

Abstract

Tissue transglutaminase (TG2) is a multifunctional enzyme involved in the cross-linking of extracellular matrix proteins, formation of complexes with fibronectin (FN) and integrins, and GTP hydrolysis. TG2 is activated in several pathological conditions, including cancer. We recently described a novel series of ligands that bind to TG2 and inhibit its interaction with FN. Because TG2 acts via multiple mechanisms, we set out to pursue a targeted protein degradation strategy to abolish TG2’s myriad functions. Here, we report the synthesis and characterization of a series of VHL-based degraders that reduce TG2 in ovarian cancer cells in a proteasome-dependent manner. Degradation of TG2 resulted in significantly reduced cancer cell adhesion and migration in vitro in scratch-wound and migration assays. These results strongly indicate that further development of more potent and in vivo efficient TG2 degraders could be a new strategy for reducing the dissemination of ovarian and other cancers.
Introduction
Tissue transglutaminase (TG2) is a 77-kD protein belonging to the transglutaminase family, which includes transglutaminases 1–7, Factor XIII, and erythrocyte protein 4.2. These related proteins have similar catalytic activity; however, their substrate preferences and patterns of expression in tissues vary. TG2 has four domains: an N-terminus β-sandwich domain, which binds to fibronectin (FN), a catalytic domain that harbors the catalytic triad C277H335D358, which carries out the acyl-transfer function, and two β-barrel domains.1,2 A GTP/GDP-binding site is located between the catalytic and the first β-barrel domain. TG2 does not have a classical switch region, characteristic of G-proteins, and it remains unclear how its binding to GTP/GDP affects signaling. TG2 was shown to interact with PLCγ through a region located in its C-terminus and thus has been implicated in adrenergic signaling.3 The functions of the protein are modulated through allosteric changes, regulated by the concentrations of GTP and Ca2+. The crystal structure of GTP-bound TG2 (PDB 1KV3) has a compact conformation with the two β-barrel domains folding over the catalytic triad, obstructing the accessibility of Cys277 and blocking the enzymatic activity. The X-ray crystal structure of TG2 obtained in the presence of Ca2+ (PDB 2Q3Z) is open and exposes the catalytic core. In this “open” state, TG2 cannot bind GTP/GDP but interacts with substrates for transamidation.
Consequently, the physiological functions of TG2 are regulated by cellular context and localization. At the plasma membrane, TG2 binds to the 42 kDa gelatin-binding domain of FN (FN42), providing a binding site for β1 and β3 integrins4−6 and other membrane receptors such as PDGFR-β7,8 and Frizzled-7.9 Mutations in the catalytic core have been shown to not affect the formation of the FN/integrin/TG2 complex, proving that this function of the protein is independent of its transamidase role.5 Within the cytosol, where Ca2+ concentrations are low, the protein assumes a closed conformation and binds to GTP and other partners, thereby altering cellular signaling.3,10 In the extracellular matrix (ECM), where Ca2+ levels are high and nucleotide concentrations are low, TG2 functions as a transamidase, facilitating Ca2+-dependent incorporation of amines into proteins and acyl-transfer between glutamine and lysine residues, leading to protein cross-linking and facilitating matrix remodeling.11 The catalytic activity of TG2 can also be inhibited by various oxidants through intramolecular disulfide bridge formation.12,13 Several matrix proteins have been shown to be TG2 substrates, including FN,14 fibrin,15 osteopontin,16 laminin,17 collagen,18 and others. These functions suggest that TG2 plays important roles in cell adhesion, migration, and stromal assembly, which are involved in fibrotic processes and cancer.
Over the past decade, TG2’s role in cancer has been firmly established. The enzyme was found to be upregulated in ovarian,19,20 pancreatic,21 lung,22 breast cancer,23 and glioblastoma.24 TG2 expression was correlated with poor clinical outcomes in ovarian,25 pancreatic,26 and lung cancer,27 suggesting that it functions as a tumor promoter. Functionally, TG2 was linked to chemotherapy resistance28 through activation of the NF-κB survival pathway29,30 and of “outside-in” signaling,26 and with metastatic progression in ovarian cancer models19,20 through induction of epithelial-to-mesenchymal transition (EMT). More recently, TG2 was reported to be highly expressed in cancer stem cells (CSCs), facilitating their interaction with the metastatic niche and activating pro-survival pathways.31 While the pro-tumorigenic role of TG2 is clear, there is still a lack of consensus regarding which function and domain of the protein is responsible and should be targeted. We and others have shown that interactions with integrins and FN at the plasma membrane activate outside-in signaling (FAK, Akt, β-catenin, EGFR)32,33 and regulate cancer metastasis and stemness. However, in other contexts, the transamidase function was shown to have pro-tumorigenic functions: IκBα is cross-linked by TG2, leading to NF-κB activation;34 RhoA is transamidated by TG2 and becomes constitutively active.35 Other studies using TG2 mutants (R580A, which is unable to bind GTP; and C277, which is enzymatically inactive) suggested that the nucleotide-binding function of TG2 is required for maintaining cancer stemness.36,37 Thus, it remains unclear how to best target TG2, and the development of effective inhibitors has been challenging. Several strategies have been attempted, including small-molecule inhibitors for the TGase activity,38,39 for the TG2/FN complex,40,41 or for the GTPase function. While some anti-oncogenic activity was observed with each of the classes of proposed inhibitors, an active in vivo anti-TG2 strategy does not currently exist. We believe that this roadblock is caused by the multifunctional properties of the protein and the need to block all its functions through a single modality.
A new therapeutic modality that overcomes many of the challenges associated with difficult-to-target proteins is the PROteolysis-TArgeting Chimera (PROTAC) drugs, which act by inducing the degradation of a target protein.42,43 PROTACs are bifunctional molecules, with one end binding to a protein of interest and the other end binding to an E3 ubiquitin ligase. The PROTAC simultaneously binds to these two proteins to bring them into close proximity and form a ternary complex which facilitates the transfer of a ubiquitin molecule to the protein of interest and subsequent proteasomal degradation. The recent development of PROTACs as a new strategy to therapeutically target proteins has begun to significantly impact translational research.44−48 There are currently more than ten different PROTACs in clinical development.44−51 Available human clinical data for these compounds have shown promising tolerability, pharmacokinetic (PK), and efficacy, further validating PROTACs as potential drugs.
There are several key advantages of PROTACs over traditional small-molecule inhibitors. First, it has been shown that degrading a target protein results in more intense and sustained inhibition of its functions.52 Second, because the compound is required to bind the target protein and the E3 ligase protein simultaneously and in the proper orientation, there is often greater selectivity regarding which proteins are degraded compared with which proteins are inhibited by the ligand. For instance, when promiscuous inhibitor warheads are used, typically, very few of the inhibited targets are degraded by the PROTACs because of the more stringent structural requirements of binding two proteins to form a ternary complex.43 Third, many proteins, including TG2, have been shown to possess functions in addition to their enzymatic/signaling aspects, such as scaffolding partners. Existing inhibitors impact either TG2 enzymatic activity, or inhibit enzyme activity and GTP binding,53 and others only suppress FN binding, while PROTAC-induced degradation can abolish all of a protein’s functions and provide more functionally relevant effects.54
Because of the potential therapeutic relevance of TG2, there has been considerable interest in developing small-molecule inhibitors. Several reports have studied covalent inhibitors, including bromodihydroisoxazole (DHI) derivatives and Michael acceptors.55 In 2014, Khosla and co-workers reported structure–activity relationship (SAR) studies on various DHI analogs, in which the proline-containing inhibitor ERW1041E was used as a starting point for lead optimization to improve upon its potency, selectivity, and PK profile (Figure 1).56 More recently, van Dam and co-workers reported that Michael acceptor BJJF078, as well as ERW1041E, potently inhibited both mouse and human TG2 (and TG1) but did not inhibit the TG2-fibronectin interaction.57 Several peptidomimetic inhibitors containing Michael acceptors have also been recently described, including ZED1227 from the biotech company Zedira, which is the first known TG2 inhibitor undergoing clinical trials.58
Figure 1.
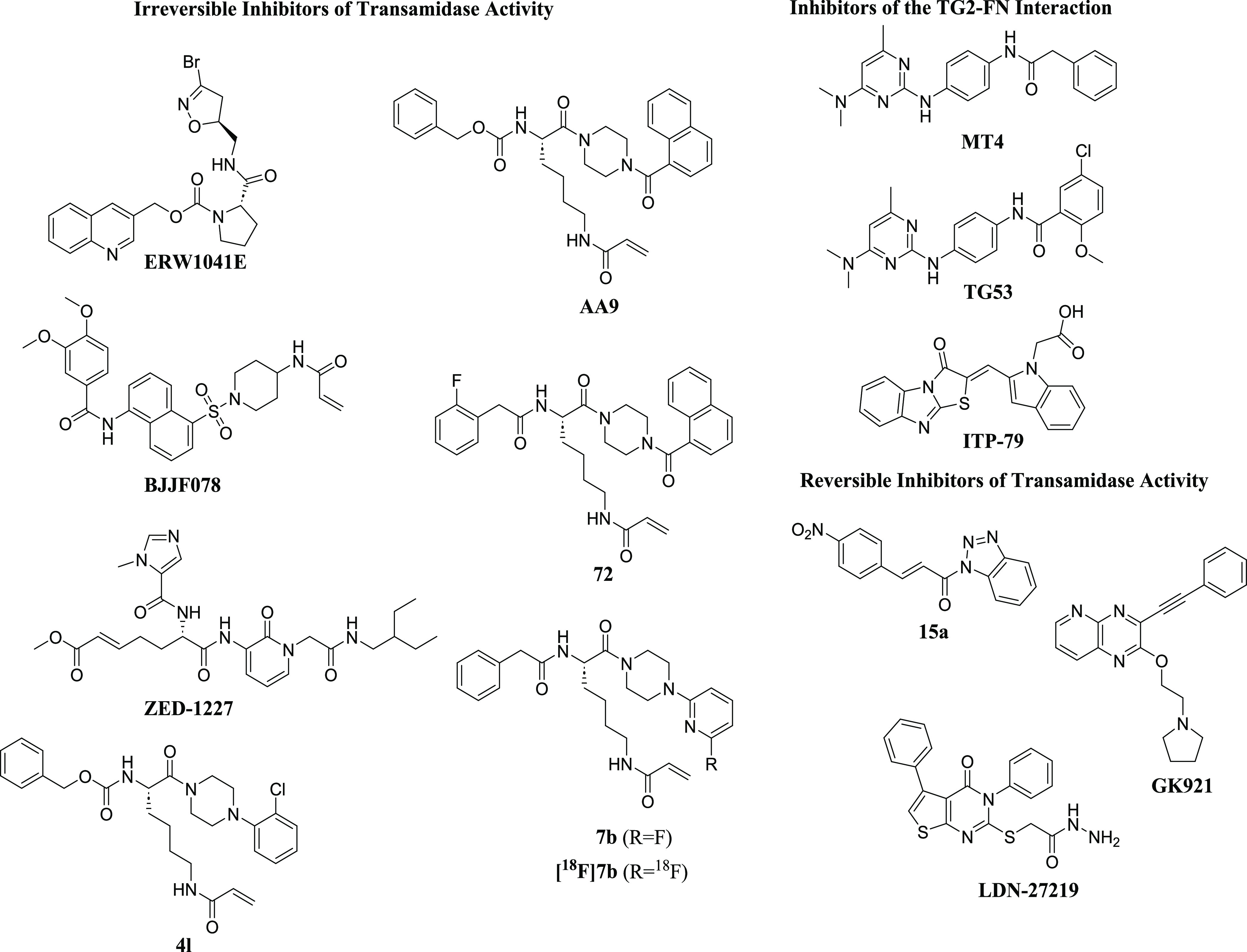
Representative structures of known TG2 inhibitors (same numbering/naming as in references).
Furthermore, there are several reports of Nε-acryloyllysine irreversible inhibitors of TG2. CHDI and Evotec reported irreversible TG2 inhibitors with an acrylamide moiety that were derived from a known lysine dipeptide.59 Compounds in the series had several favorable properties, including improved polar surface area, excellent plasma stability, and improved potency relative to the benchmark dipeptide; however, they generally suffered from high P-gp efflux and a high rate of oxidative metabolism. The structures of one of these compounds, 4l, is shown above (Figure 1). Akbar and co-workers also reported on Nε-acryloyllysine irreversible inhibitors.53 Novel inhibitors were developed that blocked both the transamidation function and GTP binding ability of hTG2. One of these compounds, AA9, was determined to have a good balance of affinity, efficiency, and physicochemical properties.
In later work, McNeil et al. reported compounds that retained the naphthoyl piperazine C-terminal functionality of AA9 but optimized the N-terminal functionality since substituents here were shown to contribute significantly to binding affinity.60 Their efforts resulted in compounds like compound 72 with improved parameters over AA9, including fewer hydrogen-bond acceptors, lower polar surface area, and fewer rotatable bonds, all of which may help improve bioavailability. Wodtke et al. described Nε-acryloyllysine piperazines in which substituents at the α-amino group and the piperazine ring were modified, resulting in compounds like 7b, which is a potent and selective TGase 2 inhibitor.61 In future work, this compound was modified to its 18F analogue to facilitate physiological studies of TGase 2 both in vitro and in vivo.62 Similarly, via a palladium-mediated [11C]CO aminocarbonylation reaction, van der Wildt et al. described the synthesis of Nε-acryloyllysine carbon-11-labeled PET tracers to further understand TG2 biology.63
The first reported reversible inhibitors of TG2 contained a thieno[2,3-d]pyrimidin-4-one core.64,65 Case and Stein later reported on compound LDN-27219, a reversible and slow-binding inhibitor that appeared to not bind to TG2’s active site but at the GTP site.66 Recent studies have also shown that competitive, reversible cinnamoyl-based inhibitors of TG2, like compound 15a, showed promising in vitro activity against TG2.67 In 2014, Kim and co-workers demonstrated that reversible TG2 inhibitor GK921 showed promise against renal cell carcinoma in xenograft models.68 There are several reports of compounds that disrupt the TG2/fibronectin interaction. Khanna et al. reported that compound ITP-79 was able to disrupt this interaction in ovarian cancer cells.69 Our group has recently reported on a novel series of TG2 inhibitors.40 This class of aminopyrimidine compounds, as exemplified by TG53 (Figure 1), inhibited the TG2/fibronectin interaction and was initially identified using an AlphaLISA-based high-throughput screen. Subsequent work modified this initial hit to improve potency and solubility and resulted in inhibitor MT4.41
Based on our previously described TG2 inhibitors,40,41 we hypothesized that we could develop a protein degrader for TG2, which would abolish all of its pro-tumorigenic functions. Here, we describe this series of novel TG2-targeting protein degraders. We prepared a small library of bifunctional molecules incorporating ligands for either cereblon (CRBN) or Von Hippel-Lindau (VHL) protein and connected them via poly(ethylene glycol) (PEG) linkers to our TG2 ligand. Screening of these compounds for TG2 degradation in ovarian cancer cells identified two molecules that caused protein degradation. We confirmed the proteasome-dependent mechanism of action of these compounds using competition assays and negative control derivatives. Finally, as an in vitro proof of concept, we showed that these PROTACs inhibit TG2-mediated ovarian cancer migration. These results demonstrate the feasibility and utility of pursuing a targeted protein degradation approach for TG2 that has the potential to ameliorate ovarian cancer growth and metastasis.
Results and Discussion
Synthesis of New TG2 PROTACs
We synthesized a small library of heterobifunctional derivatives to assess their potential to degrade TG2 (Figure 2). We chose to use our previously reported TG2 inhibitor (MT4, Figure 1) for our TG2-binding ligand,41 as this compound has better solubility than another compound (TG53, Figure 1) that we have also recently published,9 it is more potent in cellular experiments than TG53,41 and we have previously characterized its binding affinity to TG2.41 To determine an initial attachment point for the linkers of our bifunctional compounds, we first docked MT4 into the fibronectin-binding site of the crystal structure of TG2 (PDB 4PYG) to gain an understanding of the most likely solvent-accessible site (Supplementary Figure 1). Based on the docked pose, several potential interactions were identified that contribute to ligand binding, including a hydrogen bond between the amide N–H and Ser101 and a hydrogen bond between the protonated pyrimidine and Asp94 (Supplementary Figure S1A). The phenyl ring appears to be positioned in the interior of the protein in a hydrophobic region formed by Leu12, Leu14, and Trp40. A potential hydrogen bond between Thr42 and the benzyl group pi system may also be present. In contrast, the dimethylamino group appears to be positioned toward the solvent-accessible region of the binding site (Supplementary Figure S1B). Based on this, as well as the relatively straightforward nucleophilic aromatic substitution chemistry needed to install linker amines at the pyrimidine-4-position, we elected to prepare our initial set of PROTACs with linkers coming off the amine group.
Figure 2.
