

Abstract

The mitochondrial SIRT3 modulates several biological pathways such as cancer, metabolism, and hypoxia-related diseases. Recently, we discovered new 1,4-dihydropyridines, compounds 2 and 3, the latter being a SIRT3-specific activator. In the present work, a novel 2- and 3-related small series of compounds have been developed, with 3c displaying the strongest SIRT3 binding and activation, with a KD of 29 μM and 387% of enzyme activation. Differently, 3d was the best in enhancing glutamate dehydrogenase activity and deacetylating K68- and K122-acMnSOD in triple-negative MDA-MB-231 breast cancer cells. Tested in CAL-62 thyroid cancer and MDA-MB-231 cells, 3d displayed the strongest time- and dose-dependent reduction of cell viability and clonogenicity at a single-digit micromolar level, along with cell death, in both normoxia and hypoxia conditions. Moreover, 3d downregulated not only hypoxia-induced factors, such as HIF-1α, EPAS-1, and CA-IX, but also epithelial–mesenchymal transition master regulators and extracellular matrix components such as SNAIL1, ZEB1, SLUG, COL1A2, MMP2, and MMP9, markedly hampering MDA-MB-231 cell migration.
Introduction
The sirtuins family is composed of NAD+-dependent lysine deacetylases that, in humans, contains seven members (SIRT1–7) and belong to the class III histone deacetylases.1−3 The 275-amino acid catalytic core is highly conserved within the SIRT family, but their C- and N-terminal domains differ in length and sequence. Sirtuins display isoform-specific substrate affinity because of slender differences in their peptide substrate binding sites.4
Sirtuins have gained increasing attention over the past two decades due to their crucial roles in several biochemical contexts, including cell cycle progression, inflammation, energetic metabolism, apoptosis, neuro- and cardioprotection, and cancer onset and progression.5−7 Among SIRTs, SIRT1–3 mainly are deacetylating enzymes, whereas SIRT4 and SIRT6 own a substrate-specific mono-ADP-ribosyltransferase activity other their deacetylase and deacylase activities. SIRT5 can deacylate succinyl-, glutaryl-, and malonyl-lysine residues. Finally, for SIRT7, a new desuccinylase activity in addition to the previously known deacetylase one has been discovered.8
In general, sirtuins are regulated by post-translational modifications, connections with endogenous molecules and protein–protein interactions, and their physiological activity is controlled transcriptionally and by regulation of their degradation.9
In more detail, SIRT3 is predominantly located in the mitochondrial matrix and has a major role in numerous mitochondrial metabolic processes, such as the tricarboxylic acid (TCA) cycle, the urea cycle, amino acid metabolism, fatty acid oxidation, mitochondrial electron transport chain (ETC)/oxidative phosphorylation (OXPHOS), reactive oxygen species (ROS) detoxification, mitochondrial dynamics, and the mitochondrial unfolded protein response (UPR).10,11 In addition to its role in regulating metabolism-dependent diseases, SIRT3 displays a double-sided function in cancer development.12
Indeed, SIRT3 in cancer exerts a context-dependent role,13 being tumorigenic in some cancer types, tumor suppressor in others.14−16 One of the most studied oncogenic pathways concerns the regulation of hypoxia-inducible factor-1α (HIF-1α), a key transcription factor activating several glycolytic genes involved in the “Warburg effect”. SIRT3 regulates the HIF-1α activity by direct deacetylation and activation of prolyl hydroxylase (PHD), thus leading to HIF-1α ubiquitination and proteasomal degradation.17 Another study proved that SIRT3 overexpression destabilized HIF-1α in hypoxic human breast cancer cells, where the SIRT3 catalytic activity was needed for the complete repression of HIF-1α target genes.15 SIRT3 can deacetylate and modulate several targets directly or indirectly involved in glycolysis regulation leading to anticancer effects. Indeed, SIRT3 has been reported to regulate the pyruvate dehydrogenase complex (PDC) in lung cancer,18 cyclophilin D in breast cancer,19 glutamate oxaloacetate transaminase 2 (GOT2) in pancreatic cancer,20 and manganese-dependent superoxide dismutase (MnSOD) in chronic lymphocytic leukemia (CLL)21 and in hepatocellular carcinoma (HCC).22
Differently, to date, rather little literature evidence and even mixed reports are available for the role of SIRT3 in differentiated thyroid cancer (DTC). First, older evidence showed that SIRT3 was highly expressed in DTC compared to benign thyroid tumors,23 while Wang et al. showed that MiR-1225-5p might trigger DTC cell proliferation and metastasis by targeting SIRT3.24 Newer findings by Yao and Wang, in a more clinical setting, indicate the opposite, showing that mRNA expression levels of SIRT3 decreased in their DTC model compared to healthy cells and that patients with high SIRT3 expression had a longer disease-free survival.25
SIRT3 is not only involved in cancer but also in infectious,26,27 heart,28 kidney,29 metabolic,30,31 and neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and Huntington’s disease, as well as in stroke and traumatic brain injury.32 A recent study by Yan et al. stated that SIRT3 is very likely to play a crucial role in the development of neuropathic pain.33
On these bases, SIRT3 activation would be a fascinating approach for the treatment of several diseases, including cancer.
To date, several activators of sirtuins have been developed, with particular emphasis on SIRT1.34 Among these, resveratrol (Figure 1A) has been shown to activate sirtuins and demonstrated promising results in a phase II clinical trial involving patients with type 2 diabetes and coronary heart disease.35 Its effects are limited by the poor bioavailability, but different studies are underway to try to improve it.36 Due to the polyphenolic nature of resveratrol, the biological activity of this molecule may be correlated with a wide range of pleiotropic modes of action, which has spurred the identification and development of more potent sirtuin-activating compounds (STACs), such as SRT1720 and SRT2104 (Figure 1A). Among STACs, SRT2104 is the most extensively studied SIRT1 activator, with various clinical trials demonstrating its anti-inflammatory, anticoagulant,37 anti-cholesterol,38 and antipsoriasis39 effects.
Figure 1.

Known human sirtuins activators. (A) SIRT1 activators; (B) SIRT6 activators; and (C) SIRT3 activators.
The discovery that the free 14–18 carbon-containing fatty acids act as weak SIRT6 activators has spurred the development of SIRT6 activators.43,44 UBCS039 (Figure 1B) activates SIRT6 in the low-micromolar range and inhibits the proliferation of HCC cells, as well as suppresses tumor growth in a rodent xenograft model.45 MDL-800 (Figure 1B) inhibits the proliferation of 12 non-small-cell lung cancer cell lines46 and suppresses tumor growth in a rodent xenograft adenocarcinoma model.47 MDL-811 (Figure 1B) is another SIRT6 activator that exhibits twice the activity of MDL-800 and has antiproliferative effects in colorectal cancer (CRC) cells. Additionally, MDL-811 inhibits CRC growth in patient-derived organoids, in a patient-derived xenograft, and in a spontaneous CRC mouse model, enhancing vitamin D3 anticancer activity.48
As briefly outlined above, small synthetic molecules have been mainly described as activators of SIRT134,35,37−41 and SIRT6,43,45−51 while the development of potent small molecules as SIRT3 activators is still in its infancy. Different positive modulators of SIRT3 have been reported in literature, able to induce SIRT3 expression or its enzymatic activity. Noteworthy, the natural lignan honokiol (Figure 1C) has been extensively studied as a SIRT3 activator and was reported to SIRT3-dependently block cardiac fibroblast proliferation and differentiation, and finally to ameliorate cardiac hypertrophy in mice.52 Moreover, honokiol proved to stimulate SIRT3 activation to hamper the NF-κB/TGF-β1/Smad-regulated inflammation and fibrosis signaling in a renal fibrosis mouse model.53 In addition, metformin, a well-known AMPK activator used for the first-line therapy of type 2 diabetes, has been proven to SIRT3-dependently reduce atherosclerosis in type 2 diabetes patients.54 Adjudin (Figure 1C) proved to protect rodent cochlear hair cells against gentamicin ototoxicity via the SIRT3-ROS pathway in both in vitro and in vivo studies.55 The serotonin-derived pineal hormone melatonin has been found to increase SIRT3 expression and to play a protective role in heart disease, liver, and atherosclerosis.56 Lastly, caffeine was also described to interact with SIRT3 and induce its enzymatic activity, thus leading to K68 and K122 MnSOD deacetylation and activation.57 At the end of 2021, through a structure-guided design and high-throughput screening, the Liu’s research group discovered an amiodarone-derived small-molecule, compound 33c (Figure 1C), as a specific activator of SIRT3. Indeed, proliferation and migration of human triple-negative breast cancer MDA-MB-231 cells were hampered in vitro and in vivo by compound 33c through SIRT3-mediated autophagy/mitophagy signaling cascades.58
Results and Discussion
Since 2009 our research group has been working on the 1,4-dihydropyridine (1,4-DHP) scaffold as SIRT activators,40−42 identifying compound 1 (MC2562, Figure 2) as a first hit compound. Very recently, we have described the characterization of novel 1,4-DHP-based compounds that act as specific activators of SIRT3 or SIRT5.59 The latter bind to the catalytic cores independently from the NAD+ co-factor and the relative acetylated substrates and lead to an increase of the peptide or protein substrate turnover. Among them, compound 2 (MC2971, Figure 2), bearing at N1-DHP position a benzoyl moiety, proved a mixed SIRT1/3 activation providing stronger activation of SIRT3 (over 300% at 100 μM) than SIRT1 (below 200% at 100 μM), whereas compound 3 (MC2789, Figure 2), bearing a 3,4,5-trimethoxybenzoyl moiety, displayed the highest SIRT3 specific activation (over 400% at 100 μM).59 The SIRT3 activation by 3 was also confirmed by an MS-based assay, and both 2 and 3 exhibited SIRT3-driven cellular effects.59 Indeed, in MDA-MB-231 cells, 2 and 3 increased the activity of glutamate dehydrogenase (GDH), a SIRT3 substrate activated by deacetylation, and 2 proved to deacetylate the acK68 residue of MnSOD59 in the same cell line.
Figure 2.
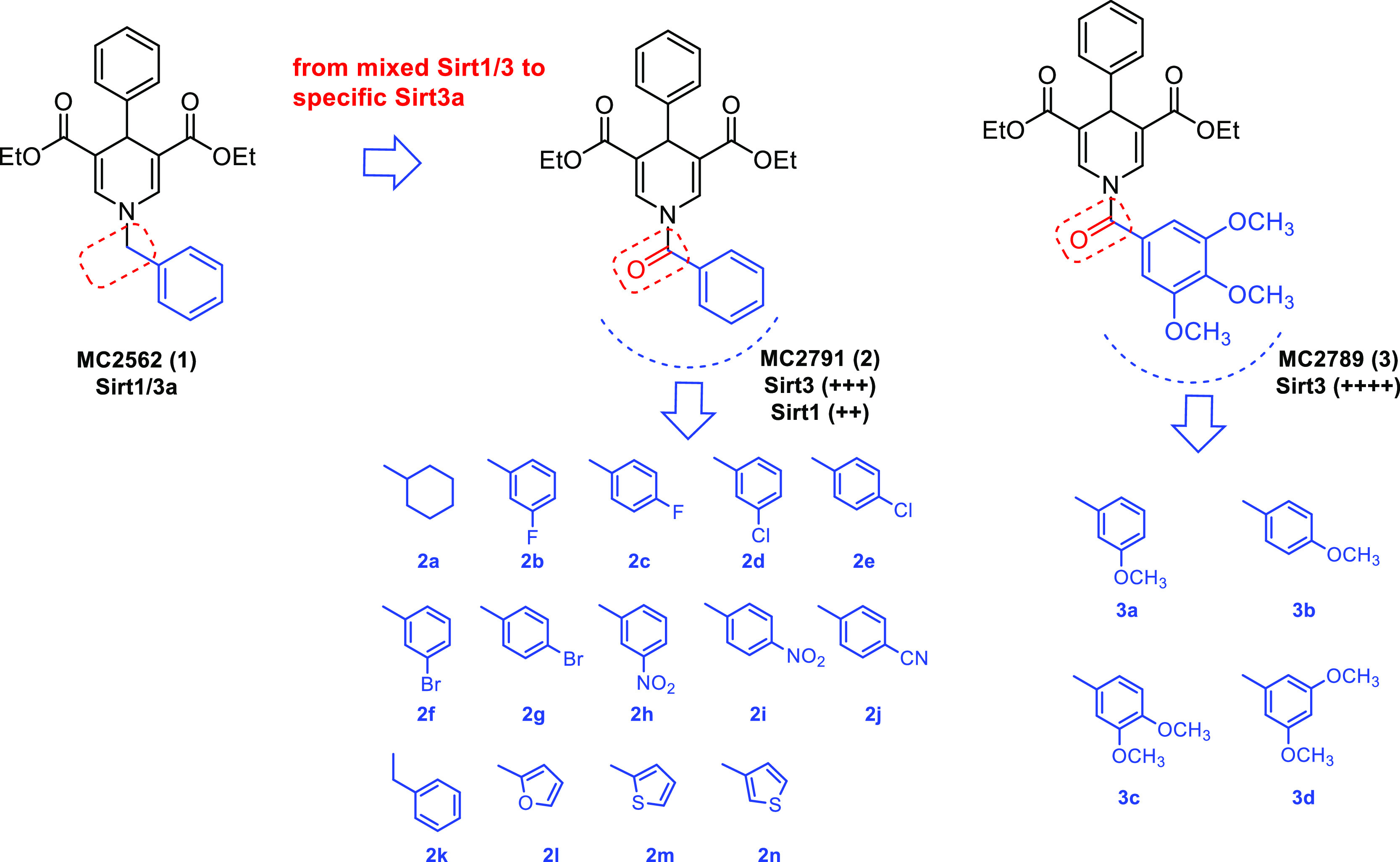
Design of novel 1,4-DHP-based SIRT3 activators 2a–n and 3a–d starting from the first identified hit 1.
