

Abstract
MICU1 is a Ca2+-binding protein that inhibits the mitochondrial Ca2+ uniporter channel complex (mtCU) and mitochondrial Ca2+ uptake. MICU1 knockout mice display disorganized mitochondrial architecture, a phenotype that is distinct from that of mice with deficiencies in other mtCU subunits and thus is likely not explained by changes in mitochondrial matrix Ca2+ content. Utilizing proteomic techniques and cell lines, we found that MICU1 localized to the mitochondrial contact site and cristae organizing system (MICOS) and directly interacted with the MICOS components MIC60 and CHCHD2 independently of the mtCU. We demonstrated that MICU1 was essential for proper MICOS complex formation and that MICU1 ablation resulted in altered cristae organization, mitochondrial ultrastructure, mitochondrial membrane dynamics and cell death signaling. Together, our results suggest that MICU1 is an intermembrane space Ca2+ sensor that modulates mitochondrial membrane dynamics independently of matrix Ca2+ content. This system enables Ca2+ signaling in the mitochondrial matrix and at the intermembrane space to modulate cellular energetics and cell death in a concerted manner.
Introduction
Calcium (Ca2+) is an essential second messenger that regulates numerous cellular functions by binding to distinct Ca2+-sensing domains or motifs present on numerous proteins (1–3). Most Ca2+ sensors contain more than one Ca2+ binding domain, often with varied binding affinities, resulting in diverse and graded regulation of numerous cellular processes (1–4). The Ca2+ concentration varies greatly between different cellular compartments, and Ca2+ sensors are strategically localized for subcellular or organelle specific signaling (1, 5, 6). Mitochondria actively regulate their Ca2+ concentration by a tightly controlled exchange system and contain Ca2+ sensors to mediate anterograde and retrograde signaling (1, 5). Examples include mitochondrial Rho GTPases (MIROs) localized to the outer mitochondrial membrane (OMM) and mitochondrial Ca2+ uptake proteins (MICUs) localized to the intermembrane space (IMS) side of the IMM (1, 7–9). MIRO Ca2+ sensing is essential for mitochondrial trafficking and structural homeostasis (1, 7, 10–12), whereas MICUs gate the mitochondrial Ca2+ uniporter channel complex (mtCU) and regulate open probability (9, 13–16).
The mtCU is a highly selective Ca2+ channel necessary for acute Ca2+ uptake into the mitochondrial matrix (5, 17–20). The mtCU consists of multiple subunits, including the pore-forming components, mitochondrial Ca2+ uniporter (MCU) and its homolog MCUB; the regulatory scaffolds, MCU regulator 1 (MCUR1) and essential MCU regulator (EMRE); and the Ca2+ sensors, mitochondrial Ca2+ uptake proteins 1, 2, and 3 (MICU1, MICU2 and MICU3) (8, 9, 17, 18, 21–25). MICU1 regulates mtCU activity by directly binding to MCU and EMRE, and its expression correlates with tissue-dependent differences in mitochondrial Ca2+ uptake (8, 9, 13, 14, 16, 23, 26–28).
Loss-of-function mutations in MICU1 induce proximal myopathy, learning difficulties, movement disorder, fatigue, and lethargy in humans (29, 30) and deletion of Micu1 in mouse models causes perinatal lethality (15, 31). In Drosophila, a MICU1 loss-of-function mutation results in lethality, which cannot be rescued by a concurrent MCU loss-of-function mutation that completely ablates mitochondrial Ca2+ (mCa2+) uptake and subsequent mitochondrial permeability transition (32). This observation suggests that the lethal phenotype of MICU1-null flies is not due to aberrant mtCU-dependent Ca2+ uptake or matrix Ca2+ overload. These findings indicate that MICU1 has mtCU-independent functions, which are vital for mitochondrial function and survival. Indeed, MICU1 knockout models show distinct abnormalities in mitochondrial ultrastructure that are not observed when other components of the mtCU are deleted (15, 20, 24, 33). Additionally, MICU1 is highly mobile within the IMM as compared to the MCU (34), suggesting that MICU1 could be associated with other complexes in the mitochondria. These observations led us to hypothesize that MICU1 regulates other essential mitochondrial processes beyond Ca2+ uptake.
To discover mtCU-independent functions of the MICU1, we utilized a proximity-based biotinylation approach by constructing a MICU1-BioID2 fusion protein. BioID2 is a highly efficient promiscuous biotin ligase which enables the detection of protein-protein interactions in living cells (35). We reconstituted MICU1−/− HEK293T cells with MICU1-BioID2-HA to identify the MICU1 interactome. We also expressed MICU1-BioID2 in MCU−/− HEK293T cells to define mtCU-independent MICU1 interactions. By comparing mass spectrometry analyses from these cell systems, we identified proteins whose interaction with MICU1 was unaffected by the loss of the mtCU complex. Here, we report that MICU1 directly interacted with the mitochondrial contact site and cristae organizing system (MICOS) components MIC60 and CHCHD2 in an MCU-independent manner. Our results suggest that MICU1 confers Ca2+ sensing to the MICOS for cell signaling-dependent changes in cristae structure and function.
Results
MICU1 localization can be independent of the mtCU
To define the mtCU-independent molecular functions of the MICU1, we utilized size-exclusion chromatography to characterize the native organization of MICU1-containing protein complexes. Total cell lysates prepared from WT and MCU−/− HEK293T cells were fractionated under non-reducing conditions by fast protein liquid chromatography (FPLC) and immunoblotted for MICU1 protein (Fig. 1A, 1B). MICU1 formed distinct high-molecular weight (MW) protein complexes ranging from ~200-kD to ~700-kD (Fig. 1A, 1B). The loss of MCU did not have a substantial effect on the overall distribution of MICU1-containing high-molecular weight (MW) protein complexes (Fig. 1A, 1B). To corroborate this result, we also performed blue-native PAGE assays after crosslinking of mitochondrial native protein complexes with succinimidyl 6-beta-maleimidopropionamido hexanoate (SMPH) in WT and MCU−/− HEK293T cells. Immunoblotting of crosslinked samples showed the presence of high-molecular weight (~1200 kD) native protein complexes containing MICU1, which were unaltered by the loss of MCU (Figure S1A). Next, we examined sub-mitochondrial localization of native MCU and FLAG-tagged MICU1 by immunofluorescent detection in Micu1−/− mouse embryonic fibroblasts (MEFs) (Fig. 1C). The deletion of MICU1 in MEFs was confirmed by Western blotting (fig. S1B). Line-scan analysis of the mitochondrial network showed that MICU1 colocalized with MCU but also distributed to sub-mitochondrial regions lacking MCU (Fig. 1C, 1D). These results suggest that MICU1 is present in mitochondrial protein complexes that lack the mtCU.
Fig. 1. MICU1 is found in mitochondrial multimeric protein complexes that do not contain MCU subunits.
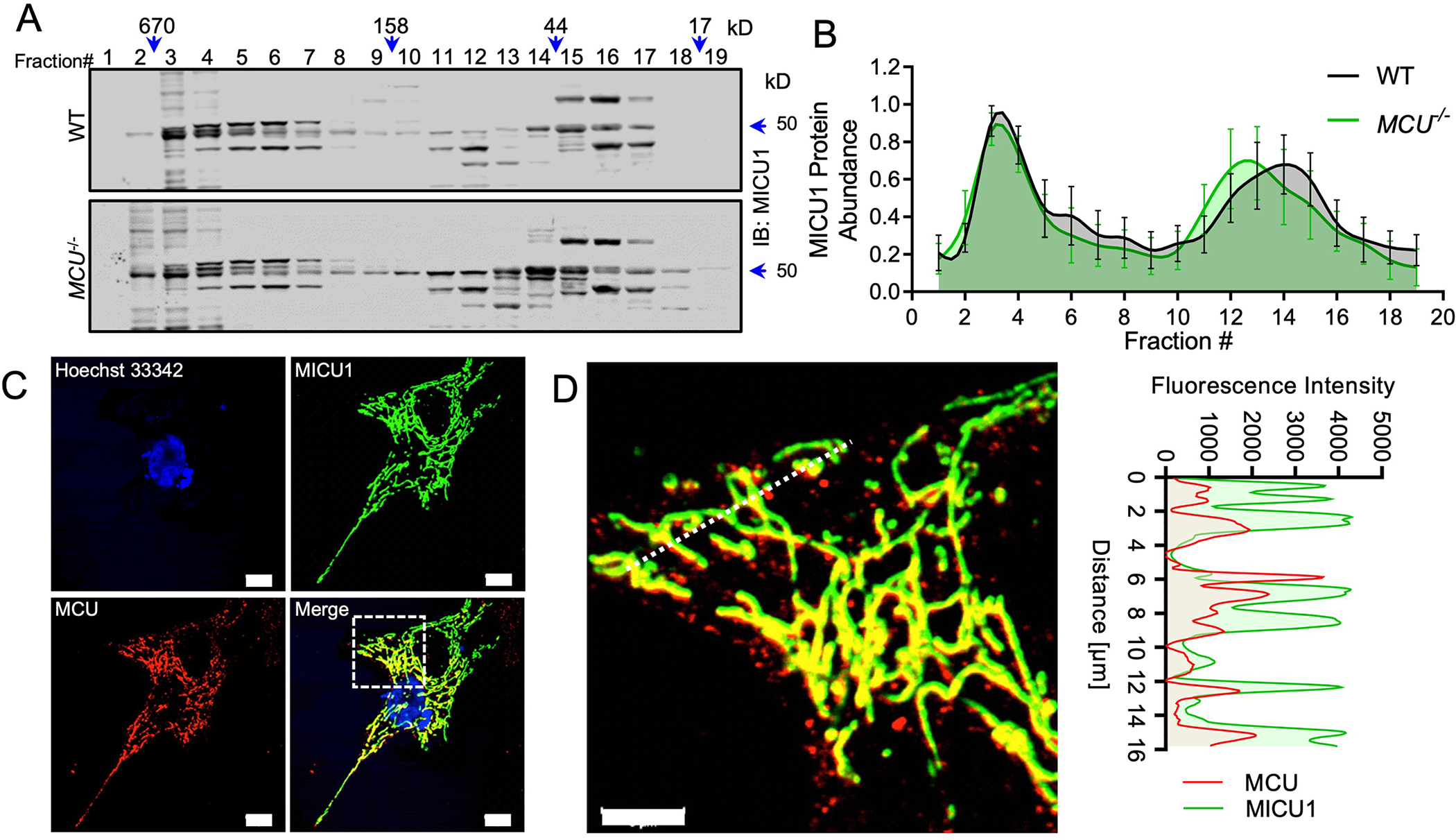
(A) Cell lysates isolated from WT and MCU−/− HEK293T cells were fractionated by FPLC size-exclusion chromatography. Protein fractions ranging from ~10kD to ~900kD were collected, concentrated, and subjected to immunoblotting for MICU1. n= 4 independent experiments. (B) Densitometry was performed to quantify MICU1 abundance in the fractions shown in Fig. 1A. Error bar= SEM. p > 0.05 in all fractions. n= 4 independent experiments. (C-D) MEFs stably expressing MICU1-FLAG were imaged for FLAG (green) and MCU (red) and a line scan for MCU and MICU1 was performed. Scale bar = 10μm (C) or 5μm (D). Images are representative of 3 independent experiments and 30 images.
