

Abstract
We report the development of drug-encapsulating nanoparticles that bind endogenous albumin upon intravenous injection, and evaluate their in vivo performance in a murine as well as canine animal model. The gene encoding a protein-G derived albumin binding domain (ABD) was fused to that of a chimeric polypeptide (CP), and the ABD-CP fusion was recombinantly synthesized by bacterial expression of the gene. Doxorubicin (DOX) was conjugated to the C-terminus of the ABD-CP fusion, and conjugation of multiple copies of the drug to one end of the ABD-CP triggered its self-assembly into ~100 nm diameter spherical micelles. ABD-decorated micelles exhibited sub-micromolar binding affinity for albumin and also preserved their spherical morphology in the presence of albumin. In a murine model, albumin-binding micelles exhibited dose-independent pharmacokinetics, while naked micelles exhibited dose-dependent pharmacokinetics. In addition, in a canine model, albumin binding micelles resulted in a 3-fold increase in plasma half-life and 6-fold increase in plasma exposure as defined by the area under the curve (AUC) of the drug, compared with naked micelles. Furthermore, in a murine colon carcinoma model, albumin-binding nanoparticles demonstrated lower uptake by the reticuloendothelial system (RES) system organs —the liver and spleen— that are the main target organs of toxicity for nanoparticulate delivery systems and higher uptake by the tumor than naked micelles. The increased uptake by s.c. C26 colon carcinoma tumors in mice translated to a wider therapeutic window of doses ranging from 20–60 mg equivalent of DOX per kg body weight (mg DOX Equiv.kg−1 BW) for albumin-binding ABD-CP-DOX micelles, as compared to naked micelles that were only effective at their maximum tolerated dose of 40 mg DOX Equiv.kg−1 BW.
Keywords: micelles, endogenous albumin, elastin like polypeptide, cancer, drug delivery, canine model
Graphical Abstract:

Schematic of the design of a recombinant albumin binding polypeptide micelle, with doxorubicin sequestered in the core and with an albumin binding protein domain presented on the corona of the micelles. Intravenous administration leads to spontaneous formation of an albumin coated corona due to the high affinity of the albumin binding domain with endogenous albumin in blood.
Many small molecule cancer drugs suffer from poor systemic exposure, resulting from their low water solubility and a short plasma half-life. Hydrophilic –colloidally stable– nanocarriers with a size above the renal filtration cutoff of 5–6 nm1 can solve these problems by solubilizing hydrophobic drugs via chemical conjugation or via physical entrapment of drugs in their core2, and by improving drug pharmacokinetics and tissue distribution, leading to enhanced accumulation of the drug in tumors compared to the free drug through the enhanced permeation and retention (EPR) effect3.
However, a vexing problem faced by most systemically administered nanoparticles is that they can be preferentially taken up by macrophages that are prevalent in the reticuloendothelial system—mainly in the liver and spleen, causing a high level of accumulation in these organs that can cause significant off-target toxicity4,5,6,7. Coating nanoparticles with albumin has been proposed as a strategy to prevent association of other serum proteins including opsonins, on the surface of nanoparticles to reduce macrophage uptake and to improve their circulation in blood8–9, which is generally thought to be a prerequisite to improving their accumulation in solid tumors by the EPR effect. Achieving this ex vivo requires incubation of the nanoparticles with albumin, which is currently produced by fractionation of plasma obtained from blood donors10. However, this method requires extra processing steps during nanoparticle synthesis that can cause conformational changes in the structure of albumin, leading to potential immunogenicity11.
An alternative approach, and one that we pursue in this paper, is to coat nanoparticles in vivo by exploiting the selective binding of endogenous albumin to a nanoparticle, and thereby creating an albumin corona on systemically injected nanoparticles. This is a simple and attractive strategy given the ~millimolar concentration of albumin in blood12, and the availability of small molecule ligands and peptides that bind albumin with high affinity and specificity13–14. To our knowledge, with the exception of a report that focused on the development of albumin-binding nanoparticles for vaccine delivery15 and a recent report that showed the extended half-life of albumin binding nanoparticles, but did not report on their therapeutic efficacy16, there are no reports of molecular engineering of nanoparticles that are rationally designed to load a defined payload of small molecule drugs in their interior, bind endogenous albumin on their surface upon systemic administration to create albumin-coated nanoparticles, and whose efficacy has been reported. Motivated by this striking lacuna in the field, we describe herein, a genetically engineered polypeptide nanoparticle that contains a defined number of covalently conjugated drug molecules in its core, and that binds endogenous serum albumin with high affinity on its corona to create an albumin-coated nanoparticle upon in vivo administration.
The recombinant polypeptide nanoparticle system that we designed to achieve this goal has three components that are encoded at its molecular —sequence— level (Figure 1A). The first component is an elastin like polypeptide (ELP). ELPs are a class of genetically encoded and thermally responsive polypeptides composed of the pentapeptide repeat Val-Pro-Gly-Xaa-Gly, where the guest residue (Xaa) can be any amino acid except proline. ELPs exhibit lower critical solution temperature (LCST) phase behavior in aqueous solution, where below a critical solution temperature —termed the inverse transition temperature (Tt) or cloud point temperature— ELPs are soluble, but above the Tt, they desolvate and phase separate into an ELP rich, coacervate phase, and an ELP poor, aqueous phase. Because ELPs are polypeptides and are hence genetically encodable, they can be recombinantly synthesized as monodisperse polymers with a precisely specified molecular weight and composition, and are biodegradable and non-toxic, attributes that are useful for drug delivery17. In addition, their thermal responsiveness allows easy, and chromatography-free purification at a large scale and low cost by a purification method known as inverse transition cycling18. To ensure that the drug conjugate has sufficient amphiphilicity to self-assemble into micelles upon drug conjugation, we chose a hydrophilic ELP that consists of 160 repeats of the Val-Pro-Gly-Xaa-Gly where Xaa is the hydrophilic amino acid, Ala.
Figure 1.
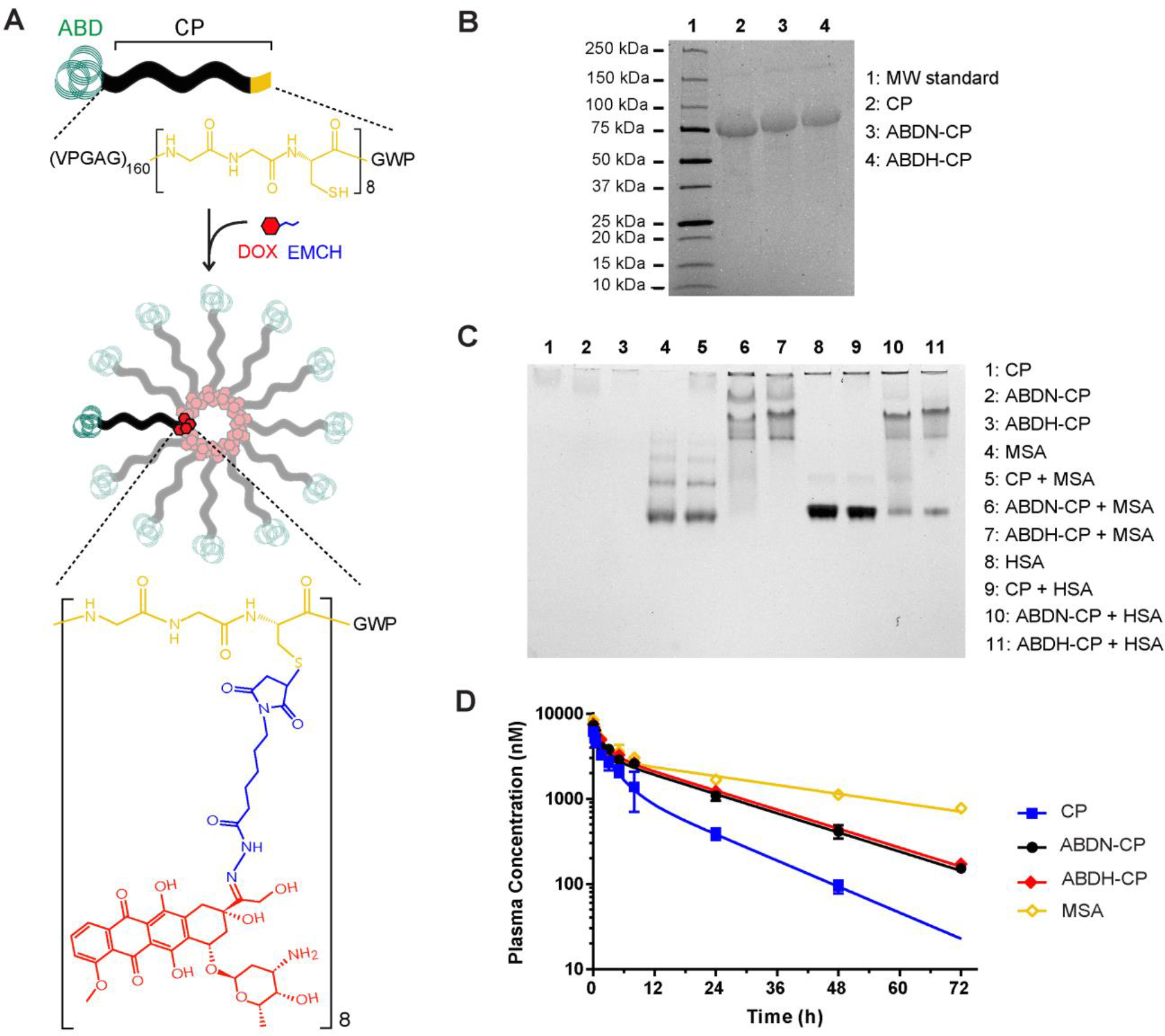