Based on these findings, we developed a novel 1,4-DHP-based series as analogues of the SIRT3 activators 2 and 3 through the following approaches: (a) by adding different substituents including halogens, nitro, and cyano groups at meta or para (ortho was known to be unfavorable by previous SAR data40,41,59) position of the N1-benzoyl ring; (b) by replacing the phenyl ring of the N1-benzoyl moiety with its sp3 carbon analogue (cyclohexane) or some its isosteres (furan and thiophene); and (c) by introducing a methylene spacer into the N1-benzoyl moiety to provide the corresponding N1-phenylacetyl derivative (see compounds 2a–n, Figure 2). Also, some simplified analogues of 3 obtained by removing one by one the methoxy substituents from the 3,4,5-trimethoxybenzoyl moiety were prepared (3a–d, Figure 2). All novel compounds were tested against human SIRT3, and the selected 3a, 3c, and 3d were also tested against human SIRT1, -2, and -5 to assess their SIRT3-specificity. Cellular functional assays for proving SIRT3 activation were performed by detecting GDH activation and changes in MnSOD acetylation levels in MDA-MB-231 cells. Moreover, the effects of selected SIRT3 activators on MDA-MB-231 and thyroid CAL-62 cancer cell viability and colony formation, as well as modulation of hypoxia-induced targets (i.e., HIF-1α, EPAS-1, and CA-IX) upon normoxia and hypoxia conditions were assessed. Furthermore, the expression of epithelial–mesenchymal transition (EMT) master regulators and of extracellular matrix (ECM) components, along with the cell migration, were analyzed upon MDA-MB-231 cells treatment with the selected compounds.
Chemistry
The key intermediate diethyl 4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate (4) has been prepared via a multicomponent cyclocondensation between benzaldehyde, ethyl propiolate, and ammonium acetate at 80 °C in glacial acetic acid as previously described by us.41 Next, compound 4 underwent N1-acylation using triethylamine and the appropriate acyl chloride in dry dichloromethane to furnish the final compounds 2a–n and 3a–d (Scheme 1).
Scheme 1.

Reagents and conditions: (a) glacial acetic acid, 80 °C, 5 h; (b) Et3N, acyl chloride, dry dichloromethane, rt, overnight.
Sirtuins Biochemical Assay
Nicotinamidase (PncA)/GDH-Coupled Deacylation Assays for Sirt1, -2, -3, and -5 Activation
To avoid artifacts and other non-specific effects, instead of the “Fluor-de-Lys” (FdL) substrate, we used a PncA/GDH-coupled deacylation assay by measuring the NADPH oxidation to NADP+ that relies on isoform-specific non-modified physiological acetyl or succinyl substrates [p53-acK381 for SIRT1, α-tubulin-acK25 for SIRT2, acetyl-CoA synthetase 2 (ACS2)-acK642 for SIRT3, and CPS1-succK537 for SIRT5].59 The assays were carried out using the 1,4-DHP derivatives 2a–n and 3a–d against SIRT3, including the prototype compounds 2 and 3 as positive controls.
As shown in Figure 3, all the novel compounds 2a–n and 3a–d displayed SIRT3 activation at 10 μM with values from 120% (cyclohexanoyl derivative, 2a) to 176% (phenylacetyl derivative, 2k), thus showing generally similar potency as the reference compounds 2 (157%) and 3 (128%) at this dose. When tested at 100 μM, the prototypes 2 and 3 confirmed their strong SIRT3 activation (188 and 269%, respectively). Among the 2a–n analogues, 2a, 2b, 2d–g, 2k, and 2n displayed activation potencies similar as or higher than 2 at 100 μM, with 2d–f being the most potent, highlighting the importance of halogen insertion at the meta (preferred) or para position of the benzoyl moiety. Indeed, while the insertion of a fluorine (2b, 196%) or bromine (2f, 214%) atom at the meta position provided a major potency than the para position (2c, 142% or 2g, 184%, respectively) in activating SIRT3, a chlorine atom displayed an opposite behavior (meta2d, 204% vs para2e, 237%), the para-chloro analogue 2e being the most potent among all halogens-containing compounds 2b–g.
Figure 3.

SIRT3 biochemical activation by novel 1,4-DHP derivatives 2a–n and 3a–d at 10 and 100 μM. Compounds 2 and 3 were used as positive controls.
Moreover, the replacement of the phenyl with the 3- (but not 2-) thienyl ring at N1 furnished 2n with a slight improvement of the potency with respect to 2. The systematic removal of methoxy groups by 3 (compounds 3a–d) generally provided a decrease of SIRT3 activation at 100 μM respect to the reference compound 3, with the sole exception of compound 3c, bearing the 3,4-dimethoxy substitution, which intriguingly exhibited the strongest SIRT3 activation (387%), thus being 1.4-fold more potent than the reference 3 (269%) at the same concentration. Among the other methoxy analogues, 3a (the 3-methoxybenzoyl derivative)—despite its lower potency with respect to 3 and 3c—was able to activate SIRT3 up to 200%.
Control reactions for compound effects on the processing of the sirtuin catalysis product nicotinamide (NAM) by the coupled enzymes PncA/GDH showed negligible effects (Figure S1 in the Supporting Information), confirming that the effects of the compounds are due to direct SIRT3 activation.
Next, we selected compounds 3a, 3c, and 3d to be tested against SIRT1, -2, and -5, as well, to assess their SIRT3-specificity.
The graph in Figure 4 clearly shows that when tested at 10 μM, 3a, 3c, and 3d displayed specific SIRT3 activation (145, 158, and 149%, respectively) with negligible effects on SIRT1, -2, and -5. At 100 μM, 3a exerted 217% activation of SIRT3, joined to lower activation of SIRT1, -2, and -5 (164, 130, and 144%, respectively), while 3c induced huge activation (387%) of SIRT3 and definitely lower activation of the other SIRT isoforms (156% (SIRT1), 136% (SIRT2), and 160% (SIRT5)), confirming a SIRT3-specific behavior. Differently, compound 3d proved to be a mixed SIRT1/3 selective activator at 100 μM since it exhibited 130% (SIRT1) and 178% (SIRT3) enzyme activation, however displaying a major activation potency toward SIRT3.
Figure 4.

SIRT1, -2, -3, and -5 isoforms biochemical activation profile by 1,4-DHP 3a, 3c, and 3d at 10 and 100 μM.
SIRT3 Biophysical Binding Assay
Surface Plasmon Resonance Assay
Next, surface plasmon resonance (SPR) experiments were conducted with selected SIRT3 activators 2e and 3a–d to ascertain their biophysical interaction with the human SIRT3 protein. Their parental compounds 2 and 3, together with honokiol, were used as positive controls. The experiments were carried out twice at different concentrations (5, 10, 20, 40, and 80 μM), by injecting the compounds at increasing doses (Figure 5). The resonance units (RU) increase with respect to baseline indicates the formation of the SIRT3/ligand complex, the plateau region represents the steady-state phase of the interaction (RUeq, RU at equilibrium) and the decrease in RU after 180 s represents dissociation of analytes from the immobilized SIRT3 protein after injection of the buffer.
Figure 5.

Sensorgrams of the interaction between selected SIRT3 activators 2e, 3a–d, reference compounds 2, 3, and honokiol, and SIRT3 protein immobilized onto a COOH5 sensor chip and analytes injected at the following concentrations: 5 (blue), 10 (orange), 20 (gray), 40 (yellow), and 80 μM (light blue).
Scatchard analysis showed that parental compounds 2 and 3 as well as honokiol interact with SIRT3 with KD values ranging from 40 to 47 μM (Figure 5). Compounds 2e and 3a–d showed affinities in the dual digit micromolar range, with 3c binding to the protein with the highest affinity (KD = 29 μM) (Figure 5), consistently with its highest biochemical SIRT3 activation (387%, Figure 3).
Activation of SIRT3 with Physiological Substrates and in Cellular Systems
Selected compounds 3a–d were tested in triple-negative breast cancer MDA-MB-231 cells to evaluate their effects on SIRT3 physiological substrates. First, the effect on the activity of GDH (an established SIRT3 substrate activated through deacetylation60−62) was investigated by a coupled enzyme assay. The treatment of MDA-MB-231 cells with 50 μM 3a, 3c, and 3d caused an increase in GDH activity in cell lysates, detected after 4 h and consistent with SIRT3 activation, while 3b exerted only a negligible effect, along with its weak biochemical SIRT3 activation. These effects of SIRT3 activators clearly phenocopied those observed in the SIRT3-overexpressing MDA-MB-231 cells (SIRT3+) (Figure 6). In detail, 3d displayed a degree of GDH activation similar to the reference compound 3 (around 175%), and 3, 3a, and 3d were even more effective than the SIRT3-overexpressing cells in increasing GDH activity (Figure 6).
Figure 6.

GDH activity upon 4 h treatment with SIRT3 activators 3a–d used at 50 μM. Compounds 2 and 3 and SIRT3 overexpressing (SIRT3+) MDA-MB-231 cells were used as positive controls.
Effects on Manganese-Dependent Superoxide Dismutase
The post-translational acetylation/deacetylation of MnSOD controlled by SIRT3 is demonstrated to modulate the MnSOD functions in different biological pathways.63−67 Among all the MnSOD lysine residues identified as targets for SIRT3 deacetylation, mainly K68 and K122 were proved to contribute to MnSOD activation.65 K68-MnSOD acetylation promotes a transformation-permissive phenotype, leading to a chemotherapy-resistant breast cancer cell model using cisplatin and doxorubicin.68 Thus, we investigated the ability of 3d, the most potent compound to induce the SIRT3-driven GDH activation, to deacetylate the acK68- and acK122-MnSOD marks in CAL-62 and MDA-MB-231 cell lines. The specific SIRT3 inhibitor 3-TYP was utilized as negative control. As depicted in the Western blot analyses (Figure 7A,C) and relative densitometries (Figure 7B,D), 3d at 50 μM and after 4 h treatment led to a decrease of the ratio acK68- or acK122-MnSOD/MnSOD in both the two tested cell lines, and it was able to display significant acK122 deacetylation already at 10 μM in MDA-MB-231 cells. As expected, in both cell lines, the SIRT3 inhibitor 3-TYP markedly increased the acetylation levels of both K68 and K122 residues.
Figure 7.

Western blotting of MnSOD acK68 and acK122 marks in CAL-62 (A) and MDA-MB-231 (C) cell lines and their respective densitometric analysis (B,D) upon treatment with 10 and 50 μM 3d for 4 h. p-values were obtained using Student’s t-test (*p < 0.05) for three independent experiments.
Effects on Cell Viability and Colony Formation in CAL-62 and MDA-MB-231 Cells
Afterward, the selected compounds 3a–d were assayed at 50 μM for 24 and 48 h in CAL-62 and MDA-MB-231 cells to assess their ability to impair the cell viability and colony formation in either normoxia or hypoxia-induced condition, the latter obtained by adding a 200 μM cobalt chloride solution, as described in previous reports69 (Figure 8). Most cancer cells, mainly solid types, are known to have an altered metabolism due to hypoxia, a non-physiological level of oxygen tension, which contributes to therapy resistance by causing cellular quiescence.70 Moreover, hypoxic cancer cells can induce several factors that lead to cell proliferation and colony formation.71
Figure 8.
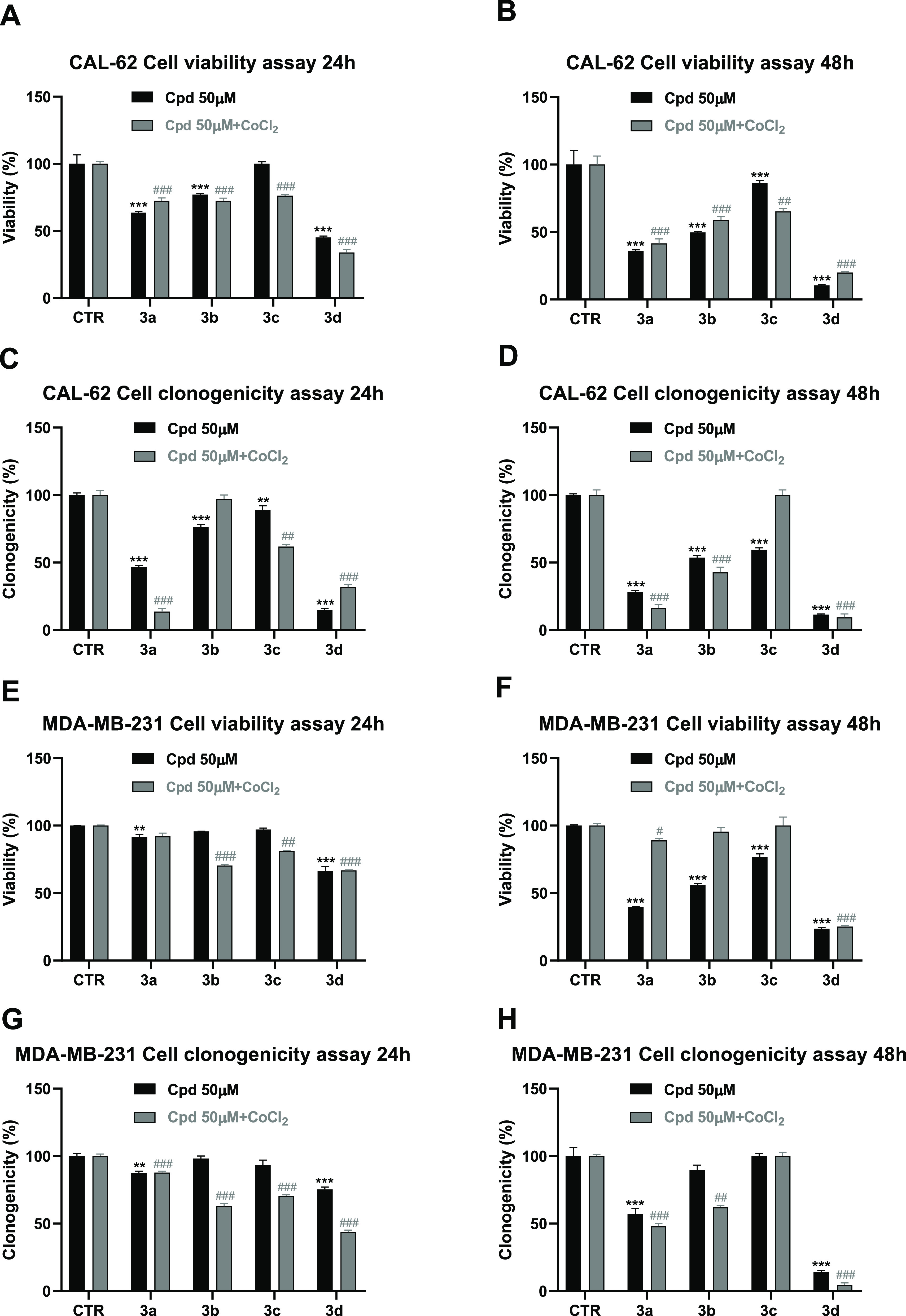
Viability and clonogenic assay under normoxia and hypoxia conditions in CAL-62 (A–D) and MDA-MB-231 (E–H) cancer cells treated with the compounds 3a–d at 50 μM for 24 and 48 h. p-values were obtained using a Student’s t-test (*p < 0.05, **p < 0.01, and ***p < 0.001; #p < 0.05, ##p < 0.01, and ###p < 0.001) for three independent experiments.