Discovery of MICU1 interactors that are independent of the mtCU
Next, we generated a MICU1-BioID2-HA fusion protein to enable the biotinylation of MICU1 interactors (<10-nm) in WT and MCU−/− HEK293T cells to distinguish between the mtCU-dependent and mtCU-independent MICU1 interactions (Fig. 2A). Expression, biotin ligase activity, and sub-mitochondrial localization of the MICU1-BioID2-HA fusion protein and the ability of this protein to regulate mCa2+ uptake were confirmed in HEK293T MICU1−/− cells expressing the MICU1-BioID2-HA fusion protein (Fig. 2B, 2C, fig. S1C). These data confirmed that our fusion construct was properly localized to the IMM and that mtCU-dependent Ca2+ uptake was not altered in our discovery system. The transient expression of the MICU1 fusion proteins used in our study was validated by immunoblotting (fig S1D). Next, we expressed the MICU1-BioID2 or BioID2 control in MICU1−/− (referred to here as MCU+/+ because MICU1 was reconstituted) and MCU−/− HEK293T cells (Fig. 2D). MICU1-BioID2-HA protein expression and biotin ligase activity were confirmed by Western blotting (Fig. 2D). Next, biotinylated proteins were captured from cell lysates with streptavidin-conjugated magnetic beads, trypsinized, and subjected to LC-MS (35). Comparing MICU1 proximal proteins in MCU+/+ and MCU−/− cells identified the MICOS components MIC60, CHCHD3, CHCHD2, APOO, and APOOL as candidate MICU1 interactors, and their proximity to MICU1 was unaltered in MCU−/− cells (Fig. 2E, Data Files S1–S5).
Fig. 2. Identification of mtCU-independent MICU1 interactors.
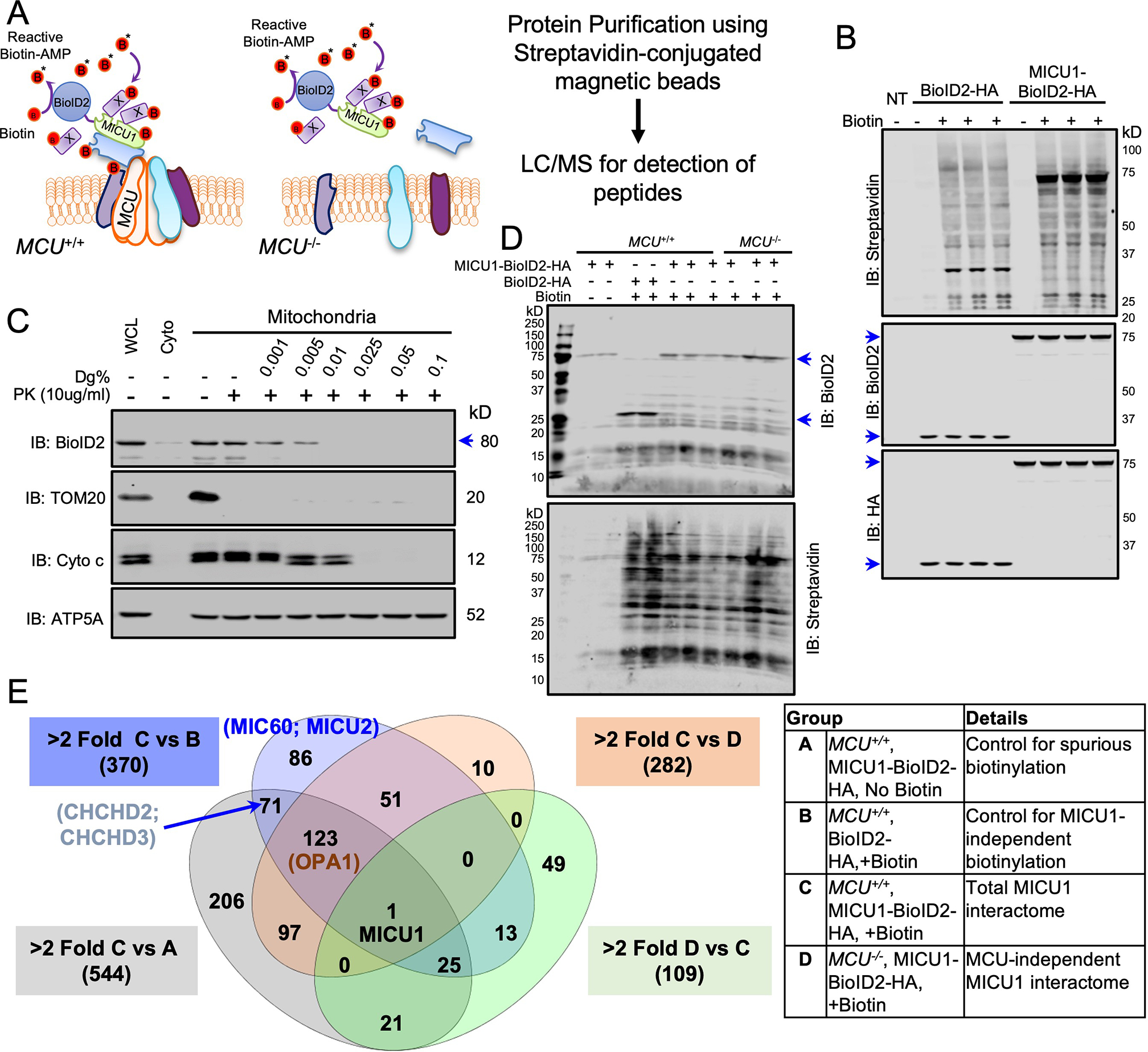
(A) Experimental scheme for identifying mtCU-independent binding partners for MICU1 by biotin-based proximity labeling using the MICU1-BioID2-HA fusion protein. (B) MICU1−/− HEK293T cells expressing BioID2-HA or MICU1-BioID2-HA were cultured with biotin (50μM) for 16h and lysates were Western blotted with the indicated antibodies. n= 2 independent experiments. (C) Mitochondrial fractions from MICU1−/− HEK293T cells reconstituted with MICU1-BioID2-HA were subjected to increasing digitonin (Dg) concentrations to permeabilize the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM). Proteinase K (PK) treatment was performed to cleave exposed proteins, and mitochondrial fractions were probed with the indicated antibodies. Western blots are representative of four independent experiments. (D) MCU−/− HEK293T cells expressing BioID2-HA or MICU1-BioID2-HA or not were cultured with biotin (50μM) for 16h and lysates were Western blotted for BioID2 and streptavidin. Western blots are representative of 2 independent experiments. (E) Streptavidin pull-downs from protein samples from 2–3 biological replicates per group were analyzed by LC-MS/MS. Estimated protein abundance after global sample normalization was used to compare different groups. These interactions are shown in the Venn diagram and the details of the experimental groups are given in the table.
MICU1 directly interacts with MIC60 and CHCHD2 in the MICOS complex
Although OPA1, which is also involved in cristae organization like the MICOS components, emerged as a MICU1 interactor in our proteomic screen, the loss of MCU resulted in loss of the interaction of MICU1 and OPA1, but not that of MICU1 with the core MICOS components (Fig. 2E, Data File S5). This observation suggests that MICU1 could be an integral component of the MICOS complex and involved in mitochondrial cristae organization independently of the mtCU and mCa2+ uptake. We sought to determine whether MICU1 bound to MIC60, CHCHD2, and CHCHD3 with coimmunoprecipitation assays. Only FLAG-tagged MIC60 or FLAG-tagged CHCHD2 were pulled-down with HA-tagged MICU1 (Fig. 3A). Conversely, MICU1-FLAG coimmunoprecipitated with endogenous MIC60 and CHCHD2, but not with CHCHD3 (Fig. 3B). These findings suggest that MICU1 may directly interact with MIC60 and CHCHD2. We also performed immunofluorescence labeling and imaging to examine sub-mitochondrial localization (Fig. 3C–F). The confocal line-scan profile showed distinct pixels with spectral overlap of MICU1 with MIC60 and of MICU1 with CHCHD2 (Fig. 3D, 3F). Together, these data suggest that MICU1 directly interacts with two core MICOS components.
Fig. 3. MICU1 directly interacts with MICOS components.
