE = family-wise error; ND = nondominant; PSP = progressive supranuclear palsy; TDP = TAR DNA-binding protein 43.
For each neuropathologic variant, available FDG-PET scans were visually categorized. Representative FDG-PET images (Figure 3) show widespread hypometabolism in bilateral posterior temporoparietal areas in CBS-AD, with more focal involvement in other disease categories. Hypometabolism in the prefrontal cortex, thalamus, and putamen was observed in CBS-PSP and in CBS-CBD exemplars. As can be seen in the CBS-DLBD/AD images, a predominantly parietal involvement is apparent, with sparing of the visual cortices.
Figure 3. FDG-PET Across CBS Pathology Categories.

Panels A–E show Z scores, relative to a normative database, of pons intensity normalized FDG-PET scans for each individual displayed on stereotactic surface projections using Cortex ID (GE Healthcare, Waukesha, WI). Red color indicates greater hypometabolism. AD = Alzheimer disease; CBD = corticobasal degeneration; CBS = corticobasal syndrome; DLBD = diffuse Lewy body dementia; FDG = 18F fluorodeoxyglucose; PSP = progressive supranuclear palsy; TDP = TAR DNA-binding protein 43.
The structural brainstem biomarker of midbrain/pons ratio differed according to disease category (Figure 4) and was smallest in CBS-PSP (median [IQR]: 0.21 [0.04], p = 0.012). Midbrain/pons ratio was larger for patients with CBS-AD, patients with CBS-DLBD/AD, and patients with CBS-FTLD-TDP (0.25 [0.07], 0.28 [0.06], and 0.24 [0.07], respectively).
Figure 4. Boxplot Showing Midbrain/Pons Ratio Differences Across Pathology Category.
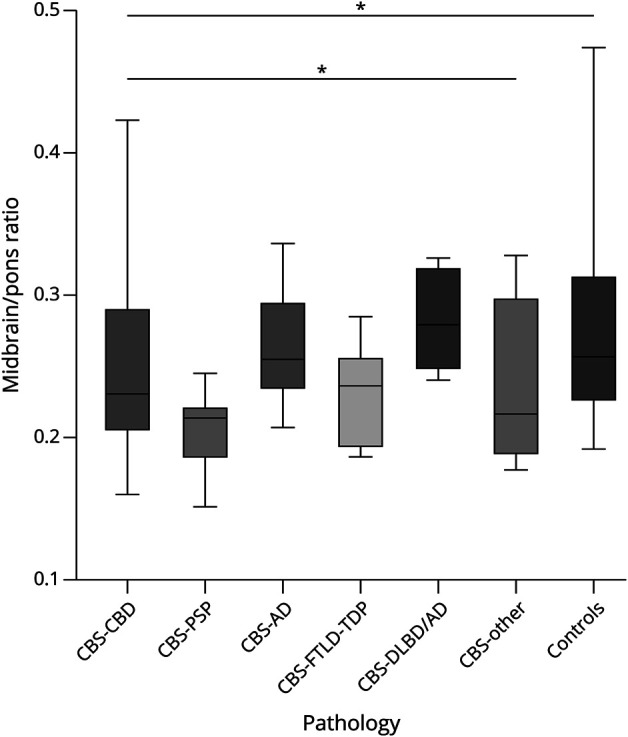
The boxes indicate the median and interquartile range of the distributions while the horizontal lines extending from the boxes stop at the most extreme data points. p Values were calculated using the Kruskal-Wallis test (p = 0.012 across all groups, p = 0.028 without controls). N = 103 (24 CBD, 13 AD, 11 PSP, 7 FTLD-TDP, 4 DLBD/AD, 4 other, 40 controls). AD = Alzheimer disease; CBD = corticobasal degeneration; CBS = corticobasal syndrome; DLBD = diffuse Lewy body dementia; FTLD = frontotemporal lobar degeneration; PSP = progressive supranuclear palsy; TDP = TAR DNA-binding protein 43.
