

Abstract
Instrumental resolution of FT-CDMS instruments with electrostatic ion trap detection of individual ions depends on the precision with which ion energy is determined. Energy can be selected using ion optic filters or from harmonic amplitude ratios (HAR) that provide Fellgett’s advantage and eliminate the necessity of ion transmission loss to improve resolution. Unlike the ion energy filtering method, the resolution of the HAR method increases with charge (improved S/N) and thus with mass. An analysis of the HAR method with current instrumentation indicates that higher resolution can be obtained with the HAR method than the best resolution demonstrated for instruments with energy selective optics for ions in the low MDa range and above. However, this gain is typically unrealized because the resolution obtainable with molecular systems in this mass range are limited by sample heterogeneity. This phenomenon is illustrated with both TMV (0.6 – 2.7 MDa) and AAV9 (3.7 – 4.7 MDa) samples where mass spectral resolution is limited by the sample, including salt adducts, and not by instrument resolution. Nevertheless, the ratio of full to empty AAV9 capsids and the included genome mass can be accurately obtained in a few minutes from 1x PBS buffer solution and an elution buffer containing 300+ mM non-volatile content despite extensive adduction and lower resolution. Empty and full capsids adduct similarly indicating that salts encrust the complexes during late stages of droplet evaporation and that mass shifts can be calibrated in order to obtain accurate analyte masses even from highly salty solutions.
Introduction
Analysis of intact biomolecules and macromolecular complexes by mass spectrometry (MS), often termed ‘native MS’, has become an important tool in structural biology to determine protein subunit stoichiometries, the extent of ligand binding, and the number and stabilities of domains in large biomolecules and complexes.1–5 In conventional native MS, the mass-to-charge ratios (m/z) of an ensemble of analyte ions are measured. The charges are determined from the ensemble data either by the spacing of isotope peaks in a single charge state or by deconvolving the different charge states of a single analyte. For large heterogeneous molecules or complexes, the charge states may not be resolved due to peak broadening and overlapping charge-state distributions resulting in the inability to accurately obtain the masses of individual constituents directly from the mass spectrum.6,7 Thus, native MS of larger biomolecular complexes typically requires relatively pure samples of the analyte of interest that have been exchanged into volatile buffers, such as ammonium acetate, that minimize the number of gas-phase adducts and concomitant peak broadening. Analyses of several different purified virus capsids up to ~18 MDa in mass have been demonstrated using conventional MS instrumentation.8,9 However, the challenge presented by sample heterogeneity grows with molecular size such that even with highly purified samples, most conventional native MS experiments are limited to molecular complexes with masses below a few MDa.7,10,11 Even when successful mass measurements are made for MDa-sized analytes, dynamic range suffers as a consequence of the increased heterogeneity because broadened peaks obscure low abundance components.
Charge detection mass spectrometry (CDMS) is an alternative to conventional MS techniques in which both the m/z and charge of an individual ion are measured simultaneously to obtain the ion mass.12–18 Results from repeated individual ion mass measurements lead to a mass histogram where the dynamic range is determined by the number of individual ions that are measured. Similar measurements can be made in Orbitrap instruments where multiple individual ion measurements are also possible.19–21 For smaller proteins and protein complexes under 200 kDa, the mass resolution achievable using current state-of-the-art CDMS instrumentation is typically inferior to that obtainable with conventional mass spectrometers. However, for much larger protein complexes and viruses weighing 500+ kDa, the resolution achieved by conventional MS methods begins to be limited by the intrinsic heterogeneity of the sample.6,7,10 Extrinsic factors, such as the extent of adduction of salts and/or solvent left over from the ionization process have been shown to often add ~1% or more to the overall mass of ions in this size range, further complicating accurate mass determinations.22 As molecular size extends into the MDa regime and intrinsic heterogeneity increases, conventional MS techniques begin to fail due to m/z peak congestion, making individual ion techniques, such as CDMS, the only option for rapid mass measurements.23–27 CDMS has been used to analyze the masses of often complex mixtures of analytes with masses ranging from a few thousand Da to 500+ MDa.16,26,28–30
CDMS instruments with electrostatic ion trap (EIT)-based detection are typically equipped with energy selective ion optics prior to the EIT to limit the range of kinetic energies of ions that are admitted into the trap.15,31 The m/z determined for each individual ion depends on ion energy so the resolution of measurements in these devices is typically limited by the width of the energy passband of the filter.32 Improved resolution comes at a cost of decreased ion transmission.33 An alternative method to measure m/z is to simultaneously measure the frequencies and energies of individual ions throughout the entire trapping period.31,34 The periodic signal generated by an ion inside of an EIT depends on the ion energy and the energy of each individual ion can be obtained from different harmonic amplitude ratios (HAR).34 This HAR method has the important advantage that ions with the exact same m/z but with different energies can be distinguished because their oscillation frequencies are different.34,35 This method makes it possible to multiplex individual ion measurements to a significantly greater extent by reducing the probability that the signals of any two ions, even those with the exact same m/z, will overlap.35,36 It also does not require attenuation of the ion signal in an energy filter in order to improve resolution, improving sensitivity for minor components in samples. Here, factors that affect the ultimate resolution of CDMS measurements that use both energy selective optics and the HAR method are explored for analytes with masses between 600 kDa and 5 MDa. These results have important implications for the mass spectrometer performance that is actually necessary to obtain useful information from high mass analytes.
Experimental
CDMS Instrumentation.
The home-built CDMS instrument used in this study has been described in detail elsewhere,31 but a brief description of the technique and parameters used are given here. Ions were generated by nanoelectrospray ionization using borosilicate capillaries that were pulled to an inner diameter of ~1.5 μm, except where otherwise specified, using a Flaming/Brown P-87 micropipette puller (Sutter Instruments, Novato, CA). An electrospray voltage of 1.5–2.1 kV (relative to the entrance skimmer potential of 350 V) was used with a modified Waters Z-spray source heated to 80 °C (Waters Corporation, Milford, MA). Ions are introduced into the instrument and transit a region containing two RF quadrupole ion guides (Ardara Technologies, Ardara, PA). Ions are accumulated in the second ion guide and then pulsed into the electrostatic ion trap at a pressure of ~3 × 10−9 Torr. A CoolFET (Amptek, Bedford, MA) charge sensitive preamplifier with a disabled Peltier cooler initially amplifies the ion induced signal, which is then conditioned by an additional custom amplification and bandpass filter stage. Data are recorded at 1.0 MHz and analyzed using short-time Fourier transforms (STFT) with a 50 ms segment length stepped across the time domain signal in 5 ms increments. Only ion signals that persisted for the entire trapping period that was selected (>80% for all analytes and acquisition times used here) were analyzed to determine m/z, charge, and mass. Typically, multiple individual ion signals, ranging from 0 to a maximum of 30 in these experiments, were recorded and analyzed in a single transient. Generally, ion current was optimized to yield an average of ~2 – 4 ions per trapping event to minimize occurrences of overlapping ion frequencies. The signals for ions with overlapping/interfering frequencies are discarded when the ions have frequencies within 80 Hz of each other.
Expression and purification of tobacco mosaic virus (TMV) proteins.
TMV was expressed and purified according to modified literature procedure.37–39 For recombinant TMV (rTMV), BL21-Codonplus (DE3)-RIL cells were transformed with the pET-rTMV vector, and colonies were selected for inoculation of Terrific Broth cultures. Cultures were induced with 30 μM isopropyl-β-D-thiogalactopyranoside (IPTG) at an OD600 of 0.6–0.8 and allowed to grow 14–18 h at 20 °C before harvesting cell pellets and storing them at −20 °C. For circularly permuted TMV (cpTMV), DH10B cells were co-transformed with the pBAD-cpTMV-S65* and pDule-pAF vectors, and colonies were selected for inoculation in autoinduction media. At an OD600 of 0.6–0.8, 1 mM p-amino-L-phenylalanine (pAF) was added and the culture was allowed to grow 18 h at 37 °C. For both rTMV and cpTMV, cell pellets were collected at 8000 rpm for 30 min and stored at −20 °C until purification. For purification, cell pellets were resuspended in 10 mL lysis buffer, 20 mM triethanolamine (TEA) pH 7.2 and lysed by sonication with a 2 s on, 4 s off cycle for a total of 10 min using a standard disruptor horn at 65% amplitude. The resulting lysate was cleared at 14,000 rpm for 30 min. The supernatant was treated with 30–40% volume of saturated ammonium sulfate and allowed to rotate for 10 min at 4 °C. The precipitated protein was collected at 11,000 rpm for 30 min and resuspended in 10 mL lysis buffer, then dialyzed in 1 L lysis buffer overnight with at least one buffer exchange. The resulting protein solution was treated with 5 mL benzonase and 4 mg MgCl2 at room temperature for 30 minutes and purified using a DEAE column with a 0 – 180 mM NaCl gradient elution in 20 mM TEA buffer, pH 7. Further purification was performed using a Sephacryl S-500 column in 10 mM NaPhos pH 7.2 elution buffer. Purity and assembly state were confirmed by SDS-PAGE, ESI-TOF LC-MS, and HPLC-SEC. After buffer exchange into 100 mM ammonium acetate, all TMV samples were diluted to ~0.5 mg/mL (~800 nM for double-disk assembly) for CDMS analysis.
Adeno-associated virus (AAV) packaging and purification.