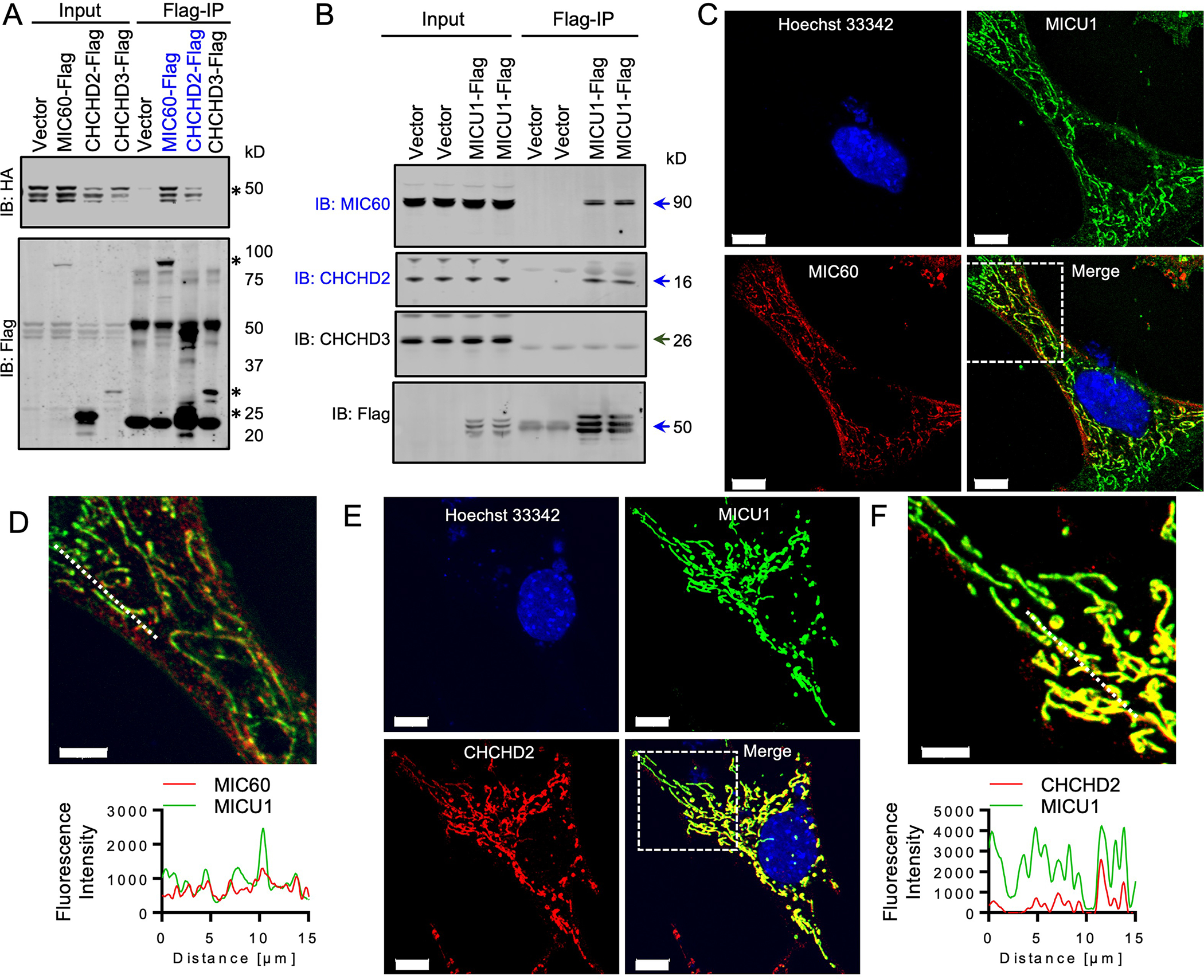
(A) MICU1-HA and FLAG-tagged MICOS components were co-expressed in MICU1−/− HEK293T cells. FLAG-immunoprecipitates (IPs) were probed with FLAG and HA antibodies to detect the interaction between MICU1 and MICOS components. Asterisk (*) indicate the bands for the specific Flag-tagged MICOS components or MICU1-HA. The blue font indicates positive interactions. Western blots are representative of 3 independent experiments. (B) FLAG immunoprecipitates from MICU1−/− HEK293T cells reconstituted with MICU1-FLAG were immunoblotted for endogenous MICOS components. Arrows indicate the specific protein bands. The blue font indicates positive interactions. Western blots are representative of 3 independent experiments. (C-F) MICU1-FLAG expressing MEFs were imaged for FLAG and MIC60 (C, D) or FLAG and CHCHD2 (E, F) and line scans of MICU1, MIC60 and CHCH2 were performed. Images are representative of 3 independent experiments. Scale bar = 10μm in (C) and (E) and 5μm in (D) and (F).
To further characterize the functional relevance of the interaction of MICU1 with MICOS components, we performed FPLC to fractionate the high-MW MICOS complex in WT, MCU−/−, and MICU1−/− HEK293T cells. Fractions showed immunoreactivity for MIC60, CHCHD2 and CHCHD3 in native protein complexes ranging from ~400–700 kD (Fig. 4A–D, fig. S2A, S2B). Genetic deletion of MCU did not affect the overall size or fraction distribution of the multi-subunit MICOS complex (Fig. 4A–D, fig. S2A, S2B). However, the loss of MICU1 resulted in a rightward shift of immunoreactive bands on immunoblots of fractionated cell lysates, indicating a decrease in the overall MW of MIC60-, CHCHD2-, and CHCHD3-containing complexes (Fig. 4A–D, fig. S2A, S2B) and suggesting that MICU1 may play an integral role in MICOS complex assembly or stability.
Fig. 4. MICU1 is essential for the formation of the MICOS complex.
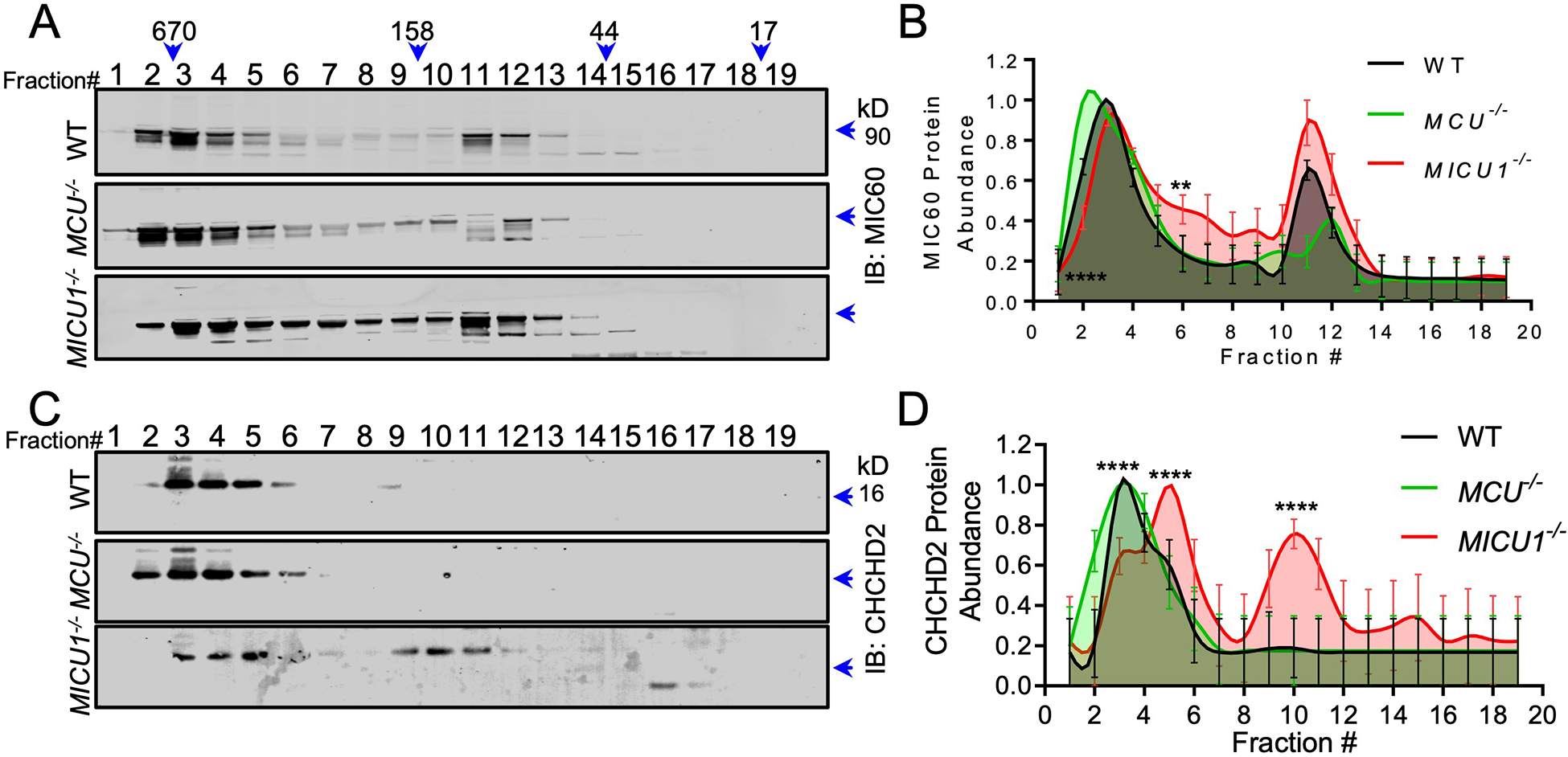
(A, C) FPLC fractions from WT, MCU−/−, and MICU1−/− HEK293T cells were subjected to Western blotting for MIC60 (A), and CHCHD2 (C). Western blots are representative of 4 independent experiments. (B, D) Densitometry was performed to quantify MICOS component distribution in different MW fractions in Fig. 4A and 4B. Error bar= SEM. ****, p<0.0001; **, p<0.01. n= 4 independent experiments.
To find a MICU1 mutant that bound to the mtCU but not the MICOS and vice versa, we mutated critical residues reported to be necessary for MICU1 function and mtCU interaction (28, 34). We expressed seven different loss-of-function point mutants of MICU1 to disrupt MCU and EMRE-binding, Ca2+-sensing, or dimer formation in Micu1−/− MEFs (Fig. 5A) (28, 34, 36–39). Coimmunoprecipitation assays revealed that loss of mtCU-binding, Ca2+-sensing, or dimer-forming ability did not disrupt the interaction of MICU1 with MIC60 (Fig. 5B). These experimental data suggest that MICU1 interacts with the MIC60 independently of known binding and functional domains. To corroborate these data, we evaluated if disrupting the reported EMRE and/or MCU binding domains in MICU1 altered the high-MW MICU1 and MIC60 complex. FPLC fractionation of Micu1−/− cells expressing the Flag-tagged WT or mutant MICU1 demonstrated that none of the MICU1 mutations disrupted high-molecular weight MICU1 containing protein complexes (fig. S3). To map the MICU1 protein region responsible for its interaction with MIC60, we generated MICU1 truncation mutants (Fig. 5C). Immunoprecipitation of Flag-tagged MICU1 truncation mutants showed that deletion of a C-terminal 77-amino acid region in MICU1 abolished its interaction with MIC60 (Fig. 5D). Together, these results support a mtCU-independent interaction of MICU1 with the MICOS.
Fig. 5. Lack mtCU interaction does not affect the MICU1-MIC60 interaction.
