

SUMMARY
Background:
Severe combined immunodeficiency (SCID) is fatal unless durable adaptive immunity is established, most commonly through allogeneic hematopoietic cell transplantation (HCT). The Primary Immune Deficiency Treatment Consortium (PIDTC) explored factors impacting survival of individuals with SCID over almost four decades, focusing on the effects of population-based newborn screening (NBS) for SCID.
Methods:
We analyzed transplant-related data from children with SCID treated at 34 PIDTC sites in the United States and Canada, using time intervals 1982–1989, 1990–1999, 2000–2009, and 2010–2018. Categorical variables were compared by chi-square test and continuous outcomes by the Kruskal-Wallis test. Overall survival (OS) was estimated by the Kaplan-Meier method and modeled using Cox regression.
Findings:
For 902 children with confirmed SCID, 5-year OS remained unchanged at 72–73% for 28 years until 2010–2018, when it increased to 87% (n=268; p<0·001). Children identified by NBS since 2010 had 92·5% OS, better than that of children identified by other means in the same interval, 79·9–85·4% (p=0·043). Multivariable analysis demonstrated active infection (HR 2·41, 95% CI 1·56–3·72; p<0·001), age at HCT ≥3·5 months (HR 2·12, 95% CI 1·38–3·24; p=0·001), Black/African-American race (2·33, 95% CI 1·56–3·46; p<0·001), and certain SCID genotypes to have lower OS during all time intervals. Moreover, after adjusting for numerous factors in this multivariable analysis, HCT after 2010 no longer conveyed a survival advantage over earlier time intervals studied (HR 0·73, 95% CI 0·43–1·26; p=0·097). This indicated that younger age and freedom from infections at HCT, both directly driven by NBS, were the main drivers for recent improvement in OS.
Interpretation:
Population-based NBS has facilitated identification of infants with SCID early in life, in turn leading to prompt HCT while avoiding infections. Public health programmes worldwide can benefit from this definitive demonstration of the benefit of NBS for SCID.
Funding:
National Institute of Allergy and Infectious Diseases, Office of Rare Diseases Research, and National Center for Advancing Translational Sciences.
INTRODUCTION
Severe combined immunodeficiency (SCID), characterized by severely impaired T- and B-cell immunity, is lethal without immune reconstitution, which can be achieved with allogeneic hematopoietic cell transplantation (HCT).1 Since 1968, when HCT for SCID was first performed successfully,2 advances in this procedure have increased the likelihood of long-term survival.3–5 Despite the favorable impact of these developments during the earliest decades of transplantation, overall survival (OS) for SCID improved only modestly from the 1990s through 2010.3,6,7 Multiple studies have shown that children transplanted at ≥3·5 months of age or in the presence of active infections have worse OS.7–12 Introduction of newborn screening (NBS) for SCID,13,14 has facilitated early diagnosis and implementation of measures to prevent infection, resulting in HCT at younger ages without concurrent infections. However, the impact of the introduction of SCID NBS on OS has not been definitively studied to date. The Primary Immune Deficiency Treatment Consortium (PIDTC) is a United States and Canadian collaborative research group that investigates factors determining outcomes for primary immunodeficiencies.15 PIDTC natural history protocols have assembled data evaluating treatments and outcomes for SCID that span almost four decades, providing the opportunity to analyze longitudinal changes in HCT practices and to identify which patient, donor, and HCT factors have influenced OS. Here, we present outcomes after HCT for SCID including the years before 2010 and from 2010–2018, when SCID NBS was being increasingly adopted across the United States and Canada.
METHODS
Study design and participants
Participants were enrolled in PIDTC natural history protocols 6901 (prospective) or 6902 (retrospective) (ClinicalTrials.gov Identifiers NCT01346150 and NCT01186913, respectively; appendix p5 Figure S1). De-identified, coded data were entered into an electronic database. Eligible patients had either “typical” or “atypical” SCID (the latter including leaky SCID, Omenn syndrome, or reticular dysgenesis).16 Study eligibility was confirmed by an expert review panel. After exclusions (appendix p5 Figure S1), the final dataset included 902 children from 34 PIDTC centers (appendix p6 Figure S2) who underwent allogeneic HCT between January 1, 1982, and December 31, 2018, inclusive of the 100 subjects published by Heimall et al.10
Procedures
Patient, transplant, and outcome variables and protocol enrollment procedures can be found in appendix p2 Document S1. Race and ethnicity were collected from the medical record. Trigger for diagnosis was defined as the single, initial reason each child received immunologic testing leading to a SCID diagnosis: 1) family history (FH; asymptomatic infant tested due to a recognized prior affected relative); 2) NBS (asymptomatic infant with abnormal population-based NBS); or 3) clinical presentation (illness, including infection or features of immune dysregulation, such as diffuse skin rash). HCT was performed according to each treating center’s standard practice.
Statistical analysis
The 36 years of this study were broken into four intervals, 1982–1989,1990–1999, 2000–2009, and 2010–2018, allowing the era of NBS initiation and expansion, 2010–2018, to be compared to other intervals. Research questions and hypotheses are listed in appendix p3 Document S2. Demographic, disease-related, and HCT-related variables were described using frequencies for categorical variables and median and inter-quartile range (IQR) for quantitative variables. The chi-square test was used to evaluate associations with categorical variables, while the Kruskal-Wallis test was used for continuous variables. The Kaplan-Meier method was used to calculate probabilities of OS after HCT, with children censored at last follow-up. Univariate comparisons of OS in select subgroups were performed using the log-rank test. Probabilities of acute and chronic graft-versus-host disease (GVHD) were summarized using the cumulative incidence method, with death considered a competing event. Confidence intervals were calculated using log-log transformation. Multivariable analysis (MVA) using Cox proportional hazards regression models17 examining risk factors for HCT outcomes were built using bi-directional stepwise selection, with p≤0·05 indicative of statistical significance. The MVA excluded HCT using human leukocyte antigen (HLA)-matched sibling donors, since this donor source facilitated consistently high rates of OS (≥92%) in all time intervals. The following variables were considered in the risk adjustment model: time interval of HCT, infection status and age at HCT, trigger for diagnosis, SCID type and genotype, race and ethnicity of the patient, non-HLA-matched sibling donor type, graft type, GVHD prophylaxis, and conditioning intensity. The proportional hazards assumption of the Cox model was assessed for each variable using graphical approaches (log(-log) plots and Schoenfeld residuals 17,18) and time-dependent covariates, and no violations of the proportional hazards assumption were identified; therefore, the impacts of covariates on time to event outcomes were summarized using hazard ratios. All two-way interactions with time interval were also assessed, but none were significant. Transplant center effects were assessed19 but were not significant; sensitivity analysis including random center effects were performed but results were similar and are not reported. Additional MVA Cox regression analyses were conducted to examine the impact of trigger for diagnosis on OS. These models adjusted for the same variables previously identified in the full-cohort MVA model evaluating OS except for age at HCT and infection history at HCT. These two variables were omitted from the risk adjustment because they are on the causal pathway from trigger for diagnosis to OS (i.e., NBS can lead to earlier age at HCT and reduced infection exposure, which then can improve OS). If they had been included, adjusting for them would then bias the estimation of the impact of NBS on OS by removing the effect of NBS that results from earlier age at HCT and reduced infection exposure. The same approach was conducted in a subgroup analysis of the NBS era (2010–2018) to further reduce confounding between trigger for diagnosis and era, since NBS was only available starting in 2010. Finally, a sensitivity analysis of the causal effect of trigger for diagnosis on OS was also conducted using a Cox model stratified on the propensity score or probability of being diagnosed by NBS vs. clinical symptoms. Propensity score was estimated using stepwise logistic regression,20 considering the following patient and disease variables in the model: SCID type and genotype, sex, race, and ethnicity of the patient. All available data was utilized; no imputation was used for missing data, but patients with missing covariates were included in survival models using a missing category for that covariate.
Role of the funding source
The funders of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. PIDTC investigators independently controlled the conduct of the study and had final approval of the manuscript.