Design of the albumin binding nanoparticle drug carrier. (A) Structure of ABD-CP unimers and ABD-CP-DOX micelles. ABD was genetically fused to the N-terminus of a hydrophilic CP and was recombinantly synthesized in E. coli. DOX was conjugated via a pH-sensitive linker (EMCH) to the thiol groups of the cysteine rich segment at the C-terminus of the CP. The amphiphilic ABD-CP-DOX conjugates self-assembled into micelles with DOX molecules sequestered in the core, and ABDs in the corona that can bind endogenous albumin. (B-D) Characterization of ABD-CP unimers: (B) SDS-PAGE analysis of purified ABDN-CP, ABDH-CP and the CP (control). Successful purification of ABDN-CP and ABDH-CP by inverse transition cycling is confirmed by SDS-PAGE. The primary band corresponds to the molecular weight of CP (63.6 kDa), ABDN-CP (68.5 kDa), and ABDH-CP (68.6 kDa) and a second faint band at higher molecular weight indicates the presence of disulfide linked polypeptide dimers. (C) Native-PAGE analysis of the interaction of human serum albumin (HSA) and mouse serum albumin (MSA) with (ABDN/H-) CP. For the samples in lanes 5–7 and lanes 9–11, polypeptides were mixed at a molar ratio of ~1:1 with MSA and HSA, respectively. (D) Pharmacokinetics of CP, ABDN-CP and ABDH-CP. Polypeptides were labeled with Alexa488 and were administered via tail vein to BALB/c mice. Error bars represent standard error of the mean (n = 5 for ABDN-CP, 6 for CP and MSA, and 7 for ABDH-CP) and plasma concentrations were measured at intervals over 72 h. The data were fitted with a two-compartment model, from which the pharmacokinetic parameters were estimated, as shown in Table 1.
The second component is a short cysteine-rich (Gly-Gly-Cys)8 peptide segment that provides multiple thiol moieties for drug attachment. We have shown that attachment of multiple copies of hydrophobic small molecules to these Cys residues imparts sufficient amphiphilicity to trigger self-assembly of the CP-drug conjugate into spherical micelles19,20. The ELP-(GGC)8 fusion is referred to as a chimeric polypeptide (CP).
The third component of this system is an albumin binding domain (ABD) that is fused at the gene level to the CP at the opposite end of the drug attachment segment. The fusion of an albumin binding domain (ABD) fused to a CP is termed ABD-CP. We used two ABDs in this study that are denoted as native ABD (ABDN) and high affinity ABD (ABDH). ABDN is a 46 amino acid, three-helix bundle derived from streptococcal protein G21, with nanomolar binding affinity for human serum albumin (HSA)22, while ABDH is an engineered variant of ABDN that has several orders of magnitude higher affinity for albumin than ABDN22. The amino acid sequence of ABDN and ABDH are in Supplementary Table S1. We chose ABDs —small protein domains that bind albumin with high affinity and specificity— instead of a small molecule ligand of albumin to create albumin coated nanoparticles, because ABDs are genetically encodable, which allows the entire system —with the exception of drug conjugation— to be synthesized by recombinant expression. Furthermore, ABDs are structurally robust and stable23, as seen by their high melting temperature24 and their ability to withstand denaturation by extremes of pH24. ABDs can also refold to their native conformation after thermal or chemical denaturation. These features are useful as they suggest that ABDs can preserve their conformation and activity under processing conditions required to purify ABD-CPs and conjugate chemotherapeutics to them, such as high temperature (55 °C) and addition of organic co-solvents. In addition, ABDs express at high levels in E. coli and have high aqueous solubility13. Although ABDs have been fused to peptide and protein drugs to extend their half-life and enhance their efficacy13,25, to the best of our knowledge, this is the first study that reports the effect of albumin binding on in vivo tumor regression in a nanoparticle drug formulation. Because the design of this nanoparticle is modular and is genetically encoded, it provides complete, molecular-level control of its properties.
We also synthesized CPs that contain the C-terminal (GGC)8 segment but that lack an ABD at their N-terminus, as negative controls for albumin binding; this version is named CP. ABDN-CP, ABDH-CP, and the CP were purified from E. coli by inverse transition cycling, a non-chromatographic method for purification of ELPs and their fusions26, with high yield (100–200 mgL−1) from shaker flask culture. All constructs were pure, as seen by the dominant single band in SDS-PAGE, and low levels of dimers were barely detectable by SDS-PAGE, presumably because of residual intermolecular disulfide formation (Figure 1B).
The binding affinity of ABDN-CP and ABDH-CP for human serum albumin (HSA) and mouse serum albumin (MSA) was analyzed qualitatively by native-PAGE and quantitatively by ITC. Due to their low charge to molecular weight ratio, ABDN-CP and ABDH-CP show no mobility on native-PAGE whereas HSA and MSA have a high negative charge, allowing them to migrate into the gel towards the cathode. However, when mixed with HSA or MSA, binding of the ABD-CPs to either species of albumin increases the apparent size and decreases the apparent charge to molecular weight ratio of HSA and MSA and consequently impedes their electrophoretic mobility. In addition, the interaction of albumin with ABD-CPs is non-covalent and dynamic, with free albumin existing in equilibrium with the ABD-CP bound albumin. The free albumin at any moment moves into the gel resulting in a smearing pattern on Native-PAGE. In contrast, mixing with the CP control affected the migration pattern of neither HSA nor MSA (Figure 1C).
The calorimetry data for HSA binding to ABDN-CP and ABDH-CP showed a typical sigmoidal shape that suggests tight binding of ABDN-CP and ABDH-CP to HSA. In contrast, the negative control, the CP lacking an ABD, showed a non-sigmoidal curve that suggests weak and nonspecific binding to HSA (Figure S2). Fitting the calorimetry data for ABDN-CP and ABDH-CP to a 1:1 binding site model showed that both ABDN-CP and ABDH-CP bind to HSA with high — nanomolar— affinity, though ABDH-CP has a 10-fold higher binding affinity for HSA (KD= 4.2 ± 1.1 nM) than ABDN-CP (KD= 48.4 ± 3.8 nM ) (Table S2). The experimental fit of the data to the model also yielded a binding stoichiometry (N) of 1.04 and 1.07 for ABDN-CP and ABDH-CP, respectively, which is consistent with the known stoichiometry of these ABDs for HSA27. Similar results were observed for the binding of ABDN-CP, ABDH-CP and the CP control to mouse serum albumin (MSA), as both ABDN-CP (Figure S3–B) and ABDH-CP (Figure S3–C) showed a sigmoidal binding curve, while the CP control showed a non-sigmoidal curve (Figure S3–A). Fitting the calorimetry titration data yielded a KD of 66.4 ± 10.7 nM for ABDN-CP and 5.2 ± 1.9 nM for ABDH-CP, respectively (Table S3).
Next, we studied the thermal behavior of ABDN-CP and ABDH-CP with albumin in the presence of other mouse serum components. To do so, the turbidity profile of ABDN-CP and ABDH-CP unimers in PBS, fetal bovine serum (FBS), and mouse serum (MS) was examined as a function of solution temperature. Fusion of ABDN or ABDH to the CP decreased the Tt of the CP from 60 °C at 100 μM concentration in PBS to 57 °C and 55 °C for ABDN-CP and ABDH-CP, respectively (Figure S1A–C). This decrease in the Tt is attributed to the hydrophobic nature of ABDs23. The Tt was however still above the body temperature and is acceptable for drug conjugation and preparing self-assembling micelles. The CP exhibited a lower Tt in MS as well as FBS than that in PBS, that is the expected consequence of macromolecular crowding in the presence of serum proteins, and is consistent with our previous results28,29. However, in marked contrast to CP unimers (Figure S1A), the ABDN-CP and ABDH-CP unimers in MS did not undergo thermally triggered phase separation into an insoluble coacervate phase up to 70°C (Figure S1B–C). The lack of thermally triggered LCST phase separation of the albumin-bound unimer corroborates that even in the presence of other serum components, MSA binds ABD-CP unimers strongly and creates a steric barrier to aggregation, which effectively abolishes their phase behavior in the temperature range of 10–70 °C.
