

Abstract
Background:
In the past decade, regulatory agencies have released guidance around risk-based management with the goal of focusing on risks to critical aspects of a research study. Several tools have been developed aimed at implementing these guidelines. We designed a risk management tool to meet the demands of our academic data coordinating center.
Methods:
We developed the Risk Assessment and Risk Management (RARM) tool on three fundamental criteria of our risk/quality program: (1) Quality by Design concepts applies to all employees, regardless of the employee’s role; (2) the RARM process must be economically feasible and dynamically flexible during the study startup and implementation process; and (3) responsibility of the RARM lay with the entire study team as opposed to a single quality expert.
Results:
The RARM tool has 20 elements for both risk assessment and risk management. The incorporation of both aspects of risk management allow for a seamless transition from identifying risks to actively monitoring risks throughout enrollment.
Conclusion:
The RARM tool achieves a simplified, seamless approach to risk assessment and risk management. The tool incorporates the concept of Quality by Design into daily work by having every team member contribute to the RARM tool. It also combines the risk assessment and risk management processes into a single tool which allows for a seamless transition from identifying risks to managing the risks throughout the life of the study. The instructions facilitate documentation of de-risking protocols early in development and the tool can be implemented in any platform and organization.
Keywords: Risk assessment, Risk management, Risk assessment categorization tool, Monitoring, Quality, RARM tool
1. Background
There has been a shift around the approach to risk management of human subjects’ research over the past decade. Previously, the most common approach included frequent on-site visits to conduct extensive source data monitoring, which potentially led to a review of all data elements (commonly referred to as 100% source data verification and review). This resource-intense approach tended to foster a reactive stance in response to issues identified during monitoring visits, audits and regulatory inspections. Research organizations, industry, and academia began moving toward a risk-based approach to study implementation in response to the increased scale, complexity, and cost of clinical trials. In 2008, the United States Food and Drug Administration (FDA) and Duke University launched the Clinical Trials Transformation Initiative, as part of FDA’s Critical Path Initiative, which focused on the value of various data verification processes and their impact on study conclusions and risk [1]. In 2011, the European Medicines Agency (EMA) and the FDA drafted a reflection paper and guidance on risk-based monitoring approaches. Shortly after, in 2012, TransCelerate was created as a non-profit organization with a mission to collaborate across the global biopharmaceutical research and development community to identify, prioritize, design, and facilitate implementation of solutions designed to drive the efficient, effective, and high-quality delivery of new medicine. The consortium published a position paper proposing a risk-based monitoring methodology in 2013 [2]. In the same year, the EMA and FDA released their final guidance for risk-based approaches [3,4] and supported TransCelerate with review of pilot risk-based monitoring plans. The ICH GCP E6 (R2) guidelines addendum [5] in 2016 outlined a modernized approach to good clinical practices to better ensure human subjects protection and reliability of trial results. This entire approach has been transformative for the industry in that it focuses on critical risks to a research study rather than a large number of lesser risks. While these guidelines outline key features of risk-based approaches, a well-designed prescription around implementation has been absent. New ICH E6 (R3) guidelines are being drafted that will hopefully advance risk-based approaches and provide additional guidance for implementation of risk assessment and risk management in studies [6].
There are several tools that have been developed aimed at implementing these guidelines. The Risk Assessment and Categorization Tool (RACT) from TransCelerate [7] laid the critical foundation for identifying and documenting risk assessment in clinical research. This tool provides a structure to identify, document, and categorize study risks (e. g., safety, complexity, technology, population, endpoints). Several prompts are included in the RACT such as documenting discussion questions, describing risk considerations, assessing risk characteristics (impact, probability, and detectability), and detailing rationale and mitigation plans [7]. Assessments are completed for all risks identified to critical aspects of the study, including when the protocol is amended to mitigate or eliminate certain risks. Once all risks are identified and categorized, a separate plan is implemented to monitor and manage risks in an ongoing fashion. A different project, the PUEKS project which was led by four pharmaceutical companies and academic institutions, included many collaborators and created risk assessment and risk management tools as part of an overall quality management system [8]. The authors discuss optimizing the clinical monitoring process and how IT plays a role in risk-based monitoring.
The RACT is comprehensive but may require significant time commitment by a dedicated person or team to accurately complete. In addition, the scores generated by the tool have potential subjectivity when different team members individually complete the risk assessments, and may be difficult to complete when a designated quality expert is not available to guide the conversation, or for a novice study team member [9]. The RACT also requires assessment of risk components when it might not be necessary to do so. For example, the RACT prompts completion of risk management elements (e.g., likelihood of occurring) when risks are eliminated from the protocol (e.g., deleting a procedure). Furthermore, the tool is designed to identify risks but does not seamlessly transition to risk monitoring and management activities. Despite these limitations, the RACT has been utilized and several new proposals for risk assessment and risk management tools have been derived from the RACT. Some proposals include monitoring organizational level risks through an overall quality management system in addition to individual trials [10]. Suprin et al. commented on the necessity of every organization having a “fit-for-purpose quality risk management program’ that identifies the most significant risks” [10]. The authors provide comments for how to successfully implement risk management in clinical development. Ciervo et al. presented the monitoring system Xcellerate [11] that is implemented through JIRA™, a commercially available project management software system, and uses third party vendors. The tool allows teams to track risks, communicate activities pertaining to risk, and monitor several processes all within Xcellerate and produces an audit trail. Many other organizations have produced their own fit-for-purpose approach to risk assessment and risk management. Even though solutions have been proposed for risk assessment and risk management, it is still difficult to implement in research. Optimal implementation strategies are still discussed at recent national consortium meetings [12,13].