Tested against CAL-62 cells, compounds 3a–d provided a time-dependent reduction of viability, 3d being the most effective (reduction of 55% at 24 h and 90% at 48 h, respectively), followed by 3a (reduction of 37% at 24 h and 64% at 48 h) (Figure 8A,B). Under hypoxia, 3a–d displayed similar effects as under normoxia, with 3d (80%) and 3a (59%) being the most potent after 48 h treatment in reducing cell viability (Figure 8A,B).
We also investigate whether our SIRT3 activators 3a–d were able to hamper the cells’ ability to form colonies. The results in Figure 8C,D clearly show that in CAL-62 cells, 3a and 3d were the most effective even in these experiments, in either normal or hypoxia conditions, at both the two-time points, with a decrease of clonogenicity of 68–91% (3d) or 53–86% (3a).
When tested against MDA-MB-231 cells, after 24 h only 3d provided weak (34%) viability impairment in both conditions. After 48 h of treatment, 3d was the most effective (75%) in both conditions, while 3a was more potent in normoxia (60% reduction viability) than in hypoxia (11% reduction viability) (Figure 8E,F). Even in this cell line, we tested the compounds 3a–d toward the colony formation. In both conditions, compound 3d displayed the strongest effect, mainly at 48 h (86 and 95% reduction of clonogenicity in normoxia and in hypoxia conditions, respectively) (Figure 8G,H). For the plate images of the clonogenic assay at 24 and 48 h, see Figure S2 in the Supporting Information.
Furthermore, prompted by the auspicious results provided by the CAL-62 and MDA-MB-231 cells treatment with the SIRT3 activator 3d, which remarkedly impaired cell viability and clonogenicity in both normal and hypoxia conditions, we decided to investigate its dose-dependent effects in the same cell lines by evaluating cell viability, cell death, and clonogenicity from 1 to 50 μM after 24 (Figure S4 in the Supporting Information) and 48 h of treatment (Figure 9), and by determining the relative IC50 or EC50 values. Compound 3d generally displayed single-digit to low dual-digit micromolar potency in both cell lines and conditions. In detail, in CAL-62 cells, 3d proved higher efficacy (4.8-fold) in cell viability reduction and cell death induction in hypoxia (IC50 and EC50 values around 3.5 μM) than in normoxia conditions (IC50 and EC50 values around 16.7 μM). Differently, in MDA-MB-231 cells, 3d displayed greater potency in normoxia (IC50: 3.64 μM) than in hypoxia (IC50: 10.56 μM) in reducing cell viability, and very similar EC50 values between the two conditions toward cell death induction. Intriguingly, in both cell lines and in both conditions, 3d provided single-digit micromolar IC50 values against colonies formation by displaying around 2.5 μM (CAL-62) and 2.2 μM (MDA-MB-231) values in normoxia and similar results in hypoxia.
Figure 9.
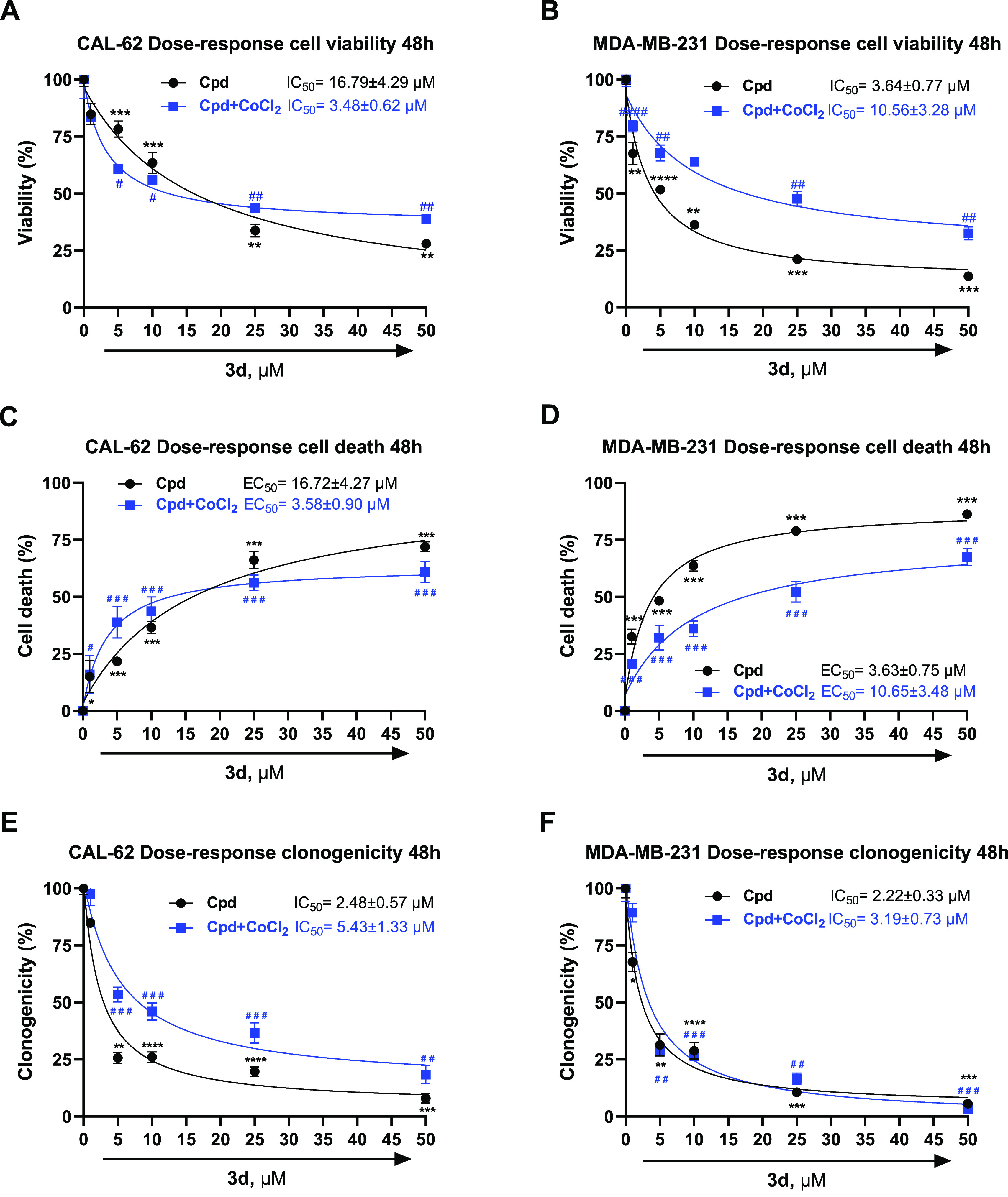
Dose-dependent effects and relative IC50/EC50 of compound 3d on cell viability, cell death, and clonogenicity in CAL-62 (A,C,E) and MDA-MB-231 (B,D,F) cells upon 48 h of treatment. P-values were obtained using a Student’s t-test (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001) (#p < 0.05, ##p < 0.01, ###p < 0.001, and ####p < 0.0001) for three independent experiments.
The relative plate images of the dose-dependent clonogenic assay at 24 and 48 h are reported in Figure S3 in the Supporting Information.
Moreover, to assess their anticancer-specific effect, we tested 3c and 3d, the most potent SIRT3 activator in biochemical and in cellular assays, respectively, against healthy HaCaT cells at 24 and 48 h, using both compounds at 5, 10, and 25 μM. As reported in Figure S5 and Table S4, the compound 3c displayed negligible toxicity at the tested doses and times, whereas the activator 3d provided some degree of cell viability reduction and cell death only after 48 h treatment and at higher concentrations (10 and 25 μM) than its antiproliferative IC50 values provided in CAL-62 and MDA-MB-231 cancer cells (Figure 9).
Effects on HIF-1α, EPAS-1 (HIF-2α), and CA-IX Expression
In a previous study, SIRT3 was proven to be able to reprogram metabolism by destabilizing HIF-1α, a transcription factor that controls glycolytic gene expression.72 SIRT3 loss-dependent increase of the ROS production was resulted in HIF-1α stabilization. Furthermore, reduced SIRT3 expression in human breast cancer correlated with the increased expression of HIF-1α target genes, whereas SIRT3 overexpression impaired glycolysis and proliferation in breast cancer cells, thus unveiling a metabolic mechanism to suppress tumor growth.15 Moreover, a recent study showed that EPAS-1 (HIF-2α) promotes tumor growth and metastasis via EMT induction in in vitro and in vivo breast cancer models.73 In addition, differently from targeting HIF-1α molecules directly, another approach to hamper metastasis could be to target the carbonic anhydrase IX (CA-IX), an important hypoxia-inducible enzyme widely present in tumors downstream of HIF-1α. CA-IX is a major regulator of intra- and extra-cellular pH with a critical influence on increased survival and invasion of cancer cells.74 Notably, pre-clinical studies in breast cancer models showed that CA-IX inhibition led to a decrease in tumor growth and metastasis progression.74
Based on these findings, we decided to assess the ability of the SIRT3 activators 3a–d to impair HIF-1α, EPAS-1, and CA-IX protein expression in CAL-62 (Figure 10A–F) and MDA-MB-231 (Figure 11A–F) cell lines treated with the compounds at 50 μM for 48 h and under hypoxia conditions. As shown by the western blotting performed on CAL-62 cells (Figure 10), compounds 3d and, to a lesser extent, 3a were able to decrease HIF-1α, whereas EPAS-1 protein was downregulated by 3c and 3d, and the CA-IX protein was significantly downregulated by compounds 3a, 3b, and 3d. Overall, compound 3d was confirmed to be the most effective in the modulation of all three proteins. Moreover, the SIRT3 inhibitor 3-TYP displayed negligible effects in all the experiments described above.
Figure 10.

Western blotting of HIF-1α (A), EPAS-1 (HIF-2α) (C), and carbonic anhydrase IX (CA-IX) (E) and their respective densitometric analysis (B,D,F) in CAL-62 cell line upon treatment with 10 and 50 μM compounds 3a–d for 48 h. Compound 3-TYP was used as a negative control. p-values were obtained using Student’s t-test (*p < 0.05) for three independent experiments.
Figure 11.

Western blotting of HIF-1α (A), EPAS-1 (HIF-2α) (C), and carbonic anhydrase IX (CA-II) (E) and their respective densitometric analysis (B,D,F) in the MDA-MB-231 cell line upon treatment with 50 μM compounds 3a–d for 48 h. Compound 3-TYP was used as a negative control. p-values were obtained using Student’s t-test (*p < 0.05, **p < 0.01) for three independent experiments.
Next, we evaluated the effects of 3a–d on HIF-1α, EPAS-1, and CA-IX in MDA-MB-231 cells, as well (Figure 11A–F). Compounds 3c and 3d significantly reduced HIF-1α and EPAS-1 protein levels, with 3d being the most effective and providing stronger effects when compared to CAL-62 cells. Even against the CA-IX protein expression, 3d proved to be the most potent giving a downregulation similar to that observed in CAL-62 cells. Also in this cell line, 3-TYP did not show any activity.
Epithelial to Mesenchymal Transition Modulation by SIRT3 Activators
Afterward, based on the double localization described for SIRT3, which was primarily found into the mitochondria but translocated into the nucleus in stressful conditions,75 the effects of SIRT3 activators on gene expression regulation were evaluated in MDA-MB-231 cells. Specifically, literature data relate SIRT3 inactivation to EMT, leading to plastic and mobile phenotype by overexpressing transcription factors such as SLUG, SNAIL, and ZEB,76 and prove the involvement of SIRT3 in counteracting EMT.77 These findings suggest a potential effect of SIRT3 activation in limiting cell migration properties. Hence, the expression of some EMT master regulator genes (i.e., SNAIL, SLUG, and ZEB1) and of ECM components (COLLAGEN and MMP2/9) was analyzed upon 48 h of treatment with selected compounds 3c and 3d used at 10 (Figure S5 in the Supporting Information) and 50 (Figure 12) μM.
Figure 12.

qRT-PCR analysis for the indicated transcripts in MDA-MB-231 treated with the compounds 3c,d at 50 μM for 48 h. CTR (control) represent the cells treated with the vehicle (DMSO). The values are calculated by the 2(−ΔCt) method, expressed as fold of expression vs the control (arbitrary value = 1) and shown as mean ± SEM. Statistically significant differences are reported (*, p < 0.05; **, p < 0.01) for three independent experiments.
Notably, both compounds 3c and 3d were able to reduce the SNAIL, COL1A2, and MMP2/9 expression levels, but only 3d reduced the ZEB1 expression, whereas the SLUG gene was not significantly affected (Figure 12). This could be due to the multiple regulation mechanisms targeting these genes that could be independent of the SIRT3 nuclear role in this cellular context. Of note, compound 3d displayed a stronger effect than 3c in all cases, providing the best downregulation for COL1A2 and MMP2/9 genes.
Intriguingly, the effectiveness of these SIRT3 activators was maintained at their lower concentration (10 μM) (Figure S5 in the Supporting Information).
Considering the previously described involvement of hypoxia in EMT induction,78 we extended our biological study by analyzing the effects of SIRT3 activators on gene expression regulation under hypoxic conditions. Hypoxia induction of EMT was verified by gene expression analysis (upregulation of SNAIL, ZEB1, COLLAGEN, MMPs, and SLUG) after 72 h of treatment with CoCl2. In these experimental conditions, we verified the effectiveness of the previously tested SIRT3 activators 3c and 3d (50 μM, 48 h) in reducing EMT genes expression. Notably, both compounds not only maintained their ability to impair SNAIL, ZEB1, COLLAGEN, and MMP2/9 gene expression but also affected SLUG expression after 48 h of treatment in hypoxic conditions (Figure 13). Overall, 3c and 3d displayed a very high and stronger efficacy in hypoxia with respect to the normoxia condition. This suggests that the contribution of SIRT3 in the regulation of EMT genes is much more evident after the hypoxia induction.
Figure 13.

qRT-PCR analysis for the indicated transcripts in MDA-MB-231 treated with the compounds at the concentration of 50 μM for 48 h. CTR (control) represent the cells treated with the vehicle (DMSO). Hypoxia was induced by the cell treatment with CoCl2, as reported in the Experimental Section. The values are calculated by the 2(−ΔCt) method, expressed as fold of expression vs the control (arbitrary value = 1) and shown as mean ± SEM. Statistically significant differences are reported (*, p < 0.05; **, p < 0.01) for four independent experiments.
