

Abstract
The cellular pathways of apoptosis have not been fully characterized; however, calpain, a cytosolic calcium-activated cysteine protease, has been implicated in several forms of programmed cell death. Reoviruses induce apoptosis both in vitro and in vivo and serve as a model for studying virus-induced cell death. We investigated the potential role of calpain in reovirus-induced apoptosis in vitro by measuring calpain activity as well as evaluating the effects of calpain inhibitors. L929 cells were infected with reovirus type 3 Abney (T3A), and calpain activity, measured as cleavage of the fluorogenic calpain substrate Suc-Leu-Leu-Val-Tyr-AMC, was monitored. There was a 1.6-fold increase in calpain activity in T3A-infected cells compared to mock-infected cells; this increase was completely inhibited by preincubation with calpain inhibitor I (N-acetyl-leucyl-leucyl-norleucinal [aLLN]), an active-site inhibitor. Both aLLN and PD150606, a specific calpain inhibitor that interacts with the calcium-binding site, inhibited reovirus-induced apoptosis in L929 cells by 54 to 93%. Apoptosis induced by UV-inactivated reovirus was also reduced 65 to 69% by aLLN, indicating that inhibition of apoptosis by calpain inhibitors is independent of effects on viral replication. We conclude that calpain activation is a component of the regulatory cascade in reovirus-induced apoptosis.
Apoptosis is a distinct mechanism of cell death, characterized by DNA fragmentation, condensation of nuclear chromatin, cell shrinkage, and membrane zeiosis (22). It plays a critical role in many physiologic processes, including normal immune system cell development and selection (24), neuronal development (33, 74), and certain pathologic conditions (71). A variety of internal and external stimuli are capable of eliciting apoptosis, as exemplified by many viral agents (reviewed in reference 55).
We have used reovirus-induced apoptosis as an experimental model system to study the viral and cellular mechanisms involved in apoptotic cell death (37). Infection of cultured fibroblasts and epithelial cells with reoviruses induces apoptosis, although reovirus strains differ in the efficiency with which they induce this cellular response (42, 69, 70). Apoptosis also occurs following reovirus infection in vivo (37, 38). Murine infection with serotype 3 reoviruses produces a lethal meningoencephalitis (68). Viral antigen, evidence of neuropathologic injury, and apoptosis colocalize to the same regions of the mouse central nervous system (38). Investigation of reovirus-induced hepatitis (27) and myocarditis (56) also demonstrates that apoptotic cell death occurs in vivo within these organs. Taken together, these data suggest that apoptosis is an important mechanism of injury in reovirus infection.
The cellular mechanisms effecting and regulating apoptosis are still incompletely characterized. Protease cascades appear to play a critical role as effectors of apoptosis (8, 30, 40, 76). Examples of proteases implicated in apoptosis include granzyme B, caspases, and calpain. Calpain is a calcium-dependent papain-like neutral cysteine protease that is distributed widely throughout the cytosol of many cell types (32). It exists in the cytosol as an inactive proenzyme in steady state with its endogenous inhibitor, calpastatin. Calpain plays physiologic regulatory roles in membrane and cytoskeletal remodelling, including mitosis (54) and platelet activation (39). Increases in cytosolic calcium levels occur in many forms of apoptosis (9, 66), an observation that led to investigation of the potential role of calpain in apoptosis. Calpain activation was demonstrated in dexamethasone-induced thymocyte apoptosis (62) and serves as an upstream regulator of apoptosis in thymocytes induced by a variety of triggers (61). Inhibitors of calpain and similar proteases reduce apoptosis in human immunodeficiency virus-infected T lymphocytes (52). Calpain is also implicated in neural cell death associated with cerebral ischemia (28) and other neural insults (19, 44, 45), neurodegenerative processes such as Alzheimer’s disease (47), myocardial ischemia (20), and cataract formation (12).
In this study, we investigated whether calpain is involved in reovirus-induced apoptosis. The results indicate that calpain activity is increased in reovirus-infected cells and that inhibition of calpain activation by two distinct classes of calpain inhibitors reduces reovirus-induced apoptosis. This study provides the first evidence calpain activity is directly linked to virus-induced cell death.
MATERIALS AND METHODS
Cells.
Murine L929 fibroblasts (L cells) (ATCC CCL 1) were passaged in spinner culture by adjusting the cell concentration daily to 5 × 105 cells/ml by dilution with Joklik’s modified Eagle’s minimum essential medium supplemented to contain 5% heat-inactivated fetal bovine serum and 2 mM l-glutamine (Gibco/BRL).
Virus. (i) P2 stocks.
Reovirus strains type 1 Lang (T1L), type 3 Dearing (T3D), and type 3 Abney (T3A) are laboratory stocks which were plaque purified and passaged (twice) in L cells to generate working stocks as previously described (67).
(ii) Purified stocks.
Purified T3A virion preparations were made by sonication and Freon extraction from P2 stock-infected L cells followed by cesium chloride gradient (density, 1.25 to 1.45 g/cm3) centrifugation and dialysis against viral dialysis buffer (VDB) (150 mM NaCl, 15 mM MgCl2, 10 mM Tris [pH 7.4]), as previously described (16). The concentrations of virions in purified preparations were determined from the equivalence 1 optical density at 260 nm unit = 2.1 × 1012 virion particles/ml (58), followed by titer determination by plaque assay.
(iii) UV-inactivated virus.
T1L and T3A were diluted to 1010 PFU/ml in VDB and exposed for 15 min to short-wave UV light (254 nm) to generate an intensity of 600 μW/cm2. Following UV irradiation, virus stocks were devoid of infectious virus as determined by plaque assay (viral titer < 10 PFU/ml).
Inhibitors.
Calpain inhibitor I (N-acetyl-leucyl-leucyl-norleucinal [aLLN]; Calbiochem, La Jolla, Calif.) is a modified peptide which competes for the active site of calpain (71). It was prepared as a 5 mM stock in dimethyl sulfoxide. PD150606, an α-mercaptoacrylic acid derivative, is a selective Ca2+-binding site blocker with high specificity for calpains (72). It was prepared immediately prior to use as a 5 mM stock in H2O-dimethylformamide (DMF) (1:1). PD145305 is an inactive analogue of PD150606. It was prepared immediately before use as a 5 mM stock in H2O-dimethyl sulfoxide (1:1). (PD150606 and PD145305 were provided by Kevin Wang, Parke-Davis Pharmaceutical Research, Ann Arbor, Mich.)
