

ABSTRACT
Despite its discovery more than 150 years ago, the cause of primary hypertension remains unknown. Most studies suggest that hypertension involves genetic, congenital or acquired risk factors that result in a relative inability of the kidney to excrete salt (sodium chloride) in the kidneys. Here we review recent studies that suggest there may be two phases, with an initial phase driven by renal vasoconstriction that causes low-grade ischemia to the kidney, followed by the infiltration of immune cells that leads to a local autoimmune reaction that maintains the renal vasoconstriction. Evidence suggests that multiple mechanisms could trigger the initial renal vasoconstriction, but one way may involve fructose that is provided in the diet (such as from table sugar or high fructose corn syrup) or produced endogenously. The fructose metabolism increases intracellular uric acid, which recruits NADPH oxidase to the mitochondria while inhibiting AMP-activated protein kinase. A drop in intracellular ATP level occurs, triggering a survival response. Leptin levels rise, triggering activation of the sympathetic central nervous system, while vasopressin levels rise, causing vasoconstriction in its own right and stimulating aldosterone production via the vasopressin 1b receptor. Low-grade renal injury and autoimmune-mediated inflammation occur. High-salt diets can amplify this process by raising osmolality and triggering more fructose production. Thus, primary hypertension may result from the overactivation of a survival response triggered by fructose metabolism. Restricting salt and sugar and hydrating with ample water may be helpful in the prevention of primary hypertension.
Keywords: autoimmune, fructose, hypertension, leptin, uric acid, vasopressin
INTRODUCTION
While the finding of cardiac hypertrophy, thickened blood vessels, and kidney disease provided clinical suspicion that some individuals suffered from high pressure of their vasculature, the first accurate documentation of high blood pressure was provided by a medical resident, Frederick Mahomed, who used the sphygmograph to measure the “pulse tension” in the general population in the 1870s [1]. However, the sphygmography was heavy and wieldy, so it was not until the invention of the blood pressure cuff and manometer by Scipione Riva Rocci in the 1890s that blood pressure could be measured easily. By the early 1900s, it was apparent that an elevated blood pressure (defined as >140/90 diastolic) could predict stroke, heart failure and kidney disease [2], and since then, blood pressure measurement has been a critical component of the standard medical examination.
In the early 1900s, the presence of elevated tension (or “hyper-tension”) in the vasculature was relatively rare, being found in less than 5% of those under the age of 65 years, although it was more common in the elderly [3]. However, during the 20th century, the prevalence climbed worldwide, such that it is currently present in nearly one-third of all adults (based on the original cutoff of 140/90 mmHg), making it arguably the most common disease in humankind [4]. Because it can be asymptomatic, it is commonly referred to as the “silent killer,” which has led to a great interest in understanding its etiology.
It became apparent that while there were clearly defined causes of hypertension, often referred to as secondary hypertension (such as from aldosterone-secreting tumors or chronic kidney disease), the vast majority of cases did not have an identifiable cause (termed primary or essential hypertension). Hemodynamically, these cases are associated with normal cardiac output but high peripheral vascular resistance, while in the kidney, the hallmark is a reduction in renal blood flow with high renal vascular resistance.
The discovery that multiple systems regulate blood pressure, including the role of sympathetic and parasympathetic nerves, circulating vasoactive mediators, high- and low-pressure baroreceptors, kidney-based mechanisms and other factors, created complexity. They led to the concept that multiple factors might be operative in the pathogenesis of hypertension (the mosaic theory of hypertension [5]), and blood pressure regulation was compared to electrical circuitry in which the kidney had the overriding dominance [6].
However, one critical—and practical—factor identified was salt (i.e. sodium chloride). Several epidemiology studies reported a general relationship between salt intake and hypertension prevalence [7, 8], and similarly, low-salt diets were found to have a modest effect on lowering blood pressure [9, 10]. Studies suggested that hypertension might be the consequence of a defect in the ability of the kidney to excrete salt and that there might be a reflex rise in blood pressure to excrete excess salt that had been ingested (“pressure natriuresis”) [6]. This led to the concept that the cause of hypertension might be a kidney disorder. Indeed, in several rat models of hypertension, hypertension could be passed to normotensive animals by kidney transplantation from the hypertensive rat [11]. Additional support came from the observation that single genetic mutations and/or polymorphisms in kidney transporters that favored sodium retention often manifested as secondary hypertension [12, 13]. It also led to the expansion of diuretics to treat hypertension while inhibiting vasoconstrictors such as renal angiotensin, and sympathetic nervous systems were used to reduce vascular tone.
These latter observations shifted the focus to what might be causing the kidney defect in sodium excretion. One possible scenario would be a net effect of genetic mutations favoring sodium retention by the kidney [12, 13], but most studies suggested that the overall contribution of genetic polymorphisms was minor. Another possibility was that it represented a congenital problem related to a low birthweight that could result in a reduction in the number of nephrons [termed pre-natal (fetal) programming] [14, 15]. However, similar to the genetic hypothesis, data suggested that low birthweight could only explain a minority of hypertension incidence [16], suggesting that there were other more important mechanisms driving the disease.
THE ROLE OF SUBTLE RENAL INJURY AND AUTOIMMUNITY
Harry Goldblatt proposed an alternative hypothesis in the 1940s [17]. He had noted that primary hypertension was almost inevitably associated with small vessel disease in the kidneys (arteriolosclerosis), and he suggested that this might lead to chronic ischemia which might lead to the release of vasoconstrictive factors such as angiotensin. Consistent with his hypothesis, early kidney biopsy studies in human hypertension identified ischemic changes in the renal tubules in more than 90% of cases [18]. His work also established the role of renal artery stenosis as a cause of secondary hypertension, but his general hypothesis that renal microvascular disease mediated primary hypertension was rejected because 10%–20% of cases with essential hypertension had normal-appearing arterioles [19].
Our group became interested in Goldblatt's hypothesis when we realized we could induce similar microvascular disease in rats by a short-term infusion of angiotensin II [20]. This manipulation would allow us to test whether such changes might increase the ability of high salt intake to cause hypertension. To test this hypothesis, we gave rats a 2-week infusion of angiotensin II, which naturally caused acute hypertension and renal microvascular changes. The angiotensin II infusion was then stopped, allowing blood pressure to return to normal, but at the expense of having microvascular disease. Then when the rats were switched to a high-salt diet, they developed hypertension [21]. In contrast, normal rats placed on a high-salt diet for the same period of time remained normotensive.
Those findings led to the idea that, typically, kidneys can excrete salt, but that subtle renal injury might trigger chronic salt retention and the development of hypertension. At this time, two collaborators, Jaime Herrera Acosta and Bernardo Rodriguez-Iturbe, provided major insights. Herrera-Acosta suggested that a critical experiment must show not the presence of microvascular disease but rather a vasoconstriction of preglomerular (afferent arteriole and interlobular artery) vessels, as this is the cardinal finding in primary hypertension. Indeed, he subsequently demonstrated that this was the case [22]. Rodriguez-Iturbe had another equally important question. How could subtle renal injury cause persistent renal vasoconstriction? There was already strong evidence that the kidney injury was accompanied by oxidative stress, impaired endothelial function and local angiotensin II [23–25]. However, it remained unknown which cells were producing these factors. One possibility, Rodriguez-Iturbe suggested, would be by local inflammatory cells. To test this hypothesis, Rodriguez-Iturbe gave the immunosuppressive drug mycophenolate (MMF), to rats during the angiotensin II infusion, which blocked the infiltration of inflammatory mononuclear cells into the kidney [26]. Notably, while it did not block hypertension induced during the acute infusion of angiotensin II, it did prevent the development of hypertension when the rats were placed on a high-salt diet after the angiotensin infusion had been stopped.
