

Summary
Globally, stroke is one of the leading causes of death and significant contributors to disability. Gaining a thorough comprehension of the underlying pathogenic processes is essential for stroke treatment and prevention. In this study, we investigated the role of p21-activated kinase 1 (PAK1) in stroke by using oxygen-glucose deprivation (OGD) and transient middle cerebral artery occlusion and reperfusion (tMCAO/R) models. We reported that focal ischemia and reperfusion affect the PAK1 expression and activity levels. We further demonstrated that PAK1 is responsible for the endothelial hyperpermeability that occurs in the early stages of ischemia and reperfusion. Additionally, inhibition of PAK1 was discovered to alleviate blood-brain barrier disruption and protect against brain injury induced by tMCAO/R. Mechanistically, we provide the evidence that PAK1 regulates the formation of stress fibers and expression of surface junctional proteins. Together, our findings reveal a pathogenic function of PAK1 in stroke.
Subject areas: Health sciences, Molecular biology, Neuroscience
Graphical abstract
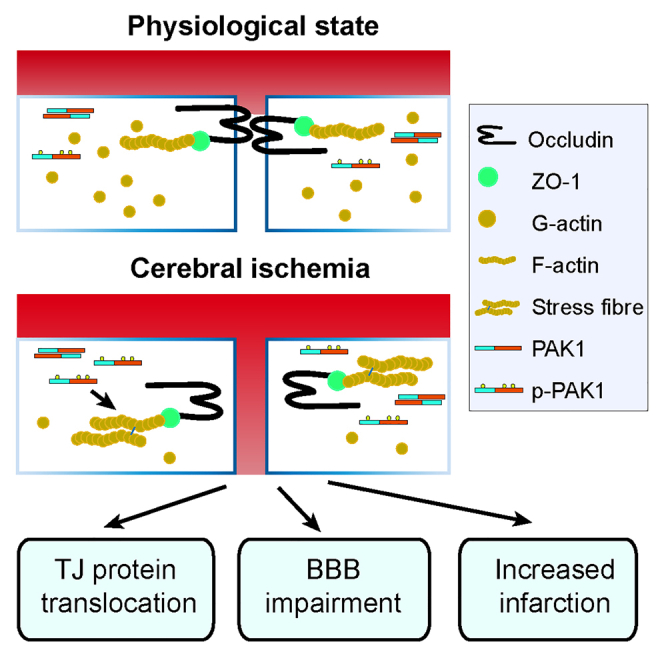
Highlights
-
•
PAK1 is responsible for the endothelial hyperpermeability at acute phase of stroke
-
•
Inhibition of PAK1 protects against tMCAO/R-induced brain injury
-
•
PAK1 regulates junctional protein expression and stress fiber formation
Health sciences; Molecular biology; Neuroscience
Introduction
Globally, stroke remains the second-leading cause of death and the third-leading cause of death and disability combined.1,2 Many survivors of acute stroke live with physical or mental disabilities, which imposing a tremendous socioeconomic burden. Moreover, the subclinical or silent brain infarcts are associated with decline in cognitive function in patients.3,4 For ischemic stroke, the currently established treatment such as the recanalization therapy is effective only to a small percentage of patients due to the narrow therapeutic window.5 Therefore, this unmet medical need for treatment of stroke undoubtedly depends on the development of new drugs and therapeutic approaches, which are based on the deep understanding the novel pathogenic mechanisms of stroke.
Blood-brain barrier (BBB) is a material barrier that controls molecule exchange between blood and brain parenchyma, which is composed of brain microvessel endothelial cells (ECs), pericyte, endfoot of astrocyte, and basement membrane.6,7,8 BBB permeability change or disruption occurs shortly after the onset of cerebral artery occlusion and persists to the chronic phases of stroke.5 BBB dysfunction is also the central genesis of hemorrhagic transformation and increased mortality after tissue plasminogen activator (tPA) treatment in stroke.9 In experimental stroke, blockade of BBB dysfunction protects parenchyma and improves functional outcome.10,11 These findings indicate that BBB dysfunction plays essential roles during ischemic brain injury and stroke recovery.
The p21-activated kinases (PAKs), belonging to the serine/threonine kinases, are the downstream effectors of Ras-related Rho GTPase Cdc42 and Rac.12 There are two groups in PAKs family including group I (PAK1, PAK2, and PAK3) and group II PAKs (PAK4, PAK5, and PAK6). PAKs participate in various biological processes, such as cell growth, motility, immune response, inflammation, apoptosis, and gene expression.13 As expected, PAKs have been proved to play critical roles in human diseases, including cancers, infectious diseases, neurological disorders, diabetes, and cardiac disorders.14,15,16,17 In addition, many small molecules or compounds have been developed as PAK inhibitors for cancers and infectious diseases treatment in preclinical research.14 Altogether, these results put forward PAKs as promising therapeutic targets for various diseases. Comparing with other members, PAK1 has been well studied for its involvement in oncogenesis and PAK1 inhibitors have been developed as potential preclinical agents for cancer therapy.18 Interestingly, PAK1/AKT signaling has been demonstrated as the important function in cardiomyocyte ischemia/reperfusion injury.19,20 Moreover, it has been reported that delayed overexpression of endothelial Ras-related C3 botulinum toxin substrate 1 (Rac1) improves cognitive and sensorimotor recovery from day 14–21 after stroke via activating PAK1.21 These results suggest theocratical roles of PAK1 during post-stroke recovery.
Here, the functions of PAK1 in stroke were explored in oxygen-glucose deprivation (OGD) model of the endothelial cells and the mouse model of brain ischemia and reperfusion. We show that focal ischemia and reperfusion increase PAK1 expression and activity in mouse brains. Our data suggest that PAK1 is responsible for destructing the BBB integrity at the acute phase of the brain ischemia and reperfusion. PAK1 inhibition reduces the stress fiber formation, stabilizes surface junctional protein expression, ameliorates the BBB disruption, and protects against transient middle cerebral artery occlusion and reperfusion (tMCAO/R) induced brain injury. Together, our findings reveal a new role of PAK1 in stroke, especially at the acute phase of the brain ischemia and reperfusion.
Results
Focal ischemia and reperfusion affect PAK1 expression in the mouse brain
To determine the role of PAK1 during brain ischemia and reperfusion, we characterized the PAK1 expression patterns in tMCAO model. The rCBF was monitored to confirm the occlusion of middle cerebral artery (Figure 1A). At different time points of reperfusion after 1 h occlusion, we measured the levels of PAK1, p-PAK1 (Ser144) in the ischemic cortex by Western blot. The resulting band corresponding p-PAK1 was more intense at 1 h reperfusion than in the sham group (Figures 1B and 1C), whereas there were no statistically significant changes for the levels of PAK1 during reperfusion (Figures 1B and 1D).
Figure 1.

Focal cerebral ischemia increases PAK1 phosphorylation
(A) Representative image of blood flow measured by laser speckle flowmeter.
(B) The time-dependent changes of PAK1 and p-PAK1 in ischemic cortex at 1 h, 6 h, 12 h, or 24 h reperfusion after 1-h transient middle cerebral artery occlusion (tMCAO). The homogenates of cortical brains from tMCAO and sham treated mice were subjected to Western blot analysis using indicated antibodies. GAPDH was used as a loading control.
(C and D) The quantitative analysis of immunoblotted p-PAK1 (Ser144) and PAK1 proteins. Data are expressed as mean ± SEM (n = 4). ∗p < 0.05 versus sham, one-way ANOVA with Dunnet’s post hoc test.
(E) Immunofluorescence images showing the colocalization of PAK1-positive (green) with microvessel markers Lectin (red) in the ipsilateral (I)/contralateral (C) cortex at 1 h reperfusion after tMCAO. White arrow shows that PAK1 is localized on blood vessels. Nuclei were stained with DAPI (blue). Scale bar = 20 μm.
See also Figure S1.