Measurement of calpain activity by fluorogenic-substrate assays.
Fluorogenic-substrate assays were performed with Suc-Leu-Leu-Val-Tyr-AMC (Suc-LLVY), a membrane-permeable calpain-specific fluorogenic substrate, as previously described (53). Proteolytic hydrolysis of the peptidyl-7-amino bond liberates the highly fluorescent 7-amino-4-methylcoumarin (AMC) moiety. Cellular fluorescence was quantified with a Cytofluor Series 4000 fluorometer with 360-nm excitation and 460-nm emission filters. Standard curves were generated with free AMC for each experiment.
For measurements of calpain activity, L929 cells were plated at 105 cells in 200 μl per well in 96-well plates (Costar, Cambridge, Mass.) and incubated for 24 h at 37°C. Medium was removed from the adherent cell layer, and the cells were then infected with 20 μl of purified T3A virus (multiplicity of infection [MOI] = 11,000) or viral dialysis buffer (mock infection). After a 1-h incubation at 37°C, 200 μl of assay buffer (115 mM NaCl, 1 mM KH2PO4, 5 mM KCl, 2 mM CaCl2, 1.2 mM MgSO4, 25 mM sodium HEPES buffer [pH 7.4]) (2) was added to all wells. To determine the maximal calpain activity under our experimental conditions, the calcium ionophore A23187 (5 μM) was added to representative wells of virus-infected and mock-infected cells. As an additional control to ensure that fluorescence activity was calpain specific, aLLN (50 μM) was added to representative wells for each condition. Other controls included aLLN alone, A23187 alone, virus alone, and VDB alone in the absence of cells. Suc-LLVY substrate (62.5 μM) was added to each well at the start of the assay. The plates were incubated at 37°C in the fluorometer, and fluorescence was measured at 5-min intervals starting immediately after the addition of substrate. Each condition was assayed in duplicate or triplicate wells.
Calpain activity was calculated as mean fluorescence ± standard error of the mean (SEM). For conditions with the calcium ionophore A23187, the inherent fluorescence of this compound (determined from wells containing A23187 in the absence of cells [mean = 5,066 fluorescence units, n = 10]) was subtracted from the total measured fluorescence to accurately represent maximal calpain activity. For comparison of calpain activity at 120 min, the net calpain activity for virus- and mock-infected conditions was calculated by subtracting the residual activity in the presence of calpain inhibitor.
Calpain inhibitor experiments.
L cells were plated at 3.7 × 104 cells/well in 500 μl in 24-well plates (Falcon, Lincoln Park, N.J.) and incubated at 37°C for 24 h to allow the formation of an adherent monolayer. For experiments with active-site inhibitor, cells were preincubated with aLLN (25 μM) or solvent for 1 to 2 h. For experiments with the calcium-binding-site inhibitor, cells were preincubated with PD150606 (25 to 50 μM), the inactive analogue PD145305 (25 to 50 μM) or solvent alone for 1 h. Medium containing inhibitor (or control) was then removed, and the cells were treated with mock solution (gel-saline) (137 mM NaCl, 0.2 mM CaCl2, 0.8 mM MgCl, 19 mM H3BO3, 0.1 mM Na2B4O7, 0.3% [wt/vol] gelatin) or P2 stocks of T3A, T3D, or T1L at an MOI of 100. After a 1-h incubation, inhibitors were added back to each well. At 48 h postinfection, apoptosis was determined as described below.
Quantification of Apoptosis:
Cells (including nonadherent cells) were harvested by gentle pipetting and trypsinization. A solution of acridine orange for determination of nuclear morphology and ethidium bromide to distinguish cell viability, at a final concentration of 1 μg/ml for each substance, was used to stain cells, as previously described (15). Following staining, cells were examined by epifluorescence microscopy (Nikon Labophot-2; B-2A filter; excitation, 450 to 490 nm; barrier, 520 nm; dichroic mirror, 505 nm). The percentage of apoptotic cells was determined by counting the number of cells containing condensed and/or marginated chromatin in a population of 100 cells.
For experiments determining the effects of calpain inhibitors on reovirus-induced apoptosis, the percent inhibition of apoptosis was calculated and reported as follows:
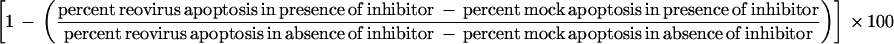 |
Determination of viral growth.
L-cells were plated at 2.5 × 104 cells/well in 100 μl in 96-well plates (Costar). At 24 h postplating, the cells were preincubated with PD150606 (50 μM), PD145305 (50 μM), or DMF-H2O (1:1). The medium was then removed, and the cells were infected with P2 stock of T3A (MOI = 100) for 1 h at 37°C. Following infection, the medium (including the inhibitor or vehicle) was replaced. At various times postinfection (0, 24, and 48 h), the cells were harvested and the viral titer determined by plaque assay as previously described (67). Viral titers are reported as log10 PFU per milliliter ± standard deviation (SD).
Statistics.
The results of fluorogenic substrate assays and apoptosis inhibition assays are reported as means ± SEM. Results of viral growth experiments are reported as means ± SD and 95% confidence intervals (CI). Means were compared using parametric two-tailed t-tests. For calpain activity slope comparisons, Wilcoxon nonparametric and parametric two-tailed t tests were used (GraphPad InSTAT, version 1.14).
RESULTS
Infection with reovirus strain T3A is associated with increased calpain activity.
To investigate whether reovirus infection is associated with changes in cellular calpain activity, cleavage of the cell-permeant fluorogenic substrate Suc-LLVY was monitored (Fig. 1). At any given time point, virus-infected cells showed greater calpain activity than did mock-infected cells, and this difference increased over time. By 2 h after adsorption, virus-infected cells achieved activation levels comparable to the calcium ionophore (A23187) positive control. Mock-infected cells showed lower levels of calpain activity, which never fully approached those in virus-infected or A23187-treated cells. The addition of the calpain inhibitor aLLN to both mock-infected and virus-infected cells markedly suppressed calpain activity to equally low levels.