PPV of Diagnostic Criteria for CBD
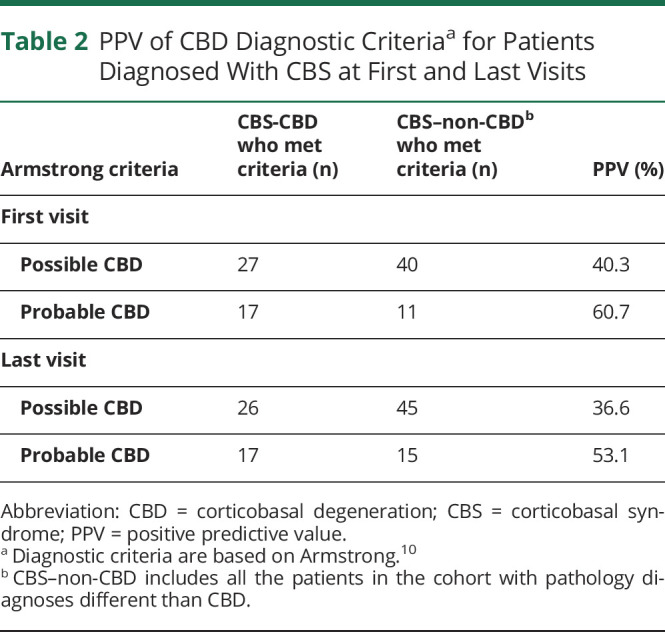
Of the 67 patients who met clinical criteria for possible CBD at presentation, 27 had CBD pathology at autopsy, yielding a PPV of 40% (Table 2). Of the 28 patients who met criteria for probable CBD at presentation, 17 had CBD at autopsy (61%). At last visit, PPV was low for both possible and probable criteria, yielding 37% and 53%, respectively. Patients with CBS-CBD who failed to meet criteria at presentation had relatively few core CBS signs on neurologic examination or they had atypical presentations, such as marked language impairment (nonfluent/agrammatic variant of PPA). Excluding the duration ≥1 year criterion slightly improved PPV, with 32 (48%) “possible” cases at presentation. Only 1 patient was excluded from the criteria because of young age at onset (42 years).
Table 2.
PPV of CBD Diagnostic Criteriaa for Patients Diagnosed With CBS at First and Last Visits
The 2 largest pathology groups contributing to the CBD mimics (i.e., meeting criteria for possible/probable CBD without CBD at autopsy) were CBS-PSP and CBS-AD, together accounting for 67.5% of possible mimics and 100% of probable mimics at presentation and 73% of possible and 80% of probable mimics at last visit, respectively. Among patients meeting the probable CBD criteria, having more positive CBS core features at presentation was not predictive of CBD pathology (p = 0.7). Detection of myoclonus was most frequent among patients with CBS-AD meeting criteria for probable CBD at presentation (6/7, 86%) compared with CBS-CBD (8/17, 47%) and CBS-PSP (0/4, 0%, p = 0.022). Detection of other CBS core features, including rigidity, dystonia, alien limb, cortical sensory deficit, or apraxia, did not differ between the pathology categories. Postural instability differentiated between the patients with CBS-PSP (4/4, 100%) who met the probable CBD criteria at presentation from CBS-AD (0/7, 0%) and CBS-CBD (6/17, 35%, p = 0.003). Midbrain/pons ratio did not differ between pathology groups among patients meeting probable CBD criteria at last visit (p = 0.221).
Three patients had symmetric CBS, but these cases did not fit in a specific pathologic category. One patient had postmortem diagnosis of DLBD/AD, 1 had NIBD-FTLD, and 1 had PSP.