AAV was packaged in HEK293 cells using standard triple transfection techniques.40 HEK293 cells were split into a 150 mm dish and transfected with rAAV9 packaging gene, transgene (ss-CAG-GFP), and pHelper using PEI. At 96 hours after transfection, AAV was harvested with a detergent-based method.40 Briefly, whole culture including cells and supernatant were treated with triton x-100 in the presence of benzonase (MilliporeSigma, St. Louis, MO) and then incubated at 37 °C for an hour with gentle shaking in every 15 min. AAV was purified from the harvest lysate and supernatant using POROS™ CaptureSelect™ AAVX Affinity Resin (Thermo Fisher Scientific, Waltham, MA) in accordance with the manufacturer’s guide. Briefly, harvested lysate and supernatant were centrifuged to remove any pellet and loaded into the column containing an AAV-binding resin. Resin-bound AAVs were collected by acidic elution buffer (pH 3.0) and then analyzed by CDMS. For buffer exchange, purified AAV was washed in Amicon Ultra Centrifugal Filter Unit (MilliporeSigma, St. Louis, MO) with 15 mL of 1x phosphate buffered saline (PBS) + 0.001% Tween or 100 mM ammonium acetate for total of 4 times and concentrated until the final volume reached ~100 μL. Concentrated AAV was quantified by qPCR via SYBR Green and Jumpstart Taq on a CFX RT-PCR machine (Bio-Rad, Hercules, CA) as described previously.41 Briefly, AAV samples were diluted and treated with proteinase K at 37 °C for an hour and deactivated at 95 °C for 20 min. PCR reactions were performed with primers targeting GFP in the transgene (5’-ACTACAACAGCCACAACGTCTATATCA-3’ and 5’-GGCGGATCTTGAAGTTCACC-3’) and standard curve was generated with plasmid DNA from 0.0001 to 1 ng/μL. AAV titers were measured at 7 × 1012, 1 × 1013, and 5 × 1012 vg/mL for the ammonium acetate, PBS, and elution buffer solutions, respectively.
Results and Discussion
Factors Affecting CDMS Instrumental Mass Resolution.
Instrumental mass resolution depends on many factors, including experimental variables as well as physical layout of the instrument and performance of constituent components. Here, we consider single ion CDMS instrumentation based on an EIT, the method used in this work and in many other studies. Ions are trapped in a region containing one14,15,18,31,33 or more16 detection electrode(s) where they produce a set of induced current pulses that are recorded over the duration of the trapping time. Fourier transform (FT) based analysis of these time-domain signals yields the amplitudes and frequencies of each individual ion that is trapped.15,42 The fundamental amplitude, A, depends primarily on ion charge (eq. 1), but both trap geometry and ion energy per charge, E, also significantly affect this value. The frequency at which an ion oscillates, f, is related to the m/z, through a calibration value, C(E), that depends on trap design and ion energy (eq. 2). Because both m/z and
| 1) |
| 2) |
charge are required for mass determination, calculating an instrumental mass resolution requires that measurement uncertainties in frequency, amplitude, and energy be known and propagated appropriately.
Both the frequencies and amplitudes of ions trapped in an EIT can be measured and tracked as they evolve over the ion’s lifetime in the trap using short-time Fourier transform (STFT) methods.26,36 Ion frequencies are typically determined with sufficiently high accuracy (5–6 significant digits) that the frequency contribution to the overall mass uncertainty is insignificant.25,35 However, the uncertainty associated with ion amplitudes can often be the limiting factor in overall mass resolution.36,43 Ion amplitude uncertainties are absolute; in other words, they do not depend on the identity of the ion but rather are determined by noise inherent to the instrumental components (especially the charge-sensitive pre-amplifier used to measure the ion-induced current), the length of the ion signal transient, and the FT analysis methods used.23 Charge-sensitive pre-amplifier configurations explored by Jarrold and co-workers include custom-built circuitry and cryogenic cooling that have demonstrated a factor of ~2 improvement in the ion amplitude signal-to-noise ratio (S/N) over commercially available pre-amplifiers used in these and other CDMS experiments.28 Increasing the ion trapping period also decreases amplitude uncertainty and scales by the square root of the length of the trapping period. Individual ion charge states resolved based purely on amplitude measurements when sufficiently long (>1.5 s) trapping periods are used have been demonstrated previously28,43 and in this work (Figure 1, inset). In these cases, direct assignments of each individual ion charge state can be made essentially unambiguously, making the charge uncertainty an insignificant source of error.43 However, the relatively long trapping periods required for unambiguous charge assignment have limited the practical use of high-accuracy charge measurements because of the long data acquisition times required to acquire a statistically meaningful sample of individual ions to constitute a mass histogram.
Figure 1.
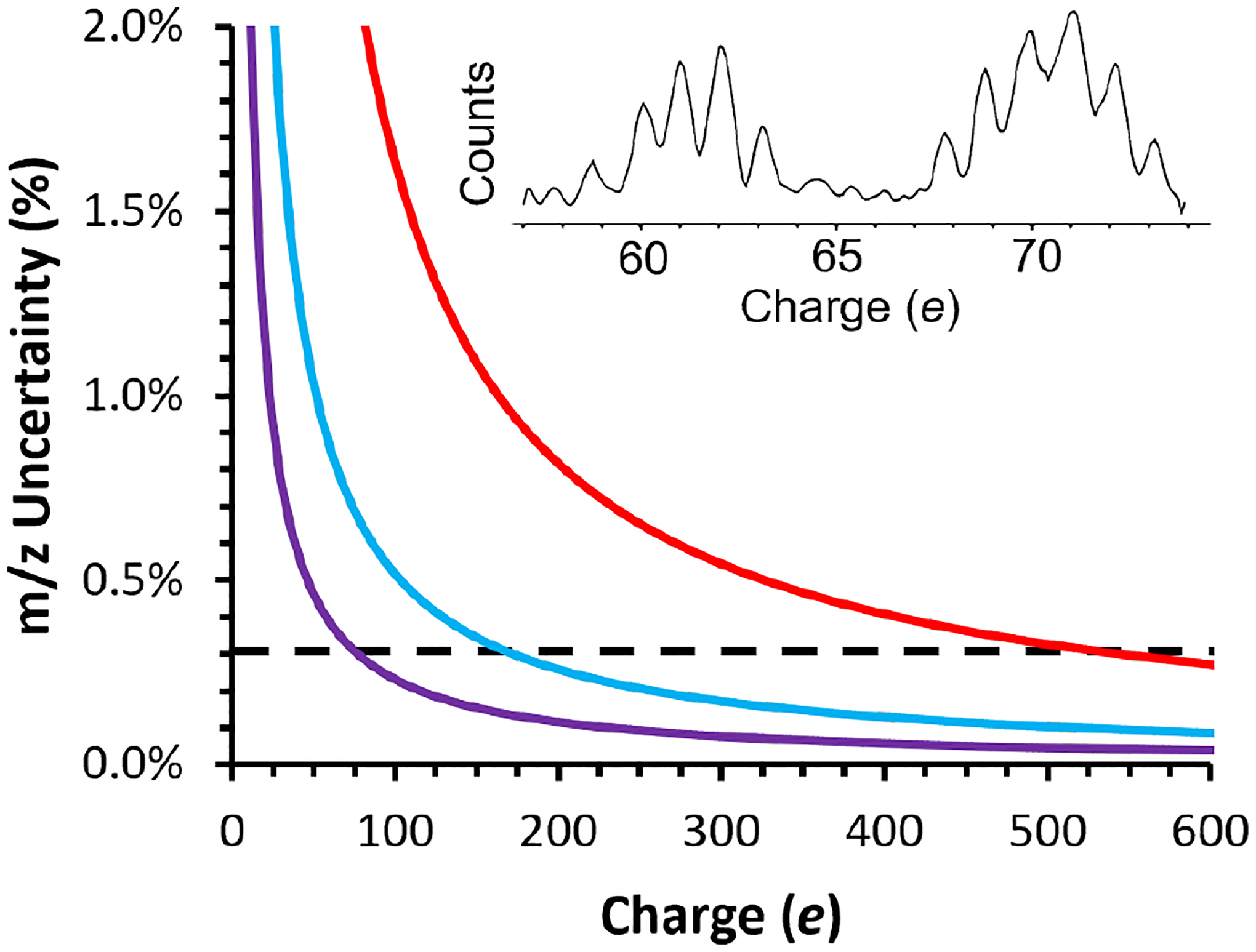
Scaling of m/z uncertainty vs. charge using different energy determination methods in CDMS. The black dashed line at ~0.3% represents m/z uncertainty with the narrowest passband filter used to date in CDMS experiments and the red, blue, and purple lines show m/z uncertainty as a function of charge for 0.1, 1.0, and 5.0 s ion trapping periods, respectively, with the HAR method used in this work. The performance of the HAR method improves and surpasses that of a physical energy filter with increasing trap time and increasing ion charge. Inset is a histogram of measured ion amplitudes (charge axis) of TMV double and triple disks (~0.60 MDa and ~0.90 MDa, respectively) trapped for 5.0 s showing resolved charge states (S/N ≈ 0.3 e).
Uncertainty in CDMS measurements of m/z depends almost entirely on uncertainty in individual ion energies (C(E) in eq. 2) and dominates the overall achievable mass resolution when charge uncertainty is low or eliminated via direct charge assignment.43 Energy selective ion filters are used in conventional CDMS instruments to pass a narrow range of ion energies into the EIT for analysis. All ion energies are then approximated as the center of the energy passband.33,42,43 The narrowest demonstrated energy bandwidth in a CDMS instrument is 0.3%; however, achieving such narrow bandwidths comes at the cost of reduced ion transmission because the energy filter naturally excludes a large fraction of the ion current that is not within this energy range.33 Nevertheless, a mass resolution of ~330 was obtained for the different assembly states of purified hepatitis B viral capsids (up to the fully assembled capsid at ~4 MDa) using a narrow ion energy passband.32 This was achieved in an EIT trap geometry where C(E) in eq. 2 is less sensitive to changes in ion energy,33 and sufficiently long transients were acquired so that charge states were amplitude resolved.32
Instead of using a filter to set ion energies in a CDMS experiment, an alternative method that uses signal harmonics to directly measure the energies of each individual ion has also been demonstrated.34 This method takes advantage of the unique pulsed signal pattern generated by ions oscillating in the EIT and its corresponding harmonics in the FT output. With a known trap geometry, a calibration between the duty cycle of the pulsed signal and the ion energy per charge can be determined with high accuracy.31,34 Differences in duty cycle corresponding to different ion energies are manifested in the FT output as different harmonic amplitude ratios (HAR). Measuring ion energies ab initio in this way eliminates the need for energy selective devices that restrict ion current enabling more ions to reach the EIT. The energy uncertainties are determined by the signal-to-noise ratios (S/N) of the two harmonic amplitudes used to determine the HAR. Because energy uncertainty is determined by S/N with this method, energy and, by extension, m/z uncertainty are a function of the ion charge.
