

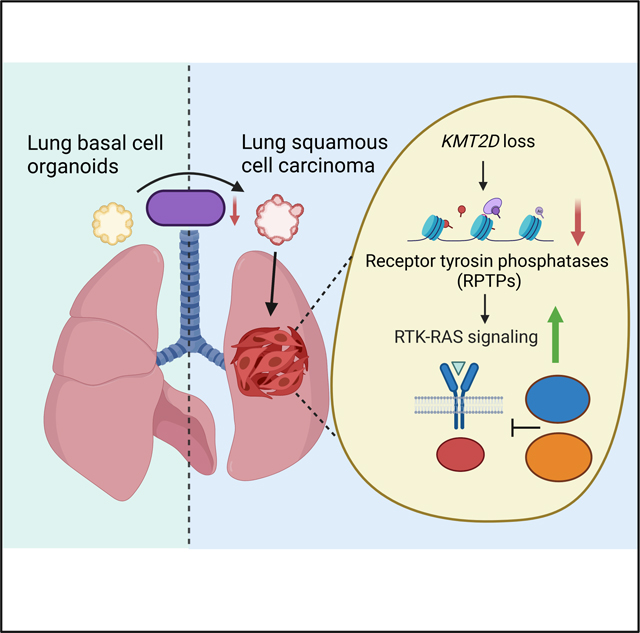
SUMMARY
Lung squamous cell carcinoma (LUSC) represents a major subtype of lung cancer with limited treatment options. KMT2D is one of the most frequently mutated genes in LUSC (>20%), and yet its role in LUSC oncogenesis remains unknown. Here, we identify KMT2D as a key regulator of LUSC tumorigenesis, wherein Kmt2d deletion transforms lung basal cell organoids to LUSC. Kmt2d loss increases activation of receptor tyrosine kinases (RTKs), EGFR and ERBB2, partly through reprogramming the chromatin landscape to repress the expression of protein tyrosine Combining SHP2 inhibitor SHP099 and pan-ERBB inhibitor afatinib inhibits lung tumor growth in Kmt2d-deficient LUSC murine models and in patient-derived xenografts (PDXs) harbouring KMT2D mutation. Our study identifies KMT2D as a pivotal epigenetic modulator for LUSC oncogenesis and suggests KMT2D loss renders LUSC therapeutically vulnerable to RTK-RAS inhibition.
Keywords: Lung squamous cell carcinoma, KMT2D, EGFR, ERBB2, SHP2, organoids
eTOC Blurb
Pan et al. identify the histone methyltransferase KMT2D as a key regulator of lung squamous cell carcinoma (LUSC) tumorigenesis. KMT2D loss triggers activation of oncogenic RTK-RAS signaling, which is partly due to epigenetic regulation of protein tyrosine phosphatases expression. KMT2D loss renders LUSC therapeutically vulnerable to SHP2 and pan-ERBB inhibition.
Graphical Abstract
INTRODUCTION
Lung cancer remains the leading cause of cancer-related death worldwide.1 Lung squamous carcinoma (LUSC) is the second most prevalent type, accounting for 20 to 30% of lung carcinoma deaths.2,3 However, unlike lung adenocarcinoma (LUAD), for which targeted therapies including EGFR and ALK inhibitors have shown significant efficacy,4 there is as of yet no first line targeted therapy approved for the treatment of patients with LUSC.5–7 Recently, immune checkpoint blockades via antibodies that block the inhibitory immune-checkpoint proteins such as programmed cell death protein 1 (PD-1) or its ligand (PD-L1), have emerged as a key component of the standard of care treatment for LUSC, but the overall response rate remains low.8,9 Therefore, identifying effective therapeutics represents an urgent unmet need for LUSC patients.
Despite extensive genomic analysis, the identification of oncogenic drivers in LUSC remains challenging.3,10 Alteration of the epigenetic landscape represents a hallmark of cancer.11 Indeed, genes encoding epigenetic regulators are frequently mutated in LUSC.10,12,13 Among the highest mutated genes is KMT2D (also known as MLL2, MLL4), which occurs in more than 20% of LUSC cases and tends to be clonal in tumorigenesis.14 The histone methyltransferase KMT2D modulates chromatin structure by promoting H3K4 methylation, which activates enhancers and gene expression involved in development, differentiation, and metabolism.15 A growing body of literature reveals that KMT2D loss induces epigenomic reprograming to rewire molecular pathways in multiple types of cancer.16–18 Recently, Kmt2d inactivation has been shown to promote KRAS-driven lung adenocarcinoma growth through activating glycolysis.19 However, the role of KMT2D in driving LUSC is poorly understood. In this study, we leverage our state-of-theart preclinical platform of organoids and murine models coupled with comprehensive transcriptomic and epigenetic profiling to investigate the function of KMT2D in LUSC oncogenesis.
RESULTS
Kmt2d deletion promotes lung organoids transformation
KMT2D is one of the most frequently mutated genes in LUSC, ranking third (24%) and second (22%) among all cancer-related genes (OncoKB Cancer Gene List) in the TCGA PanCancer Atlas 20 and Genomics, Evidence, Neoplasia, Information, Exchange (GENIE) databases,21 respectively (Figure S1A). The KMT2D protein contains 2 clusters of PHD domains in N-terminus and the enzymatic SET domain in C-terminus.22 Almost half of KMT2D mutations are truncating mutations that potentially lead to loss of its catalytic activity (Figure S1B). In addition, Genotype-Tissue Expression (GTEx 23) and TCGA data revealed that KMT2D expression is significantly lower in LUSC than the normal lung tissues 24 (Figure S1C). These data indicate that KMT2D might play an important role in LUSC oncogenesis. To investigate the function of KMT2D during this process, we utilized a mouse organoid system derived from lung basal progenitor cells,25 a hypothesized cell of origin for LUSC.26,27 Because the vast majority of KMT2D mutations co-occur with TP53 mutations (99 in 113 samples, ~87.6%) (Figure 1A), we first established mouse lung basal cell organoids from the C57BL/6J Trp53L/L mice (see STAR Methods), in which the floxed Trp53 gene allele can be conditionally inactivated by the Cre recombinase. These Trp53L/L normal lung organoids formed epithelial spheres after 7 days of culture (Figure S2A). After expansion, these organoids were subsequently infected with adenovirus-Cre (Ad-Cre-GFP), followed by flow cytometry sorting of the GFP+ cells, yielding Trp53−/− organoids (Figures 1B and 1C).
Figure 1. Kmt2d deletion promotes lung organoids transformation.

(A) OncoPrint showing frequency of KMT2D mutations and their co-occurrence with TP53 mutations in human LUSC database (TCGA, PanCancer Atlas, n=469).
(B) Schematic illustration of the workflow for establishing mutant organoids and syngeneic cell lines from parental Trp53L/L lung basal cell organoids.
(C) Western blot confirmation of P53 loss in the Trp53−/− organoids, with β-Actin as the loading control.
(D) Western blot confirmation of KMT2D loss in the Trp53−/−; Kmt2d−/− organoids, with HSP90 as the loading control.
(E) Representative images of hematoxylin and eosin (H&E) staining, and immunohistochemistry (IHC) staining of ΔNp63 in organoids with indicated genotypes. Scale bars, 100 μm.
(F) Representative images from brightfield microscopy and immunofluorescence staining of organoids after 7 days of culture. Organoids were stained with DAPI (blue), NGFR (red) and Ki-67 (green). Scale bars, 100 μm.
(G) Violin plots showing quantifications of the diameter and relative Ki-67 intensity in organoids with indicated genotypes. **p < 0.01, ***p < 0.001, ****p < 0.0001, NS, not significant (unpaired two-tailed t test).
(H) Quantifications of tumor volumes 6 weeks after implanting organoids into C57BL/6J mice. Data shown as means ± SEM. ****p < 0.0001 (unpaired two-tailed t test).
(I) (Left) Representative images of subcutaneous tumors from implanted organoids with indicated genotypes. The red circles indicate the tumors. (Right) Representative images of H&E staining and IHC staining of KRT5 and ΔNp63 in tumors derived from Trp53−/−; Kmt2d−/− and Trp53−/−; Pten−/− organoids. Scale bars, 100 μm.
See also Figures S1 and S2.
We next infected Trp53−/− organoids with CRISPR/Cas9 sgRNAs targeting Kmt2d, which were positively selected in medium with antibiotics. Mutations at Kmt2d locus were subsequently confirmed by genomic sequencing (Figure S2B) and KMT2D protein loss was further verified by western blot (Figure 1D). The morphology and histology of the genetically engineered organoids were analyzed by hematoxylin and eosin (H&E) staining and immunohistochemistry (IHC), respectively. Both Trp53L/L and Trp53−/− organoids contain multi-layered epithelium with basal cells at the periphery (Figure 1E). Strikingly, deleting Kmt2d in Trp53−/− organoids profoundly altered organoid shape and transformed the multi-layered epithelial spheres into disorganized cellular masses with characteristics of keratinization (Figures 1E, S2C–S2E). Furthermore, squamous differentiation features and a high level of basal cell marker ΔNp63 (P40) were observed in the Trp53−/−; Kmt2d−/− organoids (Figures 1E, S2D and S2E). To examine whether Kmt2d loss confers a growth advantage, we performed immunofluorescence staining of the proliferation marker Ki-67 on organoids. Compared to the Trp53L/L and Trp53−/− parental organoids, Trp53−/−; Kmt2d−/− organoids exhibited significantly higher Ki-67 expression in the basal epithelia marked by NGFR expression (Figures 1F and 1G). As a result, Trp53−/−; Kmt2d−/− organoids grew to a larger size compared to the parental controls (Figures 1F and 1G). These data indicate that Kmt2d deletion promotes overgrowth and squamous differentiation in lung basal cell organoids in vitro, consistent with early malignant transformation.
Kmt2d deletion drives LUSC in vivo
To investigate the oncogenic potential of genetically engineered organoids in vivo, we implanted the Trp53−/−; Kmt2d−/− organoids and control Trp53−/− organoids into flanks of C57BL/6J mice. In parallel, we established the Trp53−/−; Pten−/− organoids and injected them in vivo as a control for LUSC, because PTEN is frequently mutated in LUSC (Figure S2F) and PTEN inactivation promotes oncogenic tumor growth in multiple LUSC models.25,28,29 6 weeks after injection, tumors formed in mice injected with Trp53−/−; Kmt2d−/− and Trp53−/−; Pten−/− organoids, but not with the Trp53−/− organoids (up to 30 weeks of observation) (Figure 1H), suggesting Trp53 loss alone in basal cell organoids is not sufficient to generate LUSC in vivo. Histologic analysis of Trp53−/−; Kmt2d−/− and Trp53−/−; Pten−/− tumors revealed keratinization, keratin pearl formation and strong expression of KRT5 and ΔNp63, consistent with LUSC hallmarks (Figures 1I and S2G). To further evaluate whether Trp53−/−; Kmt2d−/− organoids can directly form LUSC in mouse lungs, we performed transthoracic implantation of the organoids into mouse lungs (Figure S2H). Remarkably, Trp53−/−; Kmt2d−/− organoids formed lung tumors orthotopically that mirrored histological features of LUSC, with a latency (40–50 weeks) similar to the genetically engineered mouse models (GEMMs) of LUSC.30 In summary, these findings support that Kmt2d deletion, in the absence of Trp53, can drive LUSC formation in vivo.
Establishing the Kmt2d-deficient orthotopic LUSC model
The long and variable latency of the transthoracic model renders it suboptimal for evaluation of KMT2D function and for identification of therapeutic vulnerabilities in vivo. Thus, to establish a robust orthotopic LUSC model (with a consistent and short latency) for further characterization, we first harvested the Trp53−/−; Kmt2d−/− tumor nodules and generated the syngeneic Trp53−/−; Kmt2d−/− cell lines (Figures 1B and S3A). We next inoculated Trp53−/−; Kmt2d−/− cells into B6-Albino mice (see STAR Methods)) through intravenous injection and monitored tumor growth in the lung by magnetic resonance imaging (MRI) (Figure 2A). Likewise, as an alternative LUSC model for comparison, we established the Trp53−/−; Pten−/− orthotopic model in B6-Albino mice. 6 to 8 weeks after implantation, mice injected with Trp53−/−; Kmt2d−/− or Trp53−/−; Pten−/− cells both developed lung tumors (Figure 2B). The Trp53−/−; Kmt2d−/− cells and orthotopic tumors grew slower than the Trp53−/−; Pten−/− counterparts (Figures S3B and 2B). Accordingly, the Trp53−/−; Kmt2d−/− tumor bearing mice had longer survival than the Trp53−/−; Pten−/− counterparts (Figure 2C). To confirm the lung tumor histology, we performed H&E and IHC staining of the LUSC markers including ΔNp63 and KRT5, and the LUAD marker TTF1. EGFR-T790M-L858R mouse lung tumors, the well-established model of LUAD, were used as the negative control.31 H&E staining of Trp53−/−; Kmt2d−/− tumors showed clear squamous features such as stratification and keratinization, which were also observed in the Trp53−/−; Pten−/− lung tumors (Figure 2D). Trp53−/−; Kmt2d−/− tumors strongly expressed the LUSC markers ΔNp63 and KRT5, but not the LUAD marker TTF1, similar to the Trp53−/−; Pten−/− lung tumors (Figure 2E). In contrast, EGFR-mutant LUAD tumors were positive for TTF1, but negative for ΔNp63 and KRT5. To further confirm the Kmt2d loss in Trp53−/−; Kmt2d−/− tumors, we also performed IHC staining of KMT2D in the tumor sections. As expected, KMT2D is not detected in the Trp53−/−; Kmt2d−/− tumors, whereas Trp53−/−; Pten−/− and EGFR-mutant tumors expressed KMT2D in the nucleus (Figure 2E). Thus, these orthotopic Trp53−/−; Kmt2d−/− lung tumors exhibit classic LUSC histopathology that recapitulates the human disease.