As a large data coordinating center within an academic research organization, we serve different roles than a sponsor or contract research organization. Therefore, it was appropriate for our organization to explore several tools prior to building a custom-fit-for-purpose tool to meet our unique needs. We identified several criteria for a risk management tool to meet the demands of our data coordinating center including: ease of use by all research team members, applicability to the organization’s specific role in research, ease of implementation, and finally seamless transition between the risk assessment and risk management phases. The Risk Assessment and Risk Management (RARM) tool was created to address these needs and was designed to be utilized by non-quality experts. The RARM tool can be implemented in settings without a sufficient budget to purchase a comprehensive commercial system and is particularly suited to be used in a setting that lacks a designated quality member on the study team.
2. Methods
2.1. Historic implementation of risk assessment in the DCC
The University of Utah Data Coordinating Center (DCC) is an academic research organization that provides data, statistical, and regulatory support for several governmental, philanthropically, and industry-funded clinical studies. The DCC staff is often involved in the protocol development stage which allows us to take a proactive role to assess and mitigate risk early. A typical study team consists of a PhD statistician, a Masters statistician, a data manager, a project manager (leads the day-to-day operation), and a project director (oversees the project/group of similar projects from a leadership position). In 2016, the University of Utah was awarded a Trial Innovation Center grant (U24TR001597) through the National Center for Advancing Translational Sciences (NCATS) with the goal of addressing roadblocks in clinical trials and accelerating the translation of novel interventions into life-saving therapies. An early goal of the Utah Trial Innovation Center was to create a simple and seamless RARM process that could be adopted and implemented in a variety of organizations.
2.2. Strategy behind the RARM system and tool
We based our new tool on three fundamental criteria of our risk/quality program: (1) Quality by Design concepts [14] applied to all employees, regardless of the employee’s role; (2) the RARM process must be economically feasible and dynamically flexible during the study startup and implementation process; and (3) responsibility of the RARM lay with the entire study team as opposed to one trained quality expert team member. With limited experience on quality implementation relating to a risk-based monitoring approach for the majority of employees, our guiding principle for RARM process development was to focus on three elements that teams/staff could easily remember and implement. We called this approach “Simple as 3, 3, 3”. This reinforced the following risk assessment concepts: (1) three areas of risk impact (subject protection, data reliability, operations); (2) three protocol de-risking actions that could be taken once a risk is identified (eliminate, reduce, accept); and (3) three categories a risk can fall into (critical, heightened, standard).
Delineating three distinct protocol de-risking actions was envisioned to be helpful for the study team to critically assess risks and seek opportunities to simplify the research protocol. However, a key aspect of the RARM system is the categorization of identified risks as critical, heightened, or standard. This risk categorization helps study teams easily identify and focus on the most important risks in a trial. In our tool, each risk is categorized in terms of its proportionate significance and impact on patient protection and study results. More specifically, our tool helps reduce the documentation of standard risks, i.e., risks inherent in the conduct of all trials, like consenting patients or reporting serious adverse events. Standard risks can be handled by processes that are already built into our DCC operations and no additional measures need to be put in place to monitor these risks. A heightened risk is a risk that could significantly impact the study, is sufficiently important to monitor very closely and triggers implementation of additional risk controlling activities; while a critical risk is a risk that could jeopardize the study if it occurred beyond an acceptable level and, as such, more aggressive risk controlling activities are developed and implemented to avoid, or at least minimize risk occurrence. For standard risks, our DCC routinely produces reports summarizing enrollment rates of consented subjects. We also regularly compare adverse event rates among sites to detect early any outliers and apply corrective measures promptly. Evaluating the occurrence of standard risks is a part of routine monitoring activities and additional efforts to mitigate risk are typically unnecessary. As a result, one critical simplification was to only document risks management components (e.g., mitigation plans) on critical and heightened risks that remain after risks are eliminated from the protocol. Study key risks, (combination of heightened and critical risks) are often times study specific risks where, existing standard processes are generally not sufficient to prevent and/or control risk and additional activities need to be developed and put in place to ensure that those risks do not occur or, that they do not occur beyond an acceptable frequency. The identification of a critical risk leads to the need to implement robust risk controlling activities, close monitoring, and a set a quality tolerance limits (QTL). A QTL is a breaking point limit that, when exceeded, may expose study subjects to unreasonable safety risks or critical study data may not be reliable.