Subsequently, it was shown that a common pathway for developing hypertension involved an insult that caused renal vasoconstriction and renal ischemia, followed by persistent renal inflammation which caused chronic renal vasoconstriction [27]. Even maternal malnutrition with fetal programming could cause renal inflammation and hypertension in the progeny that was blocked by MMF [28].
While the initial ischemia by a drug might be expected to elicit chemokines and an acute inflammatory response (a reactive inflammatory response), the persistence of inflammation could be less well explained unless there was more of an autoimmune response. Subsequent studies identified potential antigens such as heat shock protein 70 (HSP70) expressed by tubular cells [29] and isoketals produced by local inflammatory cells as potential autoantigens [30].
These autoantigens have an important role in stimulating T cells [31, 32], as reviewed in [33]. CD4 and CD8 T cells [31, 34], and memory T cells all play a critical role in hypertension, as do B cells [35]. Conversely, T regulatory cells [36] correct or ameliorate hypertension. Proinflammatory cytokines, such as interleukin (IL)-1, IL-6, IL-17, IL-18, IL-22 and interferon-γ, are also involved [33, 37, 38]. Clinical studies have also identified both autoantibodies and T cell reactivity to HSP70 in patients with primary hypertension [29], and pilot clinical trials suggest immune suppression can also improve blood pressure in subjects with primary hypertension [39] and may correct resistant hypertension [40]. Genetic polymorphisms have also been identified that predict primary hypertension likely by influencing the immune response [41]. In summary, multiple studies now support primary hypertension as another autoimmune disease [33].
HYPERURICEMIA AND THE INITIATION OF HYPERTENSION
The observation that hypertension may be initiated by intermittent renal ischemia which may cause episodic hypertension, which is then made constant by the development of persistent renal ischemia driven by an autoimmune inflammatory response, begs the question of what may be the initiating cause. One potential mechanism could be a hyperactive sympathetic nervous system (type A personality). Nevertheless, another common risk factor is hyperuricemia. Hyperuricemia has consistently been found to be an independent risk factor for primary hypertension [42], and its presence predicts hypertension even in lean individuals who lack any metabolic risk factors [43]. Additional support comes from Mendelian randomization studies in which genetic polymorphisms that increase uric acid levels are evaluated for their ability to predict primary hypertension. While initial studies were negative [44, 45], a recent large trial documented a significant association [46].
To evaluate uric acid as a potential risk factor, we raised serum uric acid in rats by giving a uricase inhibitor. Uricase is the hepatic enzyme that degrades uric acid, and while it is present in most mammals, uricase was lost in our primate ancestors due to a stepwise reduction in promoter activity until the enzyme was silenced entirely in the mid-Miocene [47]. When uric acid was raised, the rats developed mild hypertension, and this tended to be greatest under low-salt dietary conditions [48]. The hyperuricemic rats developed renal vasoconstriction that could be shown to be mediated by a uric acid–dependent stimulation of the renin–angiotensin system, the induction of oxidative stress and the inhibition of endothelial nitric oxide bioavailability [49–51]. Importantly, the rats developed interstitial inflammation and subtle tubular injury to the kidneys. When this happened, the rats transitioned to salt-sensitive hypertension which persisted even after the uricase inhibitor was stopped and the uric acid levels returned to normal [52].
Consistent with this hypothesis, we found a strong relationship between serum uric acid and primary hypertension in adolescents, along with increased plasma renin activity and high peripheral vascular resistance [53, 54]. Then, in a study led by Daniel Feig, adolescents with newly diagnosed primary hypertension were randomized to urate-lowering therapy in a double-blind crossover design. Of those children who lowered their uric acid to <5 mg/dL, nearly 85% became normotensive, while placebo treatment had a minimal effect [54]. Further studies showed a mild to moderate blood pressure–lowering effect in cases of early hypertension, with less benefit apparent once kidney disease developed [55–57].
SUGAR AND FRUCTOSE: A NOVEL ROLE IN HYPERTENSION
The observation that hyperuricemia may be a true risk factor for hypertension could help explain the dramatic rise in hypertension worldwide, as serum uric acid levels have risen in parallel with the rising prevalence of hypertension over the last century [58]. The next mystery was determining why serum uric acid levels were increasing. One potential explanation is that obesity, insulin resistance and diabetes were also increasing during that time, and obesity and prediabetes are highly associated with hyperuricemia [59]. Indeed, an elevated serum uric acid was initially considered part of the metabolic syndrome [60].
Certain foods can also increase serum uric acid levels, especially alcohol and purine-rich meats. Nevertheless, the intake of red meat tended to fall over the century. Fructose, the simple sugar present in honey and fruits, raises uric acid [61, 62]. Importantly, fructose is also a component in table sugar (sucrose) and in the sweetener high fructose corn syrup (HFCS). During the 20th century, these two added sugars skyrocketed such that sugar/HFCS intake can account for one-sixth of the calories we ingest [63]. Metabolic syndrome also rose from being present in just a small percentage of people to affecting one-quarter of the adult US population [64].
We therefore began studying fructose, and found that if we gave high doses of fructose to rats, they developed modest hypertension with elevated uric acid levels [65, 66]. Similar to rats with hyperuricemia, these animals also showed high glomerular pressures and reduced renal blood flow. They also developed obesity, fatty liver, insulin resistance, dyslipidemia and features of metabolic syndrome. When Takahiko Nakagawa treated the animals with allopurinol to reduce their uric acid level, he found that he could block the development of hypertension [66]. Similar results were shown when uric acid was lowered by febuxostat [67]. What was more exciting in the study by Nakagawa et al. was that the animals treated with allopurinol gained less weight and developed less insulin resistance [66].
We were puzzled, as lowering uric acid should not have affected the ability of fructose to act as a calorie, for uric acid is not generated directly from fructose itself but instead is a side-product of the first enzymatic reaction of fructose to fructose-1-phosphate by the enzyme fructokinase [68, 69]. This phosphorylation of fructose reduces ATP to ADP and AMP, and triggers the stimulation of an enzyme, AMP deaminase-2 (AMPD2), that breaks down AMP to IMP and then to uric acid [68, 69].
As we further studied this adenine nucleotide degradation pathway, we realized that the intracellular production of uric acid resulted in the recruitment of both uric acid and NADPH oxidase to the mitochondria causing stimulation of lipogenesis, a blockade in fatty acid oxidation, and a reduction in oxidative phosphorylation and ATP production [70–73]. The uric acid also inhibited AMP-activated protein kinase [74, 75]. The net result was that intracellular ATP levels fell but could not be replaced. This initiated an alarm system that energy supplies were endangered and triggered a “survival” switch (reviewed in [76]) (Fig. 1).