RESULTS
Table 1 lists baseline characteristics and median length of follow-up of surviving patients during each interval. Of all the intervals studied, 5-year OS for 2010–2018 was highest at 87% (95% CI 82·1–90·6%). In contrast, 1982–1989, 1990–1999, and 2000–2009 had OS of 72·4% (CI 63·1–79·7%), 72·6% (CI 66–78%), and 73% (CI, 67·3–77·8%), respectively (p=0·001; Figure 1A and appendix p10 Table S1). OS was high and unchanged between intervals when HCT was performed using HLA-matched sibling donors (92–100%, Figure 1B; p=0·577); however, OS after HCT from other donor sources improved only after 2010 (Figure 1C, D). The improved OS after 2010 was observed irrespective of conditioning intensity, defined as: no conditioning/immune suppression (none/IS), p=0·031; vs. reduced intensity conditioning (RIC) or myeloablative conditioning (MAC); p=0·001 (appendix p7 Figure S3). As major changes in transplant approaches have occurred over the past 20 years, a sub-analysis of OS for interval 2000–2018 was performed (appendix p8 Figure S4). In this contemporary era, no survival advantages were seen when comparing different non-HLA-matched sibling donor options (p=0·567), conditioning intensities (p=0·389), or stem cell sources (p=0·096). Causes of death, separated by the trigger for diagnosis, are described in appendix p11, Table S2.
Table 1.
Baseline SCID Patient and Transplant Characteristics, 1982–2018 (n=902)
| Time interval | |||||
|---|---|---|---|---|---|
| 1982–1989 | 1990–1999 | 2000–2009 | 2010–2018 | p value | |
| Total patients, n | 115 | 224 | 295 | 268 | |
| Race, n (%) | |||||
| American Indian/Alaska Native | 6 (5·2) | 15 (6·7) | 11 (3·7) | 7 (2·6) | 0·511 |
| Asian/Pacific Islander | 3 (2·6) | 6 (2·7) | 13 (4·4) | 17 (6·3) | |
| Black or African American | 14 (12·2) | 20 (8·9) | 30 (10·2) | 28 (10·4) | |
| White | 78 (67·8) | 154 (68·8) | 205 (69·5) | 179 (66·8) | |
| Unknown | 14 (12·2) | 29 (12·9) | 36 (12·2) | 37 (13·8) | |
| Sex, n (%) | |||||
| Male | 84 (73·0) | 166 (74·1) | 207 (70·2) | 166 (61·9) | 0·018 |
| Female | 31 (27·0) | 58 (25·9) | 88 (29·8) | 102 (38·1) | |
| Ethnicity, n (%) | |||||
| Hispanic, Latino, or Spanish origin | 11 (9·6) | 37 (16·5) | 82 (27·8) | 68 (25·4) | <0·001 |
| Not Hispanic, Latino, or Spanish origin | 73 (63·5) | 150 (67·0) | 190 (64·4) | 177 (66·0) | |
| Unknown or not reported | 31 (27·0) | 36 (16·1) | 23 (7·8) | 22 (8·2) | |
| Declined to state | 0 (0·0) | 1 (0·4) | 0 (0·0) | 1 (0·4) | |
| SCID typea, n (%) | |||||
| Typical SCID | 102 (88·7) | 211 (94·2) | 253 (85·8) | 181 (67·5) | <0·001 |
| Atypical SCID | 13 (11·3) | 13 (5·8) | 42 (14·2) | 87 (32·5) | |
| Leaky | 7 (53·8) | 7 (53·8) | 25 (59·5) | 65 (74·7) | 0·383 |
| Omenn syndrome | 5 (38·5) | 5 (38·5) | 15 (35·7) | 17 (19·5) | |
| Reticular dysgenesis | 1 (7·7) | 1 (7·7) | 2 (4·8) | 5 (5·7) | |
| SCID genotype, n (%) | |||||
| ADA | 13 (11·3) | 16 (7·1) | 15 (5·1) | 11 (4·1) | <0·001 |
| DCLRE1C/LIG4/NHEJ1 | 4 (3·5) | 12 (5·4) | 11 (3·7) | 14 (5·2) | |
| IL2RG/JAK3 | 32 (27·8) | 67 (29·9) | 109 (36·9) | 91 (34) | |
| IL7R, CD3 (any), CD45 | 3 (2·6) | 18 (8·0) | 25 (8·5) | 28 (10·4) | |
| RAG1/RAG2 | 2 (1·7) | 6 (2·7) | 35 (11·9) | 63 (23·4) | |
| Other identified genotypesb | 0 (0·0) | 1 (0·4) | 1 (0·3) | 27 (10·1) | |
| Unknown | 61 (53·0) | 104 (46·4) | 99 (33·6) | 34 (12·7) | |
| Trigger for diagnosis, n (%) | |||||
| Family history | 39 (33·9) | 72 (32·1) | 97 (32·9) | 49 (18·3) | <0·001 |
| Newborn screening | 0 (0) | 0 (0) | 0 (0) | 130 (48·5) | |
| Clinical illness | 74 (64·4) | 145 (64·7) | 197 (66·8) | 89 (33·2) | |
| Unknown | 2 (1·7) | 7 (3·1) | 1 (0·3) | 0 (0) | |
| Infection status at HCT, n (%) | |||||
| Actively infected | 68 (59·1) | 102 (45·5) | 127 (43·1) | 82 (30·6) | <0·001 |
| Resolved infection | 24 (20·9) | 66 (29·5) | 81 (27·5) | 52 (19·4) | |
| No prior history of infection | 21 (18·3) | 46 (20·5) | 77 (26·1) | 132 (49·3) | |
| Unknown infectious status | 2 (1·7) | 10 (4·5) | 10 (3·4) | 2 (0·7) | |
| Age at HCT – Categorical, n (%) | |||||
| <3·5 months old | 27 (23·5) | 62 (27·7) | 77 (26·1) | 126 (47·0) | <0·001 |
| ≥3·5 months old | 88 (76·5) | 162 (72·3) | 218 (73·9) | 142 (53·0) | |
| Median (Inter-quartile range, IQR) of follow-up for surviving patients, years | 18·1 (8·7–23·0) | 12·7 (5·5–16·4) | 5·3 (3·5–8·2) | 4·0 (2·0–5·6) | |
| Donor, n (%) | |||||
| Matched sibling | 13 (11·3) | 32 (14·3) | 40 (13·6) | 40 (14·9) | <0·001 |
| Matched other related | 8 (7·0) | 11 (4·9) | 9 (3·1) | 7 (2·6) | |
| Mismatched related (includes haploidentical related) | 91 (79·1) | 165 (73·7) | 153 (51·9) | 63 (23·5) | |
| Matched unrelated | 2 (1·7) | 4 (1·8) | 15 (5·1) | 68 (25·4) | |
| Mismatched unrelated | 1 (0·9) | 4 (1·8) | 16 (5·4) | 18 (6·7) | |
| Unrelated, unknown HLA matching | 0 (0·0) | 2 (0·9) | 4 (1·4) | 1 (0·4) | |
| Cord blood, all HLA matching | 0 (0·0) | 6 (2·7) | 58 (19·7) | 71 (26·5) | |
| Stem cell product, n (%) | |||||
| Marrow | 109 (94·8) | 200 (89·3) | 168 (56·9) | 146 (54·5) | <0·001 |
| Peripheral blood stem cells | 6 (5·2) | 18 (8·0) | 69 (23·4) | 51 (19·0) | |
| Cord blood | 0 (0·0) | 6 (2·7) | 58 (19·7) | 71 (26·5) | |
| Conditioningc, n (%) | |||||
| None | 78 (67·8) | 127 (56·7) | 127 (43·1) | 62 (23·1) | <0·001 |
| Immunosuppression | 22 (19·1) | 33 (14·7) | 46 (15·6) | 32 (11·9) | |
| Reduced-intensity conditioning (RIC) | 0 (0·0) | 10 (4·5) | 47 (15·9) | 98 (36·6) | |
| Myeloablative conditioning (MAC) | 14 (12·2) | 52 (23·2) | 73 (24·7) | 75 (28·0) | |
| Conditioned, but unknown RIC vs MAC | 1 (0·9) | 2 (0·9) | 2 (0·7) | 1 (0·4) | |
| GVHD prophylaxis, n (%) | |||||
| T cell depletion, soybean lectin/ E-rosetting | 79 (68.7) | 143 (63·8) | 79 (26.8) | 5 (1·9) | <0·001 |
| T cell depletion, other monoclonal antibody or immunotoxind | 14 (12.2) | 15 (6.7) | 2 (0.7) | 0 (0.0) | |
| T cell depletion, alpha/beta depletion | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (0.7) | |
| T cell depletion, unknown method | 0 (0.0) | 5 (2.2) | 0 (0.0) | 9 (3.4) | |
| CD34 selection ± T cell depletion | 1 (0·9) | 10 (4·5) | 73 (24·7) | 61 (22·8) | |
| Pharmacologic immunosuppressione with addition of ATG or alemtuzumab | 4 (3·5) | 28 (12·5) | 81 (27·5) | 129 (48·1) | |
| Pharmacological immunosuppressione alone | 2 (1·7) | 14 (6·3) | 47 (15·9) | 51 (19·0) | |
| None | 15 (13·0) | 9 (4·0) | 13 (4·4) | 11 (4·1) | |
Distributions of demographic, other patient-related factors, and transplant-specific characteristics compared by time interval.