To further explore the potential functions of PAK1 in ischemic stroke, we next examined the PAK1 expression in the cortex by immunofluorescent staining using the PAK1 antibodies (Figure S1). The clear PAK1 immunoreactivity was detected by a rabbit mAb (Abclonal, Cat#A19608; AB_2862697) in both sham and tMCAO/R groups (Figure 1E). Moreover, the PAK1 immunoreactivity was more obviously colocalized with the Lycopersicon esculentum (Tomato) lectin signaling, which is commonly used for labeling the endothelial cells of brain vasculature, in ipsilateral hemisphere compared with contralateral region (Figure 1E). Together, our observations suggest that endothelial PAK1 is increased at the acute phase of the brain ischemia and reperfusion injury.
PAK1 contributes to the endothelial hyperpermeability during the ischemia and reperfusion
To dissect the function of endothelial PAK1 for ischemia and reperfusion, cultured mouse cerebral endothelial cells (bEnd.3) were subjected to oxygen glucose deprivation (OGD) and reoxygenation to mimic the ischemia-reperfusion injury. Collected bEnd.3 extracts were immunoblotted with anti-PAK1 and anti-p-PAK1 (Ser144) antibodies, respectively. In consistent with our findings in mouse stroke model, a significant increase of PAK1 and p-PAK1 (Ser144) was observed in cells exposed to 1-h reoxygenation (R) after OGD (Figures 2A–2C).
Figure 2.

PAK1 inhibition by its inhibitors or siRNA reduces the hyperpermeability of bEnd.3 endothelial monolayer in OGD model
(A) Immunoblots of p-PAK1 and PAK1 with corresponding antibodies in bEnd.3 cells at reperfusion for indicated times after 2 h OGD. GAPDH was used as a loading control.
(B and C) The statistical analysis of immunoblotted p-PAK1 (Ser144) and PAK1 proteins. Data are expressed as mean ± SEM (n = 4). ∗p < 0.05, ∗∗p < 0.01 versus control groups, one-way ANOVA with Dunnet’s post hoc test.
(D) Schematic representation of the in vitro BBB model. bEnd.3 cells were planted on microporous membrane of transwell inserts till confluence. After OGD/R treatment, the fluorescence dyes were added to upper compartment and the fluorescence intensity of lower compartment was measured 30 min later.
(E and F) The transfer rate of dextran from upper compartment to lower compartment was examined to assess the endothelial monolayer permeability. Consistent diffused 4.4 kDa TRITC-dextran or 70 kDa FITC-dextran during reoxygenation after 2 h OGD was limited by FRAX486 (1 μmol/L) or IPA-3 (10 μmol/L) treatment. Data are expressed as mean ± SEM (n = 5). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus ctrl group; #p < 0.05, ##p < 0.01, ###p < 0.001 versus OGD + vehicle group, two-way ANOVA with Turkey’s post hoc test.
(G) The endothelial monolayer permeability was further expressed as coefficient of diffusion (in centimeters per second). FRAX486 (1 μmol/L) or IPA-3 (10 μmol/L) treatment significantly reduced the increased permeability of bEnd.3 monolayer to 4.4 kDa TRITC-dextran or 70 kDa FITC-dextran by exposure to OGD/R 3 h. Data are expressed as mean ± SEM (n = 5). ∗∗∗p < 0.001 versus ctrl group; #p < 0.05 versus OGD + vehicle (veh) group, one-way ANOVA with Sidak’s post hoc test.
(H) PAK1 expression in bEnd.3 cells is downregulated by specific siPAK1, not siNC. Data are expressed as mean ± SEM (n = 3). ∗∗∗p < 0.001, Student’s t test.
(I) Increased endothelial monolayer permeability of bEnd.3 cells to 4.4 kDa TRITC-dextran or 70 kDa FITC-dextran at OGD/R 3h was inhibited by siPAK1. Data are expressed as mean ± SEM (n = 5). ∗∗p < 0.01 versus ctrl group; #p < 0.05 versus OGD + siNC group, one-way ANOVA with Sidak’s post hoc test.
See also Figure S2.
As one of the major events for pathophysiological response after stroke, the disruption of the BBB continues for several days to weeks.22 The dynamic changes of BBB permeability throughout the different stages play essential roles in the development of disease and stroke recovery.5,23 We therefore interfered the PAK1 with inhibitors and siRNA to investigate its effects on endothelial permeability in the in vitro BBB model (shown in Figure 2D) challenged with OGD/R. The barrier integrity was assessed by measuring the transfer rate of fluorescent conjugated dextran from the luminal compartment to the abluminal compartment in the model. In agreement with previous studies,23 3 h reoxygenation after 2 h OGD significantly increased the diffusion rate of both 4.4 kDa TRITC-dextran and 70 kDa FITC-dextran through the monolayers of bEnd.3 cells (Figures 2E–2G). The increased diffusion of fluorescent tracers induced by OGD/R was reduced by the treatment with IPA-3 (10 μmol/L), a PAK1 inhibitor during the reoxygenation (Figures 2E–2G). This data suggest PAK1 mediates OGD/R induced hyperpermeability since PAK1 is selectively blocked by IPA-3 at 10 μmol/L concentration.24,25 Moreover, the administration of another group I PAKs inhibitor, FRAX486 (1 μmol/L) showed the similar effect (Figures 2E–2G). While cell viability test showed that 2h OGD followed by 3h reoxygenation, as well as PAK1 inhibitors did not alter the survival of bEnd.3 cells (Figure S2), suggesting that paracellular hyperpermeability during OGD/R is not due to cell death. To further confirm the role of PAK1 for the paracellular hyperpermeability during OGD/R, we used predesigned synthetic siRNA targeting PAK1 to knockdown its expression and examined the endothelial permeability during OGD/R. Indeed, PAK1 knockdown blocked the elevated tracer diffusion induced by OGD/R (Figures 2H and 2I). Taken together, these data reveal that PAK1 contributes to hyperpermeability during oxygen-glucose deprivation and reoxygenation in endothelial cells.
bEnd.3 cells is a common in vitro model that was utilized for studying BBB integrity or dysfunction. Our observations suggest that PAK1 plays a key role in maintaining the integrity of endothelial barrier. Thus, to further illuminate the significance of PAK1 function in ischemic stroke, we tested whether inhibition of PAK1 could ameliorate the blood-brain barrier disruption during the ischemia and reperfusion in mouse stroke model. The BBB integrity was measured by using 4.4 kDa TRITC-dextran and 70 kDa FITC-dextran at 3 h and 24 h after reperfusion. Intraperitoneally administration of FRAX486 or IPA3 decreased the infiltration of fluorescent tracer at 3 h and 24 h (Figures 3A–3D), suggesting that PAK1 is truly responsible for BBB hyperpermeability during tMCAO/R.
Figure 3.

PAK1 inhibition by FRAX486 or IPA-3 alleviates BBB disruption during focal cerebral ischemia/reperfusion
(A) Representative fluorescence micrographs of brain cryosections from the mice subjected to MCAO/R for 3 h or 24 h. The TRITC-dextran (4.4 kDa) and FITC-dextran (70 kDa) leakage under different conditions (+vehicle, + FRAX486, + IPA-3). Scale bar = 5 mm.
(B and C) The quantitative analysis of the TRITC-dextran (4.4 kDa) and FITC-dextran (70 kDa) leakage volume. The TRITC-dextran (4.4 kDa) and FITC-dextran (70 kDa) leakage were lessened by FRAX486 (2 μmol/kg weight) or IPA-3 (10 μmol/kg weight).
(D) Representative image of microvessel leakage in striatum at 3 h reperfusion after MCAO. The vessels were filled with 70 kD FITC-dextran and the white arrow shows the leakage site on microvessels. Scale bar = 50 μm. Data are expressed as mean ± SEM (n = 3). ∗p < 0.05 ∗∗p < 0.01, ∗∗∗p < 0.001 versus vehicle group, one-way ANOVA with Dunnet’s post hoc test.