The m/z uncertainty as a function of charge for the CDMS instrument used in this work is shown in Figure 1. The horizontal black line at 0.3% represents the best performance yet achieved using energy filtering optics.28 This value does not depend on ion charge. In contrast, the m/z uncertainty with the HAR method does depend on both ion trap time and ion charge because the uncertainty of the HAR depends on the S/N of the amplitude measurement of both the fundamental and second harmonic frequencies. The red, blue, and purple lines are the calculated m/z uncertainty using the HAR-based technique for transient lengths of 0.1 s, 1.0 s, and 5.0 s, respectively, and are obtained from experimental measurements of S/N. The method for calculating m/z uncertainty for the HAR-based technique is detailed in the Supporting Information. Inset in Figure 1 is a charge histogram measured with 5.0 s trapping time showing individually resolved charge states from signal amplitudes for tobacco mosaic virus (TMV) double and triple disks with ~60 – 70 charges with a S/N of ~0.3 e. Crossover points where the HAR-based technique results in lower overall m/z uncertainty compared to using the best demonstrated energy filtering method are at 531, 168, and 75 charges for the 0.1 s, 1.0 s, and 5.0 s trap times, respectively. These crossover points are estimated by finding the charge value at which HAR-based measurements of different acquisition lengths have an m/z uncertainty of 0.3%, equal to the lowest m/z uncertainty demonstrated to date using energy filtering optics in CDMS.28 The charging of large macromolecular complexes in native mass spectrometry depends on molecular size, and is often close to that calculated from the Rayleigh limit for an aqueous droplet of similar size.26,44 For spherical ions with a density of 0.998 g/mL formed from aqueous solutions that are charged to the Rayleigh limit, these charges correspond to ion masses of 44.8, 4.5, and 0.90 MDa, respectively. Although other factors, such as molecular shape and solution composition also play a role in molecular charging in native mass spectrometry,45–47 the Rayleigh limit approximation serves as guide that should be generally applicable to most analysis. The points at which the performance of the two types of analysis cross over is only applicable to m/z measurement performance. If charge states are not resolved in amplitude space, the measurement of the charge of individual ions also has uncertainty that contributes to the overall mass uncertainty for both the energy filtering and HAR-based methods.
An important outcome of this analysis is that the charge at which the HAR method outperforms the best energy selective ion optics decreases linearly with increasing S/N. These data are obtained with an uncooled, commercially available preamplifier. The best resolution achieved with energy filter optics was obtained with a cryogenically cooled preamplifier to increase the S/N of the amplitude measurement.32 Based on the performance reported for this cooled preamplifier (S/N of 0.174 e for a 3 s acquisition period), we estimate that the crossovers in performance between energy selective optics and the HAR method would occur at charges of 168, 76, and 24, with 0.1 s, 1.0 s and 5.0 s trap times, respectively, corresponding to masses of 4.5, 0.90, and 0.092 MDa using the same method described above. Combined with long transient acquisitions that also allow direct charge state assignment, this approach makes it possible to determine individual ion masses with arbitrarily high accuracy at the corresponding theoretical instrumental resolution, albeit at the cost of acquisition speed.
Sample Limited Resolution of High Mass Analytes.
While the HAR method enables high instrumental mass resolution for larger ions, improvements to instrumental resolution in CDMS are only valuable if they provide additional information about the analyte(s) of interest. Despite the impressive CDMS resolution of ~330 reported for highly purified hepatitis B viral capsid assemblies at 3–4 MDa,32 we are not aware of examples of such high resolution for analytes with masses above ~5 MDa and there are few examples in the low MDa range. This leads to the question of how much mass resolution is sufficient for samples with constituent masses ranging upward from the high kDa range. Figure 2 shows CDMS mass histograms for recombinant TMV (rTMV) ‘double disk’ structures composed of two 17-mer disks (Figure 2a, 602 kDa, ~60 e) measured using different trapping times. A Gaussian peak shape was used to fit the peaks in the mass histograms and the width of the Gaussian fit at 50% peak height was used to determine the mass resolution. Increasing the trapping period from 0.1 s to 1.0 s (Figure 2b and 2c, respectively) results in an increase in resolution from 23 to 76 corresponding to slightly over a 3-fold improvement. This is close to the theoretical gain in resolution expected for a 10-fold increase in measurement time. This indicates that the resolution is largely instrument-limited at these trap times for this sample. Increasing the trap time to 5 s (Figure 2d) increases the measured resolution to 108, but this improvement is less than the theoretical value of (expected peak shape shown by the red dotted line), indicating that inherent ion heterogeneity has become a significant contributor to the observed peak width in Figure 2d. Conventional mass spectra for the rTMV subunit proteins indicate that the proteins are homogeneous (Figure S1) but some salt adduction is observed. These results indicate that the ion heterogeneity observed for these complexes is a result of adduction of salts, although incomplete desolvation may contribute as well. Peak broadening and shifts to higher mass has been observed in conventional native mass spectrometry measurements for other large complexes in this size range.7,22 The peak centroids for all three sets of data are the same within 0.1 kDa. For rTMV, no similar mass species are expected or observed, and the higher mass resolution does not yield additional information.
Figure 2.
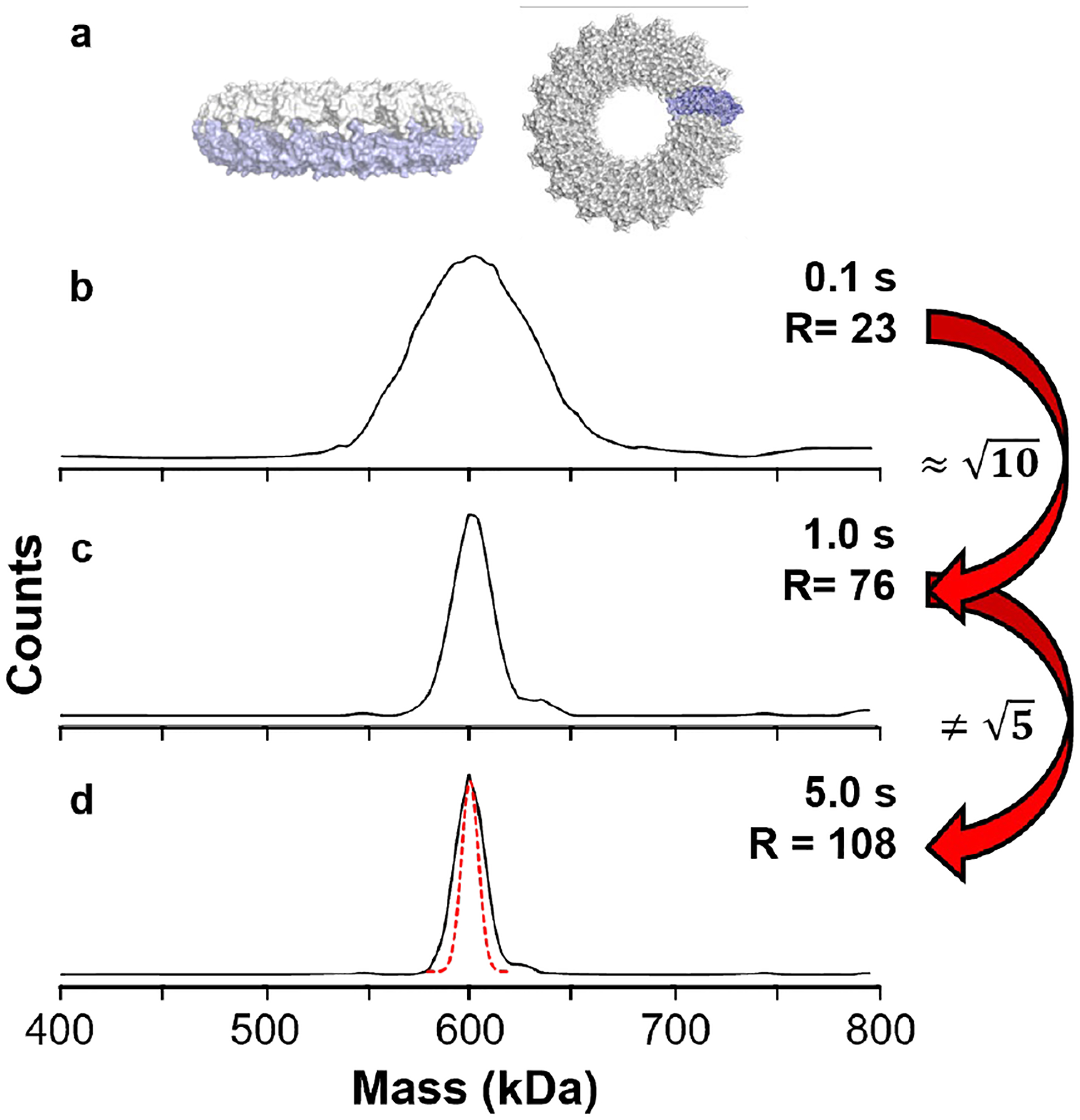
The mass resolution achievable for rTMV double disk ions (a) as a function of the ion trapping period for 0.1, 1.0, and 5.0 s and the resulting mass histograms in (b), (c), and (d), respectively. A 10-fold increase in trapping time between (b) and (c) yields the expected increase in resolution whereas the 5x increase in trapping time between (c) and (d) yields much less than a increase in resolution (expected peak shape shown by the red dotted line), suggesting that sample heterogeneity is a limiting factor in the achievable resolution.