Figure 2. Kmt2d deletion drives LUSC in vivo.

(A) Schematic illustration for the orthotopic LUSC model from tumor-derived syngeneic cells. Tumor growth was monitored by magnetic resonance imaging (MRI).
(B) Representative mouse lung MRI images at indicated times after injecting cells with indicated genotypes. The red arrows indicate lung tumors.
(C) Kaplan-Meier curves of tumor bearing mice with the indicated genotypes. (n = 8 for Trp53−/−; Kmt2d−/− and n = 8 for Trp53−/−; Pten−/−).
(D) H&E staining of Trp53−/−; Kmt2d−/− and Trp53−/−; Pten−/− lung tumors showing squamous carcinoma histology.
(E) Representative images of IHC staining of ΔNp63, KRT5, TTF1, and KMT2D from lung tumors with the indicated genotypes. Scale bars, 100 μm.
(F) Heatmap and hierarchical clustering of differentially expressed transcripts from normal mouse lung tissues, LUAD (KrasG12D; Trp53−/−) and LUSC (Trp53−/−; Kmt2d−/− and Trp53-/−; Pten−/−).
(G) Heatmap showing LUSC and LUAD marker gene expression in normal mouse lung tissues, LUAD (KrasG12D; Trp53−/−) and LUSC (Trp53−/−; Kmt2d−/− and Trp53−/−; Pten−/−). Genes shown were in “Keratins”, “Transcription factors (or TFs)”, “Secreted factors”, “Cell surface” and “Enzymes” categories.
See also Figure S3.
To examine whether the Trp53−/−; Kmt2d−/− and Trp53−/−; Pten−/− tumors recapitulate the molecular signature of LUSC, bulk RNA sequencing (RNA-seq) was performed to comprehensively evaluate the transcriptomic features. In parallel, we also analyzed the gene expression profiles of LUAD tumors (KrasG12D; Trp53−/−, KP) and normal lung tissues for comparison.32,33 Principal component analysis (PCA) revealed that Trp53−/−; Kmt2d−/− and Trp53−/−; Pten−/− tumors were clustered together and separated from the KP tumors and normal lung tissues (Figure S3C). Unsupervised hierarchical clustering of differentially expressed genes also revealed a high level of similarity between Trp53−/−; Kmt2d−/− and Trp53−/−; Pten−/− tumors, which are distinct from KP tumors and normal lung tissues (Figure 2F). Expression of LUSC hallmark genes was evidently upregulated and levels of LUAD associated genes were decreased in Trp53−/−; Kmt2d−/− and Trp53−/−; Pten−/− tumors, compared with the KP tumors and normal lung tissues (Figure 2G). For example, levels of cytokeratin genes such as Krt5 and Krt14 and transcriptional factor genes Sox2 and Trp63 were elevated, whereas expressions of LUAD marker genes including Nkx2–1, Sftpa1, Sftpb, Sftpc and Sftpd were decreased. Additionally, expressions of genes encoding secreted factors such as Wnt (Wnt4, Wnt7b), Bmp (Bmp6, Bmp7) and interleukin superfamilies (Il1a, Il1f9, Il1rn), transcriptional factors (Pax9), enzymes (Arg1, Serpinb1a, Serpinb2, Serpinb11), and cell surface proteins (Ngfr, Lgr4, Egfr, Itgb4) were upregulated in Trp53−/−; Kmt2d−/− and Trp53−/−; Pten−/− tumors, similar to the gene expression profiles of LUSC GEMMs and human LUSC.28,29,33 Taken together, these findings suggest that Trp53−/−; Kmt2d−/− tumors strongly recapitulate the histologic and molecular signatures of human LUSC.
Kmt2d deletion activates RTK-Ras signaling in LUSC
We next sought to characterize the molecular features that are unique to Trp53−/−; Kmt2d−/− (Kmt2d KO, used hereafter), in comparison to those of the Kmt2d wild-type (Kmt2d WT, Trp53−/− or Trp53−/−; Pten−/−) counterparts. Gene set enrichment analysis (GSEA) of the transcriptomic data revealed that KRAS signaling was one of the most positively enriched signatures in the Kmt2d KO cell lines, compared to the Kmt2d WT cell lines (Figures 3A and 3B). Many KRAS signaling related genes, such as Etv1, Etv5, Spry2 and Ereg, were upregulated upon Kmt2d loss (Figure 3C). In addition to the activation of KRAS signaling, several other key cancer hallmark signatures were significantly enriched, including “unfolded protein response”, “cholesterol homeostasis”, “Myc targets V1”, and “TNFa signaling via NFKB” in the Kmt2d KO cells (Figure 3A). Consistently, GSEA of differentially expressed genes revealed that KRAS signaling is also significantly enriched in the Kmt2d KO organoids, compared to the Kmt2d WT organoids (Figure S3D). Analysis of the TCGA LUSC database further confirmed that KRAS signaling was enriched in human KMT2D low LUSC versus KMT2D high LUSC samples (Figure 3D). Moreover, in line with KRAS signaling activation, phospho-ERK level was much higher in Kmt2d KO cell lines, compared to the Kmt2d WT cells (Figure 3E).
Figure 3. Kmt2d deletion activates RTK-RAS signaling in LUSC.

(A) Dot plots showing positively enriched pathways (NOM P < 0.05 and FDR q < 0.25) in Gene Set Enrichment Analysis (GSEA) comparing Kmt2d KO (Trp53−/−; Kmt2d−/−) versus the Kmt2d WT (Trp53−/−; Pten−/−) tumor-derived cell lines. “KRAS signaling up” ranks the second among positively enriched pathways.
(B) GSEA analysis showing the significantly enriched KRAS signaling from Figure 3A.
(C) Heatmap showing genes that were significantly upregulated (Log2FC >1) in the “KRAS signaling up” gene set from Figure 3B.
(D) GSEA analysis showing the significantly enriched KRAS signaling pathway comparing KMT2D low versus KMT2D high LUSC tumors (TCGA LUSC dataset).
(E) Western blot showing ERK, pERK and β-Actin in Kmt2d KO (Trp53−/−; Kmt2d−/−) and Kmt2d WT (Trp53−/−; Pten−/−) cells and quantifications of pERK/ERK. Data shown as means ± SEM. *p < 0.05 (unpaired two-tailed t test).
(F-H) Phospho-receptor tyrosine kinase arrays for Kmt2d KO and Kmt2d WT organoids (F, Trp53−/− vs Trp53−/−; Kmt2d−/−), cell lines (G, Trp53−/−; Pten−/− vs Trp53−/−; Kmt2d−/−) and tumor nodules (H, Trp53−/−; Pten−/− vs Trp53−/−; Kmt2d−/−). pEGFR and pERBB2 are highlighted by the arrows.
(I) Quantifications of pEGFR and pERBB2 in Kmt2d KO and the Kmt2d WT organoids, cell lines and tumor nodules as indicated above. Data shown as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 (unpaired two-tailed t test).
(J) Scatter plots showing a negative correlation between KMT2D mRNA level and phospho-EGFR expression in human TCGA LUSC dataset. r, Pearson’s correlation coefficient.
(K) Violin plots showing the relative phospho-EGFR protein expression in KMT2D mutant LUSC tumors and their paired normal lung tissues from Satpathy et al.37 *p < 0.05 (unpaired two-tailed t test).
See also Figure S3.
We next sought to identify potential upstream regulators responsible for the aberrant KRAS signaling upon Kmt2d loss. Given that receptor tyrosine kinases (RTKs) are well-characterized activators of the KRAS signaling in cancer,34 we performed a comprehensive phospho-RTK array that interrogates the phosphorylation of 39 RTKs (Figures 3F–3I). Notably, a significant increase in EGFR and ERBB2 phosphorylation was observed in Kmt2d KO organoids and cell lines when compared to the Kmt2d WT counterparts (Figures 3F, 3G and 3I). Consistently, Kmt2d KO tumors also had significantly higher levels of EGFR and ERBB2 phosphorylation, compared to the Kmt2d WT tumors (Figures 3H and 3I). Western blot further confirmed the increased phosphorylation of EGFR and ERBB2 in the Kmt2d KO tumors (Figure S3E). Therefore, our data suggest that RTK-RAS signaling is activated upon Kmt2d loss in LUSC.
We further investigated whether the increased levels of EGFR phosphorylation are associated with KMT2D loss in human LUSC. Utilizing the DepMap dataset (https://depmap.org/portal/),35 we investigated the correlation between KMT2D expression and levels of phospho-EGFR in a panel of 19 human LUSC cell lines. Consistently, KMT2D expression was negatively correlated with phospho-EGFR levels, measured by reverse phase protein arrays (RPPA) (Figure S3F). Exploiting the TCGA Pan-Cancer Atlas dataset,20,36 we next analyzed the correlation between KMT2D RNA levels with phosphorylated EGFR in human LUSC specimens. KMT2D mRNA abundance was negatively correlated with the level of phospho-EGFR (Figure 3J), further supporting our observations in the cell lines and mouse tumors. To validate this finding in separate cohorts, we expanded our analyses on a large-scale collection of a proteomics dataset of LUSC tumors and paired normal adjacent tissues.37 Our analysis revealed that KMT2D mutant LUSC tumors exhibited elevated phospho-EGFR compared to the paired normal lung tissues (Figure 3K). Finally, we knocked out KMT2D in human LUSC cell lines (EBC1 and HCC95) using CRISPR/Cas9 to evaluate the impact on phospho-EGFR. Notably, KMT2D loss led to an increase in the levels of phospho-EGFR in human LUSC cells (Figures S3G and S3H).
In summary, our data suggest that Kmt2d deletion promotes oncogenic RTK-RAS signaling through activating EGFR and ERBB2 in both murine and human LUSC.
KMT2D loss confers hypersensitivity to SHP2 and pan-ERBB inhibition in vitro
We next sought to identify potential therapeutic vulnerabilities of KMT2D-deficient LUSC. Based upon the aforementioned findings, we reasoned that Kmt2d KO LUSC would be hypersensitive to inhibitors that target oncogenic RTK-RAS signaling. SHP2 (encoded by PTPN11) is a protein tyrosine phosphatase that mediates KRAS activation downstream of RTKs.38 Targeting SHP2 with the allosteric inhibitor SHP099 is effective in tumors with elevated RTK-RAS signaling.39–42 Afatinib is a pan-ERBB family receptor tyrosine kinase inhibitor, which has been approved for the second-line treatment in LUSC 43,44. Thus, we hypothesized that SHP099 and afatinib would inhibit KMT2D-deficient LUSC growth (Figure 4A). We first investigated the effects of SHP099 and afatinib on cell viability in three different Kmt2d KO LUSC cell lines, Kmt2d WT LUSC cell lines, and the KP LUAD cell line. Notably, all three Kmt2d KO LUSC cell lines were hypersensitive to SHP099 treatment, with the IC50 of 0.559 μM, 0.310 μM and 1.165 μM respectively (Figure 4B). In comparison, the IC50 of SHP099 in Kmt2d WT LUSC cell lines were 9.429 μM and 4.79 μM, whereas the IC50 in KP cells was higher than 20 μM. The hypersensitivity in the Kmt2d KO cells and specificity to SHP2 inhibition were further confirmed by knocking out SHP2 using CRISPR/Cas9 and another SHP2 selective inhibitor TNO155 45 (Figures S4A and S4B). Furthermore, afatinib was also highly effective in inhibiting cell viability in the Kmt2d KO LUSC cell lines, with a much lower IC50 (<0.02 μM) (Figure 4C), in comparison to >1 μM in Kmt2d WT LUSC and KP cells. In line with this, Kmt2d KO LUSC cell lines were also more sensitive to another pan-ERBB inhibitor neratinib, when compared with Kmt2d WT LUSC and KP cells (Figure S4C). Additionally, utilizing the DepMap dataset,46 our analysis revealed that lower KMT2D levels were associated with a higher sensitivity to afatinib and other pan-ERBB inhibitors including neratinib, lapatinib and poziotinib (Figure S4D). To evaluate the long-term drug treatment effect on cell survival, we performed a 7-day colony formation assay with SHP099, afatinib alone or in combination in Kmt2d KO cells. SHP099 and afatinib alone dramatically inhibited the colony formation in Kmt2d KO cells, whereas combining SHP099 and afatinib led to a further reduction of colonies (Figure 4D). These findings suggest that Kmt2d KO cells are hypersensitive to SHP2 and pan-ERBB inhibitors, such as SHP099 and afatinib, and this inhibitory effect is further enhanced when combining SHP099 and afatinib.
Figure 4. Kmt2d-deficient LUSC is hypersensitive to SHP2 and pan-ERBB inhibition.

(A) Schematic illustration of targeting RTK-RAS signaling through SHP2 inhibitor SHP099 and pan-ERBB inhibitor afatinib.
(B and C) Cell viability assays of Kmt2d KO LUSC cell lines, Kmt2d WT LUSC cell lines, and LUAD (KP) cell line treated with SHP099 (B) and afatinib (C) for 72h. Data presented as mean ± SD (n = 3). The calculated IC50 values of SHP099 and afatinib are shown on the right.
(D) Colony formation assay of Kmt2d KO cells treated with vehicle, SHP099, afatinib, and combination of SHP099 and afatinib for 7 days.
(E) Western blot of ERK, pERK and β-Actin on Kmt2d KO (Trp53−/−; Kmt2d−/−) tumors treated with vehicle, SHP099, afatinib and combination of SHP099 and afatinib for 3 days.