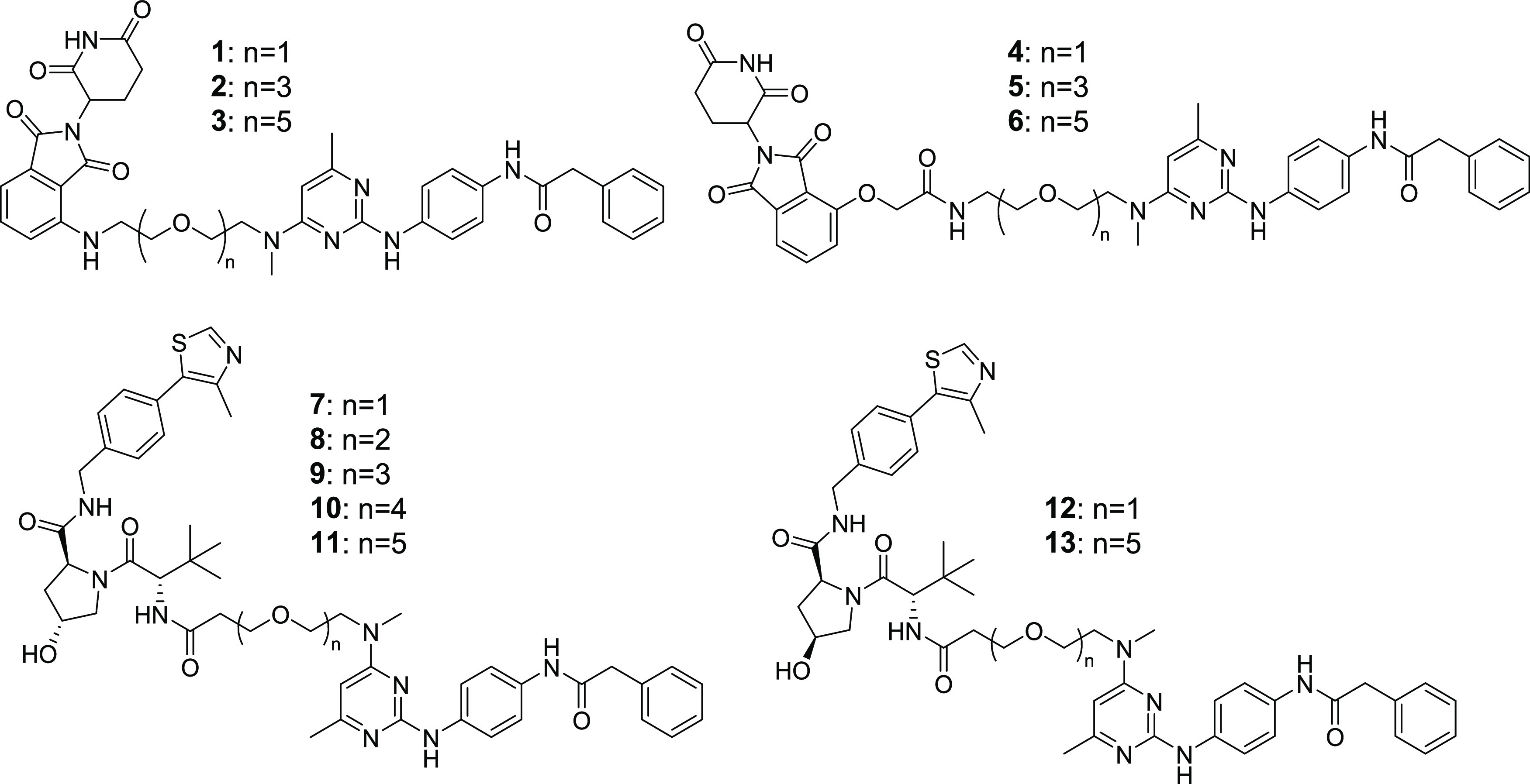
Structures of TG2 PROTACs.
Our compound set included CRBN-binding ligands attached via either amines or ethers, as well as a VHL ligand attached by its terminal amine. All compounds used poly(ethylene glycol) (PEG) linkers of varying lengths. The synthesis of our CRBN-based compounds began with reacting 2,4-dichloro-6-methylpyrimidine (14) with amino-PEGn=1–3 linkers to give 4-amine-linked derivatives 15–17 in 20–39% yields (Scheme 1). Following this amination, displacement of the chlorine in the 2-position of the pyrimidine ring was done using commercially available N-(4-aminophenyl)-2-phenyl-acetamide in the presence of acetic acid in isopropanol to afford 2,4-diaminopyrimidines 18–20 in good-to-moderate yields. The hydroxyl group was tosylated, followed by a reaction with sodium azide to give azides 24–26 in good yields. Hydrogenation of the azide to amines 27–29 followed by a nucleophilic aromatic substitution reaction with 2-(2,6-dioxopiperidin-3-yl)-4-fluoroisoindoline-1,3-dione gave final compounds 1–3. Alternatively, an amide coupling reaction between amines 27–29 and 2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetic acid gave compounds 4–6. For VHL-based PROTACs (Scheme 2), the commercially available methylamino-PEGn-t-butyl esters were reacted with 2,4-dichloro-6-methylpyrimidine (14) to give the PEGn=1–5 pyrimidines 30–34 in 32–82% yields. Nucleophilic aromatic displacement of the pyrimidines with N-(4-aminophenyl)-2-phenyl-acetamide in refluxing isopropanol delivered the 2,4-diamino-substituted intermediates 35–39. The t-butyl ester was deprotected using HCl to give the carboxylic acids (40–44) in good yield. This was followed by an amide coupling with the appropriate VHL ligand to give desired compounds 7–11 as shown in Scheme 2. To prepare the negative control versions of active degraders 7 and 11, in which the stereochemical configuration of the C4 center on the VHL ligand pyrrolidine ring was inverted, we carried out a 2-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU)-promoted amide coupling of acids 40 and 44 with the commercially available S,S,S-isomer to give negative control compounds 12 and 13 in moderate yields.
Scheme 1. Synthesis of New CRBN-Based TG2 Degrader Compounds.
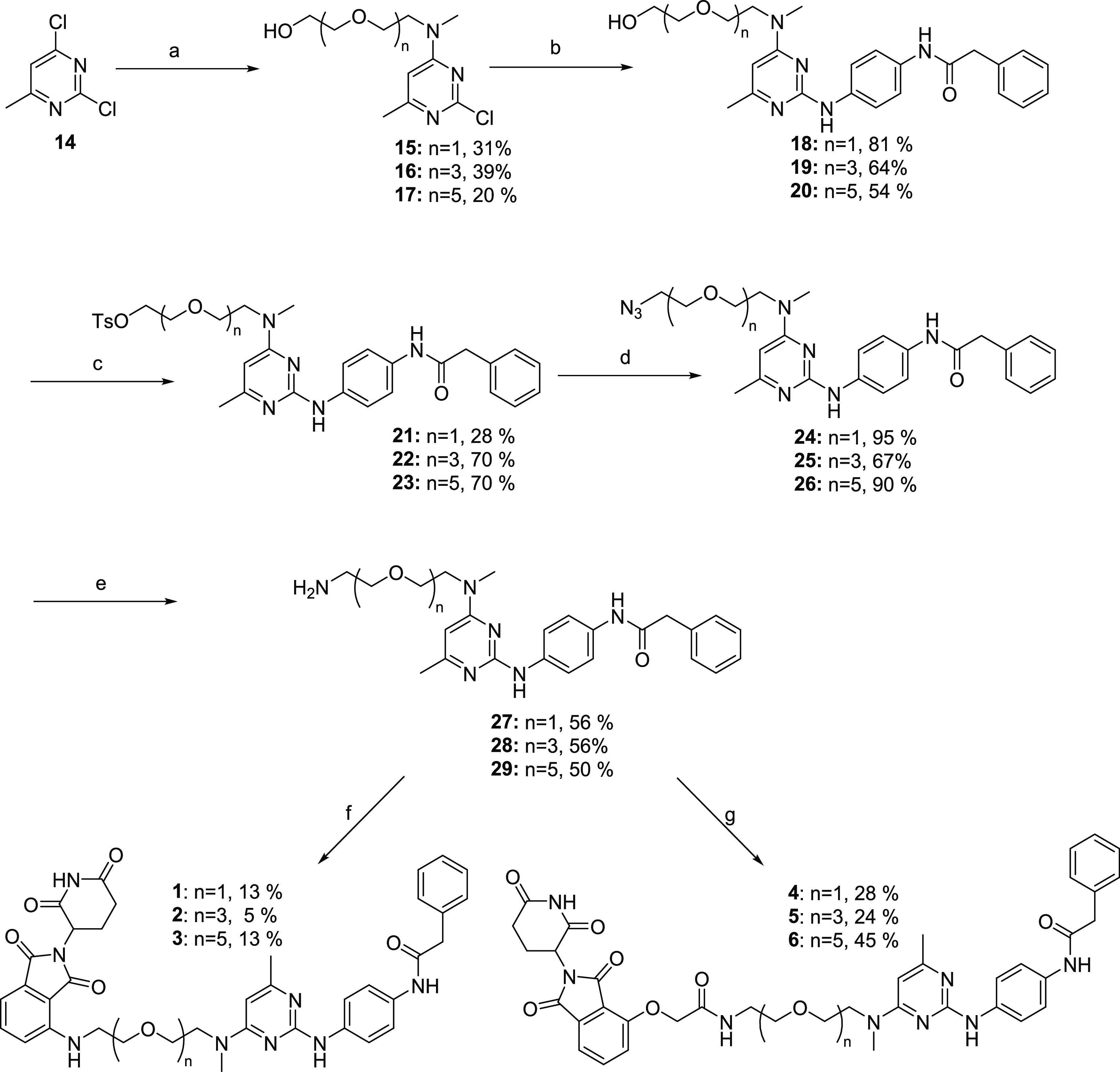
Reagents and conditions: (a) diisopropylamine (DIPEA), tetrahydrofuran (THF), methylamino-PEGn-OH, 20 °C, 12 h; (b) N-(4-aminophenyl)-2-phenyl-acetamide, AcOH, Isopropanol (IPA), 80 °C, 12 h; (c) TosCl, triethylamine (TEA), dichloromethane (DCM), 20 °C, 8 h; (d) NaN3, dimethylformamide (DMF), 50 °C, 12 h; (e) H2, Pd/C, NH4OH, MeOH, 20 °C, 12 h; (f) 2-(2,6-dioxopiperidin-3-yl)-4-fluoroisoindoline-1,3-dione, DIPEA, dimethylsulfoxide (DMSO), 60 °C, 12 h; and (g) 2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetic acid, HATU, DIPEA, DMF, 20 °C, 12 h.
Scheme 2. Synthesis of New VHL-Based TG2 Degrader Compounds.
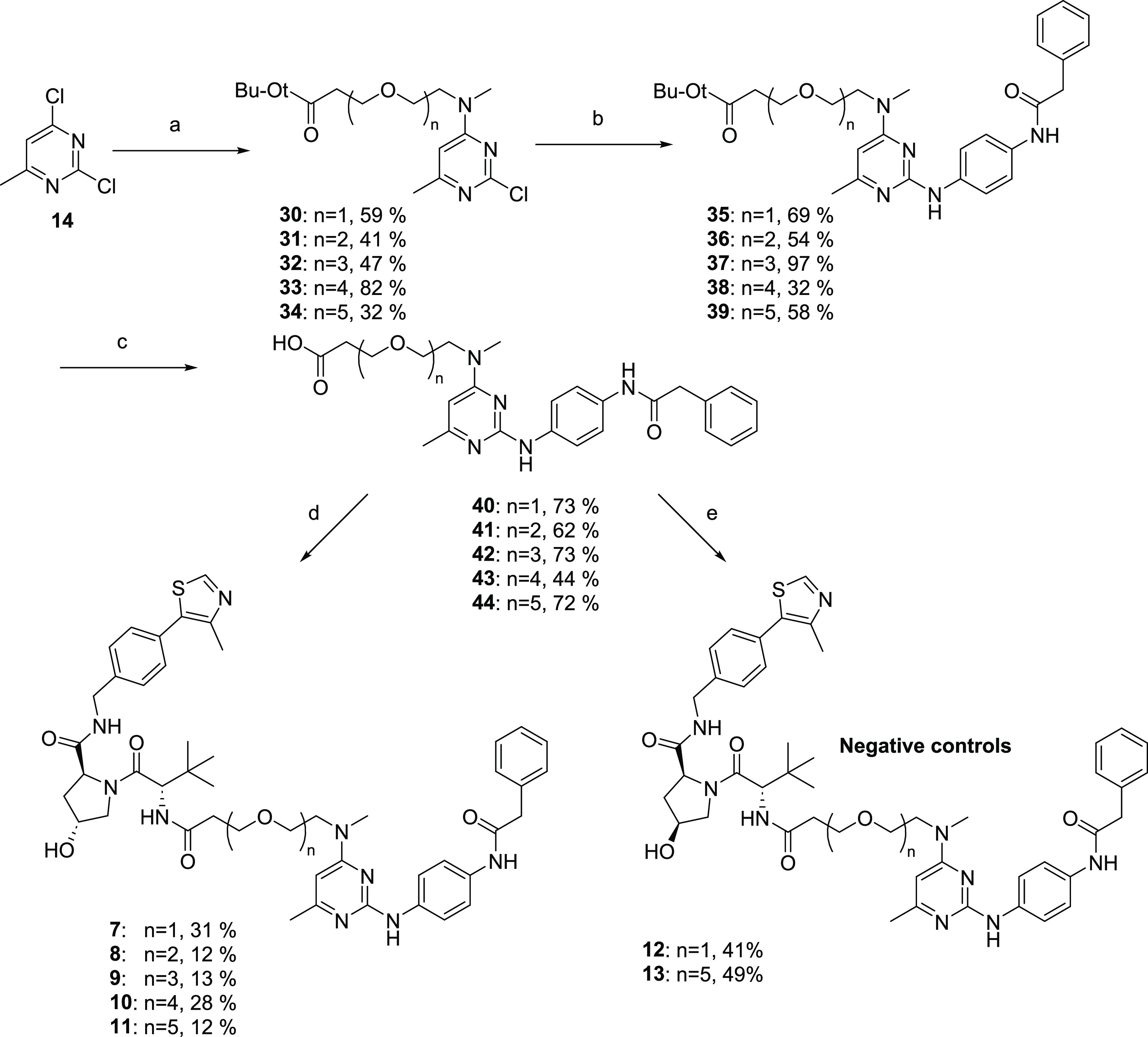
Reagents and conditions: (a) DIPEA, THF, methylamino-PEGn-t-butyl ester, 0 to 15 °C, 10 h; (b) N-(4-aminophenyl)-2-phenyl-acetamide, AcOH, IPA, 80 °C, 12 h; (c) HCl in ethyl acetate (4 M), 25 °C, 12 h; (d) (S,R,S)-AHPC, HATU, DIPEA, DMF, 25 °C, 12 h; and (e) (S,S,S)-AHPC hydrochloride, HATU, DIPEA, DMF, 25 °C, 12 h.
Screening for TG2 Degradation in Ovarian Cancer Cell Lines
We tested whether the synthesized compounds induced TG2 degradation in ovarian cancer cell lines OVCAR5 and SKOV3 using concentration range (0.1–10 μM) and time course (6–24 h) experiments. The limited solubility of the compounds precluded their evaluation at concentrations above 10 μM. Among this set of compounds, 7 and 11 both significantly reduced the level of TG2 within 6 h, in a concentration-dependent manner in both OVCAR5 and SKOV3 cells (Figure 3A,B and E,F), whereas no TG2 degradation was obtained after treatment with the other compounds (data not included). However, longer (24 h) treatment with compounds 7 and 11 did not induce reduction in TG2 levels (Figure 3C,D and G,H). To further study the dynamics of TG2 degradation induced by our active degraders, SKOV3 cells were treated with different concentrations of compounds 7 and 11 at different time points. Using western blotting analysis, we observed a concentration-dependent response (0.1, 0.3, 1, 3, 10, 30 μM) after 2-, 4-, 6-, and 12-hour treatment with 7 (Figure 3I, n = 3 replicates, Supplementary Figure S2) and 11 (Figure 3J, n = 3 replicates, Supplementary Figure S2). Both compounds induced TG2 degradation in a time-dependent manner, with maximal degradation observed at 6 h using higher concentrations (10 and 30 μM). After this degradation maximum, the TG2 levels started to increase at 12 h, returning to levels close to control at 24 h. However, repeated dosing with compounds 7 or 11 over a 24-hour period (every 6–8 h) resulted in persistent and dose-dependent decreased TG2 levels at 24 h in both OVCAR5 and SKOV3 cells (Figure 3K,L). To exclude the possibility that the observed decreased TG2 levels induced by our degraders were related to cell death, viability assays were carried out. For this, we treated OVCAR5 and SKOV3 cells with compounds 7 and 11 every 6–8 h for a period of 5 days. No significant cell death was observed after treatment with any concentrations of compounds 7 (Figure 3M) or 11 (Figure 3N).
Figure 3.

Characterization of TG2 degradation in OVCAR5 and SKOV3 cells. (A) Western blot analysis of TG2 in OVCAR5 cells treated with compounds 7 and 11 at 0.1, 1, and 10 μM at 6 h. DMSO was used as a control. (B) Western blot analysis of TG2 in SKOV3 cells treated with compounds 7 and 11 for 6 h at 1 and 10 μM. DMSO was used as a control. (C) Western blot analysis of TG2 in OVCAR5 cells treated with compounds 7 and 11 at 0.1, 1, and 10 μM at 24 h. DMSO was used as a control. (D) Western blot analysis of TG2 in SKOV3 cells treated with compounds 7 and 11 for 24 h at 1 and 10 μM. DMSO was used as a control. (E) Quantification of western blots is shown in (A) (n = 3 experimental replicates; *p < 0.05; ***p < 0.001; ****p < 0.0001). (F) Quantification of western blots is shown in (B) (n = 3 experimental replicates; **p < 0.01). (G) Quantification of western blots shown in (C) (n = 3 experimental replicates; n.s.). (H) Quantification of western blot shown in (D) (n = 3 experimental replicates; n.s.). (I) Kinetics of TG2 degradation with compound 7 based on western blot analysis in SKOV3 cells. Western blot for TG2 was performed at 0, 2, 6, 12, and 24 h using 0.1, 0.3, 1, 3, 10, and 30 μM of Compound 7. Maximum inhibition was observed at 6 h using the highest concentrations (n = 3 experimental replicates included in Supplementary Figure S2; ***p < 0.001; ****p < 0.0001). (J) Kinetics of TG2 degradation with compound 11 based on western blot analysis in SKOV3 cells. Western blot for TG2 was performed at 0, 2, 6, 12, and 24 h using 0.1, 0.3, 1, 3, 10, and 30 μM of Compound 11. Maximum inhibition was observed at 6 and 12 h using the highest concentrations (n = 3 experimental replicates included in Supplementary Figure S2; ***p < 0.001; ****p < 0.0001). (K) Western blot analysis of compounds 7 and 11 dosed every 6–8 h over 24 h in OVCAR5 cells at doses of either 1 or 10 μM. (L) Western blot analysis of compounds 7 and 11 dosed every 6–8 h over 24 h in SKOV3 cells at doses of either 1 or 10 μM. (M) The cell viability assay of OVCAR5 and SKOV3 cells treated with compound 7; no cell death was observed in either cell line when using 0.1, 0.3, 1, 3, 10, and 30 μM over the course of 5 days, dosed every 6–8 h (n = 3 replicates; n.s.). (N) The cell viability assay of OVCAR5 and SKOV3 cells treated with compound 11; no cell death was observed in either cell line using 0.1, 0.3, 1, 3, 10, and 30 μM over the course of 5 days, dosed every 6–8 h (n = 3 replicates; n.s.).