In some experiments, higher instrumental resolution may be necessary to resolve meaningful components of a sample, especially for analytes on the lower end of the practical mass range of CDMS. Circularly permuted TMV (cpTMV) forms ‘double disk’ structures similar to rTMV and has been engineered to contain residues amenable to conjugation with synthetic dyes as a platform for modeling energy transfer processes in photosynthetic light harvesting.37,38,48 A peripheral mutation from serine to the non-canonical amino acid p-aminophenylalanine (cpTMV-S65-pAF) results in populations of both 16-mer and 17-mer disk and the corresponding 32-mer and 34-mer ‘double disk’ complexes. Mass histograms of ~5,000 cpTMV-S65-pAF ions measured with different trapping times are shown in Figure 3. A 0.1 s trapping time resulted in a 3.2 min. total data acquisition time with an average of 2.6 ions per trapping event. This extent of multiplexing largely eliminates ion-ion frequency overlaps so that discrimination of predominant ions should not occur even in relatively pure samples.35 A broad peak is observed with a width significantly greater than the instrument resolution suggesting that there are multiple components (Figure 3a). The 32- and 34-mer complexes are resolved (R ~50) with a 0.5 s trap time corresponding to a total data acquisition time of 13.3 min. (Figure 3b). These two species are more clearly resolved with a 1 s trap time (Figure 3c) corresponding to a data acquisition time of 25.1 min. for ~5,000 individual ion measurements. This results in a mass resolution of ~70. Thus, for relatively low mass, low charge, and low heterogeneity analytes, such as TMVs, longer trapping periods and higher instrumental resolution can be required to reach a meaningful information threshold.
Figure 3.
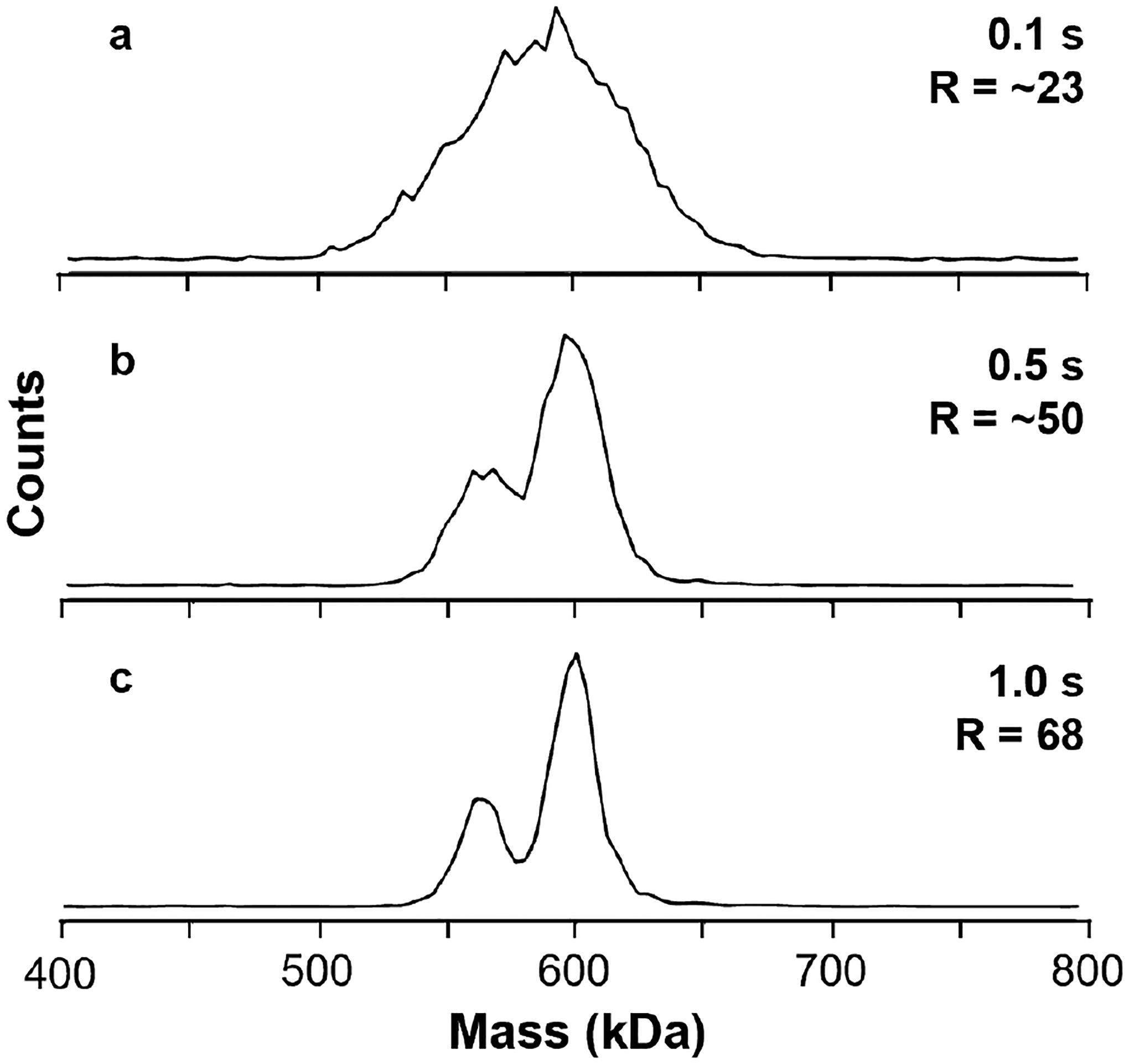
Mass histograms of cpTMV-S65-pAF for ions acquired with 0.1 s (a), 0.5 s (b), and 1.0 s (c) trapping periods. The two peaks corresponding to the 32- and 34-mer species are not well-resolved in (a) but increasing the trapping period to 0.5 s (b) sufficiently increases the resolution to make it possible to distinguish the two species. Further increasing the period to 1.0 in (c) yields two well-defined peaks that can be straightforwardly quantified.
In these experiments, the masses of multiple individual ions are measured simultaneously. For example, an average of 3.2 ions are measured in each of the 1 s trapping periods used to acquire the Figure 3c data. If these measurements were restricted to just one ion per measurement, as is commonly done in other CDMS experiments,23 data acquisition for 5000 ions using this same trap period would require a minimum of ~217 minutes under ideal ion injection conditions where trapping just a single ion is most probable (~37% of all spectra containing zero, one, or multiple ions). Thus, the multiplexing of individual ion measurements36 results in over an 8-fold gain in data acquisition speed for these measurements with no loss of effective mass resolution.
The potential for molecular heterogeneity increases with molecular size suggesting that sample-limited resolution will become increasingly relevant as molecular size increases into the MDa size range. Adeno-associated viruses (AAVs) are of interest as delivery vehicles in gene therapy and their effectiveness in the treatment of rare genetic diseases has been demonstrated.49–51 These types of samples are well suited to analysis by CDMS because of their high mass and heterogeneity. CDMS mass histograms for AAV9 empty capsid (~3.7 MDa) and genome-containing virus ions (~4.7 MDa) measured from a 100 mM ammonium acetate solution using different trapping period lengths are shown in Figure 4. The much higher average charge of AAV9 ions (~150 e) relative to TMV ions means that higher instrumentation resolutions are achieved even when the trapping period is the same (Figure 1). At 0.1 s (Figure 4a), peaks for the empty AAV9 capsid at 3.7 MDa and the ‘full’ AAV9 at 4.7 MDa are easily resolved with a resolution of 43. The short trapping period, combined with previously reported multiplexing methods, made it possible to acquire the ~2,000 individual ion mass measurements in 1.9 minutes (~1.8 ions per trapping period). A longer 0.5 s trap time (Figure 4b) increases the resolution to 65. This 1.5 gain in resolution is significantly less than the theoretical factor of = 2.2 gain in instrument resolution demonstrated for the smaller TMVs. The longer trap time increases the data acquisition time necessary for 2,000 ions to 9.2 min. or a 4.8x increase in time (the value is slightly lower than 5 because the ion current varies slightly throughout these measurements). Increasing the trapping period to 1 s (Figure 4c) or even 5 s (Figure 4d) continues to increase the total time required for a ~2,000 ion acquisition (17.4 mins and 100 mins, respectively), but yields essentially no improvement in the observed resolution (69 and 67, respectively), despite expected improvements in instrumental resolution. The observed full/empty capsid ratio varies from a low of 0.20 in Figure 4a to 0.32 in Figure 4b. This range is greater than the statistical variation (1 – 2%) expected from the number of ions analyzed. The origin of this variation is unclear, but it does not appear to depend on trapping time and the associated accumulation time in an ion guide prior to mass analysis. From these observations, it is evident that the intrinsic heterogeneity of the AAV9 sample, including that from salt adducts, dominates the observable resolution at trapping periods as short as 0.5 s and is still a relevant contributor in periods as short as 0.1 s.
Figure 4.