SCID type is defined as “typical” or “atypical” SCID16. Typical SCID is defined as having either low numbers of CD3 T cells (<300/µL) with low T cell function (<10% normal as measured by PHA mitogen stimulation) or having presence of T cells of maternal origin. Atypical SCID can be characterized by several criteria defining Leaky SCID, Omenn Syndrome, or Reticular Dysgenesis. Leaky SCID is defined by reduced numbers of CD3 T cells based on age and absence of maternal engraftment. Omenn Syndrome is defined by generalized skin rash with absence of maternal engraftment and CD3 T cells ≥300/µL and absent or low T cell function, defined as <30% normal as measured by PHA mitogenic stimulation; if PHA mitogen stimulation was not performed, at least 4 of 10 additional criteria must be met, including 1) hepatomegaly, 2) splenomegaly, 3) lymphadenopathy, 4) increased IgE level, 5) increased absolute eosinophil count, with at least one of the following included in the 4 criteria, 6) oligoclonal T cells, 7) having >80% of CD3 or CD4 T cells being CD45RO, 8) having proliferation to PHA mitogen stimulation <30% normal, 9) having proliferative response to mixed leukocyte reaction <30% normal, or 10) having a mutation in a known SCID-causing gene. Reticular dysgenesis is defined by absence or very low CD3 T cells (<300/µL), no or very low T cell function (<10% normal as measured by PHA mitogen stimulation), severe neutropenia (absolute neutrophil count <200/μL), and sensorineural deafness and/or absence of granulopoiesis at bone marrow examination and/or a deleterious AK2 mutation.
RMRP (n=9), AK2 (n=6), PNP (n=4), ZAP70 (n=4), MSN (n=2), BCL11B (n=1), MAN2B2 (n=1), ORAI1 (n=1), TTC7A (n=1).
As defined by Pai et al.· NEJM 20142· Specifically, children received 1) no conditioning (none); 2) immunosuppression (regimens containing one or more of the following: fludarabine, cyclophosphamide, ATG, or alemtuzumab); 3) reduced-intensity conditioning (defined as regimens containing melphalan, anti-CD45 antibodies, 2–4 Gray of total body irradiation, or busulfan at a total dose of <12 mg per kilogram of body weight); and 4) myeloablative conditioning (regimens containing busulfan at a total dose ≥12 mg per kilogram)·
Other monoclonal antibody or immunotoxin-based T cell depletion methods include anti-OKT12 (n=13), ricin immunotoxin (n=6), anti-Leu (n=2), anti-CD6/CD8 (n=1), anti-CD5 (n=1), anti-OKT3 (n=1), and unknown antibody, complement, or elutriation method (n=7).
Medications used for GVHD prophylaxis, such as calcineurin inhibitors or methotrexate·
Abbreviations: ATG, antithymocyte globulin; GVHD, graft-versus-host disease; HCT, hematopoietic cell transplantation; HLA, human leukocyte antigen; PHA, phytohaemagglutinin; SCID, severe combined immunodeficiency
Figure 1. Overall Survival by Donor Source.
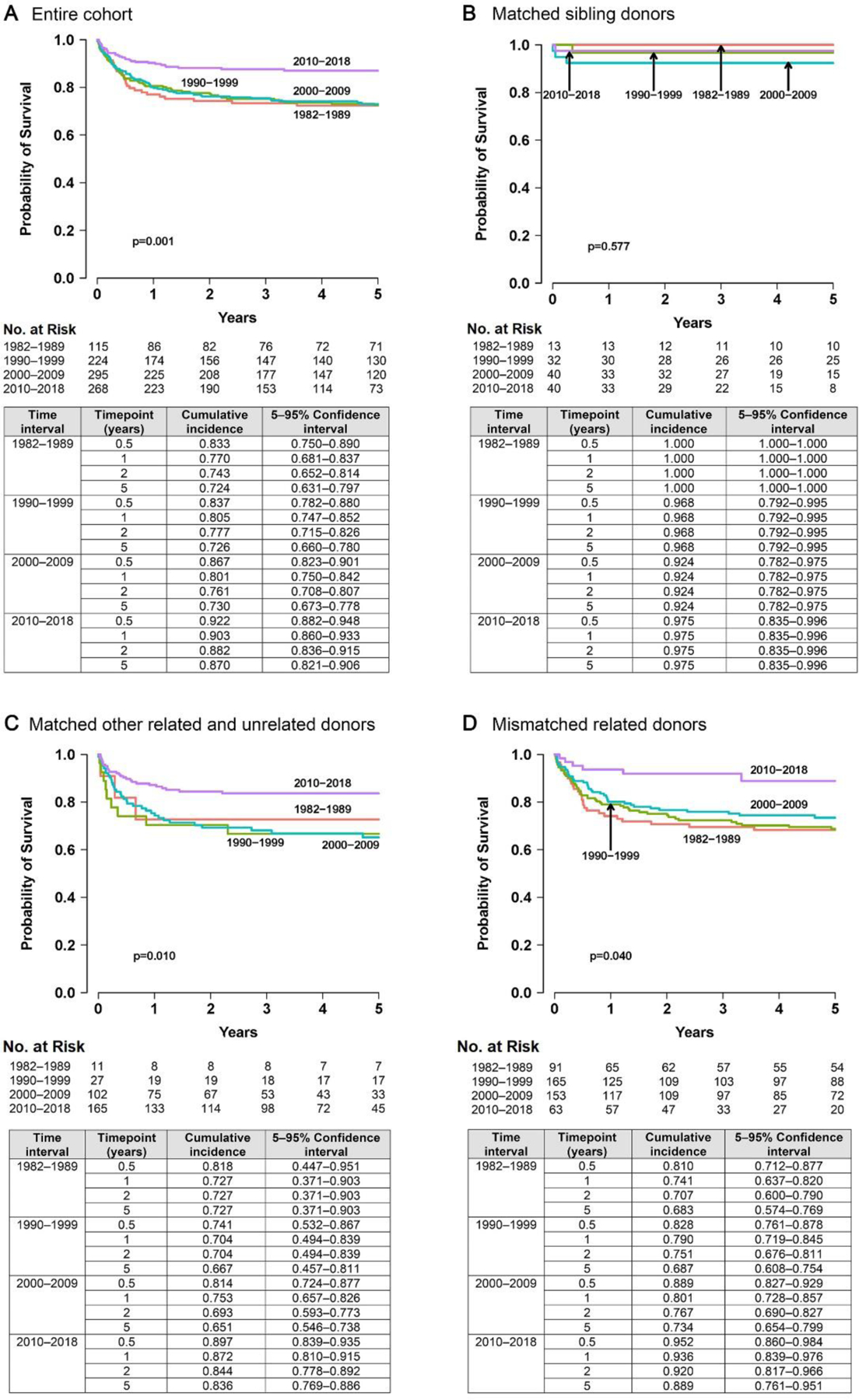
A, Entire cohort; B, matched sibling donors; C, matched other related and unrelated donors; and D, mismatched related donors. All donor sources other than HLA-matched siblings showed improvement in survival during 2010–2018. HLA-matched sibling transplantation showed superior survival throughout all time intervals.