This “Simple as 3, 3, 3” approach was used to drive the development and adoption of the RARM tool. To simplify and increase the likelihood of implementation, the RARM tool was built in one system instead of separating risk assessment and risk management documentation. This was achieved by having both risk assessment elements (e.g., what was done to de-risk the protocol) and risk management elements (e.g., likelihood of occurrence and contingency plans) captured within the same tool.
2.3. RARM implementation
Once the tool was developed and pilot tested in eight studies (seven randomized controlled trials and one observational study), it was implemented in SharePoint®. SharePoint® allowed all team members to access and contribute to the RARM tool. An organizational change management plan was created to address implementation of the RARM tool DCC-wide. This involved identifying an implementation team and Change Champions, providing proactive communications, determining the commitment needed and addressing anticipated barriers identified by each stakeholder group (e.g., project managers, leadership), integrating the new tool with IT processes, developing resource materials, and planning training. We conducted several 2-h training sessions for all impacted DCC members. Training included a demonstration of the SharePoint® tool, details of how to manage data entry, and how to change user permissions. We created an interactive quiz to be administered during the training in order to measure knowledge assessment by attendees.
A survey was sent after the training sessions to gain additional feedback on the training from attendees. Study project managers were responsible for ensuring the implementation of the RARM tool within individual projects, although all team members were expected to contribute. A Standard Operating Procedure was produced outlining the requirements for completing the RARM tool within studies, and a working guideline document was created that outlined steps and provided instructions for completion. Quality leadership was available to study teams to assist in completing the RARM or functional group meetings to answer RARM questions.
3. Results
3.1. RARM tool
The RARM tool consists of up to 20 data elements for each risk (9 for risk assessment; 11 for risk management). Each element of the tool has a label (the data element), a field type, and a description for how to complete the field (Table 1). Fig. 1 shows a visual representation of the RARM tool in SharePoint®. Several aspects of the RACT were adopted in the RARM, but some items are discussed below.
Table 1.
Risk assessment and risk management (RARM) tool.
| Label | Field Type | Description |
|---|---|---|
| Risk Short Name | Free text | Enter a short name that best captures the risk for quick reference. |
| Research Component | Choice option: 01. Regulatory Profile 02. Study Design 03. Study Population 04. Recruitment Environment/Setting (e.g., hospital, clinic, school, home, jail, etc.) 05. Eligibility Criteria 06. Screening and Enrollment 07. Randomization 08. Interventions and/or Procedures 09. Investigational Product 10. Endpoint(s) 11. Data Analysis 12. Data Management 13. Protection of Human Subjects 14. Study Training, Personnel Experience and Expertise 15. Other |
Choose ONE answer from the drop-down menu. Risks will often overlap into multiple research components; however, report the identified risk only once and select the most appropriate research component. |
| Risk Description | Free text | Concisely describe the heightened or critical risk and its potential impact. |
| Initial Risk Category | Choice option • Critical • Heightened |
Choose ONE answer from the drop-down menu. Risks listed here must be either heightened or critical. Do not enter standard risks. If uncertain whether the risk is standard or heightened, it is best to be conservative and to initially report the risk as heightened. The team member who initially identifies/captures the risk, determines the risk category. Subsequently, expert key team members decide on the best protocol de-risking approach for the risk identified and determine the final risk category. |
| De-Risking Decision | Choice option • Eliminate • Reduce • Accept • Risk not H/C |
Choose ONE answer from the drop-down menu. Expert key team members decide on the best protocol de-risking approach for the risk identified. • Eliminate (preferred option when possible): Process/data deleted from the protocol. • Reduce: Process/data modified in the protocol to reduce risk occurrence. • Accept: The protocol was not changed and the identified risk remains unchanged. • Risk not H/C: Risk downgraded from heightened or critical to standard. |
| De-Risking Summary | Free text | Briefly summarize significant team decisions and/or edits to the protocol/amendment. If the risk was accepted, briefly summarize the rational for this decision. |
| Final Risk Category | Choice option • Critical • Heightened • Standard • Risk Eliminated |
Choose ONE answer from the drop-down menu. When a H/C risk has either been reduced or downgraded from H/C to STD then, the appropriate choice from the drop-down menu is “Standard”. When a H/C risk has been eliminated (deleted from the protocol) then, the appropriate choice from the drop-down menu is “Risk Eliminated”. |
| Study Conduct Status | Choice option |
Choose ONE answer from the drop-down menu. |
| • Pre Study Conduct • During Study Conduct |
• Pre-Study Conduct: Choose this option when the risk is identified before study conduct has begun, i.e., during the study pre-enrollment period up until when the first subject is enrolled or the first chart review is initiated for registry studies. • During Study Conduct: Always choose this option when the risk is identified any time after the first subject was enrolled in the study or the first chart review was initiated. |