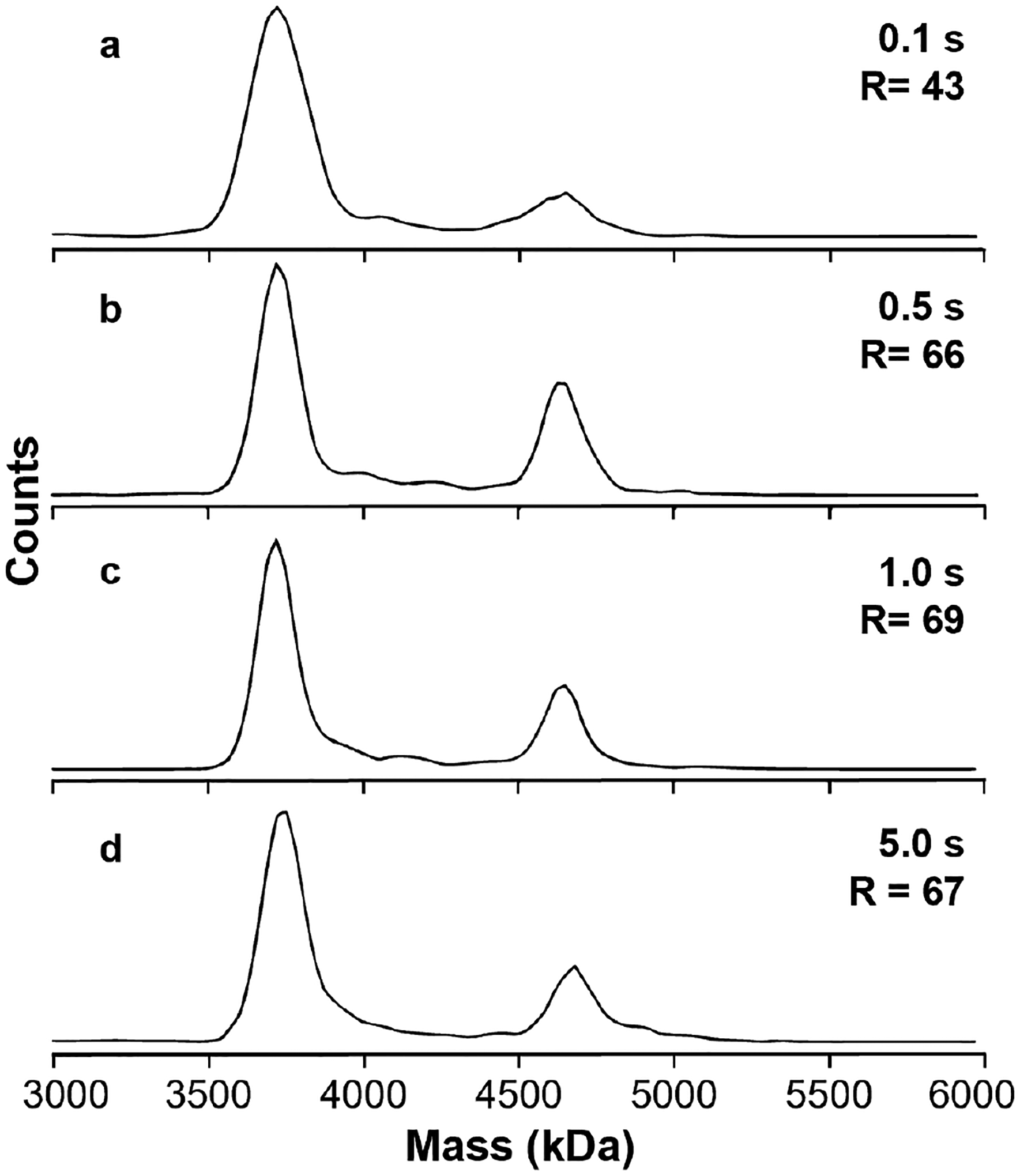
Mass histograms of AAV9 ions measured with different trap times. With the shortest period of 0.1 s (a), the empty and full capsid species are well-resolved (R = 43); at 0.5 s (b), the observed resolution (R = 68) is improved, but by less than the expected factor of ; at 1.0 s (c) or 5.0 s (d), the observed resolution does not improve, indicating that the resolution is entirely sample dependent for trapping periods >~0.5 s.
Other CDMS measurements of AAVs by Jarrold and co-workers52,53 as well as Heck and co-workers20,54 have obtained similar peak widths for different AAV samples despite differences in CDMS instrument type, configuration, experimental conditions, and trapping period length, providing additional evidence that the observable resolution for AAVs is generally sample limited. Worner et al.9 have also used Orbitrap-UHMR operating in conventional m/z-only mode to analyze highly purified wild-type AAV capsids and were able to resolve peaks corresponding to nearly isobaric combinations of the VP1, VP2, and VP3 viral proteins that compose the capsid. However, while the isobaric coincidence of the different VP protein combinations allows for some finer peak structure to be resolved with an apparent resolution of ~500, the heterogeneity of the AAV capsid still prohibits resolution of any individual subunit combination despite the very high theoretical instrumental resolution (>20,000) that is achievable using the Orbitrap-UHMR in the relevant m/z range.9
Weighing AAVs from Biochemically Relevant Solutions.
Many of the MDa-sized analytes amenable to CDMS analysis are large biomolecular complexes that exist natively in complex solution matrices consisting of various ions and osmolytes. However, most conventional and CDMS-based ‘native’ experiments are performed using volatile buffer solutions, such as ammonium acetate, to reduce adduction and resulting loss of resolution that can occur. Moreover, ammonium acetate is not an effective buffer at pH ~7, the pH of initial solutions used in most native mass spectrometry measurements.55 Unlike conventional m/z-based MS techniques, which require resolved charge states for mass determination, the individual ion mass measurements of CDMS make it possible to produce a mass histogram even with high concentrations of non-volatile solutes in solution.26 However, most CDMS experiments are still performed using ammonium acetate to avoid mass shifts and decreased resolution due to adduction, despite evidence that different buffer solution can affect the stabilities and structures of proteins and larger complexes.56
To address the viability of measuring AAVs directly from biochemically relevant buffer solutions, mass histograms of AAV9 originating from solutions of 100 mM ammonium acetate (Figure 5a), 1x PBS (Figure 5b; ~150 mM non-volatile salt content), and an elution buffer used in the purification process (Figure 5c; 300+ mM non-volatile content, including ~100 mM NaCl, ~150 mM Tris-HCl, and ~50 mM glycine) were acquired. Submicron electrospray emitters (~600 nm tip diameter) were used to attenuate the extent of adduction. The application of these nanoscale emitters to high-salt solutions is described elsewhere.57 A trapping period of 1 s was used based on the Figure 4 results, which show that the resolution for AAV9 capsids is sample limited at this trapping time. Each mass histogram in Figure 5 has two clearly resolved peaks corresponding to the empty and full AAV9 capsids. The centroid masses measured from the ammonium acetate solution in (a) are 3.73 and 4.72 MDa, respectively, corresponding well with the theoretical calculated capsid mass of 3.72 MDa and the expected genome mass of 0.987 MDa. The centroid masses of both the empty and full capsids are shifted upward in mass by ~120 kDa and ~520 kDa from 1x PBS and from the elution buffer, respectively, but retain constant spacing of ~1.0 MDa corresponding to the genome mass.
Figure 5.
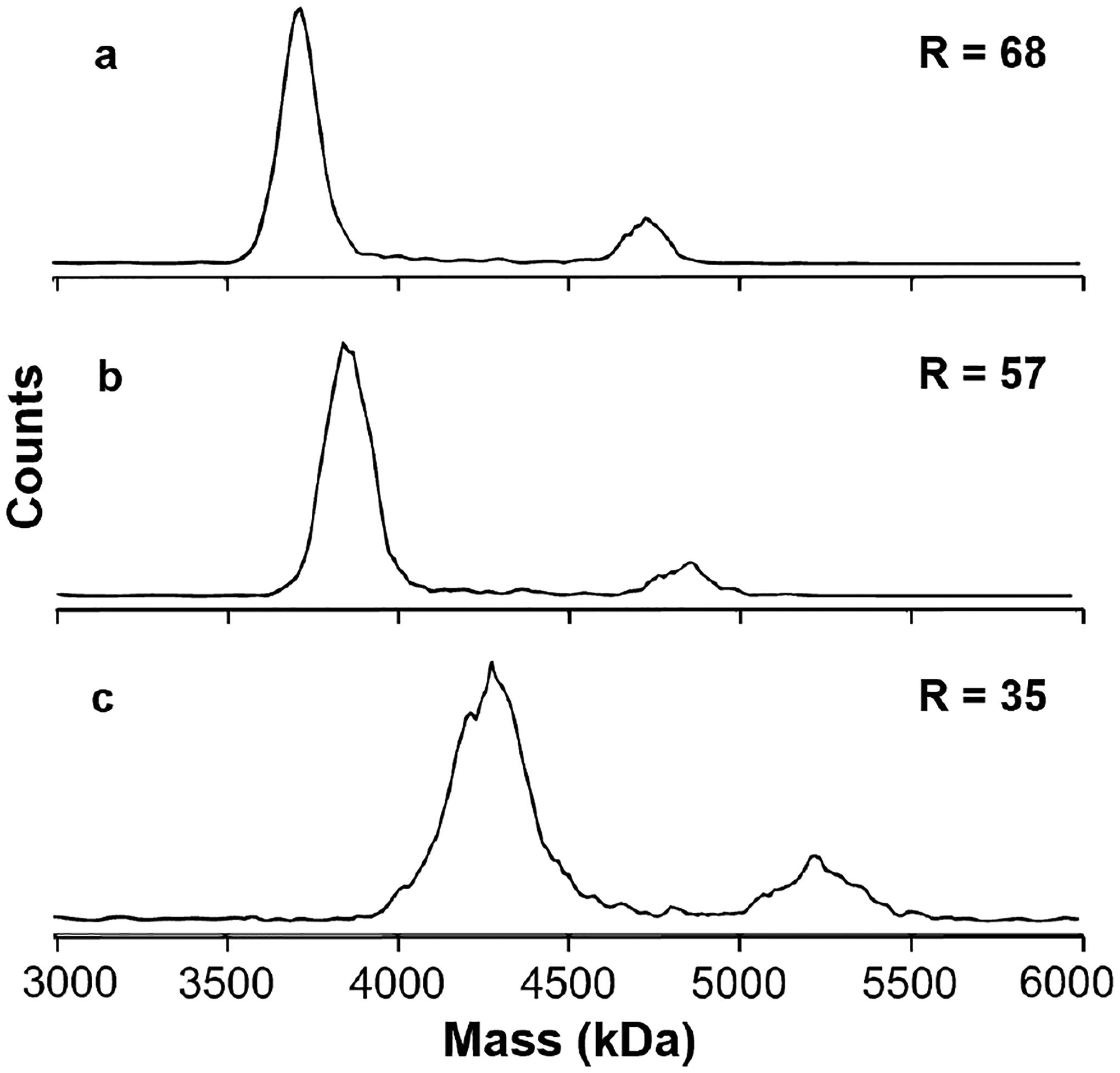
Mass histograms for AAV9 measured from an ammonium acetate solution (a), 1x PBS buffer (b) or a column elution buffer (c) containing high concentrations of non-volatile solutes (~150 mM and ~300 mM, respectively). Ammonium acetate results in the highest resolution and the measured full and empty capsids are close to the theoretical values (3.72 and 4.72 MDa, respectively). Ions formed from 1x PBS buffer and column elution buffer are highly adducted, leading to mass shifts of ~120 and ~520 kDa, respectively, but the difference in mass between the empty and full capsids remains constant in all solutions, indicating that the mass of the genome contents can be accurately measured from these solutions.