To understand factors contributing to improvement in survival since 2010, a full summary of demographic, clinical, and transplant-related factors with the potential to impact survival were collected and analyzed. There were no differences in race distribution (p=0·511) between time intervals, but a higher proportion of patients of Hispanic/Latino ethnicity enrolled after 2000 (about 25%) compared to earlier intervals (10–17%; p<0·001) (Table 1). Sex differences in each interval (p=0·018) reflected the contribution males with X-linked, IL2RG-deficient SCID, the most prevalent genotype. Although most patients were classified as typical SCID (83%, n=747), the percentage diagnosed with atypical SCID increased from 11·3% in the 1980s to 32·5% after 2010 (p<0·001; Table 1). Importantly, SCID genotype distribution changed over time (p<0·001). As previously described,21 the predominant genotype group, IL2RG and JAK3, made up one-third of this cohort, but more patients were recently identified with RAG1 and RAG2 mutations, which increased from 1·7% in the 1980s to 23·4% after 2010. This was associated with an increased proportion of patients identified as having atypical SCID in the most recent time interval (Table 1). Conversely, unknown genotypes decreased from 53% in the 1980s to 12·7% after 2010.
Prior to 2010, 32·1–32·9% of SCID diagnoses were made after pre-emptive testing due to FH and 64·7–66·8% due to presenting with clinical illness (Table 1). By contrast, between 2010–2018, 48·5% of patients were diagnosed after a positive SCID NBS vs. 33·2% due to clinical illness (p<0·001). Indeed, by 2015–2018, detection by NBS accounted for 65·5% of cases (p<0·001; appendix p12 Table S3).
Infection status also improved over time (p<0·001) (Table 1). From 1982–1989, 59·1% of the patients were transplanted with active infections, which by 2010–2018 dropped to 30·6%. The proportion of patients with no prior history of infections at HCT also rose, from 18·3% during 1982–1989 to 49·3% during 2010–2018. Active infections at HCT were remarkably fewer in patients diagnosed by NBS or FH (Table 2) (p<0·001). Over half of the children who presented to medical attention due to clinical illness did not clear their infections prior to HCT, in contrast to those diagnosed by FH (24·1%) or NBS (20·8%) (Table 2). Furthermore, within the group identified due to clinical illness, infection status at HCT was unchanged throughout the 36 years of this study (p=0·269; appendix p13 Table S4). In contrast, more children diagnosed by FH or NBS underwent HCT with no prior history of infections (51·8% and 66·2%, respectively), compared to only 11·1% of children brought to medical attention based on clinical illness (Table 2). For 2010–2018, most children diagnosed by FH (87·8%) or NBS (79·2%) proceeded to HCT without active infections (Table 2).
Table 2.
Infection Status and Age at Transplant, by Trigger for Diagnosis
| Time interval | Variable | Trigger for diagnosisa | p value | ||
|---|---|---|---|---|---|
| Family history | Newborn screening | Clinical illnessb | |||
| 1982–2018 | Total patients (n) | 257 | 130 | 505 | |
| Infection status at HCT, n (%) | <0·001 | ||||
| Infected/Active | 62 (24·1) | 27 (20·8) | 284 (56·2) | ||
| Infected/Resolved | 57 (22·2) | 17 (13·1) | 146 (28·9) | ||
| Infected/Unknownc | 5 (1·9) | 0 (0·0) | 18 (3·6) | ||
| No infection | 133 (51·8) | 86 (66·2) | 56 (11·1) | ||
| Unknown | 0 (0·0) | 0 (0·0) | 1 (0·2) | ||
| Age at HCT – actual | |||||
| Days of life, median (IQR) | 67·0 (29·0–151·0) | 92·5 (67·0–114·0) | 222·0 (167·0–311·0) | <0·001 | |
| Age at HCT – categorical n (%) | |||||
| <3·5 months old | 161 (62·6) | 87 (66·9) | 42 (8·3) | <0·001 | |
| ≥3·5 months old | 96 (37·4) | 43 (33·1) | 463 (91·7) | ||
| 2010–2018 | Total patients (n) | 49 | 130 | 89 | |
| Infection status at HCT, n (%) | |||||
| Infected/Active | 6 (12·2) | 27 (20·8) | 49 (55·1) | <0·001 | |
| Infected/Resolved | 11 (22·5) | 17 (13·1) | 24 (27·0) | ||
| Infected/Unknownc | 0 (0·0) | 0 (0·0) | 1 (1·1) | ||
| No Infection | 32 (65·3) | 86 (66·1) | 14 (15·7) | ||
| Unknown | 0 (0·0) | 0 (0·0) | 1 (1·1) | ||
| Age at HCT – actual | |||||
| Days of life, median (IQR) | 76·0 (49·0–132·0) | 92·5 (67·0–114·0) | 252·0 (152·0–461·0) | <0·001 | |
| Age at HCT – categorical, n (%) | |||||
| <3·5 months old | 31 (63·3) | 87 (66·9) | 8 (9·0) | <0·001 | |
| ≥3·5 months old | 18 (36·7) | 43 (33·1) | 81 (91·0) | ||
Infection status at HCT was evaluated based on trigger for diagnosis, with a sub-analysis (2010–2018) for the era when NBS became available.
Ten patients had an unknown single trigger for diagnosis identified and are therefore excluded from this analysis.
Typically related to infections, autoimmunity, and/or failure to thrive.
An infection pre-HCT was documented, but whether it was still active or had resolved at time of HCT was not recorded.
Abbreviations: FH, family history; IQR, interquartile range; NBS, newborn screening
Age at transplant was stable from 1982 to 2009, with median ages of 193 (1982–1989), 190 (1990–1999), and 187 (2000–2009) days (appendix p14 Table S5). However, in the era of NBS (2010–2018), the median HCT age dropped to 111 days (p<0·001) and even lower (94 days) for those with typical SCID (p<0·001). Patients diagnosed by FH had median age of 67 days (n=257), compared to 92·5 (n=130) and 222 (n=505) days for diagnoses triggered by NBS or clinical illness, respectively (p<0·001) (Table 2). The same differences were observed when the analysis was limited to the 2010–2018 time interval (p<0·001) (Table 2).
Transplant-related factors, including donor choice, stem cell source, conditioning intensity, T-cell depletion strategies and prophylaxis to prevent graft-versus-host disease (GVHD), as well as GVHD outcomes were analyzed (Table 1). While use of HLA-matched sibling donors remained stable over 36 years, transplants using HLA-mismatched related donors facilitated by extensive T-cell depletion dropped from 79·1% to 23·5%. Stem cell products diversified over time, with bone marrow predominantly used from 1982–89 (94·6%) decreasing to 54·5% after 2010, with concomitant increases after 2010 in cord blood (26·3%) and peripheral blood stem cells (19%). Measures to minimize GVHD were predominantly graft manipulation (e.g., T-cell depletion with soybean lectin, 67%) from 1982–89, with prophylactic immune suppressive medications and antibodies targeting T-cells primarily used after 2010 (66·1%). Historically, HCT was more commonly performed with no conditioning (67·8% from 1982–1989); however, by 2010–2018, this approach was utilized in only 23·1% of patients. Reduced-intensity conditioning, which did not exist in the earliest time interval studied, was the most common preparative regimen employed by 2010–2018 (36·6%). Finally, while the incidence of chronic GVHD was generally unchanged, severe grade 3–4 acute GVHD decreased by 2010–18 (p=0·025; p9 Figure S5).