Figure 1:
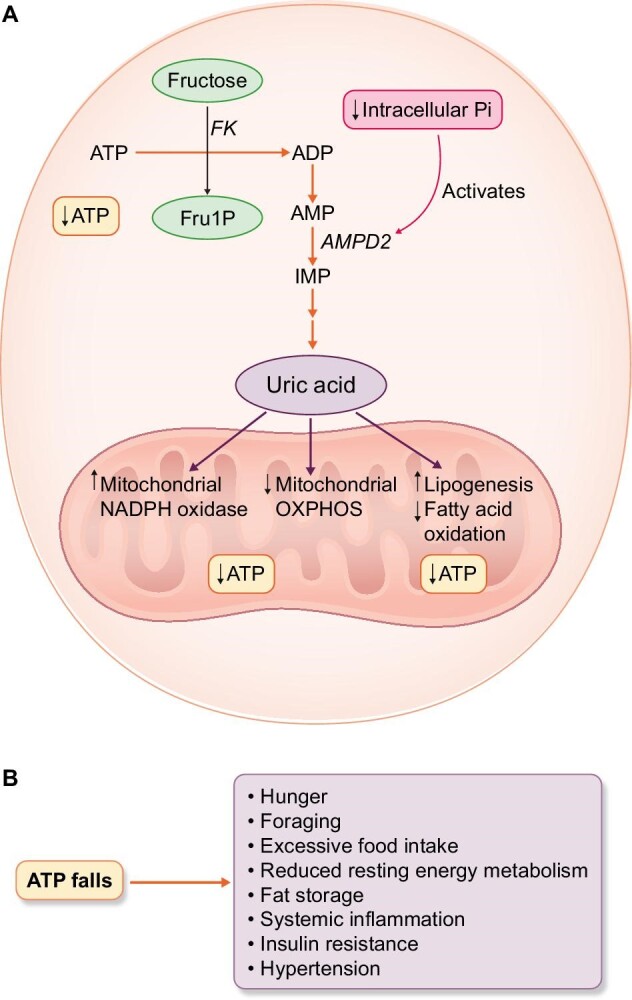
Fructose metabolism results in intracellular ATP depletion and triggers a survival response. (A) The metabolism of fructose by fructokinase (FK) results in rapid ATP consumption with a fall in ATP and intracellular phosphate (Pi) in the cell. The low intracellular phosphate activates the enzyme AMP deaminase-2 (AMPD2) which removes the AMP substrate to make inosine monophosphate (IMP) and eventually uric acid. The rise in intracellular uric acid results in the translocation of the NADPH oxidase to the mitochondria resulting in oxidative stress that suppresses oxidative phosphorylation. This is also associated with an increase in lipogenesis and a block in fatty acid oxidation. (B) As ATP levels fall, the survival response is stimulated as noted by hunger, foraging, excessive food intake, reduced resting energy metabolism, fat storage, insulin resistance, systemic inflammation and hypertension.
Consistent with these findings, we found that if we gave a soft drink to human volunteers, blood pressure and uric acid levels increased within 15 min [77]. Others also found that the acute intake of 60 g of fructose caused an immediate rise in blood pressure, whereas water ingestion did not [78]. Finally, in a study in which fructose was given in large doses (200 g/day) for 2 weeks, we found that fructose ingestion caused a marked rise in ambulatory blood pressure, but this was blocked in the group that had the rise in uric acid prevented by taking allopurinol [79].
Interestingly, the administration of glucose (60 g) does not raise blood pressure acutely in humans and is similar to what is observed with water [78]. However, elevations in fasting glucose do predict hypertension, but this likely reflects the development of insulin resistance [80].
Epidemiological studies also documented that the intake of soft drinks and fruit juices is associated with the development of hypertension [81]. Interestingly, this was not observed with fruit intake, likely because fruit contains antioxidants (vitamin C), flavonols (such as epicatechin), potassium and fiber [82].
Our early studies focused on dietary fructose as a trigger for hypertension and metabolic syndrome. However, we recognized that fructose can also be produced in the body and that the sole mechanism was the conversion of glucose to fructose via the polyol pathway, in which glucose is first converted to sorbitol via aldose reductase (the rate-limiting enzyme), followed by its conversion to fructose via sorbitol dehydrogenase [83]. While this pathway was well-recognized to occur in diabetes in association with high intracellular and blood glucose levels, we found that it could also be initiated by high glycemic foods [84]. We and others also found that other stimuli could stimulate fructose production, including increased serum osmolality, ischemia and hypoxia [85–88]. Indeed, ischemia could also stimulate the induction and expression of the fructokinase enzyme, and it appears that the classic ischemia–reperfusion syndrome that results in oxidative stress may act in part via the production and metabolism of fructose [86, 89].
These studies also provide an insight into why the uricase mutation provided a natural survival advantage during the Miocene, for the uricase mutation amplifies the increase of uric acid in response to fructose [90]. In turn, this amplifies the effect of fructose to activate the survival switch [47, 91]. During the mid-Miocene, global cooling led to seasonal starvation and the near extinction of ancestral apes [92]. It is likely that when the mutation occurred, it provided a survival advantage. In today's society, however, it may act like a thrifty gene, increasing our risk for obesity (reviewed in [93]).
It should be mentioned that fructose metabolism has also been shown to have a role in hypertension via other mechanisms. First, the ingestion of fructose has been shown to stimulate sodium absorption in the gut [94], sodium reabsorption in the proximal tubule by stimulating the Na–H exchanger [95, 96] and also in the distal tubule by stimulating the sodium–chloride cotransporter [97]. Fructose also induces hypothalamic leptin resistance, leading to obesity [98, 99]. Beautiful studies by Hall, De Silva and others have shown that while leptin resistance results in an impairment of leptin to quell hunger, the high levels of leptin still act via the melanocortin system to stimulate the sympathetic nervous system and contribute to hypertension that occurs [100, 101]. Fructose metabolism may also result in NAD consumption, and deficiency of its co-factor, sirtuin 1, which might have a role in driving inflammation [102]. Finally, rats given fructose develop tubulointerstitial injury and arteriolopathy similar to that observed in hyperuricemia, potentially leading to salt-sensitive hypertension [65, 103].
IMPAIRED PRESSURE NATRIURESIS AND THE MECHANISM BY WHICH SALT CAUSES HYPERTENSION
The impairment in pressure natriuresis that is observed in primary hypertension is associated with a relative reduction in the ability to excrete salt [104]. One of the classic hypothesized mechanisms for how the impaired excretion of salt causes hypertension was proposed by Guyton et al., who suggested that the high-salt diet leads to extracellular volume expansion that increases arterial pressure to excrete the salt. This is the primary mechanism driving hypertension [6].
However, the concept that salt may act via expanding the extracellular volume is now being challenged, as more and more data suggest that the retention of salt acts primarily by an effect on serum osmolality [105–108]. Indeed, recent data suggest that salt triggers inflammatory, metabolic and vascular effects primarily by increasing serum osmolality and that these effects can be shown in the brain and target tissues, such as the lymphocytes [109, 110]. When a high-salt diet is ingested, vasopressin is activated and water conservation occurs [111, 112], which minimizes but does not prevent the increase in serum osmolality. While some natriuresis occurs, sodium can also be concentrated in interstitial sites such as the skin [111–113].
