

Abstract
Perifornical hypothalamus (PFH) orexin glucose-inhibited (GI) neurons that facilitate arousal have been implicated in hypoglycemia awareness. Mice lacking orexin exhibit narcolepsy, and orexin mediates the effect of the antinarcolepsy drug modafinil. Thus, hypoglycemia awareness may require a certain level of arousal for awareness of the sympathetic symptoms of hypoglycemia (e.g., tremors, anxiety). Recurrent hypoglycemia (RH) causes hypoglycemia unawareness. We hypothesize that RH impairs the glucose sensitivity of PFH orexin GI neurons and that modafinil normalizes glucose sensitivity of these neurons and restores hypoglycemia awareness after RH. Using patch-clamp recording, we found that RH enhanced glucose inhibition of PFH orexin GI neurons in male mice, thereby blunting activation of these neurons in low-glucose conditions. We then used a modified conditioned place preference behavioral test to demonstrate that modafinil reversed hypoglycemia unawareness in male mice after RH. Similarly, modafinil restored normal glucose sensitivity to PFH orexin GI neurons. We conclude that impaired glucose sensitivity of PFH orexin GI neurons plays a role in hypoglycemia unawareness and that normalizing their glucose sensitivity after RH is associated with restoration of hypoglycemia awareness. This suggests that the glucose sensitivity of PFH orexin GI neurons is a therapeutic target for preventing hypoglycemia unawareness.
Introduction
All individuals with type 1 diabetes mellitus (T1DM) and >30% of those with type 2 diabetes mellitus (T2DM) require intensive insulin therapy to manage their blood glucose levels. This therapy comes at a great price: increased incidence of iatrogenic insulin-induced hypoglycemia (1). Insulin-induced hypoglycemia is not only acutely life threatening, recurrent hypoglycemia (RH) impairs the ability of the brain to detect and correct subsequent hypoglycemia. Declining glucose first triggers the hormonal (e.g., glucagon, epinephrine) counterregulatory response to stimulate gluconeogenesis. Further glucose decline leads to the behavioral response known as hypoglycemia awareness, which includes neurogenic (e.g., palpitations, anxiety) and neuroglycopenic (e.g., tiredness, confusion) symptoms that alert the individual of hypoglycemia and stimulate behaviors such as feeding to raise glucose levels. RH impairs these hormonal and behavioral responses, leading to hypoglycemia-associated autonomic failure and hypoglycemia unawareness, respectively (2–6). Although hypoglycemia-associated autonomic failure has been studied extensively, little is known about the mechanisms underlying hypoglycemia awareness/unawareness.
This lack of information has been due to a lack of an appropriate animal model of hypoglycemia awareness. Although it has been used, the feeding response to insulin-induced hypoglycemia is not impaired in rodents after RH (7,8). Similarly, humans with T1DM who use insulin therapy report no changes in hunger in response to insulin-induced hypoglycemia (9). However, these patients exhibit an impaired awakening response to nocturnal hypoglycemia (2), which suggests that RH impairs the arousal needed to recognize hypoglycemia and awaken to eat. Recently, researchers in the Levin laboratory modified the behavioral test known as conditioned place preference (CPP) to model hypoglycemia awareness in rats (7). In this test, the animal learns to associate a food cue with a visually discriminated chamber of a CPP box and develops a preference for that chamber. Normally, when insulin-induced hypoglycemia occurs in a food cue–associated chamber, the animal is aware of the aversive stimulus and no longer prefers that chamber of the CPP box. If, however, the animal is exposed to RH in its home cage prior to experiencing insulin-induced hypoglycemia in the food-associated chamber, it is not aware of the hypoglycemia and its preference for that chamber remains intact. This novel behavioral test provides the first true animal model of hypoglycemia awareness and unawareness.
Using their model, the Levin group demonstrated that the orexin neurons of the perifornical hypothalamus (PFH) were necessary for hypoglycemia awareness (7). Approximately half of the orexin neurons are glucose-inhibited (GI) neurons (10,11). Activation of PFH orexin neurons activates the adrenal sympathetic response to hypoglycemia (12–14). The PFH orexin neurons are also involved in arousal, and mice lacking orexin exhibit narcolepsy (15–18). Moreover, PFH orexin neurons are the target of antinarcolepsy drugs such as modafinil (19–22). In this study, we tested the hypothesis that RH impairs activation of PFH orexin GI neurons in low-glucose conditions and that modafinil normalizes glucose sensing by these neurons and restores hypoglycemia awareness after RH. Consistent with our hypothesis, RH significantly blunted the activation of PFH orexin GI neurons in low glucose levels. After translating the rat model of hypoglycemia unawareness to mice, we found that modafinil completely restored hypoglycemia awareness after RH. Modafinil also restored normal glucose sensitivity to PFH orexin GI neurons. These data suggest that PFH orexin GI neurons play a role in hypoglycemia unawareness after RH. Moreover, drugs that target these neurons may be of therapeutic use in treating hypoglycemia unawareness in patients with T1DM and advanced T2DM who are using intensive insulin therapy.
Research Design and Methods
Animals
Male mice, 8–12 weeks old, expressing green fluorescent protein (GFP) on the hypocretin (Hcrt; gene nomenclature for orexin) promoter (an in-house colony of mice) or C57BL/6J mice (Jackson Laboratory) were group housed on a 12:12 light:dark cycle with water and standard chow provided ad libitum in the Research Animal Facility at Rutgers, The State University of New Jersey, New Jersey Medical School. Our orexin–GFP colony was derived from founder males provided by Dr. Gina Leinninger at Michigan State University, and the colony was maintained by heterozygote breeding to female C57BL/6J mice purchased from Jackson Laboratories. Generation of these mice is described by Yamanaka et al. (23). Male mice were used because we have observed sexual dimorphism and estrus cycle effects in ventromedial hypothalamus GI neurons (24,25). All procedures were in accordance with the Rutgers University Institutional Animal Care and Use Committee.
Drugs and Chemicals
Insulin (1.2 μL of Humulin R/U100; Eli Lilly and Company) was added to 1 mL of saline for a stock solution of 1.2 U/mL. Modafinil (150 mg/kg; Sigma Aldrich) was dissolved in 0.3% carboxymethylcellulose in saline. This dose of modafinil was chosen because it induced orexin-dependent histamine release in vivo (20,26). Tetrodotoxin (TTX) was obtained from Tocris Bioscience and all other chemicals were obtained from Sigma Aldrich.