Our results suggest the elevated PAK1 expression may participate in early BBB opening during I/R injury. Substantial evidence support that the junctional proteins, including occludin, ZO-1, claudin-5, and ve-cadherin were important for maintaining BBB integrity. Moreover, in OGD/R-treated endothelial cells, the expression of junctional proteins in cellular membrane was shown to be reduced.26 Similarly, we also observed the decreased level of the surface biotinylated occludin and ve-cadherin (Figures 4A–4C). However, the expressions of total occludin and ve-cadherin were not changed significantly after 2h OGD and 3h reoxygenation (Figure 4C), which was different from the reduced expression after long term OGD/R. Importantly, the administration of PAK1 inhibitors FRAX486 and IPA-3 could partially reverse the decreased level of surface occludin and ve-cadherin (Figures 4A and 4B). These data suggest that PAK1 may be involved in early internalization of junctional proteins. Indeed, immunocytochemical studies in bEnd.3 cells showed increased perinuclear location of ZO-1 in some cells after OGD/R, but the number of cells with this phenomenon is low in FRAX486 or IPA-3-treated group (Figure 4D). These observations together suggest that PAK1 may contribute to early BBB hyperpermeability during OGD/R via regulating the junctional protein internalization.
Figure 4.

PAK1 inhibition partially reverses the reduction in surface expression of junction proteins during OGD/R
(A) Immunoblots of occludin and ve-cadherin in bEnd.3 cells after OGD 2 h and reoxygenation 3 h. The biotinylated proteins were shown as surface protein and the proteins in whole cell lysates were shown as total protein. GAPDH was used as a loading control.
(B) The statistical analysis of surface/total occludin and ve-cadherin. Relative levels are normalized to respective control groups and expressed as mean ± SEM (n = 4). ∗p < 0.05, ∗∗∗p < 0.001 versus control groups; #p < 0.05, ##p < 0.01, ###p < 0.001 versus OGD+vehicle group. One-way ANOVA with Sidak’s post hoc test.
(C) The statistical analysis of total occludin and ve-cadherin. Relative levels are normalized to respective control groups and expressed as mean ± SEM (n = 4).
(D) Immunofluorescence of ZO-1 in bEnd.3 cells with different treatment (vehicle, FRAX486, IPA-3) after OGD/R. White arrow shows internalized ZO-1. Scale bar = 20 μm.
PAK1 inhibition protect against tMCAO/R-induced brain injury in mice
To ensure the role of PAK1 in brain injury during the brain ischemia and reperfusion, we tested whether the PAKs inhibitors have any protection effect in mouse model of tMCAO/R. TTC staining was used to determine the infarct size induced by tMCAO/R or with treatments (Figure 5A). Infarct tissue was not detected in the sham group, while large volumes of infarct tissue were observed in the I/R with vehicle group. Remarkably, IPA-3 (10 μmol/kg weight) or FRAX486 (2 μmol/kg weight) treatment significantly decreased the percentage of infarct volumes (vehicle, 39.5 ± 3.45; FRAX486, 10.4 ± 2.63; IPA-3, 19.6 ± 5.20) compared to the vehicle-treated I/R group (Figure 5B). We also evaluated the neuroprotective effect of PAK inhibitors by examining the cerebral edema and found that brain edema induced by I/R was suppressed by the treatment of IPA-3 and FRAX486 (Figure 5C). Finally, the outcome of PAK1 inhibition was also accessed by measuring the extent of the neurological deficit. In vehicle-treated I/R group, the neurological deficit score was 2.78 ± 0.34, indicating a severe neurological injury. Strikingly, inhibition of PAK1 significantly attenuated the neurological deficit compared to vehicle-treated I/R group (Figure 5D). Taken together, these results demonstrated that PAK1 inhibition could protect against tMCAO/R induced brain injury.
Figure 5.

PAK1 inhibitors attenuate the ischemic insults in mice
(A) Representative TTC-stained coronal sections.
(B) Quantification of infarct volume showing that PAK1 inhibitors protect against brain damage induced by MCAO/R 24h. Data are expressed as mean ± SEM (n = 6–7). ∗∗p < 0.01, ∗∗∗p < 0.001 versus vehicle group, one-way ANOVA with Dunnet’s post hoc test.
(C) Statistic analysis of brain edema in ischemic hemisphere. Data are expressed as mean ± SEM (n = 6–7). ∗p < 0.05, ∗∗∗p < 0.001 versus vehicle group, one-way ANOVA with Dunnet’s post hoc test.
(D) PAK1 inhibitors exert behavior relief as measured by Zea Longa score. Data are expressed as mean ± SEM (n = 9–10). ∗p < 0.05 versus vehicle group, Kruskal-Wallis test with Dunn’s post hoc test.
PAK1 inhibition reduces the formation of stress fiber in endothelial cell during OGD/R
Previous studies have reported that the formation of contractile cross-linked F-actin, i.e., stress fiber leads to dislocation of tight junction proteins,26 which may contribute to matrix metalloproteinases (MMPs)-independent barrier impairment. PAK1 has also been shown to mediate actin network rearrangement through different substrate like filamin,27 and LIMK128,29 by activating the endogenous downstream signaling molecules, such as Cdc42 and Rac1. Moreover, PAK1 is essential for spatial and temporal coordination of cytoskeletal dynamics with the function in regulation of the cellular adhesion and contraction.30,31 These results together may lead to a hypothesis that PAK1 may promote stress fiber formation of endothelial cells during OGD/R, which could be one of the molecular mechanisms underlying the endothelial hyperpermeability. In agreement with this idea, immunostaining revealed a dramatic increase of F-actin positive signals at 3 h reoxygenation after 2 h OGD in bEnd.3 (Figures 6B, 6E, and 6F), suggesting an obvious stress fiber formation was induced by OGD/R. More importantly, either PAK1 inhibitors or siRNA considerably decrease the relative MFI of F-actin compared to vehicle-treated OGD/R group. These manifest that inhibition of PAK1 causes a reduction of stress fiber formation. This phenotype and similar effects were also observed in human cerebral microvessel endothelial (hCMEC/D3) cells (Figures 6A, 6C, and 6D), which is another widely used endothelial cell model with the better morphological characteristics. Together, our discoveries suggest that PAK1 may promote the formation of stress fiber during the early stage of OGD/R.
Figure 6.

PAK1 contributes to the stress fiber formation in hCMEC/D3 and bEnd.3 cells during OGD/R
(A and B) Representative fluorescence image of F-actin staining in hCMEC/D3 and bEnd.3 cells with or without PAK1 inhibition (FRAX486, 1 μmol/L or IPA-3, 10 μmol/L). Scale bar = 20 μm.
(C and D) Statistic analysis showing reduced mean fluorescence intensity (MFI) of F-actin with PAK1 inhibitors or siRNA in hCMEC/D3 cells. Data are expressed as mean ± SEM (n = 4).
(E and F) Statistic analysis showing reduced MFI of F-actin with PAK1 inhibitors or siRNA in bEnd.3 cells. Data are expressed as mean ± SEM (n = 5). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus ctrl (C, E) or ctrl + siNC (D, F) group, #p < 0.05, ##p < 0.01, ###p < 0.001 versus OGD + vehicle (C, E) or OGD + siNC (D, F) group. All analyzed by one-way ANOVA with Sidak’s post hoc test.
PAK1 inhibition reduces the stress fiber formation during the ischemia and reperfusion in the mouse brain
Our findings about PAK1 functions in vitro BBB model and stress fiber formation imply its potential roles in blood-brain barrier disruption in stroke. We therefore examined the stress fiber formation induced by ischemia and reperfusion and tested whether PAK1 inhibition could reduce the elicited formation in the mouse brain. F-actin positive signals co-stained with isolectin B4 were first identified in the endothelial cells within the ipsilateral and contralateral areas (Figure 7A). As shown in Figures 7B and 7C, the more intensive F-actin signals were observed in the ipsilateral brain area than contralateral hemisphere of stroke mice with vehicle treatment. More strikingly, there was less F-actin positive signals in coronal sections from ischemic animals intraperitoneally administrated with IPA-3 (10 μmol/kg weight) or FRAX486 (2 μmol/kg weight) compared with tMCAO/R animals with vehicle. Together, these results suggest that PAK1 contributes to the formation of stress fiber during focal ischemia/reperfusion.