One of the more striking findings is that the increase in serum osmolality can stimulate the production of aldose reductase in the polyol pathway, leading to fructose generation [114]. Our group has found that a high-salt diet stimulates fructose production in the liver and brain, associated with increased hepatic uric acid levels, even though serum uric acid does not change [85]. Over time a high-salt diet is associated not only with the development of hypertension and cardiac hypertrophy but also with the development of obesity, insulin resistance and other features of the metabolic syndrome. Surprisingly, mice unable to metabolize fructose (fructokinase knockout mice) are protected from metabolic syndrome and developing hypertension and cardiac hypertrophy [85]. This suggests that a high-salt diet may act as an amplification loop that triggers additional fructose and uric acid generation that can compound the hypertensive state. One of the mechanisms may be via the stimulation of vasopressin, as both high-salt diets and fructose stimulate vasopressin production [115]. While the vasopressin 2 receptor is important in driving the urinary concentration mechanism, the vasopressin 1a can stimulate vasoconstriction and hypertension, and stimulation of the vasopressin 1b receptor appears to have a role in driving the metabolic features of fructose-induced metabolic syndrome [115].
These results imply that it is not the amount of salt we eat that is critical but rather the effect of salt intake on osmolality. This would require looking at the balance of salt and water intake. To evaluate this principle, we gave soup containing high amounts of salt to human volunteers, some of whom were supplemented with extra drinking water [116]. The group that only received the high-salt diet developed an acute rise in serum osmolality, blood pressure and vasopressin levels, but those who were supplemented with water maintained serum osmolality in the normal range and did not develop a hypertensive response [116]. These findings align with numerous studies that reported that an increase in the sodium concentration of 2–4 mOsm/L in serum and cerebrospinal fluid, while still in the normal range of sodium concentration, is associated with hypertension [117].
To further address this relationship of salt with blood pressure, we performed an epidemiological study in a healthy Japanese population. We found that serum sodium was a better predictor for the development of hypertension than the actual amount of sodium ingested [118]. This observation might also explain why thiazide diuretics (which tend to lower serum sodium) are more effective than loop diuretics (which tend to raise serum sodium) in treating hypertension, even though loop diuretics are more effective at stimulating sodium excretion.
These new findings may also explain why aldosterone levels are sometimes elevated in patients with metabolic syndrome. Indeed, it is known that some individuals will develop resistant hypertension associated with elevated aldosterone levels independent of the renin–angiotensin system or potassium levels (often called “aldosterone breakthrough”). We have suggested that this may reflect chronic adrenocorticotrophic hormone (ACTH) release from the vasopressin 1b receptor stimulation from fructose metabolism [119]. It is even possible that the development of primary aldosteronomas may relate to chronic ACTH stimulation in genetically predisposed individuals, possibly similar to the development of tertiary hyperparathyroidism in subjects with chronic kidney disease [119].
SUMMARY
It has been 150 years since Mahomed discovered primary hypertension, and the cause of this condition remains under investigation. Our studies suggest that a primary stimulus is diets high in fructose-containing sugar, salt and high glycemic carbohydrates that generate endogenous production of fructose and uric acid. Other dietary factors are very likely important, such as alcohol and purine-containing foods that can raise uric acid. Caffeine can raise blood pressure acutely, although it has minimal effects on habitual coffee drinkers. Interestingly, vitamin C (ascorbate) may help counter fructose and uric acid effects and lower blood pressure [120]. Other factors that may increase the risk for hypertension include genetic polymorphisms that modulate sodium handling in the kidney or immune function, and congenital factors associated with low birth weight are important, as well as psychological factors (Type A personality) and drugs (such as cyclosporine). However, our data would suggest that the fructose–uric acid pathway is the major mechanism initiating disease and is largely responsible for the dramatic rise in the prevalence of hypertension in the 20th century.
We hypothesize that initially, these external stimuli drive renal vasoconstriction, leading to subtle renal injury and inflammation that reduces salt excretion. As salt is retained, there is an increase in serum osmolality, which then triggers an amplifying response linked with activation of sympathetic central nervous system, stimulation of the renin–angiotensin–aldosterone system and sodium retention, leading to a persistently hypertensive state (Fig. 2). So, in many respects, hypertension can be viewed as the consequence of persistent activation of an evolutionary survival pathway (the fructose survival system). This defense system has gone awry in a society where foods that can both contain fructose and can stimulate fructose production are plentiful.
Figure 2:

Proposed pathogenic mechanism for how fructose could cause primary hypertension. Fructose can be provided directly in the diet (such as from added sugars) or endogenously produced via the polyol pathway from high glycemic foods that provide the substrate, or from high-salt diet or uric acid (from umami foods) that stimulate aldose reductase, the rate-limiting enzyme that converts the glucose to fructose. The net effect of fructose metabolism and elevated intracellular uric acid is the activation of a “survival switch” that drives multiple metabolic responses. However, included in this response is an increase in salt absorption by the gut and kidney, a rise in leptin that activates sympathetic central nervous system, a stimulation of vasopressin with its vasoconstrictive properties, and a uric acid–dependent stimulation of the renin–angiotensin–aldosterone system (RAAS), the stimulation of oxidative stress and a fall in endothelial nitric oxide (NO) availability. Systemic and renal vasoconstriction follow with a rise in blood pressure. Initially this is labile and intermittent, but over time the recurrent ischemia to the kidney stimulates the expression of de novo antigens, HSP70, that induce an autoimmune response that maintains the renal vasoconstriction and elevated blood pressure. The renal vasoconstriction then results in impaired sodium excretion, leading to a rise in salt concentration and a reactivation of the pathway via a positive feedback system which helps maintain the elevation in blood pressure. Other factors, including genetic and congenital factors, can influence this pathway.
Contributor Information
Laura G Sánchez-Lozada, Department of Cardio-Renal Physiopathology, Instituto Nacional de Cardiología “Ignacio Chavez”, Mexico City, Mexico.
Magdalena Madero, Division of Nephrology, Department of Medicine, Instituto Nacional de Cardiología “Ignacio Chavez”, Mexico City, Mexico.
Marilda Mazzali, Division of Nephrology, University of Campinas, São Paulo, Brazil.
Daniel I Feig, Division of Pediatric Nephrology, University of Alabama, Birmingham, AL, USA.
Takahiko Nakagawa, Department of Nephrology, Rakuwakai-Otowa Hospital, Kyoto, Japan.
Miguel A Lanaspa, Department of Medicine, University of Colorado Anschutz Medical Center, Aurora, CO, USA.
Mehmet Kanbay, Department of Medicine, Koc University School of Medicine, Istanbul, Turkey.
Masanari Kuwabara, Depart of Cardiology, Toranomon Hospital, Tokyo, Japan.
Bernardo Rodriguez-Iturbe, Department of Nephrology and Mineral Metabolism, Instituto Nacional de Ciencias Médicas y Nutrición “Salvador Zubirán”, Mexico City.