Scratch Assay
Next, a scratch assay on MDA-MB-231 cells after treatment with 3c and 3d at 50 μM for 48 h was performed to assess their effect on cell migration (Figure 14). The obtained data clearly show that compound 3d, even in these experiments, displayed the highest effect by impairing over 75% of the MDA-MB-231 migration rate, consistently with the gene modulation effect discussed above.
Figure 14.

Scratch assay on MDA-MB-231 cells treated with the indicated compounds at the concentration of 50 μM for 48 h. CTR (control) represent the cells treated with the vehicle (DMSO). Left panel: phase contrast micrographs of cells at two different time points. Right panel: quantification of cell migration. Data are shown as the mean ± S.E.M. of three independent experiments. Significant results are indicated: p < 0.05 (*); p < 0.01 (**).
Conclusions
Very recently, our group reported that structurally different modifications on the 1,4-DHP scaffold allowed the discovery of specific SIRT3 activators, such as compounds 2 and 3, confirmed by functional tests in cellular settings.59 Here, we describe the development of novel 2 and 3 analogues, compounds 2a–n and 3a–d, obtained by structural modification at the N1-benzoyl moiety and their biological validation as novel SIRT3-specific activators. Among the new compounds, the 3,4-dimethoxy substitution at the N1-benzoyl moiety provided the compound 3c as the most potent and selective SIRT3 activator in biochemical activation assays (387% at 100 μM) as well as in biophysical experiments (SPR assay), with a KD of 29 μM. Afterward, to assess the cellular target engagement, we tested compound 3c and its analogues 3a, 3b, and 3d, along with their parental compounds 2 and 3, in triple-negative MDA-MB-231 breast cancer cells to evaluate their SIRT3-driven GDH activation. After 4 h of treatment at 50 μM, the most effective compound was 3d, bearing the 3,5-dimethoxybenzoyl moiety at the 1,4-DHP N1 position, affording around 175% GDH activation similar to the reference compound 3, and more effective than the SIRT3-overexpressing cells. Then, we tested 3d for evaluating its effects on acetylation level of MnSOD, another target of SIRT3. This physiological substrate was deacetylated at the known specific residues K68 and K122 after 4 h and 50 μM treatment, markedly in breast MDA-MB-231 cells, whereas the SIRT3 inhibitor 3-TYP provided increased acetylation of the above lysine residues, as expected.
Furthermore, when 3a–d were tested in thyroid CAL-62 and breast MDA-MB-231 cancer cells, 3a and 3d displayed the highest time-dependent activity reducing the cell viability and colony formation in both normoxia and hypoxia environments. Compound 3d displayed the most potent effect in all experiments, achieving a massive impairment upon 48 h treatment in both the tested cell lines and conditions. Based on these promising results, we investigated the dose-dependent (1–50 μM) effects of 3d in the same cell lines, wherein it displayed a nice dose–response curve reducing cell viability along the cell death, as well as the clonogenic activity, showing IC50/EC50 values range of 2.5–16.8 μM (normoxia) and 3.5–5.4 μM (hypoxia) in CAL-62 cells, 2.2–3.6 μM (normoxia) and 3.2–10.6 μM (hypoxia) in MDA-MB-231 cells. Intriguingly, the anti-clonogenic effect was the most remarkable. Moreover, since reduced SIRT3 expression in human breast cancer has been reported to lead to HIF-1α stabilization and upregulation of HIF-1α target genes, we explored the effects of compounds 3a–d in modulation of HIF-1α, EPAS-1 (HIF-2α) and CA-IX expression in both CAL-62 and MDA-MB-231 cells after 48 h of treatment. Even in these experiments, the SIRT3 activator 3d displayed the strongest effect, with a significant downregulation of all the three proteins mentioned above, and mainly in MDA-MB-231 cells.
Furthermore, the modulation by 3c and 3d of the EMT master regulator genes’ expression, such as SNAIL, SLUG, and ZEB1, and of ECM components, such as COLLAGEN and MMPs, in MDA-MB-231 cells was investigated under normoxia and hypoxia conditions. Both compounds were able to reduce SNAIL, ZEB1, COL1A2, MMP2, and MMP9 gene expression after 48 h of treatment at 50 μM, with compound 3d being the strongest even in this investigation, while SLUG gene expression was downregulated only in hypoxia conditions. Intriguingly, compounds 3c and 3d were effective even at 10 μM, in agreement with the reduction of cell viability, clonogenicity, and induction of cell death observed at the same dosage. Moreover, their effects were generally and clearly stronger in hypoxia cells, underlining the important role of SIRT3 in regulating the expression of genes at least partly dependent on the hypoxia. In complete agreement with this, compound 3c and mainly 3d were also able to nicely reduce the MDA-MB-231 cell migration after 48 h of treatment at 50 μM.
In conclusion, the present work highlights that our SIRT3 activators are endowed with anticancer properties and could represent a tool for future in-depth studies of the SIRT3 roles and functions in cancer pathogenesis. Moreover, the DHP scaffold could be worthy of further med-chem optimization for improving its SIRT3 activation potency and selectivity.
Experimental Section
Chemistry
Melting points were determined on a Buchi 530 melting point apparatus and are uncorrected. 1H NMR and 13C spectra were recorded at 400 MHz on a Bruker AC 400 spectrometer; chemical shifts are reported in δ (ppm) units relative to the internal reference tetramethylsilane (Me4Si). All compounds were routinely checked by TLC, 1H, and 13C NMR. IR spectra were recorded on a PerkinElmer Spectrum 100 FT-IR. TLC was performed on aluminum-backed silica gel plates (Merck DC, Alufolien Kieselgel 60 F254) with spots visualized by UV light. All solvents were reagent grade and, when necessary, were purified and dried by standard methods. Concentration of solutions after reactions involved the use of a rotary evaporator operating at a reduced pressure of ca. 20 Torr. The purity of the final compounds 2a-n and 3a-d was analyzed by elemental analysis and HPLC. The elemental analyis has been performed on Thermo Fisher FlashSmart CNHS/O. The HPLC system consisted of a Dionex UltiMate 3000 UHPLC (Thermo Fisher) system equipped with an automatic injector and column heater and coupled with a Diode Array Detector DAD-3000 (Thermo Fisher). The analytical controls were performed on a Hypersil GOLD C18 Selectivity 5 μm (4.6 × 250 mm) HPLC Column (Thermo Fisher) in gradient elution. Eluents: (A) H2O/CH3CN, 95/5 (v/v) + 0.1% TFA; (B) CH3CN/H2O, 95/5 (v/v) + 0.1% TFA. A 20 min linear gradient elution from 30% to 100% solvent B was followed by 5 min at 100% B. The flow rate was 1.0 mL/min, and the column was kept at a constant temperature of 30 °C. Samples were dissolved in solvent B at a concentration of 0.25 mg/mL, and the injection volume was 10 μL. Analytical results are within ±0.40% of the theoretical values. All chemicals were purchased from Sigma-Aldrich Chemistry, Milan (Italy), Flurochem, Manchester (UK), or from AlfaAesar, Karlsruhe (Germany), and were of the highest purity.
The intermediate diethyl 4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate (4) has been prepared according to the literature.41
The final compounds 2a–n and 3a–d possess a purity >95% purity confirmed by 1H and 13C NMR as well as elemental analysis and HPLC.
General Procedure for the Synthesis of the Final Compounds 2a–n and 3a–d
Example: Synthesis of Diethyl 1-(Cyclohexanecarbonyl)-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate (2a)
To a solution of diethyl 4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate (0.33 mmol, 0.100 g) in DCM dry (3 mL) was added TEA (1.33 mmol, 0.185 mL) and then, dropwise at 0 °C, cyclohexanecarbonyl chloride (0.36 mmol, 0.053 g) was dissolved in DCM dry (3 mL). The reaction was stirred overnight at RT. The solvent was then evaporated, and the remaining residue was purified by column chromatography (silica gel) using EtOAc/hexane (1:2) as the eluent. The product was isolated as a white solid (0.023 g, 17.0%).
2a, MC4199, Diethyl 1-(cyclohexanecarbonyl)-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate
mp 92–93 °C; yield: 17.0%; 1H NMR (CDCl3, 400 MHz, δ, ppm): δ 1.13 (t, 6H, J = 7.2 Hz, 2× −OCH2CH3), 1.18–1.36 (m, 4H, cyclohexane protons), 1.52–1.55 (m, 5H, cyclohexane protons), 1.79–1.86 (m, 2H, cyclohexane protons), 3.98–4.1 (m, 4H, 2× −OCH2CH3), 4.80 (s, 1H, ArCH−), 7.09–7.19 (m, 5H, aromatic protons), 8.03 (s, 2H, dihydropyridine protons) ppm; 13C NMR (100 MHz, CDCl3): δ 14.10 (2C), 25.41, 25.61, 29.18 (2C), 38.62, 41.11, 60.78 (2C), 115.46, 126.94, 128.18 (3C), 128.55 (3C), 129.62 (2C), 143.77, 165.96 (2C), 172.99 ppm.
2b, MC4158, Diethyl 1-(3-fluorobenzoyl)-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate
mp 124–125 °C; yield: 85.2%; 1H NMR (CDCl3, 400 MHz, δ, ppm): δ 1.21 (t, 6H, J = 7.2 Hz, 2×-OCH2CH3), 4.06–4.20 (m, 4H, 2× −OCH2CH3), 4.96 (s, 1H, ArCH−), 7,25–7,21 (m, 1H, aromatic proton), 7.28–7.44 (m, 5H, aromatic protons), 7.40–7.44 (m, 2H, aromatic protons), 7.52–7.57 (m, 1H, aromatic proton), 8.08 (s, 2H, dihydropyridine protons) ppm; 13C NMR (100 MHz, CDCl3): δ 14.08 (2C), 38.92, 60.90 (2C), 116.12, 116.49, 119.68, 124.50, 127.00, 127.13 (3C), 128.37, 128.53 (3C), 130.70 (2C), 143.44, 161.43, 165.56 (3C) ppm.
2c, MC4168, Diethyl 1-(4-fluorobenzoyl)-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate
mp 123–124 °C; yield: 93.6%; 1H NMR (CDCl3, 400 MHz, δ, ppm): δ 1.12 (t, 6H, J = 7.2 Hz, 2× −OCH2CH3), 3.97–4.11 (m, 4H, 2× −OCH2CH3), 4.88 (s, 1H, ArCH−), 7.14–7.27 (m, 7H, aromatic protons), 7.61–7.65 (m, 2H, aromatic protons), 8.00 (s, 2H, dihydropyridine protons) ppm; 13C NMR (100 MHz, CDCl3): δ 14.10 (2C), 38.88, 60.87 (2C), 115.79, 116.25 (2C), 116.47 (2C), 127.10, 128.36, 128.50 (2C), 131.04, 131.72 (3C), 131.82, 143.60, 161.43, 165.64 (3C) ppm.
2d, MC4172, Diethyl 1-(3-chlorobenzoyl)-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate
mp 154–155 °C; yield: 61.8%; 1H NMR (CDCl3, 400 MHz, δ, ppm): δ 1.21 (t, 6H, J = 7.2 Hz, 2× −OCH2CH3), 4.06–4.20 (m, 4H, 2× −OCH2CH3), 4.96 (s, 1H, ArCH−), 7.22–7.25 (m, 1H, aromatic proton), 7.30–7.36 (m, 4H, aromatic protons), 7.47–7.52 (m, 2H, aromatic protons), 7.60–7.63 (m, 1H, aromatic proton), 7.69 (s, 1H, aromatic proton), 8.07 (s, 2H, dihydropyridine proton) ppm; 13C NMR (100 MHz, CDCl3): δ 14.07 (2C), 38.92, 60.90 (2C), 116.18, 126.76, 127.14, 128.37, 128.37, 128.55 (4C), 129.25, 130.19, 130.63, 132.68, 133.55, 135.44, 143.42, 152.89, 165.53, 166.56 ppm.
2e, MC4166, Diethyl 1-(4-chlorobenzoyl)-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate
mp 122–123 °C; yield: 65.0%; 1H NMR (CDCl3, 400 MHz, δ, ppm): δ 1.22 (t, 6H, J = 7.2 Hz, 2× −OCH2CH3), 4.08–4.19 (m, 4H, 2× −OCH2CH3), 4.96 (s, 1H, −ArCH−), 7.22–7.36 (m, 5H, aromatic protons), 7.54 (d, 2H, J = 8.8 Hz, aromatic protons), 7.63 (d, 2H, J = 8.8 Hz, aromatic protons), 8.07 (s, 2H, dihydropyridine protons) ppm; 13C NMR (100 MHz, CDCl3): δ 14.10 (2C), 38.90, 60.90 (2C), 115.97, 127.12, 128.37 (3C), 128.51 (3C), 129.39 (3C), 130.13, 130.54 (2C), 130.87, 139.19, 143.52, 165.60, 166.96 ppm.
2f, MC4181, Diethyl 1-(3-bromobenzoyl)-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate
mp 154–155 °C; yield: 68.7%; 1H NMR (CDCl3, 400 MHz, δ, ppm): δ 1.21 (t, 6H, J = 7.2 Hz, 2× −OCH2CH3), 4.06–4.20 (m, 4H, 2× −OCH2CH3), 4.96 (s, 1H, ArCH−), 7.22–7.25 (m, 1H, aromatic proton), 7.30–7.36 (m, 4H, aromatic protons), 7.43 (t, 1H, J = 8.0 Hz, aromatic proton), 7.56 (d, 1H, J = 7.6 Hz, aromatic proton), 7.77 (d, 1H, J = 7.6 Hz, aromatic proton), 7.85 (s, 1H, aromatic proton), 8.07 (s, 2H, dihydropyridine protons) ppm; 13C NMR (100 MHz, CDCl3): δ 14.09 (2C), 38.91, 60.93 (2C), 116.17, 123.32, 128.38 (3C), 128.56 (3C), 130.36 (3C), 132.14, 133.71, 135.62, 143.41, 161.42, 165.53 (2C), 166.43 ppm.
2g, MC4185, Diethyl 1-(4-bromobenzoyl)-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate
mp 140–141 °C; yield: 71.6%; 1H NMR (CDCl3, 400 MHz, δ, ppm): δ 1.22 (t, 6H, J = 7.2 Hz, 2× −OCH2CH3), 4.06–4.21 (m, 4H, 2× −OCH2CH3), 4.96 (s, 1H, ArCH−), 7.22–7.25 (m, 1H, aromatic proton), 7.32–7.36 (m, 4H, aromatic protons), 7.55 (d, 2H, J = 8.4 Hz, aromatic protons), 7.69 (d, 2H, J = 8.4 Hz, aromatic protons), 8.07 (s, 2H, dihydropyridine protons) ppm; 13C NMR (100 MHz, CDCl3): δ 14.11 (2C), 38.91, 60.90 (2C), 116.00 (2C), 127.13, 127.65, 128.37 (3C), 128.51 (3C), 130.61 (3C), 130.83, 132.36, 143.51, 165.59 (2C), 167.05 ppm.
