

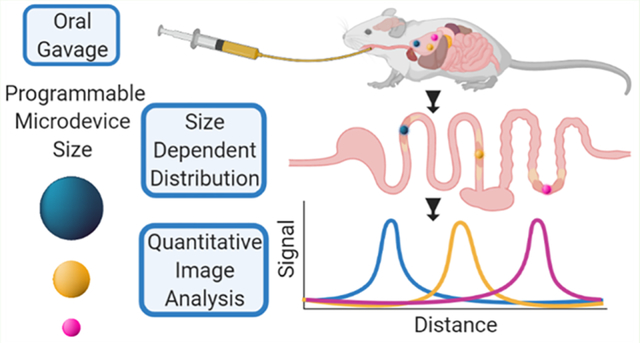
Abstract
Oral protein delivery technologies often depend on encapsulating or enclosing the protein cargo to protect it against pH-driven degradation in the stomach or enzymatic digestion in the small intestine. An emergent methodology is to encapsulate therapeutics in microscale, asymmetric, planar microparticles, referred to as microdevices. Previous work has shown that, compared to spherical particles, planar microdevices have longer residence times in the GI tract, but it remains unclear how specific design choices (e.g., material selection, particle diameter) impact microdevice behavior in vivo. Recent advances in microdevice fabrication through picoliter printing have expanded the range of device sizes that can be fabricated in a rapid manner. However, relatively little work has explored how device size governs their behavior in the intestinal environment. In this study, we probe the impact of geometry of planar microdevices on their transit and accumulation in the murine GI tract. Additionally, we present a strategy to label, image, and quantify these distributions in intact tissue in a continuous manner, enabling a more detailed understanding of device distribution and transit kinetics than previously possible. We show that smaller particles (194.6 ± 7 μm.diameter) tend to empty from the stomach faster than midsize (293.2 ± 7 μm.diameter) and larger devices (440.9 ± 9 μm.diameter) and that larger devices distribute more broadly in the GI tract and exit slower than other geometries. In general, we observed an inverse correlation between device diameter and GI transit rate. These results inform the future design of drug delivery systems, using particle geometry as an engineering design parameter to control device accumulation and distribution in the GI tract. Additionally, our image analysis process provides greater insight into the tissue level distribution and transit of particle populations. Using this technique, we demonstrate that microdevices act and translocate independently, as opposed to transiting in one homogeneous mass, meaning that target sites will likely be exposed to devices multiple times over the course of hours post administration. This imaging technique and associated findings enable data-informed design of future particle delivery systems, allowing orthogonal control of transit and distribution kinetics in vivo independent of material and cargo selection.
Keywords: enteric materials, gastrointestinal distribution, fluorescent tracking, microdevices, additive manufacturing
Graphical Abstract
1. INTRODUCTION
Throughout the last two decades, several groups have approached using micro- and nanomaterials as a means to improve the bioavailability of macromolecular therapeutics via the oral route.1–3 These technologies are often designed with the dual goal of protecting the encapsulated active pharmaceutical ingredient (API) from degradation in the low pH of the stomach or digestion via intestinal proteases and facilitating the increased absorption of API across the intestinal epithelium.4 Another potential benefit of micro- and nanoscale material approaches is the ability to incorporate targeting into the system design, which has been demonstrated using lectins, micromotors, chemically responsive materials, and other physiochemical approaches.5–9 One strategy has been to adapt microfabrication techniques to generate planar materials that incorporate asymmetry.8,10,11 Previous studies suggest the asymmetric reservoir design of these particles enables controlled drug release or mucoadhesion.10 This design also facilitates asymmetric release of the loaded compound, which allows for increased concentration of the API and associated formulation agents at the mucosal surface. Importantly, unlike similar systems using microspheres, planar materials have a greater surface-area-to mass ratio, rendering them unable to roll across mucosal surfaces and conferring an inherent adhesive property. It has been previously demonstrated on a tissue level that these materials tend to have longer residence times than equivalent microspheres.8 However, there has been little work to understand how microscale properties and design choices impact planar microdevice transit and accumulation in the gastrointestinal (GI) tract.