Figure 7.

Stress fiber formation on ischemic microvessels is decreased by the administration of PAK1 inhibitors
(A) Immunofluorescence images showing the colocalization of F-actin (green) with microvessel markers IB4 (red) in ipsilateral and contralateral hemisphere from control mice. Scale bar = 20 μm.
(B) The F-actin fluorescence of the ipsilateral and contralateral hemisphere in the same coronal sections from ischemic mice with or without PAK1 inhibition (FRAX486, 2 μmol/kg weight or IPA-3, 10 μmol/kg weight). To show a length of full vessel, the 7 μm thick z stack was acquired and processed maximum intensity projection. Scale bar = 20 μm.
(C) Quantitative analysis showing the much more obvious signal of F-actin MFI on region of interest (ROI) in ipsilateral hemisphere compared to contralateral hemisphere. FRAX486 (2 μmol/kg weight) or IPA-3 (10 μmol/kg weight) treatment reduced F-actin MFI on ipsilateral hemisphere. Data are expressed as mean ± SEM (n = 3). ∗∗∗p < 0.001 versus contralateral + vehicle; ##p < 0.01, ###p < 0.001 versus ipsilateral + vehicle, two-way ANOVA with Turkey’s post hoc test.
Discussion
Better understanding of the molecular mechanisms of stroke is the key for developing the new treatment strategies. Although limited evidence implies the potential role of PAK1 in post-stroke recovery, here by using tMCAO/R mouse and in vitro endothelial model, combined with a series biochemical, immunohistochemical and molecular assays, we demonstrated that PAK1 is responsible for the endothelial hyperpermeability in the acute phase of stroke potentially through regulating the junctional protein membrane localization and stress fiber formation. Our data suggest a key pathogenic role of PAK1 in regulating the BBB permeability during the early stage of stroke insults.
BBB alterations play a key role in the pathogenesis of many central nervous system diseases, such as ischemic stroke, tumors, HIV-1 encephalitis, multiple sclerosis, Alzheimer’s disease, and Parkinson’s disease.6,7,32 As one of the series of dynamically pathophysiological events, BBB disruption occurs soon after the initial blood flow interruption or severe reduction and lasts through the different progression and recovery stages. Consistent with previous findings,26,33 the BBB hyperpermeability or disruption was detected in the acute stage of reperfusion in our tMCAO/R mouse model. We have also found that PAK1 inhibition by either inhibitors or siRNA reduces endothelial hyperpermeability. Thus, our observations further support the idea that blocking BBB open could prevent BBB dysfunction and protect against the brain injury during the stroke.34
In mammals, PAK1 is widely expressed in the brain, muscle, spleen, pancreas, and other organs.35,36 Moreover, emerging reports suggest that PAK1 expression extends to neurons and oligodendrocytes as well.37,38 Here, our data suggests that endothelial PAK1 contributes to the increased permeability of blood vessels in the endothelium upon OGD/R, potentially by regulating the localization of junctional proteins. Importantly, we observed that inhibition of PAK1 could ameliorate the blood-brain barrier disruption during the ischemia and reperfusion in mouse stroke model. Although the positive impact of PAK1 inhibitors may not solely be a result of inhibiting PAK1 in the blood vessels, our observations suggest that PAK1 plays a role in this process. These results are in line with our findings from in vitro experiments. Additionally, our further analysis is consistent with the PAK1 function for cytoskeletal regulation. Together, these discoveries support the notion that endothelial PAK1 could play a role during early phase of brain injury after cerebral ischemia via regulating the blood-brain barrier permeability. Further experiments are necessary to validate this point and dissect its underlying cellular mechanisms.
As one of the primary autophosphorylation sites that control enzymatic activity, the PAK1 ser-144 phosphorylation prevents auto inhibitory domain packing against kinase domain, which plays pivotal roles in PAK1 activation.39,40 In our study, we found a rapid increase of p-PAK1 (Ser144) in both in vivo and in vitro conditions, which suggests that PAK1 is activated at the early stage of reperfusion. This type of phosphorylation within the first 250 amino acids could prevent the kinase from reverting to an inactive conformation. Future study that can measure the details of PAK1 phosphorylation and activity during the different stages of stoke is essential to deeply understand the PAK1 function. In cultured mouse cerebral endothelial cells (bEnd.3), we observed acute upregulation of PAK1 protein following OGD and reoxygenation. This may suggest the PAK1 level and function changes occur early during the processes of stroke insults. However, the PKA1 protein level had no significant changes during reperfusion after focal ischemia in mice. One possible explanation for the discrepancy between in vitro and in vivo study could be the relatively lower percentages of PAK1 positive cells within examined tissues, this observed phenomenon has also been reported in studies involving stroke patients and rats undergoing middle cerebral artery occlusion (MCAO).41,42 Our findings align with these studies, providing additional support for the significant role of PAK1 in the context of stroke. Although the exact effects of PAK1 inhibitors require further investigation in future studies, our preliminary data provide a piece of evidence that the reduction of the endothelial hyperpermeability by PAK1 inhibition may be responsible for the amelioration of the brain injury in mice induced by tMCAO/R. Collectively, our findings further suggest the interesting roles of endothelial PAK1 during the brain injury and recovery.
Our data suggest that stress fiber formation at the early stage is involved in the dynamic changes of endothelial permeability or the barrier integrity. These findings are consistent with the known PAK1 function as an important regulator of cytoskeleton.31 In this regard, as a downstream kinase of multiple extracellular stimuli, PAK1 mainly regulate microfilament through various substrates.27,43,44 Additionally, this role of PAK1 could be one of the potential molecular mechanisms for PAK1 inhibition, which attenuates the BBB alteration during the ischemia and reperfusion. Based on these findings, the next interesting work will be to explore the potential substrates after PAK1 activation which increase the stress fiber formation during ischemia and reperfusion.
Considering the multiple biological functions of PAK1 in cellular process and the pathogenic roles in various diseases, targeting PAKs is becoming a really promising therapeutic strategy. Natural PAKs blockers has been extracted from plants and studied.45,46,47 Moreover, many PAKs inhibitors have been identified or developed for preclinical investigation and clinical trials. However, there are still some important gaps in the process of comprehensive understanding and applying the PAKs inhibition approaches for human disease treatment. In this regard, it is crucial to clarify the exact function and detailed molecular mechanisms in biological and pathological processes for most PAKs. In this work, we identified a possible pathological role of PAK1 at the very early stage of stroke. Further efforts are required to gain a better understanding of the independent roles of PAK1 in different stages of stroke, given the diversity of the roles of Rac1 and the long duration of post-stroke recovery. Moreover, development of the specific PAK inhibitors remains significant challenges on account of the high homology of PAKs at the kinase domain. This fact may also cause pan-PAK inhibition and the off-target effects. Similarly, the effects of PAK1 inhibitors in our cellular and mouse model of stroke may extend far beyond any single function of PAK1, such as BBB protection, glucose metabolism, inflammation, autophagy regulation, apoptosis, and neurotransmission. However, with the in vitro results our findings informed the therapeutic potentials of PAK1 inhibition, future investigation and test are needed to rigorously explore this idea.