FIG. 1.

Calpain activity over time in reovirus-infected cells. L929 cells (105) were infected with T3A reovirus (MOI = 11,000) or mock infected with VDB. The calcium ionophore A23187 was used as a positive control. The calpain inhibitor aLLN (50 μm) was added to representative wells for all conditions. Following addition of Suc-LLVY (62.5 μM), fluorescence was monitored. Data was derived from 10 replicates per condition, and curves were generated from data points at 15-min intervals. Error bars indicate SEM.
Since virus-infected cells showed levels of calpain activity equal to those of the positive control (A23187) by 2 h after adsorption, this was taken as a time point for comparison between all conditions. Virus-infected cells had a mean calpain activity of 13,235 ± 1,029 fluorescene units compared to mock-infected cells, which had a mean activity of 8,262 ± 686 fluorescence units (P < 0.001). This represents a 1.6-fold increase in calpain activity for virus-infected cells compared to mock-infected cells.
Analysis of the slopes of the calpain activity curves (representing the velocity of calpain activity within each experimental condition) also revealed significant differences between virus- and mock-infected cells. The highest velocity of calpain activity was seen in virus-infected cells, was observed early in the assay (as soon as 15 min following the addition of substrate), and was sustained throughout the experiment. The mean velocity of calpain activity for each condition was calculated by averaging the velocity of calpain activity for all time segments after the initial 15-min equilibration period. Virus-infected cells exhibited an increased velocity of calpain activity (2,106 ± 86 fluorescence units/15 min) compared to mock-infected cells (1,418 ± 31 units/15 min; P < 0.0001 parametric, P < 0.02 nonparametric).
Calpain Inhibitor I inhibits reovirus-induced apoptosis.
To determine whether blocking calpain activity inhibits reovirus-induced apoptosis, we tested the effect of calpain inhibitor I (aLLN), a selective active site inhibitor of calpain, on apoptosis induced by three different strains of reovirus (T3A, T3D, and T1L). Baseline levels of apoptosis induced by mock-infected cells (1.7% ± 0.4%) and mock-infected cells treated with aLLN (10% ± 1.7%) were minimal. Infection with all three strains of reovirus resulted in increased levels of apoptosis compared to mock infection: 46% ± 4% for T3A, 50% ± 3% for T3D, and 29% ± 2% for T1L. The addition of aLLN significantly inhibited apoptosis induced by all three strains of reovirus (Fig. 2a). For T3A, apoptosis was reduced to 14% ± 2% (P < 0.0001); for T3D, apoptosis was decreased to 20% ± 1% (P < 0.001); for T1L, apoptosis was reduced to 12% ± 1% (P = 0.0001). The reductions in apoptosis attributable to aLLN were 91% for T3A, 79% for T3D, and 93% for T1L.
FIG. 2.

(a) Effect of aLLN on reovirus-induced apoptosis. L929 cells were preincubated with aLLN (50 μM) for 1 h and then infected with T3A, T3D, or T1L strains (MOI = 100) adsorbed at 37°C for 1 h. Apoptosis was determined, as described in Materials and Methods, after 48 h and compared to that in cells which were infected but not preincubated with aLLN. Data was derived from 3 to 10 replicates per condition. ∗, statistically significant decrement in apoptosis compared to untreated cells (P = 0.0001 for T1L, P < 0.0001 for T3A, and P < 0.001 for T3D). Error bars indicate SEM. (b) Effect of PD150606 on reovirus-induced apoptosis. L929 cells were preincubated with PD150606 or PD145305 (25 to 50 μM) for 1 h and then infected with T3A, T3D, or T1L strains (MOI = 100) adsorbed at 37°C for 1 h. Apoptosis was determined after 48 h, as described in Materials and Methods. Data was derived from two to four replicates per condition (except that only one replicate was used for T3A without inhibitor). ∗, statistically significant decrement in apoptosis compared to untreated cells (P = 0.001 for T3D). For T3A, comparison was made to inactive analogue (P = 0.002). ∗∗, significant decrement in apoptosis (P = 0.02) compared to untreated cells but not to inactive analogue (P > 0.05). Error bars indicate SEM.
Calpain inhibitor PD 150606 inhibits reovirus-induced apoptosis.
Although aLLN is one of the most specific active-site calpain inhibitors, it also inhibits certain other papain-like cysteine proteases. To verify that the aLLN effect of decreasing reovirus-induced apoptosis was calpain specific, we tested a novel calpain inhibitor, PD150606, which acts by blocking the Ca2+-binding sites of calpain (72), as well as its inactive analogue, PD145305 (Fig. 2b). Baseline levels of apoptosis induced by mock infection (2% ± 1%), and mock infection in the presence of PD150606 (4% ± 3%) and PD145305 (3% ± 1%) were minimal. Again, all three strains of reovirus induced significant increases in apoptosis compared to mock infection (T3A, 43%; T3D, 41% ± 1%; T1L 23% ± 2%). PD145305, the inactive analogue, had minimal effect on virus-induced apoptosis (T3A, 41% ± 4%; T3D 41% ± 2%; T1L 16% ± 3% [P = 0.2]). In sharp contrast, PD150606 significantly inhibited virus-induced apoptosis for all three strains. T3A-induced apoptosis was decreased to 20% ± 0.3%; T3D-induced apoptosis was decreased to 22% ± 1%; (P = 0.001); T1L-induced apoptosis was decreased to 11% ± 1% (P = 0.02). The reductions in reovirus-induced apoptosis attributable to PD150606 were 62% for T3A, 54% for T3D, and 67% for T1L.
Inhibition of apoptosis by calpain inhibitors is not a result of impaired viral growth.