Degradation of TG2 is Proteasome-Dependent
PROTACs act by engaging proteasome-dependent protein degradation mechanisms. To determine whether the compounds required proteasome activity for the degradation of TG2, we tested their effects in the presence or absence of the proteasome inhibitor MG132. Treatment with MG132 prevented TG2 degradation in both OVCAR5 (Figure 4A,B) and SKOV3 (Figure 4C,D) cells, supporting the proteasome-dependent nature of TG2 degradation. To further confirm this mechanism, we tested whether treatment with the von Hippel-Lindau (VHL) ligand (VHL-L) would compete with the bifunctional degraders for VHL binding, thereby abrogating their effects. Indeed, treatment with VHL-L did not alter levels of TG2, while co-treatment of VHL-L with either 7 or 11 prevented degradation of TG2 in both OVCAR5 (Figure 4E,F) and SKOV3 (Figure 4G,H) cells. To further establish that degradation by compounds 7 and 11 is proteasome-dependent, we prepared negative control versions of these degraders. These compounds, 12 and 13 (Scheme 2), have inverted configurations of the C4 stereocenter of the pyrrolidine ring and are unable to bind VHL,70,71 and therefore do not induce proteasome-dependent degradation. Treatment of OVCAR5 cells (Figure 4I,K) or SKOV3 cells (Figure 4J,L) with compounds 12 and 13 for 6 h did not induce TG2 degradation at either 0.1, 1, or 10 μM in three separate replicates, demonstrating that recruitment of the VHL E3 ligase complex by 7 and 11 is necessary for the degradation of TG2.
Figure 4.

TG2 degradation is proteasome-dependent. (A) Western blot analysis for TG2 in OVCAR5 cells treated with 10 μM of compounds 7 and 11 for 6 h and the proteasome inhibitor MG132 (20 μM) for 8 h. (B) Quantification of the western blot is shown in (A). Bars represent band intensity for each condition relative to control (DMSO) (n = 3 replicates; ****p < 0.0001). (C) Western blot analysis for TG2 in SKOV3 cells using the same conditions as in (A). (D) Quantification of the western blot is shown in (C) (n = 3 replicates; ****p < 0.0001). (E) Western blot analysis for TG2 in OVCAR5 cells treated with 10 μM of Compounds 7 and 11 for 6 h along with 10 μM of the VHL ligand as a competitive inhibitor. (F) Quantification of the western blot is shown in (E) (n = 3 replicate; **p < 0.01). (G) Western blot analysis for TG2 in SKOV3 cells treated using the same conditions as in (E). (H) Quantification of the western blot is shown in (G) (n = 3 replicates; **p < 0.01; ***p < 0.001). (I) Western blot analysis using negative control compounds 12 and 13 in OVCAR5 cells. (J) Western blot analysis using negative control compounds 12 and 13 in SKOV3 cells. (K–L) Quantification of the western blot is shown in (I) and (J) (n = 3 replicates; n.s.).
Degradation of TG2 Inhibits Ovarian Cancer Cell Migration
After demonstrating that compounds 7 and 11 effectively degrade TG2 in OC cell lines, we tested their functional effects. It has been established that the genetic knockdown of TG2 using shRNAs reduces the migration and invasion of cancer cells by blocking TG2-induced EMT.20,41 We confirmed that TG2 knockdown in OVCAR5 cells decreased cell migration, as measured through a scratch assay (Supplementary Figure S3), supporting the role of TG2 in cell motility. We therefore tested the effects of the TG2 degraders on cell migration and invasion by using wound-healing and transwell migration assays. Degraders 7 and 11 each potently inhibited the migration of OVCAR5 cells in both wound-healing assays (Figure 5A, p < 0.05) and transwell migration assays (Figure 5B, p < 0.05). We found that compounds 7 and 11 similarly inhibited the migration of SKOV3 cells (Figure 5C, p < 0.05 and Figure 5D, p < 0.05) in wound-healing and transwell migration assays, respectively. As TG2 is involved in the adhesion of cells on fibronectin,41,72 we also tested the effects of the TG2 degraders on the adhesion of OVCAR5 and SKOV3 cells to fibronectin. Treatment with 7 and 11 significantly reduced the adhesion of cells to fibronectin (Figure 5E, p < 0.05). On the other hand, treatment of both OVCAR5 and SKOV3 cells with inactive degraders 12 and 13 did not affect cell migration (Supplementary Figure 4A,B) and adhesion (Supplementary Figure 4C). Altogether, these results indicate that targeted protein degradation of TG2 has significant functional effects on the phenotype of ovarian cancer cells, affecting migration, invasion, and cell adhesion to the matrix.
Figure 5.

Compounds 7 and 11 reduce the migration and adhesion capability of cancer cells. Cell migration was analyzed on both OVCAR5 and SKOV3 cells. (A) The wound-healing assay of the OVCAR5 cells treated with compounds 7 and 11, including quantification (n = 3 replicates; *p < 0.05). (B) The transwell migration assay of the OVCAR5 cells treated with compounds 7 and 11, including quantification (n = 3 replicates; *p < 0.05). (C) The wound-healing assay of the SKOV3 cells treated with compounds 7 and 11 and quantification (n = 3 replicates; ****p < 0.0001). (D) The transwell migration assay of the SKOV3 cells (n = 3; *p < 0.05). (E) The solid phase assay measures the adhesion of OVCAR5 and SKOV3 cells to fibronectin in the presence of control or of compounds 7 and 11 (n = 3 replicates; ***p < 0.001; ****p < 0.0001).
MT4 and PROTAC Bind to TG2
To confirm the direct binding of our PROTAC with TG2 and evaluate the effect of modifying our inhibitor into a bifunctional degrader on affinity, we carried out binding studies using isothermal calorimetry (ITC). We first measured the affinity of our parental inhibitor, MT4, toward full-length TG2.40 Titration of MT4 into a solution of TG2 showed an equilibrium dissociation constant KD = 7.8 μM (Figure 6A), which is in close agreement with the result we recently published for this compound (5.1 μM).41 We then tested the binding affinity of the two active PROTACs 7 and 11 to TG2. Compound 11, with a PEG5 linker, showed a KD = 68.9 μM, which was a decrease of 8.8-fold relative to the parental compound (Figure 6B). Compound 7 did not show a significant dose-dependent saturation under these experimental conditions, indicating its KD > 100 μM or a >12.8-fold decrease in affinity (Supplementary Figure S8).
Figure 6.

Isothermal calorimetry (ITC) binding studies of compounds to TG2. (A) Calorimetric titrations of 100 μM MT4 into 2 μM TG2 at 30 °C; each peak corresponds to a single injection of 2 μL. Binding of MT4 to TG2 shows a KD = 7.8 μM. The c value for this experiment is 0.26. (B) Calorimetric titrations of 200 μM 11 into 20 μM TG2 at 30 °C; each peak corresponds to a single injection of 2 μL. The binding of 11 to TG2 shows a KD = 68.9 μM. The c value for this experiment is 0.29.
Conclusions
Over the last 10–15 years, there has been increasing awareness that TG2 plays a key role in a variety of tumorigenic and metastatic processes. We and others have made important discoveries showing that TG2 exerts several functions that drive pro-tumorigenic activity and has therefore been an attractive target for drug discovery.
However, in spite of significant efforts, no clinically effective TG2-targeting small molecules have been developed to date for cancer models. Given the multitude of functions exerted by this complex protein, it is likely that disrupting only one particular domain will be insufficient to provide efficacy. Because of the limited ability of traditional small-molecule inhibitors to target multiple protein functions simultaneously, we initiated a PROTAC strategy to target TG2. One of the key applications of targeted degradation approaches is for proteins that possess multiple functions or may serve as scaffold proteins. These targets have traditionally been very difficult to address with small-molecule enzymatic or protein–protein inhibitors. Our hypothesis is that degrading the TG2 protein will serve to abolish its myriad functions and thereby provide more robust and meaningful efficacy in cancers where TG2 plays a key role, such as ovarian cancer.
To test this approach, we used the inhibitor scaffold we have previously described as the PROTAC ligand for our protein of interest.40,41 This compound was attached via PEG linkers of varying lengths to known ligands for CRBN and VHL E3 ligases. This set of compounds was then tested in multiple ovarian cancer cell lines that express TG2 for their ability to degrade the protein. We found that two VHL-containing bifunctional degraders induced the robust reduction of the TG2 protein in multiple ovarian cancer cell lines. Mechanism of action studies using co-treatment with the proteasome inhibitor MG132 or with the excess VHL ligand confirmed the proteasome-dependent nature of degradation. We also synthesized negative control versions of our active degraders that were unable to bind VHL and found that these compounds failed to degrade TG2, further confirming the E3 ligase-dependent nature of degradation. We further evaluated the kinetics of degradation and found that the maximum reduction of TG2 occurred at 6 h, with recovery by 24 h. Importantly, in SKOV3 and OVCAR5 cells, the proposed TG2 PROTACs significantly reduced in vitro migration as assessed by both wound-healing and scratch assays. These effects were not seen with PROTAC-negative control compounds. These “chemical knockdown” experiments excitingly confirmed that TG2 is necessary for the migration of ovarian cancer cells and supported the results of previous studies using genetic methods to deplete TG2.31
To confirm the direct binding of our PROTACs with TG2 and further correlate the functional activity of our degraders to their binding and degradation of TG2, we carried out binding studies using ITC. Encouragingly, our results with the parental TG2 inhibitor MT4 were very close to the value we recently reported.41 The affinity of PROTAC 11 with TG2 showed a clear binding effect, and the KD was measured to be 68.9 μM, which is a 8.8-fold decrease in affinity versus MT4. The decrease in the affinity of the PROTAC relative to the parental inhibitor is not unexpected, as many others have observed this as well, including with BRD4,70,73 where they found a 7.8-fold decrease in affinity for the PROTAC versus the inhibitor for BRD4-BD1, as well as when looking at PROTACs for a large set of kinases.43 This ITC result demonstrates that PROTAC 11 binds TG2 and further supports the proposed proteasome-mediated (i.e., PROTAC) mechanism. This result is also consistent with data presented for inactive control compound 13, which failed to induce TG2 degradation or ovarian cancer cell migration. Data for degrader 7 did not show a binding effect under the experimental conditions we used, which could only show binding affinity up to a KD < 100 μM,74 suggesting that this compound’s affinity is >100 μM. We also observed a strong sensitivity of the TG2 protein to DMSO concentration, which limited our use of DMSO to 1%. The ITC data for 7 could also reflect this compound’s poorer solubility than compound 11 in low DMSO concentrations, as it has a single PEG group in the linker, while 11 has a PEG5 unit, which presumably gives it better solubility.
The TG2 PROTACs described here are the first known targeted degraders of TG2 and provide a unique and useful new set of molecular tools to study the functions of this important protein. Importantly, our approach uses a series of TG2-targeting compounds as the PROTAC ligands that are assumed to bind to the fibronectin-binding site. This site is separate from the transamidase enzymatic site, which is conformationally regulated by the presence of Ca2+ and GTP/GDP binding. By targeting the less-dynamic FN-binding site, we expect our PROTACs to be capable of degrading TG2 under a wider range of conditions. While our results clearly show the dependency of TG2 on migratory phenotypes, further work to optimize their potency, efficacy, and pharmaceutical properties will be necessary to develop them into compounds usable in in vivo cancer models. Work to modify the TG2-binding moiety, the E3 ligase ligand chemistry, and linker composition is ongoing with the goal of optimizing agents to help further define the role of TG2 in disease and potentially as a new therapeutic strategy for a variety of cancers.
Experimental Section
General Chemical Methods
All chemical reagents were obtained from commercial suppliers and used without further purification unless otherwise stated. Anhydrous solvents were purchased from Sigma-Aldrich and dried over 3 Å molecular sieves when necessary. Normal-phase flash column chromatography was performed using Biotage KP-Sil 50 μm silica gel columns and ACS grade solvents on a Biotage Isolera flash purification system. Reverse phase prep conditions are detailed for each compound in the experimental write-up. Analytical thin-layer chromatography (TLC) was performed on EM Reagent 0.25 mm silica gel 60 F254 plates and visualized by UV light. Proton (1H) and carbon (13C) NMR spectra were recorded on a 500 MHz Bruker Avance III with a direct cryoprobe spectrometer. Chemical shifts were reported in ppm (δ) and were referenced using residual nondeuterated solvent as an internal standard (CDCl3 at 7.24 ppm for 1H-NMR and 77.0 ppm for 13C-NMR; CD3OD at 3.33 ppm for 1H-NMR and 47.6 ppm for 13C-NMR; DMSO-d6 at 2.52 ppm for 1H-NMR and 39.9 ppm for 13C-NMR). Proton coupling constants are expressed in hertz (Hz). The following abbreviations were used to denote spin multiplicity for proton NMR: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, brs = broad singlet, dd = doublet of doublets, dt = doublet of triplets, quin = quintet, and tt = triplet of triplets. Low-resolution liquid chromatography/mass spectrometry (LCMS) was performed on a Waters Acquity-H UPLC/MS system with a 2.1 mm × 50 mm, 1.7 μm, reversed-phase BEH C18 column, and LCMS grade solvents. Gradient elution from 95% water + 0.1% formic acid/5% acetonitrile + 0.1% formic acid to 95% acetonitrile + 0.1% formic acid/5% water + 0.1% formic acid over 2 min plus a further minute continuing this mixture at a flow rate of 0.85 mL/min was used as the eluent. Total ion current traces were obtained for electrospray positive and negative ionization (ESI+/ESI−). Reactions were monitored via the integration of the appropriate UV traces, which corresponded to the desired mass of the product. High-resolution mass spectra were obtained using an Agilent 6230 LC-TOF spectrometer in the positive ion mode using electrospray ionization with an Agilent G1312A HPLC pump and an Agilent G1367B autoinjector at the Integrated Molecular Structure Education and Research Center (IMSERC), Northwestern University. All final compounds tested in biological assays are >95% pure by HPLC analysis. HPLC chromatograms of all final compounds are provided in the Supporting Information (SI).
N-(4-((4-((2-(2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide (1)
To a mixture of 2-(2-(methylamino)ethoxy)ethan-1-ol (1.10 g, 9.2 mmol) in tetrahydrofuran (10 mL) were added 2,4-dichloro-6-methylpyrimidine, 14 (1 g, 6.13 mmol), and DIPEA (1.59 g, 2.14 mL). The mixture was stirred at 20 °C for 12 h. LCMS showed that all starting materials were consumed, and around 50% of the desired product could be detected. The reaction mixture was diluted with water (10 mL) and extracted with ethyl acetate (15 mL × 2). The combined organic layers were washed with aqueous NaCl (20 mL × 2), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, dichloromethane/methyl alcohol = 100:0 to 2:1) to give 2-(2-((2-chloro-6-methylpyrimidin-4-yl)(methyl)amino)ethoxy)ethan-1-ol, 15 (0.5 g, yield 31%) as a light yellow solid. 1H-NMR: (400 MHz, CDCl3): δ 1.86–2.01 (m, 1H), 2.35 (s, 3H), 3.11 (br s, 3H), 3.54–3.62 (m, 2H), 3.67–3.85 (m, 6H), 6.15–6.25 (m, 1H).
To a mixture of N-(4-aminophenyl)-2-phenylacetamide (920 mg, 4.07 mmol) in isopropan-2-ol (5 mL) were added 2-(2-((2-chloro-6-methylpyrimidin-4-yl)(methyl)amino)ethoxy)ethan-1-ol, 15 (0.5 g, 2.03 mmol), and acetic acid (8.64 mg, 143.79 μmol, 8.22 μL). The mixture was stirred at 80 °C for 12 h. LCMS showed that all starting materials were consumed, and 60% of the desired compound was detected. The reaction mixture was diluted with water (15 mL) and extracted with ethyl acetate (20 mL). The combined organic layers were washed with aqueous NaCl (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, dichloromethane/methyl alcohol = 100:0 to 3:1) to give N-(4-((4-((2-(2-hydroxyethoxy)ethyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 18 (0.8 g, yield 81%), as a light yellow solid. 1H-NMR: (400 MHz, DMSO-d6) δ 1.24–1.30 (m, 1H), 2.25–2.31 (m, 3H), 3.10–3.19 (m, 3H), 3.39–3.50 (m, 4H), 3.61–3.64 (m, 4H), 3.67–3.83 (m, 2H), 4.49–4.68 (m, 1H), 6.29–6.41 (m, 1H), 7.21–7.27 (m, 1H), 7.29–7.38 (m, 4H), 7.44–7.52 (m, 2H), 7.53–7.64 (m, 2H), 9.67–9.81 (m, 1H), 10.19–10.30 (m, 1H).
A mixture of N-(4-((4-((2-(2-hydroxyethoxy)ethyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 18 (0.5 g, 1.1 mmol), TosCl (262 mg, 1.13 mol), and TEA (232 mg, 2.3 mmol) in DCM (5 mL) was degassed and purged with N2 3 times at 0 °C, and the mixture was stirred at 20 °C for 8 h. LCMS showed that all starting materials were consumed, and 60% of the desired compound was detected. The reaction mixture was diluted with water (20 mL) and extracted with DCM (10 mL). The organic layers were washed with aqueous NaCl (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, dichloromethane/methyl alcohol = 100:0 to 3:1) to give 2-(2-(methyl(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)amino)ethoxy)ethyl-4-methylbenzenesulfonate, 21 (0.2 g, yield 28%), as a light yellow solid. 1H-NMR: (400 MHz, CDCl3): δ 2.21–2.28 (m, 3H), 2.39–2.47 (m, 3H), 2.98–3.06 (m, 3H), 3.52–3.77 (m, 8H), 4.12 (s, 2H), 5.72–5.84 (m, 1H), 6.82–6.90 (m, 1H), 7.29–7.38 (m, 7H), 7.40–7.47 (m, 3H), 7.49–7.55 (m, 2H), 7.73–7.82 (m, 2H).
To a mixture of 2-(2-(methyl(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)amino)ethoxy)ethyl-4-methylbenzenesulfonate, 21 (0.2 g, 0.3 mmol), in DMF (2 mL) was added sodium azide (26 mg, 0.46 mmol). The mixture was stirred at 50 °C for 12 h. LCMS showed that all starting materials were consumed, and 62% of the desired product could be detected. The reaction solution was diluted with water (10 mL) and extracted with ethyl acetate (20 mL). The combined organic layers were washed with aqueous NaCl (5 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by prep-TLC (SiO2, dichloromethane/methyl alcohol = 100:0 to 3:1) to give N-(4-((4-((2-(2-azidoethoxy)ethyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 24 (0.15 g, yield 95%), as a yellow oil. 1H-NMR: (400 MHz, CDCl3): δ 2.14–2.20 (m, 3H), 3.00–3.09 (m, 3H), 3.34–3.40 (m, 2H), 3.56–3.73 (m, 8H), 5.90–6.02 (m, 1H), 7.18–7.35 (m, 5H), 7.39–7.48 (m, 2H), 7.58–7.71 (m, 2H), 8.85–8.98 (m, 1H), 9.94–10.01 (m, 1H).