Limitations of the study
Our study suggests that endothelial PAK1 plays a role during early phase of brain injury after cerebral ischemia via regulating the blood-brain barrier permeability. Further studies are essential to dissect the molecular mechanism of which PAK1 regulates blood-brain barrier integrity and the roles of PAK1 at the different stages of disease. Since the expression and function of PAK1 is complicated, it is worth investigating the effects of PAK1 inhibition on other cell types involved in stroke pathophysiology, such as neurons and glial cells. To establish the potential therapeutic benefits of targeting PAK1 for stroke treatment, it is crucial to assess the long-term impact of PAK1 inhibition on stroke recovery and functional outcomes. Additionally, it is imperative to investigate whether PAK1 inhibition elicits any side effects or off-target effects.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-PAK1 | Cell Signaling Technology | Cat#2602; RRID: AB_330222 |
| Rabbit polyclonal anti-p-PAK1 (Ser144) | Cell Signaling Technology | Cat#2606; RRID: AB_2299279 |
| Mouse anti-GAPDH | Proteintech | Cat# 60004; RRID: AB_2107436 |
| Rabbit monoclonal anti-PAK1 | Abclonal | Cat#A19608; RRID: AB_2862697 |
| Rabbit anti-ve-cadherin | Abclonal | Cat#A0734; RRID: AB_2757370 |
| Rabbit anti-occludin | Invitrogen | Cat#71-1500; RRID: AB_2533977 |
| Rabbit anti-ZO-1 | Invitrogen | Cat#40-2200; RRID: AB_2533456 |
| HRP conjugated goat anti-rabbit | Millipore | Cat# AP307P, RRID: AB_92641 |
| HRP conjugated goat anti-mouse | Millipore | Cat# AP308P, RRID: AB_11215796 |
| Alexa Fluor 488 conjugated goat anti-rabbit | Invitrogen | Cat#A32731; RRID: AB_2633280 |
| Chemicals, peptides, and recombinant proteins | ||
| DyLight 594-conjugated lectin | Vector labs | Cat#DL-1177-1 |
| DyLight 594-conjugated GSL-isolectin B4 | Vector labs | Cat#DL1207 |
| FITC-dextran, 70 kDa | Sigma-Aldrich | Cat#53471 |
| TRITC-dextran, 4.4 kDa | Sigma-Aldrich | Cat#T1037 |
| Fluorescein (FITC)-phalloidin | Sigma-Aldrich | Cat#P5282 |
| 2,3,5-triphenyltetrazolium hydrochloride (TTC) | Sigma-Aldrich | Cat#T8877 |
| FRAX486 | Selleck | Cat#S7807 |
| IPA-3 | Selleck | Cat#S7093 |
| Critical commercial assays | ||
| IP lysis buffer | Beyotime | Cat#P0013 |
| BCA protein assay kit | Beyotime | Cat#P0010 |
| Protease inhibitor cocktails | Absin | Cat#ABS9161 |
| Phosphatase inhibitor cocktails | Absin | Cat#ABS9162 |
| Sulfo-NHS-SS-Biotin | Thermo Fisher Scientific | Cat#21331 |
| Neutravidin agarose | Thermo Fisher Scientific | Cat#29202 |
| Lipo8000 | Beyotime | Cat#C0533 |
| Cell counting kit 8 (CCK8) | APExBIO | Cat#K1018 |
| Experimental models: Cell lines | ||
| bEnd.3 | ATCC | N/A |
| hCMEC/D3 | ATCC | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: wild type: C57BL/6J | Xuzhou Medical University | N/A |
| Oligonucleotides | ||
| siRNA targeting PAK1 | Synthetized by GenePharma | 5’-GCAUCAAUUCC UGAAGAUUTT-3’ |
| Negative control of siRNA | Synthetized by GenePharma | 5’-UUCUCCGAACG UGUCACGUTT-3’ |
| Software and algorithms | ||
| Prism 7.0 | Graphpad | http://www.graphpad.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
Resources availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dejun Yang (dejun.yang@xzhmu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Date and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Animals
All animal experimental procedures were reviewed and approved (Approval ID: L20210226149) by the Laboratory Animal Center of Xuzhou Medical University (License key: SYXK (su) 2020-0048). C57BL/6J mice are purchased from the Animal Center of Xuzhou Medical University and housed in a 12 h dark/light cycle with free access to water and food before surgery. Male mice between 10 to 14 weeks of age were used for following experiments. Totally, 79 mice were used, including 6 sham-operated mice and 65 mice for MCAO, also 8 mice were excluded in analysis under the following situations: rCBF decreasing was less than 70%, died from cerebral thrombosis or hemorrhage, no behavior deficits after awakening.
Method details
Middle cerebral artery occlusion and reperfusion model
The focal cerebral ischemia was induced by transient middle cerebral artery occlusion (tMCAO).21,48 Briefly, after anaesthetized with 1.5% pentobarbital sodium (dissolved in saline, 6 mL/kg weight), the mice were fixed on a plate and further surgery was performed under stereoscope (SZ61, Olympus, Tokyo, Japan). After a midline neck incision, the right common carotid artery (CCA), external carotid artery (ECA) and internal carotid artery (ICA) were separated and exposed. While the proximal CCA and ECA were clamped, a 6-0 nylon monofilament with silicone gel coated tip (Jialing, Guangzhou, China) was inserted through CCA, and advanced via ICA gently till resistance. The tip of the filament was about 1 cm away from the bifurcation of internal and external carotid artery. Regional cerebral blood flow (rCBF) was measured during occlusion using Laser speckle flowmeter (FLPI-2, Moor instruments, UK). Vehicle (2% ethanol in saline, 10 mL/kg weight), FRAX486 (2 μmol/kg weight) and IPA-3 (10 μmol/kg weight) were injected immediately after successful surgical operation. After 60 min, the mice were anesthetized, and the occluding filament was removed to allow reperfusion. Sham-operated mice received the same surgical procedures except for the occlusion of the ICA. The mice were excluded in further analysis under the following situations: rCBF decreasing was less than 70%, died from cerebral thrombosis or hemorrhage, no behavior deficits after awakening.
Neurological deficit score assessment
The behavior deficits were assessed at 24 h of tMCAO/R by an observer who was blind to the experimental design. The scoring system includes five evaluation indicators as described49: 0, no visible behavior changes; 1, failed to fully extend the left forelimb; 2, spontaneous circling; 3, collapse to left side or spontaneous rotation progression into barreling; 4, unable to move freely or loss of consciousness. Higher scores indicate more severe behavioral deficits.
Infarct volume and brain edema
TTC staining was performed to evaluate the cerebral infarct volume. After neurological deficit scores were assessed, the mice were sacrificed under anesthesia. Brains were harvested and cut into 1 mm thick coronal sections on mice brain mold (RWD, Shenzhen, China). Then the sections were stained with 2% TTC for 30 min at 37 °C. For the convenience of taking pictures, the stained brain sections were fixed with 4% paraformaldehyde (PFA) overnight. The infarct areas and brain edema were calculated using ImageJ software. Infarct volume was calculated as ∑ (contralateral hemisphere area-ipsilateral normal area)/∑contralateral hemisphere area×100%. Brain edema was calculated as ∑ (ipsilateral hemisphere area- contralateral hemisphere area)/∑contralateral hemisphere area×100%.
Evaluation of BBB permeability in vivo
To evaluate the BBB permeability, 70 kDa FITC-dextran (1.5 mg per mice) and 4.4 kDa TRITC-dextran (0.5mg per mice) were used as a marker according to a previous study.23 In brief, the fluorescent dye (diluted in saline, 0.1 mL per mice) was injected at 3 h or 24 h after reperfusion via the tail vein. The mice were sacrificed 30 min later under anesthesia, and the collected brains were embedded rapidly. Six coronal sections of each brain were prepared with 50 μm thick and 1 mm interval. The fluorescence images of each brain section were acquired by fluorescent microscope (Observer Z1, Zeiss, Oberkochen, Germany) with tile scan mode. The FITC or TRITC leakage in each section was measured by using Image J software. The total leakage volume of fluorescent dye was further calculated following the next equation: leakage volume (%) =∑leakage area /∑ hemisphere area×100%.