Discussion
Clinical, demographic, and regional structural imaging findings in patients diagnosed antemortem with CBS differed according to pathologic diagnoses. Patients with CBS-DLBD/AD had the longest disease duration and were the youngest at death. Posterior cortical signs and myoclonus were more characteristic of patients with CBS-AD and patients with CBS-DLBD/AD. Not surprisingly, patients with CBS-DLBD/AD had more clinical features of Lewy body disease compared with other patients with CBS, while typical PSP features (falls, vertical gaze features) were more frequent among patients with CBS-PSP. Widespread cortical gray matter loss was characteristic of CBS-AD, while CBS-CBD and CBS-PSP predominantly involved premotor regions, and they had greater white matter loss. Smaller midbrain/pons ratio was useful in distinguishing CBS-PSP from other CBS subtypes. PPV analysis revealed that current diagnostic criteria for antemortem clinical diagnosis of CBD in patients with CBS are not optimal.
Our results confirm the established view that CBS is a clinical phenotype caused by a diverse group of neurodegenerative pathologies, with CBD and PSP (4R tauopathies) together accounting for about two-thirds of cases, with AD the third most common pathology.
When detected, certain clinical features can be used as “diagnostic pearls” in conjunction with CBS core features for predicting underlying pathology. These include myoclonus and posterior cortical signs suggesting AD pathology and core LBD features indicating contributing Lewy body pathology. The absence of additional signs resulting in the “classic” motor form of the CBS at presentation suggests CBD or PSP pathology as the underlying cause. Typical PSP features, including supranuclear gaze palsy and postural instability, were more frequent in CBS-PSP (although these associations were not significant, possibly because of cohort size). Based on the data presented here, the CBS-AD variant overlaps closely with the atypical-AD posterior cortical atrophy (PCA) variant,36,37 both clinically and structurally. Depending on whether motor or visual-spatial features present first may determine whether a patient receives a diagnosis of CBS or PCA. CJD or other underlying pathologies should be considered when disease progression is rapid (1 year or less). Clinical diagnostic clues may appear later in the disease process and emphasize the importance of longitudinal evaluation of patients with CBS.
Differences in gray matter atrophy patterns in patients with CBS differed according to pathologic category. In agreement with previous studies,2,3,6 patients with CBS-CBD had gray matter loss in premotor cortex, supplemental motor area, and insula, while patients with CBS-AD had a more widespread pattern of posterior cortical loss, and patients with CBS-FTLD-TDP had predominant loss in the prefrontal cortex. Differences were also observed in white matter findings, with white matter loss a striking feature of both CBS-CBD and CBS-PSP. Few regions of white matter loss were observed in the other groups, with most of the findings reflecting changes in the periventricular region. Previous studies have identified white matter changes in both CBD and PSP,38 with studies suggesting greater white matter pathology in 4R tauopathies compared with FTLD-TDP.39-41 In CBS-PSP, midbrain atrophy was also observed, and the smallest midbrain/pons ratio may have diagnostic value in differential diagnosis of CBS. On the other hand, it may not be as helpful as in patients with PSP presenting as Richardson syndrome because the measured values were higher than expected.17 Overall, imaging outcomes suggest that both gray and white matter findings with MRI may be useful in prediction of underlying pathology in CBS. In addition, FDG-PET may detect hypometabolism patterns of CBS variants at the individual patient level, which correspond to gray matter loss observed in structural imaging.