To a mixture of N-(4-((4-((2-(2-azidoethoxy)ethyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 24 (0.15 g, 0.3 mmol), in methanol (3 mL) and ammonium hydroxide (1 mL) was added Pd/C (60.38 mg, 20%). The mixture was stirred at 20 °C for 12 h under H2 (15 Psi). LCMS showed that all starting materials were consumed, and 67% of the desired compound was detected. The product was filtered, and the filtrate was concentrated under high vacuum at 40 °C to give a white solid. The crude product N-(4-((4-((2-(2-aminoethoxy)ethyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 27 (0.4 g, yield 56%), was used in the next step without further purification. 1H-NMR: (400 MHz, DMSO-d6) δ 2.15–2.18 (m, 3H), 2.59–2.64 (m, 2H), 3.01 (br s, 3H), 3.15–3.19 (m, 2H), 3.35–3.36 (m, 1H), 3.55–3.62 (m, 4H), 3.63–3.73 (m, 2H), 4.05–4.13 (m, 1H), 5.87–6.02 (m, 1H), 7.19–7.27 (m, 1H), 7.29–7.34 (m, 4H), 7.39–7.46 (m, 2H), 7.61–7.67 (m, 2H), 8.81–8.99 (m, 1H), 9.90–10.01 (m, 1H).
To a solution of N-(4-((4-((2-(2-aminoethoxy)ethyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 27 (50 mg, 115.07 μmol), and 2-(2,6-dioxopiperidin-3-yl)-4-fluoroisoindoline-1,3-dione (38.14 mg, 138.08 μmol) in DMSO (1 mL) was added DIPEA (29.74 mg, 230.13 μmol, 40.08 μL). The reaction mixture was stirred at 60 °C for 12 h. LCMS showed that all starting materials were consumed, and 25% of the desired compound was detected. The reaction mixture was filtered, and the filtrate was concentrated under high vacuum at 40 °C. The residue was purified by prep-HPLC (Phenomenex Luna C18 100 mm × 40 mm × 5 μm, mobile phase: [water (0.1%TFA) – ACN]: 25–53%, 8 min) to give N-(4-((4-((2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide (1, 10 mg, yield 13%) as a yellow solid. 1H-NMR (400 MHz, DMSO-d6): δ 1.99 (br s, 1H), 2.18–2.30 (m, 3H), 2.56–2.62 (m, 2H), 2.85 (br s, 1H), 2.94–3.04 (m, 1H), 3.09–3.19 (m, 3H), 3.52 (br s, 2H), 3.63 (s, 6H), 3.75 (br s, 1H), 5.03 (br s, 1H), 6.30–6.56 (m, 1H), 7.01 (d, J = 7.03 Hz, 1H), 7.09 (br s, 1H), 7.21–7.28 (m, 1H), 7.29–7.36 (m, 4H), 7.43 (br s, 2H), 7.48–7.55 (m, 1H), 7.60 (br d, J = 8.82 Hz, 2H), 9.75 (br s, 1H), 10.19 (s, 1H), 11.04–11.17 (m, 1H), 12.33 (br s, 1H). HRMS (ESI+): m/z calcd for C37H38N8NaO6: 713.2807 [M + Na]+; found: 713.2813, [M + Na]+.
2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)-N-(2-(2-(methyl(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)amino)ethoxy)ethyl)acetamide (4)
To a mixture of 2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetic acid (40 mg, 0.11 mmol) and N-(4-((4-((2-(2-aminoethoxy)ethyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 27 (36 mg, 0.11 mmol), in DMF (1 mL) were added DIPEA (23 mg, 191.34 μmol) and HATU (52 mg, 143.50 μmol). The mixture was stirred at 20 °C for 12 h. LCMS showed that all starting materials were consumed, and 25% of the desired compound was detected. The reaction mixture was filtered, and the filtrate was concentrated to give a residue, which was purified by prep-HPLC (Phenomenex Luna C18 100 mm × 40 mm × 5 μm, mobile phase: [water (0.1%TFA) – ACN]: 25–53%, 8 min) to give 2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)-N-(2-(2-(methyl(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)amino)ethoxy)ethyl)acetamide (4, 19 mg, yield 28%), as a white solid. 1H-NMR (400 MHz, DMSO-d6): δ 2.01 (br d, J = 10.76 Hz, 1H), 2.25 (s, 3H), 2.58 (br s, 2H), 2.77–2.95 (m, 2H), 3.14 (br s, 3H), 3.30 (br s, 2H), 3.39–3.43 (m, 2H), 3.62 (s, 4H), 3.74 (br s, 1H), 4.74 (s, 2H), 5.10 (dd, J = 12.90, 5.20 Hz, 1H), 6.21–6.56 (m, 1H), 7.20–7.28 (m, 1H), 7.32 (d, J = 4.77 Hz, 3H), 7.36–7.45 (m, 3H), 7.49 (br d, J = 6.97 Hz, 1H), 7.59 (br d, J = 8.80 Hz, 2H), 7.75–7.83 (m, 1H), 7.89 (br s, 1H), 9.68 (br s, 1H), 10.18 (s, 1H), 11.12 (br s, 1H).
HRMS (ESI+): m/z calcd for C39H41N8O8: 749.3042 [M + H]+; found: 749.3045 [M + H]+.
N-(4-((4-((2-(2-(2-(2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethoxy)ethyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide (2)
To a mixture of 2-((1-(2-methoxyethoxy)-3-(methylamino)propan-2-yl)oxy)ethan-1-ol (2.86 g, 13.80 mmol) in tetrahydrofuran (28 mL) were added 2,4-dichloro-6-methylpyrimidine, 14 (1.5 g, 9.20 mmol), and DIPEA (2.38 g, 18.40 mmol, 3.21 mL). The mixture was stirred at 20 °C for 12 h. LCMS showed that all starting materials were consumed, and around 50% of the desired product could be detected. The reaction mixture was diluted with water (20 mL) and extracted with ethyl acetate (20 mL × 2). The combined organic layers were washed with aqueous NaCl (20 mL × 2), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, dichloromethane/methyl alcohol = 100:0 to 10:1) to give 2-(2-chloro-6-methylpyrimidin-4-yl)-5,8,11-trioxa-2-azatridecan-13-ol, 16 (1.34 g, yield 39%), as a light yellow solid. 1H-NMR: (400 MHz, CDCl3) δ 2.27 (s, 3H), 3.04 (br s, 3H), 3.52–3.57 (m, 9H), 3.57–3.62 (m, 5H), 3.63–3.67 (m, 3H), 6.14 (br s, 1H).
To a mixture of N-(4-aminophenyl)-2-phenylacetamide (1.30 g, 5.76 mmol) in isopropan-2-ol (13 mL) were added 2-(2-chloro-6-methylpyrimidin-4-yl)-5,8,11-trioxa-2-azatridecan-13-ol, 16 (0.48 g, 1.44 mmol), and acetic acid (8.64 mg, 143.79 μmol, 8.22 μL). The mixture was stirred at 80 °C for 12 h. LCMS showed that all starting materials were consumed, and 60% of the desired compound was detected. The reaction mixture was diluted with water (15 mL) and extracted with ethyl acetate (20 mL). The combined organic layers were washed with aqueous NaCl (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, dichloromethane/methyl alcohol = 100:0 to 10:1) to give N-(4-((4-((2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 19 (1.5 g, yield 64%), as a light yellow solid. 1H-NMR: (400 MHz, DMSO-d6) δ 2.31 (br d, J = 8.07 Hz, 3H), 3.18 (br d, J = 7.95 Hz, 3H), 3.38 (br d, J = 5.01 Hz, 4H), 3.43–3.48 (m, 11H), 3.65 (s, 3H), 6.38–6.51 (m, 1H), 7.25 (br d, J = 6.85 Hz, 1H), 7.29–7.37 (m, 4H), 7.40–7.49 (m, 2H), 7.63 (br d, J = 8.07 Hz, 2H), 10.10–10.18 (m, 1H), 10.34 (br s, 1H), 12.66 (br s, 1H).
To a mixture of N-(4-((4-((2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 19 (1.5 g, 2.86 mmol), in dichloromethane (15 mL) were added tosyl chloride (655.38 mg, 3.44 mmol) and triethylamine (579.74 mg, 5.73 mmol, 797.45 μL). The mixture was stirred at 20 °C for 12 h. LCMS showed that all starting materials were consumed, and 60% of the desired compound was detected. The reaction mixture was diluted with water (20 mL) and extracted with ethyl acetate (20 mL). The organic layers were washed with aqueous NaCl (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, dichloromethane/methyl alcohol = 100:0 to 10:1) to give 2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11-trioxa-2-azatridecan-13-yl 4-methylbenzenesulfonate, 22 (1.5 g, yield 70%), as a light yellow solid. 1H-NMR: (400 MHz, CDCl3): δ 2.24–2.29 (m, 1H), 2.27 (s, 2H), 2.43 (s, 3H), 3.10 (s, 3H), 3.57 (d, J = 2.93 Hz, 8H), 3.65–3.69 (m, 4H), 3.73 (s, 4H), 5.84 (s, 1H), 7.30–7.43 (m, 11H), 7.52 (br d, J = 8.68 Hz, 2H), 7.78 (d, J = 8.19 Hz, 2H).
To a mixture of 2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11-trioxa-2-azatridecan-13-yl 4-methylbenzenesulfonate, 22 (1 g, 1.48 mmol), in DMF (10 mL) was added sodium azide (115.09 mg, 1.77 mmol). The mixture was stirred at 50 °C for 12 h. LCMS showed that all starting materials were consumed, and 64% of the desired product could be detected. The reaction solution was diluted with water (10 mL) and extracted with ethyl acetate (20 mL). The combined organic layers were washed with aqueous NaCl (5 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by prep-TLC (SiO2, dichloromethane/methyl alcohol = 100:0 to 5:1) to give N-(4-((4-((2-(2-(2-(2-azidoethoxy)ethoxy)ethoxy)ethyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 25 (0.6 g, yield 67%), as a dark brown solid. 1H-NMR: (400 MHz, CDCl3) δ 2.28 (s, 3H), 3.11 (s, 3H), 3.35–3.39 (m, 2H), 3.58–3.62 (m, 1H), 3.61 (br d, J = 4.40 Hz, 3H), 3.64 (s, 4H), 3.65–3.70 (m, 4H), 3.74 (s, 4H), 5.84 (s, 1H), 7.11 (br s, 1H), 7.32–7.44 (m, 8H), 7.52 (br d, J = 8.80 Hz, 2H).
To a mixture of N-(4-((4-((2-(2-(2-(2-azidoethoxy)ethoxy)ethoxy)ethyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 25 (0.6 g, 1.09 mmol), in methanol (9 mL) and ammonia hydrate (3 mL) was added Pd/C (116.38 mg, 20%). The mixture was stirred at 20 °C for 12 h under H2 (15 Psi). LCMS showed that all starting materials were consumed, and 70% of the desired compound was detected. The product was filtered, and the filtrate was concentrated under high vacuum at 40 °C to give a white solid. The crude product N-(4-((4-((2-(2-(2-(2-aminoethoxy)ethoxy)ethoxy)ethyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 28 (0.4 g, yield 56%), was used in the next step without further purification. 1H-NMR: (400 MHz, CDCl3) δ 2.20 (br s, 1H), 2.26 (s, 3H), 2.34–2.42 (m, 1H), 2.86 (t, J = 5.14 Hz, 2H), 3.08 (s, 3H), 3.49–3.53 (m, 2H), 3.60 (br s, 8H), 3.64–3.69 (m, 2H), 3.74 (s, 4H), 5.82 (s, 1H), 7.30–7.45 (m, 8H), 7.47–7.55 (m, 3H).
To a mixture of N-(4-((4-((2-(2-(2-(2-aminoethoxy)ethoxy)ethoxy)ethyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 28 (100 mg, 191.34 μmol), in DMSO (1 mL) were added 2-(2,6-dioxopiperidin-3-yl)-4-fluoroisoindoline-1,3-dione (63.42 mg, 229.60 μmol) and DIPEA (49.46 mg, 382.67 μmol, 66.65 μL). The mixture was stirred at 60 °C for 12 h. LCMS showed that all starting materials were consumed, and 30% of the desired compound was detected. The reaction mixture was filtered, and the filtrate was concentrated under high vacuum at 40 °C. The residue was purified by prep-HPLC (Phenomenex Luna C18 100 mm × 40 mm × 5 μm, mobile phase: [water (0.1%TFA) – ACN]: 25–53%, 8 min) to give N-(4-((4-((2-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethoxy)ethyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide (2, 8 mg, yield 5%) as a yellow solid. 1H-NMR: (400 MHz, DMSO-d6) 2.01 (br s, 1H), 2.32 (br s, 3H), 2.59 (br s, 2H), 2.80–2.92 (m, 1H), 3.11–3.18 (m, 3H), 3.48 (br d, J = 12.23 Hz, 10H), 3.58 (br t, J = 5.32 Hz, 4H), 3.61–3.77 (m, 5H), 5.04 (dd, J = 12.90, 5.69 Hz, 1H), 6.33–6.51 (m, 1H), 7.03 (d, J = 6.97 Hz, 1H), 7.10 (d, J = 8.56 Hz, 1H), 7.25 (br d, J = 3.18 Hz, 1H), 7.29–7.36 (m, 4H), 7.43 (br s, 2H), 7.52–7.64 (m, 3H), 9.67–9.90 (m, 1H), 10.20 (s, 1H), 11.09 (s, 1H), 12.26 (s, 1H). HRMS (ESI+): m/z calcd for C41H47N8O8: 779.3512 [M + H]+; found: 779.3533 [M + H]+.
2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)-N-(2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11-trioxa-2-azatridecan-13-yl)acetamide (5)
To the mixture of 2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetic acid (38.14 mg, 114.80 μmol, 1.2 equiv) and N-(4-((4-((2-(2-(2-(2-aminoethoxy)ethoxy)ethoxy)ethyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 28 (50 mg, 95.67 μmol, 1 equiv), in DMF (1 mL) were added DIPEA (24.73 mg, 191.34 μmol, 2 equiv) and HATU (54.56 mg, 143.50 μmol, 1.5 equiv). The mixture was stirred at 20 °C for 12 h. LCMS showed that all starting materials were consumed, and 30% of the desired compound was detected. The reaction mixture was filtered, and the filtrate was concentrated to give a residue, which was purified by prep-HPLC (Phenomenex Luna C18 100 mm × 40 mm × 5 μm, mobile phase: [water (0.1%TFA) – ACN]: 25–53%, 8 min) to give 2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)-N-(2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11-trioxa-2-azatridecan-13-yl)acetamide, 5 (20 mg, yield 24%), as a white solid. 1H-NMR (400 MHz, DMSO-d6): 2.04 (br s, 1H), 2.30 (br s, 3H), 2.56 (br s, 1H), 2.59–2.62 (m, 1H), 2.59–2.62 (m, 1H), 2.60 (br s, 1H), 2.83–2.94 (m, 1H), 3.15 (s, 3H), 3.29 (br d, J = 5.14 Hz, 1H), 3.46–3.48 (m, 9H), 3.58 (br s, 2H), 3.63 (s, 3H), 3.72 (br d, J = 16.26 Hz, 2H), 4.77 (s, 2H), 5.07–5.19 (m, 1H), 6.38–6.51 (m, 1H), 7.25 (br s, 1H), 7.33 (br d, J = 3.42 Hz, 4H), 7.38 (br d, J = 8.31 Hz, 2H), 7.42–7.46 (m, 1H), 7.49 (br d, J = 7.34 Hz, 1H), 7.61 (br d, J = 7.70 Hz, 2H), 7.77–7.82 (m, 1H), 7.97 (br s, 1H), 9.69 (br s, 1H), 10.20 (s, 1H), 11.11 (s, 1H), 12.14 (br s, 1H). HRMS (ESI+): m/z calcd for C43H49N8O10: 837.3567 [M + H]+; found: 837.3579 [M + H]+.
N-(4-((4-((17-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12,15-pentaoxaheptadecyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide (3)
To a mixture of 13-((methylamino)methyl)-2,5,8,11,14-pentaoxahexadecan-16-ol (2.9 g, 9.82 mmol) in tetrahydrofuran (29 mL) were added 2,4-dichloro-6-methylpyrimidine, 14 (1.5 g, 9.82 mmol), and DIPEA (2.38 g, 19.64 mmol, 3.42 mL). The mixture was stirred at 20 °C for 12 h. LCMS showed that all starting materials were consumed, and around 50% of the desired product could be detected. The reaction mixture was diluted with water (20 mL) and extracted with ethyl acetate (20 mL × 2). The combined organic layers were washed with aqueous NaCl (20 mL × 2), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, dichloromethane/methyl alcohol = 100:0 to 10:1) to give 2-(2-chloro-6-methylpyrimidin-4-yl)-5,8,11,14,17-pentaoxa-2-azanonadecan-19-ol, 17 (0.89 g, yield 20.41%), as a light yellow solid. 1H-NMR: (400 MHz, DMSO-d6) δ ppm 2.23 (s, 3H), 3.04 (br s, 3H), 3.37–3.43 (m, 3H), 3.47–3.51 (m, 17H), 3.57–3.67 (m, 4H), 4.56 (t, J = 5.32 Hz, 1H), 6.50–6.59 (m, 1H).
To a mixture of N-(4-aminophenyl)-2-phenylacetamide (0.477 g, 2.11 mmol) in isopropan-2-ol (10 mL) were added 2-(2-chloro-6-methylpyrimidin-4-yl)-5,8,11,14,17-pentaoxa-2-azanonadecan-19-ol, 17 (0.89 g, 2.11 mmol), and acetic acid (12.67 mg, 0.211 mmol). The mixture was stirred at 80 °C for 12 h. LCMS showed that all starting materials were consumed, and 60% of the desired compound was detected. The reaction mixture was diluted with water (15 mL) and extracted with ethyl acetate (20 mL) three times. The combined organic layers were washed with aqueous NaCl (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, dichloromethane/methyl alcohol = 100:0 to 10:1) to give N-(4-((4-((17-hydroxy-3,6,9,12,15-pentaoxaheptadecyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 20 (1 g, yield 54.25%), as a light yellow solid. 1H-NMR: (400 MHz, DMSO-d6) δ ppm 2.31 (br s, 3H), 3.17 (br s, 4H), 3.45–3.50 (m, 20H), 3.59–3.65 (m, 4H), 6.34–6.54 (m, 1H), 7.21–7.35 (m, 5H), 7.43–7.51 (m, 2H), 7.63 (br d, J = 8.55 Hz, 2H), 10.11 (br s, 1H), 10.34 (s, 1H).