(F) Heatmap showing the changes in KRAS signaling downstream gene expression by RNA-seq in Kmt2d KO tumors treated as indicated in Figure 4E.
(G) Plots showing top negatively enriched pathways in GSEA comparing combination of SHP099 and afatinib (combo) treated versus vehicle treated Kmt2d KO tumors.
(H) GSEA analysis showing top negatively enriched pathways “E2F targets”, “G2M checkpoint” and “Myc targets V1” comparing combo treated tumors versus the vehicle treated tumors.
(I) IHC analysis of Ki-67 and cleaved caspase-3 from Kmt2d KO tumors with indicated treatment. Scale bars, 100 μm.
(J) Quantifications of IHC score of Ki-67 and cleaved caspase-3 of indicated treatment. Data shown as means ± SEM. **p < 0.01, ****p < 0.0001 (unpaired two-tailed t test).
See also Figure S4.
SHP099 and afatinib attenuate RTK-RAS signaling in Kmt2d-deficient LUSC
We next examined whether SHP099 and afatinib alone, and in combination, would inhibit RTK-RAS signaling in Kmt2d KO LUSC. Treating Kmt2d KO LUSC cells in vitro with SHP099 or afatinib alone robustly reduced pERK levels (Figure S4E). Combining SHP099 with afatinib led to the most significant reduction in pERK levels. To further examine the downstream effects in vivo, we established the orthotopic Kmt2d KO LUSC model (Figure 2A) and randomized mice into four groups: vehicle, SHP099 (75mpk, QD), afatinib (10mpk, QD), and combined SHP099 with afatinib (combo). Tumor nodules were collected after 3 days of treatment to examine the pharmacodynamics on downstream signaling. As expected, western blot revealed that in comparison to the vehicle, SHP099 and afatinib monotherapy substantially reduced the levels of pERK, while the combination therapy led to a greater decrease (Figure 4E). In line with these results, transcriptomic analysis of tumor nodules by RNA-seq showed that the levels of KRAS-dependent genes were substantially downregulated upon SHP099 and afatinib monotherapy, and combinational treatment (Figure 4F). Furthermore, GSEA of differentially expressed genes revealed that the combination treatment negatively affected genes associated with “E2F targets”, “G2M checkpoint” and “Myc targets” (Figures 4G and 4H). To further characterize the antiproliferative impact of SHP099 and afatinib, we performed IHC staining of the proliferation marker Ki-67 and apoptotic marker cleaved caspase-3 on the treated Kmt2d KO tumors. SHP099 and afatinib alone significantly decreased Ki-67 expression and increased cleaved caspase-3 levels in vivo, whereas the combination treatment led to the most significant effects (Figures 4I and 4J). Collectively, SHP099 and afatinib alone significantly inhibits RTK-RAS signaling in Kmt2d KO tumor in vitro and in vivo, which is further enhanced when in combination.
SHP099 and afatinib diminish KMT2D-deficient LUSC in vivo
We next determined whether targeting SHP2 and ERBB would inhibit tumor growth and prolong survival in Kmt2d KO LUSC in vivo. Upon confirmation of tumor burden, mice were randomized into vehicle, SHP099 and afatinib as single agents and combinational treatment (combo), and the tumor growth were monitored via MRI (Figures 5A and S5A). An additional group of mice were enrolled to chemotherapy (carboplatin plus paclitaxel) as the standard-of-care regimen for comparison. No significant weight loss was observed in all treatment groups (Figure S5B). All vehicle-treated mice displayed aggressive disease, with tumor volumes doubled after a 2-week period (Figures 5B–5D). While chemotherapy showed no effect in inhibiting tumor growth, SHP099 or afatinib alone significantly attenuated Kmt2d KO LUSC growth. Importantly, combining SHP099 and afatinib led to the most dramatic decrease of tumor volumes, with a reduction observed in all treated mice (n=9), including two-thirds (6 of 9) of the mice having >50% reductions (Figures 5B–5D). Long term MRI follow-up revealed that tumors began to develop resistance to SHP099 and afatinib monotherapy after 4 weeks of treatment, and by 6 weeks, most mice in the monotherapy groups had progressive disease (Figure 5E). This contrasts with the mice in combo treatment, which had a better response, with 7 of 9 (77.78%) mice still undergoing tumor shrinkage after 6 weeks of treatment.
Figure 5. SHP099 and afatinib diminish KMT2D-deficient LUSC in vivo.

(A) Schematic showing in vivo dosing schedule. After inoculating LUSC cells into mice, lung tumor burden was confirmed by MRI. Mice were then randomized and treated with vehicle, chemotherapy (chemo, carboplatin + paclitaxel), SHP099 (75mpk, 5 days per week), afatinib (10mpk, 5 days per week) alone or combined SHP099 with afatinib. Tumor growth was measured by MRI and survival was recorded.
(B and C) Waterfall plot (B) and dot plot (C) of changes in tumor volumes after 2 weeks of treatment in Kmt2d KO (Trp53−/−; Kmt2d−/−) LUSC model: vehicle (n=9), chemo (n=6), SHP099 (n=8), afatinib (n=9), and combo (n=9).
(D) Representative MRI images of Kmt2d KO lung tumor at baseline (0 week), 2 weeks, and 4 weeks after treatment initiation. The red arrows indicate lung tumors.
(E) Tumor volume changes of Kmt2d KO LUSC tumors treated as indicated in Figure 5A.
(F) Kaplan-Meier survival curve for the Kmt2d KO LUSC model after indicated treatment. Vehicle (n=9), chemo (n=6), SHP099 (n=9), afatinib (n=8), and combo (n=9). *p < 0.05, **p < 0.01, ****p < 0.0001 (log-rank test).
(G) Tumor volume changes of Trp53−/−; Pten−/− (n=7–8) and Trp53−/−; Pten−/−; Kmt2d−/− (n=6–8) allografts with indicated treatment.
(H) Tumor volume changes of KMT2D mutant LUSC PDX (PDX-1, LX-515) following treatments with vehicle (n=4), SHP099 (n=5), afatinib (n=3) and combined SHP099 with afatinib (n=7). Representative images of H&E and IHC staining of KMT2D are shown. Scale bars, 100 μm.
(I) Tumor volume changes of KMT2D WT LUSC PDX (PDX-2, LX-640) following treatments with vehicle (n=6), SHP099 (n=4), afatinib (n=5) and combined SHP099 with afatinib (n=6). Representative images of H&E and IHC staining of KMT2D are shown. Scale bars, 100 μm.
(J) Tumor volume changes of human HARA-sgCtrl xenografts following treatments with vehicle (n=14), and combined SHP099 with afatinib (n=14), as well as HARA-sgKMT2D xenografts following treatments with vehicle (n=16), and combined SHP099 with afatinib (n=15).
(K) Waterfall plot showing changes in tumor volumes after 3 weeks of treatment (as indicated in Figure 5J) in HARA-sgCtrl and HARA-sgKMT2D LUSC models.
In (B), (C) and (K), data shown as means ± SEM, **p < 0.01, ***p < 0.001 ****p < 0.0001, NS, not significant (unpaired two-tailed t test). In (G), (H), (I) and (J), data shown as means ± SEM, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS, not significant (ANOVA).
See also Figure S5.
We next examined whether the efficacy of SHP2 and pan-ERBB inhibition can also prolong the survival of Kmt2d KO LUSC tumor-bearing mice. As expected, compared with the vehicle group, chemotherapy failed to prolong overall survival (OS) (Figure 5F). Afatinib monotherapy moderately prolonged animal survival, but the benefit was not statistically significant. Notably, SHP099 treatment markedly increased the median OS to 64 days, compared to the 41 days in vehicle group. Most importantly, combining SHP099 and afatinib dramatically extended the OS of tumor-bearing mice in comparison to either SHP099 or afatinib alone. Compared with the vehicle, the combination treatment led to more than a 2-fold increase in median OS (84 days versus 41 days), with an added median OS benefit of 43 days. By contrast, the Kmt2d WT LUSC (Trp53−/−; Pten−/−) appeared to be less sensitive to SHP099 and afatinib treatment (Figure S5C), consistent with the in vitro cell viability results (Figures 4B and 4C). To further validate the drug sensitivity is due to Kmt2d loss, we generated the Trp53−/−; Pten−/−; Kmt2d−/− model and directly compared the in vivo response to SHP2 and pan-ERBB inhibition between Trp53−/−; Pten−/−; Kmt2d−/− and Trp53−/−; Pten−/− LUSC. The Trp53−/−; Pten−/−; Kmt2d−/− LUSC grew at a similar speed compared to the Trp53−/−; Pten−/− LUSC in vivo. Remarkably, Trp53−/−; Pten−/−; Kmt2d−/− LUSC was particularly sensitive to the combination treatment (Figures 5G and S5D). All tumors (7/7, 100%) exhibited a volume reduction after 3 weeks of treatment (Figure S5D). Thus, loss of Kmt2d in murine Trp53−/−; Pten−/− LUSC enhances their response to SHP2 and pan-ERBB inhibition.
We next sought to evaluate whether SHP2 and pan-ERBB inhibition might also be effective in human LUSC with KMT2D mutation in vivo. LK2, a human LUSC cell line with KMT2D nonsense mutation, was subcutaneously implanted into the flank of nude mice, which were then randomized into treatments with vehicle, SHP099, afatinib or in combination. SHP099 or afatinib alone moderately delayed LK2 tumor growth, whereas combining SHP099 with afatinib had the most significant antitumor effects (Figure S5E). In line with this, the combinational therapy led to the best OS benefit in LK2 tumor bearing mice with a median OS of 33.5 days from treatment initiation (Figure S5F). By contrast, vehicle, SHP099 and afatinib treated mice had a median OS of 14, 28 and 19 days, respectively. Additionally, we generated four patient-derived xenografts (PDXs, PDX-1 and 3 are KMT2D mutant; PDX-2 and 4 are KMT2D WT) from LUSC patient specimens to further assess the responses to SHP099 and afatinib treatment. Of note, no mutations in the ERBB family genes were detected in these four PDXs. In line with the murine data, SHP099 or afatinib monotherapy dramatically reduced tumor growth compared to the vehicle control in the KMT2D mutant PDX-1 (Figures 5H). Combining SHP099 and afatinib offered a superior antitumor effect compared with the monotherapy, leading to more than 30% decrease of tumor volumes in all treated mice (n=7). For the KMT2D mutant PDX-3, SHP099 or afatinib alone did not significantly inhibit tumor growth, although there is a trend towards tumor reduction (Figures S5G). Nonetheless, combining SHP099 and afatinib significantly attenuated tumor growth compared to the vehicle. These data support that PDXs with KMT2D mutations are hypersensitive to RTK-Ras inhibition (SHP099 + afatinib). By contrast, the KMT2D WT PDXs were less sensitive to SHP099 and afatinib treatment (Figures 5I and S5H). The KMT2D WT PDX-4 did not respond to SHP099 and afatinib treatment (Figure S5H). Interestingly, we observed a tumor growth reduction in the KMT2D WT PDX-2 in response to SHP099 and afatinib treatment compared to the vehicle (Figure 5I), raising the possibility that other underlying mutations might also contribute to the sensitivity to RTK-RAS inhibition in LUSC. Taken together, these findings support that human LUSC with KMT2D loss is hypersensitive to RTK-RAS inhibition.
We next examined whether KMT2D loss would increase the sensitivity to SHP2 and pan-ERBB inhibition in human LUSC cell line xenografts. We generated a pair of isogenic cell lines in the KMT2D WT human LUSC, namely HARA-sgCtrl and HARA-sgKMT2D. HARA- sgCtrl and HARA-sgKMT2D tumors grew at a similar rate in mice (Figure 5J). Notably, HARA-sgKMT2D tumors were hypersensitive to the combined SHP099 and afatinib treatment compared to the vehicle control, while HARA-sgCtrl tumors were not significantly responding to this treatment (Figures 5J and 5K), highlighting again that KMT2D loss renders LUSC tumors hypersensitive to RTK-RAS inhibition in vivo.
In summary, our extensive in vivo therapeutic studies demonstrate that SHP099 or afatinib alone significantly inhibits tumor growth and prolongs survival in multiple murine and human KMT2D-deficient LUSC models. Moreover, combining SHP099 with afatinib offers superior antitumor efficacy and survival benefits.
KMT2D loss reprograms epigenetic landscape in LUSC
KMT2D-mediated H3K4 methylation is a prerequisite for activation of distal enhancers marked by H3K27 acetylation (H3K27ac).47–49 Indeed, KMT2D loss has been associated with decreased H3K27ac at distal enhancers and reduced expression of their associated genes.48–50 To define how KMT2D loss affects gene enhancers in facilitating LUSC oncogenesis, we performed H3K27ac chromatin immunoprecipitation sequencing (ChIP-seq) in Kmt2d KO and Kmt2d WT cell lines. The resulting H3K27ac sites (or “peaks”) were classified into lost, gained and unaffected peaks in Kmt2d KO versus Kmt2d WT cells (Figures 6A and 6B). As expected, the majority of H3K27ac sites (26,835 sites, 76.57%) were not significantly perturbed. Notably, we found 5,301 H3K27ac sites lost (15.11%) and 2,938 gained (8.37%) in Kmt2d KO cells. The finding of more lost sites than gained ones is consistent with previous work, supporting that KMT2D primarily functions as an activator of H3K27ac.15
Figure 6. Kmt2d loss reprograms epigenetic landscape in LUSC.

(A) Heatmaps showing the H3K27ac ChIP-seq signal in Kmt2d WT (Trp53−/−; Pten−/−) and Kmt2d KO (Trp53−/−; Kmt2d−/−) cell lines. Based on the ChIP-seq signal changes, H3K27ac sites were categorized into three groups: Kmt2d KO -lost, -gained and -unaffected.