Having confirmed the tight binding of the two ABD-CP fusions to MSA, next, an in vivo pharmacokinetic study was performed in mice by labeling ABDN-CP, ABDH-CP and the CP control with Alexa488, and measuring the concentration of the polypeptides as a function of time following intravenous administration to mice. MSA was also labeled with Alexa488 and was administered as a positive control. The data were fit to a two-compartment model (Figure 1D) to compute the pharmacokinetic parameters (Table S4). ABD-CP fusions displayed a longer plasma half-life and a greater plasma exposure, measured by calculating the area under the curve (AUC), than the unmodified CP (P < 0.01). However, despite the difference in their affinity for albumin, there was no significant difference between the pharmacokinetic profiles of ABDN-CP and ABDH-CP (Table 1). This can be attributed to the fact that the concentration of albumin in serum is ~1 mM, while the albumin-binding affinity of the ABDs are in the nM regime, so that both ABDs exist in an albumin bound state. This finding is consistent with previous studies that demonstrated that the affinity for albumin has only a minor effect on half-life extension of ABD protein fusions30. The half-life of MSA was found to be 34.6±1.9 h, which is in good agreement with the previously measured half-life of endogenous MSA31.
Table 1.
Pharmacokinetic parameters of CP, ABDN-CP, ABDH-CP, and MSA.
| Pharmacokinetic Parameter | ABDN-CP | ABDH-CP | CP | MSA |
|---|---|---|---|---|
|
| ||||
| Elimination half-life [h] | 16.8 ± 0.5 | 16.9 ± 0.3 | 11.2 ± 0.6 | 34.6 ± 1.9 |
| Area under curve [μM. h] | 80.2 ± 1.9 | 88.3 ± 1.3 | 38.8 ± 0.8 | 157.3 ± 4.3 |
Values are shown as mean ± SD (n = 5 for ABDN-CP, 6 for CP and MSA, and 7 for ABDH-CP). The elimination half -life and the area under curve for ABDN-CP and ABDH-CP are statistically different from that of the CP (P < 0.01) and MSA (P < 0.001).
Next, doxorubicin (DOX) was conjugated to the eight cysteines in the (Gly-Gly-Cys)8 segment appended at the C-terminus of the ABDN-CP and ABDH-CP via a pH-sensitive hydrazone bond, as described previously19. The conjugation ratio was 5.1 ± 0.5, 4.6 ± 0.6, and 4.7 ± 0.3 DOX molecules per polypeptide for CP-DOX, ABDN-CP-DOX and ABDH-CP-DOX, respectively.
Self-assembly of CP-DOX conjugates into micelles was confirmed by light scattering and cryogenic transmission electron microscopy (cryo-TEM). At 22 °C, the hydrodynamic radius (Rh) of CP-DOX, ABDN-CP-DOX, and ABDH-CP-DOX micelles as measured by dynamic light scattering (DLS) was 36.3 nm, 59.0 nm, and 51.3 nm, respectively (Table 2). As the temperature was raised to 37°C, the Rh of CP-DOX, and ABDN-CP-DOX decreased to 22.3 nm and 45.6 nm, respectively, suggesting that with increasing temperature, the micelles desolvate and the CP chains adopt a more compact structure, resulting in a smaller size, an observation that is consistent with previous studies of CP micelles32,33. At 37°C, aggregates were detected in the ABDH-CP-DOX sample in PBS, presumably because of the initiation of the inverse phase transition of ABDH-CP-DOX micelles that has a Tt of ~40 °C.
Table 2.
Light scattering data for (ABDN/H-) CP-DOX micelles in the presence and absence of albumin.
| Sample | Temperature (°C) | Nagg | Rg (nm) | Rh (nm) | ρ = Rg/Rh |
|---|---|---|---|---|---|
|
| |||||
| CP-DOX | 22 | 11 | 28.0 | 36.3 | 0.77 |
| 37 | 17 | 18.7 | 22.3 | 0.84 | |
|
|
|||||
| CP-DOX : MSA (1:1) | 22 | — | a ND | 36.2 | b NC |
| 37 | — | a ND | 30.2 | b NC | |
|
|
|||||
| ABDN-CP-DOX | 22 | 35 | 37.0 | 59.0 | 0.62 |
| 37 | 42 | 35.0 | 45.6 | 0.77 | |
|
|
|||||
| ABDN-CP-DOX : MSA (1:1) | 22 | 59 | 38.4 | 50.2 | 0.77 |
| 37 | 58 | 29.5 | 38.2 | 0.78 | |
|
|
|||||
| ABDH-CP-DOX | 22 | 17 | 34.6 | 51.3 | 0.69 |
| 37 | c Agg | - | - | - | |
|
|
|||||
| ABDH-CP-DOX : MSA (1:1) | 22 | 31 | 37.5 | 47.3 | 0.79 |
| 37 | 36 | 36.8 | 46.9 | 0.78 | |
ND: not detectable
NC: not calculated
Agg: aggregated.
Static light scattering (SLS) provided the radius of gyration (Rg) and aggregation number (Nagg) of the micelles (Table 2). At 37 °C, the Rg and Nagg were 18.7 nm and 17 respectively, for CP-DOX conjugates, and 35.0 nm and 42 respectively, for the ABDN-CP-DOX conjugate. The Rg and Nagg could not be determined for ABDH-CP-DOX conjugate at 37 °C in PBS due to aggregation. Combining the SLS and DLS data also allowed quantification of the shape factor (ρ= Rg/Rh) that is characteristic of the morphology of the micelles. The shape factor was in the range of 0.7–0.8 for CP-DOX and ABDN-CP-DOX micelles at 37 °C, which is close to theoretical value for solid spheres (0.775) and suggests that these micelles have a spherical morphology 34.
Light scattering analysis in the presence of equimolar concentration of MSA showed that binding to albumin did not disrupt the micelle structure of ABDN-CP-DOX (Table 2). In addition, for ABDN/H-CP-DOX micelles, binding to albumin increased the Nagg and decreased the Rh of ABDN-CP-DOX micelles, assuming that each albumin molecule was bound to an ABD. For ABDH-CP-DOX micelles, no aggregates were detected in the presence of mouse albumin at 37 °C (Table 2); this observation is consistent with the thermal profile data where binding to albumin prevented the phase transition and aggregation of ABDN/H-CP-DOX micelles (Figure S1).
To independently verify their morphology by imaging, CP-DOX and ABDN-CP-DOX micelles were vitrified at 37 °C and imaged by cryo-TEM in their native, hydrated state (Figure 2A). The cryo-TEM images corroborated the mean size and morphology obtained by DLS. Due to low electron density of ELP chains, only the hydrophobic and electron-dense DOX-containing core of ABDN-CP-DOX and CP-DOX nanoparticles is visualized by cryo-TEM. The core-to-core distance was used to determine the average nanoparticle radius (RTEM) and was measured as 18.3 ± 0.6 nm and 14.6 ± 0.6 nm (n=40) for ABDN-CP-DOX and CP-DOX micelles, respectively, that is smaller than the Rh determined by DLS. This discrepancy is likely due to the compression of micelles during the vitrification process that can lead to interpenetration of the nanoparticles at their corona when compared to the dilute concentrations necessary for light scattering.
Figure 2.