On univariate analysis, 5 year OS for those presenting with clinical illness, FH, or NBS was 80% (95% CI, 69·5%−87·0%), 85·4% (95% CI, 71·8%−92·8%), and 92·5% (95% CI, 85·8%−96·1%), respectively (p=0·043) (Figure 2). In pairwise comparisons, the only significant difference in OS was between NBS and presentation with clinical illness (p=0·012); there were no differences in OS between NBS and FH (p=0·184) or FH and clinical illness (p=0·491). To further establish factors underlying the improvement of OS after 2010, MVA was performed using patient and transplant-related variables found to be significant on univariate analysis (Table 3). Upon adjusting for active infection at HCT (p<0·001), age ≥3·5 months at HCT (p=0·001), genotypes with inferior OS (ADA, DNA repair defects, plus rarely identified and unknown genes) compared to the most frequent genotype, IL2RG/JAK3 (p<0·001), and Black/African-American race (p<0·001), the decade in which HCT occurred was no longer a significant determinant of OS (p=0·097) (Table 3). Since active infection at HCT and age at HCT are strongly associated with trigger for diagnosis as previously discussed, we further examined the independent impact of trigger for diagnosis on OS without adjusting for active infection and age at HCT. In MVA adding trigger for diagnosis but removing age and infection status at HCT, we found that NBS, along with a positive family history, impacted OS [HR 0.32 (95% CI, 0.15–0.67); p=0.003 and HR 0.52 (95% CI, 0.37–0.74); p <0.001, respectively] (appendix p8 Table S6). To verify that the impact of NBS was not attributable to the confounding variable of time interval, since NBS is available only since 2010, a subgroup MVA was performed on the 2010–2018 cohort. This analysis confirmed that compared to clinical illness [n=70; HR 2·96 (95% CI, 1·32–6·65); p=0·008], NBS as a trigger for diagnosis improved OS after HCT but was similar to those presenting by FH [n=40; HR 1·70 (95% CI, 0·59–4·86); p=0·322]. Additionally, a sensitivity analysis was performed on this subgroup using propensity scoring to further reduce the effects of confounding bias that can occur in observational datasets. This analysis again confirmed that from 2010–2018, presentation with clinical illness had a detrimental effect on OS [n=70; HR 2·55 (95% CI, 1·12–5·80); p=0·026]. Finally, having typical SCID and Hispanic ethnicity were significant predictors for being identified by NBS (odds ratio (OR) for atypical vs. typical = 0·41, 95% CI 0·21–0·80; p=0·009; OR for Hispanic vs. Non-Hispanic=2·45, 95% CI 1·15–5·24, p=0·020), the latter highlighting that population-based NBS has improved detection of SCID in infants from this disadvantaged ethnic background.
Figure 2. Overall Survival 2010–2018, Based on Trigger for Diagnosis.
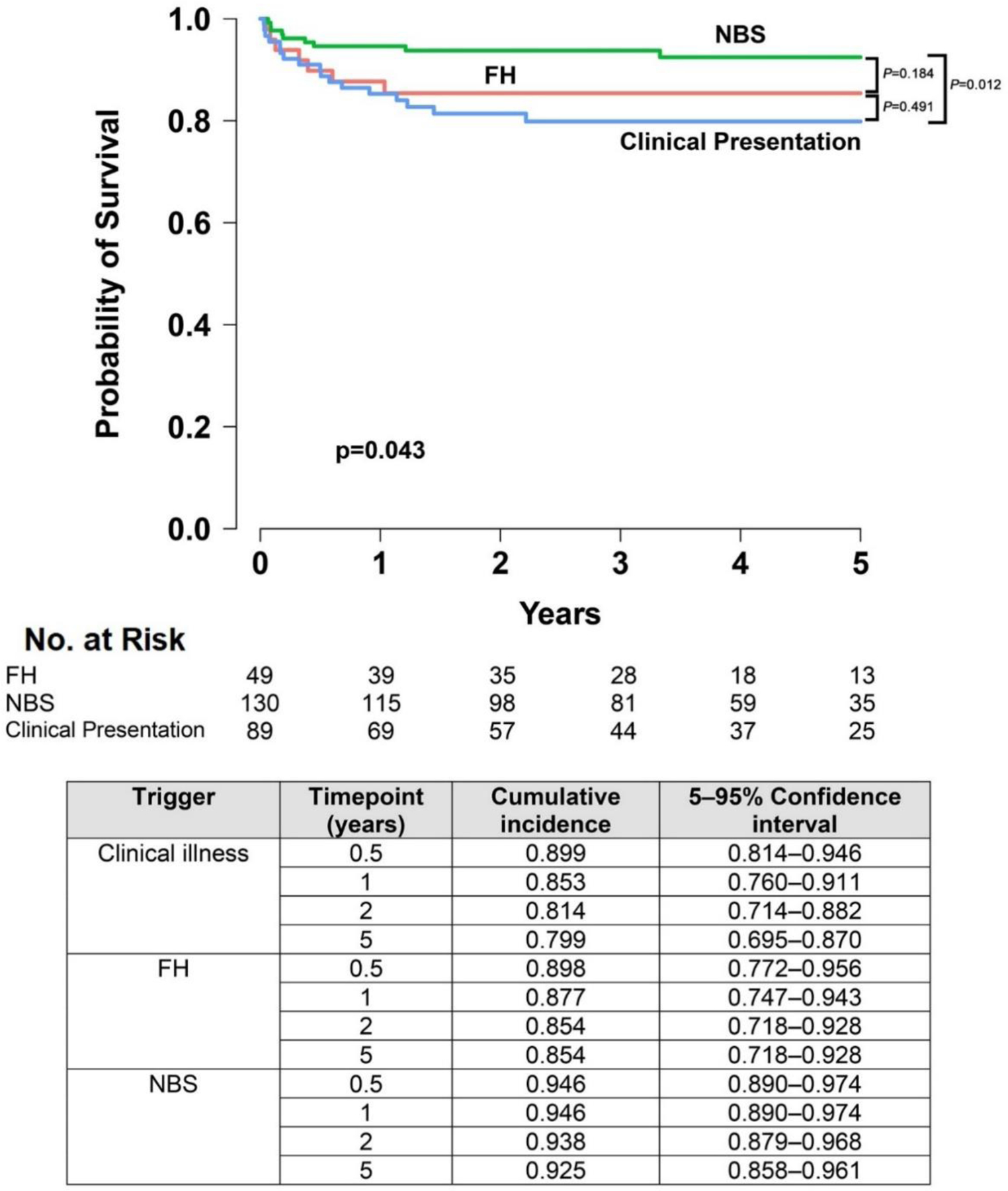
Survival was evaluated more closely during the time interval when NBS became available. The global significance of improved survival with NBS (P=0.043) was driven by differences between NBS and the clinical illness group (P=0.012) rather than differences in survival between NBS and FH (P=0.184).
Abbreviations: FH, family history; NBS, newborn screening.
Table 3.
Multivariable Analysisa of Independent Factors Having Significant Detrimental Impact on Overall Survival (1982–2018), Including Subgroup Analysis (2010–2018)
| Categories and variables | Frequency of event (n)b | Hazard ratio estimatec (95% confidence interval) | P value | Overall p valued |
|---|---|---|---|---|
| Total Group Analysis (1982–2018) | ||||
| Infection at time of transplant | ||||
| No infection | 241 | 1·00 | – | <0·001 |
| Actively infected | 324 | 2·41 (1·56–3·72) | <0·001 | |
| Resolved infection | 189 | 1·24 (0·76–2·03) | 0·382 | |
| Unknown infection status | 20 | 1·81 (0·78–4·23) | 0·169 | |
| Age at transplant | ||||
| <3·5 months | 238 | 1·00 | 0·001 | |
| ≥3·5 months | 536 | 2·12 (1·38–3·24) | 0·001 | |
| Genotype | ||||
| IL2RG/JAK3 | 274 | 1·00 | – | <0·001 |
| ADA | 46 | 2·22 (1·23–4·01) | 0·008 | |
| DCLRE1C/LIG4/NHEJ1 | 31 | 3·67 (1·83–7·35) | <0·001 | |
| IL7R, CD3 (any), CD45 | 66 | 1·26 (0·68–2·34) | 0·464 | |
| RAG1, RAG2 | 83 | 1·72 (0·96–3·09) | 0·069 | |
| Other identified genotypes | 25 | 3·32 (1·48–7·45) | 0·004 | |
| Unknown/not reported | 249 | 2·50 (1·77–3·54) | <0·001 | |
| Race | ||||
| White | 545 | 1·00 | – | <0·001 |
| Native American/Native Alaskan | 31 | 0·92 (0·45–1·85) | 0·810 | |
| Asian/Pacific Islander | 31 | 1·84 (0·98–3·44) | 0·058 | |
| Black or African American | 72 | 2·33 (1·56–3·46) | <0·001 | |
| Unknown/Not declared | 95 | 1·56 (1·05–2·30) | 0·026 | |
| Time interval of transplant | ||||
| 1982–1989 | 102 | 1·00 | – | 0·097 |
| 1990–1999 | 192 | 1·20 (0·79–1·81) | 0·388 | |
| 2000–2009 | 255 | 1·26 (0·83–1·91) | 0·276 | |
| 2010–2018 | 225 | 0·73 (0·43–1·26) | 0·261 | |
| Subgroup Analysis (2010–2018)e | ||||
| Trigger for Diagnosis | ||||
| NBS | 115 | 1·00 | 0·031 | |
| FH | 40 | 1·70 (0·59–4·86) | 0·322 | |
| Clinical Illness | 70 | 2·96 (1·32–6·65) | 0·008 |
From 2010–2018, active infection, older age, genotypes other than IL2RG/JAK3, and Black/African American race had detrimental impact for survival, while decade of transplant had no impact. A subgroup analysis (2010–2018) confirmed that the impact of NBS on survival may be attributed to earlier age and fewer infections at HCT rather than time interval of HCT.