An atypical subgroup of patients with CBS caused by DLBD/AD pathology is described in this study. Lewy body pathology is rarely considered the cause of CBS, although it has been described in autopsy studies.42,43 CBS-DLBD had more Lewy bodies in the motor cortex than patients with typical DLBD.42 Our results show that although Lewy body core clinical features were uncommon in this cohort, when present they may be indicative of Lewy body pathology. By contrast, findings on structural MRI or hypometabolism on FDG-PET for this subgroup were not typical of DLBD.44 Volumetric imaging analysis showed little atrophy in this subgroup, despite most cases having Braak stage of VI (5/7 participants), but this might be due to decreased statistical power because of small group size. There was no evidence of a cingulate island sign or predominant occipital hypometabolism in CBS-DLBD in the available FDG-PET scan.29
We evaluated the PPV of the Armstrong CBD clinical diagnostic criteria10 for possible or probable CBS. Because we focused on clinical CBS, and neuropathologically proven CBD cases without an antemortem clinical CBS diagnosis were not included, specificity and sensitivity calculations were deferred. Based on the results presented here, diagnostic precision can be improved for suspected CBD pathology both at presentation and at follow-up. While Alexander et al.11 previously demonstrated that CBD mimics are most often AD, our results show that both CBS-PSP and CBS-AD contributed to CBD mimics. Among CBD mimics, myoclonus was more frequent in CBS with AD pathology, and postural instability was more frequent in those with PSP pathology. Addition of AD biomarkers to the diagnostic evaluation would improve diagnostic performance, as previously suggested.45 Negative amyloid biomarkers would make AD pathology unlikely and increase the possibility of 4R-tauopathy. In this cohort of patients with CBS, none of the clinical features in the criteria were more common in patients with CBD pathology than in CBD mimics, although apraxia of speech was generally frequent in CBD. While maintaining core LBD features as exclusion criteria seems to be beneficial in recognizing some of the patients with CBS-DLBD/AD, the 1-year duration criterion may need to be reconsidered to increase predictive value at presentation for clinical trial enrollment. Clinical and imaging features may not be sufficient to diagnose patients accurately and include them in clinical trials until one or more biomarkers associated with CBD are validated. Identifying 4R-tauopathy biomarkers such as CSF MTBR-tau,46 shown to distinguish CBD from controls, should be the focus of future research.
The strengths of this study include the large sample size, pathology-proven diagnoses, and the large proportion of patients with volumetric imaging and with comprehensive and longitudinal clinical follow-up. A deep clinical phenotyping process reveals associations that were novel, for example the description of the CBS-DLBD/AD subgroup. Given that our cohort included patients from all over the United States with many different underlying pathologies and data were collected over a longer period of time, our results would likely generalize to all CBS cases. This study was performed in a tertiary referral center, so there may be a disproportionate number of atypical/diagnostically difficult cases, which may bias the PPV results. Other potential weaknesses include the merging of the diverse “CBS-other” subgroup and the use of univariate comparisons for clinical features and neuropathology.
Glossary
- AD
Alzheimer disease
- AGD
argyrophilic grain disease
- ARTAG
aging-related tau astrogliopathy
- CBS
corticobasal syndrome
- CBD
corticobasal degeneration
- CJD
Creutzfeldt-Jakob disease
- DLBD
diffuse Lewy body disease
- FDG
18F fluorodeoxyglucose
- FTLD
frontotemporal lobar degeneration
- FUS
fused in sarcoma
- IQR
interquartile range
- LBD
Lewy body dementia
- NIBD
neurofilament inclusion body disease
- PCA
posterior cortical atrophy
- PPA
primary progressive aphasia
- PPV
positive predictive value
- PSP
progressive supranuclear palsy
- RBD
REM sleep behavior disorder
- SPM
statistical parametric mapping
- STMS
Short Test of Mental Status
- TDP
TAR DNA-binding protein 43
- VBM
voxel-based morphometry
Appendix. Authors
Study Funding
This study was supported by the following resources: NRG ATA (R01AG050603) NRG SLD (R01-DC010367 and R01-DC14942) NGR PSP (R01-NS89757) ADRC (P30 AG062677) CurePSP ALLFTD (U19 AG063911) ARTFL (U54 NS092089) LEFFTDS (U01 AG045390) MEM YOAD (R01 AG075802) HpSp AD subtypes (R01 AG054449) LEADS (U01 AG057195).