Richard J Johnson, Department of Medicine, University of Colorado Anschutz Medical Center, Aurora, CO, USA.
DATA AVAILABILITY STATEMENT
No new data were generated or analysed in support of this research.
CONFLICT OF INTEREST STATEMENT
R.J.J. has consulted with Horizon Pharma, and he and M.A.L. and L.G.S.-L. have equity with Colorado Research Partners LLC. R.J.J. and T.N. also have stocks with XORTX therapeutics. All others disclose no conflicts of interest.
REFERENCES
- 1. Mahomed FA. The etiology of Bright's disease and the prealbuminuric state. Med Chir Trans 1874;MCT-57:197–228. 10.1177/095952877405700118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Medico-Actuarial Mortality Investigation . Association of Life Insurance Medical Directors and the Actuarial Society of America 1912;1. [PubMed] [Google Scholar]
- 3. Janeway TC. The Clinical Study of Blood Pressure. New York, NY:Appleton and Company, 1907. [Google Scholar]
- 4. Kearney PM, Whelton M, Reynolds Ket al. Global burden of hypertension: analysis of worldwide data. Lancet North Am Ed 2005;365:217–23. 10.1016/S0140-6736(05)17741-1. [DOI] [PubMed] [Google Scholar]
- 5. Page IH. The mosaic theory of arterial hypertension—its interpretation. Perspect Biol Med 1967;10:325–33. 10.1353/pbm.1967.0031. [DOI] [PubMed] [Google Scholar]
- 6. Guyton AC, Coleman TG, Cowley AV Jret al. Arterial pressure regulation. Overriding dominance of the kidneys in long-term regulation and in hypertension. Am J Med 1972;52:584–94. 10.1016/0002-9343(72)90050-2. [DOI] [PubMed] [Google Scholar]
- 7. Dahl LK. Possible role of salt intake in the development of essential hypertension. In: Cottier P, Bock Ks, eds. Essential Hypertension - an International Symposium. Berlin: Springer, 1960, 53–65. [DOI] [PubMed] [Google Scholar]
- 8. Hayasaki T, Ishimoto T, Doke Tet al. Fructose increases the activity of sodium hydrogen exchanger in renal proximal tubules that is dependent on ketohexokinase. J Nutr Biochem 2019;71:54–62. 10.1016/j.jnutbio.2019.05.017. [DOI] [PubMed] [Google Scholar]
- 9. Allen FM, Sherrill JW. The treatment of arterial hypertension. J Metabol Research 1922;2:429–545. [Google Scholar]
- 10. Kempner W. Radical dietary treatment of hypertensive and arteriosclerotic vascular disease, heart and kidney disease, and vascular retinopathy. GP 1954;9:71–92. [PubMed] [Google Scholar]
- 11. Dahl LK, Heine M. Primary role of renal homografts in setting chronic blood pressure levels in rats. Circ Res 1975;36:692–6. 10.1161/01.RES.36.6.692. [DOI] [PubMed] [Google Scholar]
- 12. Lifton RP. Genetic dissection of human blood pressure variation: common pathways from rare phenotypes. Harvey Lect 2004;100:71–101. [PubMed] [Google Scholar]
- 13. Ji W, Foo JN, O'Roak BJet al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet 2008;40:592–9. 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brenner BM, Garcia DL, Anderson S. Glomeruli and blood pressure. Less of one, more the other? Am J Hypertens 1988;1(4 Pt 1):335–47. 10.1093/ajh/1.4.335. [DOI] [PubMed] [Google Scholar]
- 15. Barker DJ, Osmond C, Golding Jet al. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 1989;298:564–7. 10.1136/bmj.298.6673.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eriksson J, Forsen T, Tuomilehto Jet al. Fetal and childhood growth and hypertension in adult life. Hypertension 2000;36:790–4. 10.1161/01.HYP.36.5.790. [DOI] [PubMed] [Google Scholar]
- 17. Goldblatt H. The renal origin of hypertension. Physiol Rev 1947;27:120–65. 10.1152/physrev.1947.27.1.120. [DOI] [PubMed] [Google Scholar]
- 18. Sommers SC, Relman AS, Smithwick RH. Histologic studies of kidney biopsy specimens from patients with hypertension. Am J Pathol 1958;34:685–715. [PMC free article] [PubMed] [Google Scholar]
- 19. Johnson RJ, Rodriguez-Iturbe B, Schreiner GFet al. Hypertension: a microvascular and tubulointerstitial disease. J Hypertens Suppl 2002;20:S1–7. [PubMed] [Google Scholar]
- 20. Johnson RJ, Alpers CE, Yoshimura Aet al. Renal injury from angiotensin II-mediated hypertension. Hypertension 1992;19:464–74. 10.1161/01.HYP.19.5.464. [DOI] [PubMed] [Google Scholar]
- 21. Lombardi D, Gordon KL, Polinsky Pet al. Salt-sensitive hypertension develops after short-term exposure to Angiotensin II. Hypertension 1999;33:1013–9. 10.1161/01.HYP.33.4.1013. [DOI] [PubMed] [Google Scholar]
- 22. Franco M, Tapia E, Santamaria Jet al. Renal cortical vasoconstriction contributes to development of salt-sensitive hypertension after angiotensin II exposure. J Am Soc Nephrol 2001;12:2263–71. 10.1681/ASN.V12112263. [DOI] [PubMed] [Google Scholar]
- 23. Wilcox CS. Oxidative stress and nitric oxide deficiency in the kidney: a critical link to hypertension? Am J Physiol Regul Integr Comp Physiol 2005;289:R913–35. 10.1152/ajpregu.00250.2005. [DOI] [PubMed] [Google Scholar]
- 24. Baylis C. Nitric oxide deficiency in chronic renal disease. Eur J Clin Pharmacol 2006;62:123–30. 10.1007/s00228-005-0003-0.16408225 [DOI] [Google Scholar]
- 25. Navar LG, Kobori H, Prieto-Carrasquero M. Intrarenal angiotensin II and hypertension. Current Hypertens Rep 2003;5:135–43. 10.1007/s11906-003-0070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rodriguez-Iturbe B, Pons H, Quiroz Yet al. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int 2001;59:2222–32. 10.1046/j.1523-1755.2001.00737.x. [DOI] [PubMed] [Google Scholar]
- 27. Rodriguez-Iturbe B, Pons H, Quiroz Yet al. The immunological basis of hypertension. Am J Hypertens 2014;27:1327–37. 10.1093/ajh/hpu142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stewart T, Jung FF, Manning Jet al. Kidney immune cell infiltration and oxidative stress contribute to prenatally programmed hypertension. Kidney Int 2005;68:2180–8. 10.1111/j.1523-1755.2005.00674.x. [DOI] [PubMed] [Google Scholar]
- 29. Pons H, Ferrebuz A, Quiroz Yet al. Immune reactivity to heat shock protein 70 expressed in the kidney is cause of salt-sensitive hypertension. Am J Physiol Renal Physiol 2013;304:F289–99. 10.1152/ajprenal.00517.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kirabo A, Fontana V, de Faria APet al. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest 2014;124:4642–56. 10.1172/JCI74084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guzik TJ, Hoch NE, Brown KAet al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 2007;204:2449–60. 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. De Miguel C, Das S, Lund Het al. T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 2010;298:R1136–42. 10.1152/ajpregu.00298.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rodriguez-Iturbe B, Pons H, Johnson RJ. Role of the immune system in hypertension. Physiol Rev 2017;97:1127–64. 10.1152/physrev.00031.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McMaster WG, Kirabo A, Madhur MSet al. Inflammation, immunity, and hypertensive end-organ damage. Circ Res 2015;116:1022–33. 10.1161/CIRCRESAHA.116.303697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chan CT, Sobey CG, Lieu Met al. Obligatory role for B cells in the development of angiotensin II-dependent hypertension. Hypertension 2015;66:1023–33. 10.1161/HYPERTENSIONAHA.115.05779. [DOI] [PubMed] [Google Scholar]