Non-significant variables that were also included in the MVA were type of SCID (typical vs atypical24), trigger for diagnosis (FH, NBS, or clinical illness), ethnicity, donor type, stem cell source, conditioning regimen, GVHD prophylaxis, and use of T cell depletion. This MVA did not include HCT from HLA-matched siblings, who had consistently high rates of OS in all time intervals. Including the HLA-matched sibling group would have hampered the ability to detect differences between the trigger for diagnosis groups in the non-sibling donor categories.
“n” is the number of subjects in the category of the variable; the “n event” is defined by death.
“Hazard ratio estimate” is the hazard ratio, with 95% confidence interval, for the event of death.
“Overall p value” provides the overall significance of the variable in the model.
Subgroup analysis (2010–2018) was performed looking at the effect of trigger for diagnosis in the era of NBS, adjusted for genotype and race as in overall model (1982–2018). Note that infection status and age at transplant were omitted from the variables adjusted for in the subgroup analysis, since these are on the causal pathway from trigger for diagnosis to survival.
Abbreviations: FH, family history; GVHD, graft-versus-host disease; HCT, hematopoietic cell transplantation; HLA, human leukocyte antigen; MVA, multivariable analysis; NBS, newborn screening; OS, overall survival; SCID, severe combined immunodeficiency.
DISCUSSION
The PIDTC’s 36-year experience of transplanting 902 children with SCID, the largest natural history study of SCID transplants published to date, provides an exceptional opportunity to study the impact of NBS on survival after HCT. Contrary to a steady improvement in OS that might have been expected with incremental advances in care, only during the most recent interval, 2010–2018, did our data demonstrate stark improvement in OS (Figure 1A). To confirm our hypothesis that NBS would be associated with improved OS, MVA was performed. Infection status, age at HCT, genotype, and race remained independently significant. A second MVA was also performed which definitively confirmed that NBS was independently associated with improvement in OS. To do this, both age and infection status at HCT were removed from the analysis. Being that both are strongly linked to NBS, it was not appropriate to include them within the model as independent predictors. Notably in both MVA models, being transplanted after 2010 no longer had an impact (Table 3 and Supplementary Table S6). Since NBS is strongly linked to time interval as it has been available only from 2010 onward, a MVA subanalysis focused only on trigger for diagnosis between 2010–2018 was performed (Table 3). This confirmed that the effect of NBS on improved OS was independent of time interval. Thus, advances in HCT approaches did not explain the survival advantage seen after 2010, but instead this advantage was due to infants receiving HCT earlier in life and without active infections. While HCT early in life and without active infection was not unique to the post-2010 time interval, both circumstances became more prevalent as population-based NBS for SCID became more widespread (appendix p6 Figure S2).
NBS for SCID first became possible after 200522 with the development of a DNA-based dried blood spot quantification of TRECs,23 a biomarker for T-cell development, absence of which identifies nearly all cases of SCID. All 50 states24 and four Canadian provinces have been performing NBS for SCID since the end of 202025 (personal communication, Paul Van Caeseele, M.D. FRCPC; Zaiping Liu, M.D, FRCPC; Huiming Yang, M.D., FRCPC; appendix p6 Figure S2). The PIDTC dataset is well-poised to evaluate the impact of SCID NBS on survival after HCT. Its longitudinal database not only encompasses years before and during implementation of SCID NBS, but also reflects extensive efforts in the attribution of a single, initial trigger for how children with SCID were first brought to clinical attention. This information cannot be simply assumed based on an imputed system of birth date, birth state, and onset of a state’s NBS programme,14 and publications from even large individual states26 or small countries27 lack non-screened control data.
While resolution of infections prior to HCT has been a universally favorable predictor for OS,9,10 a study by the Stem Cell Transplant for Primary Immune Deficiencies in Europe (SCETIDE) Registry found no survival advantage based on younger age at HCT.12 In their cohort of 338 patients with SCID transplanted between 2006–2014, age less than or >3·5 months at HCT had similar 2-year OS (87·8% vs 82·0%, p=0·15). It is unclear why this age cut-off observed in other studies did not significantly impact OS in the SCETIDE analysis; however, infants who died due to being too sick to undergo HCT were not included. It is also possible that transplant age and infection status were linked, a circumstance that could have masked an effect of age alone in this cohort of smaller sample size than the PIDTC cohort.
Two other factors that influenced OS on MVA in the PIDTC dataset were race and genotype. African American/Black children, about 10% of the cohort, had the highest risk of death (HR 2·33; 95% CI, 1·56–3·46; p<0·001). While NBS should diminish racial disparities by universally identifying SCID in the newborn period, other racial inequities in HCT outcomes have been reported.28 Our data highlight the need to identify contributors to this finding in the SCID population. Finally, we confirmed our prior findings that certain genotypes had worse OS, including those causing DNA repair defects (DCLRE1C/LIG4/NHEJ1) and ADA SCID.7 This also contrasts with data from the smaller SCETIDE cohort in which OS was similar between genotypes.12 NBS also improved detection of children with RAG1/2 mutations, whose diagnosis would otherwise have been delayed due to their hypomorphic phenotype; identification of infants with these and other rarer genotypes correlated with a decrease in the proportion of patients with unknown gene defects in the last decade.
During the last four decades, approaches to HCT for SCID have evolved. Prior to the introduction of NBS, many infants presented with severe infections and were transplanted as quickly as possible, often in the face of active infections and using HLA-mismatched related donors.1,6,9,10 In our dataset, OS using HLA-matched sibling donors had the best survival throughout all decades, while OS when using non-sibling donors improved significantly only after 2010 (Figure 1). Furthermore, HCTs were often performed without conditioning, as graft rejection was low in the absence of T cells,1 but this practice has also changed with most now receiving conditioning. Acute GVHD, an important complication of HCT, showed declines in incidence of grades 3–4 since 2010, while chronic GVHD remained unchanged, another important area for future studies.
Several limitations exist for this study. Due to the nature of this diverse patient population and intervention being studied, there are innate biases introduced such as lead-time bias. This is common in studies evaluating screening programmes and makes it statistically challenging to show their direct benefit; however, earlier diagnosis and increased likelihood of being infection-free at HCT demonstrate the advantages of NBS. Furthermore, it is not possible to directly evaluate survival in infants “exposed” to NBS versus not. We were best able to overcome this limitation by studying the role of the trigger for diagnosis as it related to transplant survival, thereby isolating those brought to medical attention by NBS compared to other means. For the MVA, since factors on the causal pathway could impact results, a sensitivity analysis was performed which confirmed the original MVA results. Additionally, since this study included only those children who were transplanted, those who may have been identified by NBS but died prior to HCT were not assessed. Race and ethnicity data were obtained from medical records and may not have been self-reported. Moreover, factors specifically addressing why race impacted HCT survival, including whether there were differences in diagnostic trigger or timely access to treatment, could not be ascertained and will need future study. Selection biases in this patient population may also exist, partially addressed by requiring participating centers to offer enrollment to all patients with SCID treated at their sites; but not all SCID transplants are performed at PIDTC sites.