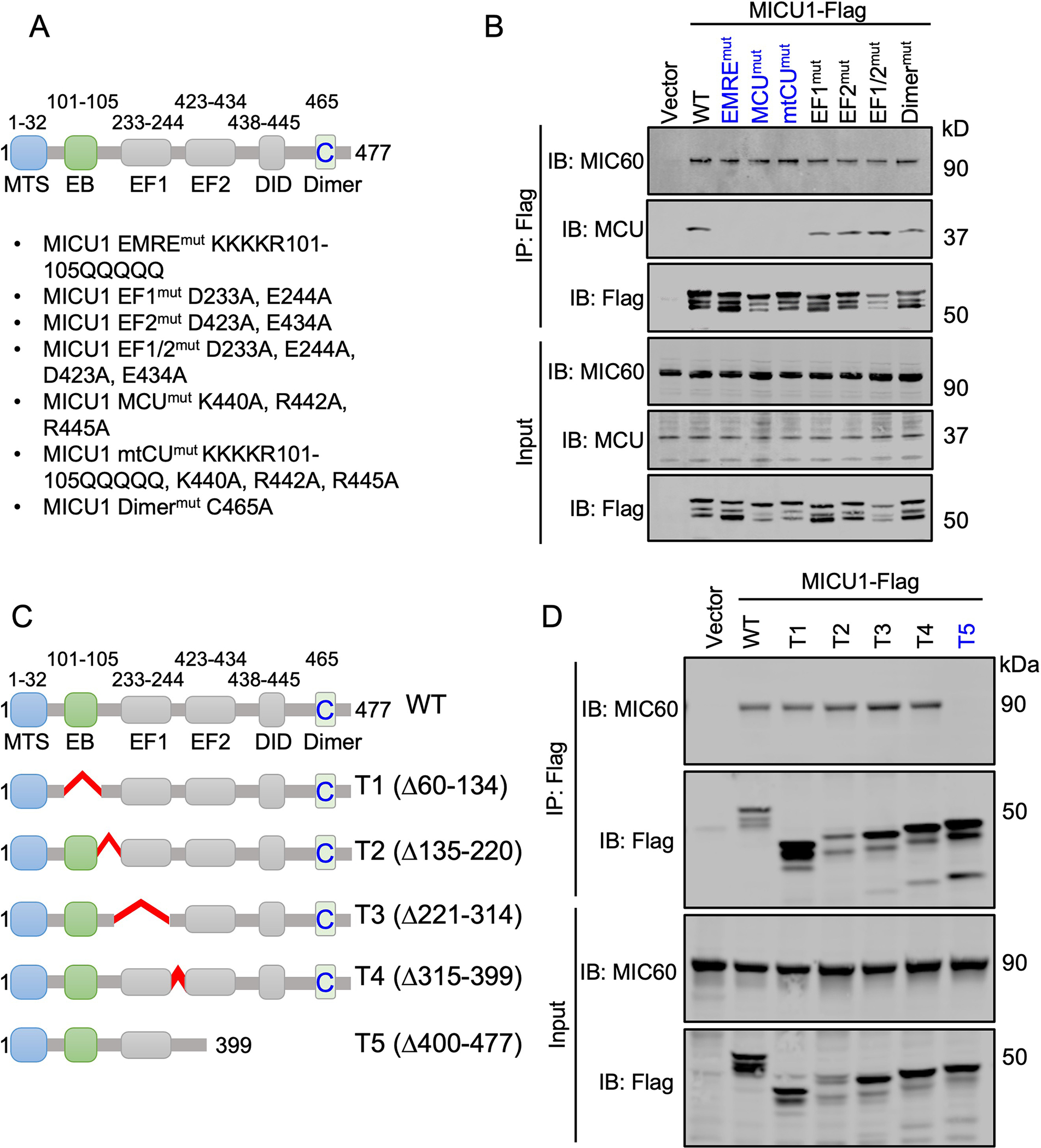
(A) Schematic representation of MICU1 domains and the point mutants utilized in the present study. (B) Micu1−/− MEFs were reconstituted with Flag-tagged MICU1 WT, MICU1 EMREmut, MICU1 MCUmut, MICU1 mtCUmut, MICU1 EF1mut, MICU1 EF2mut, MICU1 EF1/2mut, and MICU1 dimermut mutants. FLAG immunoprecipitates were probed for FLAG, MIC60, and MCU to detect MICU1-MIC60 and MICU1-MCU interactions. Western blots are representative of 3 independent experiments. (C) Schematic for the MICU1 truncation mutants utilized in the present study. (D) Flag-tagged MICU1 WT, T1, T2, T3, T4, and T5 mutants were reconstituted in Micu1−/− MEFs. FLAG immunoprecipitates were probed for FLAG and MIC60 to detect MICU1-MIC60 interactions. Western blots are representative of 3 independent experiments.
MICU1 is essential for the maintenance of mitochondrial ultrastructure and cristae organization
The MICOS is essential for maintenance of mitochondrial membrane topology and cristae bottleneck formation (40–43). The MICOS is localized at the intersection of the IMM and OMM and facilitates the formation of membrane contact sites at cristae junctions (40–43). Ca2+ modulates cristae structure (44, 45), but no Ca2+-sensing protein has yet been identified as an essential component of the MICOS. MICU1 has been reported to be localized at distinct cristae regions through an unknown mechanism (46). To discern if MICU1 serves as a conduit for Ca2+-dependent regulation of the MICOS, we examined if genetic loss of MICU1 affected mitochondrial ultrastructure and cristae junctions. In agreement with previous reports (15), transmission electron microscopy (TEM) revealed gross changes in mitochondrial ultrastructure of cells lacking MICU1 but no major changes in MCU−/− cells (Fig. 6A–G). Quantitative analysis of TEM images showed that mitochondrial Feret diameter (the distance between the two parallel planes restricting the object perpendicular to that direction) (Fig. 6B) and aspect ratio (Fig. 6C) were significantly reduced in MICU1−/− cells. An oversimplified interpretation of these results is that mitochondria were less filamentous in MICU1−/− cells. Next, we analyzed the inter-cristae junction or distance between cristae (Fig. 6D–E), is reported to be directly proportional to cristae density (47), and the cristae junction width (Fig. 6D–E) or distance between IMM of the same cristae. MICU1−/− cells displayed a significant increase in both the inter-cristae junction distance and cristae junction width, as compared to WT cells (Fig. 6F–G). To further validate the alterations in cristae structure, we performed super-resolution imaging of cells stained with the mitochondrial membrane potential (ΔΨm) dye tetramethylrhodamine methyl ester (TMRM) (Fig. 6H, fig. S4A). ΔΨm is reported to be spatially distributed in regular intervals along cristae (48), but deletion of MICU1 resulted in abnormal ΔΨm hotspots as evident by reduced patterning of TMRM peaks along the mitochondrial filaments (Fig. 6I).
Fig. 6. MICU1−/− cells display altered cristae structure and increased cytochrome c release.
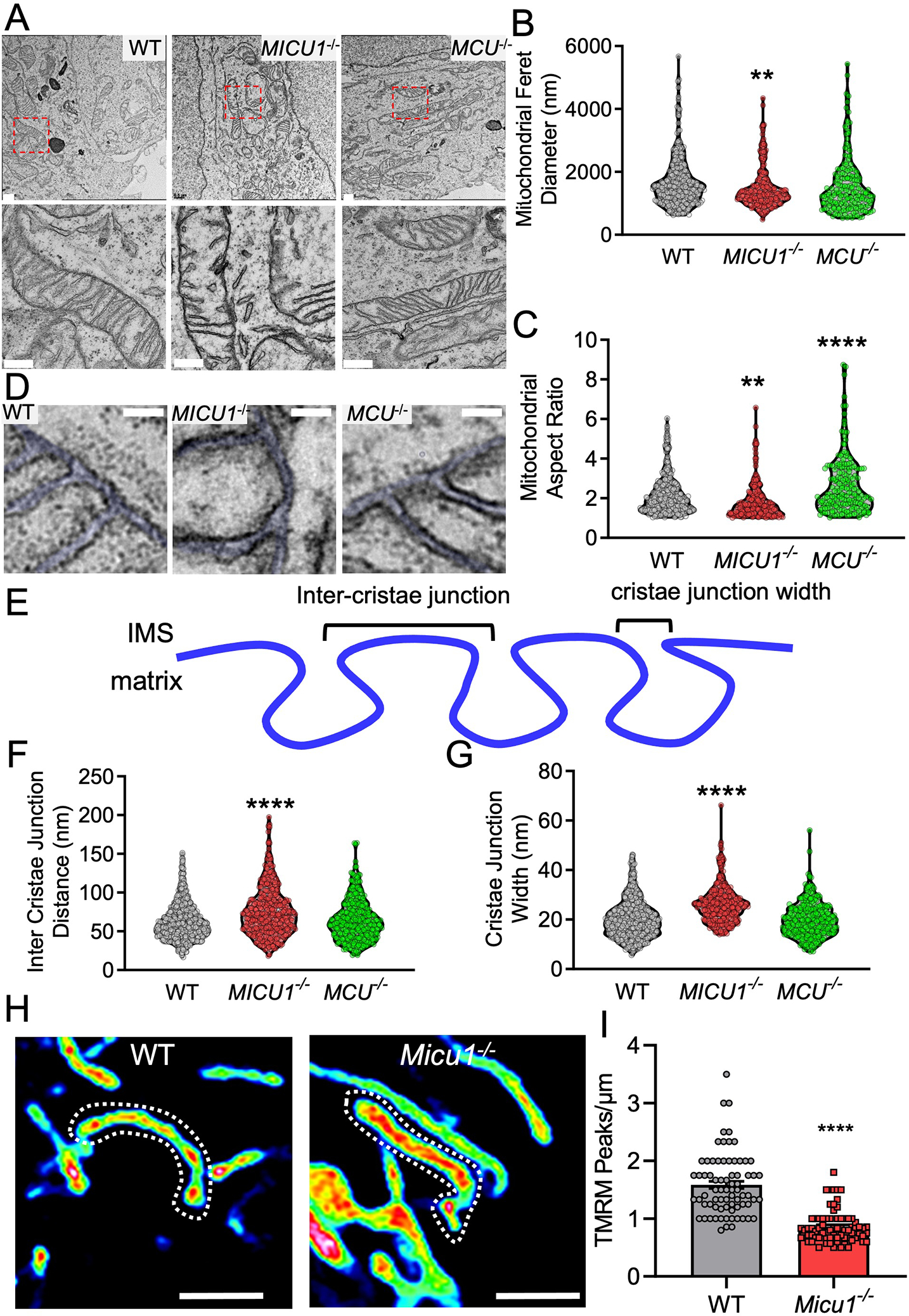
(A) WT, MICU1−/−, and MCU−/− HEK293 cells were imaged by TEM. Scale bar = 500nm. (B-C and F, G) TEM images were analyzed and quantitated using Image J Fiji. Mitochondrial feret diameter (B), aspect ratio (C), inter-cristae junction distance (F), and cristae junction width (G) were plotted. Statistical significance was determined using Welch’s t-test. ****, p<0.0001; **, p<0.01. n=200–300 mitochondria from 50 images acquired from n= 2 independent biological replicates per group. (D) Cristae ultrastructure in WT, MICU1−/−, and MCU−/− HEK293 cells imaged by TEM. Scale bar = 50nm. (E) Schematic depiction for the cristae junction width and inter-cristae junction distance. (H) ΔΨm distribution along the mitochondrial filaments in WT and Micu1−/− MEFs loaded with TMRM. Scale bar = 2μm. Images are representative of 3 independent experiments. (I) Mitochondrial filaments were quantified for the ΔΨm distribution (TMRM peaks/μm). Statistical significance was determined using Welch’s t-test. ****, p<0.0001. n=70–75 mitochondria from 2 independent experiments.