|
| Risk Status | Choice option |
Choose ONE answer from the drop-down menu. |
| • RM • F/Up • END |
• RM (risk management): The final risk category is HEIGHTENED or CRITICAL and the risk must be further assessed and managed during study conduct. Complete the rest of the questions below. • F/Up (follow-up): The final risk category has not yet been determined and requires team decision follow-up. Once the final risk category is determined, the risk status will need to be changed to RM or END. Skip all remaining questions below. • END: The final risk category is STANDARD or RISK or ELIMINATED |
|
| Likelihood | Choice option • 1 Improbable • 2 Rare • 3 Probable • 4 Very Probable • 5 Frequent |
Choose ONE likelihood score from the drop-down menu. The likelihood of errors occurring (risk occurrence) during the conduct of the trial. |
| Impact | Choice option • 1 Low • 2 Low-moderate • 3 Moderate • 4 Moderate-high • 5 High |
Choose ONE impact score from the drop-down menu. The impact of such errors (risk occurrence) on human subject protection (safety, confidentiality, rights), reliability of trial results (e.g., data integrity of the primary and secondary outcome or regulatory compliance), or other components (costs, timelines, reputation of the institution, etc.). |
| Detectability | Choice option • 5 Not at All Detectable • 4 Slightly Detectable • 3 Moderately Detectable • 2 Very Detectable • 1 Highly Detectable |
Choose ONE detectability score from the drop-down menu. The extent to which such errors (risk occurrence) would be detectable by the Study Team in light of planned study management activities. |
| Risk Score | Automatically derived | The risk score will be calculated automatically based on the likelihood, impact, and detectability scores. |
| Key Risk Indicator | Free text | Enter the KRI defined for the risk. KRI: A metric used to monitor risk exposure over time of H/C risks, (e.g., measurement of a specific data element over a meaningful data element total, over time) Ex.: KRI = #LTFU/Total Enrolled (Key risks are heightened and critical risks.) When a KRI cannot be defined, describe how the risk will be monitored during study conduct. |
| Mitigation Plan | Free text | Succinctly list all risk mitigating activities. The mitigation plan includes all activities done to prevent the identified risk from occurring; it’s the Plan A. |
| Threshold(s) & QTL | Free text | Enter the threshold value(s) and, for critical risks only, the QTL value. Ex.: T1 = 15%LTFU, T2 = 20%LTFU, T3 = 25%, QTL = 30% LTFU Threshold = predefined value of the KRI that triggers contingency plan activity(ies) to reduce/control further risk occurrence; one can define multiple escalating threshold values with corresponding escalating risk control activities (T1, T2, T3, etc.) QTL = also a predefined value of the KRI measured at the study level that triggers contingency plan activity(ies) to reduce/control further risk occurrence; however, there is only ONE quality tolerance limit (QTL) per risk and QTLs apply to critical risks only. A QTL is basically a threshold upper limit, above which a critical process or data is outside of quality. Reaching a QTL triggers a QTL Contingency Plan. |
| Threshold(s) Contingency Plan(s) |
Free text | Succinctly list all risk reducing/controlling activities. The contingency plan includes all activities done to reduce/control further occurrence of the risk; it’s the Plan B. The contingency plan includes actions that are curative when Plan A (the mitigation plan) failed, i.e., the mitigation plan was insufficient to control risk occurrence and, as such, additional actions are required to avoid and/or reduce further risk occurrence. Contingency plans may be pre-defined or developed as new data or knowledge is available. As such, the activities included here can be updated during the conduct of the trial. |
| QTL Contingency Plan (s) | Free text | QTL contingency plan(s) apply to critical risks only. Leave blank for heightened risks or enter N/A or Not applicable, if preferred. When a QTL is reached, the first step is always to assess data to verify that a systemic issue is present, as critical risks and QTLs apply only to systemic issues. The QTL contingency plan(s) should be predefined but new plan(s) can be defined as new data is available. Examples of contingency plans include: - Redefine QTL (justification must be documented) - Protocol Amendment, e.g., change study outcome, study power, etc. - Stop the study if subject protection is jeopardized beyond acceptable levels - Stop the study if the reliability of trial results is compromised beyond an interpretable level, and no other trial conclusions or tendencies would be possible |
| Responsible Party | Free text | Enter the name of the team member who is responsible for ensuring management of the risk during study conduct. The responsible party can delegate certain activities that support the overall management of the risk but remains the responsible and accountable point person for the particular risk, informing team members of risk realization and risk control activities during study conduct. |
| Measured in DB? | YES | Choose YES or NO from the drop-down menu. |
| NO | The question asks if the data elements in both the numerator and denominator of the KRI are included in the study database (DB). If “NO”, the Study Team may recommend that the data elements be included in the DB. Alternatively, the Study Team will need to define how the KRI will be monitored during study conduct. When a KRI cannot be defined, the correct answer is also “NO”. The Study Team will need to determine how the risk will be monitored during study conduct. |
H/C: Heightened/Critical; STD: Standard; QTL: Quality Tolerance Limit.
Fig. 1.