(B) Averaged H3K27ac ChIP-seq signal, centered at the Kmt2d KO-lost, -gained, and -unaffected H3K27ac sites.
(C) RNA-seq results showing downregulated (left upper corner) and upregulated (right upper corner) genes in Kmt2d KO cell lines (FDR<0.05; Fold Change>1.5). Genes that were associated with lost and gained H3K27ac sites (genes with the closest distances to the sites) are highlighted by red and blue, respectively.
(D) The comparison of lost H3K27ac sites-associated genes versus RNA-seq downregulated genes in Kmt2d KO cells (up). And the comparison of gained H3K27ac sites-associated genes versus RNA-seq upregulated genes in Kmt2d KO cells (down).
(E) The percentage of genes associated with Kmt2d KO -gained, -lost and -unaffected H3K27ac sites that were downregulated (left) or upregulated (right) based on RNA-seq results.
(F) Averaged ATAC-seq signal, centered at the Kmt2d KO-lost, -gained, and -unaffected ATAC-seq sites (left). Pie graft showing number of Kmt2d KO -lost, -gained, and -unaffected ATAC-seq sites (right).
(G) Overlap of H3K27ac lost sites-associated genes, ATAC lost sites-associated genes, and RNA-seq downregulated genes in Kmt2d KO cells.
(H) Gene ontology (GO) analysis enriched pathways in “molecular function”, based on overlapped genes in (G).
(I) Heatmap of RPTPs gene expression (RNA-seq) in Kmt2d KO and Kmt2d WT cells.
We then explored the relationship between Kmt2d loss-affected H3K27ac peaks and gene expression changes (Figure 6C). In particular, we assigned the closest genes to the affected H3K27ac peaks and examined their expression changes in response to Kmt2d loss. The results showed that genes associated with H3K27ac lost sites had significant overlap (n= 796) with genes that are downregulated in Kmt2d KO cells (Figures 6C–6E). On the other hand, genes associated with H3K27ac gained sites exhibited significant overlap (n=564) with genes that are upregulated in Kmt2d KO cells (Figures 6C–6E). These data suggest that KMT2D loss reprograms enhancer activity to affect gene expression in LUSC cells.
KMT2D loss suppresses receptor tyrosine phosphatase expression that potentiates RTK-RAS signaling
Given that KMT2D loss is associated with lost H3K27ac sites and reduced gene transcription, we sought to further characterize the alterations in chromatin organization upon Kmt2d loss. We performed the assay of transposase accessible chromatin-sequencing (ATAC-seq) to profile the genome-wide chromatin accessibility in Kmt2d KO and Kmt2d WT cells. Similar to the H3K27ac ChIP-seq data, ATAC-seq analysis revealed that there were 35,883 unaffected sites, 3,721 lost sites, and 3,276 gained sites in Kmt2d KO cells compared to Kmt2d WT cells (Figure 6F). To define the potential target genes downregulated by Kmt2d loss, we combined RNA-seq downregulated genes, H3K27ac lost sites-associated genes, and ATAC-seq lost sites-associated genes in Kmt2d KO cells, resulting in 359 high-confidence candidate Kmt2d-target genes (Figure 6G, Table S1). Gene ontology (GO) analysis of the 359 genes showed marked enrichment in the “Phosphoric ester hydrolase activity” and “Phosphatase activity” molecular functions (Figure 6H). Most notable among the phosphatase genes were members of receptor-like protein tyrosine phosphatases (RPTPs) (Figure 6I). RPTP family genes are frequently mutated in multiple types of cancers and mainly act as tumor suppressors.51,52 RPTPs have been reported to directly dephosphorylate ERBB to repress RTK-RAS signaling.52,53 In line with the reduction in gene expression, H3K27ac levels were significantly downregulated at the enhancer regions of Ptprb, Ptprf, Ptprs and Ptpru (Figures 7A–7D). Consistently, ATAC-seq analysis revealed that overall chromatin accessibility was also reduced at these regions. We next performed CUT&Tag profiling to examine the alterations of H3K4me1 and H3K4me3, which are known direct targets of KMT2D (Figures S6A–S6D). Likewise, H3K4me1 levels were significantly reduced at the enhancer regions of Ptprb, Ptprf, Ptprs and Ptpru, whereas a notable reduction in H3K4me3 signals at promoter regions of Ptprs and Ptpru, to a lesser extent at Ptprb or Ptprf, was observed (Figures 7A–7D). Together, these findings highlight KMT2D as an important epigenetic regulator in RPTPs expression.
Figure 7. KMT2D loss suppresses the expression of receptor tyrosine phosphatases.

(A-D) Representative H3K27ac, H3K4me1 and H3K4me3 and ATAC-seq signal at loci of Ptprb (A), Ptprf (B), Ptprs (C) and Ptpru (D) in Kmt2d WT (Trp53−/−; Pten−/−) and Kmt2d KO (Trp53−/−; Kmt2d−/−) cells.
(E-H) Scatterplots showing positive correlations of KMT2D mRNA levels with PTPRB (E), PTPRF (F), PTPRS (G) and PTPRU (H) mRNA levels in human TCGA LUSC dataset. r, Pearson’s correlation coefficient.
(I and J) qRT-PCR analysis of PTPRB, PTPRF, PTPRS, and PTPRU gene expression in KMT2D KO and KMT2D WT mouse LUSC cells (I) and human HARA cells (J). Data shown as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (unpaired two-tailed t test).
(K) Western blot of pEGFR, EGFR, pERK, ERK and β-Actin in Kmt2d WT (Trp53−/−; Pten−/−) cells with knockdown of Ptprb, Ptprf, Ptprs and Ptpru using shRNA.
(L) Schematic showing the proposed model of how KMT2D loss promotes LUSC tumorigenesis and hypersensitivity to RTK-RAS inhibition by SHP099 and afatinib.
See also Figure S6.
To examine the association between KMT2D and RPTPs in human LUSC samples, we analyzed the expression of KMT2D versus the RPTPs in TCGA LUSC dataset. KMT2D expression was significantly and positively correlated with the expression of PTPRB, PTPRF, PTPRS and PTPRU (Figures 7E–H). To further confirm KMT2D regulates RPTPs expression in LUSC, we performed qRT-PCR on KMT2D KO and KMT2D WT cells. Consistent with the RNA-seq and epigenetic analysis, Kmt2d loss significantly reduced the expression of Ptprb, Ptprf, Ptprs and Ptpru in Kmt2d KO murine and human LUSC cells (Figures 7I, 7J and S6E).
In order to elucidate the specific RPTP(s) responsible for the elevated RTK-RAS signaling in LUSC, we knocked down each of the RPTPs (Ptprb, Ptprf, Ptprs or Ptpru) individually in the Kmt2d WT LUSC cells (Figures S6F–S6I). Western blot showed that both shRNA targeting Ptprf led to a robust increase in pEGFR and pERK levels, phenocopying the loss of KMT2D (Figure 7K). Interestingly, knocking down Ptprb and Ptpru increased pERK but not pEGFR levels, suggesting that Ptprb and Ptpru might contribute to the activated RAS signaling through other RTKs.
Collectively, our findings indicate that KMT2D loss leads to decreased expression of RPTPs, which in turn activate the oncogenic RTK-RAS signaling to promote tumorigenesis in LUSC.
Functional analysis revealed that Ptprf plays an important role in regulating the RTK-RAS signaling in LUSC oncogenesis.
DISCUSSION
In LUSC, KMT2D is the most frequently mutated epigenetic modifier with unknown function. Leveraging the mouse lung basal cell organoids, we established the Kmt2d-deficient murine models representing human LUSC and comprehensively characterized the function of KMT2D in driving LUSC. We showed that Kmt2d deletion transforms the mouse lung basal cell organoids into LUSC. More importantly, our study revealed that KMT2D loss promotes activation of EGFR and ERBB2, which is partly due to the repressed RPTPs expression. This leads to the potentiation of the RTK-RAS signaling, facilitating tumor growth. Targeting KMT2D-deficient LUSC with SHP2 and pan-ERBB inhibitors effectively reduces murine and human LUSC tumor growth in vivo. Our study establishes KMT2D as a key epigenetic modulator for driving LUSC oncogenesis and suggests KMT2D loss renders LUSC therapeutically vulnerable to SHP2 and pan-ERBB inhibition (Figure 7L).
KMT2D is frequently mutated in multiple types of cancer and its function in tumor development is poorly understood. Several studies using GEMMs suggested that KMT2D serves as a tumor suppressor in lymphoma,54,55 medulloblastoma,56 melanoma,50 and lung adenocarcinoma.19 By contrast, other studies using established tumor cell lines revealed conflicting roles (pro- or anti-tumor) of KMT2D in cancers.57–62 These findings highlight that KMT2D’s function could be context-dependent (tumor initiation versus progression) and vary in different tumor types. In LUSC, KMT2D mutations occur frequently and tend to be clonal,14 while the role of KMT2D loss in LUSC oncogenesis has not been explored. In this study, we established that Kmt2d deletion alone is sufficient to drive mouse Trp53−/− lung basal cell organoids to LUSC in vivo. Our comprehensive histologic and molecular characterizations further demonstrated that Kmt2d-deficient tumors recapitulate key hallmarks of LUSC.
Our findings highlight a tumor suppressive role of KMT2D in controlling epigenetic and transcriptomic machinery during LUSC tumorigenesis. KMT2D loss suppresses the expression of multiple RPTPs, including PTPRB, PTPRF, PTPRS and PTRPU. In the normal basal epithelia, KMT2D promotes expression of the RPTPs, and thus maintain RTK-RAS signaling at physiological level. KMT2D loss reprograms the epigenetic landscape that alters the chromatin accessibility, enhancer H3K4me1/H3K27ac levels and gene expression of RPTPs. The reduced expression of RPTPs in turn leads to the elevation of oncogenic RTK-RAS signaling and tumor growth. A prior study showed PTPRB inhibits EGFR activity by directly dephosphorylating EGFR 63. Similarly, PTPRS loss promotes EGFR signaling in head and neck cancer,64 whereas PTPRF can also modulate EGFR phosphorylation and activity,65,66 and PTPRU was shown to interact with EGFR and ERBB2.63 Our functional analysis demonstrated that Ptprf plays an important role in maintaining RTK-RAS signaling during LUSC oncogenesis. Future studies are needed to evaluate whether these RPTPs could function together to regulate RTK-RAS signaling in the development of KMT2D-deficient LUSC.
Targeted therapies in LUSC have largely failed in the clinics.7 Thus far, inhibitors against putative oncogenic drivers such us FGFR1 and PI3K have led to poor clinical benefits.9 Although the mutations in ERBB family genes are rare in squamous carcinoma, these tumors appeared to be selectively dependent on ERBB family genes.67 Furthermore, pan-ERBB inhibitor afatinib has provided modest clinical benefits in patients and was approved for the second line treatment of LUSC.43 Our findings in this study offer direct translational significance that patients with KMT2D loss might be particularly sensitive to RTK-RAS signaling inhibition, such as pan-ERBB inhibitor afatinib. Future clinical studies involving pan-ERBB inhibitors should consider stratifying LUSC patients based on KMT2D mutations. Furthermore, SHP2 (PTPN11) has been identified as a promising target on inhibiting RTK-RAS signaling in cancer.68 Cancer cells that depend on RTK signaling for survival respond particularly well to the allosteric SHP2 inhibitor SHP099 in vitro and in vivo.41 In line with this, we demonstrate that KMT2D-deficient LUSC cells are sensitive to SHP099 alone, and this antiproliferative effect is further enhanced when combining with afatinib. Of note, this combination reduces tumor growth and improves overall survival in murine LUSC models and human LUSC allografts and PDXs with KMT2D mutations. Nevertheless, blocking RTK-RAS signaling does not always lead to tumor shrinkage in KMT2D-deficient LUSC in our study. Therefore, a combination with other agents (e.g. chemotherapy) may offer further benefits. In summary, our work identifies KMT2D as a pivotal epigenetic modulator for LUSC oncogenesis and serves as the basis for targeting KMT2D-deficient LUSC through RTK-RAS signaling inhibition. With multiple SHP2 inhibitors currently testing in clinical trials, and afatinib already clinically available, our work warrant evaluation of these therapies in KMT2D-deficient LUSC patients in future clinical studies.
STAR METHODS
Lead contact
Correspondence and requests regarding this manuscript should be sent to and will be fulfilled by the lead investigator Kwok-Kin Wong (Kwok-Kin.Wong@nyulangone.org).