Many pharmaceutical systems protect their cargo in the GI tract via encapsulation with enteric polymers. Enteric polymers are pH-sensitive materials designed to erode and solubilize at a specific pH, enabling targeting and modified release profiles in the GI tract.12,13 Enteric materials exist in a number of form factors, and have been widely incorporated into many FDA-approved products, making them an excellent choice for the development of oral drug delivery systems.6,14,15 Previously, we demonstrated a technique to generate planar microdevices using a noncontact picoliter droplet printer and layer-by-layer additive manufacturing techniques.16 This process enables device fabrication, protein loading, and capping using a single instrument. These particles are composed entirely of Eudragit brand enteric polymers (Evonik, Essen Germany), which are widely used in clinical products to enable extended-release formulations and as solubility enhancers for solid dispersions.13 The printer-based fabrication also enables rapid iteration of device design, enabling production of microdevices that can range in diameter by 3–4-fold in an on-demand, programmatic fashion. Rapid device iteration is an achievement that would be challenging to replicate with conventional photolithography. This design flexibility enables further study of device diameter as a design parameter without the need for alternate materials or fabrication processes. Previously, other groups have explored how nanoscale materials of varying sizes and geometries distributed through and were taken up by GI tissue, demonstrating how material choices impact organ targeting and delivery.7,17–19 Because these materials are intended to reach systemic circulation, we expect that the extent of their interaction with GI tissue is likely very different than larger scale materials, which are more prone to gravitational sedimentation and cannot be taken up through transporter or diffusive mechanisms. Despite progress, it remains unclear how low-aspect-ratio materials, such as planar microdevices, will distribute and accumulate in the GI tract over time. Studies that have approached tracking the distribution of microscale materials generally have examined spherical particles and have been limited by the resolution of their tracking methodologies.20,21
Applying the flexibility of the printed microdevice system, we sought to understand the downstream impact of design choices on the in vivo behavior of these materials. Although progress has been made to understand how materials on the nanometer and millimeter scale behave in the GI tract, relatively little progress has been made to understand the GI transit and distribution kinetics of nonspherical materials on the order of hundreds of micrometers in diameter. Because of the unique capabilities of the microdevice printing platform, we generated monodisperse particle populations of distinct sizes, using the same base materials and body geometry. This consistency in fabrication is unique in comparison to other systems that have been used to examine the impact of particle size on GI transit, which have used distinct formulations for each particle size or were restricted in the range of generatable particle sizes.7 Here, we demonstrate the fabrication and use of fluorescently labeled, planar microdevices as a model to understand the transit behavior of planar materials through the murine GI tract. Because the printed microdevices are on the order of hundreds of micrometers in diameter, they were unable to be absorbed through transcellular or paracellular processes, enabling transit throughout the entire length of the GI tract. The introduction of a covalently bound near-IR fluorophore into the backbone of the enteric materials used in our microdevice bodies allowed for direct tracking of devices in intact tissue via IVIS imaging. We have also developed an accompanying image analysis workflow to understand how these devices distribute in tissue at a finer resolution than previously possible. This computational image analysis workflow allowed us to extract continuous distance vs fluorescence signals and provided refined insight into impact of microdevice size on transit speed. This generation of a continuous signal allowed us to explore how microdevices traffic as populations and how they interact with each other in vivo and demonstrated an inverse correlation between device diameter and transit speed. Taken together, this study confirms that microdevice size can be used as an engineering design parameter to control distribution kinetics and retention time in the GI tract and validates an image analysis approach to quantify the continuous distribution of materials in the GI tract, enabling a finer degree of understanding that may be lost in regionalized approaches.
2. MATERIALS AND METHODS
Materials.
Eudragit FS 30 D and S 100 were kindly provided by Evonik Industries (Essen, Germany). Alexa-Fluor 647 cadaverine (A30679), and simulated gastric fluid (710832) were acquired from Thermo Fisher (Waltham, MA). Trichloro(1H,1H,2H,2H-perfluorooctyl)silane (448931), HPLC Acetonitrile (34998), trifluoroacetic acid (T6508), simulated intestinal fluid (55331), sulfuric acid (258105), 30% (w/v) hydrogen peroxide solution (216763), potassium phosphate (P3786/P0662), potassium chloride (P3911), N,N-dimethylformamide (270547), N-hydroxysuccinimide (130672), N,N′-dicyclohexylcarbodiimide (D80002), triethyl amine (471283), 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid, N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES) (H3375), sodium hydroxide (221465), triethyl citrate (27500), and poly(vinyl alcohol) (9002–89–5) were acquired from Sigma-Aldritch (St. Louis, MO). Low autofluorescence animal feed was acquired from LabDiet (5v75, St. Louis, MO). Silicon wafers were obtained from Addison Engineering (3 in ⟨100⟩ p-type boron doped silicon) (San Jose, California).
Fluorescent Labeling of Eudragit Polymer.