To a mixture of N-(4-((4-((17-hydroxy-3,6,9,12,15-pentaoxaheptadecyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 20 (1 g, 2.86 mmol), in dichloromethane (10 mL) was added tosyl chloride (655.38 mg, 3.44 mmol) and triethylamine (579.74 mg, 5.73 mmol). The mixture was stirred at 20 °C for 12 h. LCMS showed that all starting materials were consumed, and 60% of the desired compound was detected. The reaction mixture was diluted with water (20 mL) and extracted with ethyl acetate (20 mL). The organic layers were washed with aqueous NaCl (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, dichloromethane/methyl alcohol = 100:0 to 10:1) to give 2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11,14,17-pentaoxa-2-azanonadecan-19-yl 4-methylbenzenesulfonate, 23 (1.5 g, yield 70%), as a light yellow solid. 1H-NMR: (400 MHz, CDCl3) δ ppm 2.29 (s, 3H), 2.44 (s, 3H), 3.12 (s, 3H), 3.50 (s, 2H), 3.56 (s, 7H), 3.62 (br s, 8H), 3.66 (br d, J = 5.14 Hz, 4H), 3.73 (s, 4H), 4.12–4.16 (m, 2H), 5.85 (s, 1H), 7.33–7.42 (m, 10H), 7.52 (br d, J = 8.56 Hz, 2H), 7.79 (d, J = 8.19 Hz, 2H).
To a mixture of 2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11,14,17-pentaoxa-2-azanonadecan-19-yl 4-methylbenzenesulfonate, 23 (1 g, 1.31 mmol), in DMF (10 mL) was added sodium azide (101.85 mg, 1.57 mmol). The mixture was stirred at 50 °C for 12 h. LCMS showed that all starting materials were consumed, and 64% of the desired product could be detected. The reaction solution was diluted with water (10 mL) and extracted with ethyl acetate (20 mL) three times. The combined organic layers were washed with aqueous NaCl (5 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by prep-TLC (SiO2, dichloromethane/methyl alcohol = 100:0 to 5:1) to give N-(4-((4-((17-azido-3,6,9,12,15-pentaoxaheptadecyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 26 (1 g, yield 90.21%), as a dark brown solid. 1H-NMR: (400 MHz, CDCl3) δ ppm 2.29 (s, 3H), 3.11 (br s, 3H), 3.35–3.40 (m, 2H), 3.58 (br s, 4H), 3.65 (br d, J = 5.99 Hz, 16H), 3.74 (br s, 4H), 5.84 (s, 1H), 7.30–7.55 (m, 11H).
To a mixture of N-(4-((4-((17-azido-3,6,9,12,15-pentaoxaheptadecyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 26 (1 g, 1.18 mmol), in methanol (15 mL) and ammonia hydrate (5 mL) was added Pd/C (100 mg, 10%). The mixture was stirred at 20 °C for 12 h under H2 (15 Psi). LCMS showed that all starting materials were consumed, and 70% of the desired compound was detected. The product was filtered, and the filtrate was concentrated under high vacuum at 40 °C to give a white solid. The crude product N-(4-((4-((17-amino-3,6,9,12,15-pentaoxaheptadecyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 29 (360 mg, yield 50.1%), was used in the next step without further purification.1H-NMR: (400 MHz, CDCl3) δ ppm 2.26 (s, 3H), 3.07 (s, 3H), 3.49–3.68 (m, 24H), 3.72–3.78 (m, 4H), 5.81 (s, 1H), 7.29–7.41 (m, 6H), 7.42–7.56 (m, 4H).
To a mixture of N-(4-((4-((17-amino-3,6,9,12,15-pentaoxaheptadecyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 29 (50 mg, 69.59 μmol), in DMSO (0.5 mL) were added 2-(2,6-dioxopiperidin-3-yl)-4-fluoroisoindoline-1,3-dione (23.07 mg, 83.50 μmol) and DIPEA (17.99 mg, 139.17 μmol). The mixture was stirred at 60 °C for 12 h. LCMS showed that all starting materials were consumed, and 30% of the desired compound was detected. The reaction mixture was filtered, and the filtrate was concentrated under high vacuum at 40 °C. The residue was purified by prep-HPLC (Phenomenex Luna C18 100 mm × 40 mm × 5 μm, mobile phase: [water (0.1%TFA) – ACN]: 25–53%, 8 min) to give N-(4-((4-((17-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12,15-pentaoxaheptadecyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide (3, 8 mg, yield 12.86%) as a yellow solid. 1H-NMR: (400 MHz, DMSO-d6) δ ppm 2.01 (br d, J = 10.76 Hz, 1H), 2.29 (br s, 3H), 2.55–2.57 (m, 2H), 2.81–2.90 (m, 1H), 3.15 (s, 3H), 3.44 (br s, 9H), 3.52 (br d, J = 7.09 Hz, 11H), 3.57–3.64 (m, 10H), 3.69 (br s, 5H), 5.05 (dd, J = 13.02, 5.20 Hz, 1H), 6.36–6.52 (m, 1H), 6.55–6.61 (m, 1H), 7.03 (d, J = 6.97 Hz, 1H), 7.12 (d, J = 8.56 Hz, 1H), 7.24 (br s, 1H), 7.29–7.35 (m, 4H), 7.43 (br s, 2H), 7.53–7.65 (m, 3H), 9.73 (s, 1H), 10.20 (s, 1H), 11.09 (s, 1H), 12.14–12.81 (m, 1H). HRMS (ESI+): m/z calcd for C45H55N8O10: 867.4036 [M + H]+; found: 867.4054 [M + H]+.
2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)-N-(2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11,14,17-pentaoxa-2-azanonadecan-19-yl)acetamide (6)
To the mixture of 2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetic acid (27.75 mg, 83.50 μmol) and N-(4-((4-((17-amino-3,6,9,12,15-pentaoxaheptadecyl)(methyl)amino)-6-methylpyrimidin-2-yl)amino)phenyl)-2-phenylacetamide, 29 (50 mg, 69.59 μmol), in DMF (0.5 mL) were added DIPEA (17.99 mg, 139.17 μmol) and HATU (39.69 mg, 104.38 μmol). The mixture was stirred at 20 °C for 12 h. LCMS showed that all starting materials were consumed, and 30% of the desired compound was detected. The reaction mixture was filtered, and the filtrate was concentrated to give a residue, which was purified by prep-HPLC (Phenomenex Luna C18 100 mm × 40 mm × 5 μm, mobile phase: [water (0.1%TFA) – ACN]: 25–53%, 8 min) to give 2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)-N-(2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11,14,17-pentaoxa-2-azanonadecan-19-yl)acetamide (6, 30 mg, yield 44.74%), as a white solid. 1H-NMR: (400 MHz, DMSO-d6) δ ppm 1.98–2.06 (m, 1H), 2.29 (br s, 3H), 2.51–2.62 (m, 2H), 2.81–2.94 (m, 1H), 3.14 (s, 3H), 3.26–3.32 (m, 2H), 3.41–3.48 (m, 17H), 3.61 (s, 6H), 4.76 (s, 2H), 5.10 (dd, J = 13.01, 5.29 Hz, 1H), 6.37–6.50 (m, 1H), 7.20–7.26 (m, 1H), 7.28–7.34 (m, 4H), 7.35–7.50 (m, 4H), 7.60 (br d, J = 8.60 Hz, 2H), 7.79 (t, J = 7.83 Hz, 1H), 7.99 (br t, J = 5.51 Hz, 1H), 9.89 (br s, 1H), 10.21 (s, 1H), 11.11 (s, 1H), 12.44 (s, 1H). HRMS (ESI+): m/z calcd for C47H57N8O12: 925.4091 [M + H]+; found: 925.4097 [M + H]+.
(2S,4R)-1-((S)-3,3-Dimethyl-2-(3-(2-(methyl(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)amino)ethoxy)propanamido)butanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (7)
To a solution of tert-butyl 3-(2-(methylamino)ethoxy)propanoate (3.45 g, 9.20 mmol, 1.5 equiv) and DIPEA (2.38 g, 18.39 mmol, 3.21 mL, 3 equiv) in THF (50 mL) was added 2,4-dichloro-6-methyl-pyrimidine, 14 (1 g, 6.13 mmol, 1 equiv), at 0 °C under N2. After addition, the reaction mixture was stirred at 15 °C for 10 h. TLC (petroleum ether/ethyl acetate = 3:1) showed that the starting material was consumed, and the desired product was obtained. The reaction mixture was concentrated and purified by column chromatography on silica gel (pet. ether/ethyl acetate = 100:1 to 100:20) to give tert-butyl 3-(2-((2-chloro-6-methylpyrimidin-4-yl)(methyl)amino)ethoxy)propanoate (30, 1.2 g, 3.64 mmol, yield 59.4%) as a colorless oil. 1H-NMR: (400 MHz CDCl3) δ = 1.43 (s, 9H), 2.33 (s, 3H), 2.50–2.39 (m, 2H), 3.08 (br s, 3H), 3.74–3.52 (m, 6H), 6.17 (br s, 1H). The regioselectivity of this reaction was confirmed by NOE (see the SI).
To a solution of N-(4-aminophenyl)-2-phenyl-acetamide (792.39 mg, 3.50 mmol, 1.05 equiv) and AcOH (200.28 mg, 3.34 mmol, 190.74 μL, 1 equiv) in i-PrOH (50 mL) was added tert-butyl 3-(2-((2-chloro-6-methylpyrimidin-4-yl)(methyl)amino)ethoxy)propanoate, 30 (1.1 g, 3.34 mmol, 1 equiv), at 0 °C under N2. After addition, the reaction mixture was stirred at 80 °C for 10 h. LCMS showed that the starting material was consumed, and the desired product was obtained. The reaction mixture was cooled to 25 °C and filtered to obtain tert-butyl 3-(2-(methyl(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)amino)ethoxy)propanoate (35, 1.2 g, 2.31 mmol, yield 69.2%) as a brown solid. LCMS (ESI+): m/z 520.1 (M + H)+. 1H-NMR (400 MHz, CDCl3): δ = 1.34−1.43 (m, 9H), 2.29–2.46 (m, 5H), 3.08–3.20 (m, 3H), 3.52–3.68 (m, 5H), 3.72–3.82 (m, 3H), 5.81 (s, 1H), 5.93 (s, 1H), 7.20–7.44 (m, 1H), 7.21–7.34 (m, 4H), 7.36–7.42 (m, 1H), 7.55 (d, J = 8.8 Hz, 2H), 8.55–8.79 (m, 1H), 9.91–10.15 (m, 1H), 13.23–13.52 (m, 1H).
To a solution of tert-butyl 3-(2-(methyl(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)amino)ethoxy)propanoate, 35 (600 mg, 987.27 μmol, 1 equiv), in ethyl acetate (20 mL) was added HCl/EtOAc (4 M, 6 mL), and the reaction mixture was stirred at 25 °C for 12 h. The reaction mixture was concentrated to give 3-(2-(methyl(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)amino)ethoxy)propanoic acid (40, 400 mg, 73.5% yield) as a brown solid, which was used in the next step without further purification.
To a solution of 3-(2-(methyl(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)amino)ethoxy)propanoic acid, 40 (60 mg, 129.44 μmol, 1 equiv), (2S,4R)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide ((S,R,S)-AHPC, CAS 1448297-52-6 from Bide Pharmatech Ltd.) (55.73 mg, 129.44 μmol, 1 equiv), and DIPEA (66.92 mg, 517.77 μmol, 90.19 μL, 4 equiv) in DMF (4 mL) was added HATU (98.44 mg, 258.88 μmol, 2 equiv) at 0 °C. After addition, the reaction mixture was stirred at 25 °C for 12 h. LCMS showed that the starting material was consumed and the desired product was detected. The reaction mixture was concentrated and purified by prep-HPLC (method: column-Phenomenex Gemini-NX C18 75 mm × 30 mm × 3 μm; mobile phase: [water (0.05% NH3H2O + 10 mM NH4HCO3) – MeCN]; 35–55%, 8 min) to give (2S,4R)-1-((S)-3,3-dimethyl-2-(3-(2-(methyl(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)amino)ethoxy)propanamido)butanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (7, 35 mg, 39.95 μmol, 30.9% yield, 100% purity) as a white solid. 1H-NMR: (400 MHz, DMSO-d6) δ 0.93 (s, 9H), 1.90 (ddd, J = 4.5, 8.6, 12.8 Hz, 1H), 1.99–2.08 (m, 1H), 2.16 (s, 3H), 2.34–2.41 (m, 1H), 2.42–2.45 (m, 3H), 2.54–2.63 (m, 1H), 2.94–3.08 (m, 3H), 3.29–3.32 (m, 1H), 3.51–3.71 (m, 10H), 4.21 (dd, J = 5.4, 15.9 Hz, 1H), 4.35 (br s, 1H), 4.39–4.47 (m, 2H), 4.56 (d, J = 9.2 Hz, 1H), 5.14 (d, J = 3.1 Hz, 1H), 5.94 (s, 1H), 7.18–7.26 (m, 1H), 7.28–7.45 (m, 10H), 7.63 (d, J = 8.8 Hz, 2H), 7.95 (d, J = 9.4 Hz, 1H), 8.57 (t, J = 6.0 Hz, 1H), 8.90 (s, 1H), 8.97 (s, 1H), 9.97 (s, 1H). 13C-NMR (126 MHz, DMSO-d6) δ 16.39, 24.21, 26.76, 35.84, 36.07, 38.41, 42.06, 43.69, 48.94, 56.71, 56.88, 59.15, 67.40, 67.46, 68.40, 69.32, 93.20, 118.87, 119.88, 126.89, 127.83, 128.71, 129.07, 129.50, 130.05, 131.60, 132.63, 136.68, 137.57, 139.94, 148.14, 151.92, 159.53, 162.79, 165.44, 168.90, 169.96, 170.38, 172.39. HRMS (ESI+): m/z calcd for C47H58N9O6S: 876.4226 [M + H]+; found: 876.4227 [M + H]+.
(2S,4S)-1-((S)-3,3-Dimethyl-2-(3-(2-(methyl(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)amino)ethoxy)propanamido)butanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (12)
To a mixture of 3-(2-(methyl(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)amino)ethoxy)propanoic acid (40, 30.0 mg, 1 equiv, 64.7 μmol) in DMF (1 mL) was added DIPEA (25.1 mg, 33.8 μL, 3 equiv, 194 μmol) after which HATU (36.9 mg, 1.5 equiv, 97.1 μmol) was added. The mixture was stirred for 10 min after which (2S,4S)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide hydrochloride (30.2 mg, 1 equiv, 64.7 μmol, VHL negative control (S,S,S)-AHPC hydrochloride from MedChemExpress; Cat # HY-125845A) was added. After 2 h, LCMS indicated the product. The mixture was stirred for another 1 h, after which it was purified by RP HPLC (Phenomenex Gemini-NX C18, 110 Å, 150 mm × 21.2 mm × 5 μm). Eluting with a gradient of 15 to 90% acetonitrile in water with 0.1% formic acid, a flow rate of 20 mL/min, a gradient time of 30 min, and a detection wavelength of 200 nm to give (2S,4S)-1-((S)-3,3-dimethyl-2-(3-(2-(methyl(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)amino)ethoxy)propanamido)butanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (12, 25 mg, 41%). 1H-NMR (500 MHz, CD3OD) δ 1.00 (s, 9H), 1.98 (dt, J = 13.2, 4.6 Hz, 1H), 2.36–2.446 (m, 4H), 2.47 (s, 4H), 2.56 (q, J = 10.7, 7.0 Hz, 1H), 3.26 (s, 3H), 3.63–3.77 (m, 8H), 3.79–3.91 (m, 3H), 3.99 (dd, J = 10.6, 5.1 Hz, 1H), 4.28–4.60 (m, 5H), 6.11 (d, J = 1.3 Hz, 1H), 6.61 (s, 2H), 7.23–7.29 (m, 1H), 7.31–7.39 (m, 4H), 7.40–7.43 (m, 2H), 7.46 (d, J = 8.3 Hz, 2H), 7.61 (s, 4H), 7.65 (s, 2H), 7.94 (d, J = 8.1 Hz, 1H), 8.90 (s, 1H). 13C-NMR (126 MHz, CD3OD) δ 14.39, 17.59, 25.48, 25.58, 34.65, 35.49, 36.48, 42.35, 43.27, 56.10, 57.89, 59.62, 66.87, 67.85, 70.04, 97.18, 120.11, 121.86, 126.60, 127.63, 128.23, 128.76, 128.93, 128.96, 130.08, 132.03, 133.57, 135.37, 135.63, 138.65, 147.45, 151.58, 153.00, 153.93, 160.27, 170.86, 170.99, 172.41, 173.44. HRMS (ESI+): m/z calcd for C47H58N9O6S: 876.4226 [M + H]+; found: 876.4234 [M + H]+.
(2S,4R)-1-((S)-13-(tert-Butyl)-2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-11-oxo-5,8-dioxa-2,12-diazatetradecan-14-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (8)
A mixture of tert-butyl 3-(2-(2-(methylamino)ethoxy)ethoxy)propanoate (4 g, 16.2 mmol), DIPEA (6.27 g, 48.5 mmol), and 2,4-dichloro-6-methyl-pyrimidine, 14 (2.64 g, 16.2 mmol), in THF (40 mL) was degassed and purged with N2 3 times and then stirred at 20 °C for 12 h under a N2 atmosphere. The reaction mixture was diluted with water (250 mL) and then extracted with ethyl acetate (150 mL × 3). The combined organic layers were washed with aqueous sodium chloride, dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography on silica gel (eluted with petroleum ether ethyl acetate = 50:1 to 0:100) to give tert-butyl 3-(2-(2-((2-chloro-6-methylpyrimidin-4-yl)(methyl)amino)ethoxy)ethoxy)propanoate, 31 (2.5 g, yield 41%), as a colorless oil. 1H-NMR (400 MHz, CDCl3) δ 1.44 (s, 9H), 2.33 (s, 3H), 2.48 (t, J = 6.47 Hz, 2H), 3.10 (br s, 3H), 3.53–3.60 (m, 4H), 3.63–3.82 (m, 6H), 6.19 (br s, 1H).
A mixture of tert-butyl 3-(2-(2-((2-chloro-6-methylpyrimidin-4-yl)(methyl)amino)ethoxy)ethoxy)propanoate, 31 (1.50 g, 4.01 mmol), acetic acid (120 mg, 2.01 mmol), and N-(4-aminophenyl)-2-phenyl-acetamide (907 mg, 4.01 mmol) in isopropanol (20 mL) was degassed and purged with N2 3 times and then stirred at 80 °C for 12 h under a N2 atmosphere. The reaction mixture was cooled to 20 °C, filtered, and concentrated under reduced pressure to give a residue, which was washed with isopropanol (50 mL) to give tert-butyl 3-(2-(2-(methyl(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)amino)ethoxy)ethoxy)propanoate, 36 (1.3 g, yield 54%), as a yellow solid. 1H-NMR (400 MHz, CDCl3): δ 1.41–1.46 (m, 9H), 2.34–2.41 (m, 3H), 3.17–3.23 (m, 3H), 3.48–3.72 (m, 9H), 3.76–3.83 (m, 3H), 5.83–6.00 (m, 1H), 7.24–7.30 (m, 1H), 7.31–7.37 (m, 3H), 7.37–7.45 (m, 3H), 7.49–7.58 (m, 2H), 8.42–8.63 (m, 1H), 10.05–10.16 (m, 1H), 13.40–13.55 (m, 1H).