RESOURCE AVAILABILITY
All unique reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Materials availability
Materials and reagents used in this study are listed in the key resources table. Reagents generated in our laboratory in this study or previous studies are available upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit p53 | Cell Signaling | Cat#: 32532; RRID: AB_2757821 |
| Rabbit KMT2D | Sigma-Aldrich | Cat#: HPA035977; RRID: AB_10670673 |
| Ki-67 Monoclonal Antibody | Thermo Fisher Scientific | Cat#: 14–5698-82; RRID: AB_10854564 |
| Purified anti-Keratin 5 Antibody | BioLegend | Cat#: 905504; RRID: AB_2616956 |
| Anti-p40 DeltaNp63 Antibody | Abcam | Cat#: ab203826 |
| p44/42 MAPK (Erk1/2) Antibody | Cell Signaling | Cat#: 9102; RRID: AB_330744 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) Antibody | Cell Signaling | Cat#: 9101; RRID: AB_331646 |
| Phospho-HER2/ErbB2 (Tyr1221/1222) Antibody | Cell Signaling | Cat#: 2243; RRID: AB_490899 |
| HER2/ErbB2 (29D8) Rabbit mAb | Cell Signaling | Cat#: 2165; RRID: AB_10692490 |
| EGF Receptor Antibody | Cell signaling | Cat#: 2232; RRID: AB_331707 |
| Phospho-EGF Receptor (Tyr1068) (D7A5) XP® Rabbit mAb | Cell signaling | Cat#: 3777; RRID: AB_2096270 |
| Anti-Histone H3 (mono methyl K4) antibody | Abcam | Cat#: ab8895; RRID: AB_306847 |
| Anti-Histone H3 (acetyl K27) antibody | Abcam | Cat#: ab4729; RRID: AB_2118291 |
| Anti-Histone H3 (tri methyl K4) antibody | Abcam | Cat#: ab8580; RRID: AB_306649 |
| Anti-p75 NGF Receptor antibody | Abcam | Cat#: ab8875; RRID: AB_306828 |
| Monoclonal Anti-Acetylated Tubulin antibody | Sigma | Cat#: T7451; RRID: AB_609894 |
| CC10 Antibody (E-11) | Santa Cruz | Cat#: sc-365992; RRID: AB_10915481 |
| HSP90 (C45G5) | Cell signaling | Cat#: 4877; RRID: AB_2233307 |
| Mll4 polyclonal antibody | Diagenode | Cat#: C15310100 |
| Monoclonal Anti-β-Actin antibody | Sigma | Cat#: A5441; RRID: AB_476744 |
| Recombinant Anti-TTF1 antibody | Abcam | Cat#: ab133638; RRID: AB_2734144 |
| IRDye 800CW Donkey anti-Rabbit IgG Secondary Antibody | LICOR | Cat#: 926–32213; RRID: AB_621848 |
| IRDye 680RD Donkey anti-Mouse IgG Secondary Antibody | LICOR | Cat#: 926–68072; RRID: AB_10953628 |
| Goat anti-Mouse Secondary Antibody, Alexa Fluor 488 | Thermo Fischer Scientific |
Cat#: A-11001; RRID: AB_2534069 |
| Goat anti-Rabbit Secondary Antibody, Alexa Fluor Plus 555 |
Thermo Fischer Scientific |
Cat#: A32732; RRID: AB_2633281 |
| Bacterial and virus strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Invitrogen | Cat#: C737303 |
| NEB® 5-alpha F’Iq Competent E. coli | NEB | Cat#: C2992H |
| Biological samples | ||
| Mouse sample | This manuscript | N/A |
| Patient-derived xenografts (PDX), PDX-1 (LX-515) and PDX-2 (LX-640) | Preclinical Therapeutics Program NYU Langone Health |
N/A |
| Patient-derived xenografts (PDX), PDX-3 and PDX-4 | Chinese Academy of Sciences | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase D | Roche | Cat#: 11088866001 |
| DNase I | Roche | Cat#: 10104159001 |
| Protease and phosphatase inhibitor cocktail | Thermo Fisher Scientific | Cat#: 78440 |
| RIPA Lysis and Extraction Buffer | Thermo Fisher Scientific | Cat#: 89900 |
| Blocking buffer | LICOR | Cat#: 927–50003 |
| DAPI | BioLegend | Cat#: 422801 |
| CCK-8 | Dojindo | Cat#: CK04 |
| Fluorescence Mounting Medium | DAKO | Cat#: S3023 |
| PowerUp™ SYBR™ Green Master Mix | Applied Biosystems | Cat#: 4367659 |
| Corning® Matrigel® Growth Factor Reduced (GFR) Basement Membrane Matrix |
Corning | Cat#: 354230 |
| Corning® Matrigel® Basement Membrane Matrix | Corning | Cat#: 354234 |
| PEG-it Virus Precipitation Solution | System Biosciences | Cat#: LV810A-1 |
| Advanced DMEM/F12 | Thermo Fisher Scientific | Cat#: 12634010 |
| DMEM/F-12 with 15 mM HEPES | StemCell Technologies | Cat#: 36254 |
| N2 Supplement (100x) | Life Technologies | Cat#: 17502048 |
| B27 Supplement 50x | Life Technologies | Cat#: A1895601 |
| N-Acetylcysteine | Thermo Fisher Scientific | Cat#: A15409.14 |
| h-EGF | Sigma Aldrich | Cat#: E9644 |
| Y-27632 | Selleck | Cat#: S1049 |
| BAMBANKER freeze medium | Fisher Scientific | Cat#: NC9582225 |
| Lipofectamine 3000 | Invitrogen | Cat#: L3000008 |
| Dynabeads™ Protein G for Immunoprecipitation | Thermo Fisher Scientific | Cat#: 10003D |
| Paraformaldehyde | Electron Microscopy Sciences | Cat#: 15714 |
| Afatinib | MCE | Cat#: HY-10261 |
| SHP099 | Wuxi AppTec | Customized synthesis |
| Critical commercial assays | ||
| Proteome Profiler Mouse Phospho-RTK Array Kit | R&D system | Cat#: ARY014 |
| Pierce™ BCA protein assay kit | Thermo Fisher Scientific | Cat#: 23225 |
| CUT&Tag-IT Assay Kit | Active Motif | Cat#: 53160 |
| Universal Mycoplasma Detection Kit |
ATCC | Cat#: 30–1012K |
| High-Capacity RNA-to-cDNA™ Kit | Thermo Fisher Scientific | Cat#: 4387406 |
| Deposited data | ||
| Data files for Omics data (RNA-seq, ChIP-seq, CUT&Tag and ATAC-seq) | This manuscript, GEO | GSE200505 |
| Public RNA-seq data of KrasG12D; Trp53−/− tumors | Deng et al,32 GEO | GSE137396 |
| Public RNA-seq data of normal mouse lung tissues | Mollaoglu et al,33 GEO | GSE118246 |
| Public RNA-seq data of human LUSC | TCGA | https://portal.gdc.cancer.gov |
| Experimental models: Cell lines | ||
| Trp53L/L organoids | This manuscript | N/A |
| Trp53−/− organoids | This manuscript | N/A |
| Trp53−/−; Kmt2d−/− organoids | This manuscript | N/A |
| Trp53−/−; Pten−/− organoids | This manuscript | N/A |
| Trp53−/−; Kmt2d−/− cells | This manuscript | N/A |
| Trp53−/−; Pten−/− cells | This manuscript | N/A |
| Trp53−/−; Pten−/−; Kmt2d−/− cells | This manuscript | N/A |
| Trp53−/−; Lkb1−/−; Pten−/− cells | This manuscript | N/A |
| HEK-293T | ATCC | Cat#: CRL-1573, RRID: CVCL_0063 |
| L-WRN | ATCC | Cat#: CRL-3276 |
| HARA | CCLE | RRID: CVCL_2914 |
| HCC-95 | CCLE | RRID: CVCL_5137 |
| EBC-1 | CCLE | RRID: CVCL_2891 |
| LK2 | CCLE | RRID: CVCL_1377 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664; RRID: IMSR_JAX:000664 |
| Mouse: B6(Cg)-Tyrc-2J/J (B6-Albino) | The Jackson Laboratory | JAX: 000058; RRID: IMSR_JAX:000058 |
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) | The Jackson Laboratory | JAX: 005557; RRID: IMSR_JAX:005557 |
| Mouse: Crl:NU-Foxn1nu (Nude) | Charles River | Cat#: 088 |
| Mouse: LSL-EGFR-T790M-L858R | Li et al.31 | N/A |
| Mouse: B6.129P2-Trp53tm1Brn/J (p53loxp) |
The Jackson Laboratory | JAX: 008462; RRID: IMSR_JAX:008462 |
| Oligonucleotides | ||
| See Table S2 | ||
| Recombinant DNA | ||
| psPAX2 | Addgene | Cat#: 12260 |
| pMD2.G | Addgene | Cat#: 12259 |
| lentiCRISPR v2-Blast | Addgene | Cat#: 83480 |
| lentiCRISPR v2-Blast-sgPten | This manuscript | N/A |
| lentiCRISPR v2-Blast-sgKmt2d-1 | This manuscript | N/A |
| lentiCRISPR v2-Blast-sgKmt2d-2 | This manuscript | N/A |
| pSpCas9(BB)-2A-GFP | Addgene | Cat#: 48138 |
| pX458-sgSHP2 | Fedele et al.42 | N/A |
| lentiCas9-Blast | Addgene | Cat#: 52962 |
| pLV-sgControl | This manuscript | N/A |
| pLV-sgKMT2D-1 | This manuscript | N/A |
| pLV-sgKMT2D-2 | This manuscript | N/A |
| Software and algorithms | ||
| GraphPad Prism V9 | GraphPad Software Inc. | http://www.graphpad.com |
| R 4.0.4 | R | https://www.r-project.org/ |
| R Studio | RStudio | https://www.rstudio.com/ |
| Fiji v2.0.0 | ImageJ | https://imagej.net/software/fiji/ |
| Snapgene | Snapgene | https://www.snapgene.com/ |
| GEPIA2 | Tang et al.24 | http://gepia2.cancer-pku.cn |
| DESeq2 | Love et al.69 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Gene Set Enrichment Analysis (GSEA) | Broad Institute.70 | http://software.broadinstitute.org/gsea/index.jsp |
| Cbioportal | Cbioportal.71, 72 | https://www.cbioportal.org/ |
| Bowtie2 | Langmead et al.73 | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
Data and code availability
Data are available upon reasonable request. The data sets used and/analyzed during the current study are available from the corresponding author upon reasonable request. The accession number for the raw and processed omics sequencing data (RNA-seq, ChIP-seq, CUT&Tag and ATAC-seq) is GSE200505.
Public RNA-seq for TCGA LUSC dataset were downloaded from Genomic Data Commons (GDC) Data Portal (https://portal.gdc.cancer.gov). Public RNAseq data of KrasG12D; Trp53−/− tumors and normal mouse lung tissues were downloaded from GEO, with the accession number of GSE137396 and GSE118246, respectively.
Expression of KMT2D mRNA in LUSC tumor and normal tissues was analyzed using GEPIA2 (http://gepia2.cancer-pku.cn/). Expression of KMT2D mRNA, phospho-EGFR, and drug sensitivity data in human LUSC cell lines were obtained from DepMap (https://depmap.org/portal/).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mouse work was reviewed and approved by the Institutional Animal Care and Use Committee at NYU School of Medicine and the Center for Excellence in Molecular Cell Science, Chinese Academy of Sciences. p53LoxP mice (JAX: 008462; RRID: IMSR_JAX:008462) were used to generate the Trp53L/L lung basal cell organoids. C57BL/6J (JAX: 000664; RRID: IMSR_JAX:000664) and B6(Cg)-Tyrc-2J/J (B6-Albino, JAX: 000058; RRID: IMSR_JAX:000058) mice were used for murine organoids or LUSC cell lines allograft studies. NU-Foxn1nu (Nude, Cat#: 088) and NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG, JAX: 005557; RRID: IMSR_JAX:005557) were used for the human LUSC cell lines or PDXs xenografts. Both male and female mice of 6–10 weeks old were used, and all mice were maintained in accordance with the respective NYULH and CAS on the care, welfare, and treatment of laboratory animals. All experiments met or exceeded the standards of the Association for the Assessment and Accreditation of Laboratory Animal Care, International (AAALAC), the United States Department of Health and Human Services, and all local and federal animal welfare laws.
Cell lines
Mouse LUSC cell lines (Trp53−/−; Kmt2d−/−, Trp53−/−; Pten−/− and Trp53−/−; Pten−/−; Kmt2d−/−) and human LUSC cell lines (HARA, HCC95, EBC1, and LK2) were maintained in Advanced DMEM/F12 (Thermo Fisher Scientific, Cat#: 12634010) supplemented with 10% Fetal Bovine Serum (FBS, Sigma-Aldrich), GlutaMAX™ Supplement (Gibco, Cat#: 35050061) and 1x Antibiotic-Antimycotic (Gibco, Cat#: 15240062). HEK-293T (ATCC, Cat#: CRL-1573) cells were cultured in Dulbecco’s Modified Eagle Medium (Gibco, Cat#: 11965118), 10% FBS and 1x Antibiotic-Antimycotic. All cell lines used in this study were cultured in an incubator at 37 °C (with 5% CO2) and tested as Mycoplasma-negative using the Universal Mycoplasma Detection Kit (ATCC, Cat#: 30–1012K).
METHOD DETAILS
Organoid culture and manipulation
Trp53L/L basal cell lung organoids were generated from 8–10 weeks Trp53L/L mice of the C57BL/6J background. In brief, the trachea and main bronchi were dissected from mouse and washed 2 times with phosphate buffered saline (PBS). The tissues were minced by scissors and then digested in collagenase D and DNase I in Hank’s Balanced Salt Solution (HBSS) at 37°C for 30 minutes. After incubation, the digested tissues were passed through a 70 μm cell strainer to obtain single-cell suspensions. After spinning down for 350g 5min, cells were resuspended in organoid media: DMEM/F-12 with 15 mM HEPES (StemCell Technologies, Cat#: 36254) supplemented with GlutaMAX™ Supplement (Gibco, Cat#: 35050061), 1x Antibiotic-Antimycotic (Gibco, Cat#: 15240062), N2 Supplement (Gibco, Cat#: 17502048), B27 supplement (Gibco, Cat#: A1895601), 1 mmol/L N-Acetylcysteine (Thermo Fisher Scientific, Cat#: A15409.14), 50 ng/mL human recombinant EGF (Sigma-Aldrich, Cat#: E9644), and 3% conditioned media from L-WRN cells containing Wnt3a, Noggin, and R-spondin). Using a 1:2 ratio of organoid media and growth factor reduced Matrigel (Corning, Cat#: 354230), lung epithelial organoids were maintained for successive passages.