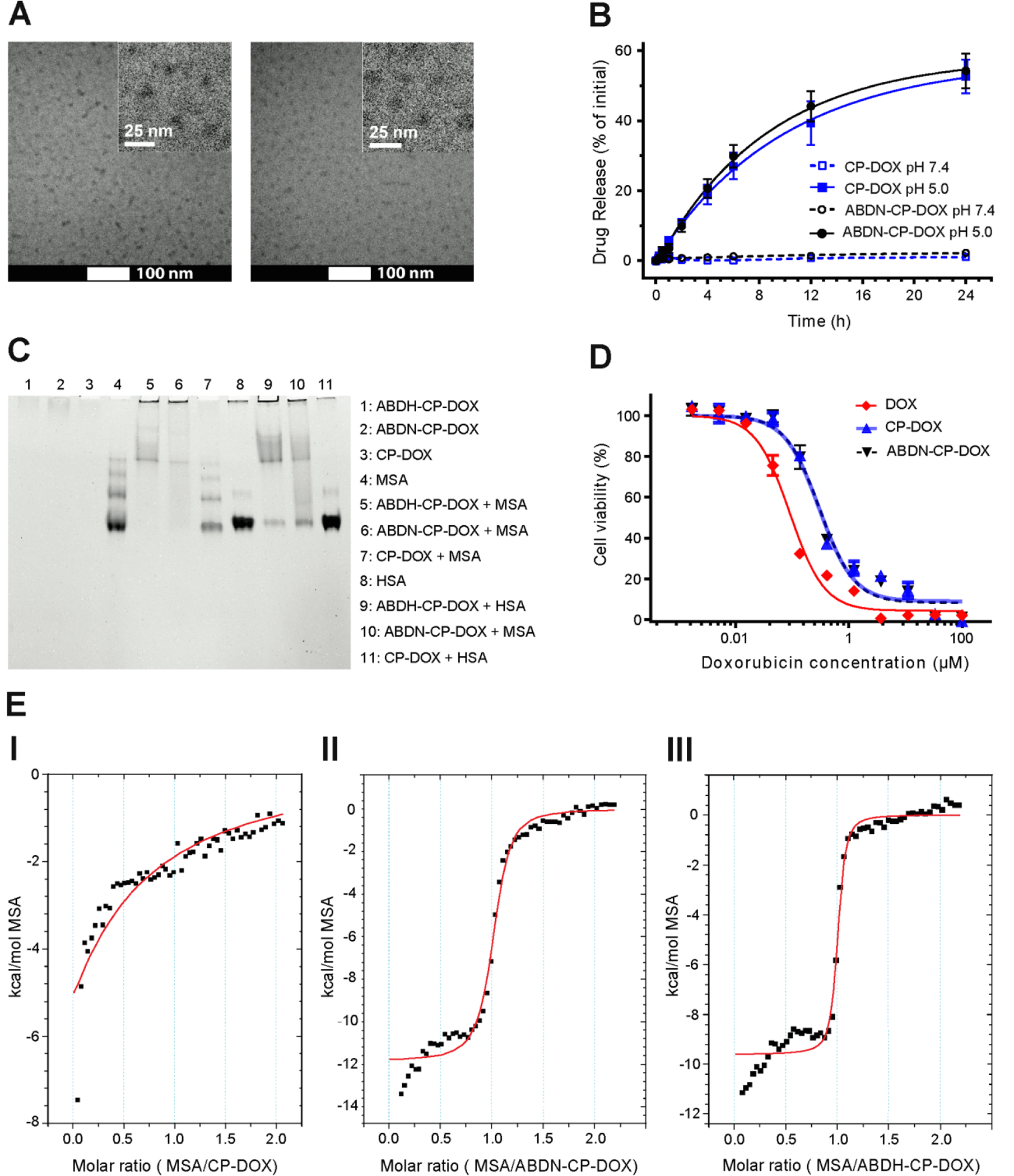
In vitro characterization of ABD-CP-DOX micelles: (A) Cryo-TEM micrographs of CP–DOX and ABDN-CP-DOX micelles (inset: magnified view); (I) ABDN-CP–DOX micelles (II) CP-DOX micelles. (B) In vitro drug release profile from CP-DOX and ABDN-CP-DOX micelles.
The micelles released the loaded DOX under acidic conditions at pH 5.0 corresponding to the pH of late endosomes, but remained stable at pH 7.4 corresponding to vascular and extracellular space. Error bars represent standard error of the mean (n=3). (C) Native-PAGE analysis of interaction of human serum albumin (HSA) and mouse serum albumin (MSA) with ABDN-CP-DOX and ABDH-CP-DOX. For lanes 5–7 and lanes 9–11, polypeptide carriers were mixed at a molar ratio of 1:1 with MSA and HSA, respectively. (D) Cytotoxicity of ABDN-CP-DOX vs. CP-DOX and free DOX in C26 cells after 72 h incubation. The IC50 was 0.09 μM for free DOX, 0.30 μM for CP-DOX, and 0.31 μM for ABDN-CP-DOX. Error bars represent standard error of the mean (n=3). (E) Calorimetric titration of ABDN-CP-DOX and ABDH-CP-DOX micelles with MSA. The experiments were performed in phosphate buffered saline (pH 7.4) at 37 °C. The solid red line indicates the best-fit binding of the binding isotherm. MSA at 500 μM was titrated into the sample cell containing 50 μM of CP-DOX (I), ABDN-CP-DOX (II), and ABDH-CP-DOX (III). The integrated heat data were fit to a single site binding model and the binding stoichiometry (N) and dissociation constant (KD) were calculated, as shown in Table 3.
As with the ABD-CP unimers, the albumin binding by the ABDN-CP-DOX and ABDH-CP-DOX micelles was analyzed by native-PAGE and ITC. Similar to the ABD-CP unimers, incubation of ABDN-CP-DOX and ABDH-CP-DOX with HSA and MSA altered the migration pattern of albumin that is apparent by a smearing effect in the gel, while the construct lacking the ABD domain had no impact on the electrophoretic mobility of albumin (Figure 2C). The binding of albumin by ABDN-CP-DOX and ABDH-CP-DOX was confirmed by ITC. Analysis of the ITC data revealed that DOX conjugated ABDN-CP displayed a 1:1 albumin:CP binding stoichiometry and sub-μM affinity for MSA, with a KD of 318±54 nM for ABDN-CP-DOX and a KD of 66 ± 20 nM for ABDH-CP-DOX. Interaction of ABDN-CP-DOX with MSA was further corroborated by studying the thermal behavior of both micelles in mouse serum.
Monitoring the turbidity profile of CP-DOX, ABDN-CP-DOX, and ABDH-CP-DOX micelles as a function of solution temperature showed that DOX conjugation markedly changed the phase behavior of the micelles relative to their unimer controls — CP, ABDN-CP and ABDH-CP. The Tt of the micelles —the temperature at which the micelles separate into an insoluble cocervate phase— occurred at a lower temperature relative to the Tt of the unimer transitioning ino a coacervate phase and showed a far lower concentration dependence than their unconjugated counterparts (Figure S1A–C). The lower Tt and weaker concentration dependence of the Tt of the micelles, as compared to the CP unimers is consistent with previous studies that showed that the micelle-aggregate transition shows a far weaker concentration dependence than the unimer-aggregate transition, presumably because of the high local concentration of the CP in the micelles35.
As with CP unimers, and consistent with previous reports36, CP-DOX micelles exhibited a lower Tt in MS and FBS than that in PBS (Figure S1A), while ABDN-CP-DOX and ABDH-CP-DOX micelles showed no thermally triggered phase transition in MS up to the highest temperature —60 °C— that was investigated by temperature-programmed turbidimetry (Figure S1B–C). The lack of phase separation is consistent with the fact that these micelles are rapidly decorated with an albumin corona upon exposure to MSA, as seen by native-PAGE and ITC. In addition, as with unimers, the fusion of an ABD decreased the micelle-macroscopic aggregate Tt of CP-DOX micelles. In case of ABDH-CP-DOX, the Tt was ~40 °C with some aggregation at ~37 °C. Because any aggregation of micelles at body temperature is undesirable and the fact that the pharmacokinetics of ABDN-CP-DOX and ABDH-CP-DOX micelles were similar, ABDH-CP-DOX was abandoned at this stage, and ABDN-CP-DOX micelles were carried forward for in vivo studies.
Next, the release of free DOX from ABDN-CP-DOX micelles and the CP-DOX control was studied at the physiological pH of 7.4 and at pH 5.0, a pH that corresponds to the acidic environment of late endosomes37. These two pH values were chosen to understand the stability of the hydrazone linker between the drug and CP in circulation, and to investigate whether the drug would be released in endosomes and lysosomes that CP-DOX nanoparticles are known to be trafficked into upon uptake by cells19,38. For both CP-DOX, and ABDN-CP-DOX micelles, drug release was negligible at pH 7.4, which corresponds to the pH in the vascular compartment and the extracellular space of normal tissues. The micelles however released over 50% of DOX after 24 h at pH 5.0, a pH that corresponds to that in late endosomes (Figure 2B)37, which is attributed to the hydrolytic breakdown of the hydrazone linkage between DOX and the EMCH linker at acidic pH39. The release data at pH 5.0 were fit to a first order model from which the maximum extent of drug release was calculated as 57.4 ± 1.3% and 59.4 ± 3.6% for CP-DOX and ABDN-CP-DOX micelles, respectively. In addition, from the model, 50% of total DOX release at pH 5.0 was also calculated to occur within 6.9 h and 5.9 h for CP-DOX, and ABDN-CP-DOX micelles, respectively.