To ensure that the reductions in reovirus-induced apoptosis attributed to the calpain inhibitors were not due to direct inhibition of viral growth, the yields of T3A and T3D in L929 cells were assessed in the presence and absence of aLLN, PD145305, and PD150606 (Fig. 3). For T3A, the mean viral titer at 48 h postinfection in the absence of inhibitor (8.3 ± 0.3 log10 PFU/ml; 95% CI = 7.7 to 8.9) was slightly greater than the titer in the presence of aLLN (7.6 ± 0.2 log10 PFU/ml; 95% CI = 7.2 to 8.0) (P = 0.02). A similar effect was also observed for growth of T3D: the mean viral titer at 48 h in the absence of inhibitor was 8.0 ± 0.3 log10 PFU/ml 3 (95% CI = 7.4 to 8.6) compared to the titer in the presence of aLLN, which was 6.9 ± 0.6 log10 PFU/ml (95% CI = 5.7 to 8.1) (P = 0.05). Although these differences in growth were statistically significant, in both cases the 95% CI overlapped. PD150606 and PD145305 had no effect on viral growth. For T3A, the mean viral titer at 24 h in the absence of inhibitors was 7.0 ± 0.3 log10 PFU/ml, which was no different from that in the presence of PD150606 (6.8 ± 0.2 log10 PFU/ml; P = 0.3) or PD 145305 (6.7 ± 0.3 log10 PFU/ml; P = 0.2).
FIG. 3.

Effect of calpain inhibitors on viral growth. L929 cells were preincubated with aLLN (active-site inhibitor) for 1 h prior to infection with T3A and T3D. Viral growth was compared by measuring the plaque assay titer at 24 h postinfection to that in L929 cells infected without preincubation in inhibitor. The experiment was then repeated with 50 μM PD150606 (Ca2+ site inhibitor, active), 50 μm PD145305 (inactive analogue) or DMF-H2O (diluent of PD150606). Data was derived from three replicates per condition for aLLN and seven to nine replicates per condition for the PD compounds. Error bars indicate SD.
Apoptosis induced by UV-inactivated (replication-incompetent) virus is inhibited by aLLN and PD150606.
The lack of a major effect by calpain inhibitors on viral growth indicated that their capacity to inhibit apoptosis was not due to inhibition of viral replication. To independently confirm this result, we tested the effects of these inhibitors on UV-inactivated virus, which induces apoptosis at high MOI in the absence of a requirement for viral replication (70). Experiments with aLLN and PD150606 were performed by UV-inactivated T3D infection of L-cells (Fig. 4). UV-T3D induced increased apoptosis (82% ± 2%) compared to mock infection (2% ± 1%) at 48 h. The addition of aLLN significantly reduced UV-inactivated T3D-induced apoptosis to 37% ± 6% (P = 0.02). Treatment with PD150606 reduced apoptosis to 48% ± 4% (P = 0.02), whereas treatment with PD145305 (inactive analogue) had minimal effect (77% ± 2%; P = 0.2). Thus, even in apoptosis triggered by UV-inactivated virus, both inhibitors reduced apoptotic death by 68% (aLLN) and 45% (PD150606). Apoptosis induced by UV-T1L (data not shown) was also inhibited by a similar magnitude with the use of aLLN (65% reduction) and PD150606 (67% reduction).
FIG. 4.

Effect of aLLN and PD150606 on UV-inactivated T3D-induced apoptosis. L929 cells were preincubated in aLLN, PD150606, or PD145305 (inactive analogue) prior to infection with UV-inactivated T3D (MOI = 8,650 to 15,000). Apoptosis was determined at 48 h, as described in Materials and Methods, and compared to that in UV-inactivated T3D-infected cells which were not preincubated with inhibitors. Data was derived from two to four replicates per condition. ∗, statistically significant decrement with comparison to cells treated with no inhibitors at P = 0.02 for PD150606 and P = 0.02 for aLLN. Error bars indicate SEM.
DISCUSSION
In this study, we provide evidence that calpain is involved in reovirus-induced apoptosis, demonstrating that reovirus-infected cells exhibit increased levels of calpain activity and that inhibition of this activity reduces apoptotic cell death. This is the first report that directly links increased activity of this protease with virus-induced apoptosis.
Calpain activity increases following reovirus infection.
The fluorogenic substrate assay permits monitoring of cellular processes in living cells over time in response to a variety of experimental manipulations (2, 43). By this method, we demonstrated a statistically significant increase in calpain activity in reovirus-infected cells compared to mock-infected cells, both in absolute terms (peak activity) and in the rate of increase of activity. The magnitude of the increased activity following viral infection (1.6-fold increase) is similar to that seen in other signaling pathways involving protease activation (26, 62). The calpain inhibitor aLLN dramatically reduced the increase in fluorogenic substrate signal under all conditions tested, providing further support that this increase reflected calpain activity.
There are several possible explanations for the increased calpain activity observed following reovirus infection. Calpain could be acting as an effector of the late morphologic changes characteristic of apoptotic cell death. However, since the increase occurred so rapidly following viral adsorption, this suggests an earlier, more integral role for calpain as part of a regulatory cascade that ultimately leads to apoptosis. A similar role for calpain has been suggested in apoptotic signaling pathways in neurons and immune system cells (34, 35, 60, 65). Such a cascade has been described with regard to the caspases (49); perhaps calpain participates in that pathway.
Potential mechanisms of reovirus-induced increases in calpain activity.
The process by which calpain becomes activated within any cell is not completely understood, although it is thought to occur primarily at the cell membrane in association with membrane phospholipids and with an absolute requirement for calcium ions (59, 65). There are several plausible mechanisms by which reovirus infection could trigger cellular increases in calpain activity. First, it is possible that viral attachment initiates a calcium flux, leading to a rapid increase in intracellular calcium levels, as has been demonstrated for rotavirus, a closely related virus (14). This flux would initiate autocatalysis of the large subunit of the inactive proenzyme, which is required for protease activity. Similarly, autocatalysis of the small subunit results in facilitated interaction with phospholipids at the cell membrane, which is thought to reduce the calcium requirements for autocatalytic activation as well as to accelerate the protease activity of the active species. Second, it is possible that reovirus attachment triggers changes in growth factor levels that facilitate calpain activation. Studies have suggested that growth factors might regulate calpain-calpastatin expression and translocation to the membrane for interaction with lipids for enzyme activation (4, 36). Upregulation of epidermal growth factor receptor on certain cell types has been shown to increase the cytopathicity following reovirus infection (63); perhaps these or other receptors could activate calpain. Finally, a variety of endogenous calpain activator proteins have been found in human hematopoietic cells (41, 48, 57) as well as human and bovine brain cells (13). It is possible that reovirus attachment is linked to upregulation of these proteins and thus to increased calpain activation.