Immunofluorescence
Anesthetized mice were transcardially perfused with PBS followed by 4% PFA (dissolved in PBS). Harvested brains were incubated in 4% PFA at 4°C overnight and then dehydrated in 30% sucrose till sedimentation. Brain blocks were sliced (40 μm) with cyrostat microtome (CM1850, Leica), and stored in anti-freezing buffer (30% ethylene glycol, 20% glycerol in PBS) at 4°C. Sections were permeabilized with 0.3% Triton X-100 and blocked with blocking buffer (10% goat serum, 300 mM glycine, 0.3% Triton X-100, dissolved in PBS) for 1 hour, then incubated with rabbit anti-PAK1 antibody (1:50, Abclonal) for 3 h at room temperature (RT). The sections were washed 3 times with tris-buffered saline for 5 min each time. Then incubated with Alexa Flour 488-conjugated goat anti-rabbit antibody (1:800) for 1 h at RT, washed 5 times with tris-buffered saline for 10 min each time. The brain microvessel were stained with DyLight 594-conjugated GSL-isolectin B4 (IB4, 1:50) or DyLight 594-conjugated lectin (1:200). Finally, the nucleus was stained with 4,6-diamidino-2-phenylindole dihydrochloride (DAPI, 1:1000). The fluorescent images were captured with confocal microscope (LSM 710, Zeiss).
Oxygen-glucose deprivation (OGD)
Mouse brain microvascular endothelial cell line bEnd.3 cells (ATCC, Rockefeller, Maryland, USA) and human microvascular endothelial cell line hCMEC/D3 cells (ATCC) were cultured in high glucose DMEM supplemented with 10% fetal bovine serum in an incubator containing 5% CO2 (Thermo Scientific) at 37 °C. The cultured cells were washed twice, replaced medium to no-glucose DMEM, and then transferred into an incubator pre-equilibrated with 95% N2 and 5% CO2. After 2 hours deprivation, the medium was replaced by standard culture medium, and cultures were returned to the normoxic incubator. Control groups were washed twice, and medium was replaced with high glucose DMEM for the same time, then cultured in the normoxic incubator. The PAK1 inhibitors FRAX486 (1 μmol/L) and IPA-3 (10 μmol/L) were added during and after OGD.
Endothelial cell monolayer permeability assay
The bEnd.3 were seeded 3×104 per well on the upper side of transwell PET membranes (0.4 μm pore, 6.5 mm diameter; Corning, Lowell, Massachusetts, USA), which inserted into 24-well culture plate. Cells were maintained in DMEM supplemented with 10% fetal bovine serum in 5% CO2 incubators for 4 days to reach confluence. After OGD treatment, 4.4 kDa TRITC-dextran and 70 kDa FITC-dextran were added into the luminal chamber (insert) at the concentration of 0.2 g/L in a total 100 μL media. The abluminal chamber (lower) was filled with 1 mL phenol red free DMEM. To assess the tracer diffusion, 100 μL media were acquired from abluminal chamber at 1, 3 and 6 hours after OGD/reoxygenation (OGD/R). And the fluorescent intensity in abluminal chamber was measured by fluorescence microplate reader (Glomax Discover, Promega, Madison, Wisconsin, USA). The concentrations of tracers were calculated from a standard curve, which fitted by using known concentrations of same tracers separately. To assess the diffusion rate from the luminal to the abluminal chamber through endothelial monolayers at 3 h reoxygenation, the tracers were added at 3 h after OGD/R and the tracer concentration in abluminal chamber was measured 0.5 h later. The diffusion rate was represented as apparent diffusion coefficient, which calculated as described previously50: Apparent diffusion coefficient (cm∗s-1) = CabVab/(C0∗S∗T). Where Cab is the concentration of tracer in abluminal chamber, Vab is the volume of medium in abluminal chamber, C0 is the primary concentration of tracer in luminal chamber, S is the area of microporous membrane, T is the duration time from adding tracers to moving medium for detection.
F- actin staining
After OGD treatment, the cells were washed twice with PBS, blocked with 3% BSA, and incubated with FITC-phalloidin (1 μg/mL) for 0.5 hour. For brain sections, slices were blocked with 3% BSA dissolved in PBS for 1 hour, then incubated with FITC-phalloidin (5 μg/mL) at 4°C overnight. The coverslips or sections were counter stained with DAPI (1:1000) before the fluorescent images were captured with confocal laser scanning microscope (LSM 710, Zeiss). To analyze the fluorescence intensity on cells, at least three regions of interest (ROI) were randomly selected on each coverslip and the mean fluorescent intensity (MFI) was calculated with Image J. Brain blocks were cut into 7 μm sections for measuring the fluorescence intensity on blood vessels. Stained slices were imaged with confocal laser scanning microscope (LSM 710, Zeiss) with Z-stack method, ROI was randomly selected on microvessels shown in maximum intensity projection images and MFI was calculated using Image J. For analyzing the MFI of F-actin on blood vessels, 3 sections were chosen for each mouse, and at least six ROI were selected on each brain section.
Western blotting
Used a similar approach for separating the ischemic cortex,51,52 a 4mm thick coronal block was cut from the mouse brain, which containing infarct core area (has color difference with surrounding tissues). In ischemic side, the cortex at an angle of about -60 to 30 degrees horizontally from the center of the block was separated as infarct cortex. Cells or brain tissues were homogenized in ice-cold IP lysis buffer (Beyotime) containing protease and phosphatase inhibitors. The protein concentration was determined using the BCA protein assay kit. Protein samples were separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and then electrotransferred to nitrocellulose membrane (0.22 μm). After blocking with 3 % BSA for about 2 h, the membrane was incubated with the indicated primary antibodies and then with horseradish peroxidase-conjugated secondary antibodies. The membrane was washed three times and detected by ECL detection solution (Millipore). The bands were visualized in an automatic chemiluminescence imaging system (Protein Simple, Pluorchem E) and analyzed with Image J software.
Biotinylation and isolation of cell surface proteins
Briefly, Cells cultured in 60 mm dishes were washed twice with ice-cold PBS before biotinylation. The cells were incubated with Sulfo-NHS-SS-Biotin (0.4mg, dissolved in PBS) at 4°C for 30 min. Then the cells were washed with 50 mM Tris (pH 8.0) to quench non-reacted biotin and washed twice with PBS. After biotinylation, the cells lysate was prepared as described above and incubated with Neutravidin agarose resin at 4°C overnight. After washed with 0.2% tween-20, the biotinylated surface proteins were eluted with SDS-PAGE sample buffer, then analyzed by Western blotting.
RNA interference of PAK1
The bEnd.3 cells at 70% confluence were transfected with PAK1 siRNA and siNC (synthetized by GenePharma) using Lipo8000 (Beyotime) according to the manufacturer’s instructions. After 48 h of transfection, the cells were treated with OGD or other experiment. The sequences are as follows: PAK1 siRNA: 5’-GCAUCAAUUCCUGAAGAUUTT-3’; Negative control (NC): 5’-UUCUCCGAACGUGUCACGUTT-3’.
Cell viability
Cell viability of bEnd.3 after OGD/R were measured by cell counting kit-8 (CCK-8, APExBIO, Cat#K1018) following instruction of manuals. Before the test, cells were cultured on 96-well plate and subjected to OGD as previously mentioned. After oxygenation for 3 h or 24 h, 10 μL CCK-8 reagent was added in 100 μL medium per well. After incubation in 37°C for 1 h, OD value was detected on spectrophotometric measurements at 450 nm.
Quantification and statistical analysis
All data were statistically analyzed using GraphPad Prism 7 (GraphPad software, San Diego, CA, USA). The results were represented as mean ± SEM. The normality of the data was evaluated by a Shapiro-Wilk test. Comparisons between two groups were evaluated by Student's t test. Comparisons between three or more groups were evaluated by one-way analysis of variance (ANOVA) with Dunnett’s or Sidak’s post hoc test. Comparisons with two independent variables were evaluated by two-way ANOVA with Turkey’s post hoc test. Particularly, non-parametric data (neurological score) or non-normality data were evaluated by Kruskal-Wallis test with Dunn’s post hoc test. Statistical significance was considered as appropriate p value (∗p < 0.05, ∗∗p < 0.01, ∗∗∗ p < 0.001).