The pharmacokinetics of ABDN-CP-DOX micelles was studied at two doses of 10 and 20 mg DOX Equiv.kg−1 BW in healthy BALB/c mice. The DOX concentration in blood as a function of time post i.v. injection of ABDN-CP-DOX and CP-DOX micelles was measured by quantifying the intrinsic fluorescence of DOX, and the data were fit to a two-compartment model (Figure 4A). A complete list of the pharmacokinetic parameters is provided in Table S5. At a lower dose of 10 mg DOX Equiv.kg−1 BW, ABDN-CP-DOX micelles displayed a half-life of 18.3 ± 0.6 h and an AUC of 1400.4 ± 34.2 μM.h, and both these parameters were two-fold greater than the half-life and AUC of CP-DOX micelles of 9.7 ± 0.6 h and 724.82 ± 33.1 μM.h, respectively (P < 0.001).
Figure 4.
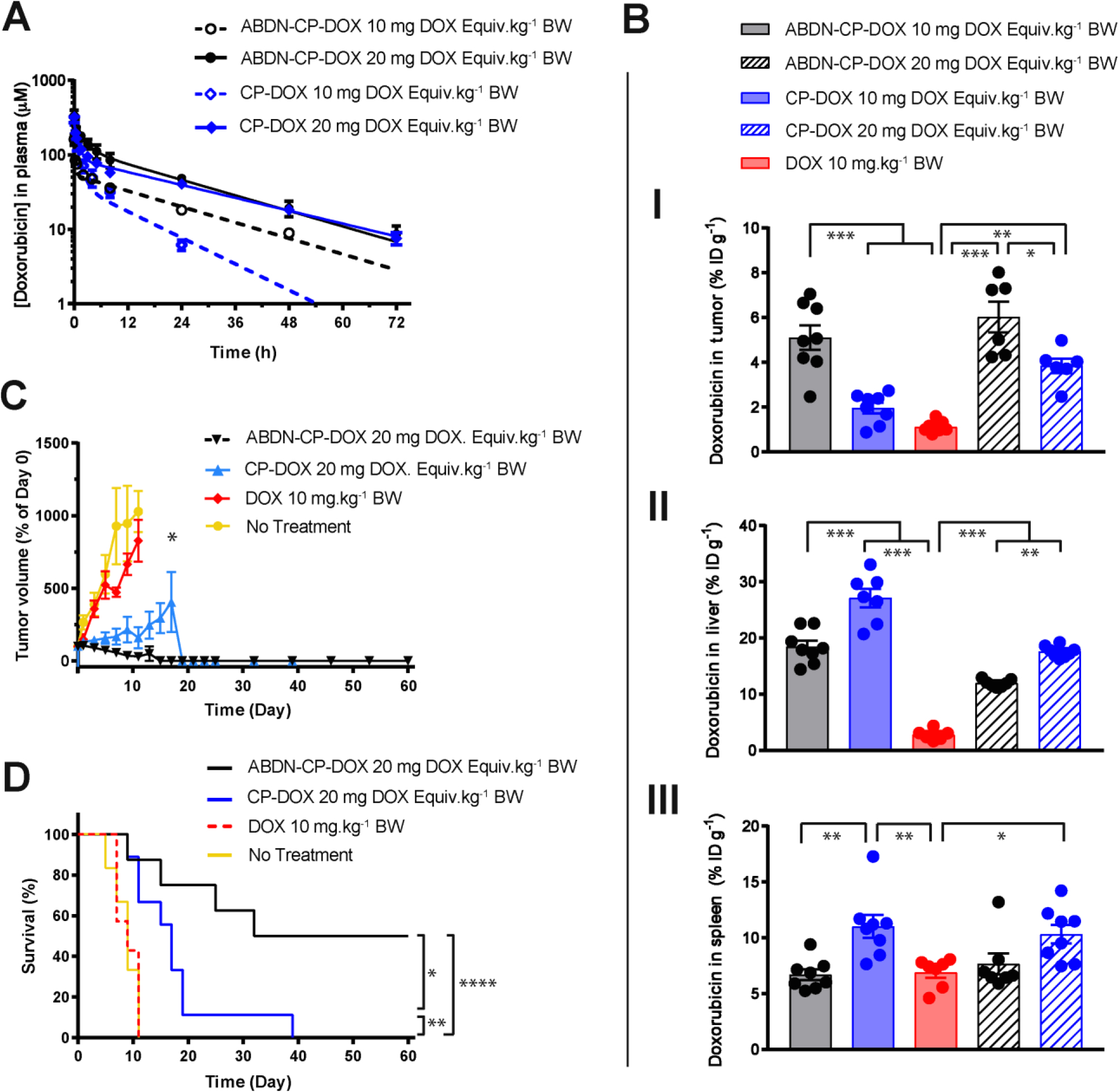
In vivo characterization of ABD-CP-DOX micelles: (A) Pharmacokinetics of ABDN-CP-DOX micelles vs CP-DOX micelles. CP-DOX and ABDN-CP-DOX micelles were administered via tail vein to BALB/c mice at 10 and 20 mg DOX Equiv.kg−1 BW, and plasma DOX concentration was measured at intervals over 72 h. Error bars represent standard error of the mean (n=5–6). The data was fit to a two-compartment model from which pharmacokinetic parameters were estimated as shown in Table 5. (B) Biodistribution of ABDN-CP-DOX micelles, CP-DOX micelles and free DOX at 24 h post administration in the (I) tumor, (II) liver, and (III) spleen. C26 tumor cells were implanted subcutaneously and allowed to grow to approximately 75–100 mm3. Mice were treated with free DOX at 10 mg.kg−1 BW, and CP–DOX, and ABDN-CP-DOX both at 10 and 20 mg DOX Equiv. kg−1 BW. The DOX concentration was measured in tumor and normal tissues at 24 h post-administration. Error bars represent standard error of the mean (n=6–8). (C and D) Anti-tumor activity of (ABDN-)CP–DOX micelles. C26 tumor cells were implanted subcutaneously and allowed to grow to approximately 75–100 mm3. Mice were treated on day 0 with free DOX (10 mg.kg−1 BW), CP–DOX (20 mg DOX Equiv.kg−1 BW), and ABDN-CP-DOX (20 mg DOX Equiv.kg−1 BW). (C) Tumor volume up to day 60 (mean ± SEM.; n=7 for DOX, 8 for ABDN-CP-DOX, and 9 for CP-DOX). (D) Cumulative survival of mice up to day 60 (n=7 for DOX, 8 for ABDN-CP-DOX, and 9 for CP-DOX). * for P < 0.05, ** for P < 0.01, and *** for P < 0.001.
In addition, at a higher dose of 20 mg DOX Equiv.kg−1 BW, we found no significant difference between the half-life of ABDN-CP-DOX as compared to CP-DOX micelles (17.5 ± 0.8 h for ABDN-CP-DOX and 20.1 ± 0.9 h for CP-DOX). The AUC of ABDN-CP-DOX at 20 mg DOX Equiv.kg−1 BW was measured as 3328.5 ± 100.3 μM.h and is about 10% greater than that of CP-DOX (2994 ± 59.0 μM.h) that is statistically (P < 0.001), but not therapeutically relevant in our opinion (Table 4). These data show that the pharmacokinetics of ABDN-CP-DOX is linear and dose-independent, as its half-life is independent of the dose and its AUC linearly scales with the injection dose. In contrast, the pharmacokinetics of CP-DOX is non-linear and dose-dependent40. This altered pharmacokinetics behavior as a result of albumin binding was previously reported for Abraxane (ABI-007) by Desai et al.41; they observed that paclitaxel formulated with Cremophor (Taxol) showed nonlinear pharmacokinetics42, whereas the pharmacokinetics of Abraxane was linear. The linear pharmacokinetics of ABD-CP-DOX micelle is significant from a clinical perspective, as dosing drugs with non-linear pharmacokinetics is difficult and introduces an element of unpredictability in terms of adverse reactions of the drug.
Table 4:
Murine pharmacokinetic parameters of ABDN-CP-DOX, and CP-DOX.
| Pharmacokinetic Parameter | CP-DOX (10 mg DOX Equiv.kg−1 BW) | ABDN-CP-DOX (10 mg DOX Equiv.kg−1 BW) | CP-DOX (20 mg DOX Equiv.kg−1 BW) | ABDN-CP-DOX (20 mg DOX Equiv.kg−1 BW) |
|---|---|---|---|---|
|
| ||||
| Elimination half-life [h] | 9.7 ± 0.6 | 18.3 ± 0.6 | 20.1 ± 0.9 | 17.5 ± 0.8 |
| Area under curve [μM. h] | 724.8 ± 33.1 | 1400.4 ± 34.2 | 2994.5 ± 59.0 | 3328.5 ± 100.3 |
Values are shown as mean ± SD (n=5–6). The elimination half -life for ABDN-CP-DOX is statistically different from that of the CP-DOX at a dose of 10 mg DOX Equiv.kg−1 BW (P < 0.001), but not at 20 mg DOX Equiv.kg−1 BW. The area under the curve for ABDN-CP-DOX was about two-fold greater than that of CP-DOX at both 10 DOX Equiv.kg−1 BW (P < 0.001), but not at 20 mg DOX Equiv.kg−1 BW.