The bottleneck structure of cristae is essential for maintenance of the mitochondrial electron transport complexes and efficient respiration (40, 41). Disorganization and cristae remodeling are associated with the release of the electron shuttle cytochrome c, which subsequently initiates cytosolic apoptotic signaling (49). To define the role of MICU1 in cristae regulation, we monitored tBid-induced cytochrome c release in Micu1−/− MEFs. The assessment of tBid-induced cytochrome c release and the spatial distribution of ΔΨm examined by super-resolution microscopy were selected as functional assays because both are directly impacted by cristae organization and function (48, 50–53). The loss of MICU1 resulted in increased basal cytochrome c release, which was potentiated by application of tBID (fig. S4B–C). Next, we monitored if the increased cytochrome c release phenomenon in Micu1−/− cells depended upon changes in matrix Ca2+ levels. We treated cells with the mtCU inhibitor Ru360 to acutely inhibit mCa2+ uptake or with the mitochondrial permeability transition pore (MPTP) inhibitor cyclosporine A (CsA) to increase matrix Ca2+ retention capacity (Fig. 7A–B). Cytochrome c release was potentiated by both Ru360 and CsA in WT cells (Fig. 7A–B) and neither Ru360 nor CsA suppressed the elevated cytochrome c release observed in Micu1−/− cells (Fig. 7A–B). To further resolve the involvement of matrix Ca2+ content in MICU1 mediated cytochrome c release, we evaluated the tBid-induced cytochrome c release in Mcu−/− and Mcu−/− Micu1−/− double knockout MEFs (Fig. 7C–D). The loss of MCU did not significantly affect tBid-induced cytochrome c release (Fig. 7C–D). However, double knockout of Mcu and Micu1 potentiated tBid-induced cytochrome c release similar to Micu1 knockout (Fig. 7C–D). The Mcu−/− Micu1−/− MEFs were validated for the loss of MCU protein by immunoblotting (fig. S5A) as well as loss of MCU mediated Ca2+ flux (fig. S5B). We also evaluated the spatial distribution of ΔΨm in Mcu−/− and Mcu−/− Micu1−/− MEFs (fig. S5C). The quantification of TMRM peaks along the mitochondrial filaments showed that loss of MCU did not alter the spatial distribution of ΔΨm (fig. S5C, S5D). However, Mcu−/− Micu1−/− MEFs showed alterations in ΔΨm distribution (loss of sinusoidal patterning) along mitochondrial filaments similar to those seen in Micu1−/− MEFs (fig. S5C, S5D). These data suggest that MICU1 regulates cristae organization, and that cytochrome c release is not influenced by changes in matrix Ca2+ content. Next, to delineate the involvement of MICU1 mediated Ca2+ sensing in cytochrome c release, we reconstituted WT MICU and forms of MICU1 with mutations in the Ca2+ sensing EF1 and EF2 hand domains in Micu1−/− MEFs (Fig. 7E). Micu1−/− MEFs reconstituted with MICU1 WT and the MICU1 EF2 mutant showed reduced cytochrome c release in comparison to those reconstituted with the EF2 mutant, which displayed equivalent cytochrome c release in response to tBid as compared to vector control (Fig. 7E–F). These data suggest that the EF1 domain of MICU1 is essential for Ca2+ regulation of cristae function.
Fig. 7. Loss of MICU1 results in increased cytochrome c release.
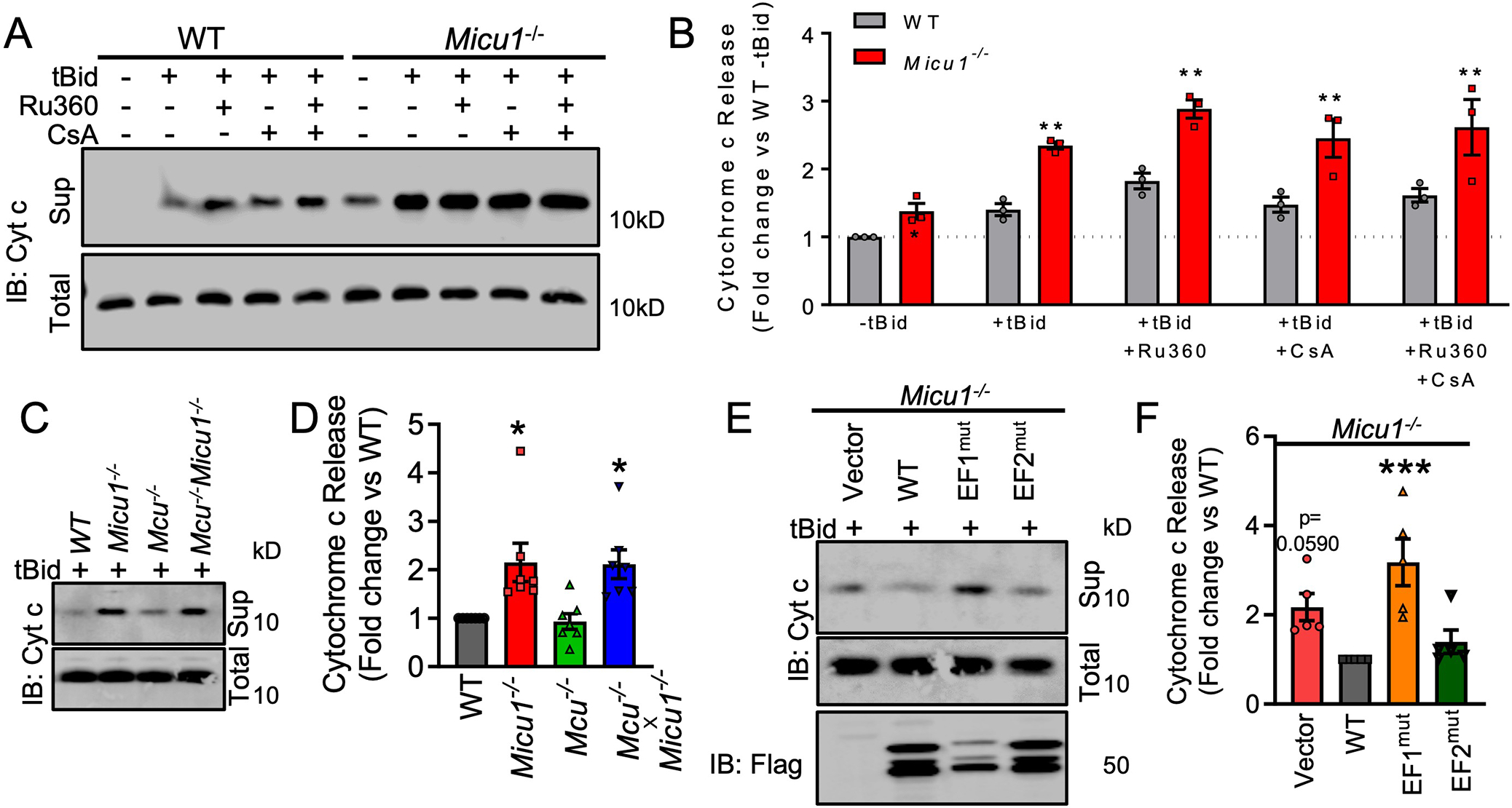
(A) Immunoblot showing MICU1- regulated cytochrome c release in WT and Micu1−/− MEFs treated with the MCU inhibitor Ru360 (1μM) or the CypD/MPTP inhibitor cyclosporine A (CsA, 1μM) to alter matrix Ca2+. Western blots are representative of 3 independent experiments. (B) Densitometry was performed to quantify cytochrome c release in Fig. 7A. Statistical significance was determined using 2-Way ANOVA with Tukey’s multiple comparisons test. **, p<0.01. n=3 independent experiments. (C) Immunoblot showing cytochrome c release in WT MEFs or MEFs deficient in Micu1 and/or Mcu. Western blots are representative of 7 independent experiments. (D) Densitometry was performed to quantify cytochrome c release in Fig. 7C. Statistical significance was determined using 1-way ANOVA with Dunnett’s multiple comparisons test. *, p<0.05. n=7 independent experiments. (E) Immunoblot showing cytochrome c release in Micu1−/− cells reconstituted with WT MICU1 WT, EF1 mutant, or EF2 mutant. Western blots are representative of 5 independent experiments. (F) Densitometry was performed to quantify cytochrome c release in Fig. 7E. Statistical significance was determined using 1-way ANOVA with Dunnett’s multiple comparisons test. ***, p<0.001. n=5 independent experiments.
To further rule out possible indirect effects of MICU1 modulation of mtCU-mediated Ca2+ uptake on cristae structure, we examined mCa2+ uptake kinetics in Chchd2−/− MEFs. CHCHD2 is a core MICOS component and its genetic deletion results in abnormal cristae organization (54). WT (Chchd2+/+) and Chchd2−/− MEFs were monitored for mCa2+ uptake independent of plasma membrane and ER Ca2+ transport using the ratiometric Ca2+ sensor Fura-FF. Chchd2−/− cells were indistinguishable from WT cells in all measurements of mCa2+ uptake, suggesting that altered cristae structure alone is insufficient to impact mtCU-dependent mCa2+ uptake (fig. S6A–B). Further, we found that loss of CHCHD2 did not affect mitochondrial Ca2+ efflux as measured by the rate of matrix Ca2+ efflux following Ru360 inhibition of the mtCU (fig. S6A, S6C). FPLC-based protein fractionation revealed no changes in MCU distribution in high-MW mtCU complexes in Chchd2−/− MEFs (fig. S6D, S6E). Together, these observations suggest that MICU1-dependent alterations in cristae remodeling do not depend on matrix Ca2+ flux and that altered cristae structure alone is not sufficient to induce changes in mCa2+ exchange. These results bolster our hypothesis that MICU1 regulates cristae structure and function independently of its role in gating the mtCU.
Discussion
In this report, we characterized the MICU1 interactome and identified a distinct involvement of MICU1 in cristae organization independent of the mtCU and mitochondrial Ca2+ uptake. We experimentally validated the presence of MICU1 high-MW protein complexes in MCU−/− cells suggesting that MICU1 is part of mtCU independent protein complexes at the IMM. Using an unbiased proteomics approach and an improved promiscuous BirA biotin ligase (BioID2) fused to MICU1, we found that multiple MICOS components were interacting partners of MICU1 (Data File S5). We validated that MICU1 directly interacted with core MICOS component MIC60 and accessory subunit CHCHD2 (Fig. 3B). Our study reveals a direct interaction between MICU1 and core MICOS components and shows that this interaction is essential to form the functional MICOS complex and maintain mitochondrial membrane structure and function.
We propose that MICU1 modulates mtCU mCa2+ and MICOS activity independently of each other to fine-tune mitochondrial function. For example, mCa2+ regulates dehydrogenase activity and TCA cycle flux to augment the generation of reducing equivalents (NADH) for the electron transport chain (ETC) (55–58). However, the ETC does not contain direct sites of Ca2+ control (59) and therefore regulation of the ETC may be secondary to changes in cristae structure or function regulated by MICU1 sensing of IMS Ca2+. This would provide a mechanism for independent Ca2+ microdomains (matrix and IMS) to regulate cellular energetics. Further, our results showed that MICU1 was essential for bioenergetic homeostasis and cell death signaling events (specifically, MPTP opening as opposed to apoptogen release from cristae bottlenecks). These effects could explain the lethal phenotype observed in MICU1 knockout mice and fly models (15, 31, 32), because our results suggest that loss of MICU1 could induce cell death signaling through both the necrotic and apoptotic pathways and would also explain why MICU1 mutations and/or genetic loss are linked to severe phenotypes (29, 30, 32, 60–63).