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NSFC; 82171348 to DY), the Research Foundation of Xuzhou Medical University (DY and MH), and an Open Competition Grant of Xuzhou Medical University (DY).
Author contributions
Conceptualization, M.H., Q.Z., and D.Y.; Methodology, M.H., Q.Z., and D.Y.; Investigation, M.H., J.Z., M.L., and H.C.; Resource, M.H., Q.Z., and D.Y.; Writing—Original Draft, M.H. and D.Y.; Writing—Review & Editing, M.H., Q.Z., and D.Y.; Funding Acquisition, M.H. and D.Y.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: July 11, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107333.
Contributor Information
Qiuju Zhu, Email: zhuqiuju@xzhmu.edu.cn.
Dejun Yang, Email: dejun.yang@xzhmu.edu.cn.
Supplemental information
References
- 1.GBD 2016 Lifetime Risk of Stroke Collaborators. Feigin V.L., Nguyen G., Cercy K., Johnson C.O., Alam T., Parmar P.G., Abajobir A.A., Abate K.H., Abd-Allah F., et al. Global, Regional, and Country-Specific Lifetime Risks of Stroke, 1990 and 2016. N. Engl. J. Med. 2018;379:2429–2437. doi: 10.1056/NEJMoa1804492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campbell B.C.V., Khatri P. Stroke. Lancet. 2020;396:129–142. doi: 10.1016/S0140-6736(20)31179-X. [DOI] [PubMed] [Google Scholar]
- 3.Vermeer S.E., Prins N.D., den Heijer T., Hofman A., Koudstaal P.J., Breteler M.M.B. Silent brain infarcts and the risk of dementia and cognitive decline. N. Engl. J. Med. 2003;348:1215–1222. doi: 10.1056/NEJMoa022066. [DOI] [PubMed] [Google Scholar]
- 4.Lei C., Deng Q., Li H., Zhong L. Association Between Silent Brain Infarcts and Cognitive Function: A Systematic Review and Meta-Analysis. J. Stroke Cerebrovasc. Dis. 2019;28:2376–2387. doi: 10.1016/j.jstrokecerebrovasdis.2019.03.036. [DOI] [PubMed] [Google Scholar]
- 5.Bernardo-Castro S., Sousa J.A., Brás A., Cecília C., Rodrigues B., Almendra L., Machado C., Santo G., Silva F., Ferreira L., et al. Pathophysiology of Blood-Brain Barrier Permeability Throughout the Different Stages of Ischemic Stroke and Its Implication on Hemorrhagic Transformation and Recovery. Front. Neurol. 2020;11 doi: 10.3389/fneur.2020.594672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sweeney M.D., Zhao Z., Montagne A., Nelson A.R., Zlokovic B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019;99:21–78. doi: 10.1152/physrev.00050.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liebner S., Dijkhuizen R.M., Reiss Y., Plate K.H., Agalliu D., Constantin G. Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol. 2018;135:311–336. doi: 10.1007/s00401-018-1815-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langen U.H., Ayloo S., Gu C. Development and Cell Biology of the Blood-Brain Barrier. Annu. Rev. Cell Dev. Biol. 2019;35:591–613. doi: 10.1146/annurev-cellbio-100617-062608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jickling G.C., Liu D., Stamova B., Ander B.P., Zhan X., Lu A., Sharp F.R. Hemorrhagic transformation after ischemic stroke in animals and humans. J Cerebr Blood F Met. 2014;34:185–199. doi: 10.1038/jcbfm.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao M., Xiao Z.J., Yang B., Lan Z., Fang F. Blood-Brain Barrier: More Contributor to Disruption of Central Nervous System Homeostasis Than Victim in Neurological Disorders. Front. Neurosci. 2020;14:764. doi: 10.3389/fnins.2020.00764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J., Wang K., Qi J., Cao X., Wang F. The Hsp90 Inhibitor 17-DMAG Attenuates Hyperglycemia-Enhanced Hemorrhagic Transformation in Experimental Stroke. BioMed Res. Int. 2021;2021 doi: 10.1155/2021/6668442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morrice N.A., Gabrielli B., Kemp B.E., Wettenhall R.E. A Cardiolipin-Activated Protein-Kinase from Rat-Liver Structurally Distinct from the Protein-Kinases-C. J. Biol. Chem. 1994;269:20040–20046. doi: 10.1016/s0021-9258(17)32124-5. [DOI] [PubMed] [Google Scholar]
- 13.Yao D., Li C., Rajoka M.S.R., He Z., Huang J., Wang J., Zhang J. P21-Activated Kinase 1: Emerging biological functions and potential therapeutic targets in Cancer. Theranostics. 2020;10:9741–9766. doi: 10.7150/thno.46913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu H., Liu K., Dong Z. The Role of p21-Activated Kinases in Cancer and Beyond: Where Are We Heading? Front. Cell Dev. Biol. 2021;9:641381. doi: 10.3389/fcell.2021.641381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Q., Wu X., Guo L., Shi J., Li J. MicroRNA-7-5p induces cell growth inhibition, cell cycle arrest and apoptosis by targeting PAK2 in non-small cell lung cancer. FEBS Open Bio. 2019;9:1983–1993. doi: 10.1002/2211-5463.12738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee J.S., Mo Y., Gan H., Burgess R.J., Baker D.J., van Deursen J.M., Zhang Z. Pak2 kinase promotes cellular senescence and organismal aging. Proc. Natl. Acad. Sci. USA. 2019;116:13311–13319. doi: 10.1073/pnas.1903847116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meng J., Meng Y., Hanna A., Janus C., Jia Z. Abnormal long-lasting synaptic plasticity and cognition in mice lacking the mental retardation gene Pak3. J. Neurosci. 2005;25:6641–6650. doi: 10.1523/JNEUROSCI.0028-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Radu M., Semenova G., Kosoff R., Chernoff J. PAK signalling during the development and progression of cancer. Nat. Rev. Cancer. 2014;14:13–25. doi: 10.1038/nrc3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu C., Tang M., Zhang X., Li J., Cao G. Knockdown of miR-665 Protects Against Cardiomyocyte Ischemia/Reperfusion Injury-Induced ROS Accumulation and Apoptosis Through the Activation of Pak1/Akt Signaling in Myocardial Infarction. Int. Heart J. 2020;61:347–354. doi: 10.1536/ihj.19-416. [DOI] [PubMed] [Google Scholar]
- 20.Egom E.E.A., Mohamed T.M.A., Mamas M.A., Shi Y., Liu W., Chirico D., Stringer S.E., Ke Y., Shaheen M., Wang T., et al. Activation of Pak1/Akt/eNOS signaling following sphingosine-1-phosphate release as part of a mechanism protecting cardiomyocytes against ischemic cell injury. Am. J. Physiol. Heart Circ. Physiol. 2011;301:H1487–H1495. doi: 10.1152/ajpheart.01003.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bu F., Min J.W., Munshi Y., Lai Y.J., Qi L., Urayama A., McCullough L.D., Li J. Activation of endothelial ras-related C3 botulinum toxin substrate 1 (Rac1) improves post-stroke recovery and angiogenesis via activating Pak1 in mice. Exp. Neurol. 2019;322:113059. doi: 10.1016/j.expneurol.2019.113059. ARTN 113059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang W., Zhu L., An C., Wang R., Yang L., Yu W., Li P., Gao Y. The blood brain barrier in cerebral ischemic injury – Disruption and repair. Brain Hemorrhages. 2020;1:34–53. doi: 10.1016/j.hest.2019.12.004. [DOI] [Google Scholar]