Reovirus-induced apoptosis is inhibited by calpain inhibitors.
Since calpain is linked to apoptosis and since reoviruses are known to induce apoptosis, the rise in calpain activity seen in virus-infected cells suggested a biologically significant role for calpain in reovirus infection. We addressed this question by demonstrating the efficacy of two calpain inhibitors with distinctly different mechanisms of inhibition in reducing viral-induced apoptosis. Currently available inhibitors of calpain include modified peptides, which compete for the active site of the protease (71), or non-active-site-binding inhibitors, whose selectivity comes from their unique interaction with the calcium-binding domains of calpain, a feature not found in other protease inhibitors (72). In our experiments, both classes of calpain inhibitor resulted in significant reduction of apoptosis induced by the prototype reovirus strains T3A, T3D, and T1L. To ensure that these reductions were attributable to inhibition of calpain activity rather than to a detrimental effect on viral growth, we tested the effect of these compounds on viral growth in L cells. PD150606 (and the inactive analogue) had no effect on viral growth. There was a small decrease in viral yield in the presence of aLLN. The possibility that this small decrement accounted for the effects of aLLN appears unlikely, given that apoptosis induced by UV-inactivated virus (which cannot replicate) was still blocked by both aLLN and PD150606. Lysosomal and proteosomal proteases are not inhibited by PD150606 (72). Taken together, these observations indicate that the inhibition of apoptosis observed in these experiments is attributable to specific inhibition of calpain activity and that the increased calpain activity seen following reovirus infection of these cells has biological importance with regard to apoptosis in these cells.
It is interesting that although reovirus T1L is known to cause apoptosis less efficiently than type 3 strains (70), treatment with both calpain inhibitors resulted in equal reductions (on a percentage basis) of apoptosis induced by both viral serotypes. Differences in the capacity of T1L and T3D to induce apoptosis are determined by the S1 gene, which encodes the viral attachment protein ς1. The mechanism by which binding of reovirus leads to apoptosis has not been defined, but it has been previously suggested that reovirus binding activates a receptor-linked signaling pathway that results in inhibition of DNA synthesis and apoptosis (17). Our results suggest that despite strain-specific differences in the efficiency with which apoptosis occurs, calpain is subsequently involved in both systems. Moreover, since calpain inhibitors block apoptosis induced by both type 1 and type 3 strains, it is likely that the signaling pathways for reovirus-induced apoptosis converge at some point following attachment. This could theoretically be as late as 24 h after adsorption, but since activation experiments indicated an earlier role for calpain (increased activity was seen 1 h postadsorption), it is probable that if critical strain-specific differences in signaling exist, they act soon after attachment but before calpain activity increases. Alternatively, the pathways could be identical but could result in different levels of apoptosis based on quantitative differences in signaling and protease activation.
Possible mechanisms of reovirus-induced apoptosis via calpain signaling.
Although calpain appears to be implicated in reovirus-induced apoptosis, it is not clear what constitutes the upstream and downstream components of a signaling cascade, within which calpain might fit, either during reovirus infection or in other systems where calpain is involved. Unlike the proteasome, the ability of calpain to process substrates into only a few fragments suggests a more suitable role for calpain in the modulation of other enzymatic components of the apoptotic pathway (59), including other proteases. Known substrates which calpain degrades in vivo include proto-oncogenes, steroid hormone receptors, protein kinases, and cytoskeletal elements (11). Calpain also may play a physiologic role in the regulation of a variety of cellular transcription factors and cell cycle-regulating factors including c-Jun, c-Fos, and NF-κB (3, 5, 29, 46, 73). Recently, p53, which is thought to play essential roles in tumor suppression, G1 cell cycle arrest, and some forms of apoptosis, has been shown to be proteolyzed in vitro by calpain (18, 25, 59).
There are several possible links between the effect of calpain on signal transduction pathways as well as transcriptional and cell cycle regulatory factors that may be relevant to mechanisms of reovirus-induced apoptosis. First, reovirus-induced apoptosis is associated with inhibition of cellular proliferation (69). Aberrant cell cycle signals trigger apoptosis in many cell systems (6, 23, 31). Second, reovirus infection may be associated with activation of NF-κB (7, 10), a key transcription factor that participates in regulation of the expression of genes involved in the apoptotic process (1). Finally, reovirus infection is associated with and influenced by perturbation of cellular kinase signaling pathways (50, 51, 63, 64). Differential activation of mitogen-activating kinase signaling cascades has been shown to induce or inhibit apoptosis in a variety of experimental systems (21, 75).
Potential therapeutic efficacy of calpain inhibitors.
Once the intermediary biochemical signaling pathways leading to apoptosis have been more clearly delineated and ordered, it may be possible to construct therapeutic interventions which could prevent or limit cell death and minimize damage to the host organism. Our data suggest that calpain plays an important role in reovirus-induced cell death and could therefore serve as a potential target in preventing reovirus-induced pathologic changes. Development of novel interventions is especially necessary with regard to serious viral infections such as meningoencephalitis and myocarditis, which result in irreversible cell damage and loss, since highly effective and specific antiviral therapies are currently unavailable. Further studies with the reovirus model may shed light on potential therapeutic interventions for these infections and other types of viral and nonviral damage.
ACKNOWLEDGMENTS
We thank Kevin Wang of the Parke-Davis Research Center for providing calpain inhibitors and their inactive analogues. The University of Colorado Cancer Center provided core tissue culture and media facilities.
This work was supported by Public Health Service grant 1RO1AG14071 from the National Institute of Aging, Merit and REAP grants from the Department of Veterans Affairs, and a U.S. Army Medical Research and Material Command grant (USAMRMC 98293015) (K.L.T.). This work was also supported by Public Health Service grant AI38296 from the National Institute of Allergy and Infectious Diseases and by the Elizabeth B. Lamb Center for Pediatric Research (T.S.D.).