A mixture of tert-butyl 3-(2-(2-(methyl(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)amino)ethoxy)ethoxy)propanoate, 36 (0.8 g, 1.42 mmol), and trifluoroacetic acid (1 mL) in dichloromethane (10 mL) was degassed and purged with N2 three times and then stirred at 25 °C for 12 h under a N2 atmosphere. The reaction mixture was concentrated under reduced pressure to give 3-(2-(2-(methyl(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)amino)ethoxy)ethoxy)propanoic acid, 41 (500 mg, yield 62%), as a yellow solid. 1H-NMR (400 MHz, T = 273 + 80K, DMSO-d6) δ 2.33 (s, 3H), 2.41 (t, J = 6.28 Hz, 2H), 3.19 (s, 3H), 3.49 (br d, J = 4.63 Hz, 4H), 3.57–3.69 (m, 6H), 3.72–3.82 (m, 2H), 7.21–7.26 (m, 1H), 7.28–7.38 (m, 4H), 7.46 (br d, J = 8.38 Hz, 2H), 7.63 (d, J = 8.82 Hz, 2H), 10.12 (s, 1H), 10.33 (s, 1H).
A mixture of (2S,4R)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide ((S,R,S)-AHPC, CAS 1448297-52-6 from Bide Pharmatech Ltd.) (140 mg, 325 μmol), 3-(2-(2-(methyl(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)amino)ethoxy)ethoxy)propanoic acid, 41 (150 mg, 295 μmol), HATU (168 mg, 443 μmol), and DIPEA (76.4 mg, 591 μmol,) in DMF (2 mL) was degassed and purged with N2 three times and then stirred at 25 °C for 12 h under a N2 atmosphere. The reaction mixture was concentrated under reduced pressure to give a residue. The residue was purified by Prep-HPLC (Gilson 281 semi-preparative HPLC system; mobile phase: A: TFA/H2O = 0.075% v/v; B: acetonitrile; column: Phenomenex Luna C18 100 mm × 40 mm × 5 μm; flow rate: 50 mL/min; monitoring wavelength: 220 and 254 nm) to give (2S,4R)-1-((S)-13-(tert-butyl)-2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-11-oxo-5,8-dioxa-2,12-diazatetradecan-14-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (8, 35 mg, yield 12%) as a white solid. 1H-NMR (500 MHz, DMSO-d6) δ 0.94 (s, 9H), 1.91 (ddd, J = 4.5, 8.7, 13.1 Hz, 1H), 2.05 (dt, J = 5.0, 9.5 Hz, 1H), 2.24–2.39 (m, 4H), 2.45 (s, 3H), 3.18 (m, 3H), 3.37–3.83 (m, 11H), 4.23 (dd, J = 5.5, 15.9 Hz, 1H), 4.37 (q, J = 3.2 Hz, 1H), 4.40–4.49 (m, 2H), 4.57 (d, J = 9.3 Hz, 1H), 6.46 (m, 1H), 7.26 (dtt, J = 2.3, 4.3, 6.1 Hz, 1H), 7.34 (d, J = 6.5 Hz, 4H), 7.38–7.54 (m, 6H), 7.64 (dd, J = 5.5, 8.8 Hz, 2H), 7.97 (d, J = 9.3 Hz, 1H), 8.63 (t, J = 6.1 Hz, 1H), 9.01 (s, 1H), 10.27 (s, 1H), 10.34 (d, J = 10.1 Hz, 1H), 12.92 (s, 1H). HRMS (ESI+): m/z calcd for C49H62N9O7S: 920.4488 [M + H]+; found: 920.4503 [M + H]+.
(2S,4R)-1-((S)-16-(tert-Butyl)-2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-14-oxo-5,8,11-trioxa-2,15-diazaheptadecan-17-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (9)
To a solution of tert-butyl 5,8,11-trioxa-2-azatetradecan-14-oate (4.27 g, 9.20 mmol, 1.5 equiv) and DIPEA (7.14 g, 55.20 mmol, 3.0 equiv) in tetrahydrofuran (50 mL) was added 2,4-dichloro-6-methyl-pyrimidine, 14 (1 g, 6.13 mmol, 1 equiv), at 20 °C under N2. After addition, the reaction mixture was stirred at 25 °C for 10 h. The reaction mixture was concentrated and purified by column chromatography on silica gel (petro ether:ethyl acetate = 100:1 to 40:100) to give tert-butyl-2-(2-chloro-6-methylpyrimidin-4-yl)-5,8,11-trioxa-2-azatetradecan-14-oate, 32 (1.2 g, yield 46.8%), as a colorless oil. 1H-NMR: (400 MHz CDCl3) δ 1.45 (s, 9H), 2.34 (s, 3H), 2.50 (t, J = 6.5 Hz, 2H), 3.11 (br s, 3H), 3.55–3.64 (m, 8H), 3.65–3.74 (m, 5H), 6.09–6.29 (m, 1H).
To a solution of tert-butyl 2-(2-chloro-6-methylpyrimidin-4-yl)-5,8,11-trioxa-2-azatetradecan-14-oate, 32 (1.13 g, 2.70 mmol, 1 equiv), and N-(4-aminophenyl)-2-phenyl- acetamide (642.39 mg, 2.70 mmol, 1 equiv) in isopropanol (10 mL) was added acetic acid (11.87 mg, 0.1 equiv) at 0 °C under N2. After addition, the reaction mixture was stirred at 80 °C for 12 h. The reaction mixture was cooled to 25 °C and filtered to obtain tert-butyl-2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11-trioxa-2-azatetradecan-14-oate, 37 (1.6 g, yield 97.37%), as a brown solid. LCMS (ESI+): m/z 608.5 (M + H)+.
To a solution of tert-butyl 2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11-trioxa-2-azatetradecan-14-oate, 37 (600 mg, 987.27 μmol, 1 equiv), in ethyl acetate (2 mL) was added ethyl acetate/HCl (4 M, 6 mL) and the reaction mixture was stirred at 25 °C for 12 h. The reaction mixture was concentrated to give 2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11-trioxa-2-azatetradecan-14-oic acid, 42 (400 mg, 73.45% yield), as a brown solid, which was used without further purification. LCMS (ESI+): m/z 552.4 (M + H)+.
To a solution of 2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11-trioxa-2-azatetradecan-14-oic acid, 42 (100 mg, 160.11 μmol, 1 equiv), and (2S,4R)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide ((S,R,S)-AHPC, CAS 1448297-52-6 from Bide Pharmatech Ltd.) (68.94 mg, 160.11 μmol, 1 equiv) in DMF (2 mL) were added HATU (121.76 mg, 320.23 μmol, 2 equiv) and DIPEA (82.7761 mg, 640.46 μmol, 4 equiv) at 0 °C. After addition, the reaction mixture was stirred at 25 °C for 12 h. LCMS showed that the starting material was consumed. The reaction mixture was treated with water (3 mL) and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine (3 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain a residue, which was purified by prep-HPLC (Phenomenex Gemini-NX C18 75 mm × 30 mm × 3 μm; mobile phase: [water (0.05%NH3H2O + 10 mM NH4HCO3) – MeCN]; B%: 30–60%, 8 min) to give (2S,4R)-1-((S)-16-(tert-butyl)-2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-14-oxo-5,8,11-trioxa-2,15-diazaheptadecan-17-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (9, 20 mg, yield 12.93%) as a white solid. 1H-NMR: (400 MHz, DMSO-d6) δ 0.93 (s, 9H), 1.90 (ddd, J = 12.84, 8.49, 4.53 Hz, 1H), 1.98–2.08 (m, 1H), 2.16 (s, 3H), 2.30–2.36 (m, 1H), 2.44 (s, 3H), 2.53–2.57 (m, 1H), 3.02 (s, 3H), 3.43–3.51 (m, 8H), 3.54–3.70 (m, 10H), 4.21 (dd, J = 15.85, 5.36 Hz, 1H), 4.35 (br s, 1H), 4.38–4.48 (m, 2H), 4.55 (d, J = 9.42 Hz, 1H), 5.12 (d, J = 3.58 Hz, 1H), 5.95 (s, 1H), 7.20–7.26 (m, 1H), 7.29–7.34 (m, 4H), 7.36–7.45 (m, 6H), 7.64 (d, J = 8.94 Hz, 2H), 7.91 (d, J = 9.42 Hz, 1H), 8.56 (t, J = 6.02 Hz, 1H), 8.90 (s, 1H), 8.97 (s, 1H), 9.96 (s, 1H). HRMS (ESI+): m/z calcd for C51H66N9O8S: 964.4750 [M + H]+; found: 964.4755 [M + H]+.
(2S,4R)-1-((S)-19-(tert-Butyl)-2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-17-oxo-5,8,11,14-tetraoxa-2,18-diazaicosan-20-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (10)
To a solution of tert-butyl 5,8,11,14-tetraoxa-2-azaheptadecan-17-oate (1.8 g, 3.49 mmol, 65% purity) and DIPEA (901.60 mg, 6.98 mmol) in THF (18 mL) was added 2,4-dichloro-6-methyl-pyrimidine, 14 (379.04 mg, 2.33 mmol), at 20 °C. The reaction mixture was stirred at 20 °C for 16 h. The reaction mixture was concentrated to a residue, which was purified by column chromatography on silica gel (petro ether:ethyl acetate = 100:1 to 1:2) to give tert-butyl 2-(2-chloro-6-methylpyrimidin-4-yl)-5,8,11,14-tetraoxa-2-azaheptadecan-17-oate, 33 (1.1 g, yield 81.92%, 80% purity), as a colorless oil. 1H-NMR: (400 MHz CDCl3) δ 1.37 (s, 9H), 2.22–2.30 (m, 3H), 2.39–2.46 (m, 2H), 3.03 (br s, 3H), 3.50–3.57 (m, 12H), 3.57–3.70 (m, 6H), 6.13 (br s, 1H).
To a solution of tert-butyl 2-(2-chloro-6-methylpyrimidin-4-yl)-5,8,11,14-tetraoxa-2-azaheptadecan-17-oate, 33 (1.1 g, 2.38 mmol), and AcOH (14.30 mg, 238.11 μmol) in isopropanol (11 mL) was added N-(4-aminophenyl)-2-phenyl-acetamide (538.77 mg, 2.38 mmol) at 20 °C. The reaction mixture was stirred at 80 °C for 12 h. LCMS showed that the reaction was complete, and the desired compound was detected. The reaction mixture was diluted with H2O (20 mL) and extracted with ethyl acetate (3 × 25 mL). The organic layer was concentrated under reduced pressure to give tert-butyl 2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11,14-tetraoxa-2-azaheptadecan-17-oate, 38 (0.5 g, yield 32.22%), as a white solid. 1H-NMR: (400 MHz CDCl3) δ 1.42 (d, J = 1.32 Hz, 9H), 2.24 (s, 3H), 3.08 (s, 3H), 3.51–3.73 (m, 21H), 5.79 (s, 1H), 7.29–7.38 (m, 6H), 7.45–7.56 (m, 3H), 8.35–8.51 (m, 1H).
To a solution of tert-butyl-2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11,14-tetraoxa-2-azaheptadecan-17-oate, 38 (0.5 g, 767.12 μmol), in ethyl acetate (5 mL) was added ethyl acetate/HCl (4 M, 5 mL) at 25 °C, and the reaction mixture was stirred at 25 °C for 12 h. The reaction mixture was concentrated to give 2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11,14-tetraoxa-2-azaheptadecan-17-oic acid, 43 (0.2 g, yield 43.77%), as a brown solid, which was used without further purification.
To a solution of 2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11,14-tetraoxa-2-azaheptadecan-17-oic acid, 43 (0.1 g, 167.87 μmol), and (2S,4R)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide ((S,R,S)-AHPC, CAS 1448297-52-6 from Bide Pharmatech Ltd.) (72.28 mg, 167.87 μmol) in DMF (1 mL) was added HATU (127.66 mg, 335.75 μmol) and DIPEA (65.09 mg, 503.62 μmol) at 25 °C. The reaction mixture was stirred at 25 °C for 12 h. LCMS showed that the starting material was consumed, and LCMS indicated the desired product. The reaction mixture was treated with water (3 mL) and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine (3 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain a residue, which was purified by prep-HPLC (Phenomenex Gemini-NX C18 75 mm × 30 mm × 3 μm; mobile phase: [water (0.05%NH3H2O + 10 mM NH4HCO3) – MeCN]; B%: 30–60%, 8 min) to give (2S,4R)-1-((S)-19-(tert-butyl)-2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-17-oxo-5,8,11,14-tetraoxa-2,18-diazaicosan-20-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (10, 48 mg, yield 28.36%), as a white solid. 1H-NMR: (400 MHz, DMSO-d6) δ 0.93 (s, 9H), 1.90 (ddd, J = 12.78, 8.44, 4.58 Hz, 1H), 1.98–2.08 (m, 1H), 2.16 (s, 3H), 2.25–2.42 (m, 2H), 2.44 (s, 3H), 2.52–2.56 (m, 1H), 3.02 (br s, 3H), 3.46 (s, 7H), 3.49 (br d, J = 0.98 Hz, 4H), 3.53–3.72 (m, 10H), 4.22 (dd, J = 15.77, 5.38 Hz, 1H), 4.35 (br s, 1H), 4.39–4.48 (m, 2H), 4.55 (d, J = 9.41 Hz, 1H), 5.12 (d, J = 3.42 Hz, 1H), 5.95 (s, 1H), 7.20–7.27 (m, 1H), 7.28–7.35 (m, 4H), 7.36–7.47 (m, 6H), 7.64 (d, J = 8.80 Hz, 2H), 7.91 (d, J = 9.41 Hz, 1H), 8.56 (br t, J = 5.93 Hz, 1H), 8.90 (s, 1H), 8.97 (s, 1H), 9.96 (s, 1H). HRMS (ESI+): m/z calcd for C53H70N9O9S: 1008.5012 [M + H]+; found: 1008.5015 [M + H]+.
(2S,4R)-1-((S)-22-(tert-Butyl)-2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-20-oxo-5,8,11,14,17-pentaoxa-2,21-diazatricosan-23-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (11)
To a solution of tert-butyl 5,8,11,14,17-pentaoxa-2-azaicosan-20-oate (3.5 g, 9.22 mmol, 1.5 equiv) and DIPEA (2.38 g, 18.45 mmol, 3 equiv) in tetrahydrofuran (30 mL) was added 2,4-dichloro-6-methyl-pyrimidine (14, 2.06 g, 6.15 mmol, 1 equiv) at 20 °C under N2. After addition, the reaction mixture was stirred at 20 °C for 16 h. The reaction mixture was concentrated and purified by column chromatography on silica gel (petro ether:ethyl acetate = 100:1 to 40:100) to give tert-butyl-2-(2-chloro-6-methylpyrimidin-4-yl)-5,8,11,14,17-pentaoxa-2-azaicosan-20-oate (34, 1 g, yield 32.1%) as a colorless oil. 1H-NMR: (400 MHz CDCl3) δ 1.45 (s, 9H), 2.35 (s, 3H), 2.51 (t, J = 6.56 Hz, 2H), 3.11 (br s, 3H), 3.60–3.66 (m, 17H), 3.68 (br d, J = 4.65 Hz, 2H), 3.71 (br t, J = 6.62 Hz, 3H), 6.20 (br s, 1H).
To a solution of tert-butyl-2-(2-chloro-6-methylpyrimidin-4-yl)-5,8,11,14,17-pentaoxa-2-azaicosan-20-oate, 34 (1 g, 1.98 mmol, 1 equiv), and N-(4-aminophenyl)-2-phenyl-acetamide (447.15 mg, 1.98 mmol, 1 equiv) in isopropanol (10 mL) was added acetic acid (11.87 mg, 0.1 equiv) at 0 °C under N2. After addition, the reaction mixture was stirred at 80 °C for 12 h. Then, the reaction mixture was cooled to 25 °C and filtered to obtain tert-butyl-2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11,14,17-pentaoxa-2-azaicosan-20-oate (39, 800 mg, yield 58.18%) as a brown solid. 1H-NMR: (400 MHz CDCl3) δ 1.45 (s, 9H), 2.25 (s, 3H), 2.49 (t, J = 6.56 Hz, 2H), 3.08 (s, 3H), 3.56–3.67 (m, 19H), 3.67–3.75 (m, 6H), 5.82 (s, 1H), 6.84 (br s, 1H), 7.32–7.42 (m, 7H), 7.52 (d, J = 8.94 Hz, 2H).
To a solution of tert-butyl 2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11,14,17-pentaoxa-2-azaicosan-20-oate (39, 300 mg, 431.13 μmol, 1 equiv) in ethyl acetate (1 mL) was added HCl/EtOAc (4 M, 5 mL), and the reaction mixture was stirred at 25 °C for 2 h. The reaction mixture was concentrated to give 2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11,14,17-pentaoxa-2-azaicosan-20-oic acid (44, 200 mg, yield 72.51%) as a brown solid, which was used without further purification. LCMS(ESI+): m/z 640.4 (M + H)+.