- 23.Shi Y., Jiang X., Zhang L., Pu H., Hu X., Zhang W., Cai W., Gao Y., Leak R.K., Keep R.F., et al. Endothelium-targeted overexpression of heat shock protein 27 ameliorates blood-brain barrier disruption after ischemic brain injury. Proc. Natl. Acad. Sci. USA. 2017;114:E1243–E1252. doi: 10.1073/pnas.1621174114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nuche-Berenguer B., Ramos-Álvarez I., Jensen R.T. The p21-activated kinase, PAK2, is important in the activation of numerous pancreatic acinar cell signaling cascades and in the onset of early pancreatitis events. Biochim. Biophys. Acta. 2016;1862:1122–1136. doi: 10.1016/j.bbadis.2016.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deacon S.W., Beeser A., Fukui J.A., Rennefahrt U.E.E., Myers C., Chernoff J., Peterson J.R. An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem. Biol. 2008;15:322–331. doi: 10.1016/j.chembiol.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi Y., Zhang L., Pu H., Mao L., Hu X., Jiang X., Xu N., Stetler R.A., Zhang F., Liu X., et al. Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat. Commun. 2016;7 doi: 10.1038/ncomms10523. ARTN 10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vadlamudi R.K., Li F., Adam L., Nguyen D., Ohta Y., Stossel T.P., Kumar R. Filamin is essential in actin cytoskeletal assembly mediated by p21-activated kinase 1. Nat. Cell Biol. 2002;4:681–690. doi: 10.1038/ncb838. [DOI] [PubMed] [Google Scholar]
- 28.Bernard O. Lim kinases, regulators of actin dynamics. Int. J. Biochem. Cell Biol. 2007;39:1071–1076. doi: 10.1016/j.biocel.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 29.Bernstein B.W., Bamburg J.R. ADF/cofilin: a functional node in cell biology. Trends Cell Biol. 2010;20:187–195. doi: 10.1016/j.tcb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar R., Gururaj A.E., Barnes C.J. p21-activated kinases in cancer. Nat. Rev. Cancer. 2006;6:459–471. doi: 10.1038/nrc1892. [DOI] [PubMed] [Google Scholar]
- 31.Rane C.K., Minden A. P21 activated kinases: structure, regulation, and functions. Small GTPases. 2014;5 doi: 10.4161/sgtp.28003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sweeney M.D., Sagare A.P., Zlokovic B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018;14:133–150. doi: 10.1038/nrneurol.2017.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simpkins A.N., Dias C., Leigh R., National Institutes of Health Natural History of Stroke Investigators Identification of Reversible Disruption of the Human Blood-Brain Barrier Following Acute Ischemia. Stroke. 2016;47:2405–2408. doi: 10.1161/STROKEAHA.116.013805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang X., Andjelkovic A.V., Zhu L., Yang T., Bennett M.V.L., Chen J., Keep R.F., Shi Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018;163-164:144–171. doi: 10.1016/j.pneurobio.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hofmann C., Shepelev M., Chernoff J. The genetics of Pak. J. Cell Sci. 2004;117:4343–4354. doi: 10.1242/jcs.01392. [DOI] [PubMed] [Google Scholar]
- 36.Kelly M.L., Chernoff J. Mouse models of PAK function. Cell. Logist. 2012;2:84–88. doi: 10.4161/cl.21381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown T.L., Hashimoto H., Finseth L.T., Wood T.L., Macklin W.B. PAK1 Positively Regulates Oligodendrocyte Morphology and Myelination. J. Neurosci. 2021;41:1864–1877. doi: 10.1523/jneurosci.0229-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bu F., Munshi Y., Furr J.W., Min J.W., Qi L., Patrizz A., Spahr Z.R., Urayama A., Kofler J.K., McCullough L.D., Li J. Activation of neuronal Ras-related C3 botulinum toxin substrate 1 (Rac1) improves post-stroke recovery and axonal plasticity in mice. J. Neurochem. 2021;157:1366–1376. doi: 10.1111/jnc.15195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chong C., Tan L., Lim L., Manser E. The mechanism of PAK activation - Autophosphorylation events in both regulatory and kinase domains control activity. J. Biol. Chem. 2001;276:17347–17353. doi: 10.1074/jbc.M009316200. [DOI] [PubMed] [Google Scholar]
- 40.Mayhew M.W., Jeffery E.D., Sherman N.E., Nelson K., Polefrone J.M., Pratt S.J., Shabanowitz J., Parsons J.T., Fox J.W., Hunt D.F., Horwitz A.F. Identification of phosphorylation sites in betaPIX and PAK1. J. Cell Sci. 2007;120:3911–3918. doi: 10.1242/jcs.008177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitsios N., Saka M., Krupinski J., Pennucci R., Sanfeliu C., Wang Q., Rubio F., Gaffney J., Kumar P., Kumar S., et al. A microarray study of gene and protein regulation in human and rat brain following middle cerebral artery occlusion. BMC Neurosci. 2007;8:93. doi: 10.1186/1471-2202-8-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li M., Wen Y., Zhang R., Xie F., Zhang G., Qin X. Adenoviral vector-induced silencing of RGMa attenuates blood-brain barrier dysfunction in a rat model of MCAO/reperfusion. Brain Res. Bull. 2018;142:54–62. doi: 10.1016/j.brainresbull.2018.06.010. [DOI] [PubMed] [Google Scholar]
- 43.Edwards D.C., Sanders L.C., Bokoch G.M., Gill G.N. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat. Cell Biol. 1999;1:253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- 44.Zhang H., Webb D.J., Asmussen H., Niu S., Horwitz A.F. A GIT1/PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. J. Neurosci. 2005;25:3379–3388. doi: 10.1523/JNEUROSCI.3553-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ryu B.J., Kim S., Min B., Kim K.Y., Lee J.S., Park W.J., Lee H., Kim S.H., Park S. Discovery and the structural basis of a novel p21-activated kinase 4 inhibitor. Cancer Lett. 2014;349:45–50. doi: 10.1016/j.canlet.2014.03.024. [DOI] [PubMed] [Google Scholar]
- 46.Shahinozzaman M., Ishii T., Ahmed S., Halim M.A., Tawata S. A computational approach to explore and identify potential herbal inhibitors for the p21-activated kinase 1 (PAK1) J. Biomol. Struct. Dyn. 2020;38:3514–3526. doi: 10.1080/07391102.2019.1659855. [DOI] [PubMed] [Google Scholar]
- 47.Qin Q., Wu T., Yin W., Sun Y., Zhang X., Wang R., Guo J., Zhao D., Cheng M. Discovery of 2,4-diaminopyrimidine derivatives targeting p21-activated kinase 4: Biological evaluation and docking studies. Arch. Pharm. 2020;353 doi: 10.1002/ardp.202000097. [DOI] [PubMed] [Google Scholar]
- 48.Tuo Q.Z., Lei P., Jackman K.A., Li X.L., Xiong H., Li X.L., Liuyang Z.Y., Roisman L., Zhang S.T., Ayton S., et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry. 2017;22:1520–1530. doi: 10.1038/mp.2017.171. [DOI] [PubMed] [Google Scholar]
- 49.Zhang B., Zhang H.X., Shi S.T., Bai Y.L., Zhe X., Zhang S.J., Li Y.J. Interleukin-11 treatment protected against cerebral ischemia/reperfusion injury. Biomed. Pharmacother. 2019;115 doi: 10.1016/j.biopha.2019.108816. ARTN 108816. [DOI] [PubMed] [Google Scholar]
- 50.Tominaga N., Kosaka N., Ono M., Katsuda T., Yoshioka Y., Tamura K., Lötvall J., Nakagama H., Ochiya T. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood-brain barrier. Nat. Commun. 2015;6:6716. doi: 10.1038/ncomms7716. ARTN 6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shu L., Chen B., Chen B., Xu H., Wang G., Huang Y., Zhao Y., Gong H., Jiang M., Chen L., et al. Brain ischemic insult induces cofilin rod formation leading to synaptic dysfunction in neurons. J. Cerebr. Blood Flow Metabol. 2019;39:2181–2195. doi: 10.1177/0271678x18785567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bonova P., Burda J., Danielisova V., Nemethova M., Gottlieb M. Development of a pattern in biochemical parameters in the core and penumbra during infarct evolution after transient MCAO in rats. Neurochem. Int. 2013;62:8–14. doi: 10.1016/j.neuint.2012.10.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.