RH Hypoglycemia Protocol
For electrophysiological and behavioral studies of RH, mice were fasted for 5 h and injected with either insulin (1.2 IU/kg; RH group) or an equivalent volume of saline (RS; control group) once daily for 3 consecutive days. Blood glucose level was measured every 15 min for 2 h after injection by distal tail amputation method using a glucometer (Bayer Contour). Only mice whose blood glucose level declined below 60 mg/dL after insulin injection were used for analysis. Food was returned after 2 h. Animals exhibiting seizures or extreme torpor were given glucose intraperitoneally (i.p.) and euthanized.
CPP
The behavioral test for hypoglycemia awareness/unawareness was derived from the report by Otlivanchik et al. (7) and modified for mice. Verification of this model is presented in Supplementary Figure 1.
Hypoglycemia Awareness
The CPP apparatus was a two-chamber box with a removable divider separating the chambers. CPP involves establishing preference for the chamber associated with a food reward. The chambers were discriminated visually by either striped or checked walls and texturally by either a ribbed or smooth floor texture. Mice were habituated to the box, with the divider removed for 30 min daily for 2–4 days. After habituation, mice were placed in the open box and the time spent on each side of the chamber was video recorded and measured by digital detection software (written by author O.I.) at 15, 20, and 30 min. The optimal time point when the mice finished exploration of the two chambers and initiated grooming or sleeping was determined to be 20 min. This time point was used for all data comparisons. For 6–8 days (conditioning), the mice were restricted for 30 min on alternating days to the chamber of the box in which they initially spent the least amount of time with a food reward or to the chamber where they initially spent the most time without a food reward. After conditioning, mice were again placed in the open box for 30 min, and the time spent in each chamber was measured. Although rats in the Levin et al. studies (7) developed a CPP for chocolate drops (i.e., they spent more time in the chocolate-associated chamber), we found that the mice would only develop a CPP when sweetened cereal (Froot Loops; Kellogg Co.) was used as a reward. Mice that increased their time in the cereal-associated chamber (initially the less preferred side) were considered to have developed CPP. The mice were then restricted to the cereal-associated chamber and exposed to insulin-induced hypoglycemia. The mice were removed from the chamber 60 min after insulin injection and placed in their home cage with food. After 1–2 days, the mice were placed in the open box and the time in each chamber was recorded.
Hypoglycemia Unawareness
After conditioning, the mice were subjected to RH or RS for 3 consecutive days in their home cage. After 1–2 days later, the mice were exposed to insulin-induced hypoglycemia in the cereal-associated chamber of the CPP box followed by measurement of the time in each chamber.
Electrophysiology
On the day of the experiment, mice were anesthetized and transcardially perfused with ice-cold oxygenated (95% O2/5% CO2) perfusion solution (composition [in mmol/L]: 2.5 KCl, 7 MgCl2, 1.25 NaH2PO4, 28 NaHCO3, 0.5 CaCl2, 7 glucose, 1 ascorbate, and 3 pyruvate) of ∼300 mOsm and pH 7.4. Brains were rapidly removed and placed in ice-cold (slushy) oxygenated perfusion solution as previously described (27–29). We made 300-μm coronal sections (containing PFH orexin neurons) through the hypothalamus on a vibratome (Vibroslice, Camden Instruments). The brain sections were maintained in oxygenated artificial cerebrospinal fluid (in mmol/L: 126 NaCl, 1.9 KCl, 1.2 KH2PO4, 26 NaHCO3, 2.5 glucose, 1.3 MgCl2, and 2.4 CaCl2; ∼300 mOsm; pH 7.4) for at least 1 h at room temperature prior to recording.
Whole-cell current clamp recordings were made on visually identified GFP-expressing cells in the PFH at 28–30°C, as previously described (11,30). Borosilicate pipettes (4–6 mol/LΩ resistance; Sutter Instruments) were filled with an intracellular solution containing (in mmol/L): 128 K-gluconate, 10 KCl, 4 KOH, 10 HEPES, 4 MgCl2, 0.5 CaCl2, 5 EGTA, 2 or 5 Na2ATP, and 0.4 Na2GTP; pH 7.2. Osmolarity was adjusted with sucrose to 300 mOsm. The recording pipette contained 2 or 5 mmol/L ATP as described in RESULTS and in provided figures. Lucifer Yellow (250 μg/mL) was included in the pipette for postrecording visualization of the neuron. Neurons with membrane potentials (MPs) more negative than −45 mV in 2.5 mmol/L glucose and action potentials that overshot 0 mV were considered viable for recording. Pipette access resistance <20 mol/LΩ with <20% change during the time course of the experiment was considered acceptable. Whole-cell resistance (R) was calculated from the membrane voltage responses to a −10 to −30 pA hyperpolarizing pulse (500-ms duration). Because glucose acts on nonsynaptic ion channels, it predominantly affects R. R is a sensitive measure of changes in neuronal excitability. Thus, treatment effects were quantified using the percentage change in MP and R as extracellular glucose levels decreased (29,31).
Data and Resource Availability
The data sets generated and/or analyzed during this study are available from the corresponding author upon reasonable request. No applicable resources were generated or analyzed during the study.
Results
Electrophysiological Characterization of PFH Orexin GI Neurons
Like ventromedial hypothalamic (VMH) GI neurons (24), we observed two different patterns of activation among PFH orexin GI neurons as glucose was reversibly decreased from 2.5 to 0.1 mmol/L. Some PFH orexin GI neurons (sustained GI [sGI]) maintained their increased activity for the duration of time in low glucose conditions, becoming inhibited only after return to 2.5 mmol/L glucose concentration (Fig. 1A). In contrast, a different subpopulation of orexin GI neurons, adapting (AdGI) neurons, were transiently activated in low glucose conditions but became inhibited again before glucose concentration was returned to 2.5 mmol/L (Fig. 1B). As seen for VMH AdGI neurons, orexin AdGI neurons also showed a small-magnitude transient depolarization upon return to 2.5 mmol/L glucose concentration.
Figure 1.