Disclosure
D.S. Knopman serves on a Data Safety Monitoring Board for the DIAN study. He served on a Data Safety monitoring Board for a tau therapeutic for Biogen but receives no personal compensation. He is a site investigator in the Biogen aducanumab trials. He is an investigator in a clinical trial sponsored by Lilly Pharmaceuticals and the University of Southern California. He serves as a consultant for Samus Therapeutics, Roche, and Alzeca Biosciences but receives no personal compensation. R.C. Petersen serves as a consultant for Biogen, Inc., Roche, Inc., Merck, Inc., Genentech Inc. (DSMB), Nestle, Inc., and Eisai, Inc.; receives publishing royalties from Mild Cognitive Impairment (Oxford University Press, 2003) and UpToDate. B.F. Boeve, D.W. Dickson, K.A. Josephs, and J.L. Whitwell received research funding from the NIH and declare no competing financial interests. M.E. Murray is a consultant for AVID Radiopharmaceuticals. She receives support from the NIH/NIA and State of Florida. N.R. Graff-Radford receives royalties from UpToDate; has participated in multicenter therapy studies sponsored by Biogen, TauRx, AbbVie, Novartis, and Lilly; and he receives research support from NIH. J. Graff-Radford serves on the editorial board for Neurology® and receives research support from the NIH. The other authors declare no financial or other conflict of interest. Go to Neurology.org/N for full disclosures.
References
- 1.Svenningsson P. Corticobasal degeneration: advances in clinicopathology and biomarkers. Curr Opin Neurol. 2019;32(4):597-603. doi: 10.1097/wco.0000000000000707 [DOI] [PubMed] [Google Scholar]
- 2.Lee SE, Rabinovici GD, Mayo MC, et al. Clinicopathological correlations in corticobasal degeneration. Ann Neurol. 2011;70(2):327-340. doi: 10.1002/ana.22424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whitwell JL, Jack CR, Boeve BF, et al. Imaging correlates of pathology in corticobasal syndrome. Neurology. 2010;75(21):1879-1887. doi: 10.1212/wnl.0b013e3181feb2e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ling H, O'Sullivan SS, Holton JL, et al. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain. 2010;133(7):2045-2057. doi: 10.1093/brain/awq123 [DOI] [PubMed] [Google Scholar]
- 5.Boxer AL, Gold M, Feldman H, et al. New directions in clinical trials for frontotemporal lobar degeneration: methods and outcome measures. Alzheimers Dement. 2020;16(1):131-143. doi: 10.1016/j.jalz.2019.06.4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Josephs KA, Whitwell JL, Boeve BF, et al. Anatomical differences between CBS-corticobasal degeneration and CBS-Alzheimer's disease. Mov Disord. 2010;25(9):1246-1252. doi: 10.1002/mds.23062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kouri N, Murray ME, Hassan A, et al. Neuropathological features of corticobasal degeneration presenting as corticobasal syndrome or Richardson syndrome. Brain. 2011;134(11):3264-3275. doi: 10.1093/brain/awr234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Litvan I, Agid Y, Goetz C, et al. Accuracy of the clinical diagnosis of corticobasal degeneration: a clinicopathologic study. Neurology. 1997;48(1):119-125. doi: 10.1212/wnl.48.1.119 [DOI] [PubMed] [Google Scholar]
- 9.Koga S, Josephs KA, Aiba I, Yoshida M, Dickson DW. Neuropathology and emerging biomarkers in corticobasal syndrome. J Neurol Neurosurg Psychiatry. 2022;93(9):919-929. doi: 10.1136/jnnp-2021-328586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80(5):496-503. doi: 10.1212/wnl.0b013e31827f0fd1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alexander SK, Rittman T, Xuereb JH, Bak TH, Hodges JR, Rowe JB. Validation of the new consensus criteria for the diagnosis of corticobasal degeneration. J Neurol Neurosurg Psychiatry. 2014;85(8):925-929. doi: 10.1136/jnnp-2013-307035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ouchi H, Toyoshima Y, Tada M, et al. Pathology and sensitivity of current clinical criteria in corticobasal syndrome. Mov Disord. 2014;29(2):238-244. doi: 10.1002/mds.25746 [DOI] [PubMed] [Google Scholar]