2h, MC4165, Diethyl 1-(3-nitrobenzoyl)-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate
mp 147–148 °C; yield: 63.1%; 1H NMR (CDCl3, 400 MHz, δ, ppm): δ 1.21 (s, 6H, J = 7.2 Hz, 2× −OCH2CH3), 4.08–4.19 (m, 4H, 2× −OCH2CH3), 4.98 (s, 1H, ArCH−), 7.23–7.28 (m, 1H, aromatic proton), 7.31–7.49 (m, 4H, aromatic protons), 7.78 (t, 1H, J = 8.4 Hz, aromatic proton), 7.96 (d, 1H, J = 8.4 Hz, aromatic proton), 8.04 (s, 2H, dihydropyridine protons), 8.50 (d, 1H, J = 8.4 Hz, aromatic proton), 8.58 (s, 1H, aromatic proton) ppm; 13C NMR (100 MHz, CDCl3): δ 14.07 (2C), 38.98, 61.04 (2C), 116.89, 124.31, 126.98, 127.27, 128.43 (3C), 128.55 (3C), 130.09, 130.26, 133.66, 134.13, 143.08, 148.52, 165.34 (2C), 202.37 ppm.
2i, MC4167, Diethyl 1-(4-nitrobenzoyl)-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate
mp 109–110 °C; yield: 65.6%; 1H NMR (CDCl3, 400 MHz, δ, ppm): δ 1.21 (t, 6H, J = 7.2 Hz, 2× −OCH2CH3), 4.08–4.19 (m, 4H, 2× −OCH2CH3), 4.97 (s, 1H, ArCH−), 7.23–7.28 (m, 1H, aromatic proton), 7.31–7.34 (m, 4H, aromatic protons), 7.85 (d, 2H, J = 8.4 Hz, aromatic protons), 8.02 (s, 2H, dihydropyridine protons), 8.42 (d, 2H, J = 8.4 Hz, aromatic protons) ppm; 13C NMR (100 MHz, CDCl3): δ 14.08 (2C), 38.99, 61.06 (2C), 116.92, 120.48, 124.25 (2C), 127.28, 128.43 (3C), 128.53 (3C), 130.01 (3C), 137.63, 143.04, 149.92, 165.33, 165.89 ppm.
2j, MC4177, Diethyl 1-(4-cyanobenzoyl)-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate
mp 155–156 °C; yield: 60.4%; 1H NMR (CDCl3, 400 MHz, δ, ppm): δ 1.12 (t, 6H, J = 6.8 Hz, 2× −OCH2CH3), 3.97–4.11 (m, 4H, 2× −OCH2CH3), 4.87 (s, 1H, ArCH−), 7.17–7.17 (m, 1H, aromatic proton), 7.19–7.21 (m, 2H, aromatic protons), 7.23–7.24 (m, 2H, aromatic protons), 7.68–7.70 (d, 2H, aromatic protons), 7.77–7.79 (d, 2H, aromatic protons), 7.93 (s, 2H, dihydropyridine protons) ppm; 13C NMR (100 MHz, CDCl3): δ 14.09 (2C), 38.96, 61.05 (2C), 116.21, 117.50, 120.47, 127.27, 128.43 (3C), 128.52 (3C), 129.53 (2C), 132.79 (2C), 135.95, 143.11, 161.42, 165.38 (2C), 166.13 ppm.
2k, MC4208, Diethyl 4-phenyl-1-(2-phenylacetyl)-1,4-dihydropyridine-3,5-dicarboxylate
mp 153–154 °C; yield: 14.1%; 1H NMR (CDCl3, 400 MHz, δ, ppm): δ 1.22 (t, 6H, J = 7.2 Hz, 2× −OCH2CH3), 1.57 (s, 2H, −CH2Ar), 4.05–4.18 (m, 4H, 2× −OCH2CH3), 4.85 (s, 1H, ArCH−), 7.16–7.26 (m, 5H, aromatic protons), 7.32–7.37 (m, 3H, aromatic protons), 7.40–7.43 (m, 2H, aromatic protons), 8.14–8.18 (s, 2H, dihydropyridine protons) ppm; 13C NMR (100 MHz, CDCl3): δ 14.10 (2C), 38.52, 41.30, 60.79 (2C), 126.96, 127.80, 128.18 (3C), 128.51 (3C), 128.91 (3C), 129.19 (3C), 132.32, 143.35, 161.43, 165.62, 167.93 ppm.
2l, MC4204, Diethyl 1-(furan-2-carbonyl)-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate
mp 125–126 °C; yield: 12.2%; 1H NMR (CDCl3, 400 MHz, δ, ppm): δ 1.15 (t, 6H, J = 7.2 Hz, 2× −OCH2CH3), 3.99–4.14 (m, 4H, 2× −OCH2CH3), 4.88 (s, 1H, ArCH−), 6.57–6.59 (m, 1H, furan proton), 7.10–7.14 (m, 1H, aromatic proton), 7.18–7.22 (m, 2H, aromatic proton), 7.24–7.26 (m, 2H, furan and aromatic protons), 7.32–7.34 (m, 1H, aromatic proton), 7.65 (s, 1H, furan proton), 8.31 (s, 2H, dihydropyridine protons) ppm; 13C NMR (100 MHz, CDCl3): δ 14.12 (2C), 38.83, 60.84 (2C), 112.58, 116.05, 120.48, 121.59, 127.00, 128.27 (3C), 128.57 (3C), 130.83, 143.51, 145.39, 146.99, 156.15, 165.80 ppm.
2m, MC4188, Diethyl 4-phenyl-1-(thiophene-2-carbonyl)-1,4-dihydropyridine-3,5-dicarboxylate
mp 142–143 °C; yield: 56.4%; 1H NMR (CDCl3, 400 MHz, δ, ppm): δ 1.23 (t, 6H, J = 7.2 Hz, 2× −OCH2CH3), 4.08–4.23 (m, 4H, 2× −OCH2CH3), 4.98 (s, 1H, ArCH−), 7.21–7.24 (m, 2H, aromatic protons), 7.28–7.32 (m, 2H, thiophene and aromatic protons), 7.35–7.37 (m, 2H, aromatic and thiophene protons), 7.67 (d, 1H, J = 4.0 Hz, aromatic proton), 7.76 (d, 1H, J = 4.0 Hz, thiophene proton), 8.29 (s, 2H, dihydropyridine protons) ppm; 13C NMR (100 MHz, CDCl3): δ 14.13 (2C), 38.85, 60.86 (2C), 115.81, 127.07, 128.01, 128.36 (3C), 128.46 (3C), 131.34, 133.32, 133.80, 134.64, 143.59, 161.09, 165.73 (2C) ppm.
2n, MC4186, Diethyl 4-phenyl-1-(thiophene-3-carbonyl)-1,4-dihydropyridine-3,5-dicarboxylate
mp 114–115 °C; yield: 51.2%; 1H NMR (CDCl3, 400 MHz, δ, ppm): δ 1.22 (t, 6H, J = 7.2 Hz, 2× −OCH2CH3), 4.07–4.22 (m, 4H, 2× −OCH2CH3), 4.97 (s, 1H, ArCH−), 7.20–7.36 (m, 5H, thiophene and aromatic protons), 7.45–7.50 (m, 2H, thiophene and aromatic protons), 7.97–7.98 (s, 1H, thiophene proton), 8.21 (s, 2H, dihydropyridine protons) ppm; 13C NMR (100 MHz, CDCl3): δ 14.12 (2C), 38.88, 60.83 (2C), 115.67, 127.05, 127.24, 127.97, 128.33 (3C), 128.48 (3C), 131.10, 132.44, 133.21, 143.68, 161.43, 162.60, 165.75 ppm.
3a, MC4164, Diethyl 1-(3-methoxybenzoyl)-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate
mp 134–135 °C; yield: 58.2%; 1H NMR (CDCl3, 400 MHz, δ, ppm): δ 1.21 (t, 6H, J = 7.2 Hz, 2× −OCH2CH3), 3.89 (s, 3H, −OCH3), 4.07–4.18 (m, 4H, 2× −OCH2CH3), 4.96 (s, 1H, ArCH−), 7.15–7.24 (m, 4H, aromatic protons), 7.28–7.29 (m, 1H, aromatic proton), 7.31–7.36 (m, 3H, aromatic protons), 7.43–7.47 (m, 1H, aromatic proton), 8.12 (s, 2H, dihydropyridine protons) ppm; 13C NMR (100 MHz, CDCl3): δ 14.11 (2C), 38.88, 55.56, 60.82 (2C), 114.25, 115.59, 118.72, 120.97, 127.05, 128.33 (3C), 128.54 (3C), 130.00, 131.16, 132.98, 143.71, 159.98, 165.73 (2C), 167.86 ppm.
3b, MC4182, Diethyl 1-(4-methoxybenzoyl)-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate
mp 131–132 °C; yield: 17.9%; 1H NMR (CDCl3, 400 MHz, δ, ppm): δ 1.21 (t, 6H, J = 7.2 Hz, 2× −OCH2CH3), 3.92 (s, 3H, −OCH3), 4.06–4.19 (m, 4H, 2× −OCH2CH3), 4.97 (s, 1H, ArCH−), 7.04 (d, 2H, J = 8.2 Hz, aromatic protons), 7.21–7.25 (t, 1H, aromatic proton), 7.28–7.38 (m, 4H, aromatic protons), 7.68 (d, 2H, J = 8.2 Hz, aromatic protons), 8.12 (s, 2H, dihydropyridine protons) ppm; 13C NMR (100 MHz, CDCl3): δ 14.12 (2C), 38.85, 55.60, 60.76 (2C), 114.30, 115.04 (2C), 120.47, 123.62, 126.99, 128.32 (2C), 128.49 (2C), 131.64 (2C), 131.73 (2C), 143.98, 161.42, 163.27, 165.87, 167.62 ppm.
3c, MC4175, Diethyl 1-(3,4-dimethoxybenzoyl)-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate
mp 174–175 °C; yield: 27.6%; 1H NMR (CDCl3, 400 MHz, δ, ppm): δ 1.24 (t, 6H, J = 7.2 Hz, 2× −OCH2CH3), 3.95 (s, 3H, −OCH3), 3.99 (s, 3H, −OCH3), 4.06–4.21 (m, 4H, 2× −OCH2CH3), 4.97 (s, 1H, ArCH−), 6.97 (d, 1H, J = 6.8 Hz, aromatic proton), 7.21–7.38 (m, 7H, aromatic protons), 8.15 (s, 2H, dihydropyridine protons) ppm; 13C NMR (100 MHz, CDCl3): δ 14.15 (2C), 38.84, 56.19, 60.79 (2C), 110.44, 112.43, 115.06, 123.25, 123.69, 127.02, 128.33 (3C), 128.46 (3C), 131.79 (2C), 143.98, 149.39, 152.98, 165.85 (2C), 167.64 ppm.
3d, MC4176, Diethyl 1-(3,5-dimethoxybenzoyl)-4-phenyl-1,4-dihydropyridine-3,5-dicarboxylate
mp 159–160 °C; yield: 64.3%; 1H NMR (CDCl3, 400 MHz, δ, ppm): δ 1.22 (t, 6H, J = 6.8 Hz, 2× −OCH2CH3), 3.85 (s, 6H, 2× −OCH3), 4.07–4.18 (m, 4H, 2× −OCH2CH3), 4.95 (s, 1H, ArCH−), 6.67–6.68 (m, 1H, aromatic proton), 6.76 (d, 2H, J = 8.0 Hz, aromatic protons), 7.22–7.24 (m, 1H, aromatic proton), 7.28–7.36 (m, 4H, aromatic protons), 8.12 (s, 2H, dihydropyridine protons); 13C NMR (100 MHz, CDCl3): δ 14.10 (2C), 38.89, 55.68 (2C), 60.79 (2C), 104.49, 106.79 (2C), 115.63, 127.04, 128.31 (3C), 128.53 (3C), 131.07, 133.51, 143.70, 160.97 (2C), 165.70 (2C), 167.78 ppm.
Biochemistry and Biology
Protein Production and Purification
N-terminally his-tagged human sirtuins were expressed in Escherichia coli using constructs in pET15b (Sirt1) or pET151D/Topo (Sirt3-114-380 and Sirt5-34-302).4,59,61 Sirt2-43-356 was expressed from a modified pET19b containing a TEV protease cleavage site and an N-terminal SUMO-tag.59,79 Proteins were expressed and purified as described elsewhere.59,61,79−81 In brief, proteins were expressed in E. coli, crude extracts were cleared by centrifugation, and the proteins were affinity purified using NiNTA beads. After tag removal through proteolytic cleavage and reverse affinity chromatography, sirtuin proteins were subjected to size exclusion chromatography in 20 mM Tris/HCl pH 7.5–8.5 and 150 mM NaCl.
In Vitro Sirtuins Activity Assays
Sirtuin deacylation activities were analyzed by using a coupled enzymatic assay as described in.59,82 For initial modulation screenings, substrate peptide specificity analysis, and compound titrations, 1.5 μM sirtuin was incubated with 50 μM substrate peptide and 100 μM NAD+ in absence or presence of 10 μM, 100 μM or the indicated concentration of 1,4-DHP at a final DMSO concentration of 5%. For kinetic experiments, 1.5 μM sirtuin was incubated with saturating concentrations of either substrate peptide (0.4 mM) or NAD+ (2 mM), with the second substrate’s concentration being varied in absence or presence of 200 μM 1,4-DHP at a final concentration of 5% DMSO. Potential 1,4-DHP effects on the coupled enzymes GDH and nicotinamidase were tested in control assays containing 50 μM NAM instead of the substrate peptide.
SPR Experiments
SPR experiments were carried out using a SensiQ Pioneer system (SensiQ, Oklahoma City, OK, USA), essentially as in Genovese et al. 202084 and in Battista et al. 2021,85 with some modifications, as follows.
The sensor chip (COOH5) was chemically activated by a 35 μL injection of a 1:1 mixture of N-ethyl-N′-3-(diethylaminopropyl) carbodiimide (200 mM) and N-hydroxysuccinimide (50 mM) at a flow rate of 5 μL/min. Sirtuin3 (ligand) was immobilized on activated sensor chips via amine coupling. Immobilization was carried out in 20 mM sodium acetate at pH 4.0; unreacted groups were blocked by injecting 1 M ethanolamine hydrochloride (35 μL). Sirtuin3 immobilization level obtained was 1500–4000 RU.