The mass shift observed for both the empty and genome-containing species is due to adduction of non-volatile solutes onto these large complexes. The empty and full capsids have nearly identical surface areas as evidenced by their similar extent of charging (Figure S2). The similar extents of adduction to both the empty and full capsids appears to be related to the surface area and not the masses of these complexes. This suggests that the majority of the solutes are adducting at the surfaces of these complexes as a result of solvent evaporation and subsequent envelopment of the complexes with residual non-volatile content. This result is important in that although the masses of these ions increase significantly as a result of being formed from salty solutions, the ability to measure the genome-containing species inside these complexes is not adversely affected. The salt adduction does reduce the mass resolution, from 68 in ammonium acetate (Figure 5a) to 57 and 35 in 1x PBS and the elution buffer, Figures 5b and 5c, respectively. This is due to the heterogeneity inherent to increasing numbers of adducts. This experiment demonstrates that genome masses and empty/full/intermediate capsid ratios can still be determined from biochemically relevant solutions even if exact capsid/virion masses are increased by adduction. In principle, the unadducted masses of complexes formed from 1x PBS or other formulation buffers could be obtained from calibrations of mass shifts as a function of salt concentration similar to the mass correction that is routinely done for large complexes formed from ammonium acetate.22 These results also indicate that increased heterogeneity inherent to the use of biochemically relevant formulations further limits the achievable resolution and decreases the instrumental resolution requirements required to reach key information thresholds.
The higher viral protein antigen load associated with empty capsids has been implicated in host immune responses to AAV gene therapies, such that it is important to develop analytical methods to accurately measure their presence.58 The rapid characterization of empty/full capsid ratios of AAVs demonstrated here (<2 min.) and by others9,20,52–54 (~10–120 min.) using CDMS methods has the potential to accelerate the development process of AAV-based gene therapies in both basic research and in quality assurance testing stages. However, the formulation buffers used for the final products typically contain significant concentrations (100 mM+) of non-volatile content. Characterization directly from formulation buffers is highly desirable both to identity solution-dependent behavior and to demonstrate fidelity of the AAVs in the administered solutions, an important step in obtaining regulatory approvals.59 Although the ratio of empty to full capsid does not differ significantly in these different buffers, a result most likely related to their high stabilities, this may not be the case where multiple forms of a complex exist that have low barriers to inter-conversion.56
Practical CDMS Resolution.
Molecular complexity and heterogeneity naturally increase with mass. Figure 6 illustrates this phenomenon by comparing the theoretical resolution with the HAR method and the observed resolution of analytes acquired with 1 s trapping periods as a function of charge. The blue points correspond to the observed resolution and average charge for different size ‘stacks’ of the rTMV 17-mer disk, starting from the ‘double disk’ shown in Figure 2 to the 9-disk stack species. A mass histogram showing all rTMV stacking species is included in the Supporting Information (Figure S3). The black points are observed resolution and charge for AAV9 and are labeled with the sample buffer solution used. Finally, the red squares indicate the theoretical expected instrumental resolution at matching values of charge for each species observed experimentally. For the lower mass rTMV species that have fewer charges, the observed and expected resolutions are comparable, suggesting that the observed resolution is mostly instrument limited. The observed resolution improves as charge and mass increase but at a slower rate than expected resolution, indicating that a transition to sample limited resolution occurs. For AAV9, it is clear that the resolution is entirely sample limited because the observed resolution falls well below the expected resolution and decreases further when non-volatile buffer solutions are used.
Figure 6.
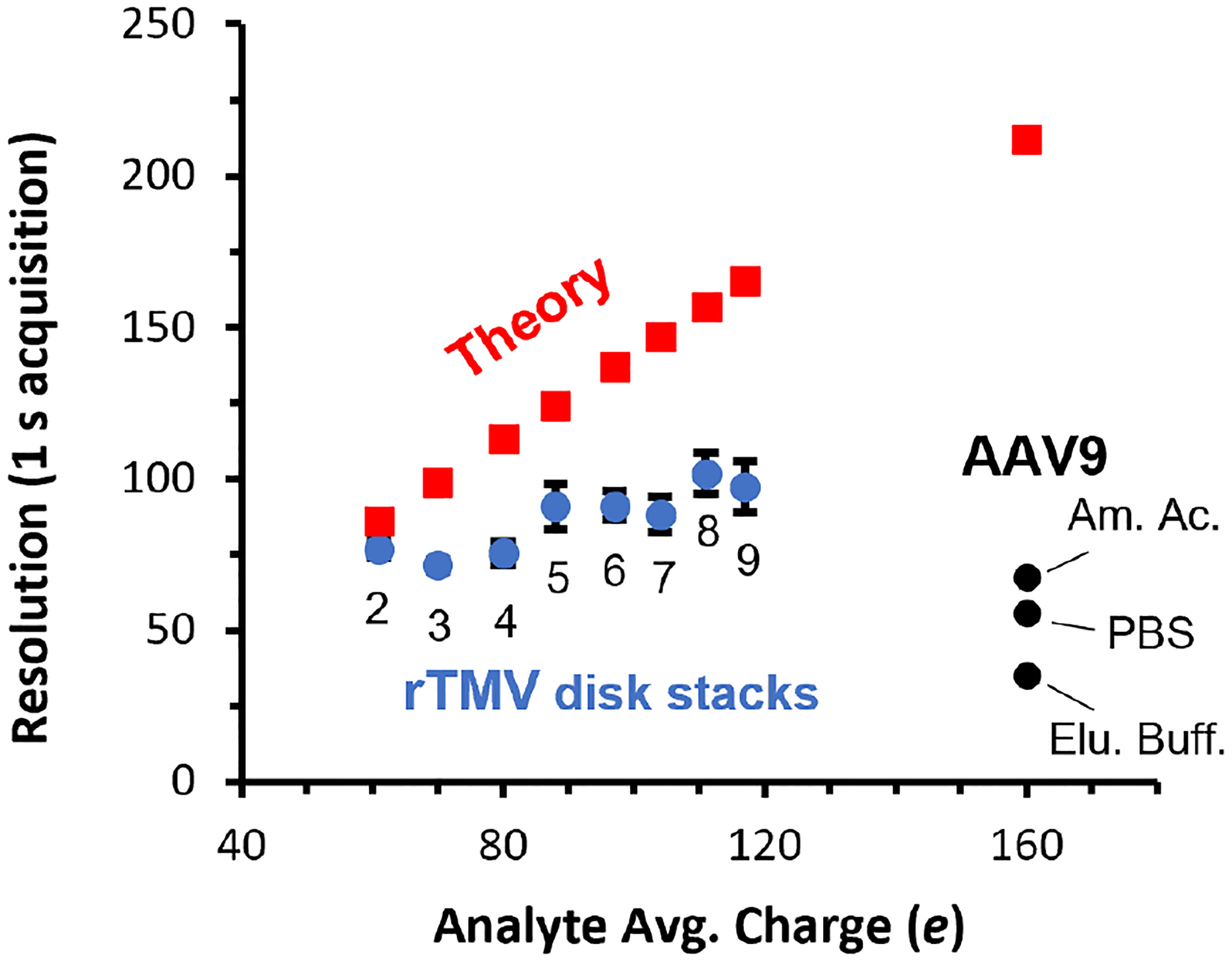
Mass resolution as a function of analyte charge for a 1 s trapping period using the HAR-based method. The experimentally observed resolution for rTMV disk stacks ranging in mass from ~600–2700 kDa (blue points) and observed resolutions for AAV9 formed from three different solutions (black points) show an increasing deviation from the theoretical instrument resolution (red points) with increasing mass and nonvolatile salt concentration, indicating that sample heterogeneity can limit mass resolution for analyte even in the low MDa mass range.
The resolution at which meaningful information thresholds are achieved decreases as mass increases. Conversely, CDMS instrument resolution using the HAR method improves with increased mass, charge, and measurement time, resulting in a ‘crossover’ point in mass and charge where instrumental resolution clearly exceeds the inherent sample resolution, as demonstrated for AAV9 in this work. The exact crossover point varies depending on the inherent heterogeneity of the analyte, instrument performance, and trapping time but the results presented here and by others in state-of-the-art CDMS experiments indicate this crossover typically occurs in the 1–10 MDa, ~75–250 e range (assuming Rayleigh charge and a density of 1.0 g/mL) when typical trapping periods (0.1–1 s) are used. For especially heterogeneous samples, such as those measured directly from formulation or in other complex matrices, crossover into the sample limited resolution regime may even occur at lower mass. In these cases, the speed of data acquisition, i.e., trapping periods, should be optimized to match the theoretical instrumental resolution with the ‘information threshold’ resolution determined by the sample. Doing so significantly decreases the overall acquisition time required, enables time-dependent experiments on faster timescales, and accelerates the characterization of analytes by CDMS.