- 13.Constantinides VC, Paraskevas GP, Paraskevas PG, Stefanis L, Kapaki E. Corticobasal degeneration and corticobasal syndrome: a review. Clin Park Relat Disord. 2019;1:66-71. doi: 10.1016/j.prdoa.2019.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burrell JR, Hornberger M, Villemagne VL, Rowe CC, Hodges JR. Clinical profile of PiB-positive corticobasal syndrome. PLoS One. 2013;8(4):e61025. doi: 10.1371/journal.pone.0061025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sha SJ, Ghosh PM, Lee SE, et al. Predicting amyloid status in corticobasal syndrome using modified clinical criteria, magnetic resonance imaging and fluorodeoxyglucose positron emission tomography. Alzheimers Res Ther. 2015;7:8. doi: 10.1186/s13195-014-0093-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Massey LA, Jager HR, Paviour DC, et al. The midbrain to pons ratio: a simple and specific MRI sign of progressive supranuclear palsy. Neurology. 2013;80(20):1856-1861. doi: 10.1212/wnl.0b013e318292a2d2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grijalva RM, Pham NTT, Huang Q, et al. Brainstem biomarkers of clinical variant and pathology in progressive supranuclear palsy. Mov Disord. 2022;37(4):702-712. doi: 10.1002/mds.28901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whitwell JL, Jack CR, Parisi JE, et al. Midbrain atrophy is not a biomarker of progressive supranuclear palsy pathology. Eur J Neurol. 2013;20(10):1417-1422. doi: 10.1111/ene.12212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pardini M, Huey ED, Spina S, et al. FDG-PET patterns associated with underlying pathology in corticobasal syndrome. Neurology. 2019;92(10):E1121-E1135. doi: 10.1212/wnl.0000000000007038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lawton M, Kasten M, May MT, et al. Validation of conversion between mini-mental state examination and montreal cognitive assessment. Mov Disord. 2016;31(4):593-596. doi: 10.1002/mds.26498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Townley RA, Syrjanen JA, Botha H, et al. Comparison of the short test of mental status and the Montreal cognitive assessment across the cognitive spectrum. Mayo Clin Proc. 2019;94(8):1516-1523. doi: 10.1016/j.mayocp.2019.01.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kokmen E, Smith GE, Petersen RC, Tangalos E, Ivnik RC. The short test of mental status: correlations with standardized psychometric testing. Arch Neurol. 1991;48(7):725-728. doi: 10.1001/archneur.1991.00530190071018 [DOI] [PubMed] [Google Scholar]
- 23.Dickson DW, Bergeron C, Chin SS, et al. Office of rare diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002;61(11):935-946. doi: 10.1093/jnen/61.11.935 [DOI] [PubMed] [Google Scholar]
- 24.Hauw JJ, Daniel SE, Dickson D, et al. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology. 1994;44(11):2015-2019. doi: 10.1212/wnl.44.11.2015 [DOI] [PubMed] [Google Scholar]
- 25.Roemer SF, Grinberg LT, Crary JF, et al. Rainwater Charitable Foundation criteria for the neuropathologic diagnosis of progressive supranuclear palsy. Acta Neuropathol. 2022;144(4):603-614. doi: 10.1007/s00401-022-02479-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging. 1997;18(4 suppl):S1-S2. [PubMed] [Google Scholar]
- 27.Mackenzie IRA, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119(1):1-4. doi: 10.1007/s00401-009-0612-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47(5):1113-1124. doi: 10.1212/wnl.47.5.1113 [DOI] [PubMed] [Google Scholar]