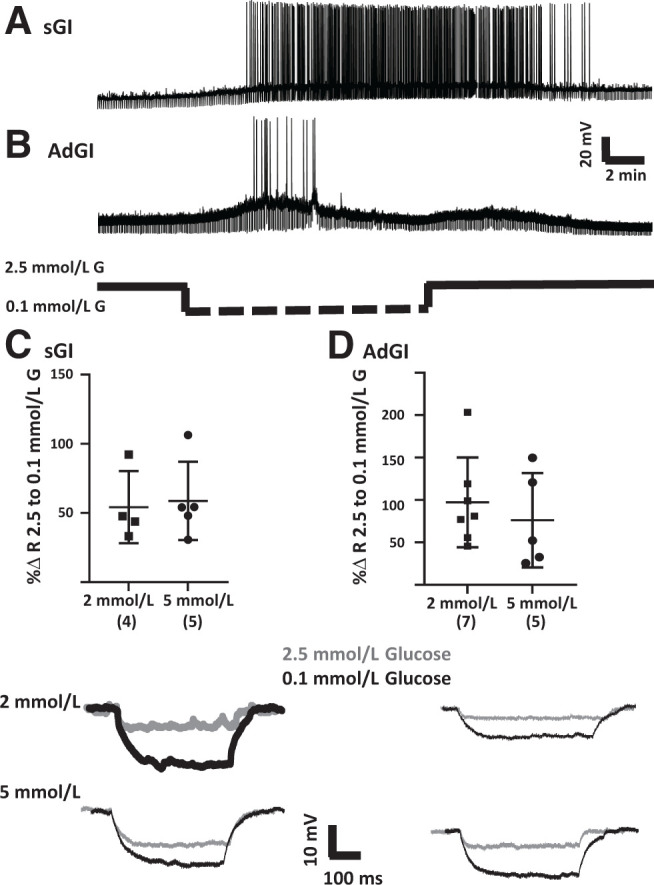
Representative whole-cell current clamp recordings of PFH orexin GFP neurons in brain sections. A: Example of a sustained response to decreased glucose level (sGI). This neuron depolarized and increased action potential frequency and R as extracellular glucose (G) decreased from 2.5 to 0.1 mmol/L. This response was sustained throughout the time in 0.1 mmol/L glucose and only reversed after return to 2.5 mmol/L glucose. B: Example of AdGI. This neuron depolarized and increased action potential frequency and R as extracellular glucose concentration decreased from 2.5 to 0.1 mmol/L. However, in contrast to the sGI neuron, this AdGI neuron hyperpolarized and decreased action potential frequency and R back to levels seen in 2.5 mmol/L glucose in the presence of 0.1 mmol/L extracellular glucose. As we have seen for VMH AdGI neurons, there is a small rebound depolarization upon return to 2.5 mmol/L glucose (24). The percent change (%Δ) in R for both sGI (C) and AdGI (D) neurons in response to decreased glucose from 2.5 to 0.1 mmol/L was similar with either 2 or 5 mmol/L ATP in the recording pipette (P > 0.05, Student unpaired t test; individual values shown with mean and SD). Top panels show the bar graphs and bottom panels give examples of individual voltage pulses in response to a hyperpolarizing current pulse. Sample size is shown in parentheses on the x-axis.
Unlike VMH GI neurons (32), lateral hypothalamus (LH) orexin GI neurons sense the glucose molecule per se and do not respond to glucose metabolism or changes in intracellular ATP concentration (11,33). Glucose sensing by PFH orexin GI neurons was also independent of intracellular ATP concentration. There was no significant difference in the increase in R of either PFH orexin sGI (Fig. 1C) or AdGI (Fig. 1D) neurons as glucose concentration decreased from 2.5 to 0.1 mmol/L when the neurons were dialyzed with either 2 or 5 mmol/L ATP in the recording pipette. There was also no significant difference in MP in 2.5 mmol/L glucose at either ATP concentration for sGI neurons (2 mmol/L: −73.8 ± 3.7 mV [± SD], n = 4; 5 mmol/L: −70.8 ± 2.6 mV, n = 5; P = 0.52), although AdGI neurons showed a trend toward depolarization with 5 mmol/L ATP (2 mmol/L: −72.9 ± 1.4 mV, n = 7; 5 mmol/L: −67.9 ± 2.1 mV, n = 5; P = 0.06). There were no differences in the R in 2.5 mm glucose at either ATP concentration for sGI (MΩ in 2 mmol/L: 289 ± 35, n = 4; in 5 mmol/L: 357 ± 73, n = 5; P = 0.46) or AdGI (MΩ in 2 mmol/L: 499 ± 110, n = 7; in 5 mmol/L: 550 ± 182, n = 5; P = 0.80) neurons. On the basis of these results, data obtained using 2 or 5 mmol/L ATP in the recording pipette were combined in subsequent experiments.
The nonmetabolizable glucose analog 2 deoxyglucose (2DG) inhibits rather than activates LH orexin GI neurons (11,33). Similarly, we found that 2.5 mmol/L 2DG reversed the activation of PFH orexin sGI neurons in low glucose conditions and blocked activation of PFH orexin AdGI neurons at low glucose levels (Fig. 2A). The response to decreased glucose level for both sGI (n = 3) and AdGI (n = 6) neurons persisted in the presence of the sodium channel blocker TTX (Fig. 2B). Together, these data suggest that these patterns of glucose sensing are intrinsic to PFH orexin GI neurons and that these neurons directly sense the glucose molecule (as opposed to a presynaptic or metabolic effect). Interestingly, the change in R of sGI and AdGI neurons in response to decreased glucose levels was significantly enhanced in the presence of TTX (Fig. 2B). However, TTX had no effect on MP and R of sGI or AdGI neurons in 2.5 mmol/L glucose. This suggests that PFH orexin GI neurons receive presynaptic input that specifically blunts their activation at low glucose levels without affecting baseline properties in 2.5 mmol/L glucose.
Figure 2.