Conclusions
Instrumental resolution in CDMS is ultimately limited by the measurement of ion energy. Energy resolution for instruments with energy selective optics can be improved by reducing the energy passband, but this comes at a cost of decreased ion transmission. Instrumental resolution with the HAR method is limited by S/N, which improves with charge and hence molecular mass. For larger, more highly charged ions, the instrument resolution with the HAR method has the potential to be significantly higher than that obtained with energy selective optics to date. Because ions are not filtered for a specific energy, ions with a broader range of energies can be analyzed. This Fellgett’s advantage means that higher sensitivity can be obtained when the ion count for a minor constituent is low, as can often be the case for 10+ MDa complexes. The HAR method has the additional advantage that ions with the exact same m/z but with different energies can be readily resolved. This significantly reduces the probability of signal overlap when the mass of many ions inside the trap are measured simultaneously for higher measurement speed.
With state-of-the-art cooled amplifiers, the mass resolution using the HAR method should be ~400+ for analytes with masses at or above about 1 MDa. This exceeds the highest resolution achieved for high mass analytes measured with CDMS instruments that use energy selective optics to improve mass resolution. However, an important outcome of this work is that resolution for large macromolecular assemblies is not typically limited by the instrumental resolution but rather the intrinsic heterogeneity of the sample. Although extensive purification and the use of non-physiologically relevant solutions, such as ammonium acetate, have been used to extend the mass of range of conventional mass measurements to the low tens of MDa, the extension of these methods to analytes with even higher mass is challenging. Moreover, many analytes, such as AAVs, can have high intrinsic heterogeneity that limits the achievable resolution for full capsids to under 100, as reported for different CDMS and Orbitrap instruments to date.9,20,52–54 Heterogeneity is increased when ions are formed from 1x PBS or other formulation buffers due to the adduction of non-volatile solutes. However, this work demonstrates that the mass of the genome contained in the capsid can still be accurately determined from these solutions because the extent of adduction to the empty and full capsids is nearly the same. This also indicates that most of the adduction occurs at late stages of ion formation as a result of concentrated salts forming a ‘crust’ on the outside of these assemblies. The ability to characterize AAVs and other high mass therapeutics directly from phosphate or other formulation buffers extends “native” mass spectrometry to solutions that are more biochemically relevant.
Supplementary Material
Acknowledgements.
The authors are grateful for financial support from the National Institutes of Health (5R01GM139338 for C.C.H., Z.M.M. and E.R.W), a Bakars Fellows Award (to D.V.S.). A.J.B. thanks a Chemical Biology Training Grant from the NIH (T32 GM066698) and the NSF Graduate Fellowship Program (DGE 1752814 and 2146752) for financial support. A.J.B. and M.B.F. thank the Director, Office of Science, Chemical Sciences, Geosciences, and Biosciences Division, of the U.S. Department of Energy under Contract No. DEAC02-05CH1123, for financial support.
Footnotes
Supporting Information.
Supplementary experimental section for TMV preparation and characterization including conventional mass spectra, detailed methods for error propagation in HAR-based m/z determination, 2D histogram of mass and charge of AAV9, and extended 1D histogram for rTMV.
References
- (1).Van Den Heuvel RHH; Heck AJR Native Protein Mass Spectrometry: From Intact Oligomers to Functional Machineries. Curr. Opin. Chem. Biol 2004, 8 (5), 519–526. [DOI] [PubMed] [Google Scholar]
- (2).Hernández H; Robinson CV Determining the Stoichiometry and Interactions of Macromolecular Assemblies from Mass Spectrometry. Nat. Protoc 2007, 2 (3), 715–726. [DOI] [PubMed] [Google Scholar]
- (3).Loo JA Electrospray Ionization Mass Spectrometry: A Technology for Studying Noncovalent Macromolecular Complexes. Int. J. Mass Spectrom 2000, 200 (1–3), 175–186. [Google Scholar]
- (4).Kintzer AF; Sterling HJ; Tang II; Abdul-Gader A; Miles AJ; Wallace BA; Williams ER; Krantz BA Role of the Protective Antigen Octamer in the Molecular Mechanism of Anthrax Lethal Toxin Stabilization in Plasma. J. Mol. Biol 2010, 399 (5), 741–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Han L; Hyung SJ; Mayers JJS; Ruotolo BT Bound Anions Differentially Stabilize Multiprotein Complexes in the Absence of Bulk Solvent. J. Am. Chem. Soc 2011, 133 (29), 11358–11367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Weiss VU; Bereszcazk JZ; Havlik M; Kallinger P; Gösler I; Kumar M; Blaas D; Marchetti-Deschmann M; Heck AJR; Szymanski WW; Allmaier G Analysis of a Common Cold Virus and Its Subviral Particles by Gas-Phase Electrophoretic Mobility Molecular Analysis and Native Mass Spectrometry. Anal. Chem 2015, 87 (17), 8709–8717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lössl P; Snijder J; Heck AJR Boundaries of Mass Resolution in Native Mass Spectrometry. J. Am. Soc. Mass Spectrom 2014, 25 (6), 906–917. [DOI] [PubMed] [Google Scholar]
- (8).Snijder J; Rose RJ; Veesler D; Johnson JE; Heck AJR Studying 18 MDa Virus Assemblies with Native Mass Spectrometry. Angew. Chemie - Int. Ed 2013, 52 (14), 4020–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wörner TP; Bennett A; Habka S; Snijder J; Friese O; Powers T; Agbandje-McKenna M; Heck AJR Adeno-Associated Virus Capsid Assembly Is Divergent and Stochastic. Nat. Commun 2021, 12 (1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Aquilina JA; Benesch JLP; Bateman OA; Slingsby C; Robinson CV Polydispersity of a Mammalian Chaperone: Mass Spectrometry Reveals the Population of Oligomers in AB-Crystallin. Proc. Natl. Acad. Sci. U. S. A 2003, 100 (19), 10611–10616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Snijder J; Van De Waterbeemd M; Damoc E; Denisov E; Grinfeld D; Bennett A; Agbandje-Mckenna M; Makarov A; Heck AJR Defining the Stoichiometry and Cargo Load of Viral and Bacterial Nanoparticles by Orbitrap Mass Spectrometry. J. Am. Chem. Soc 2014, 136 (20), 7295–7299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Shelton H; Hendricks CD; Wuerker RF Electrostatic Acceleration of Microparticles to Hypervelocities. J. Appl. Phys 1960, 31 (7), 1243–1246. [Google Scholar]
- (13).Barney BL; Daly RT; Austin DE A Multi-Stage Image Charge Detector Made from Printed Circuit Boards. Rev. Sci. Instrum 2013, 84 (11), 114101. [DOI] [PubMed] [Google Scholar]
- (14).Benner WH A Gated Electrostatic Ion Trap to Repetitiously Measure the Charge and m/z of Large Electrospray Ions. Anal. Chem 1997, 69 (20), 4162–4168. [Google Scholar]
- (15).Contino NC; Jarrold MF Charge Detection Mass Spectrometry for Single Ions with a Limit of Detection of 30 Charges. Int. J. Mass Spectrom 2013, 345–347, 153–159. [Google Scholar]
- (16).Elliott AG; Merenbloom SI; Chakrabarty S; Williams ER Single Particle Analyzer of Mass: A Charge Detection Mass Spectrometer with a Multi-Detector Electrostatic Ion Trap. Int. J. Mass Spectrom 2017, 414, 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Miller MEC; Mezher M; Continetti RE Tapered Image Charge Detector for Measuring Velocity Distributions of Submicrometer Particle Scattering. Rev. Sci. Instrum 2020, 91 (6), 063305. [DOI] [PubMed] [Google Scholar]
- (18).Doussineau T; Yu Bao C; Clavier C; Dagany X; Kerleroux M; Antoine R; Dugourd P Infrared Multiphoton Dissociation Tandem Charge Detection-Mass Spectrometry of Single Megadalton Electrosprayed Ions. Rev. Sci. Instrum 2011, 82 (8), 084104. [DOI] [PubMed] [Google Scholar]
- (19).Kafader JO; Melani RD; Durbin KR; Ikwuagwu B; Early BP; Fellers RT; Beu SC; Zabrouskov V; Makarov AA; Maze JT; Shinholt DL; Yip PF; Tullman-Ercek D; Senko MW; Compton PD; Kelleher NL Multiplexed Mass Spectrometry of Individual Ions Improves Measurement of Proteoforms and Their Complexes. Nat. Methods 2020, 17 (4), 391–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wörner TP; Snijder J; Bennett A; Agbandje-McKenna M; Makarov AA; Heck AJR Resolving Heterogeneous Macromolecular Assemblies by Orbitrap-Based Single-Particle Charge Detection Mass Spectrometry. Nat. Methods 2020, 17 (4), 395–398. [DOI] [PubMed] [Google Scholar]
- (21).Kafader JO; Melani RD; Senko MW; Makarov AA; Kelleher NL; Compton PD Measurement of Individual Ions Sharply Increases the Resolution of Orbitrap Mass Spectra of Proteins. Anal. Chem 2019, 91 (4), 2776–2783. [DOI] [PubMed] [Google Scholar]