Ca2+-induced changes in mitochondrial ultrastructure (44) have been hypothesized to be primarily due to the matrix Ca2+ overload or due to the effect of elevated cytosolic Ca2+ on mitochondrial fission or fusion and trafficking events (11, 64–69). The Ca2+ concentration in the IMS could be directly involved in the regulation of mitochondrial ultrastructure by modulating cristae organization (45, 46). However, no Ca2+ sensor at the MICOS complex or cristae junctions has been identified that would provide Ca2+ detection. Gottschalk et al. (46) reported the involvement of MICU1 in regulating the cristae ultrastructure, linking this function to mCa2+ uptake by the mtCU. These authors hypothesized that MICU1 may be a physical linker between the mtCU and MICOS. However, our results suggest that MICU1 interaction with the MICOS does not depend on the mtCU complex, mCa2+ uptake, or matrix Ca2+ overload. Therefore, a reappraisal of the MICU1/mtCU literature is warranted because some of the reported phenotypes may be a result of alterations in MICOS and cristae organization, rather than merely changes in mCa2+ uptake. Further research is needed to define the precise interaction of MICU1 with MICOS components to identify tools to dissect the mtCU-dependent and independent functions of MICU1 in mitochondrial biology. Ongoing studies will explore if MICU1-mediated Ca2+ sensing at the MICOS is a prominent mechanism contributing to the pathogenesis of diseases featuring alterations in mitochondrial membrane structure and dynamics. In summary, we identified an IMS Ca2+ sensor that regulates the MICOS complex independently of matrix Ca2+ flux (Fig. 8). This study provides a paradigm to understand Ca2+-dependent regulation of mitochondrial structure and function and may help explain the cell signaling underlying mitochondrial remodeling that is reported in many disease states.
Fig. 8. Schematic representation of the two different modes of MICU1-dependent regulation of Ca2+-mediated regulation of mitochondrial structure and function.
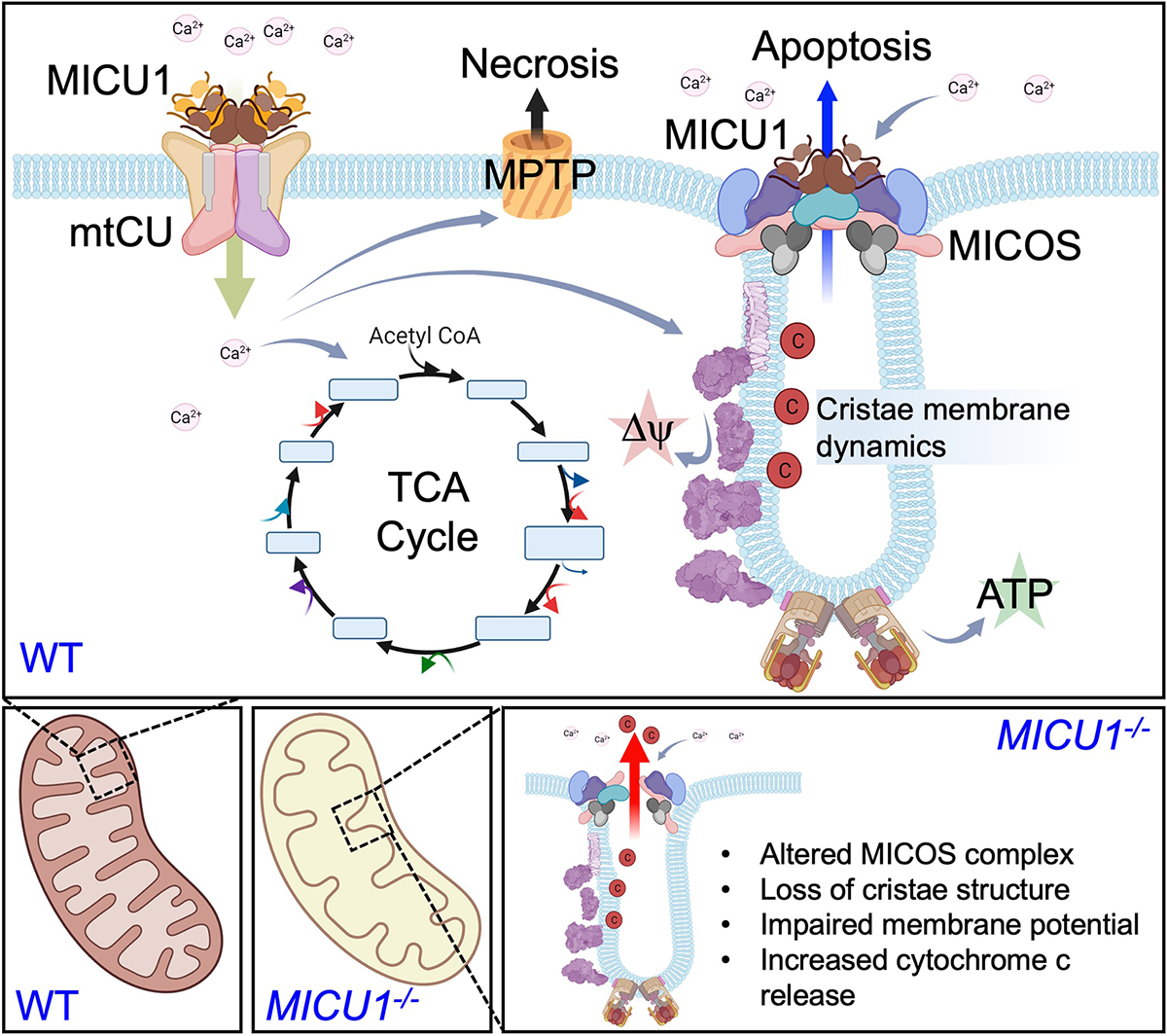
MICU1 regulates the gating of the mitochondrial Ca2+ uniporter channel (mtCU) to control matrix Ca2+ levels and modulate dehydrogenase activity and also impacts mitochondrial permeability transition during matrix Ca2+ overload. Separately, MICU1 associates with the MICOS complex to regulate cristate structure and function and therein provides a mechanism for IMS Ca2+ to regulate mitochondrial membrane potential and the release of apoptogens, independently of matrix Ca2+ signaling. This hypothesis proposes two modes of MICU1-dependent Ca2+ signaling (IMS vs. matrix) to regulate cellular energetics and cell death programs.
Materials and Methods
Plasmid Construction
To generate the BioID2-HA plasmid, BioID2 was PCR-amplified from the MCS-BioID2-HA plasmid (Addgene #74224) using primers designed to introduce an ATG start codon immediately downstream of the BamHI restriction site of the MCS. The PCR product was cut by BamHI and HindIII and cloned into the MCS-BioID2-HA plasmid (Addgene #74224). To generate the MICU1-BioID2-HA, MICU1 was PCR amplified from the hMICU1-Myc-DDK plasmid using primers to introduce a 5’ AgeI and a 3’ BamHI restriction site. The PCR product was cut by AgeI and BamHI and cloned into the MCS-BioID2-HA plasmid (Addgene #74224). The MICU1-HA plasmid was generated by cleaving the MICU1 fragment from the MICU1-FLAG plasmid (Origene # MR207652) using the SgfI-MluI restriction sites and inserted into the same sites in pCMV6-AC-HA vector (Origene # PS100004). Flag-tagged Mouse MICU1 (NM_144822) mutants; EMREmut KKKKR101-105QQQQQ; EF1mut D233A, E244A; EF2mut D423A, E434A; EF1/2mut D233A, E244A, D423A, E434A; MCUmut K440A, R442A, R445A; mtCUmut KKKKR101-105QQQQQ, K440A, R442A, R445A; Dimermut C465A; Truncation 1 (T1): Δ60 – 134; Truncation 2 (T2): Δ135 – 220; Truncation 3 (T3): Δ221 – 314; Truncation 4 (T4): Δ315 – 399; Truncation 5 (T5): Δ400 – 477 were custom cloned by Vector Builder Inc. Plasmids were confirmed by restriction digestion and DNA sequencing. Specific details of plasmid sources are provided in table S1.
Cell culture
HEK293T WT, HEK293T MCU−/− and HEK293T MICU1−/− cells were grown in Dulbecco’s Modification of Eagle’s Medium with 4.5 g/L glucose, L-glutamine, and sodium pyruvate (Corning Cellgro, Cat#10-013-CV) supplemented with 10% fetal bovine serum (Peak Serum, Cat#PS-FB3), 1% penicillin/streptomycin (Sigma-Aldrich, Cat# P0781-100ML) at 37°C in the presence of 5% CO2. MEFs from Micu1fl/fl and Mcufl/fl mice (20) were immortalized by infecting the cells with SV40 large T antigen-expressing adenovirus. The immortalized Micu1fl/fl MEFs served as WT control cells. Micu1−/− and Mcu−/− MEFs were generated by transducing the corresponding fl/fl MEFs with adenovirus encoding Cre-recombinase (Ad-Cre). Double knockout Mcu−/− Micu1−/− MEFs were generated by CRISPR/Cas9 mediated genetic modification of Mcu−/− MEFs. In brief, Mcu−/− MEFs were transfected with pRP[2CRISPR]-hCas9-U6 mMicu1 encoding two guide RNAs targeting Micu1 (g1: CGAAGTGTTCATGACTCCGC; g2: CAGAACGTAAGTTGCTAGCG). Loss of MCU and MICU1 was validated in transfected cells. Single clones were established validated for the loss of MICU1 and clone C3 which showed deletion of MICU1 was used for further experiments. Loss of MCU and MICU1 was further validated in MEFs utilizing a permeabilized cell system to assess mCa2+ flux. MEFs were grown in Dulbecco’s Modification of Eagle’s Medium with 4.5 g/L glucose, L-glutamine, and sodium pyruvate (Corning Cellgro, Cat#10-013-CV) supplemented with 10% fetal bovine serum (Peak Serum, Cat#PS-FB3), 1% Gibco® MEM Non-Essential Amino Acids (Thermo Fisher Scientific, Cat# 11-140-050), 1% penicillin/streptomycin (Sigma-Aldrich, Cat# P0781-100ML), at 37°C in the presence of 5% CO2. Chchd2+/+ and Chchd2−/− MEFs were cultured as previously described (54). To exogenously express MICU1, MIC60, CHCHD3, and CHCHD2, cells were transfected with the Fugene HD transfection reagent (Promega, Cat#E2311) as per the manufacturer’s instructions. To generate MEFs stably expressing MICU1-FLAG, immortalized WT MEFs were transfected with MICU1-FLAG plasmid (OriGene Technologies, Cat#MR207652) using the Fugene HD transfection reagent (Promega, Cat#E2311). 24h post-transfection, culture media was replaced with media supplemented with 500 μg/mL G418 (Thermo Fisher Scientific, Cat#10131035). Fresh culture media supplemented with G418 was replaced at two-day intervals until all dead cells were cleared. After incubation for two weeks, the cells were maintained in DMEM supplemented with 200 μg/mL G418. Protein expression was validated by Western blotting and immunofluorescence. Specific details of cell line sources are provided in table S1.