Visual representation of the Risk Assessment and Risk Management (RARM) Tool in SharePoint® (first four data elements only).
The research component categorizes risk into common buckets that can be impacted through research. This categorization also allows for the identification of common areas of risk across multiple studies. Additional resources can be invested into de-risking common protocol components across like studies. After initial risk categorization into heightened or critical, study teams and sponsors can evaluate potential protocol de-risking options (Simple as 3,3,3’ de-risking options for the protocol: eliminate, reduce, accept). The protocol can be altered to reduce or eliminate the risk. If the risk is accepted and the protocol does not change, then the risk can be monitored throughout enrollment and follow-up. After further discussion with the study team and sponsor, the risk might be relabeled as a standard risk. Labeling the risk as not heightened or critical, and keeping it within the RARM tool, allows for the team to more easily tag items for future discussion, if needed.
The final risk category has the three risk levels for study teams to choose among critical, heightened and standard levels. After the risk assessment section is completed, the study team indicates when the risk was identified (pre-study conduct or during study conduct). This helps quantify how many risks were reduced or eliminated proactively as a result of risk assessment during the early study development phase (i.e., after study funding, preferably before finalizing and submitting the protocol to the IRB, but prior to first patient in).
Each risk has a metric (key risk indicator – KRI) that is used to quantify the risk throughout enrollment. The thresholds and QTL outline various levels of unacceptable risk concern, while the contingency plans describe the specific mitigating actions that the study team will implement based on respective threshold reached. This reduces iterative conversations when observing a risk mid-study and reduces unnecessary delays in responses. Thresholds are chosen based on collaborative input between the study investigators and the study team (includes the lead statistician). These thresholds may be statistically, clinically, or safety driven depending on the risk. Even though the RARM tool is completed during study startup, it is intended to be a living document that should be revisited throughout the trial as new information is learned about the identified risks and new risks are discovered.
3.2. Example risk
We provide two hypothetical examples which demonstrate how the RARM tool can be used to de-risk studies during the protocol development phase as well as monitor risk throughout a clinical trial. In this first example (Table 2), the primary study outcome was a change in an individual’s gait assessment quantifying how much faster a person could walk compared to baseline after receiving one of two types of surgery. After the surgery, the individual was supposed to use any walking assistance that was used at baseline (e.g., a cane). During site qualification, it became apparent that standard-of-care gait assessments at participating sites varied substantially from the gait assessment outlined in the protocol. To reduce the risk of the variability in the primary outcome assessments across sites, the protocol was revised and the sites were required to video record each gait assessment. A quality reviewer viewed all videos to assure sites adhered to the gait standards outlined in the protocol and to verify the timed gait assessment values, eliminating the risk of error to the primary outcome.
Table 2.
Example gait assessment risk.
| Label | Value |
|---|---|
| Risk Short Name | Inaccurate gait assessment |
| Research Component | 10. Endpoint(s) |
| Risk Description | There is variability in the way sites perform gait assessments as standard of care. This could lead to a different quantification of the primary outcome. |
| Initial Risk Category | Critical |
| De-Risking Decision | Eliminate |
| De-Risking Summary | The protocol was updated to require a video of the primary outcome be captured. This allows for an independent reviewer to confirm primary outcome calculation |
| Final Risk Category | Standard |
| Study Conduct Status | Pre-study Conduct |
| Risk Status | End |
In this second risk example (Table 3), a secondary outcome was being collected from the subjects at 6 months. The protocol allowed for a +/− 2-week window to conduct the visit. The study team categorized this visit as a heightened risk because it did not affect the primary outcome or patient safety. After consultation with the investigators, the study team accepted this as a risk and determined the follow-up rate needed to be properly monitored. After internal discussions and consultation with the investigative team, the team developed a likelihood, impact, and detectability score. Based on the importance of follow-up, the study team decided to be more conservative in the definition of the KRI and flag a subject as potential lost to follow-up as soon as the follow-up window begins (instead of waiting until the end of the follow-up window when it is too late). This allows the study team to act on low follow-up rates while subjects are still in the follow-up windows and preempt the issue. Follow-up rates can be calculated and monitored based on data in the database.
Table 3.
Example lost-to-follow-up risk.