- 36. Barhoumi T, Kasal DA, Li MWet al. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension 2011;57:469–76. 10.1161/HYPERTENSIONAHA.110.162941. [DOI] [PubMed] [Google Scholar]
- 37. Wang W, Lu Y, Hu Xet al. Interleukin-22 exacerbates angiotensin II-induced hypertensive renal injury. Int Immunopharmacol 2022;109:108840. 10.1016/j.intimp.2022.108840. [DOI] [PubMed] [Google Scholar]
- 38. Thomas JM, Ling YH, Huuskes Bet al. IL-18 (interleukin-18) produced by renal tubular epithelial cells promotes renal inflammation and injury during deoxycorticosterone/salt-induced hypertension in mice. Hypertension 2021;78:1296–309. 10.1161/HYPERTENSIONAHA.120.16437. [DOI] [PubMed] [Google Scholar]
- 39. Herrera J, Ferrebuz A, Macgregor EGet al. Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol 2006;17(12 Suppl 3):S218–25. 10.1681/ASN.2006080918. [DOI] [PubMed] [Google Scholar]
- 40. Rodriguez-Iturbe B. Autoimmunity in the pathogenesis of hypertension. Hypertension 2016;67:477–83. 10.1161/HYPERTENSIONAHA.115.06418. [DOI] [PubMed] [Google Scholar]
- 41. Rodriguez-Iturbe B, Johnson RJ. Genetic polymorphisms in hypertension: are we missing the immune connection? Am J Hypertens 2019;32:113–22. 10.1093/ajh/hpy168. [DOI] [PubMed] [Google Scholar]
- 42. Feig DI, Madero M, Jalal DIet al. Uric acid and the origins of hypertension. J Pediatr 2013;162:896–902. 10.1016/j.jpeds.2012.12.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kuwabara M, Niwa K, Hisatome Iet al. Asymptomatic hyperuricemia without comorbidities predicts cardiometabolic diseases: five-year Japanese cohort study. Hypertension 2017;69:1036–44. 10.1161/HYPERTENSIONAHA.116.08998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang Q, Kottgen A, Dehghan Aet al. Multiple genetic loci influence serum urate levels and their relationship with gout and cardiovascular disease risk factors. Circ Cardiovasc Genet 2010;3:523–30. 10.1161/CIRCGENETICS.109.934455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Palmer TM, Nordestgaard BG, Benn Met al. Association of plasma uric acid with ischaemic heart disease and blood pressure: mendelian randomisation analysis of two large cohorts. BMJ 2013;347:f4262. 10.1136/bmj.f4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gill D, Cameron AC, Burgess Set al. Urate, blood pressure, and cardiovascular disease: evidence from mendelian randomization and meta-analysis of clinical trials. Hypertension 2021;77:383–92. 10.1161/HYPERTENSIONAHA.120.16547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kratzer JT, Lanaspa MA, Murphy MNet al. Evolutionary history and metabolic insights of ancient mammalian uricases. Proc Natl Acad Sci USA 2014;111:3763–8. 10.1073/pnas.1320393111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mazzali M, Hughes J, Kim YGet al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension 2001;38:1101–6. 10.1161/hy1101.092839. [DOI] [PubMed] [Google Scholar]
- 49. Mazzali M, Kanellis J, Han Let al. Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism. Am J Physiol Renal Physiol 2002;282:F991–7. 10.1152/ajprenal.00283.2001. [DOI] [PubMed] [Google Scholar]
- 50. Sanchez-Lozada LG, Soto V, Tapia Eet al. Role of oxidative stress in the renal abnormalities induced by experimental hyperuricemia. Am J Physiol Renal Physiol 2008;295:F1134–41. 10.1152/ajprenal.00104.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sanchez-Lozada LG, Tapia E, Lopez-Molina Ret al. Effects of acute and chronic L-arginine treatment in experimental hyperuricemia. Am J Physiol Renal Physiol 2007;292:F1238–44. 10.1152/ajprenal.00164.2006. [DOI] [PubMed] [Google Scholar]
- 52. Watanabe S, Kang DH, Feng Let al. Uric acid, hominoid evolution, and the pathogenesis of salt-sensitivity. Hypertension 2002;40:355–60. 10.1161/01.HYP.0000028589.66335.AA. [DOI] [PubMed] [Google Scholar]
- 53. Feig DI, Johnson RJ. Hyperuricemia in childhood primary hypertension. Hypertension 2003;42:247–52. 10.1161/01.HYP.0000085858.66548.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA 2008;300:924–32. 10.1001/jama.300.8.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Soletsky B, Feig DI. Uric acid reduction rectifies prehypertension in obese adolescents. Hypertension 2012;60:1148–56. 10.1161/HYPERTENSIONAHA.112.196980. [DOI] [PubMed] [Google Scholar]
- 56. Gunawardhana L, McLean L, Punzi HAet al. Effect of febuxostat on ambulatory blood pressure in subjects with hyperuricemia and hypertension: a phase 2 randomized placebo-controlled study. J Am Heart Assoc 2017;6:e006683. 10.1161/JAHA.117.006683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Higgins P, Walters MR, Murray HMet al. Allopurinol reduces brachial and central blood pressure, and carotid intima-media thickness progression after ischaemic stroke and transient ischaemic attack: a randomised controlled trial. Heart 2014;100:1085–92. 10.1136/heartjnl-2014-305683. [DOI] [PubMed] [Google Scholar]
- 58. Johnson RJ, Titte S, Cade JRet al. Uric acid, evolution and primitive cultures. Semin Nephrol 2005;25:3–8. 10.1016/j.semnephrol.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 59. Hayden MR, Tyagi SC. Uric acid: a new look at an old risk marker for cardiovascular disease, metabolic syndrome, and type 2 diabetes mellitus: the urate redox shuttle. Nutr Metab (Lond) 2004;1:10. 10.1186/1743-7075-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kylin E. [Studies of the hypertension-hyperglycemia-hyperuricemia syndrome] Studien uber das Hypertonie-Hyperglykamie-hyperurikamiesyndrome. Zentralblatt fur innere Medizin 1923;44:105–27. [Google Scholar]
- 61. Perheentupa J, Raivio K. Fructose-induced hyperuricaemia. Lancet North Am Ed 1967;290:528–31. 10.1016/S0140-6736(67)90494-1. [DOI] [PubMed] [Google Scholar]
- 62. Maenpaa PH, Raivio KO, Kekomaki MP. Liver adenine nucleotides: fructose-induced depletion and its effect on protein synthesis. Science 1968;161:1253–4. 10.1126/science.161.3847.1253. [DOI] [PubMed] [Google Scholar]
- 63. Johnson RJ, Segal MS, Sautin Yet al. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am J Clin Nutr 2007;86:899–906. [DOI] [PubMed] [Google Scholar]