Immunoblotting
Cells were harvested, washed with ice-cold PBS, and lysed in 1X RIPA lysis buffer (EMD Millipore, Cat#20–188) supplemented with SIGMAFAST™ Protease Inhibitor Cocktail (Sigma-Aldrich, Cat#S8830). Protein concentrations were determined by Pierce 660nm Protein Assay (Thermo Fisher Scientific, Cat#22660) and equal amounts of protein were separated by electrophoresis on NuPAGE 4–12% Bis-Tris protein gels (Thermo Fisher Scientific, Cat#WG1402BOX), under denaturing conditions. Proteins were transferred PVDF membranes (EMD Millipore, Cat#IPFL00010). Membranes were incubated in Blocking Buffer (Rockland, Cat#MB-070) for 1h at room temperature and incubated overnight with specific primary antibodies at 4°C. Membranes were washed with TBS-T (TBS containing 0.1% Tween 20) 3 times for 10 min each and incubated with specific secondary antibodies for 1h at room temperature. Membranes were washed again and imaged on an LI-COR Odyssey system. Specific details of antibody sources are provided in table S1.
Sub-mitochondrial protein localization assay
Mitochondria were isolated as described earlier (70). Briefly, cells were grown in 150 mm2 culture dishes, washed with PBS, and resuspended in isotonic mitochondria isolation buffer (10 mM HEPES, pH 7.5, containing 200 mM mannitol, 70 mM sucrose, and 1 mM EGTA). Cell suspensions were homogenized by Dounce homogenizer and centrifuged at 500 g for 10 min at 4°C. Supernatants were collected and centrifuged at 12,000 g for 15 min at 4°C to obtain crude mitochondrial pellets. Pellets were resuspended in mitochondria isolation buffer and washed 2 times using the centrifuge at 12,000 g for 15 min at 4°C. Mitochondrial pellets were resuspended in intracellular buffer (120 mM KCl, 10 mM NaCl, 1 mM KH2PO4, 20 mM HEPES-Tris, pH 7.2) and permeabilized with varying digitonin concentrations and digested with proteinase K (10 μg/mL) for 10min at room temperature. Proteinase K digestion was stopped by adding SIGMAFAST™ Protease Inhibitor Cocktail (Sigma-Aldrich, Cat#S8830) and 2X SDS-loading dye and heating the samples at 95°C for 10min.
Biotinylation and mass spectrometry analysis
To induce BioID2-mediated protein biotinylation, cells were cultured with media supplemented with 50 μM biotin for 16h. Cells were collected, washed with PBS 2 times, and lysed in BioID2 lysis buffer (50 mM Tris, pH 7.4, 500 mM NaCl, 2% Triton X-100, 0.4% SDS, 1 mM dithiothreitol) supplemented with SIGMAFAST™ Protease Inhibitor Cocktail (Sigma-Aldrich, Cat#S8830). Cell suspensions were sonicated for 2 times each for 1 min at an output level of 40 (Vibra-Cell, Sonics). An equal volume of 50 mM Tris, pH 7.4, was added and suspensions were cleared using centrifugation at 16,500 g for 20 min. Supernatants were used for immunoblotting or streptavidin based pull-down experiments using MyOne Dynabeads Streptavidin C1. Mass spectroscopy analysis to identify biotinylated proteins was performed as previously described (35).
Fast protein liquid chromatography (FPLC), protein fractionation, and MICU1 multiprotein complex analysis
Size-exclusion gel filtration was used to separate the high-molecular-weight protein complexes using FPLC (ÄKTA Pure FPLC; GE Healthcare) (24). PBS-equilibrated Superdex 200 10/300 columns (GE Healthcare, Cat#17517501) were calibrated with a gel filtration calibration standard (Bio-Rad, Cat#1511901). Cleared cell lysates were fractionated through the FPLC at a flow rate of 0.5 mL/min. Protein fractions were collected in 0.5 mL PBS and concentrated to 50μL volume using an AMICON Ultra-0.5 Centrifugal Filter Devices (with a 3,000 kD cutoff) (EMD Millipore, Cat#UFC500396). Concentrated fractions were immunoblotted as indicated. Protein abundance in each fraction was quantified in Image J and normalized to the highest protein levels in each group. To monitor the MICU1 multiprotein complexes, isolated liver mitochondria from wild-type or global MCU knockout mice were lysed in a buffer containing 250mM sucrose, 20mM MOPS (pH7.4), 1mM EDTA and 1% CHAPS. The lysate was recovered by centrifugation (10min; 10,000xg) and was incubated in the presence or absence of the crosslinker succinimidyl-6-[ß-maleimidopropionamido]hexanoate (SMPH; 1 mM) for 30min at 25°C. The reaction was stopped by the addition of 1mM DTT and 5mM TrisHCl (pH8.0). Aliquots (15ug protein) were processed by Blue Native PAGE using commercial Novex 3–12% native gradient gels according to the manufacturer’s protocol. The gels were transferred to PVDF membranes, which were probed for MICU1.
Coimmunoprecipitation assays
To study protein-protein interactions, immunoprecipitation experiments were performed as previously described (24). Briefly, HEK293T cells were co-transfected with the indicated plasmids. 36h after transfection, cells were harvested, washed with ice-cold PBS, and lysed in 1X RIPA lysis buffer (EMD Millipore, Cat#20-188) supplemented with SIGMAFAST™ Protease Inhibitor Cocktail (Sigma-Aldrich, Cat#S8830). Protein concentrations were determined by Pierce 660nm Protein Assay (Thermo Fisher Scientific, Cat#22660) and equal proteins amounts were used for co-immunoprecipitation. Cleared cell lysates were incubated with FLAG M2 Magnetic Beads (Sigma-Aldrich) on a roller shaker overnight at 4°C. Beads were washed 3 times with RIPA buffer and 2 times with TBS-T, resuspended in 2X SDS-PAGE sample buffers, and immunoblotted as indicated.
Co-immunofluorescence and TMRM imaging
The mitochondrial localization of mtCU, MICOS, and MICU1 was analyzed by immunofluorescence using a standard protocol (70). Briefly, MEFs stably expressing MICU1-FLAG were grown on collagen-coated 35-mm dishes. Cells were washed with PBS, fixed for 20min with 4% paraformaldehyde, and permeabilized for 15min by 0.15% Triton X-100. Permeabilized cells were blocked with 10% BSA for 45min at room temperature and incubated with primary antibodies overnight at 4°C. After incubation, cells were washed 3 times with blocking reagent and incubated with Alexa Fluor-tagged secondary antibodies for 1h at room temperature. Cells were washed 3 times with PBS, and confocal images were obtained using an LSM 510 META Laser Scanning Microscope (Carl Zeiss, Inc.) at 488- and 647-nm excitations using a 63x oil objective. To assess the ΔΨm distribution along mitochondrial filaments, MEFs were grown on MatTek collagen-coated glass bottom 35 mm dishes and stained with TMRM (15nM) for 15 min. Images were acquired by Zeiss LSM 900 microscope with Airyscan 2 detector. Images were analyzed and quantitated using ZEN blue software (Carl Zeiss, Inc.) and Image J Fiji.
Transmission electron microscopy
Transmission electron microscopy (TEM) was utilized to evaluate mitochondrial ultrastructure and cristae organization. HEK293T cells of the indicated genotypes were grown to 80% confluency on 25 mm diameter Thermanox® Cover Slips (Thermo Fisher Scientific, Cat#174985PK) in 6-well plates. Culture media was removed, and cells were fixed with freshly prepared TEM fixation buffer (2% glutaraldehyde, 2% paraformaldehyde in 0.1M sodium cacodylate buffer) for 30 min at room temperature. Fixative was replaced with 0.1M sodium cacodylate buffer, and samples were processed for TEM imaging. Images were obtained using Zeiss LIBRA120 TEM equipped with Gatan UltraScan with 1000 2k x 2k CCD EFTEM and energy filtering. Images were analyzed and quantitated Image J Fiji.
Cytochrome c release assay
The cytochrome c release assay was performed as described earlier (70) with slight modifications. Briefly, MEFs grown in 150mm2 culture dishes were washed with ice-cold PBS, pH 7.4 and divided into two fractions with equal cell numbers, one to generate total cell lysate and the other for the cytochrome c release assay. For the assay, equal numbers of cells were suspended in intracellular buffer (120 mM KCl, 10 mM NaCl, 1 mM KH2PO4, 20 mM HEPES-Tris, pH 7.2) supplemented with SIGMAFAST™ Protease Inhibitor Cocktail (Sigma-Aldrich, Cat#S8830) and permeabilized with digitonin (80 μg/mL) for 5 min at room temperature. Ru360 (1μM) and CsA (1μM) were added in the permeabilization buffer. Cytochrome c release was induced by adding tBid (20nM) and incubating cell suspensions at 30°C for 30 min. Cell homogenates were spun at 16,500 g at 4°C for 10 min, and supernatants (which was the cytosolic fraction) were removed. Cell pellets from total cells were lysed in 1XRIPA buffer and centrifuged at 16,500 g at 4°C for 10 min to obtain the total cell lysate. Both total cell lysates and cytosolic fractions were immunoblotted for cytochrome c.
mCa2+ flux analysis
mCa2+ flux was analyzed as previously described (20, 24). Briefly, cells were washed in Ca2+-free DPBS (Thermo Fisher Scientific, Cat#14190235). Equal numbers of cells (7×106 cells) were resuspended and permeabilized with 40 μg/ml digitonin in 1.5 ml of intracellular medium (120 mM KCl, 10 mM NaCl, 1 mM KH2PO4, 20 mM HEPES-Tris, pH 7.2), containing 2 μM thapsigargin to block the SERCA pump and supplemented with 5 mM succinate. Fura-FF (1μM) was added to cell suspensions, and fluorescence was monitored in a multiwavelength excitation dual-wavelength emission fluorimeter (Delta RAM, PTI). Extramitochondrial Ca2+ is measured as the excitation ratio (340 nm/380 nm) of Fura-FF fluorescence. A Ca2+ bolus and the mitochondrial uncoupler FCCP (2 μM) were added at the indicated time points (8, 9). All the experiments were performed at 37°C with constant stirring.