REFERENCES
- 1.Baeuerle P A, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 2.Bronk S F, Gores G J. pH-dependent nonlysosomal proteolysis contributes to lethal anoxic injury of rat hepatocytes. Am J Physiol. 1993;264:G744–G751. doi: 10.1152/ajpgi.1993.264.4.G744. [DOI] [PubMed] [Google Scholar]
- 3.Carillo S, Pariat M, Steff A, Roux P, Etienne-Julan M, Lorca T, Piechaczyk M. Differential sensitivity of FOS and JUN family members to calpains. Oncogene. 1994;9:1679–1689. [PubMed] [Google Scholar]
- 4.Chakrabarti A K, Neuberger T, Russell T, Banik N L, DeVries G H. Immunolocalization of cytoplasmic and myelin mCalpain in transfected Schwann cells. II. Effect of withdrawal of growth factors. J Neurosci Res. 1997;47:609–616. doi: 10.1002/(sici)1097-4547(19970315)47:6<609::aid-jnr6>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 5.Chen F, Yongju L, Kuhn D C, Maki M, Shi X, Sun S, Demers L M. Calpain contributes to silica-induced IκB-α degradation and nuclear factor-κB activation. Arch Biochem Biophys. 1997;342:383–388. doi: 10.1006/abbi.1997.0132. [DOI] [PubMed] [Google Scholar]
- 6.Chiatugi V, Magnelli L, Cinelli M, Basi G. Apoptosis and the cell cycle. Cell Mol Biol. 1995;40:603–612. [PubMed] [Google Scholar]
- 7.Clarke, P. C., and K. L. Tyler. Unpublished data.
- 8.Cohen G M. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen J J, Duke R C. Glucocorticoid activation of a calcium-dependent endonuclease in thymocyte nuclei leads to cell death. J Immunol. 1984;132:38–42. [PubMed] [Google Scholar]
- 10.Connolly J L, Rodgers S E, Pike B, Clarke P, Tyler K L, Dermody T S. Reovirus-induced apoptosis requires activation of transcription factor NF-κB. 1998. p. 89. , abstr. 17-2. In Abstracts of the 17th Annual Meeting of the American Society for Virology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Croall D E, DeMartino G N. Calcium-activated neutral protease (calpain) system: structure, function and regulation. Physiol Rev. 1991;71:813–847. doi: 10.1152/physrev.1991.71.3.813. [DOI] [PubMed] [Google Scholar]
- 12.David L L, Shearer R R, Shih M. Sequence analysis of lens beta-crystallins suggest involvement of calpain in cataract formation. J Biol Chem. 1993;268:1937–1940. [PubMed] [Google Scholar]
- 13.DeMartino G N, Blumenthal D K. Identification and partial purification of a factor that stimulates calcium-dependent proteases. Biochemistry. 1982;21:4297–4303. doi: 10.1021/bi00261a019. [DOI] [PubMed] [Google Scholar]
- 14.Dong Y, Zeng C Q, Ball J M, Estes M K, Morris A P. The rotavirus enterotoxin NSP4 mobilizes intracellular calcium in human intestinal cells by stimulating phospholipase C-mediated inositol 1,4,5-triphosphate production. Proc Natl Acad Sci USA. 1997;94:3960–3965. doi: 10.1073/pnas.94.8.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duke R C, Cohen J J. Morphological and biochemical assays of apoptosis. In: Coligan J E, editor. Current protocols in immunology. New York, N.Y: John Wiley & Sons, Inc.; 1992. pp. 3.17.1–3.17.16. [Google Scholar]
- 16.Furlong D B, Nibert M L, Fields B N. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol. 1988;62:246–256. doi: 10.1128/jvi.62.1.246-256.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaulton G N, Greene M I. Inhibition of cellular DNA synthesis by reovirus occurs through a receptor-linked signalling pathway that is mimicked by antiidiotypic, antireceptor antibody. J Exp Med. 1989;169:197–211. doi: 10.1084/jem.169.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonen H, Shkedy D, Barnoy S, Kosower N S, Ciechanover A. On the involvement of calpains in the degradation of the tumor suppressor protein p53. FEBS Lett. 1997;406:17–22. doi: 10.1016/s0014-5793(97)00225-1. [DOI] [PubMed] [Google Scholar]
- 19.Hajimohammadreza I, Raser K J, Nath R, Nadimpalli R, Scott M, Wang K K W. Neuronal nitric oxide synthase and calmodulin-dependent protein kinase IIα undergo neurotoxin-induced proteolysis. J Neurochem. 1997;69:1006–1013. doi: 10.1046/j.1471-4159.1997.69031006.x. [DOI] [PubMed] [Google Scholar]
- 20.Iizuka K, Kawaguchi H, Kitabatake A. Effects of thiol protease inhibitors on fodrin degradation during hypoxia in cultured myocytes. J Mol Cell Cardiol. 1993;25:1101–1109. doi: 10.1006/jmcc.1993.1122. [DOI] [PubMed] [Google Scholar]
- 21.Johnson N L, Gardner A M, Diener K M, Lange-Carter C A, Gleavy J, Jarpe M B, Minden A, Karin M, Zon L I, Johnson G L. Signal transduction pathways regulated by mitogen-activated/extracellular response kinase kinase kinase induce cell death. J Biol Chem. 1996;271:3229–3237. doi: 10.1074/jbc.271.6.3229. [DOI] [PubMed] [Google Scholar]
- 22.Kerr J F R, Wylie A H, Currie A R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.King K L, Cidlowski J A. Cell cycle and apoptosis: common pathways to life and death. J Cell Biochem. 1995;58:175–180. doi: 10.1002/jcb.240580206. [DOI] [PubMed] [Google Scholar]
- 24.Krammer P H, Behrmann I, Daniel P, Dhein J, Debatin K M. Regulation of apoptosis in the immune system. Curr Opin Immunol. 1994;6:276–289. doi: 10.1016/0952-7915(94)90102-3. [DOI] [PubMed] [Google Scholar]
- 25.Kubbutat M H G, Vousden K H. Proteolytic cleavage of human p53 by calpain: a potential regulator of protein stability. Mol Cell Biol. 1997;17:460–468. doi: 10.1128/mcb.17.1.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwo P, Patel T, Bronk S F, Gores G J. Nuclear serine protease activity contributes to bile acid-induced apoptosis in hepatocytes. Am J Physiol. 1995;268:G613–G621. doi: 10.1152/ajpgi.1995.268.4.G613. [DOI] [PubMed] [Google Scholar]
- 27.LaHair M M, Tyler K L, Oberhaus S M. Abstracts of the Fifth Southeastern Regional Virology Conference 1998. 1998. Reovirus-induced biliary atresia-like disease in mice is associated with apoptosis; p. 9. [Google Scholar]
- 28.Lee K S, Frank S, Vanderklish P, Arai A, Lynch G. Inhibition of proteolysis protects hippocampal neurons from ischemia. Proc Natl Acad Sci USA. 1991;88:6233–7237. doi: 10.1073/pnas.88.16.7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Z, Kunimatsu M, Yang J, Ozaki Y, Sasaki M, Okamoto T. Proteolytic processing of nuclear factor-κB by calpain in vitro. FEBS Lett. 1996;385:109–113. doi: 10.1016/0014-5793(96)00360-2. [DOI] [PubMed] [Google Scholar]