| Label | Value |
|---|---|
| Risk Short Name | Lost-to-follow-up |
| Research Component | 10. Endpoint(s) |
| Risk Description | The secondary endpoint is being collected at 6 months. A high proportion of subjects lost-to-follow-up will limit data available for this analysis. |
| Initial Risk Category | Heightened |
| De-Risking Decision | Accept |
| De-Risking Summary | |
| Final Risk Category | Heightened |
| Study Conduct Status | Pre-study Conduct |
| Risk Status | RM |
| Likelihood | 3 Probable |
| Impact | 5 High |
| Detectability | 1 Highly Detectable |
| Risk Score | 15 |
| Key Risk Indicator | KRI: # Completed / # Expected A subject will count in the numerator whenever a follow-up is completed. A subject will count in the denominator (# Expected) as soon as he/she enters the follow-up window (6 months – 2 weeks). Logic: By counting subjects as expected as soon as they enter the window, our follow-up rate will be biased lower. We will take a more conservative approach to follow-up rate monitoring to ensure an adequate rate upon completion. The contingency plans for this risk will not be enacted until we have at least 30 subjects who have entered the follow-up window. |
| Mitigation Plan | Follow-up rates will be presented on every research coordinator and all-site call. In addition, we will create a report that shows site when a subject is expected to fall in the window. An incentive has been incorporated in the protocol for the subject to encourage follow-up completion. We will also send a text message the day prior to the scheduled follow-up to remind the subject of the phone call. |
| Threshold(s) & QTL | The following threshold categories have been identified: • Green: [0.9, 1.0] • Yellow: [0.8, 0.9) • Red: [0, 0.8) |
| Threshold(s) Contingency Plan(s) | Yellow: A report showing follow-up rates by site will be reviewed. Sites with follow-up rates that individually fall in the Red threshold region will have retraining provided. A detailed report will be created for the site that summarizes characteristics of subjects who do not complete the follow-up visit. Red: Sites with follow-up rates falling in the Red threshold region will be put on probationary with 6 months given to increase their follow-up rates. At the end of the 6-month probationary period, if the site is in the red, the site will be dropped and an additional site will be added. Additionally, we will increase incentive amounts for subjects to complete the 6-month follow-up across the study. |
| QTL Contingency Plan (s) | |
| Responsible Party | Statistician |
| Measured in DB? | Yes |
3.3. Monitoring risks
Once the RARM tool is completed, the risks are monitored through a report that is automatically updated based on data entered in the database. An example of a report with three risks is shown in Fig. 2. A short description of the risk is included in the first column; the trend of the summarized key risk indicator based on the data are shown in the second column, and the current value of the key risk indicator is shown in the third column. To simplify visualization and draw attention to data that stands out, color codes (i.e., green, yellow, orange, red) are utilized in the reports to more easily monitor risk realization. Each color corresponds with the pre-identified thresholds and QTLs documented in the RARM tool. For example, the green zone indicates that no action is needed because a risk threshold has not been met. However, the yellow zone would indicate that the first threshold has been met and additional action should be taken to mitigate further realization of a particular risk. Contingency plans would be enacted when a risk enters non-green regions. These reports provide the structure to have conversations with the investigative team and sites.
Fig. 2.
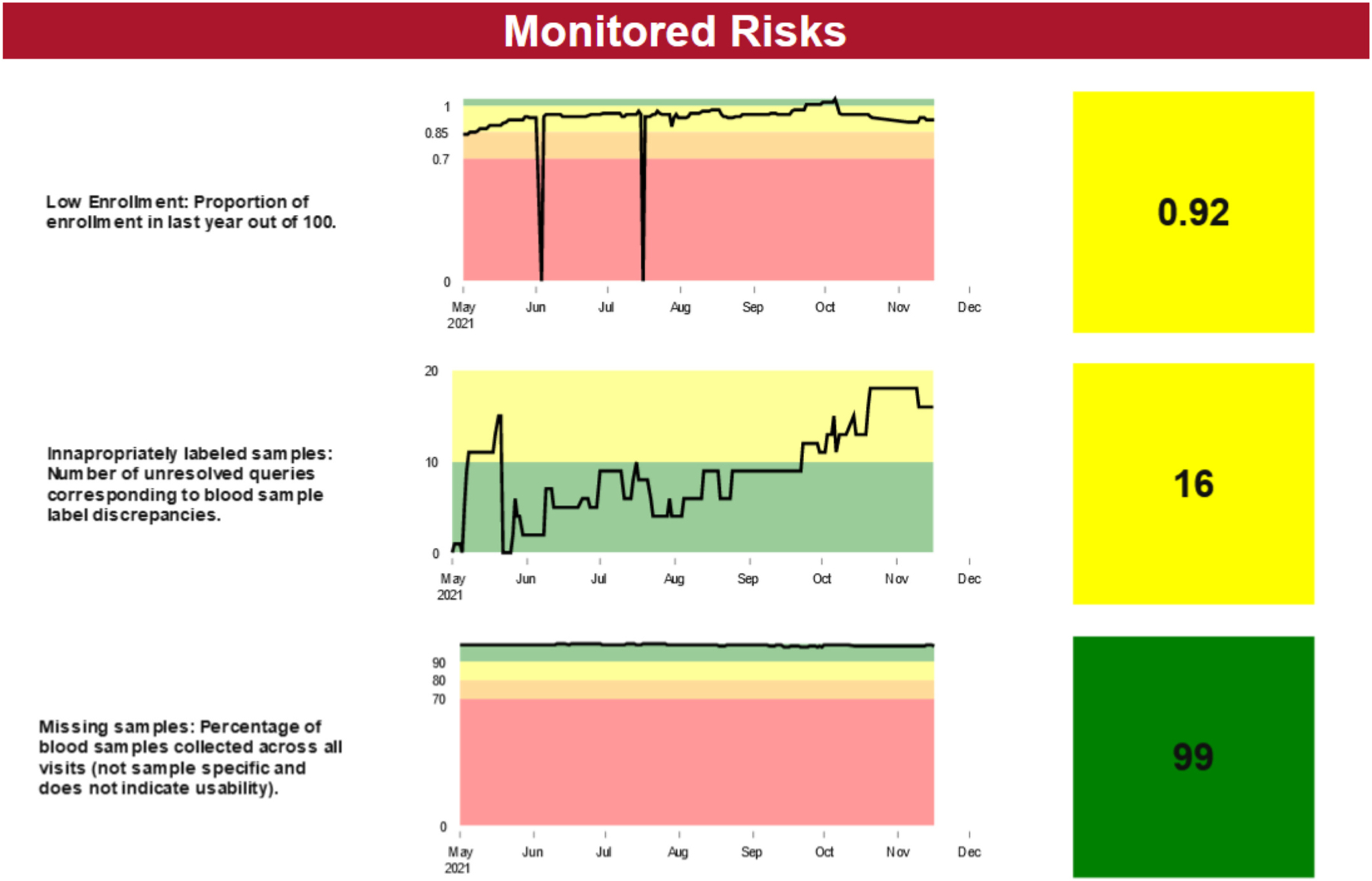
Example RARM monitoring report for three identified study risks.