- 64. Ford ES, Giles WH, Mokdad AH. Increasing prevalence of the metabolic syndrome among U.S. adults. Diabetes Care 2004;27:2444–9. 10.2337/diacare.27.10.2444. [DOI] [PubMed] [Google Scholar]
- 65. Sanchez-Lozada LG, Tapia E, Jimenez Aet al. Fructose-induced metabolic syndrome is associated with glomerular hypertension and renal microvascular damage in rats. Am J Physiol Renal Physiol 2007;292:F423–9. 10.1152/ajprenal.00124.2006. [DOI] [PubMed] [Google Scholar]
- 66. Nakagawa T, Hu H, Zharikov Set al. A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol 2006;290:F625–31. 10.1152/ajprenal.00140.2005. [DOI] [PubMed] [Google Scholar]
- 67. Sanchez-Lozada LG, Tapia E, Bautista-Garcia Pet al. Effects of febuxostat on metabolic and renal alterations in rats with fructose-induced metabolic syndrome. Am J Physiol Renal Physiol 2008;294:F710–8. 10.1152/ajprenal.00454.2007. [DOI] [PubMed] [Google Scholar]
- 68. Smith CM, Rovamo LM, Raivio KO. Fructose-induced adenine nucleotide catabolism in isolated rat hepatocytes. Can J Biochem 1977;55:1237–40. 10.1139/o77-185. [DOI] [PubMed] [Google Scholar]
- 69. van den Berghe G, Bronfman M, Vanneste Ret al. The mechanism of adenosine triphosphate depletion in the liver after a load of fructose. A kinetic study of liver adenylate deaminase. Biochem J 1977;162:601–9. 10.1042/bj1620601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sautin YY, Nakagawa T, Zharikov Set al. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am J Physiol Cell Physiol 2007;293:C584–96. 10.1152/ajpcell.00600.2006. [DOI] [PubMed] [Google Scholar]
- 71. Lanaspa MA, Sanchez-Lozada LG, Choi YJet al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J Biol Chem 2012;287:40732–44. 10.1074/jbc.M112.399899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sanchez-Lozada LG, Lanaspa MA, Cristobal-Garcia Met al. Uric acid-induced endothelial dysfunction is associated with mitochondrial alterations and decreased intracellular ATP concentrations. Nephron Exp Nephrol 2012;121:e71–8. 10.1159/000345509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sanchez-Lozada LG, Lanaspa MA, Cristobal-Garcia Met al. Uric acid-induced endothelial dysfunction is associated with mitochondrial alterations and decreased intracellular ATP concentrations. Nephron Exp Nephrol 2012;121:e71–8. 10.1159/000345509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cicerchi C, Li N, Kratzer Jet al. Uric acid-dependent inhibition of AMP kinase induces hepatic glucose production in diabetes and starvation: evolutionary implications of the uricase loss in hominids. FASEB J 2014;28:3339–50. 10.1096/fj.13-243634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lanaspa MA, Cicerchi C, Garcia Get al. Counteracting roles of AMP deaminase and AMP kinase in the development of fatty liver. PLoS One 2012;7:e48801. 10.1371/journal.pone.0048801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Johnson RJ, Stenvinkel P, Andrews Pet al. Fructose metabolism as a common evolutionary pathway of survival associated with climate change, food shortage and droughts. J Intern Med 2020;287:252–62. 10.1111/joim.12993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Le MT, Frye RF, Rivard CJet al. Effects of high-fructose corn syrup and sucrose on the pharmacokinetics of fructose and acute metabolic and hemodynamic responses in healthy subjects. Metabolism 2012;61:641–51. 10.1016/j.metabol.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Brown CM, Dulloo AG, Yepuri Get al. Fructose ingestion acutely elevates blood pressure in healthy young humans. Am J Physiol Regul Integr Comp Physiol 2008;294:R730–7. 10.1152/ajpregu.00680.2007. [DOI] [PubMed] [Google Scholar]
- 79. Perez-Pozo SE, Schold J, Nakagawa Tet al. Excessive fructose intake induces the features of metabolic syndrome in healthy adult men: role of uric acid in the hypertensive response. Int J Obes 2010;34:454–61. 10.1038/ijo.2009.259. [DOI] [PubMed] [Google Scholar]
- 80. Kuwabara M, Chintaluru Y, Kanbay Met al. Fasting blood glucose is predictive of hypertension in a general Japanese population. J Hypertens 2019;37:167–74. 10.1097/HJH.0000000000001895. [DOI] [PubMed] [Google Scholar]
- 81. Jalal DI, Smits G, Johnson RJet al. Increased fructose associates with elevated blood pressure. J Am Soc Nephrol 2010;21:1543–9. 10.1681/ASN.2009111111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nakagawa T, Lanaspa MA, Johnson RJ. The effects of fruit consumption in patients with hyperuricaemia or gout. Rheumatology (Oxford) 2019;58:1133–41. 10.1093/rheumatology/kez128. [DOI] [PubMed] [Google Scholar]
- 83. Andres-Hernando A, Johnson RJ, Lanaspa MA. Endogenous fructose production: what do we know and how relevant is it? Curr Opin Clin Nutr Metab Care 2019;22:289–94. 10.1097/MCO.0000000000000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lanaspa MA, Ishimoto T, Li Net al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat Commun 2013;4:2434. 10.1038/ncomms3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lanaspa MA, Kuwabara M, Andres-Hernando Aet al. High salt intake causes leptin resistance and obesity in mice by stimulating endogenous fructose production and metabolism. Proc Natl Acad Sci USA 2018;115:3138–43. 10.1073/pnas.1713837115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Andres-Hernando A, Li N, Cicerchi Cet al. Protective role of fructokinase blockade in the pathogenesis of acute kidney injury in mice. Nat Commun 2017;8:14181. 10.1038/ncomms14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mirtschink P, Krishnan J, Grimm Fet al. HIF-driven SF3B1 induces KHK-C to enforce fructolysis and heart disease. Nature 2015;522:444–9. 10.1038/nature14508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Park TJ, Reznick J, Peterson BLet al. Fructose-driven glycolysis supports anoxia resistance in the naked mole-rat. Science 2017;356:307–11. 10.1126/science.aab3896. [DOI] [PubMed] [Google Scholar]
- 89. Mirtschink P, Krek W. Hypoxia-driven glycolytic and fructolytic metabolic programs: pivotal to hypertrophic heart disease. Biochim Biophys Acta 2016;1863(7PtB):1822–8. 10.1016/j.bbamcr.2016.02.011. [DOI] [PubMed] [Google Scholar]
- 90. Stavric B, Johnson WJ, Clayman Set al. Effect of fructose administration on serum urate levels in the uricase inhibited rat. Experientia 1976;32:373–4. 10.1007/BF01940847. [DOI] [PubMed] [Google Scholar]