The effect of albumin binding on pharmacokinetics was further investigated in dogs, as they are large, long-lived animals that share stronger similarities in their anatomy and physiology with humans than mice and are therefore a more relevant model for assessing the clinical utility of these drug loaded nanoparticles43,44,45. All three formulations were injected at a dose that was reduced by 10% relative to the clinical therapeutic dose of DOX. Observing the relatively small animal-to-animal variation and given the high cost of these experiments and limited animal availability, the pharmacokinetic study was closed with three dogs for CP-DOX and free DOX, and two dogs for ABDN-CP-DOX. The calculated elimination half-life and area-under-curve (AUC) are shown in Table 4 and a complete list of the pharmacokinetic parameters is contained in Table S6. Free DOX formulation had an elimination half-life (t1/2) of 2.9 ± 0.8 h and an AUC of 1769.1 ± 498.0 nM.h. In contrast CP-DOX micelles exhibited a three-fold longer t1/2 of 9.8 ± 1.9 h (P < 0.05), and a 48-fold greater AUC of 84106.0 ± 7616.8 nM.h AUC compared to free DOX (P < 0.05) (Figure 3). The improved elimination half-life and plasma exposure as a result of albumin binding was more pronounced in the canine model than in mice, with ABDN-CP-DOX micelles showing a plasma half-life of 30.8 ± 2.0 h that was three-fold greater than that of CP-DOX (P < 0.001), and an AUC of 509371.7 ± 43095.2 nM.h that is six-fold larger than that of CP-DOX (P < 0.001) (Table 4). Furthermore, despite a significant increase in the plasma half-life and AUC of ABD-CP-DOX compared to CP-DOX and free DOX, no hematology or serum chemistry abnormalities were noted with ABDN-CP-DOX and CP-DOX groups one week after infusion (Table S7 and S8), suggesting that these micellar formulations do not cause dose-limiting adverse effects associated with free DOX. The significantly greater improvement in canine pharmacokinetics —between ABDN-CP-DOX and CP-DOX— as a result of albumin binding relative to murine pharmacokinetics, can, we speculate, be attributed to the intrinsically longer half-life of albumin in dogs (~8 days)46 than mice (~1.5 days)31. These data promise an even more significant pharmacokinetic enhancement for ABD-CP-DOX relative to CP-DOX in humans where albumin has a remarkably long half-life of 18–21 days47.
Figure 3.
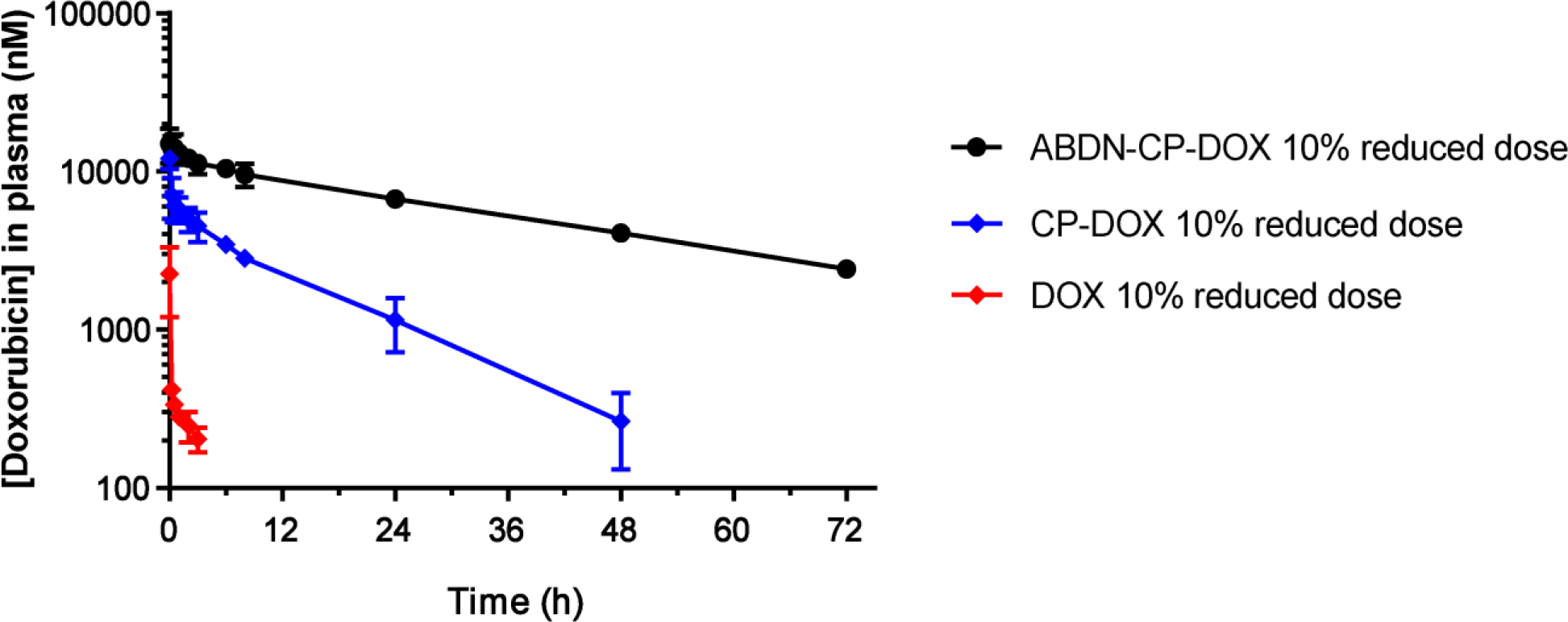
Pharmacokinetics of ABDN-CP-DOX micelles in dogs. Free DOX, CP-DOX micelles and ABDN-CP-DOX micelles were administered via the cephalic vein at the clinical dose of DOX in dogs reduced by 10% i.e. 27 mg DOX Equiv.m−2 body surface area (BSA) for dogs weighing greater than 10 kg and 0.9 mg DOX Equiv.kg−1 of body weight (BW) for dogs weighing less than 10 kg, and plasma DOX concentration was measured at intervals over 72 h. Error bars represent standard error of the mean (n=2 for ABDN-CP-DOX, and 3 for CP-DOX and DOX). The data were fit to a two-compartment model from which pharmacokinetic parameters were estimated as shown in Table 5.
Next, the in vivo tissue distribution of CP-DOX and ABDN-CP-DOX micelles was evaluated by i.v. injection of a dose of 10 and 20 mg DOX Equiv.kg−1 BW into BALB/c mice bearing s.c. C26 tumors. Free DOX was injected at a dose of 10 mg.kg−1 BW. The higher dose of 20 mg.kg−1 BW was not possible, as free DOX is known to have a maximum tolerated dose (MTD) that is ≤ 10 mg.kg−1 BW. At 24 h post-injection, free DOX showed lower accumulation in the liver than both CP-DOX and ABDN-CP-DOX micelles (P < 0.001) (Figure 4B-II), and lower accumulation in the spleen than CP-DOX micelles (Figure 4B-III). No significant difference was observed between accumulation of free DOX and ABDN-CP-DOX in the spleen (Figure 4B-III, S6–C). Furthermore, compared with CP-DOX, ABDN-CP-DOX accumulated to a significantly lower extent in the liver and spleen at 2 h (Figure S5B–C) and had significantly lower accumulation in the liver at both 10 and 20 mg DOX Equiv.kg−1 BW doses (Figure 4B-II) and in the spleen at the 10 mg DOX Equiv.kg−1 BW dose at 24 h post-injection (Figure 4B-III) indicating that albumin binding helps decrease the accumulation of micelles in RES organs i.e. liver and spleen.
The dose-dependence of the PK of naked CP-DOX micelle, wherein plasma clearance is slower at a higher dose is speculated to result from saturation of uptake mechanisms or depletion of plasma opsonins such as C3 complement protein that is known to precede phagocytosis by macrophages. Micelles are colloidal nanoparticles that are typically opsonized and sequestered by the RES organs i.e. the liver and spleen48. An albumin coating —such as in ABD-CP-DOX— prevents the adsorption of other serum proteins including opsonins and results in lower RES uptake, as reported in previous studies49,50,51, and is consistent with an increase in the plasma half-life at the higher dose. Consistent with the lower RES organ uptake, which are well known “sinks” for nanoparticles, the tumor accumulation of ABDN-CP-DOX micelles was approximately 2 fold greater than CP-DOX micelles at both doses of 10 (P < 0.001) and 20 (P < 0.01) mg DOX Equiv.kg−1 BW at 24 h post-injection (Figure 4B-I).