The response of sGI and AdGI neurons to the nonmetabolizable glucose analog 2DG or the sodium channel blocker TTX, which blocks action potentials. A: 2DG. The traces are examples of whole-cell current clamp recordings of sGI and AdGI neurons in brain sections containing the PFH. Data are quantified below as percent change (%Δ) in R. sGI neuron (top trace): glucose concentration in the perfusion solution was changed from 2.5 to 0.1 mmol/L at the beginning of the recording. Decreased glucose concentration depolarized and increased action potential frequency and R; this effect was completely reversed by 2.5 mmol/L 2DG. The percent change in R as glucose concentration decreased from 2.5 to 0.1 mmol/L was completely reversed when 2.5 mmol/L 2DG was added to 0.1 mmol/L glucose (P < 0.05, n = 5, paired Student t test). AdGI neuron (bottom trace): glucose concentration in the perfusion solution was changed from 2.5 to 0.1 mmol/L at the beginning of the recording. This neuron transiently depolarized and increased R in the presence of 0.1 mmol/L glucose; this effect was completely blocked when glucose was again decreased in the presence of 2.5 mmol/L 2DG. The percent change in R as glucose concentration decreased from 2.5 to 0.1 mmol/L was significantly reduced in the presence of 2.5 mmol/L 2DG (P < 0.001, n = 6, paired Student t test; individual values shown with mean and SD). B: TTX (blocks action potentials). Examples of whole-cell current clamp recordings of sGI (top trace) and AdGI (bottom trace) neurons in brain sections containing TTX in the perfusion media. Data are quantified below as the percent change in R. The response to decreased glucose persisted in the presence of TTX, indicating that these glucose-sensing patterns are intrinsic. The response to decreased glucose concentration from 2.5 to 0.1 mmol/L was enhanced in both sGI (P < 0.05, n = 3, paired Student t test) and AdGI (P < 0.05, n = 6) in the presence of TTX.
We then compared the MP and R of sGI and AdGI orexin neurons in 2.5 mmol/L glucose and the response to decreased glucose concentration from 2.5 to 0.1 mmol/L, using all neurons in the experiments discussed above. In 2.5 mmol/L glucose, there were no differences in the MP (sGI, −71.2 ± 1.7 mV, n = 15; AdGI, −71.1 ± 0.8 mV, n = 22; P = 0.93) or R (sGI, 366 ± 42 MΩ, n = 15; AdGI, 416 ± 50 MΩ, n = 22; P = 0.48) of sGI and AdGI neurons. The change in MP as glucose concentration decreased was also similar between these neuronal subtypes. However, although there was considerable overlap in the range of responses, the change in R as glucose decreased from 2.5 to 0.1 mmol/L was significantly greater in AdGI neurons compared with sGI neurons (Fig. 3).
Figure 3.
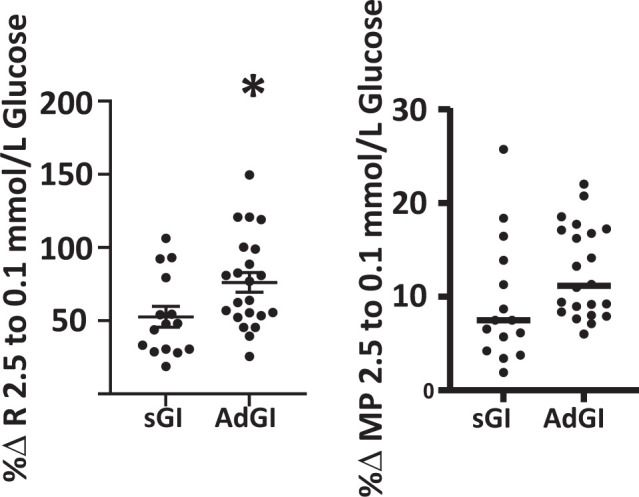
The percent change (%Δ) in R as glucose concentration decreased from 2.5 to 0.1 mmol/L was significantly greater in AdGI (n = 22) vs. sGI (n = 15) neurons (P < 0.05, Student t test, individual values shown with mean and SD). There was no difference in the percent change in MP between AdGI and sGI as glucose decreased.
Effect of RH on a Mouse Model of Hypoglycemia Awareness and the Glucose Sensitivity of PFH Orexin GI Neurons
For this study, glucose sensitivity was evaluated on the day after the last insulin or saline injection. Because baseline MP and R did not differ between sGI and adGI neurons, these values were analyzed together. RH had no effect on the MP (RS: −71.7 ± 1.6 mV, n = 9; RH: −68.2 ± 2.0, n = 11; P = 0.21) or R (RS: 376 ± 47, n = 9; RH: 316 ± 35, n = 11; P = 0.30) of orexin GI neurons in 2.5 mmol/L glucose. However, RH significantly blunted the change in R for AdGi (Fig. 4A and C) and sGI (Fig. 4B and C) neurons as glucose decreased from 2.5 to 0.1 mmol/L. RH had no effect on the MP response for either subtype.
Figure 4.
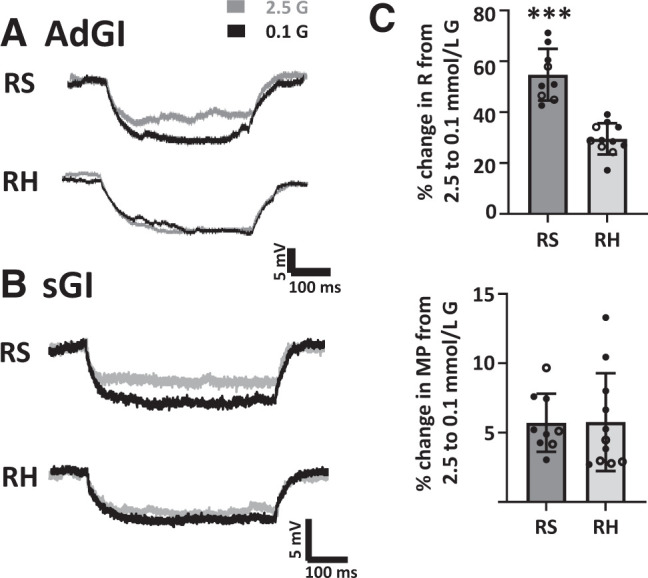
RH blunts the increase in R of PFH orexin GI neurons in response to low glucose concentration. Representative voltage responses to constant hyperpolarizing current pulses from AdGI (A) or sGI (B) neurons from RS- and RH-treated mice. The voltage response is directly proportional to the change in R. Both AdGI and sGI neurons from RS-treated mice increased the magnitude of the voltage response in 0.1 vs. 2.5 mmol/L glucose, whereas those from RH-treated mice did not. C: The percent change (%Δ) in R of sGI (open circles) and AdGI (closed circles) from RS- and RH-treated mice as glucose concentration decreased from 2.5 to 0.1 mmol/L. RH significantly decreased the percent change in R for sGI and adGI neurons, whether analyzed separately (sGI, P = 0.004, n = 3 RS and n = 4 RH; AdGI, P = 0.0003, n = 6 RS and n = 7 RH) or in combination (***P < 0.0001; C). There were no significant differences in the percent change in MP. Data analyzed by Student unpaired t test; individual values shown with mean and SD.