4. Conclusion
We designed a RARM tool to meet the needs of a data coordinating center within an academic research organization. The tool was developed through an iterative process of pilot testing and revising to achieve a simplified, seamless approach to risk assessment and risk management. There are several valuable items and concepts the RARM tool adds to the risk assessment and risk management literature. It incorporates the concept of Quality by Design [14] into daily work by having every team member contribute to the RARM tool. It also combines the risk assessment and risk management processes into a single tool which allows for a seamless transition from identifying risks to actively thinking about and managing the risks throughout the life of the study. The RARM tool has guidelines that allow end users to implement the tool regardless of past quality experience. The tool also focuses on the concept of de-risking protocols early in development by editing the protocol to reduce or eliminate potential risks. It can be implemented in any platform (e.g. Excel, SharePoint®), allowing organizations with limited IT infrastructure to utilize it. It also is applicable to all study types (e.g., registries, observational studies, interventional trials). The tool follows the regulatory guidelines by having study teams focus on heightened and critical risks to subject protection, data reliability, and operations (heightened risks only). This increases the likelihood of achieving study objectives by focusing the study team on key items compared to being diluted with standard risks.
Our tool has several elements similar to the RACT: it categorizes risk components, the impact should the event occur, probability, detectability, risk score calculation, and mitigation plans. The RARM tool aligns with Suprin et al. comments of creating a fit-for-purpose quality management system for risk assessment and risk management [10]. It has similar concepts to Xcellerate that allows for documenting how risks can be monitored after risks are identified [11]. We embodied Quality by Design principles outlined by Landray et al. [15] and our system coincides with the AVOCA process of protocol de-risking documentation [16].
We differ from the RACT tool in that we do not document standard risks due to the routine monitoring and reporting processes that are in place within the DCC and at sites. We also do not complete risk management components (e.g., developing contingency plans) for risks that are standard or are eliminated from the protocol. This approach allows study teams to focus on key risks instead of monitoring all possible risks through the RARM. Key risks can then be monitored throughout enrollment and contingency plans can be implemented when preestablished thresholds are met. Our tool can be completed by most staff members without the need for a designated quality team. Completion of the RARM tool as a team allows for consensus on risk categorization which is a limitation of the RACT [9]. We also created a seamless pathway for our teams from risk identification to mitigation. This is important because it simplifies the process and allows study team members to conceptually transition from study startup activities to ongoing monitoring activities throughout enrollment.
While the RARM tool worked well within the studies piloted testing within an DCC, there are several limitations of the tool that were identified following implementation. Once risks are identified, the study team is asked to develop key risk indicators, thresholds, and contingency plans prior to fully experiencing the implications of the risk (i.e., pre-study conduct). This process can be difficult to conceptualize what thresholds are appropriate and what contingency plans should be implemented at their respective thresholds. We can envision a situation where appropriate thresholds and contingency plans become clearer once enrollment has begun and the study team has a better understanding of study implementation. However, because this tool is intended to be a living document, these thresholds and contingency plans can and should be revisited once enrollment begins. We have found study team members have a difficult time deriving key risk indicators. This usually involves mathematical thinking about risk and how to quantify its occurrence. For example, if there is a window allowed for follow-up, the study team needs to evaluate when someone should count as having an expected follow-up when calculating follow-up rates. The team could count a visit as expected as soon as a subject enters the beginning of the follow-up window, recognizing the visit likely has not occurred yet. On the contrary, they could count a subject as expected as soon as the follow-up window has passed, but this could lead to biased, inaccurately inflated follow-up rates. This exemplifies how quantifying a key risk indicator could be conceptually difficult for study team members. SharePoint® has additional limitations such as limited programming options to prevent risk management elements from being completed when a risk was eliminated (i.e., if you eliminate the risk through risk assessment, the SharePoint® list still has the option for filling out risk management components). In addition, SharePoint® lists cannot link with actual study data to incorporate risk management with the actual monitoring reports within a single application. If the study team does not document decisions within SharePoint®, then the audit trail for actions taken when contingency plans are enacted are not automatically saved.