Other factors need to be considered in support of SCID NBS programmes both regionally and globally. Without a population-based NBS programme, babies with SCID are generally asymptomatic at birth, typically presenting with life-threatening infections within the first year of life. The economic burden of managing these sick infants, as well as additional risks and higher cost of treatment when they eventually proceed to HCT, is tremendous 29 and can shift resources away from other public health initiatives. Finally, while advocacy is strong for NBS, implementing policies in countries where there is ubiquitous use of live vaccines at birth may be challenging.30
In conclusion, the PIDTC has provided the largest longitudinal transplant dataset to date for SCID and includes the era of NBS implementation. Our findings summarize the evolution of almost four decades of transplant practices and provide direct evidence that NBS for SCID has been the primary driver of improved survival in the United States and Canada since 2010. This study can assist in supporting global health efforts to expand SCID NBS worldwide.
Supplementary Material
PANEL: RESEARCH IN CONTEXT.
Evidence before the study
We searched PubMed from inception to November 1, 2022, examining publications focused on outcomes after allogeneic hematopoietic cell transplantation (HCT) for severe combined immunodeficiency (SCID) when considering newborn screening (NBS). Keywords used included “severe combined immunodeficiency”, “hematopoietic stem cell transplantation,” and “newborn screening.” This yielded 123 articles from centers throughout Asia, Europe, North America, and South America. Review articles, book chapters, and non-clinical papers were excluded. No randomized controlled trials were identified. Risk factors for poor overall survival after HCT, including active infections at transplant, were extensively described in the literature. While several case reports, single- and multi-institutional retrospective studies, and clinical trials postulated that SCID NBS is needed to improve survival outcomes, no publications definitively measured the impact of NBS for SCID upon survival.
Added value of the study
This is the first time that comparative data have shown the benefit of SCID NBS, accounting for improved survival after allogeneic HCT for infants lacking an HLA matched related donor. Beginning in 2008, SCID NBS, accomplished by quantifying T-cell receptor excision circles DNA from newborn dried blood spots, has been adopted across the United States and increasingly in Canadian provinces, as well as in multiple countries throughout the world. By evaluating time intervals both before and after the launch of NBS and ascertaining which infants were identified by NBS in the large, longitudinal dataset of the Primary Immune Deficiency Treatment Consortium (PIDTC), we were able to demonstrate a substantial favorable impact of NBS on survival, even after adjusting for era of transplantation, when advancements in supportive care and transplant practices might also have driven improvement in survival. Since randomized controlled studies will never be possible to test the advantages of population-based SCID NBS, our analysis, which spans the years of introduction of SCID NBS, provides the strongest level of support for this public health programme.
Implications of all the available evidence
After initiation of SCID NBS, the United States and Canada have witnessed enhanced survival of patients with SCID independent of advances in preventing infections and in transplantation procedures. Despite differences in local medical practice, the ability to identify infants with SCID early through NBS and to provide allogeneic HCT prior to the onset of life-threatening infections has the potential to save lives worldwide.
Funding/Support
This work was supported by the Division of Allergy, Immunology and Transplantation, National Institute of Allergy and Infectious Diseases (NIAID); the Office of Rare Diseases Research (ORDR), National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH); Public Health Service, NIH, NIAID grant U54AI082973 (MPI: J. M. Puck, Christopher C. Dvorak, Elie Haddad); NIH, National Institute of Neurological Disorders and Stroke grant U54NS064808 and National Center for Advancing Translational Sciences (NCATS) grant U01TR001263 (PI: J. Krischer); NIH, NIAID grant R13AI094943 (PI: J.M. Puck). The PIDTC is a part of the Rare Diseases Clinical Research Network (RDCRN) of ORDR, NCATS. The collaborative work of the PIDTC with the Pediatric Transplantation and Cellular Therapy Consortium (PTCTC) is supported by the U54 grants listed here, along with support of the PBMTC Operations Center by the St. Baldrick’s Foundation and NIH, National Heart, Lung and Blood Institute (NHLBI) grant/cooperative agreement U10HL069254 (PI: M.M. Horowitz). Collaborative work of the PIDTC with the Center for International Blood and Marrow Transplant Research (CIBMTR) is supported by NIH, National Cancer Institute grant/cooperative agreement U24CA076518 (PI: B.E. Shaw), and NHLBI, and NIAID; and NIH, NHLBI grant/cooperative agreement U01HL069294; by contracts HHSH250201200016C and HHSH234200637015C with the Health Resources and Services Administration (HRSA/DHHS); and by grants N00014–13–1–0039 and N00014–14–1–0028 from the Office of Naval Research. L.D.N.is supported by the Division of Intramural Research, NIH, NIAID grant 1 ZIA AI001222–02 (PI: L.D. Notarangelo). S.Y.P. is supported by the Intramural Research Program, National Institutes of Health, National Cancer Institute, Center for Cancer Research. M.J.C is supported by the California Institute of Regenerative Medicine (CLIN2–10830 and CLIN2–09504) and NIAID grant U54AI082973. L.F.S is supported by NIAD R21 AI156583 and NCATS UG3 TR003908 and the William T. Shearer Texas Children’s Hospital Center for Human Immunobiology. M.A.P. is supported by 1U01AI126612–01A1, P30CA040214, and 2UG1HL069254. E.H. is supported by the Bank of Montreal Chair of Pediatric Immunology.
The content and opinions expressed are solely the responsibility of the authors and do not represent the official policy or position of the NIAID, ORDR, NCATS, NIH, HRSA, or any other agency of the US Government.
Footnotes
Conflicts of Interest Disclosures
M.S.T. is a member of the Scientific Advisory Board for Proteios Technology, Inc. and has been a consultant for the non-profit Infectious Disease Research Institute.
J.M.P. has received royalties from Up-To-Date, and her spouse is employed by and owns stock in Invitae (a DNA sequencing company).
M.J.C. has received royalties from Up-to-Date and is on the scientific board of Homology Medicines, Inc, a gene therapy company and is a DSMB member for Bluebird Bio, Chiesi USA, and Rocket Pharmaceuticals.
L.F.S. is a member of the adjudication committee for Orchard Therapeutics and has received consulting fees from Takeda, CSL Behring, ADMA, Grifols and Horizon.
J.H. is on an Investigator-initiated grant, CSL Behring; Consultant, ADMA; Consultant, CIRM; Clinical Trial PI (industry sponsored research), Regeneron; Consultant, Horizon.
L.M.B. has received clinical trial support through the Fred Hutchinson Cancer Research Center by Medac GmbH (including supply of treosulfan), is a member of the DSMB for clinical trials with Rocket Pharmaceuticals and Jasper Therapeutics and has served on an advisory board for Horizon Therapeutics USA.
B.D.S. has ad hoc consultancies for Sobi and Orchard Therapeutics.
S.C. is a member on the advisory board for SOBI.
J.J.B. is a member on advisory board for Sobi and Horizon Therapeutics.
G.D.E.C. has received consultancy fees from Miltenyi Biotech.
F.G. has received consultancy fees from GLG and Guidepoint Global and is on the advisory board for Omerus.
E.B.S. is on the speaker’s bureau for Sobi.
E.H.C. is an advisory board member for Pfizer.
S.P. has received funding for the conduct of sponsored trials Atara Biotherapeutics, AlloVir, and Jasper Therapeutics and is on the advisory board for ADMA and Neovii; Inventor of IP Licensed to Atara with all rights assigned to MSKCC.
K.S. has served as a consultant for Enzyvant and the Immune Deficiency Foundation.
A.P. is on the advisory boards for Orchard (MLD GT), Horizon, and Enzyvant, serves on the DSMB for ExCellThera.M.K, and is a consultant for Enzyvant (2020).
T.Q. has been on the speaker’s bureau and advisory board for Alexion Pharmaceuticals.