The tumor regression by ABDN-CP-DOX micelles was compared with CP-DOX micelles and free DOX in a s.c. mouse C26 colon cancer model. To this end, the tumoricidal activity of the ABDN-CP-DOX against C26 cancer cells was first assessed in vitro; the half-maximal inhibitory concentration (IC50), defined as the concentration of the drug required to cause 50% decrease in viable C26 cells in culture, was found to be 0.31, 0.30, and 0.09 μM for ABDN-CP-DOX, CP-OX, and free DOX, respectively (Figure 2D). These IC50 values are similar, confirming that the anticancer activity of DOX is not markedly reduced upon conjugation to ABDN-CP or the CP. Prior to initiating the in vivo tumor regression study, we carried out a dose-escalation study in healthy mice (Figure S6). Increasing concentrations of free DOX, CP-DOX micelles, and ABDN-CP-DOX micelles were administered i.v. to healthy BALB/c mice on day 0. The concentrations were 5, 10, 15, and 20 mg.kg−1 BW for free DOX and 20, 40, 50, and 60 mg DOX Equiv.kg−1 BW for CP-DOX, and ABDN-CP-DOX. Unconjugated CP and ABDN-CP carriers were also administered, as controls, at a protein concentration equivalent to the protein concentration in 60 mg Dox Equiv.kg−1 BW CP-DOX and ABDN-CP-DOX treatments. We defined the MTD as the maximal dose that does not cause animal mortality due to systemic toxicity or greater than 15% BW loss. Using this definition, and by monitoring the BW of treated mice, we established that the MTD for free DOX is 10 mg.kg−1 BW, while that of CP-DOX is 40 mg DOX Equiv.kg−1 BW. In contrast, the MTD of ABDN-CP-DOX is at least 60 mg DOX Equiv.kg−1 BW, which is above the highest dose administered in this study (Figure S6). Though the average of maximum BW loss for this treatment group was 13.4 ± 1.1 % which was below the acceptable threshold of 15%, 2 out of 5 mice exhibited a BW loss close to 15%, and therefore, the MTD for treatment with ABDN-CP-DOX is unlikely to be much higher than 60 mg DOX Equiv.kg−1 BW.
Based on these studies, we chose three doses of CP-DOX and ABDN-CP-DOX for tumor regression studies — 10, 20, and 40 mg DOX Equiv.kg−1 BW. As a control, DOX was administered at its MTD of 10 mg.kg−1 BW. At a lower dose of 10 mg DOX Equiv.kg−1 BW, drug accumulation in tumors, while higher for ABD-decorated micelles than naked micelles, was not enough to result in a statistically significant difference in tumor regression between the two micelle formulations or compared to free DOX (Figure S7A–B). At a DOX equivalent dose of 20 mg DOX Equiv.kg−1 BW, ABD-CP-DOX micelles slowed the tumor growth and prolonged the survival of tumor bearing mice as compared to CP-DOX micelles (Figure 4C–D) (P < 0.05). 50% of tumor bearing mice treated with ABD-CP-DOX survived 60 days after treatment, whereas none of the CP-DOX treated group survived more than 40 days. Mice treated with CP-DOX and ABDN-CP-DOX showed slower tumor growth and longer survival than mice treated with free DOX (P < 0.01). At a dose of 40 mg DOX Equiv.kg−1 BW, however, both the ABDN-CP-DOX and the CP-DOX treated mice survived out to 60 days (Figure S7C–D). Because the MTD of CP-DOX is 40 mg DOX Equiv.kg−1 BW, we did not examine tumor regression at higher doses, though the dose of ABDN-CP-DOX could be increased to 60 mg DOX Equiv.kg−1 BW, if needed. These results clearly show that the albumin decorated ABDN-CP-DOX micelles show a 3-fold increase in the therapeutic window (from 20–60 mg DOX Equiv.kg−1 BW) compared to undecorated micelles as they can achieve regression at a dose that is a third of the MTD, whereas the CP-DOX micelles are only effective in eradicating tumors at their MTD of 40 mg DOX Equiv.kg−1 BW.
In conclusion, the three most important findings of this study are: first, albumin binding micelles have dose-independent pharmacokinetics, suggesting predictable in vivo behavior as one transitions across dose and species52, which is important for clinical translation52. Second, in a canine model, the albumin binding micelles have significantly improved pharmacokinetics compared to micelles that do not bind albumin relative to their pharmacokinetics in mice. The improvement in pharmacokinetics afforded by albumin binding, and the lack of systemic toxicity observed in the canine model for the ABDN-CP-DOX micelles highlights its potential for translation into humans. Third, equally importantly from a clinical translation perspective, sheathing the corona of the CP-DOX micelles with an albumin corona greatly decreases their uptake by the RES organs in mice, and significantly increases the accumulation of the drug in the tumor, leading to a 3-fold increase in the therapeutic window compared to naked nanoparticles. This increase in the therapeutic window is likely to be important for clinical translation, we believe, because mice are not men —they are far more robust to the physiological insults posed by chemotherapeutics at doses likely to cause severe systemic toxicity in humans53,54, so that lowering the dose that produces a therapeutic effect below MTD augurs well for the clinical translation of ABDN-CP-DOX.
Finally, this system has several attractive features from a design standpoint that are important for scale-up and manufacturing, and are hence relevant to translation. First, the nanoparticle delivery system consists of a genetically encoded polypeptide, which allows the sequence and chain length of the “unimer” polypeptide constituent of the micelle to be readily tuned at the gene level and to be manufactured at a high expression level in E. coli. Second, the design of the delivery system is completely modular, as it consists of an albumin binding protein domain connected to a drug attachment domain via an intervening ELP that provides the bulk of the nanoparticle; each component of the delivery system can be independently modified and optimized as needed. Finally, ELPs have been in a number of clinical trials and have shown a good safety profile17. Studies are now ongoing to test this system in large animal models that more closely recapitulate human diseases, with the ultimate goal of taking this nanoparticle chemotherapeutic delivery technology into the clinic.
Supplementary Material
Table 3:
Thermodynamic binding parameters of CP-DOX, ABDN-CP-DOX, and ABDH-CP-DOX micelles with MSA.
| Thermodynamic Parameter | CP-DOX | ABDN-CP-DOX | ABDH-CP-DOX |
|---|---|---|---|
|
| |||
| Binding stoichiometry | 0.04 ± 0.09 | 1.008 ± 0.007 | 0.99 ± 0.006 |
| Dissociation constant | 91.91 ± 68.36 μM | 317.56 ± 53.55 nM | 66.09 ± 20.20 nM |
Values are shown as mean ± SD.
Table 5:
Canine pharmacokinetic parameters of ABDN-CP-DOX, CP-DOX and free DOX at a dose of 27 mg DOX Equiv.m−2 body surface area (BSA) for dogs weighing greater than 10 kg and 0.9 mg DOX Equiv.kg−1 of body weight (BW) for dogs weighing less than 10 kg.
| Pharmacokinetic Parameter | DOX | CP-DOX | ABDN-CP-DOX |
|---|---|---|---|
|
| |||
| Elimination half-life [h] | 2.9 ± 0.8 | 9.8 ± 1.9 | 30.8 ± 2.0 |
| Area under curve [nM. h] | 1769.1 ± 498.0 | 84106.0 ± 7616.8 | 509371.7 ± 43095.2 |
Values are shown as mean ± SD (n= 2 for ABDN-CP-DOX, and 3 for CP-DOX and DOX). The elimination half - life and the area under the curve (AUC) for ABDN-CP-DOX are statistically greater than that of the CP-DOX and free DOX (P < 0.001).
ACKNOWLEDGMENT
A.C. acknowledges the support of the National Institutes of Health through grants R01EB000188 and R01EB007205. A.C. and S.S. acknowledge the support of DCI/NCSU Consortium for Canine Comparative Oncology grant (2016) for the canine pharmacokinetic study.
Footnotes
Conflicts of Interest
A.C. serves as a scientific advisor and board member for PhaseBio Pharmaceuticals, Inc., which has licensed the ELP technology for drug delivery from Duke University. The other authors declare no competing financial interest.