Effects of Modafinil on Hypoglycemia Awareness and Glucose Sensitivity of PFH Orexin GI Neurons After RH
Modafinil Restores Hypoglycemia Awareness After RH
After successful place-preference conditioning, mice were exposed to RS or RH in their home cages, as described above, in Supplementary Data, and in Supplementary Figure 1. On the day of insulin-induced hypoglycemia in the food-associated chamber of the CPP box, all mice were given an injection of modafinil (150 mg/kg i.p.) or vehicle 30 min prior to the insulin injection, and preference was measured 1–2 days later, as described above. Initially, all groups of mice developed a preference for the food-associated chamber (Fig. 5A–D; postconditioning [PC] vs. preconditioning [Pre]). After insulin-induced hypoglycemia, RH vehicle–treated mice did not reverse their preference for the food-associated chamber (Fig. 5A), whereas preference for the food-associated chamber reversed to that seen before conditioning in the RS vehicle–treated mice (insulin injection PC [PC-I] vs. Pre; Fig. 5B). In contrast, both the RH modafinil– and RS modafinil–treated mice reversed their preference for the food-associated chamber after insulin-induced hypoglycemia (Fig. 5C and D). Figure 5E shows that there was a significant interaction between drug and treatment (RH vs. RS) groups, in addition to significant main effects of drug and treatment, for the percent change in time spent in the food-associated chamber before and after insulin-induced hypoglycemia (PC-I vs. PC). The amount of time spent in the food-associated chamber by RH vehicle–treated mice after experiencing insulin-induced hypoglycemia was significantly greater than the time spent by all other groups of mice. Thus, modafinil restores hypoglycemia awareness after RH.
Figure 5.
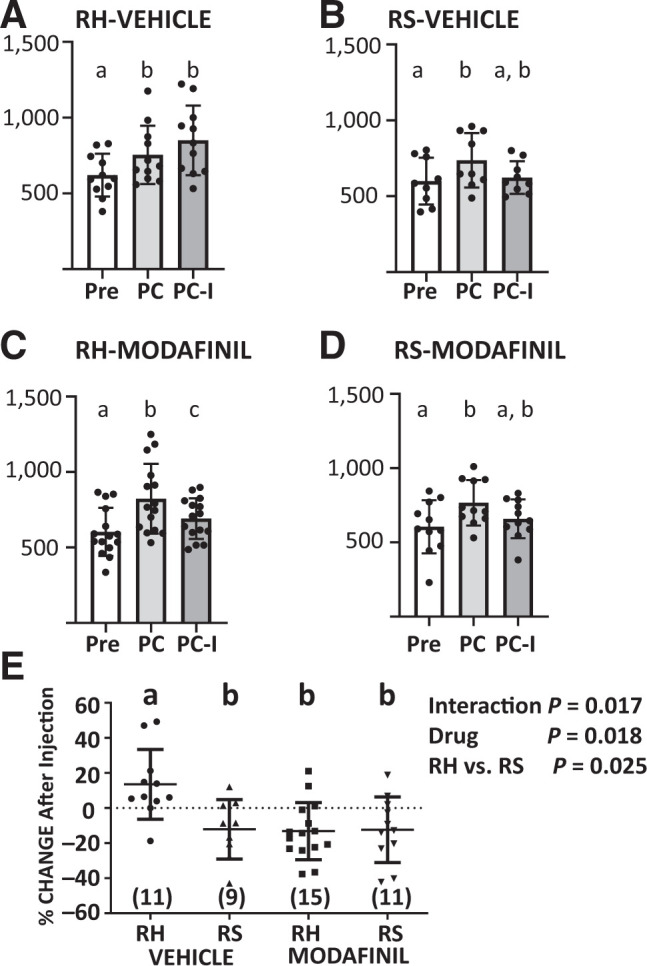
Modafinil prevents hypoglycemia unawareness. A–D: The time in seconds (s) was measured for the least-preferred chamber of the CPP box immediately following habituation to the box (Pre), after conditioning the mice with sweetened cereal in that chamber (PC), and after an injection of insulin in the food-associated chamber (PC-I). Mice received a daily injection of saline (RS) or insulin (RH) for 3 days in their home cage after conditioning and before the insulin injection in the food-associated chamber (between PC-I and PC). Modafinil (150 mg/kg i.p.) or vehicle was given 30 min prior to the insulin injection in the CPP box. Data were analyzed by repeated measures one-way ANOVA followed by Tukey’s multiple comparison test. Different letters are significantly different (P < 0.05). Successful conditioning for all groups is indicated by a significant increase in the time PC compared with Pre. RH vehicle–treated mice (A) did not reverse their preference for the food-associated chamber (PC-I vs. PC); however, all other groups (B–D) did. E: Percent change in time spent in the food-associated chamber before and after insulin injection (PC-I vs. PC). RH vehicle–treated mice spent significantly more time in the food-associated chamber than did RS vehicle– or modafinil–treated mice. RH modafinil–treated mice were not significantly different from either group of RS mice (two-way ANOVA followed by Tukey’s multiple comparisons; interaction F(1,42) = 6.147, P = 0.173; RH vs. RS: F(1,42) = 5.438, P = 0.0246; modafinil: F(1,42) = 6.011, P = 0.0185. N values given on E. All data are presented as individual values shown with mean and SD. Different letters represent statistical differences (P < 0.05).
Modafinil Restores Normal Glucose Sensitivity to PFH Orexin GI Neurons of Male Mice After RH
RH and RS mice were injected i.p. with 150 mg/kg modafinil 1 h prior to sacrifice for electrophysiological evaluation of the glucose sensitivity of PFH orexin GI neurons. There were no differences in MP (RS: −69.3 ± 2.4 mV, n = 10; RH: −70.4 ± 1.5, n = 10; P = 0.69) or R (RS: 376 ± 55, n = 10; RH: 334 ± 38, n = 10; P = 0.54) of PFH orexin GI neurons from RH modafinil– and RS modafinil–treated mice in 2.5 mmol/L glucose. Moreover, there was no significant difference in the change in R or MP of PFH orexin GI neurons of RH and RS mice that received modafinil in response to decreased glucose (Fig. 6). Thus, modafinil normalizes glucose sensitivity of PFH orexin GI neurons after RH.
Figure 6.