AlexaFluor-647 Cadaverine (AF 647-cad) was covalently bound to Eudragit S 100 using an DCC-NHS coupling reaction. Briefly, Eudragit S 100 (50 mg, ~4 nmol) was dissolved in N,N-dimethylformamide (DMF) (4 mL) on a magnetic stir plate. The solution was moved to an ice bath, and N-hydroxysuccinimide (NHS) (7.8 mg, 67.8 μmol) in DMF (2 mL) was added stirring on ice for 5 min, followed by the addition of N,N’-dicyclohexylcarbodiimide (DCC) (14 mg, 67.8 μmol) in DMF (2 mL). The reaction was allowed to proceed for 3 h stirring on ice. A stock of AlexaFluor 647-Cadaverine (AF 647-Cad) (1 mg, 1 μmol) was prepared in dimethyl sulfoxide (100 μL). AD-647 Cad stock solution (40 μL, 0.4 mg, 0.4 μmol dye) was diluted in DMF (600 μL) along with triethyl amine (TEA) (0.56 μL, 4 μmol) and allowed to react on an orbital shaker (100 rpm) for 1 min protected from light at ambient temperature. The dye solution was added to the polymer solution and allowed to react on a magnetic stir plated for 72 h while protected from light. After 72 h, the reaction solution was diluted with 45 mL of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (100 mM) adjusted to pH 8 with 1 M NaOH for 8 h on an orbital shaker (100 rpm) protected from light at ambient temperature. The resulting solution was passed through a 0.2 μm vacuum filter to remove precipitates, followed by multiple rounds of concentration using a 30K MWCO spin concentrator to remove excess small molecule dye. The resulting product was lyophilized to afford a blue powder (50 mg, 100% polymer recovery). Conjugation was validated through reverse-phase liquid chromatography (Figure S1).
Device Fabrication.
Microdevices were fabricated as previously described using a ScienIon sciFLEXARRAYER S3 (ScienIon, Berlin, Germany) noncontact, picoliter, inkjet printer (Figure 1A).16 Briefly: A 3 in. silicon ⟨100⟩ wafer was cleaned in a piranha solution (1:1 sulfuric acid: hydrogen peroxide) for 30 min and allowed to soak in deionized water for 30 min to remove excess solution. Wafers were then dried completely using dry nitrogen gas and silanized with trichloro(1H,1H,2H,2H-perfluorooctyl)silane under vacuum at room temperature for 40 min, followed by curing for 20 min on a 150 °C hot plate. Polymer solutions were prepared at 5% (w/v) total polymer. For fluorescently labeled devices, the AF 647 Eudragit S 100 was incorporated at 20% (w/w) of the total polymer mass, with Eudragit FS 30 D comprising the remaining material. Triethyl citrate was added to 0.5% (w/v) as a plasticizer. All solutions were filtered through a 0.22 μm filter prior to aspiration. The polymer solution was aspirated into the printer nozzle and dispensed in ~400 pL droplets onto the wafer surface, which was maintained at 20 °C. Fluorescently labeled devices were protected from light throughout the printing process. After printing, the devices were allowed to completely dry under ambient conditions, before being moved to a hot plate and baked at 100 °C for 20 min to remove residual moisture. Resulting device bodies were then stored in a vacuum desiccator at room temperature and protected from light until further use.
Figure 1.
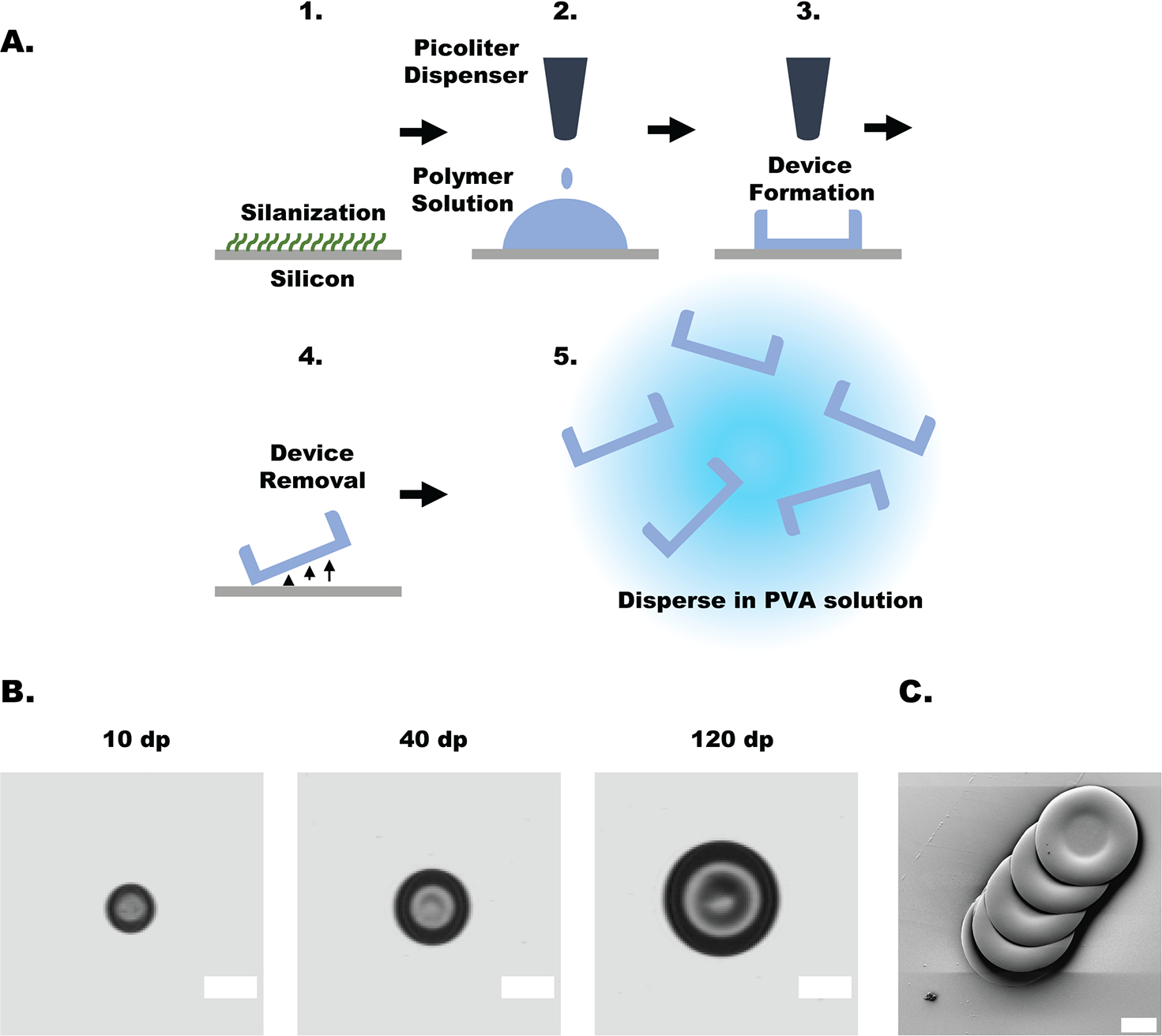