- 30.Martin S J, Green D R. Protease activation during apoptosis: death by a thousand cuts? Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- 31.Meikrantz W, Schlegel R. Apoptosis and the cell cycle. J Cell Biochem. 1995;59:160–174. doi: 10.1002/jcb.240580205. [DOI] [PubMed] [Google Scholar]
- 32.Murachi T. Calpain and calpastatin. Trends Biochem Sci. 1983;8:167–169. [Google Scholar]
- 33.Narayanan V. Apoptosis in development and disease of the nervous system. 1. Naturally occurring cell death in the developing nervous system. Pediatr Neurol. 1997;16:9–13. doi: 10.1016/s0887-8994(96)00257-3. [DOI] [PubMed] [Google Scholar]
- 34.Nath R, Raser K J, Stafford D, Hajimohammadreza I, Posner A, Allen H, Talanian R V, Yuen P, Gilbertsen R B, Wang K K W. Non-erythroid α-spectrin breakdown by calpain and interleukin 1β-converting-enzyme-like protease(s) in apoptotic cells: contributory roles of both protease families in neuronal apoptosis. Biochem J. 1996;319:686–690. doi: 10.1042/bj3190683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nath R, Raser K J, McGinnis K, Nadimpalli R, Stafford D, Wang K K W. Effects of ICE-like protease and calpain inhibitors on neuronal apoptosis. NeuroReport. 1996;8:249–255. doi: 10.1097/00001756-199612200-00050. [DOI] [PubMed] [Google Scholar]
- 36.Neuberger T, Chakrabarti A K, Russell T, DeVreis G H, Hogan E L, Banik N L. Immunolocalization of cytoplasmic and myelin mCalpain in transfected Schwann cells. I. Effect of treatment with growth factors. J Neurosci Res. 1997;47:521–530. [PubMed] [Google Scholar]
- 37.Oberhaus S M, Dermody T S, Tyler K L. Apoptosis and the cytopathic effects of reovirus. Curr Top Microbiol Immunol. 1998;233:23–49. doi: 10.1007/978-3-642-72095-6_2. [DOI] [PubMed] [Google Scholar]
- 38.Oberhaus S M, Smith R L, Clayton G H, Dermody T S, Tyler K L. Reovirus infection and tissue injury in the mouse central nervous system are associated with apoptosis. J Virol. 1997;71:2100–2106. doi: 10.1128/jvi.71.3.2100-2106.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Halloran T, Beckerle M C, Burridge K. Identification of talin as a major cytoplasmic protein implicated in platelet activation. Nature. 1985;317:449–451. doi: 10.1038/317449a0. [DOI] [PubMed] [Google Scholar]
- 40.Patel T, Gores G J, Kaufmann S H. The role of proteases during apoptosis. FASEB J. 1996;10:587–597. doi: 10.1096/fasebj.10.5.8621058. [DOI] [PubMed] [Google Scholar]
- 41.Pontremoli S, Melloni E, Michetti M, Salamino F, Sparatore B, Horecker B L. An endogenous activator of the Ca2+-dependent proteinase of human neutrophils that increases its affinity for Ca2+ Proc Natl Acad Sci USA. 1988;85:1740–1743. doi: 10.1073/pnas.85.6.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodgers S E, Barton E S, Oberhaus S M, Pike B, Gibson C A, Tyler K L, Dermody T S. Reovirus-induced apoptosis of MDCK cells is not linked to viral yield and is blocked by Bcl-2. J Virol. 1997;71:2540–2546. doi: 10.1128/jvi.71.3.2540-2546.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosser B G, Powers S P, Gores G J. Calpain activity increases in hepatocytes following addition of ATP. J Biol Chem. 1993;268:23593–23600. [PubMed] [Google Scholar]
- 44.Saatman K E, Murai H, Bartus R T, Smith D H, Hayward N J, Perri B R, McIntosh T K. Calpain inhibitor AK295 attenuates motor and cognitive deficits following experimental brain injury in the rat. Proc Natl Acad Sci USA. 1996;93:3428–3433. doi: 10.1073/pnas.93.8.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saatman K E, Bozyczko-Coyne D, Marcy V, Siman R, McIntosh T K. Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol. 1996;55:850–860. doi: 10.1097/00005072-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 46.Saido T C, Sorimachi H, Suzuki K. Calpain: new perspectives in molecular diversity and physiological-pathological involvement. FASEB J. 1994;8:814–822. [PubMed] [Google Scholar]
- 47.Saito K, Elce J S, Hamos J E, Nixon R A. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci USA. 1993;90:2628–2632. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salamino F, DeTullio R, Mengotti P, Viotti P L, Melloni E, Pontremoli S. Site-directed activation of calpain is promoted by a membrane-associated natural activator protein. Biochem J. 1993;290:191–197. doi: 10.1042/bj2900191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salvesen G S, Dixit V M. Caspases: intracellular signalling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- 50.Saragovi H U, Bhandoola A, Lemercier M M, Akbar G K M, Greene M I. A receptor that subserves reovirus binding can inhibit lymphocyte proliferation triggered by mitotic signals. DNA Cell Biol. 1995;14:653–664. doi: 10.1089/dna.1995.14.653. [DOI] [PubMed] [Google Scholar]
- 51.Saragovi H U, Rebai N, Roux E, Gagnon M, Zhang X, Robaire B, Bromberg J, Greene M I. Signal transduction and antiproliferative function of the mammalian receptor for type 3 reovirus. Curr Top Microbiol Immunol. 1998;233:155–166. doi: 10.1007/978-3-642-72092-5_7. [DOI] [PubMed] [Google Scholar]
- 52.Sarin A, Clerici M, Blatt S P, Hendrix C W, Shearer G M, Henkart P A. Inhibition of activation-induced programmed cell death and restoration of defective immune responses of HIV+ donors by cysteine protease inhibitors. J Immunol. 1994;153:862–871. [PubMed] [Google Scholar]
- 53.Sasaki T, Kikuchi T, Yumoto N, Yoshimura N, Murachi T. Comparative specificity and kinetic studies on porcine calpain I and calpain II with naturally occurring peptides and synthetic fluorogenic substrates. J Biol Chem. 1984;259:12489–12494. [PubMed] [Google Scholar]
- 54.Schollmeyer J E. Calpain II involvement in mitosis. Science. 1988;240:911–913. doi: 10.1126/science.2834825. [DOI] [PubMed] [Google Scholar]
- 55.Shen Y, Shenk T E. Viruses and apoptosis. Curr Opin Genet Dev. 1995;5:105–111. doi: 10.1016/s0959-437x(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 56.Sherry, B., and K. L. Tyler. Unpublished data.