The analytes, dissolved in 100% DMSO at 10 mM concentration, were diluted in buffer 20 mM Hepes, 150 mM NaCl, 0.005% surfactant P20 buffer (HBSP) to a concentration of 200 μM (2% final DMSO concentration), further diluted in HBSP + 2% DMSO (HBSP2%D) and injected on the sensor chip at a constant flow rate (nominal flow rate = 30 μL/min) at the following concentrations: 5, 10, 20, 40, and 80 μM.
The RU increase relative to baseline indicates complex formation (taking place at known analytes concentrations at each moment in the gradient); the plateau region represents the steady-state phase of the interaction (RUeq); and the decrease in RU after 180 s represents dissociation of analytes from the immobilized Sirtiun3 after injection of HBSP2%D. Regeneration procedures are based on two long (2000 and 500 s) injections of buffer, separated by a brief (5 s) injection of 10 mM NaOH.
Control experiments were performed in sensor chips treated as described above in the absence of an immobilized ligand. Kinetic evaluation of the sensorgrams was obtained using the SensiQ Qdat program and full fitting with 1, 2, and 3 sites; Scatchard analysis was used to determine apparent KD values.
Cell Culture and Treatments
Breast cancer cell line MDA-MB-231, anaplastic thyroid cancer cell line CAL-62 and human keratinocytes HaCaT were purchased from ATCC and were cultured in RPMI 1640 medium (R0883 Sigma Aldrich-MERCK) (MDA-MB-231 and CAL-62) or DMEM medium (11965092, Gibco) supplemented with 10% fetal bovine serum (Sigma-Aldrich-MERCK, F9665), 2 mM glutamine (G7513; Sigma-Aldrich-MERCK), 100 units/mL penicillin and 0.1 mg/mL streptomycin (P0781; Sigma-Aldrich-MERCK). Adherent cells were detached by Trypsin–EDTA solution (TA049; Sigma-Aldrich-MERCK). Cells were maintained at 37 °C in humidified incubator with 5% CO2. Cells were treated with cobalt(III) chloride hexahydrate (CoCl2) (Merck; C8661) dissolved in distilled, sterile water for 24 h to induce hypoxia (200 μM). Hypoxic and normoxic cells were also treated with 10/50 μM of the indicated compounds (dissolved in DMSO) for 48 h, maintaining the CoCl2 in the culture medium for hypoxic conditions. SIRT3 inhibitor 3-TYP was dissolved in DMSO and added to a final concentration of 1 μM for 24 and/or 48 h.
Antibodies and Western Blot
The following antibodies were used in this study: ACTB (Merck; A5316), HIF1-α (Cell Signaling; 14179), HIF2/EPAS-1 (Santa Cruz Biotechnology sc-13596), HXKII (Santa Cruz Biotechnology sc-6521), CAIX (Novus Biotechnology NB100-417), SIRT3 (Cell Signaling; 2627S), MnSOD (Abcam ab13533), MnSOD K68 (Abcam ab137037), MnSOD K122 (Abcam ab214675), peroxidase-conjugated AffiniPure Goat Anti-Rabbit IgG (H + L) (Jackson ImmunoResearch; 111-035-003), and peroxidase-conjugated AffiniPure Goat Anti-Mouse IgG (H + L) (Jackson ImmunoResearch; 115-035-062).
Cell Viability
Cell viability was assessed by the trypan blue exclusion assay. At the end of treatments, cells were harvested and stained with 0,4% trypan blue (Merck; T8154). The cell suspension was applied to a hemocytometer and counted with phase contrast microscopy (NIKON EclipseTE2000U, Nikon Netherlands, Amsterdam, The Netherlands).
GDH Assay
GDH activity in cells treated or not with the SIRT3 activators 3a–d and the reference compounds 2 and 3 was measured using a GDH activity assay kit (MAK099, Merck) following the manufacturer’s instructions. Briefly, 106 cells were lysed in 40 μL of GDH assay buffer and kept for 10 min in ice. Afterward, lysates were centrifuged to collect supernatants. Ten microliters of supernatant were added to 40 μL of GDH buffer and 100 μL of a mix containing GDH assay buffer, developer, and glutamate. The whole mix was transferred into a 96-well plate and incubated at 37 °C for 3 min. Finally, absorbance was read at 450 nm using a GloMax multi detection system (Promega, Milan, Italy). The absorbance of the assay buffer was subtracted from each experimental sample.
Clonogenic Assay
Cells were seeded in a 100 mm dish and, once at 80–90% confluence, treated as described in the Results and Discussion section. After 24 and 48 h, cells were collected, counted, and 500 cells plated in a 100 mm dish. After about 7 days for CAL62 and 10 days for MDA-MB-231, plates were washed with phosphate-buffered saline solution (PBS); (Merck; 79382), and clones were fixed with 4% formaldehyde solution in PBS (Merck; F8775) at room temperature for 15 min. After that, the dishes were washed with PBS, and clones stained for 5 min with 0.5% crystal violet (Merck; C0775). Finally, the plates were washed with distilled water and air-dried. After scanning each individual dish, the colonies were counted the following day.
Proteins Extraction and Immunoblotting
Treated and untreated cells were collected and centrifuged at 3000 rpm for 5 min. After removing the supernatant, cells were lysed in 70 μL of lysis buffer containing: 50 mM Tris-Cl (Merck; 93352), 250 mM sodium chloride (NaCl, Merck; S7653), 5 mM ethylenediaminetetraacetic acid (EDTA; Merck; E6758), 0.1% Triton X-100 and 0.1 mM dithiothreitol (Merck; D9163) plus 1 mM phenylmethylsulfonyl fluoride (PMSF, Merck; 93482), protease inhibitor cocktail (PI; Merck; P8340), 1 mM sodium orthovanadate (NA3VO4, Merck; S6508), and 10 mM sodium fluoride (NaF, Merck; 201154) (lysis buffer). After 30 min on ice, samples were centrifuged at 13,000 rpm for 10 min at 4 °C, and the supernatants collected. Protein concentration was determined by the Bradford assay (Bio-Rad; 500-0205). The equivalent of 15 or 50 μg of protein was boiled for 5 min, electrophoresed onto denaturing SDS-PAGE gel, and transferred onto a 0.45 μM nitrocellulose membrane (Bio-Rad; 162-0115). After blocking with 5% milk, membranes were incubated with the appropriate primary antibody overnight. The next day, after three washes with 0.1% Tween 20 (Merck; P9416) in PBS (PBST) for 30 min at rt, membranes were incubated with the appropriate secondary antibody for 1 h at rt. After three more washes in PBST, the detection of the relevant protein was assessed by enhanced chemiluminescence (Lite Ablot TURBO, Euro-Clone; EMP012001). Densitometric analysis of the bands, relative to ACTB, was performed using ImageJ Software v1.51 (NIH, Bethesda, MD, USA).
RNA Extraction, Reverse Transcription, and Real-Time PCR
RNAs were extracted by ReliaPrep RNA tissue miniprep system (Promega, Madison, WI, USA) and reverse transcribed with an iScriptTM c-DNA synthesis kit (Bio-Rad Laboratories Inc., Hercules, CA, USA). Real-time (RT-qPCR) analyses were performed on cDNAs that were amplified by the qPCR reaction using GoTaq qPCR Master Mix (Promega, Madison, WI, USA). Relative amounts obtained with the 2(−ΔCt) method were normalized with respect to the housekeeping gene human L32. Primer sequences are reported in Table S3. Statistical analysis was performed with GraphPad Prism 8, and differences in gene expression were considered significant with a p-value <0.05.
Scratch Assay
Cell lines were maintained in culture medium (as above) until reaching 100% confluence, then shifted to a serum-depleted culture medium to inhibit cell proliferation, as described in Magistri et al.;83 a scratch wound was created on the cell layer using a micropipette tip. Micrographs were taken at 0 and 48 h after the scratch. Cell-devoid areas at 0 and 48 h after the scratch were quantified through the Fiji ImageJ image processing package.
Acknowledgments
This work was supported by AIRC2021 (IG26172) (S.V.), PRIN2020 (2020CW39SJ) (S.V.), Ateneo Sapienza Project 2020 (RG120172B8E53D03) (S.V.), Ateneo Sapienza Project 2022 (RM1221814885E5A2) (R.F.), Sapienza SEED PNR 2021 (C.B.), Ateneo Sapienza Project 2022 (RM12218166AEFC72) (C.B.), AIRC2021 (IG26290) (M.T.), PNRR-Rome Technopole (M.T.), and FISR2019_00374 MeDyCa (A.M.). C. Castiello and C. Zwergel are thankful for the generous funding from FSE REACT-EU within the program PON “Research and Innovation” 2014–2020, Action IV.6 “Contratti di ricerca su tematiche Green”. All authors would like to thank Francesco Fiorentino and Stefan Boettcher for their excellent technical support regarding the HPLC and HRMS analysis and Ken Rory for the final proofreading.
Glossary
Abbreviations
- ACS2
acetyl-CoA synthetase 2
- AMPK
5′ adenosine monophosphate-activated protein kinase
- CA-IX
carbonic anhydrase IX
- CLL
chronic lymphocytic leukemia
- CTR
control
- DTC
differentiated thyroid cancer
- 1,4-DHP
1,4-dihydropyridine
- ECM
extracellular matrix
- EMT
epithelial–mesenchymal transition
- EPAS-1
endothelial PAS domain-containing protein 1
- ETC
electron transport chain
- FdL
“Fluor-de-Lys”
- GDH
glutamate dehydrogenase
- GOT2
glutamate oxaloacetate transaminase 2
- HBSP
HEPES-buffered saline surfactant P20
- HCC
hepatocellular carcinoma
- HIF1-α
hypoxia-inducible factor-1α
- MMP2/9
matrix metalloprotease 2/9
- MnSOD
manganese-dependent superoxide dismutase
- NAM
nicotinamide
- NiNTA
nickel(II) nitrilotriacetic acid complex
- OXPHOS
oxidative phosphorylation
- PDC
pyruvate dehydrogenase complex
- PHD
prolyl hydroxylase
- PI
protease inhibitor cocktail
- PMSF
phenylmethylsulfonyl fluoride
- rt-PCR
real-time PCR
- RU
resonance units
- RUeq
resonance units at equilibrium
- SIRT
sirtuins
- SUMO
small ubiquitin-like modifier
- TCA
tricarboxylic acid
- TEV protease
tobacco etch virus nuclear-inclusion-a endopeptidase
- UPR
unfolded protein response
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00337.