Modafinil prevents the change in glucose sensitivity of PFH GI neurons after RH. Representative voltage responses to constant hyperpolarizing current pulses from AdGI (A) or sGI (B) neurons from RS and RH mice treated with modafinil (150 mg/kg) 1 h prior to sacrifice. The voltage response is directly proportional to the change in R. C: The percent change (%Δ) in R and MP of sGI (open circles) and AdGI (closed circles) from RS and RH treated mice as glucose decreased from 2.5 to 0.1 mmol/L. There were no significant differences between sGI or AdGI neurons from RS- and RH-modafinil treated mice. This finding was independent of whether sGI and adGI neurons were analyzed separately or in combination (P > 0.05, Students unpaired t test, individual values shown with mean and SD).
Discussion
The findings of this study are consistent with our hypothesis that PFH orexin GI neurons play a critical role in hypoglycemia awareness. Furthermore, drugs that normalize their glucose sensitivity after RH may be of therapeutic value for treating hypoglycemia unawareness in patients with T1DM and those with advanced T2DM. We demonstrated that, like VMH GI neurons (28), RH increases the inhibitory effect of glucose on PFH orexin GI neurons, leading to reduced activation in low glucose concentration. The antinarcolepsy drug modafinil (22) restored normal glucose sensitivity to these neurons and restored hypoglycemia awareness after RH. Modafinil is generally considered a dopamine reuptake inhibitor, which increases synaptic dopamine levels (19). Orexin neurons express the dopamine 1 receptor (D1R) (34). Modafinil-induced increases in synaptic dopamine activate orexin neurons via the D1R (21,35). Inhibition of orexin neurons by the nonmetabolizable glucose analog 2DG suggests that these neurons possess a glucose receptor, as opposed to sensing glucose metabolism. It is thus possible that the D1R and glucose signaling pathways in orexin GI neurons interact, enabling presynaptic dopamine release to modulate glucose sensitivity. Interestingly, there are reports suggesting that modafinil improves hypoglycemia awareness in patients with diabetes (36,37). These studies attributed the effect of modafinil to decreased γ–aminobutyric acid (GABA) transmission, although this mechanism was not evaluated in this study. Modafinil and orexin might affect learning; however, neither orexin stimulation nor inhibition impairs memory (38–41). Moreover, the mice learned CPP prior to RH or modafinil. Thus, it is unlikely that effects on learning explain our results. Further studies to elucidate the mechanism(s) underlying the effect of modafinil observed here are required.
Interestingly, the response to low glucose by orexin GI neurons appears to be less robust in mice that received saline or insulin injections with and without modafinil compared with uninjected mice (compare Figs. 1–3 with Figs. 4 and 6). A potential explanation is that the stress of the injections affected glucose sensitivity. This suggests that orexin GI neurons may be sensitive to other stressors in addition to hypoglycemia. However, even if glucose sensitivity did not return to that observed in naïve mice, our observation that modafinil abolishes the difference in glucose sensitivity between RS and RH still suggests that modafinil opposes the effect of RH. Moreover, our data suggest that the stress of RH overrides the putative injection stress. Like VMH GI neurons (24), we found two patterns of glucose responses in PFH orexin GI neurons: those that remain active for the duration in low glucose conditions (sGI) and those that adapt and return to an inhibited state in low glucose conditions (AdGI). VMH sGI and AdGI neurons have distinct glucose sensitivity and showed differences in estrus cycle–phase sensitivity (24,25). Glucose sensitivity similarly differed in AdGI compared with sGI PFH orexin neurons, although the difference was much slighter in PFH than VMH neurons, as seen by the significant overlap in Fig. 3. Both subtypes exhibited 2DG inhibition and responded to glucose similarly in the presence of TTX and when intracellular ATP level was increased. In addition, both subtypes responded similarly to RH and modafinil. These data suggest that their mechanism of glucose sensing is similar. The inclusion criteria for LH orexin GI neurons in our earlier studies excluded AdGI responses, so we do not know whether these different glucose responses exist in the PFH only (11,42). Interestingly, Williams et al. (43) also reported adapting orexin GI neurons that were more common in the PFH than LH. However, in this case, the investigators characterized the AdGI response as a transient inhibition as glucose levels increased, and the response persisted in the absence of presynaptic input (i.e., in TTX). Whether these disparate responses represent distinct types of GI neurons or whether they result potentially from transient factors (e.g., different energy status of the cell within the slice) remains to be seen.
Our results with TTX suggest that PFH orexin GI neurons directly sense glucose. The caveat to the use of TTX is that it only blocks action potential dependent and not molecular presynaptic transmitter release. Interestingly, the response to low glucose levels was enhanced in the presence of the sodium channel blocker TTX. This suggests that PFH orexin GI neurons also receive presynaptic input from an upstream glucose-sensing neuron within the brain section (or an intact glucose-sensing terminal) that tonically inhibits their response to low glucose levels. LH orexin GI neurons receive input from leptin receptor–positive GABA neurons (11). Interestingly, some LH GABA neurons are also GI neurons, thus activation in low glucose conditions could specifically inhibit activation of PFH orexin GI neurons in low glucose conditions (44). Alternatively, LH melanin-concentrating neurons are glucose excited (10). If these neurons were excitatory and innervated PFH orexin GI neurons, then decreased excitation in low glucose conditions could also explain our result. The teleological advantage of sensing the glucose molecule in the brain as opposed to glucose metabolism is not clear. Our brains did not evolve under conditions in which they would be exposed to nonmetabolizable glucose sources and there would be little if any physiological impact. However, in modern society, if artificial sweeteners cross the blood-brain barrier, disparate signals from glucose sensors could sense the glucose molecule as opposed to glucose metabolism. Such inconsistent signals could potentially disrupt energy homeostasis.
Finally, our difficulties establishing CPP and hypoglycemia in C57BL/6 mice from different vendors (Supplementary Data) are consistent with an increasing body of literature regarding these “control” mice (45–47). In our study, we hypothesize that there were two factors involved: group housing and phenotypic differences in vision. First, conditioning in both strains was dependent on group housing, potentially because of anxiety in single housing. This was likely exacerbated in the Charles River C57BL/6N strain because these mice exhibit increased conditional fear (this occurs to a lesser extent in C57BL/6N mice from other vendors) (45). This increased anxiety could have also contributed to the difficulty in lowering blood glucose levels in these animals. In addition, C57BL/6N mice have a mutation associated with retinal degeneration that may have made visual discrimination of the chambers difficult (46–48). On the other hand, C57BL/6J mice possess the nnt mutation, which predisposes them to diabetes (46). For these reasons, it is very important to be deliberate and transparent in choice of strain and vendor.