To generate lentivirus, HEK-293T cells were co-transfected with lentiviral plasmids, packaging plasmids psPAX2 (Addgene, Cat#: 12260) and pMD2.G (Addgene, Cat#: 12259) using Lipofectamine 3000 (Invitrogen, Cat#: L3000008) according to the manufacturer’s instructions. Viral particles in the cell culture supernatant were filtered with 0.45-μm filters (Corning, Cat#: 431225) to remove cellular debris. Virus was concentrated by PEG-it Virus Precipitation Solution (System Biosciences, Cat#: LV810A-1).
Trp53−/− organoids were generated from Trp53L/L organoids by Ad-Cre-GFP virus infection, followed by flow cytometry sorting of GFP+ cells. To generate Trp53−/−; Kmt2d−/− and Trp53−/−; Pten−/− organoids, CRISPR/Cas9 was performed using LentiCRISPRv2-Blast vector obtained from Addgene (Cat#: 83480). Guide RNAs (gRNA) against mouse Kmt2d and Pten were cloned into lentiCRISPRv2-Blast. Lentivirus was generated by transfection of HEK-293T cells with lentiCRISPRv2-Blast (sgKmt2d or sgPten) and the packaging plasmids psPAX2 and pMD2.G using Lipofectamine 3000. CRISPR/Cas9 sgRNA guides and sequencing primers were listed in Table S2. Organoids were isolated by digesting the Matrigel with TrypLE™ Express Enzyme (Thermo Fisher Scientific, Cat#: 12605028) in culture plates for 5–10 minutes at 37°C and washed twice with PBS. Once organoids were dissociated, cells were pelleted and resuspended in 250 μL lentiviral solution. Spinoculation was performed by transferring the suspension into a 24-well plate and centrifuging the plate at 600g for 1 hour at 32°C. Plates were then incubated at 37°C for 6 hours before washing the suspension with fresh media and pelleting the cells to be embedded in fresh Matrigel media mixture. Antibiotic (blasticidin, 5μg/ml) was added to the media to select the infected organoids.
Cell line generation
To generate the syngeneic mouse LUSC Trp53−/−; Kmt2d−/− and Trp53−/−; Pten−/− cell lines. Subcutaneous Trp53−/−; Kmt2d−/− and Trp53−/−; Pten−/− tumors were harvested and washed twice in 1× PBS, and then the tumors were cut into small pieces using scissors. The shredded tissues were cultured in an incubator at 37 °C (with 5% CO2) in completed Advanced DMEM/F12 media (See above). Fresh medium was changed every other day. The cells were cultured for at least five passages to establish the stable cell lines. To generate the Trp53−/−; Pten−/−; Kmt2d−/− cell line, Trp53−/−; Pten−/− cells were infected with CRISPR/Cas9 sgRNAs targeting Kmt2d and validated by genomic sequencing. To generate the Trp53−/−; Kmt2d−/− sgControl and Trp53−/−; Kmt2d−/− sgSHP2 cell lines, Trp53−/−; Kmt2d−/− cells was transfected with pX458-sgCtrl and pX458-sgSHP2 42 followed by flow cytometry sorting of GFP+ cells.
To knock out KMT2D in human LUSC cells, HARA, EBC1 and HCC95 cells were first infected with Cas9 expressing lentivirus (lentiCas9-Blast, Addgene #52962). The HARA-Cas9 cells were then infected with lentivirus targeting human KMT2D (lentiviral vectors purchased from Vector builder). KMT2D mutations were confirmed by sequencing. CRISPR guides and sequencing primers were listed in Table S2.
To knockdown Ptprb, Ptprf, Ptprs and Ptpru in mouse Kmt2d WT (Trp53−/−; Pten−/−) cells, shRNA vectors were obtained from Sigma MISSION TRC shRNA library with clone ID as follows: shPtprb (mouse) TRCN0000029926, shPtprf-1 (mouse) TRCN0000029944, shPtprf-2 (mouse) TRCN0000029948, shPtprs-1 (mouse) TRCN0000238010, shPtprf-2 (mouse) TRCN0000257330, shPtpru-1 (mouse) TRCN0000029964 and shPtpru-2 (mouse) TRCN0000029968. Stable cell lines with Ptprb, Ptprf, Ptprs and Ptpru knockdown were generated using the lentiviral packaging system described above.
Western blot
Cells were lysed in RIPA buffer (Thermo Fisher Scientific, Cat#: 89900) containing protease/phosphatase inhibitor cocktail (Thermo Fisher Scientific, Cat#: 78440). Protein concentration was measured using the Pierce™ BCA assay (Thermo Fisher Scientific, Cat#: 23225). Equivalent amounts of each sample were loaded on 4% to 12% Bis-Tris gels (Bio-Rad), transferred to nitrocellulose membranes, and immunoblotted with antibodies directed against KMT2D (Diagenode, Cat#: C15310100), EGFR (Cell Signaling, Cat#: 2232), pEGFR (Cell Signaling, Cat#: 3777), ERBB2 (Cell Signaling, Cat#: 2165), pERBB2 (Cell Signaling, Cat#: 2243) and β-actin (Sigma, Cat#: A5441). IRDye 800CW Donkey anti-Rabbit IgG (LI-COR, Cat#: 926–32213) and IRDye 680RD Donkey anti-Mouse IgG (LI-COR, Cat#: #: 926–68072) were used as secondary antibodies, and membranes were detected with an Odyssey detection system (LI-COR Biosciences).
Phospho-RTK array
The Mouse Phospho-RTK Array Kit (R&D Systems, Cat#: ARY014) was used to determine the relative levels of tyrosine phosphorylation of 39 distinct receptor tyrosine kinase (RTK) in organoids, cell lines and tumor nodules, according to the manufacturer’s protocol. Chemiluminescent signals were captured with a Chemidoc MP Imaging System (Bio-Rad Laboratories) and images were analyzed using Image Studio Lite (LI-COR Biosciences).
Cell viability assay
Cells were seeded in 96-well plates (1000–2000 cells/well) in media and treated with drugs at indicated concentrations and time points. Cell viability was measured using the MTS-based CCK-8 assay (Dojindo, Cat#: CK04). Absorption at 450 nm was measured 3 hours after addition of CCK-8 reagent to cells using FlexStation 3 multi-mode microplate reader according to the manufacturer’s instructions.
Colony formation assay
Cells were trypsinized to produce a single-cell suspension. 2,000 cells were counted and plated in each well of a 6-well plate. Medium was changed every 2 days. After 7 days, cells were fixed with 70% ethanol for 10 minutes, and the cells were stained with 0.5% crystal violet (dissolved in 20% methanol) for 5 minutes and washed. Photos were taken and quantified using ImageJ.
Animal studies
To study whether mutated organoids can form LUSC in vivo, 6–8 weeks old C57BL/6J mice were obtained from Jackson Laboratory and subcutaneously inoculated with organoids into both flanks. Tumor length and width were measured using calipers. Tumor volumes were calculated using the formula (Length x Width2)/2. To investigate whether Kmt2d KO organoids could directly form tumors in the lung, transthoracic injection of Trp53−/−; Kmt2d−/− organoids (1 × 106) was performed under the guidance of ultrasound. Tumor burden was monitored by MRI.
To establish the orthotopic LUSC model, Trp53−/−; Kmt2d−/− or Trp53−/− Pten−/− cells were injected into B6(Cg)-Tyrc-2J/J (B6-albino) mice via tail vein injection at 1–2 × 106 cells per mice. MRI was used to monitor tumor formation and progression of LUSC. After confirming the tumor burden by MRI, mice were randomized and then treated with vehicle, chemotherapy (carboplatin 40mpk I.P. QW + paclitaxel 10mpk I.P. QW), SHP099 (75mpk, 5 days per week), afatinib (10mpk, 5 days per week) or the combination of SHP099 and afatinib. Subsequent MRI was performed every 2 weeks after treatment initiation and survival of animals were monitored. To compare the in vivo treatment efficacy of Trp53−/−; Pten−/− and Trp53−/−; Pten−/−; Kmt2d−/− LUSC, Trp53−/−; Pten−/− cells (4 × 106) and Trp53−/−; Pten−/−; Kmt2d−/− cells (4 × 106) were injected with 1:1 mixture of cell suspension and Matrigel (Corning, Cat#: 354234) subcutaneously into both flanks of C57BL/6J mice. When the tumor volume reached approximately 100–200 mm3, the animals were randomized into treatment groups and dosing was initiated on day 0 with vehicle or combined SHP099 (75mpk, 5 days per week) and afatinib (10mpk, 5 days per week).
For human patient-derived xenograft (PDX) xenograft study. PDX-1 (KMT2D mutant, LX-515), PDX-2 (KMT2D WT, LX-640), PDX-3 (KMT2D mutant), and PDX-4 (KMT2D WT) derived from primary LUSC tumor fragments were implanted subcutaneously in a single flank of 6–8 weeks old female NOD-SCID-Il2rgnull (NSG) mice (Jackson Laboratory). For human cell line xenograft study, LK2 cells (1 × 106), HARA-sgCtrl (2 × 106) and HARA-sgKMT2D (2 × 106) were injected with 1:1 mixture of cell suspension and Matrigel (Corning, Cat#: 354234) subcutaneously into both flanks of nude mice (Jackson Laboratory). For PDXs and human cell line xenograft study, when the tumor volume reached approximately 100–200 mm3, the animals were randomized into treatment groups and dosing was initiated on day 0 with vehicle, SHP099 (75mpk, 5 days per week), afatinib (10mpk, 5 days per week) or the combination of SHP099 and afatinib. Tumor size and body weight were measured twice weekly, and the tumor volumes were calculated using the (Length x Width2)/2.
MRI quantification
Animals were anesthetized with isoflurane to perform MRI of the lung field using BioSpec USR70/30 horizontal bore system (Bruker) to scan 16 consecutive sections. Tumor volumes of the whole lung were quantified using 3-D slicer software to reconstruct MRI volumetric measurements. Acquisition of the MRI signal was adapted according to cardiac and respiratory cycles to minimize motion effects during imaging.
Histology and immunohistochemistry
Lungs were perfused with 10% formalin, stored in fixative for 48h, and embedded in paraffin. 4 μm thick sections of formalin fixed tissue were used for immunoperoxidase analysis after baking at 60°C for 1 hour, deparaffinization and rehydration (100% xylene x4 for 3 minutes each, 100% ethanol x4 for 3 minutes each and running water for 5 minutes). The sections were blocked for peroxidase activity with 3% hydrogen peroxide in methanol for 10 minutes and washed under the running water for 5 minutes. The sections with pressure cooked (Biocare Medical) antigen retrieval were incubated at 120°C in Citrate Buffer (Dako Target Retrieval Solution, Cat#: S1699). The slides were cooled for 15 minutes and transferred to Tris-buffer saline (TBS). The sections were incubated with P40 (ΔNp63), TTF1, CK5, Ki-67, cleaved caspase 3, or KMT2D antibody for 40 minutes at room temperature. The secondary antibody was used Leica Novolink Polymer (Cat#: RE7161) of 30 minutes incubation. All the incubations were carried out in a humid chamber at room temperature. The slides were rinsed with TBS in between incubation. The sections were developed using 3,3’-diaminobenzidine (DAB) as substrate and counter-stained with Mayer’s Hematoxylin. IHC images were analyzed and quantified by FIJI (NIH).
Immunofluorescence staining and imaging
Organoids were fixed in 4% paraformaldehyde (diluted the 32% paraformaldehyde in PBS, Electron Microscopy Sciences, Cat#: 15714) for 10 minutes at room temperature. Cells were washed three times for 5 min with 200 mM glycine containing PBS, followed by permeabilization with 0.2% Triton X-100 in PBS for 15 min. After blocking with 5% bovine serum albumin (BSA) in PBS for 1 hour, cells were incubated with primary antibody NGFR (Abcam, Cat#: ab8875) and Ki-67 (Thermo Fisher Scientific, Cat#: 14–5698-82) diluted in a 5% BSA in PBS solution overnight at 4°C. After washing four times with PBS, cells were incubated with secondary antibodies Alexa Fluor Plus 555 (Thermo Fisher Scientific, Cat#: A32732) and Alexa Fluor Plus 488 (Thermo Fisher Scientific, Cat#: A-11001) and for 1 hour and washed three times with PBS. Cell nuclei were counterstained with DAPI (BioLengend, Cat#: 422801) for 5 min. Cells were washed two more times in PBS before mounting with Fluorescence Mounting Medium (Dako, Cat#: S3023). Images were acquired using Zeiss 880 Laser Scanning Confocal Microscope and were processed and analyzed by FIJI (NIH).
RNA extraction and RT-qPCR
Cell pellets were collected and then subjected to total RNA extraction using RNeasy Plus Mini Kit (QIAGEN, Cat#: 74136) according to the manufacturer’s instructions. The extracted RNA was reversely transcribed into cDNA using the High-Capacity RNA-to-cDNA™ Kit (Thermo Fisher Scientific, Cat#: 4387406) according to the manufacturer’s instructions. The obtained cDNA samples were diluted and used for RT-qPCR using PowerUp™ SYBR™ Green Master Mix (Thermo Fisher Scientific, Cat#: A25742). Gene specific primers with sequences listed in Table S2 were used for PCR amplification and detection on the QuantStudio 3 Real-Time PCR System (Applied Biosystems). RT-qPCR data were normalized to Actb (mouse cells) or ACTB (human cells) and presented as fold changes of gene expression in the test sample compared to the control.