- 29.McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies. Neurology. 2017;89(1):88-100. doi: 10.1212/wnl.0000000000004058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez RD, Suemoto CK, Molina M, et al. Argyrophilic Grain Disease: demographics, clinical, and neuropathological features from a large autopsy study. J Neuropathol Exp Neurol. 2016;75(7):628-635. doi: 10.1093/jnen/nlw034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braak H, Braak E. Cortical and subcortical argyrophilic grains characterize a disease associated with adult onset dementia. Neuropathol Appl Neurobiol. 1989;15(1):13-26. doi: 10.1111/j.1365-2990.1989.tb01146.x [DOI] [PubMed] [Google Scholar]
- 32.Kovacs GG, Ferrer I, Grinberg LT, et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol. 2016;131(1):87-102. doi: 10.1007/s00401-015-1509-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Josephs KA, Tang-Wai DF, Edland SD, et al. Correlation between antemortem magnetic resonance imaging findings and pathologically confirmed corticobasal degeneration. Arch Neurol. 2004;61(12):1881-1884. doi: 10.1001/archneur.61.12.1881 [DOI] [PubMed] [Google Scholar]
- 34.Quattrone A, Nicoletti G, Messina D, et al. MR imaging index for differentiation of progressive supranuclear palsy from Parkinson disease and the Parkinson variant of multiple system atrophy. Radiology. 2008;246(1):214-221. doi: 10.1148/radiol.2453061703 [DOI] [PubMed] [Google Scholar]
- 35.Oba H, Yagishita A, Terada H, et al. New and reliable MRI diagnosis for progressive supranuclear palsy. Neurology. 2005;64(12):2050-2055. doi: 10.1212/01.wnl.0000165960.04422.d0 [DOI] [PubMed] [Google Scholar]
- 36.Crutch SJ, Schott JM, Rabinovici GD, et al. Consensus classification of posterior cortical atrophy. Alzheimers Dement. 2017;13(8):870-884. doi: 10.1016/j.jalz.2017.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Graff-Radford J, Yong KXX, Apostolova LG, et al. New insights into atypical Alzheimer's disease in the era of biomarkers. Lancet Neurol. 2021;20(3):222-234. doi: 10.1016/s1474-4422(20)30440-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Josephs KA, Whitwell JL, Dickson DW, et al. Voxel-based morphometry in autopsy proven PSP and CBD. Neurobiol Aging. 2008;29(2):280-289. doi: 10.1016/j.neurobiolaging.2006.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kovacs GG, Lukic MJ, Irwin DJ, et al. Distribution patterns of tau pathology in progressive supranuclear palsy. Acta Neuropathol. 2020;140(2):99-119. doi: 10.1007/s00401-020-02158-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mimuro M, Yoshida M. Chameleons and mimics: progressive supranuclear palsy and corticobasal degeneration. Neuropathology. 2020;40(1):57-67. doi: 10.1111/neup.12590 [DOI] [PubMed] [Google Scholar]
- 41.McMillan CT, Irwin DJ, Avants BB, et al. White matter imaging helps dissociate tau from TDP-43 in frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry. 2013;84(9):949-955. doi: 10.1136/jnnp-2012-304418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kasanuki K, Josephs KA, Ferman TJ, et al. Diffuse Lewy body disease manifesting as corticobasal syndrome: a rare form of Lewy body disease. Neurology. 2018;91(3):E268-E279. doi: 10.1212/wnl.0000000000005828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haug A, Boyer P, Kluger B. Diffuse Lewy body disease presenting as corticobasal syndrome and progressive supranuclear palsy syndrome. Mov Disord. 2013;28(8):1153-1155. doi: 10.1002/mds.25368 [DOI] [PubMed] [Google Scholar]
- 44.Raji CA, Benzinger TLS. The value of neuroimaging in dementia diagnosis. Continuum (Minneap Minn). 2022;28(3):800-821. doi: 10.1212/con.0000000000001133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Litvan I, Lang AE, Armstrong M. CBD diagnostic criteria: exclusions as important as inclusions. J Neurol Neurosurg Psychiatry. 2022;94(4):328. doi: 10.1136/jnnp-2022-330564 [DOI] [PubMed] [Google Scholar]
- 46.Horie K, Barthélemy NR, Spina S, et al. CSF tau microtubule-binding region identifies pathological changes in primary tauopathies. Nat Med. 2022;28(12):2547-2554. doi: 10.1038/s41591-022-02075-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, on request.