Statistical analysis
Results are presented as means ± standard error. Statistical analysis was performed using GraphPad PRISM 7.05 (Graph Pad Software). Experiments were repeated independently at least two times. Technical and biological replicates were mentioned in figure legends. p-value analysis were performed using an unpaired, 2-tailed t-test (for 2 groups) with Welch’s correction. For grouped analyses, 2-way ANOVA with Tukey post-hoc analysis or Dunnett’s multiple comparisons test was performed. P values less than 0.05 (95% confidence interval) were considered significant.
Supplementary Material
Acknowledgments:
The authors thank to Trevor Tierney for technical and managerial assistance in the Elrod Laboratory. The authors thank Dr. Alex Rosa Campos (The Sanford Burnham Prebys Proteomics Core) for helping in BioID2 proteomics screening and Shannon Modla (Delaware Biotechnology Institute) for her help with TEM sample processing. The authors acknowledge Drs. Yuzuru Imai and Nobutaka Hattori (Juntendo University Graduate School of Medicine, Japan) for sharing the Chchd2+/+ and Chchd2−/− cells.
Funding:
This work was funded by National Institutes of Health grants (R01HL142271, R01HL136954, R01HL123966, P01HL134608, and P01HL147841 to J.W.E.; K99DK120876 to D.T.; K99AG065445 to P.J.; F32HL151146 to J.F.G.; GM132611 to S.K.J.); American Heart Association grants (17POST33660251 and 19CDA34490009 to D.T.).
Footnotes
Competing interests: J.W.E. and J.F.G. are consultants for Mitobridge, Inc. The other authors declare that they have no competing interests.
Data and materials availability: The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium by the PRIDE (71) partner repository with the dataset identifier PXD028462. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The recombinant plasmids (MICU1 mutants) and MEF lines (Mcu−/−, Micu1−/−, Mcu−/−Micu1−/−) generated in current study are available from J.W.E. upon a material transfer agreement with Temple University, USA.
References and Notes
- 1.Bagur R, Hajnoczky G, Intracellular Ca(2+) Sensing: Its Role in Calcium Homeostasis and Signaling. Mol Cell 66, 780–788 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carafoli E, The calcium-signalling saga: tap water and protein crystals. Nat Rev Mol Cell Biol 4, 326–332 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Carafoli E, Calcium signaling: a tale for all seasons. Proc Natl Acad Sci U S A 99, 1115–1122 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tadross MR, Dick IE, Yue DT, Mechanism of local and global Ca2+ sensing by calmodulin in complex with a Ca2+ channel. Cell 133, 1228–1240 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rizzuto R, De Stefani D, Raffaello A, Mammucari C, Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13, 566–578 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Rizzuto R, Pozzan T, Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev 86, 369–408 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Fransson A, Ruusala A, Aspenstrom P, Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem 278, 6495–6502 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Perocchi F et al. , MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 467, 291–296 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Plovanich M et al. , MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One 8, e55785 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nemani N et al. , MIRO-1 Determines Mitochondrial Shape Transition upon GPCR Activation and Ca(2+) Stress. Cell Rep 23, 1005–1019 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frederick RL, McCaffery JM, Cunningham KW, Okamoto K, Shaw JM, Yeast Miro GTPase, Gem1p, regulates mitochondrial morphology via a novel pathway. J Cell Biol 167, 87–98 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saotome M et al. , Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A 105, 20728–20733 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mallilankaraman K et al. , MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151, 630–644 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Csordas G et al. , MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab 17, 976–987 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu JC et al. , MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell Rep 16, 1561–1573 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patron M et al. , MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell 53, 726–737 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baughman JM et al. , Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R, A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kirichok Y, Krapivinsky G, Clapham DE, The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Luongo TS et al. , The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep 12, 23–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mallilankaraman K et al. , MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol 14, 1336–1343 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raffaello A et al. , The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J 32, 2362–2376 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sancak Y et al. , EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342, 1379–1382 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tomar D et al. , MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep 15, 1673–1685 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lambert JP et al. , MCUB Regulates the Molecular Composition of the Mitochondrial Calcium Uniporter Channel to Limit Mitochondrial Calcium Overload During Stress. Circulation 140, 1720–1733 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xing Y et al. , Dimerization of MICU Proteins Controls Ca(2+) Influx through the Mitochondrial Ca(2+) Uniporter. Cell Rep 26, 1203–1212 e1204 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Phillips CB, Tsai CW, Tsai MF, The conserved aspartate ring of MCU mediates MICU1 binding and regulation in the mitochondrial calcium uniporter complex. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paillard M et al. , MICU1 Interacts with the D-Ring of the MCU Pore to Control Its Ca(2+) Flux and Sensitivity to Ru360. Mol Cell 72, 778–785 e773 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Logan CV et al. , Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat Genet 46, 188–193 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Lewis-Smith D et al. , Homozygous deletion in MICU1 presenting with fatigue and lethargy in childhood. Neurol Genet 2, e59 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Antony AN et al. , MICU1 regulation of mitochondrial Ca(2+) uptake dictates survival and tissue regeneration. Nat Commun 7, 10955 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tufi R et al. , Comprehensive Genetic Characterization of Mitochondrial Ca(2+) Uniporter Components Reveals Their Different Physiological Requirements In Vivo. Cell Rep 27, 1541–1550 e1545 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bick AG et al. , Cardiovascular homeostasis dependence on MICU2, a regulatory subunit of the mitochondrial calcium uniporter. Proc Natl Acad Sci U S A 114, E9096–E9104 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoffman NE et al. , MICU1 motifs define mitochondrial calcium uniporter binding and activity. Cell Rep 5, 1576–1588 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim DI et al. , An improved smaller biotin ligase for BioID proximity labeling. Mol Biol Cell 27, 1188–1196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Keuren AM et al. , Mechanisms of EMRE-Dependent MCU Opening in the Mitochondrial Calcium Uniporter Complex. Cell Rep 33, 108486 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y et al. , Structural insights into the Ca(2+)-dependent gating of the human mitochondrial calcium uniporter. Elife 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang C, Jacewicz A, Delgado BD, Baradaran R, Long SB, Structures reveal gatekeeping of the mitochondrial Ca(2+) uniporter by MICU1-MICU2. Elife 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan M et al. , Structure and mechanism of the mitochondrial Ca(2+) uniporter holocomplex. Nature 582, 129–133 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van der Laan M, Horvath SE, Pfanner N, Mitochondrial contact site and cristae organizing system. Curr Opin Cell Biol 41, 33–42 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Friedman JR, Mourier A, Yamada J, McCaffery JM, Nunnari J, MICOS coordinates with respiratory complexes and lipids to establish mitochondrial inner membrane architecture. Elife 4, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harner M et al. , The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J 30, 4356–4370 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tarasenko D et al. , The MICOS component Mic60 displays a conserved membrane-bending activity that is necessary for normal cristae morphology. J Cell Biol 216, 889–899 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Greenawalt JW, Rossi CS, Lehninger AL, Effect of Active Accumulation of Calcium and Phosphate Ions on the Structure of Rat Liver Mitochondria. J Cell Biol 23, 21–38 (1964). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gottschalk B, Klec C, Waldeck-Weiermair M, Malli R, Graier WF, Intracellular Ca(2+) release decelerates mitochondrial cristae dynamics within the junctions to the endoplasmic reticulum. Pflugers Arch 470, 1193–1203 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gottschalk B et al. , MICU1 controls cristae junction and spatially anchors mitochondrial Ca(2+) uniporter complex. Nat Commun 10, 3732 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glytsou C et al. , Optic Atrophy 1 Is Epistatic to the Core MICOS Component MIC60 in Mitochondrial Cristae Shape Control. Cell Rep 17, 3024–3034 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wolf DM et al. , Individual cristae within the same mitochondrion display different membrane potentials and are functionally independent. EMBO J 38, e101056 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scorrano L et al. , A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell 2, 55–67 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Schlame M, Mitochondrial cristae as insulated transformers of metabolic energy. EMBO J 38, e103472 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kondadi AK et al. , Cristae undergo continuous cycles of membrane remodelling in a MICOS-dependent manner. EMBO Rep 21, e49776 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gottlieb E, Armour SM, Harris MH, Thompson CB, Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ 10, 709–717 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Cipolat S et al. , Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell 126, 163–175 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Meng H et al. , Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat Commun 8, 15500 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Griffiths EJ, Rutter GA, Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim Biophys Acta 1787, 1324–1333 (2009). [DOI] [PubMed] [Google Scholar]
- 56.McCormack JG, Denton RM, The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem J 180, 533–544 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Denton RM, Richards DA, Chin JG, Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem J 176, 899–906 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Denton RM, Randle PJ, Martin BR, Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem J 128, 161–163 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wescott AP, Kao JPY, Lederer WJ, Boyman L, Voltage-energized Calcium-sensitive ATP Production by Mitochondria. Nat Metab 1, 975–984 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mojbafan M et al. , Reporting a rare form of myopathy, myopathy with extrapyramidal signs, in an Iranian family using next generation sequencing: a case report. BMC Med Genet 21, 77 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Debattisti V et al. , Dysregulation of Mitochondrial Ca(2+) Uptake and Sarcolemma Repair Underlie Muscle Weakness and Wasting in Patients and Mice Lacking MICU1. Cell Rep 29, 1274–1286 e1276 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Musa S et al. , A Middle Eastern Founder Mutation Expands the Genotypic and Phenotypic Spectrum of Mitochondrial MICU1 Deficiency: A Report of 13 Patients. JIMD Rep 43, 79–83 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bhosale G et al. , Pathological consequences of MICU1 mutations on mitochondrial calcium signalling and bioenergetics. Biochim Biophys Acta Mol Cell Res 1864, 1009–1017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cereghetti GM et al. , Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A 105, 15803–15808 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Han XJ et al. , CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol 182, 573–585 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cribbs JT, Strack S, Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep 8, 939–944 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaasik A, Safiulina D, Zharkovsky A, Veksler V, Regulation of mitochondrial matrix volume. Am J Physiol Cell Physiol 292, C157–163 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Guo X et al. , The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 47, 379–393 (2005). [DOI] [PubMed] [Google Scholar]
- 69.A. P. Halestrap, P. T. Quinlan, D. E. Whipps, A. E. Armston, Regulation of the mitochondrial matrix volume in vivo and in vitro. The role of calcium. Biochem J 236, 779–787 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tomar D et al. , TRIM4; a novel mitochondrial interacting RING E3 ligase, sensitizes the cells to hydrogen peroxide (H2O2) induced cell death. Free Radic Biol Med 89, 1036–1048 (2015). [DOI] [PubMed] [Google Scholar]
- 71.Perez-Riverol Y et al. , The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47, D442–D450 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.