RNA extraction and Bulk-RNA sequencing analysis
Tumor nodules or cell pellets were subjected to total RNA extraction using RNeasy Plus Mini Kit (QIAGEN, Cat#: 74136) according to the manufacturer’s instructions. Read qualities were evaluated using FASTQC (Babraham Institute) and mapping to mm10 reference genome using STAR program 34, with default parameters. Read counts, TPM and FPKM were calculated using RSEM program 35. Identification of differentially expressed genes was performed using DESeq2 69 in R/Bioconductor (R version 4.0.4). Genes with false discovery rate (FDR) lower than 0.05 were considered significantly differentially expressed.
All plots were generated using customized R scripts. Pathway enrichment analysis was performed on all genes ranked from high to low DESeq2 estimated fold-change using the GSEAPreRanked function with enrichment statistic classic and 1000 permutations using GSEA program.70 Gene sets (Hallmark and C6) were downloaded from MsigDB 37. Differential expression genes involved in top enriched pathways were selected to generate heatmaps using pheatmap R function with default hierarchical clustering method for gene orders. Dot plots of enriched pathways, heat maps of genes, and volcano plots were generated using the pheatmap, ggplot2, and Enhanced-Volcano in R (version 4.0.4).
Comparing LUSC, LUAD and normal lung gene expression
Raw gene expression tables of LUAD (KP) 32 normal lung tissues 33 and were downloaded from gene expression omnibus (GEO) and combined with LUSC for differential expression analysis using DESeq2 as described in above. Differential expression genes for each condition were identified by comparing all samples from one condition to the rest samples. Top differential expression genes with highest log2 fold changes were selected to generate heatmap using pheatmap R package, which was also used for generating targeted gene heatmaps.
Human LUSC analysis
RNA-seq raw counts for TCGA LUSC dataset were downloaded from Genomic Data Commons (GDC) Data Portal (https://portal.gdc.cancer.gov). 249 LUSC samples with high KMT2D expression were compared with 246 LUSC samples with low KMT2D expression using DESeq2. Pathway enrichment analysis was performed on all genes ranked from high to low DESeq2 estimated fold-change using the GSEAPreRanked function with enrichment statistic classic and 1000 permutations using GSEA program.
Oncoprint and gene expression correlation data were obtained and analyzed using cBioportal for cancer genomics database (http://www.cbioportal.org ).71,72
Expression of KMT2D mRNA in LUSC tumor and normal tissues was analyzed using the online tool, GEPIA2 (http://gepia2.cancer-pku.cn/ ).24 The phospho-EGFR in LUSC tumor and normal tissues data was obtained and analyzed from Satpathy et al.37
Expression of KMT2D mRNA, phospho-EGFR, and drug sensitivity to afatinib, neratinib, lapatinib and poziotinib in human LUSC cell lines were obtained and analyzed using DepMap (https://depmap.org/portal/).
ATAC-seq and analysis
Freshly harvested cells were directly sent to NYU Langone Health Genome Technology Center for library construction and sequencing. The library was constructed with Nextera DNA library Prep Kit (Illumina, Cat#: FC-121–1030) according to the manufacturer’s instructions and was sequenced by Illumina NovaSeq 6000.
Illumina sequencing adapter was removed using Trimgalore/0.5.0 from raw sequence files in fastq format. The reads were aligned to the mm10 reference genome using Bowtie2 73 with default parameters. The aligned reads were used after removing PCR duplicates using SAMtools and filtered off an ATAC blacklist 74 for mitochondrial DNA and homologous sequences. Both fragment ends were shifted +4 nt for positive strand and −5 nt for negative strand to account for the distance from Tn5 binding and helicase activity to identify cut sites. Extended Tn5 cut sites were used for peak calling with MACS2 with parameters --nomodel --extsize 100 --shift 50 --nolambda --keep-dup all. The gained/lost peaks comparing Kmt2d KO versus Kmt2d WT cells were identified using R package DiffBind with cutoffs of a false discovery rate (FDR) ≤ 0.05. Normalized ATAC-seq as RPKM signals for each sample were visualized on Integrative Genome Viewer genome browser.75 Average signal plots were generated using plotProfile from deeptools/3.2.1.76
ChIP-seq, CUT&Tag and analysis
For H3K27ac chromatin immunoprecipitation (ChIP), cells were crosslinked with 1% formaldehyde in PBS for 10 minutes at room temperature, washed in 5 mg/mL BSA in PBS and then in just cold PBS, resuspended in lysis buffer [50 mmol/L Tris-HCl pH 8.1, 10 mmol/L EDTA, 1% SDS, 1 x protease inhibitor cocktail] and sonicated by the Diagenode Bioruptor Sonication System. Fragmented chromatin was diluted in immunoprecipitation buffer (20 mmol/L Tris-HCl pH 8.1, 150 mmol/L NaCl, 2 mmol/L EDTA, 1% Triton X-100) and incubated overnight at 4°C with protein G magnetic beads (Dynabeads, Thermo Fisher Scientific, Cat#: 10003D) that had been preincubated with anti-H3K27ac (Abcam, Cat#: ab4729). Immunoprecipitates were washed 6 times with the wash buffer (50 mmol/L HEPES pH 7.6, 0.5 mol/L LiCl, 1 mmol/L EDTA, 0.7% sodium deoxycholate, 1% IGEPAL CA-630) and twice with Tris-EDTA buffer. Immunoprecipitated DNA was treated with RNase A and Proteinase K on the beads, recovered in 1% SDS and 0.1 mol/L NaHCO3 over a period of 6 hours at 65°C, and purified with DNA clean and concentrator-25 (Zymo Research, Cat#: D4033). The DNA was sent to NYU School of Medicine Genome Technology Center for library construction and sequencing. The library was constructed with KAPA Hyper-Prep Kits (Roche, Cat#: 07962347001) according to the manufacturer’s instructions and was sequenced by Illumina NovaSeq 6000.
CUT&Tag profiling was performed using CUT&Tag-IT Assay Kit (Active Motif, Cat#: 53160) according to the manufacturer’s instructions and the library was sent to NYU School of Medicine Genome Technology Center for sequencing by Illumina NovaSeq 6000.
The sequencing reads were aligned to the mm10 reference genome using Bowtie2.73 Samtools 77 was used to sort and index the aligned reads, and MACS2 78 was used to calculate signal per million reads (SPMR) and to call significant ChIP-seq peaks (q value < 0.05) in Kmt2d KO and Kmt2d WT cells. MAnorm 79 was used to identify differential peaks between Kmt2d KO and Kmt2d WT cells. Heatmap of peaks and average signal plots were generated by Deeptools.76 To study the relationship between Kmt2d loss-affected H3K27ac peaks and gene expression changes, binding and expression target analysis (BETA) package 80 was used by combining H3K27ac ChIP-seq and RNA-seq results.
Illustration Tool
The graphical abstract image, and the schematic illustrations of Figure 1B, Figure 2A, Figure 4A and Figure 7L were created with BioRender (https://biorender.com/).
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 9 software and statistical significance was determined by p < 0.05. Data were presented as means with SEM unless otherwise specified. Statistical comparisons were performed using unpaired Student t test for two-tailed P values unless otherwise specified (* P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001). Survival was measured using the Kaplan-Meier method.
Supplementary Material
Table S1. KMT2D candidate target genes in LUSC, related to Figure 6
Table S2. CRISPR and RT-qPCR related oligo sequences, related to STAR Methods
Highlights.
Kmt2d deletion transforms lung basal cell organoids to lung squamous cell carcinoma
KMT2D loss triggers activation of oncogenic RTK-RAS signaling
KMT2D-deficient LUSC is hypersensitive to RTK-RAS inhibition
KMT2D governs RTK-RAS signaling partly through epigenetic regulation of RPTPs
ACKNOWLEDGEMENT
We thank NYU Langone Preclinical Imaging Laboratory (partially funded by P30CA016087 and P41 EB017183) for MRI, the Genome Technology Center (partially supported by P30CA016087) for library preparation and sequencing, and the Experimental Pathology Research Laboratory (partially funded by P30CA016087) for immunohisochemistry. This work is supported by U01 CA233084 (K.K.W), R01 CA219670 (K.K.W), R01 CA216188 (K.K.W), R01 CA205150 (K.K.W), R01 CA166480 (K.K.W), P01 CA154303 (K.K.W), P01 CA098101 (A.K.R, A.J.B. J.A.D. and K.K.W; and the Molecular Pathology and Biostatistics Shared Resources) and P30 CA013696 (A.K.R).
DECLARATION OF INTERESTS
K.K.W. is a founder and equity holder of G1 Therapeutics and has sponsored research agreements with Takeda, TargImmune, Bristol-Myers Squibb (BMS), Mirati, Merus, Alkermes, and consulting and sponsored research agreements with AstraZeneca, Janssen, Pfizer, Novartis, Merck, Zentalis, BridgeBio and Blueprint. A.J.B has received funding from Bayer, Novartis, Merck and Repare and is a co-founder with equity in Signet Therapeutics. Y.P., H.H., H.Z., and K.K.W. have ownership interest in a patent application.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, and Bray F.(2021). Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 71, 209–249. 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Chen Z, Fillmore CM, Hammerman PS, Kim CF, and Wong KK (2014). Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer 14, 535–546. 10.1038/nrc3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gandara DR, Hammerman PS, Sos ML, Lara PN Jr., and Hirsch FR (2015). Squamous cell lung cancer: from tumor genomics to cancer therapeutics. Clin Cancer Res 21, 2236–2243. 10.1158/1078-0432.CCR-14-3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yuan M., Huang LL., Chen JH., Wu J., and Xu Q. (2019). The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal Transduct Target Ther 4, 61. 10.1038/s41392-019-0099-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang CY, Yang JC, and Yang PC (2020). Precision Management of Advanced Non-Small Cell Lung Cancer. Annu Rev Med 71, 117–136. 10.1146/annurev-med-051718-013524. [DOI] [PubMed] [Google Scholar]
- 6.Herbst RS, Morgensztern D, and Boshoff C.(2018). The biology and management of non-small cell lung cancer. Nature 553, 446–454. 10.1038/nature25183. [DOI] [PubMed] [Google Scholar]
- 7.Lau SCM, Pan Y, Velcheti V, and Wong KK (2022). Squamous cell lung cancer: Current landscape and future therapeutic options. Cancer Cell. 10.1016/j.ccell.2022.09.018. [DOI] [PubMed] [Google Scholar]
- 8.Paik PK, Pillai RN, Lathan CS, Velasco SA, and Papadimitrakopoulou V.(2019). New Treatment Options in Advanced Squamous Cell Lung Cancer. Am Soc Clin Oncol Educ Book 39, e198–e206. 10.1200/edbk_237829. [DOI] [PubMed] [Google Scholar]
- 9.Socinski MA, Obasaju C, Gandara D, Hirsch FR, Bonomi P, Bunn PA Jr., Kim ES, Langer CJ, Natale RB, Novello S, et al. (2018). Current and Emergent Therapy Options for Advanced Squamous Cell Lung Cancer. J Thorac Oncol 13, 165–183. 10.1016/j.jtho.2017.11.111. [DOI] [PubMed] [Google Scholar]
- 10.Cancer Genome Atlas Research, N. (2012). Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525. 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flavahan WA, Gaskell E, and Bernstein BE (2017). Epigenetic plasticity and the hallmarks of cancer. Science 357. 10.1126/science.aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim Y, Hammerman PS, Kim J, Yoon JA, Lee Y, Sun JM, Wilkerson MD, Pedamallu CS, Cibulskis K, Yoo YK, et al. (2014). Integrative and comparative genomic analysis of lung squamous cell carcinomas in East Asian patients. J Clin Oncol 32, 121–128. 10.1200/JCO.2013.50.8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dotto GP, and Rustgi AK (2016). Squamous Cell Cancers: A Unified Perspective on Biology and Genetics. Cancer Cell 29, 622–637. 10.1016/j.ccell.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R, Rosenthal R, et al. (2017). Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med 376, 2109–2121. 10.1056/NEJMoa1616288. [DOI] [PubMed] [Google Scholar]