In summary, our most important findings are that RH blunted activation of PFH orexin GI neurons in low glucose conditions and that modafinil normalized glucose sensing by these neurons and restored hypoglycemia awareness after RH. Although our data are correlative and do not yet address causality, they suggest that the glucose sensitivity of PFH orexin GI neurons is a therapeutic target for treating hypoglycemia unawareness. Currently, there is no treatment for hypoglycemia unawareness. Modafinil is in clinical use for narcolepsy (49). However, modafinil is in the class of psychomotor stimulants that are schedule IV drugs. Thus, further studies into the mechanism(s) by which RH alters glucose sensing by PFH orexin GI neurons are needed to develop safe and effective therapies for patients with diabetes using intensive insulin therapy to control hyperglycemia.
Article Information
Funding. This work was funded in part by the JDRF (grant 3-SRA-2017-488-S-B to V.H.R. and grant 2017-403-A-N to V.P.), and the National Institutes of Health (grant 1 R01 DK103676-01A1 to V.H.R.).
Author Contributions. V.P. and V.H.R. conceived of and designed the research. P.S., V.P., S.B.T., P.R.H., D.M.S., and H.W. performed experiments. O.I. wrote the CPP detection software. P.S., V.P., S.B.T., and V.H.R. analyzed the data and interpreted results of experiments. P.S. and V.H.R. prepared the figures. V.H.R. drafted manuscript and produced the final version of manuscript, which was approved by all authors. All authors edited and revised the manuscript. V.H.R. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains supplementary material online at https://doi.org/10.2337/figshare.21714221.
V.P. currently is affiliated with the Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY.
V.P. and P.S. share first authorship.
See accompanying article, p. 1055.
References
- 1. Diabetes Control and Complications Trial Research Group . Effect of intensive diabetes treatment on the development and progression of long-term complications in adolescents with insulin-dependent diabetes mellitus. J Pediatr 1994;125:177–188 [DOI] [PubMed] [Google Scholar]
- 2. Schultes B, Jauch-Chara K, Gais S, et al. Defective awakening response to nocturnal hypoglycemia in patients with type 1 diabetes mellitus. PLoS Med 2007;4:e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Towler DA, Havlin CE, Craft S, Cryer P. Mechanism of awareness of hypoglycemia. Perception of neurogenic (predominantly cholinergic) rather than neuroglycopenic symptoms. Diabetes 1993;42:1791–1798 [DOI] [PubMed] [Google Scholar]
- 4. Routh VH, Donovan CM, Ritter S. 2. Hypoglycemia detection. Transl Endocrinol Metab 2012;3:47–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cryer PE. Hypoglycemia-associated autonomic failure in diabetes. Am J Physiol Endocrinol Metab 2001;281:E1115–E1121 [DOI] [PubMed] [Google Scholar]
- 6. Cryer PE. The barrier of hypoglycemia in diabetes. Diabetes 2008;57:3169–3176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Otlivanchik O, Sanders NM, Dunn-Meynell A, Levin BE. Orexin signaling is necessary for hypoglycemia-induced prevention of conditioned place preference. Am J Physiol Regul Integr Comp Physiol 2016;310:R66–R73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sanders NM, Figlewicz DP, Taborsky GJ Jr, Wilkinson CW, Daumen W, Levin BE. Feeding and neuroendocrine responses after recurrent insulin-induced hypoglycemia. Physiol Behav 2006;87:700–706 [DOI] [PubMed] [Google Scholar]
- 9. Schultes B, Oltmanns KM, Kern W, Fehm HL, Born J, Peters A. Modulation of hunger by plasma glucose and metformin. J Clin Endocrinol Metab 2003;88:1133–1141 [DOI] [PubMed] [Google Scholar]
- 10. Burdakov D, Gerasimenko O, Verkhratsky A. Physiological changes in glucose differentially modulate the excitability of hypothalamic melanin-concentrating hormone and orexin neurons in situ. J Neurosci 2005;25:2429–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sheng Z, Santiago AM, Thomas MP, Routh VH. Metabolic regulation of lateral hypothalamic glucose-inhibited orexin neurons may influence midbrain reward neurocircuitry. Mol Cell Neurosci 2014;62:30–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Otlivanchik O, Le Foll C, Levin BE. Perifornical hypothalamic orexin and serotonin modulate the counterregulatory response to hypoglycemic and glucoprivic stimuli. Diabetes 2015;64:226–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Korim WS, Bou Farah L, McMullan S, Verberne AJM. Orexinergic activation of medullary premotor neurons modulates the adrenal sympathoexcitation to hypothalamic glucoprivation. Diabetes 2014;63:1895–1906 [DOI] [PubMed] [Google Scholar]
- 14. Korim WS, Llewellyn-Smith IJ, Verberne AJM. Activation of medulla-projecting perifornical neurons modulates the adrenal sympathetic response to hypoglycemia: involvement of orexin type 2 (OX2-R) receptors. Endocrinology 2016;157:810–819 [DOI] [PubMed] [Google Scholar]
- 15. Harris GC, Aston-Jones G. Arousal and reward: a dichotomy in orexin function. Trends Neurosci 2006;29:571–577 [DOI] [PubMed] [Google Scholar]
- 16. Nevárez N, de Lecea L. Recent advances in understanding the roles of hypocretin/orexin in arousal, affect, and motivation. F1000 Res 2018;7(F1000 Faculty Rev):1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sakurai T. Roles of orexins in regulation of feeding and wakefulness. Neuroreport 2002;13:987–995 [DOI] [PubMed] [Google Scholar]
- 18. Lin J-S, Dauvilliers Y, Arnulf I, et al. An inverse agonist of the histamine H(3) receptor improves wakefulness in narcolepsy: studies in orexin-/- mice and patients. Neurobiol Dis 2008;30:74–83 [DOI] [PubMed] [Google Scholar]
- 19. Ishizuka T, Murotani T, Yamatodani A. Chapter Fourteen - Action of Modafinil Through Histaminergic and Orexinergic Neurons. In Vitamins & Hormones. Gerald L, Ed. Cambridge, MA, Academic Press, 2012, pp. 259–278 [DOI] [PubMed] [Google Scholar]
- 20. Ishizuka T, Murotani T, Yamatodani A. Modanifil activates the histaminergic system through the orexinergic neurons. Neurosci Lett 2010;483:193–196 [DOI] [PubMed] [Google Scholar]