Our DCC produced a risk assessment and risk management tool that was more easily accommodated and accepted into the daily activities of our academic research organization. It is accessible to all study members and links to the reports can be included in regular team meetings. Although, adoption of the tool was slow, we have observed widespread acceptance and understanding of risk concepts as a result of the RARM. Study teams with minimal quality training are now able to utilize the tool to not only identify risks but to also determine strategies to reduce or control risks. Our system was easily built in SharePoint®, an accessible platform for all DCC staff, but is extendable to other platforms. Future goals include the creation of a library common heightened and critical risks and have RARM reports that are viewable across multiple studies at once. Our study reports are now more informative as they provide a pulse on subject protection and reliability of study data by monitoring risk occurrence of those study-specific key risks identified through the RARM tool. Furthermore, our risk-based monitoring strategy continues to evolve with the knowledge ensuing from study risks assessment. Study teams now have a common language with which to identify risks, and teams are able to clearly describe study risks to investigators and sites. This results in risk metrics that can be assessed across sites which produces higher quality studies.
Acknowledgements
The authors would like to thank Dr. Erin Rothwell, Dr. Nael Abdelsamad, Ms. Jeri Burr, University of Utah, and Dr. Jerry M. Stein, Summer Creek Consulting, for writing assistance.
Funding sources and support
Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health [Award Number U24TR001597]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
CRediT authorship contribution statement
John M. VanBuren: Conceptualization, Methodology, Writing – original draft. Shelly Roalstad: Methodology, Writing – review & editing. Marie Kay: Methodology, Writing – review & editing. Sally Jo Zuspan: Methodology, Writing – review & editing. J. Michael Dean: Writing – review & editing, Supervision, Funding acquisition. Maryse Brulotte: Conceptualization, Methodology, Writing – review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Robb MN, David McKeever Paula, Krame Judith M., The clinical trials transformation initiative (CTTI), Monitor 23 (7) (2009) 73–75. [Google Scholar]
- [2].Position Paper, Risk-Based Monitoring Methodology, 2013.
- [3].Reflection Paper on Risk Based Quality Management in Clinical Trials, European Medicines Agency, 2013. [Google Scholar]
- [4].Oversight of Clinical Investigations — A Risk-Based Approach to Monitoring, Department of Health and Human Services Food and Drug Administration, 2013. [Google Scholar]
- [5].Integrated Addendum to ICH E6(R1), Guideline for Good Clinical Practice, ICH Harmonised Guideline, 2016.
- [6].ICH-E6(R3) Good Clinical Practice (GCP) Draft Principles, ICH Harmonised Guideline, 2021.
- [7].See TransCelerate’s RACT Tool, available at, https://www.transceleratebiopharmainc.com/assets/risk-based-monitoring-solutions/, 2022.
- [8].Johanna Schenk AA, Proupín-Pérez María Koch Martin, Fehlinger Matthias, The PUEKS Project: Process Innovation in Clinical Trial Monitoring, Applied Clinical Trials, 2017.
- [9].Moe Alsumidaie PS, Widler Beat, Andrianov Artem, Data from Global RACT Analysis Reveals Subjectivity, Applied Clinical Trials, 2016.
- [10].Suprin M, Chow A, Pillwein M, Rowe J, Ryan M, Rygiel-Zbikowska B, et al. , Quality risk management framework: guidance for successful implementation of risk Management in Clinical Development, Ther. Innov. Regul. Sci 53 (1) (2019) 36–44. [DOI] [PubMed] [Google Scholar]
- [11].Ciervo J, Shen SC, Stallcup K, Thomas A, Farnum MA, Lobanov VS, et al. , A new risk and issue management system to improve productivity, quality, and compliance in clinical trials, JAMIA Open 2 (2) (2019) 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].2020 Avoca Quality and Innovation Summit 2020, Virtual, October 14–15, 2020.
- [13].MAGI’s Clinical Research vConference, Spring; 2021, 2021 April 26–29 6. [Google Scholar]
- [14].Juran JM, Juran on Quality by Design: The New Steps for Planning Quality into Goods and Services, Maxwell Macmillan International, New York, 1992. [Google Scholar]
- [15].Landray MJ, Grandinetti C, Kramer JM, Morrison BW, Ball L, Sherman RE, Clinical trials: rethinking how we ensure quality, Drug Inform. J 46 (6) (2012) 657–660. [Google Scholar]
- [16].AVOCA, Avoca Quality Consortium: Protocol De-Risking Checklist, 2020.