S.S. is on the advisory board and provides consultancy for Graphite, Takeda, Janssen, Bristol Myers Squibb. DSMC - NHLBI and Aruvant Sciences.
H.D. NONE for this study. Advisor on a Eli Lilly Canada Scientific Advisory Board in April 2020.
J.R. Green Cross, Therapure
T.R.T. None in relation to this work.
J.W.L. In the last 12 months: Speaker and consultant for Sobi; Speaker, Consultant, and received research funding from Horizon Therapeutics; Speaker, Consultant, and Advisory Board for Pharming; Speaker and Consultant for CSL-Behring; Advisory Board : ADMA Biologisc; Site PI for Therapure and GreenCross. Employee and Shareholder at Bluebird Bio.
M.P. has served on the advisory boards of Novartis, Medexus, Equillium, Mesoblast, has received clinical study support from Adaptive and Miltenyi Biotech, and has received financial support for educational lectures for Miltenyi Biotech and Novartis.
D.B.K. is an inventor for the UC Regents on a lentiviral vector for gene therapy of ADA SCID and is a member of the DSMB for Revcovi PEG-ADA (Chiesi, USA).
E.H. has received consultancy fees from JASPERS, Takeda, CSL-Behring, Octapharma and Leadiant Biosciences.
C.C.D. is a member of the DSMB for Revcovi PEG-ADA (Chiesi, USA). Consulting for Orchard Therapeutics.
All other authors expressed no conflicts of interest to disclose.
Role of Sponsors
The sponsors had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication
Data Sharing
The study data will be available upon request from the PIDTC after the publication of the study. Individual participant data (de-identified), the coding dictionary, and other specific sets of data will be provided upon approval of a research proposal and executed data sharing agreement. The protocol, analysis plan, and informed consent documents are also available upon request.
REFERENCES
- 1.Buckley RH, Schiff SE, Schiff RI, et al. Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. N Engl J Med 1999; 340(7): 508–16. [DOI] [PubMed] [Google Scholar]
- 2.Gatti RA, Meuwissen HJ, Allen HD, Hong R, Good RA. Immunological reconstitution of sex-linked lymphopenic immunological deficiency. Lancet (London, England) 1968; 2(7583): 1366–9. [DOI] [PubMed] [Google Scholar]
- 3.Antoine C, Muller S, Cant A, et al. Long-term survival and transplantation of haemopoietic stem cells for immunodeficiencies: report of the European experience 1968–99. Lancet 2003; 361(9357): 553–60. [DOI] [PubMed] [Google Scholar]
- 4.Haddad E, Landais P, Friedrich W, et al. Long-term immune reconstitution and outcome after HLA-nonidentical T-cell-depleted bone marrow transplantation for severe combined immunodeficiency: a European retrospective study of 116 patients. Blood 1998; 91(10): 3646–53. [PubMed] [Google Scholar]
- 5.Dvorak CC, Long-Boyle J, Dara J, et al. Low Exposure Busulfan Conditioning to Achieve Sufficient Multilineage Chimerism in Patients with Severe Combined Immunodeficiency. Biol Blood Marrow Transplant 2019; 25(7): 1355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gennery AR, Slatter MA, Grandin L, et al. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: entering a new century, do we do better? J Allergy Clin Immunol 2010; 126(3): 602–10; e1–11. [DOI] [PubMed] [Google Scholar]
- 7.Haddad E, Logan BR, Griffith LM, et al. SCID genotype and 6-month posttransplant CD4 count predict survival and immune recovery. Blood 2018; 132(17): 1737–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dvorak CC, Cowan MJ, Logan BR, et al. The natural history of children with severe combined immunodeficiency: Baseline features of the first fifty patients of the Primary Immune Deficiency Treatment Consortium Prospective Study 6901. Journal of Clinical Immunology 2013; 33(7): 1156–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pai SY, Logan BR, Griffith LM, et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med 2014; 371(5): 434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heimall J, Logan BR, Cowan MJ, et al. Immune reconstitution and survival of 100 SCID patients post-hematopoietic cell transplant: a PIDTC natural history study. Blood 2017; 130(25): 2718–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dorsey MJ, Wright NAM, Chaimowitz NS, et al. Infections in Infants with SCID: Isolation, Infection Screening, and Prophylaxis in PIDTC Centers. J Clin Immunol 2021; 41(1): 38–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lankester AC, Neven B, Mahlaoui N, et al. Hematopoietic cell transplantation in severe combined immunodeficiency: The SCETIDE 2006–2014 European cohort. J Allergy Clin Immunol 2021. [DOI] [PubMed]
- 13.Kwan A, Abraham RS, Currier R, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. Jama 2014; 312(7): 729–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marsh RA, Hebert KM, Keesler D, et al. Practice pattern changes and improvements in hematopoietic cell transplantation for primary immunodeficiencies. Journal of Allergy and Clinical Immunology 2018; 142(6): 2004–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griffith LM, Cowan MJ, Kohn DB, et al. Allogeneic hematopoietic cell transplantation for primary immune deficiency diseases: current status and critical needs. J Allergy Clin Immunol 2008; 122(6): 1087–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shearer WT, Dunn E, Notarangelo LD, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the Primary Immune Deficiency Treatment Consortium experience. J Allergy Clin Immunol 2014; 133(4): 1092–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klein JP, Moeschberger ML. Survival analysis : techniques for censored and truncated data New York: Springer; 2003. [Google Scholar]
- 18.Grambsch PM, Therneau TM. Proportional hazards tests and diagnostics based on weighted residuals. Biometrika 1994; 81(3): 515–26. [Google Scholar]
- 19.Commenges D, Andersen PK. Score test of homogeneity for survival data. Lifetime Data Anal 1995; 1(2): 145–56; discussion 57–9. [DOI] [PubMed] [Google Scholar]
- 20.Austin PC. A tutorial and case study in propensity score analysis: An application to estimating the effect of in-hospital smoking cessation counseling on mortality. Multivariate Behav Res 2011; 46(1): 119–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dvorak CC, Haddad E, Buckley RH, et al. The genetic landscape of severe combined immunodeficiency in the United States and Canada in the current era (2010–2018). J Allergy Clin Immunol 2019; 143(1): 405–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan K, Puck JM. Development of population-based newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol 2005; 115(2): 391–8. [DOI] [PubMed] [Google Scholar]
- 23.Hazenberg MD, Verschuren MC, Hamann D, Miedema F, van Dongen JJ. T cell receptor excision circles as markers for recent thymic emigrants: basic aspects, technical approach, and guidelines for interpretation. J Mol Med (Berl) 2001; 79(11): 631–40. [DOI] [PubMed] [Google Scholar]
- 24.Currier R, Puck JM. SCID newborn screening: What we’ve learned. J Allergy Clin Immunol 2021; 147(2): 417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Souza A, Wolan V, Battochio A, et al. Newborn Screening: Current Status in Alberta, Canada. Int J Neonatal Screen 2019; 5(4): 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amatuni GS, Currier RJ, Church JA, et al. Newborn Screening for Severe Combined Immunodeficiency and T-cell Lymphopenia in California, 2010–2017. Pediatrics 2019; 143(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lev A, Sharir I, Simon AJ, et al. Lessons Learned From Five Years of Newborn Screening for Severe Combined Immunodeficiency in Israel. J Allergy Clin Immunol Pract 2022; 10(10): 2722–31.e9. [DOI] [PubMed] [Google Scholar]
- 28.Majhail NS, Nayyar S, Santibañez ME, Murphy EA, Denzen EM. Racial disparities in hematopoietic cell transplantation in the United States. Bone Marrow Transplant 2012; 47(11): 1385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van den Akker-van Marle ME, Blom M, van der Burg M, Bredius RGM, Van der Ploeg CPB. Economic evaluation of different screening strategies for severe combined immunodeficiency based on real-life data. Int J Neonatal Screen 2021; 7(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marciano BE, Huang CY, Joshi G, et al. BCG vaccination in patients with severe combined immunodeficiency: complications, risks, and vaccination policies. J Allergy Clin Immunol 2014; 133(4): 1134–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.