- 21. Qu W-M, Huang Z-L, Xu X-H, Matsumoto N, Urade Y. Dopaminergic D1 and D2 receptors are essential for the arousal effect of modafinil. J Neurosci 2008;28:8462–8469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Scammell TE, Estabrooke IV, McCarthy MT, et al. Hypothalamic arousal regions are activated during modafinil-induced wakefulness. J Neurosci 2000;20:8620–8628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamanaka A, Beuckmann CT, Willie JT, et al. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron 2003;38:701–713 [DOI] [PubMed] [Google Scholar]
- 24. Santiago AM, Clegg DJ, Routh VH. Estrogens modulate ventrolateral ventromedial hypothalamic glucose-inhibited neurons. Mol Metab 2016;5:823–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Santiago AM, Clegg DJ, Routh VH. Ventromedial hypothalamic glucose sensing and glucose homeostasis vary throughout the estrous cycle. Physiol Behav 2016;167:248–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ishizuka T, Murakami M, Yamatodani A. Involvement of central histaminergic systems in modafinil-induced but not methylphenidate-induced increases in locomotor activity in rats. Eur J Pharmacol 2008;578:209–215 [DOI] [PubMed] [Google Scholar]
- 27. Song Z, Levin BE, McArdle JJ, Bakhos N, Routh VH. Convergence of pre- and postsynaptic influences on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 2001;50:2673–2681 [DOI] [PubMed] [Google Scholar]
- 28. Song Z, Routh VH. Recurrent hypoglycemia reduces the glucose sensitivity of glucose-inhibited neurons in the ventromedial hypothalamus nucleus. Am J Physiol Regul Integr Comp Physiol 2006;291:R1283–R1287 [DOI] [PubMed] [Google Scholar]
- 29. Wang R, Liu X, Hentges ST, et al. The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes 2004;53:1959–1965 [DOI] [PubMed] [Google Scholar]
- 30. Hao L, Sheng Z, Potian J, Deak A, Rohowsky-Kochan C, Routh VH. Lipopolysaccharide (LPS) and tumor necrosis factor alpha (TNFα) blunt the response of Neuropeptide Y/Agouti-related peptide (NPY/AgRP) glucose inhibited (GI) neurons to decreased glucose. Brain Res 2016;1648:181–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Song Z, Routh VH. Differential effects of glucose and lactate on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 2005;54:15–22 [DOI] [PubMed] [Google Scholar]
- 32. Murphy BA, Fakira KA, Song Z, Beuve A, Routh VH. AMP-activated protein kinase (AMPK) and nitric oxide (NO) regulate the glucose sensitivity of ventromedial hypothalamic (VMH) glucose-inhibited (GI) neurons. Am J Physiol Cell Physiol 2009;297:C750–C758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. González JA, Jensen LT, Fugger L, Burdakov D. Metabolism-independent sugar sensing in central orexin neurons. Diabetes 2008;57:2569–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang Y-L, Ran X-R, Li Y, et al. Expression of dopamine receptors in the lateral hypothalamic nucleus and their potential regulation of gastric motility in rats with lesions of bilateral substantia nigra. Front Neurosci 2019;13:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alberto CO, Trask RB, Quinlan ME, Hirasawa M. Bidirectional dopaminergic modulation of excitatory synaptic transmission in orexin neurons [retracted in J Neurosci 2012;32:9116]. J Neurosci 2006;26:10043–10050 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36. Klement J, Mergelkuhl B, Born J, Lehnert H, Hallschmid M. Role of γ-aminobutyric acid signalling in the attenuation of counter-regulatory hormonal responses after antecedent hypoglycaemia in healthy men. Diabetes Obes Metab 2014;16:1274–1278 [DOI] [PubMed] [Google Scholar]
- 37. Smith D, Pernet A, Rosenthal JM, et al. The effect of modafinil on counter-regulatory and cognitive responses to hypoglycaemia. Diabetologia 2004;47:1704–1711 [DOI] [PubMed] [Google Scholar]
- 38. Stanojlovic M, Pallais Yllescas JP Jr, Mavanji V, Kotz C. Chemogenetic activation of orexin/hypocretin neurons ameliorates aging-induced changes in behavior and energy expenditure. Am J Physiol Regul Integr Comp Physiol 2019;316:R571–R583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eacret D, Grafe LA, Dobkin J, et al. Orexin signaling during social defeat stress influences subsequent social interaction behaviour and recognition memory. Behav Brain Res 2019;356:444–452 [DOI] [PubMed] [Google Scholar]
- 40. Aitta-Aho T, Pappa E, Burdakov D, Apergis-Schoute J. Cellular activation of hypothalamic hypocretin/orexin neurons facilitates short-term spatial memory in mice. Neurobiol Learn Mem 2016;136:183–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Uslaner JM, Tye SJ, Eddins DM, et al. Orexin receptor antagonists differ from standard sleep drugs by promoting sleep at doses that do not disrupt cognition. Sci Transl Med 2013;5:179ra144. [DOI] [PubMed] [Google Scholar]
- 42. Teegala SB, Sheng Z, Dalal MS, Hirschberg PR, Beck KD, Routh VH. Lateral hypothalamic orexin glucose-inhibited neurons may regulate reward-based feeding by modulating glutamate transmission in the ventral tegmental area. Brain Res 2020;1731:145808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Williams RH, Alexopoulos H, Jensen LT, Fugger L, Burdakov D. Adaptive sugar sensors in hypothalamic feeding circuits. Proc Natl Acad Sci USA 2008;105:11975–11980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Karnani MM, Szabó G, Erdélyi F, Burdakov D. Lateral hypothalamic GAD65 neurons are spontaneously firing and distinct from orexin- and melanin-concentrating hormone neurons. J Physiol 2013;591:933–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bryant CD, Zhang NN, Sokoloff G, et al. Behavioral differences among C57BL/6 substrains: implications for transgenic and knockout studies. J Neurogenet 2008;22:315–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fontaine DA, Davis DB. Attention to background strain is essential for metabolic research: C57BL/6 and the International Knockout Mouse Consortium. Diabetes 2016;65:25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Simon MM, Greenaway S, White JK, et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol 2013;14:R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mattapallil MJ, Wawrousek EF, Chan C-C, et al. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci 2012;53:2921–2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Barateau L, Lopez R, Dauvilliers Y. Management of narcolepsy. Curr Treat Options Neurol 2016;18:43. [DOI] [PubMed] [Google Scholar]