To a solution of 2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11,14,17-pentaoxa-2-azaicosan-20-oic acid (44, 67.30 mg, 156.31 μmol, 1 equiv) and (2S,4R)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide ((S,R,S)-AHPC, CAS 1448297-52-6 from Bide Pharmatech Ltd.) (100 mg, 156.31 μmol, 1 equiv) in DMF (1 mL) were added HATU (118.87 mg, 312.63 μmol, 2 equiv) and DIPEA (60.61 mg, 468.94 μmol, 81.68 μL, 3 equiv) at 0 °C. After addition, the reaction mixture was stirred at 25 °C for 12 h. TLC (dichloromethane/methanol = 10:1) showed that the starting material was consumed. The reaction mixture was treated with water (3 mL) and extracted with ethyl acetate (3 × 3 mL). The combined organic phases were washed with brine (3 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by prep-TLC (SiO2, dichloromethane/methanol = 10:1) to give (2S,4R)-1-((S)-25-(tert-butyl)-2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-23-oxo-5,8,11,14,17,20-hexaoxa-2,24-diazahexacosan-26-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (11, 20 mg, yield 12.16%) as a white solid. 1H-NMR: (400 MHz, DMSO-d6) δ 0.93 (s, 9H), 1.90 (ddd, J = 12.83, 8.33, 4.49 Hz, 1H), 1.98–2.07 (m, 1H), 2.16 (s, 3H), 2.33–2.39 (m, 1H), 2.44 (s, 2H), 3.02 (s, 3H), 3.30 (s, 1H), 3.45–3.50 (m, 17H), 3.56–3.60 (m, 6H), 3.62–3.70 (m, 4H), 4.21 (dd, J = 15.68, 5.37 Hz, 1H), 4.34 (br s, 1H), 4.39–4.47 (m, 2H), 4.55 (d, J = 9.21 Hz, 1H), 5.13 (d, J = 3.51 Hz, 1H), 5.95 (s, 1H), 7.23 (td, J = 5.75, 2.30 Hz, 1H), 7.30–7.34 (m, 4H), 7.41 (t, J = 8.77 Hz, 6H), 7.64 (d, J = 8.77 Hz, 2H), 7.92 (d, J = 9.43 Hz, 1H), 8.57 (t, J = 5.92 Hz, 1H), 8.90 (s, 1H), 8.98 (s, 1H), 9.96 (s, 1H). 13C-NMR (126 MHz, DMSO-d6) δ 16.39, 24.19, 26.75, 35.82, 36.03, 36.70, 38.39, 42.06, 43.69, 48.84, 56.71, 56.85, 59.14, 67.37, 68.43, 69.31, 69.88, 70.11, 70.16, 70.18, 70.23, 70.29, 93.17, 118.88, 119.87, 126.89, 127.83, 128.50, 128.71, 129.07, 129.29, 129.49, 130.05, 131.60, 132.65, 136.68, 137.55, 139.94, 148.14, 151.93, 159.54, 162.87, 165.43, 168.87, 169.95, 170.38, 172.39. HRMS (ESI+): m/z calcd for C55H74N9O10S: 1052.5274 [M + H]+; found: 1052.5279 [M + H]+.
(2S,4S)-1-((S)-22-(tert-Butyl)-2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-20-oxo-5,8,11,14,17-pentaoxa-2,21-diazatricosan-23-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (13)
To a vial containing 2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-5,8,11,14,17-pentaoxa-2-azaicosan-20-oic acid (44, 30.0 mg, 1 equiv, 46.9 μmol) was added DMF (1 mL) after which the mixture was cooled to zero degrees and HATU (26.7 mg, 1.5 equiv, 70.3 μmol) was added. Then, DIPEA (18.2 mg, 24.5 μL, 3 equiv, 141 μmol) was added, and the reaction mixture was stirred for 10 min after which (2S,4S)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide hydrochloride (21.9 mg, 1 equiv, 46.9 μmol, VHL negative control (S,S,S)-AHPC hydrochloride from MedChemExpress; Cat # HY-125845A) was added and the reaction mixture was stirred in the ice bath for 10 min, then at room temperature for 2 h after which the mixture was directly purified by RP HPLC (Phenomenex Gemini-NX C18, 110 Å, 150 mm × 21.2 mm; 5 μm). Eluting with a gradient of 15 to 90% acetonitrile in water with 0.1% TFA, a flow rate of 20 mL/min, a gradient time of 30 min, and a detection wavelength of 200 nm to give (2S,4S)-1-((S)-22-(tert-butyl)-2-(6-methyl-2-((4-(2-phenylacetamido)phenyl)amino)pyrimidin-4-yl)-20-oxo-5,8,11,14,17-pentaoxa-2,21-diazatricosan-23-oyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (13, 27 mg, 49%). 1H-NMR (500 MHz, CD3OD) δ 1.04 (s, 9H), 1.98 (dt, J = 13.1, 4.6 Hz, 1H), 2.38–2.42 (m, 3H), 2.40–2.47 (m, 1H), 2.49 (s, 3H), 2.56 (ddd, J = 15.0, 7.5, 5.3 Hz, 1H), 3.28 (s, 3H), 3.52–3.62 (m, 15H), 3.64–3.77 (m, 7H), 3.86 (t, J = 5.1 Hz, 2H), 4.02 (dd, J = 10.6, 5.1 Hz, 1H), 4.39 (dt, J = 9.9, 5.3 Hz, 2H), 4.46–4.58 (m, 3H), 6.12 (d, J = 1.1 Hz, 1H), 7.24–7.30 (m, 1H), 7.32–7.39 (m, 4H), 7.39–7.44 (m, 2H), 7.45–7.49 (m, 2H), 7.62 (s, 4H), 7.95 (d, J = 8.3 Hz, 1H), 8.97 (s, 1H). 13C-NMR (126 MHz, CD3OD) δ 14.40, 17.60, 25.51, 25.62, 34.79, 35.68, 35.88, 36.51, 42.36, 43.28, 50.13, 56.10, 57.74, 59.58, 66.81, 69.93, 70.02, 70.04, 70.06, 70.08, 70.13, 70.18, 97.15, 120.11, 121.89, 126.61, 127.61, 127.78, 128.24, 128.76, 128.93, 128.96, 130.05, 132.07, 133.58, 135.40, 135.67, 138.68, 147.45, 151.60, 152.92, 160.29, 170.82, 170.95, 172.54, 172.63, 173.48. HRMS (ESI+): m/z calcd for C55H74N9O10S: 1052.5274 [M + H]+; found: 1052.5276 [M + H]+.
Molecular Modeling
The TG2 crystal structure 4PYG was used for docking studies. The protein was prepared using the default parameters of the Schrödinger workflow implemented in Maestro. The protein preparation module was used to complete missing atoms, relieve torsional inconsistencies, fix protonation states at pH = 7.4, and the protein was minimized using the OPLS3 force field. A receptor grid was then generated centered on Leu102, which was assessed to be the approximate middle of the fibronectin-binding site (residues 88–106), and the grid extended 25 Å in each direction around this residue to encompass the FN-binding site. The MT4 structure was prepared for docking using the ligand preparation module to produce the low-energy conformation at pH = 7.4. Docking was performed using the standard parameters of the XP level of Schrödinger Glide, and the low-energy conformation (among 10 poses) was evaluated.
Cell Lines, Reagents, and Antibodies
SKOV3 cells were purchased from ATCC. OVCAR5 cells were obtained from Dr. Marcus Peter at Northwestern University. Cells were maintained at 37 °C in an environment of 5% CO2 and 100% humidity. The media used for culturing the cell lines are included in Supplementary Table S1. Cells lines used in all experiments were at low passages and were confirmed to be pathogen and mycoplasma-free by Charles River Animal Diagnostic Services. Cell lines were authenticated by IDEXX BioAnalytics with short tandem repeat (STR) profiling. MG132 was obtained from SelleckChem. The anti-TG2 antibody (cat#: MAB3839-I-100UG) was purchased from Millipore Sigma and the GAPDH antibody (cat#: H86504M) was purchased from Meridian Bioscience. The full-length TG2 protein used in the ITC experiments was obtained according to previously published procedures.40
Western Blotting
Briefly, SKOV3 and OVCAR5 cells were seeded on 6 cm dishes and treated with different concentrations of compounds at different time points. Proteins were extracted using radioimmunoprecipitation assay (RIPA) buffer and quantified using the Bradford assay. After gel electrophoresis and transfer, poly(vinylidene difluoride) (PDVF) membranes were blocked with TBS-Tween 5% bovine serum albumin (BSA) for 1 h and incubated with primary antibodies (1:1000 dilution, overnight at 4 °C). After incubation with the secondary antibody (anti-rabbit/mouse-horseradish peroxidase 1:1000 dilution) for 1 h at room temperature, the signal was developed using the SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific cat#: 34580) and captured with an ImageQuant LAS 4000 machine. To detect additional proteins, membranes were treated with Restore Western Blot Stripping Buffer (Thermo Fisher Scientific cat#: 21059), blocked, and then incubated with primary antibody. After Imaging, western blots were quantified using IMAGEJ, and statistical analysis was performed using GraphPad prism 8.
Cell Migration
For the scratch assay, confluent 6-well plates were scratched with a pipette tip, washed, and treated with compounds or vehicles. The plate was photographed at set time points, and the area of wound closure was quantified with ImageJ. Cell migration was assessed by using transwell inserts (8 μm pore size, Merck-Millipore, Watford, U.K.). Briefly, 15,000 cells were seeded in the upper chambers of transwells in serum-free media containing compounds or control, while media containing 10% serum was added to the lower chamber. After 18 h, cells invaded through the transwells were fixed with warm 4% PFA for 20 min at room temperature and stained with crystal violet for 3 min. The membranes were removed, mounted on cover slides, and cells were counted using a microscope at 40× magnification.
Cell Viability Assay
Cells seeded in 96-well dishes were treated with compounds at different concentrations (0.1, 0.3, 1, 3, 10, 30 μM) for 24 h. Higher concentrations of compounds were not tested as the amount of DMSO required to dissolve the compounds would be toxic to the cells. The medium was replaced after 24 h, and cells were allowed to grow for another 48 h. The numbers of viable cells were quantified by using the CCK-8 assay (ApexBio) following the manufacturer’s directions. Absorbance was measured by a spectrophotometer at 495 nm and used to estimate the viability of the cells.
Solid Phase Adhesion Assay
Briefly, wells were coated with 10 μg/mL fibronectin diluted in phosphate-buffered saline (PBS) for 1 h. Thirty thousand cells pretreated with compounds for 6 h were seeded and allowed to attach for 30 min. After washing with PBS to remove unattached cells, cells adhering to coated wells were quantified by using the CCK-8 assay (ApexBio), following the manufacturer’s protocol. Relative absorbance was measured at 496 nm and was used to estimate the relative number of cells attached to fibronectin-coated wells.
Isothermal Titration Calorimetry (ITC)
Sample Preparation
Protein dilutions in PBS with 1% DMSO were prepared from stock solutions of 108 mM TG240 in PBS. The protein dilutions were initially equilibrated on ice for 5 min, then for 20 min at 30 °C. The solutions were filtered using a 0.22 μM Millipore centrifugal device just prior to insertion in the ITC cell. The solubility of the protein solutions was confirmed independently by DLS (data not shown), and the concentration of each solution (after filtration) was measured with a NanoDrop 2000 Instrument.
For each compound, dilutions were prepared from stocks in DMSO; specifically, 40 mM stock solutions were used for compound 11 and compound 7, and a 20 mM stock solution for MT4 due to its lower solubility than the PROTAC compounds. All compound dilutions were made in PBS, and the final DMSO concentration in each solution was carefully adjusted to 1%.
ITC Experiments
All ITC experiments were performed on an Affinity Low Volume instrument (Waters/TA Instruments) equipped with a fixed 198 μL gold cell. In each experiment, 320 μL of protein solution was loaded into the ITC cell, and 200 μL of the corresponding compound was in the syringe. The data were acquired using ITCRun data acquisition software. The incremental ITC experiments consisted of multiple 2 μL injections at 50-350-second intervals with a stirring speed of 100 revolutions per minute (rpm). The Auto Equilibrate function allowed for the equilibration of the baseline to a peak-to-peak standard deviation of less than 12 nW. For each compound, separate control experiments were performed to evaluate the heat of dilution. The control experiments were set with identical parameters but performed with reaction buffer instead of protein in the ITC cell and consisted of fewer (6–10) injections.
Data Analysis
ITC data were analyzed with Nano Analyze Software (Waters/TA Instruments) using the “independent sites” model after correction for heats of dilution. Of note, because of limitations in the solubility of the compounds and the protein’s sensitivity to higher (>1%) concentrations of DMSO, these experiments were performed at c values lower than 1.
Statistical Analysis
Data are presented as means ± standard deviation (SD) with *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Statistical significance was determined by using a two-tailed Student’s t-test (Prism 8, GraphPad Software). P-values less than 0.05 were considered statistically significant. The number of biological replicates in each experiment is indicated by n in every figure caption.
Acknowledgments
This work was supported by the Office of the Assistant Secretary of Defense for Health Affairs through the Ovarian Cancer Research Program under Award Nos. W81XWH-22-1-0470 and W81XWH-22-1-0471. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense. The authors thank WuXi AppTec (China) for technical assistance with compound synthesis.
Glossary
Abbreviations Used
- AcOH
acetic acid
- ATCC
American Type Culture Collection
- CRBN
cereblon
- CSC
cancer stem cell
- DHI
bromodihydroisoxazole
- DIPEA
diisopropylamine
- DMF
dimethylformamide
- DMSO
dimethylsulfoxide
- EGFR
epidermal growth factor receptor
- EMT
epithelial–mesenchymal transition
- ESI
electrospray ionization
- EtOAc
ethyl acetate
- FN
fibronectin
- GDP
Guanosine diphosphate
- GTP
Guanosine triphosphate
- HATU
2-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HCl
hydrochloric acid
- HRMS
high-resolution mass spectrometry
- IMSERC
Integrated Molecular Structure Education and Research Center
- IPA
isopropanol
- ITC
isothermal calorimetry
- LCMS (LC/MS)
liquid chromatography/mass spectrometry
- NMR
nuclear magnetic resonance
- OC
ovarian cancer
- PDB
protein databank
- PEG
poly(ethylene glycol)
- PK
pharmacokinetics
- POI
protein of interest
- PROTAC
PROteolysis-TArgeting Chimera
- SAR
structure–activity relationship
- STR
short tandem repeat
- TG2
tissue transglutaminase
- THF
tetrahydrofuran
- TLC
thin-layer chromatography
- TOF
time-of-flight
- UPLC
ultra-high pressure liquid chromatography
- VHL
von Hippel–Lindau
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01859.
Author Contributions
This manuscript was written through contributions of all authors. All authors critically read and intellectually contributed to the manuscript. Conception and design: D.M., G.E.S.; development of methodology: A.V., P.P.V., D.M., and G.E.S.; acquisition of data: A.V., P.P.V., G.G., and E.O.; analysis and interpretation of data: A.V., P.P.V., D.M., and G.E.S.; writing, review, and/or revision of the manuscript: A.V., P.P.V., E.O., D.M., and G.E.S.; administrative, technical, or material support: D.M. and G.E.S.; study supervision: DM and GES.
The authors declare no competing financial interest.