REFERENCES
- (1).Choi HS; Liu W; Misra P; Tanaka E; Zimmer JP; Ipe BI; Bawendi MG; Frangioni JV Nat. Biotechnol. 2007, 25 (10), 1165–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Cho K; Wang X; Nie S; Chen Z. (Georgia); Shin DM. Clin. Cancer Res. 2008, 14 (5), 1310 LP–1316. [DOI] [PubMed] [Google Scholar]
- (3).Maeda H; Wu J; Sawa T; Matsumura Y; Hori KJ Control. Release 2000, 65 (1), 271–284. [DOI] [PubMed] [Google Scholar]
- (4).Nie S Nanomedicine (Lond). 2010, 5 (4), 523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Albanese A; Tang PS; Chan WCW Annu. Rev. Biomed. Eng. 2012, 14, 1–16. [DOI] [PubMed] [Google Scholar]
- (6).Rao L; Xu J-H; Cai B; Liu H; Li M; Jia Y; Xiao L; Guo S-S; Liu W; Zhao X-Z Nanotechnology 2016, 27 (8), 85106. [DOI] [PubMed] [Google Scholar]
- (7).Li S-D; Huang L Biochim. Biophys. Acta 2009, 1788 (10), 2259–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ogawara K; Furumoto K; Nagayama S; Minato K; Higaki K; Kai T; Kimura TJ Control. Release 2004, 100 (3), 451–455. [DOI] [PubMed] [Google Scholar]
- (9).Mariam J; Sivakami S; Dongre PM Drug Deliv. 2016, 23 (8), 2668–2676. [DOI] [PubMed] [Google Scholar]
- (10).Burnouf T Transfus. Med. Rev. 2007, 21 (2), 101–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Shang L; Wang Y; Jiang J; Dong S Langmuir 2007, 23 (5), 2714–2721. [DOI] [PubMed] [Google Scholar]
- (12).Peters T In Advances in Protein Chemistry; Anfinsen CB, Edsall JT, Richards F. M. B. T.-A. in P. C., Eds.; Academic Press, 1985; Vol. 37, pp 161–245. [DOI] [PubMed] [Google Scholar]
- (13).Nilvebrant J; Hober S Comput. Struct. Biotechnol. J. 2013, 6 (7), e201303009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wang P; Zhao P; Dong S; Xu T; He X; Chen M Theranostics 2018, 8 (1), 223–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Liu H; Moynihan KD; Zheng Y; Szeto GL; Li AV; Huang B; Van Egeren DS; Park C; Irvine DJ Nature 2014, 507 (7493), 519–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wang C; Zhang C; Li Z; Yin S; Wang Q; Guo F; Zhang Y; Yu R; Liu Y; Su Z Biomacromolecules 2018. [DOI] [PubMed] [Google Scholar]
- (17).MacEwan SR; Chilkoti AJ Control. Release 2014, 190, 314–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).MacEwan SR; Hassouneh W; Chilkoti AJ Vis. Exp. 2014, No. 88, 51583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).MacKay JA; Chen M; McDaniel JR; Liu W; Simnick AJ; Chilkoti A Nat. Mater. 2009, 8 (12), 993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bhattacharyya J; Bellucci JJ; Weitzhandler I; McDaniel JR; Spasojevic I; Li X; Lin C-C; Chi J-TA; Chilkoti A Nat. Commun. 2015, 6, 7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kraulis PJ; Jonasson P; Nygren P-Å; Uhlén M; Jendeberg L; Nilsson B; Kördel J FEBS Lett. 1996, 378 (2), 190–194. [DOI] [PubMed] [Google Scholar]
- (22).Jonsson A; Dogan J; Herne N; Abrahmsén L; Nygren P-Å Protein Eng. Des. Sel. 2008, 21 (8), 515–527. [DOI] [PubMed] [Google Scholar]
- (23).Rozak DA; Orban J; Bryan PN Biochim. Biophys. Acta 2005, 1753 (2), 226–233. [DOI] [PubMed] [Google Scholar]
- (24).Johansson MU; Frick I-M; Nilsson H; Kraulis PJ; Hober S; Jonasson P; Linhult M; Nygren P-Å; Uhlén M; Björck L; et al. J. Biol. Chem. 2002, 277 (10), 8114–8120. [DOI] [PubMed] [Google Scholar]
- (25).Andersen JT; Pehrson R; Tolmachev V; Daba MB; Abrahmsén L; Ekblad CJ Biol. Chem. 2011, 286 (7), 5234–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Meyer DE; Chilkoti A Nat. Biotechnol. 1999, 17 (11), 1112–1115. [DOI] [PubMed] [Google Scholar]
- (27).Nilvebrant J; Hober S Comput. Struct. Biotechnol. J. 2013, 6, e201303009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Meyer DE; Kong GA; Dewhirst MW; Zalutsky MR; Chilkoti A Cancer Res. 2001, 61 (4), 1548–1554. [PubMed] [Google Scholar]
- (29).Ellis RJ Trends Biochem. Sci. 2001, 26 (10), 597–604. [DOI] [PubMed] [Google Scholar]
- (30).Hopp J; Hornig N; Zettlitz KA; Schwarz A; Fuss N; Muller D; Kontermann RE Protein Eng. Des. Sel. 2010, 23 (11), 827–834. [DOI] [PubMed] [Google Scholar]
- (31).Chaudhury C; Mehnaz S; Robinson JM; Hayton WL; Pearl DK; Roopenian DC; Anderson CL J. Exp. Med. 2003, 197 (3), 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Garanger E; MacEwan SR; Sandre O; Brûlet A; Bataille L; Chilkoti A; Lecommandoux S Macromolecules 2015, 48 (18), 6617–6627. [Google Scholar]
- (33).Phan HTT; Zhu K; Kjøniksen A-L; Nyström B Colloid Polym. Sci. 2011, 289 (9), 993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Burchard W In Physical techniques for the study of food biopolymers; Springer, 1994; pp 151–213. [Google Scholar]
- (35).Dreher MR; Simnick AJ; Fischer K; Smith RJ; Patel A; Schmidt M; Chilkoti AJ Am. Chem. Soc. 2008, 130 (2), 687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).McDaniel JR; MacEwan SR; Dewhirst M; Chilkoti AJ Control. Release 2012, 159 (3), 362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Huotari J; Helenius A EMBO J. 2011, 30 (17), 3481–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Wang J; MacEwan SR; Chilkoti A Nano Lett. 2017, 17 (2), 1226–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Sonawane SJ; Kalhapure RS; Govender T Eur. J. Pharm. Sci. 2017, 99, 45–65. [DOI] [PubMed] [Google Scholar]
- (40).Saba C; Paoloni M; Mazcko C; Kisseberth W; Burton JH; Smith A; Wilson-Robles H; Allstadt S; Vail D; Henry C; et al. PLoS One 2016, 11 (2), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ibrahim NK; Desai N; Legha S; Soon-Shiong P; Theriault RL; Rivera E; Esmaeli B; Ring SE; Bedikian A; Hortobagyi GN; et al. Clin. Cancer Res. 2002, 8 (5), 1038–1044. [PubMed] [Google Scholar]
- (42).Gianni L; Kearns CM; Giani A; Capri G; Vigano L; Lacatelli A; Bonadonna G; Egorin MJ J. Clin. Oncol. 1995, 13 (1), 180–190. [DOI] [PubMed] [Google Scholar]
- (43).Paoloni M; Khanna C Nat. Rev. Cancer 2008, 8 (2), 147–156. [DOI] [PubMed] [Google Scholar]
- (44).Richards KL; Suter SE Inmunol Rev. 2016, 263 (1), 173–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Khanna C; Lindblad-Toh K; Vail D; London C; Bergman P; Barber L; Breen M; Kitchell B; McNeil E; Modiano JF; et al. Nat. Biotechnol. 2006, 24, 1065. [DOI] [PubMed] [Google Scholar]
- (46).Slatter Douglas H.. Textbook of Small Animal Surgery; Elsevier Health Sciences, 2003. [Google Scholar]
- (47).Boldt J Br. J. Anaesth. 2010, 104 (3), 276–284. [DOI] [PubMed] [Google Scholar]
- (48).Guo S; Huang LJ Nanomater. 2011, 2011, 11. [Google Scholar]
- (49).Furumoto K; Yokoe J-I; Ogawara K; Amano S; Takaguchi M; Higaki K; Kai T; Kimura T Int. J. Pharm. 2007, 329 (1–2), 110–116. [DOI] [PubMed] [Google Scholar]
- (50).Yokoe J; Sakuragi S; Yamamoto K; Teragaki T; Ogawara K; Higaki K; Katayama N; Kai T; Sato M; Kimura T Int. J. Pharm. 2008, 353 (1–2), 28–34. [DOI] [PubMed] [Google Scholar]
- (51).Ogawara K; Furumoto K; Nagayama S; Minato K; Higaki K; Kai T; Kimura TJ Control. Release 2004, 100 (3), 451–455. [DOI] [PubMed] [Google Scholar]
- (52).Ludden TM Clin. Pharmacokinet. 1991, 20 (6), 429–446. [DOI] [PubMed] [Google Scholar]
- (53).Erickson-Miller CL; May RD; Tomaszewski J; Osborn B; Murphy MJ; Page JG; Parchment RE Cancer Chemother. Pharmacol. 1997, 39 (5), 467–472. [DOI] [PubMed] [Google Scholar]
- (54).Teicher BA Toxicol. Pathol. 2009, 37 (1), 114–122. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.