- 15.Froimchuk E, Jang Y, and Ge K.(2017). Histone H3 lysine 4 methyltransferase KMT2D. Gene 627, 337–342. 10.1016/j.gene.2017.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rao RC, and Dou Y.(2015). Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer 15, 334–346. 10.1038/nrc3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fagan RJ, and Dingwall AK (2019). COMPASS Ascending: Emerging clues regarding the roles of MLL3/KMT2C and MLL2/KMT2D proteins in cancer. Cancer Lett 458, 56–65. 10.1016/j.canlet.2019.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhar SS, and Lee MG (2021). Cancer-epigenetic function of the histone methyltransferase KMT2D and therapeutic opportunities for the treatment of KMT2D-deficient tumors. Oncotarget 12, 1296–1308. 10.18632/oncotarget.27988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alam H., Tang M., Maitituoheti M., Dhar SS., Kumar M., Han CY., Ambati CR., Amin SB., Gu B., Chen TY., et al. (2020). KMT2D Deficiency Impairs Super-Enhancers to Confer a Glycolytic Vulnerability in Lung Cancer. Cancer Cell 37, 599–617 e597. 10.1016/j.ccell.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Research, Weinstein N, Collisson JN, Mills EA, Shaw GBKR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, and Stuart JM (2013). The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45, 1113–1120. 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Consortium APG (2017). AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov 7, 818–831. 10.1158/2159-8290.CD-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruthenburg AJ, Allis CD, and Wysocka J.(2007). Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell 25, 15–30. 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 23.Consortium GT (2013). The Genotype-Tissue Expression (GTEx) project. Nat Genet 45, 580–585. 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang Z, Kang B, Li C, Chen T, and Zhang Z.(2019). GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res 47, W556–W560. 10.1093/nar/gkz430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hai J, Zhang H, Zhou J, Wu Z, Chen T, Papadopoulos E, Dowling CM, Pyon V, Pan Y, Liu JB, et al. (2020). Generation of Genetically Engineered Mouse Lung Organoid Models for Squamous Cell Lung Cancers Allows for the Study of Combinatorial Immunotherapy. Clin Cancer Res. 10.1158/1078-0432.CCR-19-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanchez-Danes A, and Blanpain C.(2018). Deciphering the cells of origin of squamous cell carcinomas. Nat Rev Cancer 18, 549–561. 10.1038/s41568-018-0024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferone G, Lee MC, Sage J, and Berns A.(2020). Cells of origin of lung cancers: lessons from mouse studies. Genes Dev 34, 1017–1032. 10.1101/gad.338228.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu C, Fillmore CM, Koyama S, Wu H, Zhao Y, Chen Z, Herter-Sprie GS, Akbay EA, Tchaicha JH, Altabef A, et al. (2014). Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell 25, 590–604. 10.1016/j.ccr.2014.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferone G, Song JY, Sutherland KD, Bhaskaran R, Monkhorst K, Lambooij JP, Proost N, Gargiulo G, and Berns A.(2016). SOX2 Is the Determining Oncogenic Switch in Promoting Lung Squamous Cell Carcinoma from Different Cells of Origin. Cancer Cell 30, 519–532. 10.1016/j.ccell.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pan Y, Han H, Labbe KE, Zhang H, and Wong KK (2021). Recent advances in preclinical models for lung squamous cell carcinoma. Oncogene 40, 2817–2829. 10.1038/s41388-021-01723-7. [DOI] [PubMed] [Google Scholar]
- 31.Li D, Shimamura T, Ji H, Chen L, Haringsma HJ, McNamara K, Liang MC, Perera SA, Zaghlul S, Borgman CL, et al. (2007). Bronchial and peripheral murine lung carcinomas induced by T790M-L858R mutant EGFR respond to HKI-272 and rapamycin combination therapy. Cancer Cell 12, 81–93. 10.1016/j.ccr.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 32.Deng J, Thennavan A, Dolgalev I, Chen T, Li J, Marzio A, Poirier JT, Peng D, Bulatovic M, Mukhopadhyay S, et al. (2021). ULK1 inhibition overcomes compromised antigen presentation and restores antitumor immunity in LKB1 mutant lung cancer. Nat Cancer 2, 503–514. 10.1038/s43018-021-00208-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mollaoglu G, Jones A, Wait SJ, Mukhopadhyay A, Jeong S, Arya R, Camolotto SA, Mosbruger TL, Stubben CJ, Conley CJ, et al. (2018). The Lineage-Defining Transcription Factors SOX2 and NKX2–1 Determine Lung Cancer Cell Fate and Shape the Tumor Immune Microenvironment. Immunity 49, 764–779 e769. 10.1016/j.immuni.2018.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McKay MM, and Morrison DK (2007). Integrating signals from RTKs to ERK/MAPK. Oncogene 26, 3113–3121. 10.1038/sj.onc.1210394. [DOI] [PubMed] [Google Scholar]
- 35.Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, Barretina J, Gelfand ET, Bielski CM, Li H, et al. (2019). Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508. 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoadley KA., Yau C., Hinoue T., Wolf DM., Lazar AJ., Drill E., Shen R., Taylor AM., Cherniack AD., Thorsson V., et al. (2018). Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 173, 291–304 e296. 10.1016/j.cell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Satpathy S, Krug K, Jean Beltran PM, Savage SR, Petralia F, Kumar-Sinha C, Dou Y, Reva B, Kane MH, Avanessian SC, et al. (2021). A proteogenomic portrait of lung squamous cell carcinoma. Cell 184, 4348–4371 e4340. 10.1016/j.cell.2021.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chan G, Kalaitzidis D, and Neel BG (2008). The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev 27, 179–192. 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- 39.Wong GS, Zhou J, Liu JB, Wu Z, Xu X, Li T, Xu D, Schumacher SE, Puschhof J, McFarland J, et al. (2018). Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat Med 24, 968–977. 10.1038/s41591-018-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ, Araki K, Ozerdem U, Simeone DM, Miller G, et al. (2018). SHP2 Inhibition Prevents Adaptive Resistance to MEK Inhibitors in Multiple Cancer Models. Cancer Discov 8, 1237–1249. 10.1158/2159-8290.CD-18-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, Antonakos B, Chen CH, Chen Z, Cooke VG, et al. (2016). Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535, 148–152. 10.1038/nature18621. [DOI] [PubMed] [Google Scholar]
- 42.Fedele C, Li S, Teng KW, Foster CJR, Peng D, Ran H, Mita P, Geer MJ, Hattori T, Koide A, et al. (2021). SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J Exp Med 218. 10.1084/jem.20201414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soria JC, Felip E, Cobo M, Lu S, Syrigos K, Lee KH, Goker E, Georgoulias V, Li W, Isla D, et al. (2015). Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-Lung 8): an open-label randomised controlled phase 3 trial. Lancet Oncol 16, 897–907. 10.1016/S1470-2045(15)00006-6. [DOI] [PubMed] [Google Scholar]
- 44.Santos ES, and Hart L.(2020). Advanced Squamous Cell Carcinoma of the Lung: Current Treatment Approaches and the Role of Afatinib. Onco Targets Ther 13, 9305–9321. 10.2147/OTT.S250446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.LaMarche MJ, Acker M, Argintaru A, Bauer D, Boisclair J, Chan H, Chen CH, Chen YN, Chen Z, Deng Z, et al. (2020). Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J Med Chem 63, 13578–13594. 10.1021/acs.jmedchem.0c01170. [DOI] [PubMed] [Google Scholar]
- 46.Corsello SM., Nagari RT., Spangler RD., Rossen J., Kocak M., Bryan JG., Humeidi R., Peck D., Wu X., Tang AA., et al. (2020). Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat Cancer 1, 235–248. 10.1038/s43018-019-0018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang C, Lee JE, Lai B, Macfarlan TS, Xu S, Zhuang L, Liu C, Peng W, and Ge K.(2016). Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition. Proc Natl Acad Sci U S A 113, 11871–11876. 10.1073/pnas.1606857113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lai B, Lee JE, Jang Y, Wang L, Peng W, and Ge K.(2017). MLL3/MLL4 are required for CBP/p300 binding on enhancers and super-enhancer formation in brown adipogenesis. Nucleic Acids Res 45, 6388–6403. 10.1093/nar/gkx234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee JE, Wang C, Xu S, Cho YW, Wang L, Feng X, Baldridge A, Sartorelli V, Zhuang L, Peng W, and Ge K.(2013). H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife 2, e01503. 10.7554/eLife.01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maitituoheti M, Keung EZ, Tang M, Yan L, Alam H, Han G, Singh AK, Raman AT, Terranova C, Sarkar S, et al. (2020). Enhancer Reprogramming Confers Dependence on Glycolysis and IGF Signaling in KMT2 D Mutant Melanoma. Cell Rep 33, 108293. 10.1016/j.celrep.2020.108293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Östman A, Hellberg C, and Böhmer FD (2006). Protein-tyrosine phosphatases and cancer. Nature Reviews Cancer 6, 307–320. 10.1038/nrc1837. [DOI] [PubMed] [Google Scholar]
- 52.Meeusen B, and Janssens V.(2018). Tumor suppressive protein phosphatases in human cancer: Emerging targets for therapeutic intervention and tumor stratification. Int J Biochem Cell Biol 96, 98–134. 10.1016/j.biocel.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 53.Tonks NK (2006). Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol 7, 833–846. 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 54.Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, Jiang M, Hu D, Agirre X, Niesvizky I, et al. (2015). The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med 21, 1199–1208. 10.1038/nm.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K, et al. (2015). Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med 21, 1190–1198. 10.1038/nm.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dhar SS, Zhao D, Lin T, Gu B, Pal K, Wu SJ, Alam H, Lv J, Yun K, Gopalakrishnan V, et al. (2018). MLL4 Is Required to Maintain Broad H3K4me3 Peaks and Super-Enhancers at Tumor Suppressor Genes. Mol Cell 70, 825–841 e826. 10.1016/j.molcel.2018.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, Dickler MN, Scaltriti M, Leslie CS, Armstrong SA, and Baselga J.(2017). PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science 355, 1324–1330. 10.1126/science.aah6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim JH., Sharma A., Dhar SS., Le SH., G B., Cha CH., Lin HK., and Lee MG. (2014). UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res 74, 1705–1717. 10.1158/0008-5472.CAN-13-1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dawkins JB, Wang J, Maniati E, Heward JA, Koniali L, Kocher HM, Martin SA, Chelala C, Balkwill FR, Fitzgibbon J, and Grose RP (2016). Reduced Expression of Histone Methyltransferases KMT2C and KMT2D Correlates with Improved Outcome in Pancreatic Ductal Adenocarcinoma. Cancer Res 76, 4861–4871. 10.1158/0008-5472.CAN-16-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lv S, Ji L, Chen B, Liu S, Lei C, Liu X, Qi X, Wang Y, Lai-Han Leung E, Wang H, et al. (2018). Histone methyltransferase KMT2D sustains prostate carcinogenesis and metastasis via epigenetically activating LIFR and KLF4. Oncogene 37, 1354–1368. 10.1038/s41388-017-0026-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun P, Wu T, Sun X, Cui Z, Zhang H, Xia Q, and Zhang D.(2019). KMT2D inhibits the growth and metastasis of bladder Cancer cells by maintaining the tumor suppressor genes. Biomed Pharmacother 115, 108924. 10.1016/j.biopha.2019.108924. [DOI] [PubMed] [Google Scholar]
- 62.Koutsioumpa M, Hatziapostolou M, Polytarchou C, Tolosa EJ, Almada LL, Mahurkar-Joshi S, Williams J, Tirado-Rodriguez AB, Huerta-Yepez S, Karavias D, et al. (2019). Lysine methyltransferase 2D regulates pancreatic carcinogenesis through metabolic reprogramming. Gut 68, 1271–1286. 10.1136/gutjnl-2017-315690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yao Z, Darowski K, St-Denis N, Wong V, Offensperger F, Villedieu A, Amin S, Malty R, Aoki H, Guo H, et al. (2017). A Global Analysis of the Receptor Tyrosine Kinase-Protein Phosphatase Interactome. Mol Cell 65, 347–360. 10.1016/j.molcel.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morris LG, Taylor BS, Bivona TG, Gong Y, Eng S, Brennan CW, Kaufman A, Kastenhuber ER, Banuchi VE, Singh B, et al. (2011). Genomic dissection of the epidermal growth factor receptor (EGFR)/PI3K pathway reveals frequent deletion of the EGFR phosphatase PTPRS in head and neck cancers. Proc Natl Acad Sci U S A 108, 19024–19029. 10.1073/pnas.1111963108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kulas DT, Goldstein BJ, and Mooney RA (1996). The transmembrane protein-tyrosine phosphatase LAR modulates signaling by multiple receptor tyrosine kinases. J Biol Chem 271, 748–754. 10.1074/jbc.271.2.748. [DOI] [PubMed] [Google Scholar]
- 66.Du WW, Fang L, Li M, Yang X, Liang Y, Peng C, Qian W, O’Malley YQ, Askeland RW, Sugg SL, et al. (2013). MicroRNA miR-24 enhances tumor invasion and metastasis by targeting PTPN9 and PTPRF to promote EGF signaling. J Cell Sci 126, 1440–1453. 10.1242/jcs.118299. [DOI] [PubMed] [Google Scholar]
- 67.Zhou J, Wu Z, Zhang Z, Goss L, McFarland J, Nagaraja A, Xie Y, Gu S, Peng K, Zeng Y, et al. (2022). Pan-ERBB kinase inhibition augments CDK4/6 inhibitor efficacy in oesophageal squamous cell carcinoma. Gut 71, 665–675. 10.1136/gutjnl-2020-323276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Frankson R, Yu ZH, Bai Y, Li Q, Zhang RY, and Zhang ZY (2017). Therapeutic Targeting of Oncogenic Tyrosine Phosphatases. Cancer Res 77, 5701–5705. 10.1158/0008-5472.CAN-17-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Love MI, Huber W, and Anders S.(2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Subramanian A., Tamayo P., Mootha VK., Mukherjee S., Ebert BL., Gillette MA., Paulovich A., Pomeroy SL., Golub TR., Lander ES., and Mesirov JP. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T.(2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shao Z, Zhang Y, Yuan GC, Orkin SH, and Waxman DJ (2012). MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol 13, R16. 10.1186/gb-2012-13-3-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang S, Sun H, Ma J, Zang C, Wang C, Wang J, Tang Q, Meyer CA, Zhang Y, and Liu XS (2013). Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat Protoc 8, 2502–2515. 10.1038/nprot.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. KMT2D candidate target genes in LUSC, related to Figure 6
Table S2. CRISPR and RT-qPCR related oligo sequences, related to STAR Methods
Data Availability Statement
Data are available upon reasonable request. The data sets used and/analyzed during the current study are available from the corresponding author upon reasonable request. The accession number for the raw and processed omics sequencing data (RNA-seq, ChIP-seq, CUT&Tag and ATAC-seq) is GSE200505.
Public RNA-seq for TCGA LUSC dataset were downloaded from Genomic Data Commons (GDC) Data Portal (https://portal.gdc.cancer.gov). Public RNAseq data of KrasG12D; Trp53−/− tumors and normal mouse lung tissues were downloaded from GEO, with the accession number of GSE137396 and GSE118246, respectively.
Expression of KMT2D mRNA in LUSC tumor and normal tissues was analyzed using GEPIA2 (http://gepia2.cancer-pku.cn/). Expression of KMT2D mRNA, phospho-EGFR, and drug sensitivity data in human LUSC cell lines were obtained from DepMap (https://depmap.org/portal/).