Chemical and physical data of final compounds 2a–n and 3a–d including copies of the 1H and 13C NMR as well as the IR-spectra, HPLC traces and the elemental analysis data, control experiments, representative plate images of the colony formation, cell viability data, qRT-PCR analysis with the relative primers used, and quantification of living and death cells in various cancerous and non-cancerous cell lines upon 24 and 48 h of treatment relative to three independent experiments (PDF)
Molecular strings for SIRT3 (CSV)
Author Contributions
○ C.Z., M.A., and S.G. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Jing H.; Lin H. Sirtuins in Epigenetic Regulation. Chem. Rev. 2015, 115, 2350–2375. 10.1021/cr500457h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang W. The Controversial World of Sirtuins. Drug Discov. Today Technol. 2014, 12, e9–e17. 10.1016/j.ddtec.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao E.; Hou J.; Ke X.; Abbas M. N.; Kausar S.; Zhang L.; Cui H. The Roles of Sirtuin Family Proteins in Cancer Progression. Cancers 2019, 11, 1949. 10.3390/cancers11121949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauh D.; Fischer F.; Gertz M.; Lakshminarasimhan M.; Bergbrede T.; Aladini F.; Kambach C.; Becker C. F.; Zerweck J.; Schutkowski M.; Steegborn C. An Acetylome Peptide Microarray Reveals Specificities and Deacetylation Substrates for All Human Sirtuin Isoforms. Nat. Commun. 2013, 4, 2327. 10.1038/ncomms3327. [DOI] [PubMed] [Google Scholar]
- Carafa V.; Rotili D.; Forgione M.; Cuomo F.; Serretiello E.; Hailu G. S.; Jarho E.; Lahtela-Kakkonen M.; Mai A.; Altucci L. Sirtuin Functions and Modulation: From Chemistry to the Clinic. Clin. Epigenet. 2016, 8, 61. 10.1186/s13148-016-0224-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosciuk T.; Wang M.; Hong J. Y.; Lin H. Updates on the Epigenetic Roles of Sirtuins. Curr. Opin. Chem. Biol. 2019, 51, 18–29. 10.1016/j.cbpa.2019.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; He J.; Liao M.; Hu M.; Li W.; Ouyang H.; Wang X.; Ye T.; Zhang Y.; Ouyang L. An Overview of Sirtuins as Potential Therapeutic Target: Structure, Function and Modulators. Eur. J. Med. Chem. 2019, 161, 48–77. 10.1016/j.ejmech.2018.10.028. [DOI] [PubMed] [Google Scholar]
- Mautone N.; Zwergel C.; Mai A.; Rotili D. Sirtuin Modulators: Where Are We Now? A Review of Patents from 2015 to 2019. Expert Opin. Ther. Pat. 2020, 30, 389–407. 10.1080/13543776.2020.1749264. [DOI] [PubMed] [Google Scholar]
- Wang M.; Lin H. Understanding the Function of Mammalian Sirtuins and Protein Lysine Acylation. Annu. Rev. Biochem. 2021, 90, 245–285. 10.1146/annurev-biochem-082520-125411. [DOI] [PubMed] [Google Scholar]
- Samant S. A.; Zhang H. J.; Hong Z.; Pillai V. B.; Sundaresan N. R.; Wolfgeher D.; Archer S. L.; Chan D. C.; Gupta M. P. Sirt3 Deacetylates and Activates Opa1 to Regulate Mitochondrial Dynamics During Stress. Mol. Cell. Biol. 2014, 34, 807–819. 10.1128/mcb.01483-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa L.; Germain D. Correction for Papa and Germain, Sirt3 Regulates a Novel Arm of the Mitochondrial Unfolded Protein Response. Mol. Cell. Biol. 2017, 37, e00191 10.1128/MCB.00191-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Fu L. L.; Wen X.; Wang X. Y.; Liu J.; Cheng Y.; Huang J. Sirtuin-3 (Sirt3), a Therapeutic Target with Oncogenic and Tumor-Suppressive Function in Cancer. Cell Death Dis. 2014, 5, e1047 10.1038/cddis.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrens-Mas M.; Oliver J.; Roca P.; Sastre-Serra J. SIRT3: Oncogene and Tumor Suppressor in Cancer. Cancers 2017, 9, 90. 10.3390/cancers9070090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell E. L.; Emerling B. M.; Ricoult S. J.; Guarente L. SirT3 suppresses hypoxia inducible factor 1α and tumor growth by inhibiting mitochondrial ROS production. Oncogene 2011, 30, 2986–2996. 10.1038/onc.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley L. W.; Carracedo A.; Lee J.; Souza A.; Egia A.; Zhang J.; Teruya-Feldstein J.; Moreira P. I.; Cardoso S. M.; Clish C. B.; Pandolfi P. P.; Haigis M. C. SIRT3 Opposes Reprogramming of Cancer Cell Metabolism through HIF1α Destabilization. Cancer Cell 2011, 19, 416–428. 10.1016/j.ccr.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell E. L.; Guarente L. The Sirt3 Divining Rod Points to Oxidative Stress. Mol. Cell 2011, 42, 561–568. 10.1016/j.molcel.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin W. G. Jr.; Ratcliffe P. J. Oxygen Sensing by Metazoans: The Central Role of the Hif Hydroxylase Pathway. Mol. Cell 2008, 30, 393–402. 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Fan J.; Shan C.; Kang H. B.; Elf S.; Xie J.; Tucker M.; Gu T. L.; Aguiar M.; Lonning S.; Chen H.; Mohammadi M.; Britton L. M.; Garcia B. A.; Aleckovic M.; Kang Y.; Kaluz S.; Devi N.; Van Meir E.; Hitosugi T.; Seo J. H.; Lonial S.; Gaddh M.; Arellano M.; Khoury H. J.; Khuri F. R.; Boggon T. J.; Kang S.; Chen J. Tyr Phosphorylation of Pdp1 Toggles Recruitment between Acat1 and Sirt3 to Regulate the Pyruvate Dehydrogenase Complex. Mol. Cell 2014, 53, 534–548. 10.1016/j.molcel.2013.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L.; Zhou Y.; Dai Q.; Qiao C.; Zhao L.; Hui H.; Lu N.; Guo Q. L. Oroxylin a Induces Dissociation of Hexokinase Ii from the Mitochondria and Inhibits Glycolysis by Sirt3-Mediated Deacetylation of Cyclophilin D in Breast Carcinoma. Cell Death Dis. 2013, 4, e601 10.1038/cddis.2013.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Zhou L.; Shi Q.; Zhao Y.; Lin H.; Zhang M.; Zhao S.; Yang Y.; Ling Z. Q.; Guan K. L.; Xiong Y.; Ye D. Sirt3-Dependent Got2 Acetylation Status Affects the Malate-Aspartate Nadh Shuttle Activity and Pancreatic Tumor Growth. EMBO J. 2015, 34, 1110–1125. 10.15252/embj.201591041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W.; Denu R. A.; Krautkramer K. A.; Grindle K. M.; Yang D. T.; Asimakopoulos F.; Hematti P.; Denu J. M. Loss of Sirt3 Provides Growth Advantage for B Cell Malignancies. J. Biol. Chem. 2016, 291, 3268–3279. 10.1074/jbc.m115.702076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Liu Y. L.; Cheng W.; Yin X. M.; Jiang B. The Expression of Sirt3 in Primary Hepatocellular Carcinoma and the Mechanism of Its Tumor Suppressing Effects. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 978–998. [PubMed] [Google Scholar]
- Shackelford R.; Hirsh S.; Coppola D. Nicotinamide Phosphoribosyltransferase and Sirt3 Expression Are Increased in Well-Differentiated Thyroid Carcinomas. Am. J. Clin. Pathol. 2013, 140, A011. 10.1093/ajcp/140.suppl1.011. [DOI] [PubMed] [Google Scholar]
- Wang S.; Chen X.; Zhang Z.; Wu Z. Microrna-1225-5p Inhibits the Development and Progression of Thyroid Cancer Via Targeting Sirtuin 3. Pharmazie 2019, 74, 423–427. 10.1691/ph.2019.9411. [DOI] [PubMed] [Google Scholar]
- Yao L.; Wang Y. Bioinformatic Analysis of the Effect of the Sirtuin Family on Differentiated Thyroid Carcinoma. BioMed Res. Int. 2022, 2022, 1–15. 10.1155/2022/5794118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P.; Zhao T.; Zhou W. The Clinical Significance of Sirt3 in Covid-19 Patients: A Single Center Retrospective Analysis. Ann. Clin. Lab. Sci. 2021, 51, 686–693. [PubMed] [Google Scholar]
- Ren J. H.; Hu J. L.; Cheng S. T.; Yu H. B.; Wong V. K. W.; Law B. Y. K.; Yang Y. F.; Huang Y.; Liu Y.; Chen W. X.; Cai X. F.; Tang H.; Hu Y.; Zhang W. L.; Liu X.; Long Q. X.; Zhou L.; Tao N. N.; Zhou H. Z.; Yang Q. X.; Ren F.; He L.; Gong R.; Huang A. L.; Chen J. Sirt3 Restricts Hepatitis B Virus Transcription and Replication through Epigenetic Regulation of Covalently Closed Circular DNA Involving Suppressor of Variegation 3-9 Homolog 1 and Set Domain Containing 1a Histone Methyltransferases. Hepatology 2018, 68, 1260–1276. 10.1002/hep.29912. [DOI] [PubMed] [Google Scholar]
- Guo X.; Yan F.; Li J.; Zhang C.; Bu P. Sirt3 Attenuates Angii-Induced Cardiac Fibrosis by Inhibiting Myofibroblasts Transdifferentiation Via Stat3-Nfatc2 Pathway. Am. J. Transl. Res. 2017, 9, 3258–3269. [PMC free article] [PubMed] [Google Scholar]
- Morigi M.; Perico L.; Benigni A. Sirtuins in Renal Health and Disease. J. Am. Soc. Nephrol. 2018, 29, 1799–1809. 10.1681/asn.2017111218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wang K.; Huang C.; Lin D.; Zhou Y.; Wu Y.; Tian N.; Fan P.; Pan X.; Xu D.; Hu J.; Zhou Y.; Wang X.; Zhang X. Sirt3 Activation by Dihydromyricetin Suppresses Chondrocytes Degeneration Via Maintaining Mitochondrial Homeostasis. Int. J. Biol. Sci. 2018, 14, 1873–1882. 10.7150/ijbs.27746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter L. C.; Franczyk M. P.; Pietka T.; Yamaguchi S.; Lin J. B.; Sasaki Y.; Verdin E.; Apte R. S.; Yoshino J. Nad(+)-Dependent Deacetylase Sirt3 in Adipocytes Is Dispensable for Maintaining Normal Adipose Tissue Mitochondrial Function and Whole Body Metabolism. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E520–E530. 10.1152/ajpendo.00057.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra Y.; Kaundal R. K. Role of Sirt3 in Mitochondrial Biology and Its Therapeutic Implications in Neurodegenerative Disorders. Drug Discov. Today 2023, 28, 103583. 10.1016/j.drudis.2023.103583. [DOI] [PubMed] [Google Scholar]
- Yan B.; Liu Q.; Ding X.; Lin Y.; Jiao X.; Wu Y.; Miao H.; Zhou C. Sirt3-Mediated Cypd-K166 Deacetylation Alleviates Neuropathic Pain by Improving Mitochondrial Dysfunction and Inhibiting Oxidative Stress. Oxid. Med. Cell. Longev. 2022, 2022, 1–17. 10.1155/2022/4722647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentino F.; Mautone N.; Menna M.; D’Acunzo F.; Mai A.; Rotili D. Sirtuin Modulators: Past, Present, and Future Perspectives. Future Med. Chem. 2022, 14, 915–939. 10.4155/fmc-2022-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoseini A.; Namazi G.; Farrokhian A.; Reiner Z.; Aghadavod E.; Bahmani F.; Asemi Z. The Effects of Resveratrol on Metabolic Status in Patients with Type 2 Diabetes Mellitus and Coronary Heart Disease. Food Funct. 2019, 10, 6042–6051. 10.1039/c9fo01075k. [DOI] [PubMed] [Google Scholar]
- Curry A. M.; White D. S.; Donu D.; Cen Y. Human Sirtuin Regulators: The ″Success″ Stories. Front. Physiol. 2021, 12, 752117. 10.3389/fphys.2021.752117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libri V.; Brown A. P.; Gambarota G.; Haddad J.; Shields G. S.; Dawes H.; Pinato D. J.; Hoffman E.; Elliot P. J.; Vlasuk G. P.; Jacobson E.; Wilkins M. R.; Matthews P. M. A Pilot Randomized, Placebo Controlled, Double Blind Phase I Trial of the Novel Sirt1 Activator Srt2104 in Elderly Volunteers. PLoS One 2012, 7, e51395 10.1371/journal.pone.0051395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer A. J.; Scicluna B. P.; Moerland P. D.; Lin J.; Jacobson E. W.; Vlasuk G. P.; van der Poll T. The Selective Sirtuin 1 Activator Srt2104 Reduces Endotoxin-Induced Cytokine Release and Coagulation Activation in Humans. Crit. Care Med. 2015, 43, e199–e202. 10.1097/ccm.0000000000000949. [DOI] [PubMed] [Google Scholar]
- Krueger J. G.; Suarez-Farinas M.; Cueto I.; Khacherian A.; Matheson R.; Parish L. C.; Leonardi C.; Shortino D.; Gupta A.; Haddad J.; Vlasuk G. P.; Jacobson E. W. A Randomized, Placebo-Controlled Study of Srt2104, a Sirt1 Activator, in Patients with Moderate to Severe Psoriasis. PLoS One 2015, 10, e0142081 10.1371/journal.pone.0142081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai A.; Valente S.; Meade S.; Carafa V.; Tardugno M.; Nebbioso A.; Galmozzi A.; Mitro N.; De Fabiani E.; Altucci L.; Kazantsev A. Study of 1,4-Dihydropyridine Structural Scaffold: Discovery of Novel Sirtuin Activators and Inhibitors. J. Med. Chem. 2009, 52, 5496–5504. 10.1021/jm9008289. [DOI] [PubMed] [Google Scholar]
- Valente S.; Mellini P.; Spallotta F.; Carafa V.; Nebbioso A.; Polletta L.; Carnevale I.; Saladini S.; Trisciuoglio D.; Gabellini C.; Tardugno M.; Zwergel C.; Cencioni C.; Atlante S.; Moniot S.; Steegborn C.; Budriesi R.; Tafani M.; Del Bufalo D.; Altucci L.; Gaetano C.; Mai A. 1,4-Dihydropyridines Active on the Sirt1/Ampk Pathway Ameliorate Skin Repair and Mitochondrial Function and Exhibit Inhibition of Proliferation in Cancer Cells. J. Med. Chem. 2016, 59, 1471–1491. 10.1021/acs.jmedchem.5b01117. [DOI] [PubMed] [Google Scholar]
- Spallotta F.; Cencioni C.; Straino S.; Nanni S.; Rosati J.; Artuso S.; Manni I.; Colussi C.; Piaggio G.; Martelli F.; Valente S.; Mai A.; Capogrossi M. C.; Farsetti A.; Gaetano C. A Nitric Oxide-Dependent Cross-Talk between Class I and Iii Histone Deacetylases Accelerates Skin Repair. J. Biol. Chem. 2013, 288, 11004–11012. 10.1074/jbc.m112.441816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman J. L.; Baeza J.; Denu J. M. Activation of the Protein Deacetylase Sirt6 by Long-Chain Fatty Acids and Widespread Deacylation by Mammalian Sirtuins. J. Biol. Chem. 2013, 288, 31350–31356. 10.1074/jbc.c113.511261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein M. A.; Liu C.; Kuznetsov V. I.; Feltenberger J. B.; Tang W.; Denu J. M. Mechanism of Activation for the Sirtuin 6 Protein Deacylase. J. Biol. Chem. 2020, 295, 1385–1399. 10.1016/s0021-9258(17)49896-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iachettini S.; Trisciuoglio D.; Rotili D.; Lucidi A.; Salvati E.; Zizza P.; Di Leo L.; Del Bufalo D.; Ciriolo M. R.; Leonetti C.; Steegborn C.; Mai A.; Rizzo A.; Biroccio A. Pharmacological Activation of Sirt6 Triggers Lethal Autophagy in Human Cancer Cells. Cell Death Dis. 2018, 9, 996. 10.1038/s41419-018-1065-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.; Zhao J.; Deng W.; Chen Y.; Shang J.; Song K.; Zhang L.; Wang C.; Lu S.; Yang X.; He B.; Min J.; Hu H.; Tan M.; Xu J.; Zhang Q.; Zhong J.; Sun X.; Mao Z.; Lin H.; Xiao M.; Chin Y. E.; Jiang H.; Xu Y.; Chen G.; Zhang J. Identification of a Cellularly Active Sirt6 Allosteric Activator. Nat. Chem. Biol. 2018, 14, 1118–1126. 10.1038/s41589-018-0150-0. [DOI] [PubMed] [Google Scholar]