- 91. Tapia E, Cristobal M, Garcia-Arroyo FEet al. Synergistic effect of uricase blockade plus physiological amounts of fructose-glucose on glomerular hypertension and oxidative stress in rats. Am J Physiol Renal Physiol 2013;304:F727–36. 10.1152/ajprenal.00485.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Johnson RJ, Andrews P. Fructose, uricase, and the Back-to-Africa hypothesis. Evol Anthropol 2010;19:250–7. 10.1002/evan.20266. [DOI] [Google Scholar]
- 93. Johnson RJ, Sanchez Lozada LG, Nakagawa Tet al. Do thrifty genes exist? Revisiting uricase. Obesity (Silver Spring) 2022;30:1917–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Soleimani M, Alborzi P. The role of salt in the pathogenesis of fructose-induced hypertension. Int J Nephrol 2011;2011:1. 10.4061/2011/392708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Cabral PD, Hong NJ, Hye Khan MAet al. Fructose stimulates Na/H exchange activity and sensitizes the proximal tubule to angiotensin II. Hypertension 2014;63:e68–73. 10.1161/HYPERTENSIONAHA.113.02564. [DOI] [PubMed] [Google Scholar]
- 96. Hayasaki T, Ishimoto T, Doke Tet al. Fructose increases the activity of sodium hydrogen exchanger in renal proximal tubules that is dependent on ketohexokinase. J Nutr Biochem 2019;71:54–62. 10.1016/j.jnutbio.2019.05.017. [DOI] [PubMed] [Google Scholar]
- 97. Bahena-Lopez JP, Rojas-Vega L, Chavez-Canales Met al. Glucose/fructose delivery to the distal nephron activates the sodium-chloride cotransporter via the calcium-sensing receptor. J Am Soc Nephrol 2023;34:55–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Shapiro A, Mu W, Roncal Cet al. Fructose-induced leptin resistance exacerbates weight gain in response to subsequent high-fat feeding. Am J Physiol Regul Integr Comp Physiol 2008;295:R1370–5. 10.1152/ajpregu.00195.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Shapiro A, Tumer N, Gao Yet al. Prevention and reversal of diet-induced leptin resistance with a sugar-free diet despite high fat content. Br J Nutr 2011;106:390–7. 10.1017/S000711451100033X. [DOI] [PubMed] [Google Scholar]
- 100. Hall JE, da Silva AA, do Carmo JMet al. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem 2010;285:17271–6. 10.1074/jbc.R110.113175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. da Silva AA, do Carmo JM, Hall JE. Role of leptin and central nervous system melanocortins in obesity hypertension. Curr Opin Nephrol Hypertens 2013;22:135–40. 10.1097/MNH.0b013e32835d0c05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Rodriguez-Iturbe B, Johnson RJ, Lanaspa MAet al. Sirtuin deficiency and the adverse effects of fructose and uric acid synthesis. Am J Physiol Regul Integr Comp Physiol 2022;322:R347–59. 10.1152/ajpregu.00238.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Nakayama T, Kosugi T, Gersch Met al. Dietary fructose causes tubulointerstitial injury in the normal rat kidney. Am J Physiol Renal Physiol 2010;298:F712–20. 10.1152/ajprenal.00433.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hall JE, Mizelle HL, Hildebrandt DAet al. Abnormal pressure natriuresis. A cause or a consequence of hypertension? Hypertension 1990;15(6 Pt 1):547–59. 10.1161/01.HYP.15.6.547. [DOI] [PubMed] [Google Scholar]
- 105. Levy JR, Stevens W. Plasma hyperosmolality stimulates leptin secretion acutely by a vasopressin-adrenal mechanism. Am J Physiol Endocrinol Metab 2004;287:E263–8. 10.1152/ajpendo.00514.2003. [DOI] [PubMed] [Google Scholar]
- 106. Charkoudian N, Eisenach JH, Joyner MJet al. Interactions of plasma osmolality with arterial and central venous pressures in control of sympathetic activity and heart rate in humans. Am J Physiol Heart Circ Physiol 2005;289:H2456–60. 10.1152/ajpheart.00601.2005. [DOI] [PubMed] [Google Scholar]
- 107. Wenner MM, Rose WC, Delaney EPet al. Influence of plasma osmolality on baroreflex control of sympathetic activity. Am J Physiol Heart Circ Physiol 2007;293:H2313–9. 10.1152/ajpheart.01383.2006. [DOI] [PubMed] [Google Scholar]
- 108. Toney GM, Stocker SD. Hyperosmotic activation of CNS sympathetic drive: implications for cardiovascular disease. J Physiol 2010;588(Pt 18):3375–84. 10.1113/jphysiol.2010.191940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Wu C, Yosef N, Thalhamer Tet al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 2013;496:513–7. 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Rucker AJ, Rudemiller NP, Crowley SD. Salt, hypertension, and immunity. Annu Rev Physiol 2018;80:283–307. 10.1146/annurev-physiol-021317-121134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kitada K, Daub S, Zhang Yet al. High salt intake reprioritizes osmolyte and energy metabolism for body fluid conservation. J Clin Invest 2017;127:1944–59. 10.1172/JCI88532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Rakova N, Kitada K, Lerchl Ket al. Increased salt consumption induces body water conservation and decreases fluid intake. J Clin Invest 2017;127:1932–43. 10.1172/JCI88530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wiig H, Schroder A, Neuhofer Wet al. Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J Clin Invest 2013;123:2803–15. 10.1172/JCI60113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ruepp B, Bohren KM, Gabbay KH. Characterization of the osmotic response element of the human aldose reductase gene promoter. Proc Natl Acad Sci USA 1996;93:8624–9. 10.1073/pnas.93.16.8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Andres-Hernando A, Jensen TJ, Kuwabara Met al. Vasopressin mediates fructose-induced metabolic syndrome by activating the V1b receptor. JCI Insight 2021;6:e140848. 10.1172/jci.insight.140848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Kanbay M, Aslan G, Afsar Bet al. Acute effects of salt on blood pressure are mediated by serum osmolality. J Clin Hypertens 2018;20:1447–54. 10.1111/jch.13374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. de Wardener HE, He FJ, MacGregor GA. Plasma sodium and hypertension. Kidney Int 2004;66:2454–66. [DOI] [PubMed] [Google Scholar]
- 118. Kuwabara M, Kanbay M, Niwa Ket al. Hyperosmolarity and increased serum sodium concentration are risks for developing hypertension regardless of salt intake: a five-year cohort study in Japan. Nutrients 2020;12:1422. 10.3390/nu12051422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Hahn K, Rodriguez-Iturbe B, Winterberg Bet al. Primary aldosteronism: a consequence of sugar and western diet? Med Hypotheses 2022;160. [Google Scholar]
- 120. Juraschek SP, Guallar E, Appel LJet al. Effects of vitamin C supplementation on blood pressure: a meta-analysis of randomized controlled trials. Am J Clin Nutr 2012;95:1079–88. 10.3945/ajcn.111.027995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated or analysed in support of this research.