- (22).McKay AR; Ruotolo BT; Ilag LL; Robinson CV Mass Measurements of Increased Accuracy Resolve Heterogeneous Populations of Intact Ribosomes. J. Am. Chem. Soc 2006, 128 (35), 11433–11442. [DOI] [PubMed] [Google Scholar]
- (23).Keifer DZ; Pierson EE; Jarrold MF Charge Detection Mass Spectrometry: Weighing Heavier Things. Analyst 2017, 142 (10), 1654–1671. [DOI] [PubMed] [Google Scholar]
- (24).Keifer DZ; Jarrold MF Single-Molecule Mass Spectrometry. Mass Spectrom. Rev 2017, 36 (6), 715–733. [DOI] [PubMed] [Google Scholar]
- (25).Jarrold MF Applications of Charge Detection Mass Spectrometry in Molecular Biology and Biotechnology. Chem. Rev 2022, 122 (8), 7415–7441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Harper CC; Brauer DD; Francis MB; Williams ER Direct Observation of Ion Emission from Charged Aqueous Nanodrops: Effects on Gaseous Macromolecular Charging. Chem. Sci 2021, 12 (14), 5185–5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Antoine R Weighing Synthetic Polymers of Ultra-High Molar Mass and Polymeric Nanomaterials: What Can We Learn from Charge Detection Mass Spectrometry? Rapid Commun. Mass Spectrom 2020, 34 (S2), e8539. [DOI] [PubMed] [Google Scholar]
- (28).Todd AR; Alexander AW; Jarrold MF Implementation of a Charge-Sensitive Amplifier without a Feedback Resistor for Charge Detection Mass Spectrometry Reduces Noise and Enables Detection of Individual Ions Carrying a Single Charge. J. Am. Soc. Mass Spectrom 2020, 31 (1), 146–154. [DOI] [PubMed] [Google Scholar]
- (29).Doussineau T; Bao CY; Antoine R; Dugourd P; Zhang W; D’Agosto F; Charleux B Direct Molar Mass Determination of Self-Assembled Amphiphilic Block Copolymer Nanoobjects Using Electrospray-Charge Detection Mass Spectrometry. ACS Macro Lett 2012, 1 (3), 414–417. [DOI] [PubMed] [Google Scholar]
- (30).Barnes LF; Draper BE; Jarrold MF Analysis of Recombinant Adenovirus Vectors by Ion Trap Charge Detection Mass Spectrometry: Accurate Molecular Weight Measurements beyond 150 MDa. Anal. Chem 2022, 94 (3), 1543–1551. [DOI] [PubMed] [Google Scholar]
- (31).Elliott AG; Harper CC; Lin HW; Williams ER Mass, Mobility and MSN Measurements of Single Ions Using Charge Detection Mass Spectrometry. Analyst 2017, 142 (15), 2760–2769. [DOI] [PubMed] [Google Scholar]
- (32).Todd AR; Barnes LF; Young K; Zlotnick A; Jarrold MF Higher Resolution Charge Detection Mass Spectrometry. Anal. Chem 2020, 92 (16), 11357–11364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hogan JA; Jarrold MF Optimized Electrostatic Linear Ion Trap for Charge Detection Mass Spectrometry. J. Am. Soc. Mass Spectrom 2018, 29 (10), 2086–2095. [DOI] [PubMed] [Google Scholar]
- (34).Harper CC; Elliott AG; Lin HW; Williams ER Determining Energies and Cross Sections of Individual Ions Using Higher-Order Harmonics in Fourier Transform Charge Detection Mass Spectrometry (FT-CDMS). J. Am. Soc. Mass Spectrom 2018, 29 (9), 1861–1869. [DOI] [PubMed] [Google Scholar]
- (35).Harper CC; Williams ER Enhanced Multiplexing in Fourier Transform Charge Detection Mass Spectrometry by Decoupling Ion Frequency from Mass to Charge Ratio. J. Am. Soc. Mass Spectrom 2019, 30 (12), 2637–2645. [DOI] [PubMed] [Google Scholar]
- (36).Harper CC; Elliott AG; Oltrogge LM; Savage DF; Williams ER Multiplexed Charge Detection Mass Spectrometry for High-Throughput Single Ion Analysis of Large Molecules. Anal. Chem 2019, 91 (11), 7458–7468. [DOI] [PubMed] [Google Scholar]
- (37).Delor M; Dai J; Roberts TD; Rogers JR; Hamed SM; Neaton JB; Geissler PL; Francis MB; Ginsberg NS Exploiting Chromophore-Protein Interactions through Linker Engineering to Tune Photoinduced Dynamics in a Biomimetic Light-Harvesting Platform. J. Am. Chem. Soc 2018, 140 (20), 6278–6287. [DOI] [PubMed] [Google Scholar]
- (38).Ramsey AV; Bischoff AJ; Francis MB Enzyme Activated Gold Nanoparticles for Versatile Site-Selective Bioconjugation. J. Am. Chem. Soc 2021, 143 (19), 7342–7350. [DOI] [PubMed] [Google Scholar]
- (39).Hammill JT; Miyake-Stoner S; Hazen JL; Jackson JC; Mehl RA Preparation of Site-Specifically Labeled Fluorinated Proteins for 19f-Nmr Structural Characterization. Nat. Protoc 2007, 2 (10), 2601–2607. [DOI] [PubMed] [Google Scholar]
- (40).Dias Florencio G; Precigout G; Beley C; Buclez PO; Garcia L; Benchaouir R Simple Downstream Process Based on Detergent Treatment Improves Yield and in Vivo Transduction Efficacy of Adeno-Associated Virus Vectors. Mol. Ther. - Methods Clin. Dev 2015, 2, 15024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Barnes CR; Lee H; Ojala DS; Lewis KK; Limsirichai P; Schaffer DV Genome-Wide Activation Screens to Increase Adeno-Associated Virus Production. Mol. Ther. - Nucleic Acids 2021, 26 (S2), 94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Elliott AG; Harper CC; Lin HW; Susa AC; Xia Z; Williams ER Simultaneous Measurements of Mass and Collisional Cross-Section of Single Ions with Charge Detection Mass Spectrometry. Anal. Chem 2017, 89 (14), 7701–7708. [DOI] [PubMed] [Google Scholar]
- (43).Keifer DZ; Shinholt DL; Jarrold MF Charge Detection Mass Spectrometry with Almost Perfect Charge Accuracy. Anal. Chem 2015, 87 (20), 10330–10337. [DOI] [PubMed] [Google Scholar]
- (44).Keifer DZ; Motwani T; Teschke CM; Jarrold MF Acquiring Structural Information on Virus Particles with Charge Detection Mass Spectrometry. J. Am. Soc. Mass Spectrom 2016, 27 (6), 1028–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Susa AC; Mortensen DN; Williams ER Effects of Cations on Protein and Peptide Charging in Electrospray Ionization from Aqueous Solutions. J. Am. Soc. Mass Spectrom 2014, 25 (6), 918–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Susa AC; Xia Z; Tang HYH; Tainer JA; Williams ER Charging of Proteins in Native Mass Spectrometry. J. Am. Soc. Mass Spectrom 2017, 28 (2), 332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Chowdhury SK; Katta V; Chait BT Probing Conformational Changes in Proteins by Mass Spectrometry. J. Am. Chem. Soc 1990, 112 (24), 9012–9013. [Google Scholar]
- (48).Dedeo MT; Duderstadt KE; Berger JM; Francis MB Nanoscale Protein Assemblies from a Circular Permutant of the Tobacco Mosaic Virus. Nano Lett 2010, 10 (1), 181–186. [DOI] [PubMed] [Google Scholar]
- (49).Mendell JR; Al-Zaidy SA; Rodino-Klapac LR; Goodspeed K; Gray SJ; Kay CN; Boye SL; Boye SE; George LA; Salabarria S; Corti M; Byrne BJ; Tremblay JP Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther 2021, 29 (2), 464–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Ojala DS; Amara DP; Schaffer DV Adeno-Associated Virus Vectors and Neurological Gene Therapy. Neuroscientist 2015, 21 (1), 84–98. [DOI] [PubMed] [Google Scholar]
- (51).Kotterman MA; Chalberg TW; Schaffer DV Viral Vectors for Gene Therapy: Translational and Clinical Outlook. Annu. Rev. Biomed. Eng 2015, 17, 63–89. [DOI] [PubMed] [Google Scholar]
- (52).Pierson EE; Keifer DZ; Asokan A; Jarrold MF Resolving Adeno-Associated Viral Particle Diversity with Charge Detection Mass Spectrometry. Anal. Chem 2016, 88 (13), 6718–6725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).O’Connor DM; Lutomski C; Jarrold MF; Boulis NM; Donsante A Lot-to-Lot Variation in Adeno-Associated Virus Serotype 9 (AAV9) Preparations. Hum. Gene Ther. Methods 2019, 30 (6), 214–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Wörner TP; Snijder J; Friese O; Powers T; Heck AJR Assessment of Genome Packaging in AAVs Using Orbitrap-Based Charge-Detection Mass Spectrometry. Mol. Ther. - Methods Clin. Dev 2022, 24, 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Konermann L Addressing a Common Misconception: Ammonium Acetate as Neutral PH “Buffer” for Native Electrospray Mass Spectrometry. J. Am. Soc. Mass Spectrom 2017, 28 (9), 1827–1835. [DOI] [PubMed] [Google Scholar]
- (56).Xia Z; Degrandchamp JB; Williams ER Native Mass Spectrometry beyond Ammonium Acetate: Effects of Nonvolatile Salts on Protein Stability and Structure. Analyst 2019, 144 (8), 2565–2573. [DOI] [PubMed] [Google Scholar]
- (57).Susa AC; Xia Z; Williams ER Small Emitter Tips for Native Mass Spectrometry of Proteins and Protein Complexes from Nonvolatile Buffers That Mimic the Intracellular Environment. Anal. Chem 2017, 89 (5), 3116–3122. [DOI] [PubMed] [Google Scholar]
- (58).Wright JF AAV Vector Manufacturing Process Design and Scalability - Bending the Trajectory to Address Vector-Associated Immunotoxicities. Mol. Ther 2022, 30 (6), 2119–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).2008-D-0205. Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs) Guidance for Industry United States Federal Food and Drug Adminstration. 2020. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.