- Shang J. L.; Ning S. B.; Chen Y. Y.; Chen T. X.; Zhang J. Mdl-800, an Allosteric Activator of Sirt6, Suppresses Proliferation and Enhances Egfr-Tkis Therapy in Non-Small Cell Lung Cancer. Acta Pharmacol. Sin. 2021, 42, 120–131. 10.1038/s41401-020-0442-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J.; Zhu Z.; Chen Y.; Song J.; Huang Y.; Song K.; Zhong J.; Xu X.; Wei J.; Wang C.; Cui L.; Liu C. Y.; Zhang J. Small-Molecule Activating Sirt6 Elicits Therapeutic Effects and Synergistically Promotes Anti-Tumor Activity of Vitamin D(3) in Colorectal Cancer. Theranostics 2020, 10, 5845–5864. 10.7150/thno.44043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You W.; Rotili D.; Li T. M.; Kambach C.; Meleshin M.; Schutkowski M.; Chua K. F.; Mai A.; Steegborn C. Structural Basis of Sirtuin 6 Activation by Synthetic Small Molecules. Angew. Chem., Int. Ed. Engl. 2017, 56, 1007–1011. 10.1002/anie.201610082. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Chen J.; Sun X.; Yu J.; Qian Z.; Wu L.; Xu X.; Wan X.; Jiang Y.; Zhang J.; Gao S.; Mao Z. The Sirt6 Activator Mdl-800 Improves Genomic Stability and Pluripotency of Old Murine-Derived Ips Cells. Aging Cell 2020, 19, e13185 10.1111/acel.13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Li Y.; Liu Q.; Huang Y.; Li R.; Wu T.; Zhang Z.; Zhou J.; Huang H.; Tang Q.; Huang C.; Zhao Y.; Zhang G.; Jiang W.; Mo L.; Zhang J.; Xie W.; He J. Sirt6 Alleviated Liver Fibrosis by Deacetylating Conserved Lysine 54 on Smad2 in Hepatic Stellate Cells. Hepatology 2021, 73, 1140–1157. 10.1002/hep.31418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai V. B.; Samant S.; Sundaresan N. R.; Raghuraman H.; Kim G.; Bonner M. Y.; Arbiser J. L.; Walker D. I.; Jones D. P.; Gius D.; Gupta M. P. Honokiol Blocks and Reverses Cardiac Hypertrophy in Mice by Activating Mitochondrial Sirt3. Nat. Commun. 2015, 6, 6656. 10.1038/ncomms7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan Y.; Park W.; Jin J.; Kim W.; Park S. K.; Kang K. P. Sirtuin 3 Activation by Honokiol Decreases Unilateral Ureteral Obstruction-Induced Renal Inflammation and Fibrosis via Regulation of Mitochondrial Dynamics and the Renal NF-κB-TGF-β1/Smad Signaling Pathway. Int. J. Mol. Sci. 2020, 21, 402. 10.3390/ijms21020402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Morales N.; Rovira-Llopis S.; Banuls C.; Lopez-Domenech S.; Escribano-Lopez I.; Veses S.; Jover A.; Rocha M.; Hernandez-Mijares A.; Victor V. M. Does Metformin Protect Diabetic Patients from Oxidative Stress and Leukocyte-Endothelium Interactions?. Antioxid. Redox Signaling 2017, 27, 1439–1445. 10.1089/ars.2017.7122. [DOI] [PubMed] [Google Scholar]
- Quan Y.; Xia L.; Shao J.; Yin S.; Cheng C. Y.; Xia W.; Gao W. Q. Adjudin Protects Rodent Cochlear Hair Cells against Gentamicin Ototoxicity Via the Sirt3-Ros Pathway. Sci. Rep. 2015, 5, 8181. 10.1038/srep08181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Xiang H.; Liu J.; Chen Y.; He R. R.; Liu B. Mitochondrial Sirtuin 3: New Emerging Biological Function and Therapeutic Target. Theranostics 2020, 10, 8315–8342. 10.7150/thno.45922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.; Gan C.; Gao Z.; Huang Y.; Wu S.; Zhang D.; Wang X.; Sheng J. Caffeine Targets Sirt3 to Enhance Sod2 Activity in Mitochondria. Front. Cell Dev. Biol. 2020, 8, 822. 10.3389/fcell.2020.00822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Zou L.; Shi D.; Liu J.; Zhang J.; Zhao R.; Wang G.; Zhang L.; Ouyang L.; Liu B. Structure-Guided Design of a Small-Molecule Activator of Sirtuin-3 That Modulates Autophagy in Triple Negative Breast Cancer. J. Med. Chem. 2021, 64, 14192–14216. 10.1021/acs.jmedchem.0c02268. [DOI] [PubMed] [Google Scholar]
- Suenkel B.; Valente S.; Zwergel C.; Weiss S.; Di Bello E.; Fioravanti R.; Aventaggiato M.; Amorim J. A.; Garg N.; Kumar S.; Lombard D. B.; Hu T.; Singh P. K.; Tafani M.; Palmeira C. M.; Sinclair D.; Mai A.; Steegborn C. Potent and Specific Activators for Mitochondrial Sirtuins Sirt3 and Sirt5. J. Med. Chem. 2022, 65, 14015–14031. 10.1021/acs.jmedchem.2c01215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz M.; Steegborn C. Using Mitochondrial Sirtuins as Drug Targets: Disease Implications and Available Compounds. Cell. Mol. Life Sci. 2016, 73, 2871–2896. 10.1007/s00018-016-2180-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlicker C.; Gertz M.; Papatheodorou P.; Kachholz B.; Becker C. F.; Steegborn C. Substrates and Regulation Mechanisms for the Human Mitochondrial Sirtuins Sirt3 and Sirt5. J. Mol. Biol. 2008, 382, 790–801. 10.1016/j.jmb.2008.07.048. [DOI] [PubMed] [Google Scholar]
- Lombard D. B.; Alt F. W.; Cheng H. L.; Bunkenborg J.; Streeper R. S.; Mostoslavsky R.; Kim J.; Yancopoulos G.; Valenzuela D.; Murphy A.; Yang Y.; Chen Y.; Hirschey M. D.; Bronson R. T.; Haigis M.; Guarente L. P.; Farese R. V. Jr.; Weissman S.; Verdin E.; Schwer B. Mammalian Sir2 Homolog Sirt3 Regulates Global Mitochondrial Lysine Acetylation. Mol. Cell. Biol. 2007, 27, 8807–8814. 10.1128/mcb.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarsour E. H.; Kalen A. L.; Xiao Z.; Veenstra T. D.; Chaudhuri L.; Venkataraman S.; Reigan P.; Buettner G. R.; Goswami P. C. Manganese Superoxide Dismutase Regulates a Metabolic Switch During the Mammalian Cell Cycle. Cancer Res. 2012, 72, 3807–3816. 10.1158/0008-5472.can-11-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Zhang H.; Chen X.; Zou Y.; Li J.; Wang L.; Wu M.; Zang J.; Yu Y.; Zhuang W.; Xia Q.; Wang J. A Small Molecule Activator of Sirt3 Promotes Deacetylation and Activation of Manganese Superoxide Dismutase. Free Radic. Biol. Med. 2017, 112, 287–297. 10.1016/j.freeradbiomed.2017.07.012. [DOI] [PubMed] [Google Scholar]
- Lu J.; Cheng K.; Zhang B.; Xu H.; Cao Y.; Guo F.; Feng X.; Xia Q. Novel Mechanisms for Superoxide-Scavenging Activity of Human Manganese Superoxide Dismutase Determined by the K68 Key Acetylation Site. Free Radic. Biol. Med. 2015, 85, 114–126. 10.1016/j.freeradbiomed.2015.04.011. [DOI] [PubMed] [Google Scholar]
- Lone M. U.; Baghel K. S.; Kanchan R. K.; Shrivastava R.; Malik S. A.; Tewari B. N.; Tripathi C.; Negi M. P.; Garg V. K.; Sharma M.; Bhatt M. L.; Bhadauria S. Physical Interaction of Estrogen Receptor with Mnsod: Implication in Mitochondrial O(2)(.-) Upregulation and Mtorc2 Potentiation in Estrogen-Responsive Breast Cancer Cells. Oncogene 2017, 36, 1829–1839. 10.1038/onc.2016.346. [DOI] [PubMed] [Google Scholar]
- Tao R.; Coleman M. C.; Pennington J. D.; Ozden O.; Park S. H.; Jiang H.; Kim H. S.; Flynn C. R.; Hill S.; Hayes McDonald W.; Olivier A. K.; Spitz D. R.; Gius D. Sirt3-Mediated Deacetylation of Evolutionarily Conserved Lysine 122 Regulates Mnsod Activity in Response to Stress. Mol. Cell 2010, 40, 893–904. 10.1016/j.molcel.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.; Zhu Y.; Tran E. L.; Tokars V.; Dean A. E.; Quan S.; Gius D. Mnsod Lysine 68 Acetylation Leads to Cisplatin and Doxorubicin Resistance Due to Aberrant Mitochondrial Metabolism. Int. J. Biol. Sci. 2021, 17, 1203–1216. 10.7150/ijbs.51184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Sanchez J.; Chanez-Cardenas M. E. The Use of Cobalt Chloride as a Chemical Hypoxia Model. J. Appl. Toxicol. 2019, 39, 556–570. 10.1002/jat.3749. [DOI] [PubMed] [Google Scholar]
- Muz B.; de la Puente P.; Azab F.; Azab A. K. The Role of Hypoxia in Cancer Progression, Angiogenesis, Metastasis, and Resistance to Therapy. Hypoxia 2015, 3, 83–92. 10.2147/hp.s93413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. J.; Khadka D.; Seo J. H. Interplay between Solid Tumors and Tumor Microenvironment. Front. Immunol. 2022, 13, 882718. 10.3389/fimmu.2022.882718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L.; Zhou Y.; Qiao C.; Ni T.; Li Z.; You Q.; Guo Q.; Lu N. Oroxylin A inhibits glycolysis-dependent proliferation of human breast cancer via promoting SIRT3-mediated SOD2 transcription and HIF1α destabilization. Cell Death Dis. 2015, 6, e1714 10.1038/cddis.2015.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Geng Y. H.; Wang P.; Zhou Y. T.; Yang H.; Huo Y. F.; Zhang H. Q.; Li Y.; He H. Y.; Tian X. X.; Fang W. G. Extracellular ATP promotes breast cancer invasion and epithelial-mesenchymal transition via hypoxia-inducible factor 2α signaling. Cancer Sci. 2019, 110, 2456–2470. 10.1111/cas.14086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou Y.; McDonald P. C.; Oloumi A.; Chia S.; Ostlund C.; Ahmadi A.; Kyle A.; Auf dem Keller U.; Leung S.; Huntsman D.; Clarke B.; Sutherland B. W.; Waterhouse D.; Bally M.; Roskelley C.; Overall C. M.; Minchinton A.; Pacchiano F.; Carta F.; Scozzafava A.; Touisni N.; Winum J. Y.; Supuran C. T.; Dedhar S. Targeting Tumor Hypoxia: Suppression of Breast Tumor Growth and Metastasis by Novel Carbonic Anhydrase Ix Inhibitors. Cancer Res. 2011, 71, 3364–3376. 10.1158/0008-5472.can-10-4261. [DOI] [PubMed] [Google Scholar]
- Scher M. B.; Vaquero A.; Reinberg D. Sirt3 Is a Nuclear Nad+-Dependent Histone Deacetylase That Translocates to the Mitochondria Upon Cellular Stress. Genes Dev. 2007, 21, 920–928. 10.1101/gad.1527307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Antin P.; Berx G.; Blanpain C.; Brabletz T.; Bronner M.; Campbell K.; Cano A.; Casanova J.; Christofori G.; Dedhar S.; Derynck R.; Ford H. L.; Fuxe J.; García de Herreros A.; Goodall G. J.; Hadjantonakis A. K.; Huang R. Y. J.; Kalcheim C.; Kalluri R.; Kang Y.; Khew-Goodall Y.; Levine H.; Liu J.; Longmore G. D.; Mani S. A.; Massague J.; Mayor R.; McClay D.; Mostov K. E.; Newgreen D. F.; Nieto M. A.; Puisieux A.; Runyan R.; Savagner P.; Stanger B.; Stemmler M. P.; Takahashi Y.; Takeichi M.; Theveneau E.; Thiery J. P.; Thompson E. W.; Weinberg R. A.; Williams E. D.; Xing J.; Zhou B. P.; Sheng G.; Association E. M. T. I. Author Correction: Guidelines and Definitions for Research on Epithelial-Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2021, 22, 834. 10.1038/s41580-021-00428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q.; Zhou J.; Li F.; Guo S.; Zhang L.; Li J.; Qi Q.; Shi Y. The Role and Therapeutic Perspectives of Sirtuin 3 in Cancer Metabolism Reprogramming, Metastasis, and Chemoresistance. Front. Oncol. 2022, 12, 910963. 10.3389/fonc.2022.910963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hapke R. Y.; Haake S. M. Hypoxia-Induced Epithelial to Mesenchymal Transition in Cancer. Cancer Lett. 2020, 487, 10–20. 10.1016/j.canlet.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniot S.; Forgione M.; Lucidi A.; Hailu G. S.; Nebbioso A.; Carafa V.; Baratta F.; Altucci L.; Giacche N.; Passeri D.; Pellicciari R.; Mai A.; Steegborn C.; Rotili D. Development of 1,2,4-Oxadiazoles as Potent and Selective Inhibitors of the Human Deacetylase Sirtuin 2: Structure-Activity Relationship, X-Ray Crystal Structure, and Anticancer Activity. J. Med. Chem. 2017, 60, 2344–2360. 10.1021/acs.jmedchem.6b01609. [DOI] [PubMed] [Google Scholar]
- Suenkel B.; Steegborn C. Recombinant Preparation, Biochemical Analysis, and Structure Determination of Sirtuin Family Histone/Protein Deacylases. Methods Enzymol. 2016, 573, 183–208. 10.1016/bs.mie.2015.12.004. [DOI] [PubMed] [Google Scholar]
- Lakshminarasimhan M.; Curth U.; Moniot S.; Mosalaganti S.; Raunser S.; Steegborn C. Molecular Architecture of the Human Protein Deacetylase Sirt1 and Its Regulation by Aros and Resveratrol. Biosci. Rep. 2013, 33, e00037 10.1042/bsr20120121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B. C.; Hallows W. C.; Denu J. M. A Continuous Microplate Assay for Sirtuins and Nicotinamide-Producing Enzymes. Anal. Biochem. 2009, 394, 101–109. 10.1016/j.ab.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistri P.; Battistelli C.; Strippoli R.; Petrucciani N.; Pellinen T.; Rossi L.; Mangogna L.; Aurello P.; D’Angelo F.; Tripodi M.; Ramacciato G.; Nigri G. Smo Inhibition Modulates Cellular Plasticity and Invasiveness in Colorectal Cancer. Front. Pharmacol. 2018, 8, 956. 10.3389/fphar.2017.00956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese I.; Giamogante F.; Barazzuol L.; Battista T.; Fiorillo A.; Vicario M.; D'Alessandro G.; Cipriani R.; Limatola C.; Rossi D.; Sorrentino V.; Poser E.; Mosca L.; Squitieri F.; Perluigi M.; Arena A.; van Petegem F.; Tito C.; Fazi F.; Giorgi C.; Cali T.; Ilari A.; Colotti G. Sorcin Is an Early Marker of Neurodegeneration, Ca(2+) Dysregulation and Endoplasmic Reticulum Stress Associated to Neurodegenerative Diseases. Cell Death Dis 2020, 11, 861. 10.1038/s41419-020-03063-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battista T.; Pascarella G.; Staid D. S.; Colotti G.; Rosati J.; Fiorillo A.; Casamassa A.; Vescovi A. L.; Giabbai B.; Semrau M. S.; Fanelli S.; Storici P.; Squitieri F.; Morea V.; Ilari A. Known Drugs Identified by Structure-Based Virtual Screening Are Able to Bind Sigma-1 Receptor and Increase Growth of Huntington Disease Patient-Derived Cells. Int J Mol Sci 2021, 22, 1293. 10.3390/ijms22031293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.