- 57.Shiba R, Ariyoshi H, Yano Y, Kawasaki T, Sakon M, Kambayashi J, Mori T. Purification and characterization of a calpain activator from human platelets. Biochem Biophys Res Commun. 1992;182:461–465. doi: 10.1016/0006-291x(92)91754-e. [DOI] [PubMed] [Google Scholar]
- 58.Smith R E, Zweerink H J, Joklik W K. Polypeptide components of virions, top component, and cores of reovirus type 3. Virology. 1969;39:791–810. doi: 10.1016/0042-6822(69)90017-8. [DOI] [PubMed] [Google Scholar]
- 59.Sorimachi H, Ishiura S, Suzuki K. Structure and physiological function of calpains. Biochem J. 1997;328:721–732. doi: 10.1042/bj3280721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Squier M K T, Cohen J J. Calpain and cell death. Cell Death Differ. 1996;3:275–283. [PubMed] [Google Scholar]
- 61.Squier M K T, Cohen J J. Calpain, an upstream regulator of thymocyte apoptosis. J Immunol. 1997;158:3690–3697. [PubMed] [Google Scholar]
- 62.Squier M K T, Miller A C K, Malkinson A M, Cohen J J. Calpain activation in apoptosis. J Cell Physiol. 1994;159:229–237. doi: 10.1002/jcp.1041590206. [DOI] [PubMed] [Google Scholar]
- 63.Strong J E, Tang D, Lee P W K. Evidence that the epidermal growth factor receptor on host cells confers reovirus infection efficiency. Virology. 1993;197:405–411. doi: 10.1006/viro.1993.1602. [DOI] [PubMed] [Google Scholar]
- 64.Strong J E, Lee P W. The v-erbB oncogene confers enhanced cellular susceptibility to reovirus infection. J Virol. 1996;70:612–616. doi: 10.1128/jvi.70.1.612-616.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Suzuki K, Saido T C, Hirai S. Modulation of cellular signals by calpain. Ann N Y Acad Sci. 1992;674:218–27. doi: 10.1111/j.1749-6632.1992.tb27490.x. [DOI] [PubMed] [Google Scholar]
- 66.Trump B F, Berezesky I K. Calcium-mediated cell-injury and cell death. FASEB J. 1995;9:219–228. doi: 10.1096/fasebj.9.2.7781924. [DOI] [PubMed] [Google Scholar]
- 67.Tyler K L, Bronson R T, Byers K B, Fields B N. Molecular basis of viral neurotropism: experimental reovirus infection. Neurology. 1985;35:88–92. doi: 10.1212/wnl.35.1.88. [DOI] [PubMed] [Google Scholar]
- 68.Tyler K L, Fields B N. Reoviruses. In: Fields B N, Knipe D M, Howley P M, et al., editors. Virology. 3rd ed. New York, N.Y: Raven Press; 1996. pp. 1597–1624. [Google Scholar]
- 69.Tyler K L, Squier M K T, Brown A L, Pike B, Willis D, Oberhaus S M, Dermody T S, Cohen J J. Linkage between reovirus-induced apoptosis and inhibition of cellular DNA synthesis: role of the S1 and M2 genes. J Virol. 1996;70:7984–7991. doi: 10.1128/jvi.70.11.7984-7991.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tyler K L, Squier M K T, Rodgers S E, Schneider B E, Oberhaus S M, Grdina T A, Cohen J J, Dermody T S. Differences in the capacity of reovirus strains to induce apoptosis are determined by viral attachment protein ς1. J Virol. 1995;69:6972–6979. doi: 10.1128/jvi.69.11.6972-6979.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang K K W, Yuen P. Calpain inhibition: an overview of its therapeutic potential. Trends Pharmacol Sci. 1994;15:412–419. doi: 10.1016/0165-6147(94)90090-6. [DOI] [PubMed] [Google Scholar]
- 72.Wang K K W, Nath R, Posner A, et al. An alpha-mercaptoacrylic acid derivative is a selective nonpeptide cell-permeable calpain inhibitor and is neuroprotective. Proc Natl Acad Sci USA. 1996;93:6687–6692. doi: 10.1073/pnas.93.13.6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Watt F, Molloy P L. Specific cleavage of transcription factors by the thiol protease, m-calpain. Nucleic Acids Res. 1993;21:5092–5100. doi: 10.1093/nar/21.22.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Weller M, Schulz J B, Wullner U, Loschmann P A, Klockgether T, Dichgans J. Developmental and genetic regulation of programmed neuronal death. J Neural Transm. 1997;50:115–123. doi: 10.1007/978-3-7091-6842-4_12. [DOI] [PubMed] [Google Scholar]
- 75.Xia Z, Dickens M, Raingeaud J, Davis R J, Greenberg M E. Opposing effects of ERK and JNK p-38 MAP-kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 76.Zhivotovsky B, Burgess D H, Vanags D M, Orrenius S. Involvement of cellular proteolytic machinery in apoptosis. Biochem Biophys Res Commun. 1997;230:481–488. doi: 10.1006/bbrc.1996.6016. [DOI] [PubMed] [Google Scholar]