Supplementary Material
References
- Huang X.; Lin T.; Gu J.; Zhang L.; Roth J. A.; Stephens L. C.; Yu Y.; Liu J.; Fang B. Combined TRAIL and Bax gene therapy prolonged survival in mice with ovarian cancer xenograft. Gene Ther. 2002, 9, 1379–1386. 10.1038/sj.gt.3301810. [DOI] [PubMed] [Google Scholar]
- Stephens P.; Grenard P.; Aeschlimann P.; Langley M.; Blain E.; Errington R.; Kipling D.; Thomas D.; Aeschlimann D. Crosslinking and G-protein functions of transglutaminase 2 contribute differentially to fibroblast wound healing responses. J. Cell Sci. 2004, 117, 3389–3403. 10.1242/jcs.01188. [DOI] [PubMed] [Google Scholar]
- Fesus L.; Piacentini M. Transglutaminase 2: an enigmatic enzyme with diverse functions. Trends Biochem. Sci. 2002, 27, 534–539. 10.1016/S0968-0004(02)02182-5. [DOI] [PubMed] [Google Scholar]
- Akimov S. S.; Belkin A. M. Cell surface tissue transglutaminase is involved in adhesion and migration of monocytic cells on fibronectin. Blood 2001, 98, 1567–1576. 10.1182/blood.V98.5.1567. [DOI] [PubMed] [Google Scholar]
- Akimov S. S.; Krylov D.; Fleischman L. F.; Belkin A. M. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J. Cell Biol. 2000, 148, 825–838. 10.1083/jcb.148.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio E. A.; Telci D.; Okoye A.; Melino G.; Griffin M. A novel RGD-independent cel adhesion pathway mediated by fibronectin-bound tissue transglutaminase rescues cells from anoikis. J. Biol. Chem. 2003, 278, 42604–42614. 10.1074/jbc.M303303200. [DOI] [PubMed] [Google Scholar]
- Zemskov E. A.; Loukinova E.; Mikhailenko I.; Coleman R. A.; Strickland D. K.; Belkin A. M. Regulation of platelet-derived growth factor receptor function by integrin-associated cell surface transglutaminase. J. Biol. Chem. 2009, 284, 16693–16703. 10.1074/jbc.M109.010769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemskov E. A.; Mikhailenko I.; Smith E. P.; Belkin A. M. Tissue transglutaminase promotes PDGF/PDGFR-mediated signaling and responses in vascular smooth muscle cells. J. Cell Physiol. 2012, 227, 2089–2096. 10.1002/jcp.22938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condello S.; Sima L. E.; Ivan C.; Cardenas H.; Schiltz G. E.; Mishra R. K.; Matei D. Tissue transglutaminase regulates interactions between ovarian cancer stem cells and the tumor niche. Cancer Res. 2018, 78, 2990–3001. 10.1158/0008-5472.CAN-17-2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facchiano F.; Facchiano A.; Facchiano A. M. The role of transglutaminase-2 and its substrates in human diseases. Front. Biosci. 2006, 11, 1758–1773. 10.2741/1921. [DOI] [PubMed] [Google Scholar]
- Yuan Z. Q.; Sun M.; Feldman R. I.; Wang G.; Ma X.; Jiang C.; Coppola D.; Nicosia S. V.; Cheng J. Q. Frequent activation of AKT2 and induction of apoptosis by inhibition of phosphoinositide-3-OH kinase/Akt pathway in human ovarian cancer. Oncogene 2000, 19, 2324–2330. 10.1038/sj.onc.1203598. [DOI] [PubMed] [Google Scholar]
- Yi M. C.; Khosla C. Thiol-Disulfide Exchange Reactions in the Mammalian Extracellular Environment. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 197–222. 10.1146/annurev-chembioeng-080615-033553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palanski B. A.; Khosla C. Cystamine and Disulfiram Inhibit Human Transglutaminase 2 via an Oxidative Mechanism. Biochemistry 2018, 57, 3359–3363. 10.1021/acs.biochem.8b00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimov S. S.; Belkin A. M. Cell-surface transglutaminase promotes fibronectin assembly via interaction with the gelatin-binding domain of fibronectin: a role in TGFbeta-dependent matrix deposition. J. Cell Sci. 2001, 114, 2989–3000. 10.1242/jcs.114.16.2989. [DOI] [PubMed] [Google Scholar]
- Belkin A. M.; Tsurupa G.; Zemskov E.; Veklich Y.; Weisel J. W.; Medved L. Transglutaminase-mediated oligomerization of the fibrin(ogen) alphaC domains promotes integrin-dependent cell adhesion and signaling. Blood 2005, 105, 3561–3568. 10.1182/blood-2004-10-4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaartinen M. T.; El-Maadawy S.; Rasanen N. H.; McKee M. D. Tissue transglutaminase and its substrates in bone. J. Bone Miner. Res. 2002, 17, 2161–2173. 10.1359/jbmr.2002.17.12.2161. [DOI] [PubMed] [Google Scholar]
- Aeschlimann D.; Paulsson M. Cross-linking of laminin-nidogen complexes by tissue transglutaminase. A novel mechanism for basement membrane stabilization. J. Biol. Chem. 1991, 266, 15308–15317. 10.1016/S0021-9258(18)98617-5. [DOI] [PubMed] [Google Scholar]
- Jones R. A.; Kotsakis P.; Johnson T. S.; Chau D. Y.; Ali S.; Melino G.; Griffin M. Matrix changes induced by transglutaminase 2 lead to inhibition of angiogenesis and tumor growth. Cell Death Differ. 2005, 13, 1442–1453. 10.1038/sj.cdd.4401816. [DOI] [PubMed] [Google Scholar]
- Satpathy M.; Cao L.; Pincheira R.; Emerson R.; Bigsby R.; Nakshatri H.; Matei D. Enhanced peritoneal ovarian tumor dissemination by tissue transglutaminase. Cancer Res. 2007, 67, 7194–7202. 10.1158/0008-5472.CAN-07-0307. [DOI] [PubMed] [Google Scholar]
- Shao M.; Cao L.; Shen C.; Satpathy M.; Chelladurai B.; Bigsby R. M.; Nakshatri H.; Matei D. Epithelial-to-mesenchymal transition and ovarian tumor progression induced by tissue transglutaminase. Cancer Res. 2009, 69, 9192–9201. 10.1158/0008-5472.CAN-09-1257. [DOI] [PubMed] [Google Scholar]
- Iacobuzio-Donahue C. A.; Ashfaq R.; Maitra A.; Adsay N. V.; Shen-Ong G. L.; Berg K.; Hollingsworth M. A.; Cameron J. L.; Yeo C. J.; Kern S. E.; Goggins M.; Hruban R. H. Highly expressed genes in pancreatic ductal adenocarcinomas: a comprehensive characterization and comparison of the transcription profiles obtained from three major technologies. Cancer Res. 2003, 63, 8614–8622. [PubMed] [Google Scholar]
- Martinet N.; Bonnard L.; Regnault V.; Picard E.; Burke L.; Siat J.; Grosdidier G.; Martinet Y.; Vignaud J. M. In vivo transglutaminase type 1 expression in normal lung, preinvasive bronchial lesions, and lung cancer. Am. J. Respir. Cell Mol. Biol. 2003, 28, 428–435. 10.1165/rcmb.2002-0114OC. [DOI] [PubMed] [Google Scholar]
- Grigoriev M. Y.; Suspitsin E. N.; Togo A. V.; Pozharisski K. M.; Ivanova O. A.; Nardacci R.; Falasca L.; Piacentini M.; Imyanitov E. N.; Hanson K. P. Tissue transglutaminase expression in breast carcinomas. J. Exp. Clin. Cancer Res. 2001, 20, 265–268. [PubMed] [Google Scholar]
- Derynck R.; Zhang Y. E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Hwang J. Y.; Mangala L. S.; Fok J. Y.; Lin Y. G.; Merritt W. M.; Spannuth W. A.; Nick A. M.; Fiterman D. J.; Vivas-Mejia P. E.; Deavers M. T.; Coleman R. L.; Lopez-Berestein G.; Mehta K.; Sood A. K. Clinical and biological significance of tissue transglutaminase in ovarian carcinoma. Cancer Res. 2008, 68, 5849–5858. 10.1158/0008-5472.CAN-07-6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A.; Wang H.; Manavathi B.; Fok J. Y.; Mann A. P.; Kumar R.; Mehta K. Increased expression of tissue transglutaminase in pancreatic ductal adenocarcinoma and its implications in drug resistance and metastasis. Cancer Res. 2006, 66, 10525–10533. 10.1158/0008-5472.CAN-06-2387. [DOI] [PubMed] [Google Scholar]
- Jeong J. H.; Cho B. C.; Shim H. S.; Kim H. R.; Lim S. M.; Kim S. K.; Chung K. Y.; Islam S. M.; Song J. J.; Kim S. Y.; Kim J. H. Transglutaminase 2 expression predicts progression free survival in non-small cell lung cancer patients treated with epidermal growth factor receptor tyrosine kinase inhibitor. J. Korean Med. Sci. 2013, 28, 1005–1014. 10.3346/jkms.2013.28.7.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta K.; Lopez-Berestein G.; Moore W. T.; Davies P. J. Interferon-gamma requires serum retinoids to promote the expression of tissue transglutaminase in cultured human blood monocytes. J. Immunol. 1985, 134, 2053–2056. 10.4049/jimmunol.134.4.2053. [DOI] [PubMed] [Google Scholar]
- Cao L.; Petrusca D. N.; Satpathy M.; Nakshatri H.; Petrache I.; Matei D. Tissue Transglutaminase Protects Epithelial Ovarian Cancer Cells from Cisplatin Induced Apoptosis by Promoting Cell Survival Signaling. Carcinogenesis 2008, 29, 1893–1900. 10.1093/carcin/bgn158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann A. P.; Verma A.; Sethi G.; Manavathi B.; Wang H.; Fok J. Y.; Kunnumakkara A. B.; Kumar R.; Aggarwal B. B.; Mehta K. Overexpression of Tissue Transglutaminase Leads to Constitutive Activation of Nuclear Factor-{kappa}B in Cancer Cells: Delineation of a Novel Pathway. Cancer Res. 2006, 66, 8788–8795. 10.1158/0008-5472.CAN-06-1457. [DOI] [PubMed] [Google Scholar]
- Cao L.; Shao M.; Schilder J.; Guise T.; Mohammad K. S.; Matei D. Tissue transglutaminase links TGF-β, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene 2012, 31, 2521–2534. 10.1038/onc.2011.429. [DOI] [PubMed] [Google Scholar]
- Verma A.; Guha S.; Wang H.; Fok J. Y.; Koul D.; Abbruzzese J.; Mehta K. Tissue transglutaminase regulates focal adhesion kinase/AKT activation by modulating PTEN expression in pancreatic cancer cells. Clin. Cancer Res. 2008, 14, 1997–2005. 10.1158/1078-0432.CCR-07-1533. [DOI] [PubMed] [Google Scholar]
- Condello S.; Cao L.; Matei D. Tissue transglutaminase regulates beta-catenin signaling through a c-Src-dependent mechanism. FASEB J. 2013, 10.1096/fj.12-222620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A.; Guha S.; Diagaradjane P.; Kunnumakkara A. B.; Sanguino A. M.; Lopez-Berestein G.; Sood A. K.; Aggarwal B. B.; Krishnan S.; Gelovani J. G.; Mehta K. Therapeutic significance of elevated tissue transglutaminase expression in pancreatic cancer. Clin. Cancer Res. 2008, 14, 2476–2483. 10.1158/1078-0432.CCR-07-4529. [DOI] [PubMed] [Google Scholar]
- Singh U. S.; Kunar M. T.; Kao Y. L.; Baker K. M. Role of transglutaminase II in retinoic acid-induced activation of RhoA-associated kinase-2. EMBO J. 2001, 20, 2413–2423. 10.1093/emboj/20.10.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr C.; Szmacinski H.; Fisher M. L.; Nance B.; Lakowicz J. R.; Akbar A.; Keillor J. W.; Lok Wong T.; Godoy-Ruiz R.; Toth E. A.; Weber D. J.; Eckert R. L. Transamidase site-targeted agents alter the conformation of the transglutaminase cancer stem cell survival protein to reduce GTP binding activity and cancer stem cell survival. Oncogene 2017, 36, 2981–2990. 10.1038/onc.2016.452. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Kumar A.; Xu J.; Sung B.; Kumar S.; Yu D.; Aggarwal B. B.; Mehta K. Evidence that GTP-binding domain but not catalytic domain of transglutaminase 2 is essential for epithelial-to-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2012, 14, R4 10.1186/bcr3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L.; Choi K.; Khosla C.; Zheng X.; Higashikubo R.; Chicoine M. R.; Rich K. M. Tissue transglutaminase 2 inhibition promotes cell death and chemosensitivity in glioblastomas. Mol. Cancer Ther. 2005, 4, 1293–1302. 10.1158/1535-7163.MCT-04-0328. [DOI] [PubMed] [Google Scholar]
- Yuan L.; Siegel M.; Choi K.; Khosla C.; Miller C. R.; Jackson E. N.; Piwnica-Worms D.; Rich K. M. Transglutaminase 2 inhibitor, KCC009, disrupts fibronectin assembly in the extracellular matrix and sensitizes orthotopic glioblastomas to chemotherapy. Oncogene 2007, 26, 2563–2573. 10.1038/sj.onc.1210048. [DOI] [PubMed] [Google Scholar]
- Yakubov B.; Chen L.; Belkin A. M.; Zhang S.; Chelladurai B.; Zhang Z. Y.; Matei D. Small molecule inhibitors target the tissue transglutaminase and fibronectin interaction. PLoS One 2014, 9, e89285 10.1371/journal.pone.0089285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sima L. E.; Yakubov B.; Zhang S.; Condello S.; Grigorescu A. A.; Nwani N. G.; Chen L.; Schiltz G. E.; Arvanitis C.; Zhang Z. Y.; Matei D. Small Molecules Target the Interaction between Tissue Transglutaminase and Fibronectin. Mol. Cancer Ther. 2019, 18, 1057–1068. 10.1158/1535-7163.MCT-18-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y.; Ma D.; Wang Y. The PROTAC technology in drug development. Cell Biochem. Funct. 2019, 37, 21–30. 10.1002/cbf.3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Smith B. E.; Burslem G. M.; Buhimschi A. D.; Hines J.; Jaime-Figueroa S.; Wang J.; Hamman B. D.; Ishchenko A.; Crews C. M. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 2018, 25, 78–87.e5. 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K. M.; Kim K. B.; Kumagai A.; Mercurio F.; Crews C. M.; Deshaies R. J. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sc.i U.S.A. 2001, 98, 8554–8559. 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H. R.; Zheng S.; Gurpinar E.; Koehl A.; Manglik A.; Kruse A. C. Crystal structure of the human sigma1 receptor. Nature 2016, 532, 527–530. 10.1038/nature17391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd M. S.; Testa A.; Lucas X.; Chan K.-H.; Chen W.; Lamont D. J.; Zengerle M.; Ciulli A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa T. K.; Crews C. M. Chemical biology: Greasy tags for protein removal. Nature 2012, 487, 308–309. 10.1038/487308a. [DOI] [PubMed] [Google Scholar]
- Winter G. E.; Buckley D. L.; Paulk J.; Roberts J. M.; Souza A.; Dhe-Paganon S.; Bradner J. E. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa T.; Snyder L. B.; Willard R. R.; Vitale N.; Pizzano J.; Gordon D. A.; Bookbinder M.; Macaluso J.; Dong H.; Ferraro C.; et al. ARV-110: an oral androgen receptor PROTAC degrader for prostate cancer. J. Clin. Oncol. 2019, 37, 259. 10.1200/JCO.2019.37.7_suppl.259. [DOI] [Google Scholar]
- Mullard A. Targeted protein degraders crowd into the clinic. Nat. Rev. Drug Discovery 2021, 20, 247–250. 10.1038/d41573-021-00052-4. [DOI] [PubMed] [Google Scholar]
- Békés M.; Langley D. R.; Crews C. M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discovery 2022, 21, 181–200. 10.1038/s41573-021-00371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burslem G. M.; Smith B. E.; Lai A. C.; Jaime-Figueroa S.; McQuaid D. C.; Bondeson D. P.; Toure M.; Dong H.; Qian Y.; Wang J.; Crew A. P.; Hines J.; Crews C. M. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol. 2018, 25, 67–77.e3. 10.1016/j.chembiol.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbar A.; McNeil N. M. R.; Albert M. R.; Ta V.; Adhikary G.; Bourgeois K.; Eckert R. L.; Keillor J. W. Structure-Activity Relationships of Potent, Targeted Covalent Inhibitors That Abolish Both the Transamidation and GTP Binding Activities of Human Tissue Transglutaminase. J. Med. Chem. 2017, 60, 7910–7927. 10.1021/acs.jmedchem.7b01070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina K.; Lu J.; Qian Y.; Altieri M.; Gordon D.; Rossi A. M. K.; Wang J.; Chen X.; Dong H.; Siu K.; Winkler J. D.; Crew A. P.; Crews C. M.; Coleman K. G. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 7124–7129. 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M.; Hwang H.; Im C. Y.; Kim S. Y. Recent Progress in the Development of Transglutaminase 2 (TGase2) Inhibitors. J. Med. Chem. 2017, 60, 554–567. 10.1021/acs.jmedchem.6b01036. [DOI] [PubMed] [Google Scholar]
- Klöck C.; Herrera Z.; Albertelli M.; Khosla C. Discovery of potent and specific dihydroisoxazole inhibitors of human transglutaminase 2. J. Med. Chem. 2014, 57, 9042–9064. 10.1021/jm501145a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrobok N. L.; Bol J.; Jongenelen C. A.; Breve J. J. P.; El Alaoui S.; Wilhelmus M. M. M.; Drukarch B.; van Dam A. M. Characterization of Transglutaminase 2 activity inhibitors in monocytes in vitro and their effect in a mouse model for multiple sclerosis. PLoS One 2018, 13, e0196433 10.1371/journal.pone.0196433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büchold C.; Hils M.; Gerlach U.; Weber J.; Pelzer C.; Heil A.; Aeschlimann D.; Pasternack R. Features of ZED1227: The First-In-Class Tissue Transglutaminase Inhibitor Undergoing Clinical Evaluation for the Treatment of Celiac Disease. Cells 2022, 11, 1667. 10.3390/cells11101667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wityak J.; Prime M. E.; Brookfield F. A.; Courtney S. M.; Erfan S.; Johnsen S.; Johnson P. D.; Li M.; Marston R. W.; Reed L.; Vaidya D.; Schaertl S.; Pedret-Dunn A.; Beconi M.; Macdonald D.; Munoz-Sanjuan I.; Dominguez C. SAR Development of Lysine-Based Irreversible Inhibitors of Transglutaminase 2 for Huntington’s Disease. ACS Med. Chem. Lett. 2012, 3, 1024–1028. 10.1021/ml300241m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil N. M. R.; Gates E. W. J.; Firoozi N.; Cundy N. J.; Leccese J.; Eisinga S.; Tyndall J. D. A.; Adhikary G.; Eckert R. L.; Keillor J. W. Structure-activity relationships of N-terminal variants of peptidomimetic tissue transglutaminase inhibitors. Eur. J. Med. Chem. 2022, 232, 114172 10.1016/j.ejmech.2022.114172. [DOI] [PubMed] [Google Scholar]
- Wodtke R.; Hauser C.; Ruiz-Gomez G.; Jackel E.; Bauer D.; Lohse M.; Wong A.; Pufe J.; Ludwig F. A.; Fischer S.; Hauser S.; Greif D.; Pisabarro M. T.; Pietzsch J.; Pietsch M.; Loser R. N(epsilon)-Acryloyllysine Piperazides as Irreversible Inhibitors of Transglutaminase 2: Synthesis, Structure-Activity Relationships, and Pharmacokinetic Profiling. J. Med. Chem. 2018, 61, 4528–4560. 10.1021/acs.jmedchem.8b00286. [DOI] [PubMed] [Google Scholar]
- Wodtke R.; Wodtke J.; Hauser S.; Laube M.; Bauer D.; Rothe R.; Neuber C.; Pietsch M.; Kopka K.; Pietzsch J.; Loser R. Development of an (18)F-Labeled Irreversible Inhibitor of Transglutaminase 2 as Radiometric Tool for Quantitative Expression Profiling in Cells and Tissues. J. Med. Chem. 2021, 64, 3462–3478. 10.1021/acs.jmedchem.1c00096. [DOI] [PubMed] [Google Scholar]
- van der Wildt B.; Wilhelmus M. M.; Bijkerk J.; Haveman L. Y.; Kooijman E. J.; Schuit R. C.; Bol J. G.; Jongenelen C. A.; Lammertsma A. A.; Drukarch B.; Windhorst A. D. Development of carbon-11 labeled acryl amides for selective PET imaging of active tissue transglutaminase. Nucl. Med. Biol. 2016, 43, 232–242. 10.1016/j.nucmedbio.2016.01.003. [DOI] [PubMed] [Google Scholar]
- Duval E.; Case A.; Stein R. L.; Cuny G. D. Structure-activity relationship study of novel tissue transglutaminase inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 1885–1889. 10.1016/j.bmcl.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Keillor J. W.; Apperley K. Y.; Akbar A. Inhibitors of tissue transglutaminase. Trends Pharmacol. Sci. 2015, 36, 32–40. 10.1016/j.tips.2014.10.014. [DOI] [PubMed] [Google Scholar]
- Case A.; Stein R. L. Kinetic analysis of the interaction of tissue transglutaminase with a nonpeptidic slow-binding inhibitor. Biochemistry 2007, 46, 1106–1115. 10.1021/bi061787u. [DOI] [PubMed] [Google Scholar]
- Pardin C.; Pelletier J. N.; Lubell W. D.; Keillor J. W. Cinnamoyl inhibitors of tissue transglutaminase. J. Org. Chem. 2008, 73, 5766–5775. 10.1021/jo8004843. [DOI] [PubMed] [Google Scholar]
- Ku B. M.; Kim S. J.; Kim N.; Hong D.; Choi Y. B.; Lee S. H.; Gong Y. D.; Kim S. Y. Transglutaminase 2 inhibitor abrogates renal cell carcinoma in xenograft models. J. Cancer Res. Clin. Oncol. 2014, 140, 757–767. 10.1007/s00432-014-1623-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna M.; Chelladurai B.; Gavini A.; Li L.; Shao M.; Courtney D.; Turchi J. J.; Matei D.; Meroueh S. Targeting ovarian tumor cell adhesion mediated by tissue transglutaminase. Mol. Cancer Ther. 2011, 10, 626–636. 10.1158/1535-7163.MCT-10-0912. [DOI] [PubMed] [Google Scholar]
- Zengerle M.; Chan K. H.; Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy M.; Shao M.; Emerson R.; Donner D. B.; Matei D. Tissue transglutaminase regulates matrix metalloproteinase-2 in ovarian cancer by modulating cAMP-response element-binding protein activity. J. Biol. Chem. 2009, 284, 15390–15399. 10.1074/jbc.M808331200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull W. B.; Daranas A. H. On the value of c: can low affinity systems be studied by isothermal titration calorimetry?. J. Am. Chem. Soc. 2003, 125, 14859–14866. 10.1021/ja036166s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.