Schematic of device fabrication and device characterization. (A) (1) Silicon wafer silanized to enable fluid beading on the surface. (2) Programmed volume of polymer solution was dispensed onto the surface. (3) Polymer solution was allowed to fully dry, forming the device reservoir. (4) Devices were removed from the wafer surface using a razorblade. (4) Prior to oral dosing, devices were dispersed in a simulated gastric fluid solution containing 1% (w/v) poly(vinyl alcohol). (B) Light microscopy images of representative microdevices. Device measurements can be found in Table 1. Scale bars represent 200 μm. (C) Scanning electron microscopy image of devices mechanically removed from silicon wafer; scale bar represents 100 μm.
In Vitro Assessment of Conjugate Stability.
For fluorescent imaging, devices were printed onto glass coverslips using the method described above. Printed devices were incubated in either simulated intestinal fluid or simulated gastric fluid for the indicated amount of time. After incubation, devices were washed briefly in deionized water and dried at room temperature in a vacuum desiccator. Devices were then affixed to a microscope slide and imaged using an inverted Nikon 6D fluorescent microscope using a Cy-5 filter set at 20× magnification. Quantification of maximum pixel intensity was performed using imageJ.
Oral Gavage of Fluorescent Microdevices.
All animal studies were performed in accordance with the UCSF Animal Use and Care Program, under protocol no. AN180829–02A. All studies were performed with 8–12 week old female C57BL/6J mice acquired from Jackson laboratories (Bar Harbor, ME). One week prior to dosing, the diet of animals was switched to a low autofluorescence diet. Twenty-four hours before device administration, mice were fasted to reduce the residual background signal and confounding matter in the GI tract in accordance with our animal use protocol. To prepare devices for administration, they were removed from the silicon wafers mechanically using a razorblade and dispersed in a simulated gastric fluid solution containing 1% (w/v) poly(vinyl alcohol) (PVA) as a dispersant. Devices were concentrated either via natural sedimentation or low-speed benchtop centrifugation, and then loaded into a 30 mm 18 G polypropylene feeding tube. Devices were dispersed in 200–250 μL and gavaged directly into the stomach of each mouse at time 0 min. At each time point, mice were sacrificed via carbon dioxide asphyxiation and cervical dislocation. The complete GI tract of each mouse (stomach to anus) was extracted and the ends were sutured closed. The tissue was stored on ice between paper towels saturated with PBS and imaged immediately using a Xenogen IVIS Spectrum Imaging System using the preset AF 647 filter settings. To capture regional fluorescence data, we masked out nontissue areas using Living Image software, and ROIs were drawn to capture each area and limit signal cross-contamination.
Visualization of Device Transit.
Visualization of device tracking was performed using a MATLAB script in combination with an ImageJ plugin. Briefly: Images of mouse GI tracts were exported as separate monochromatic fluorescent and light photograph images (Figure S3A). Both images were identically cropped to only contain relevant areas. Next, the GI tract in the light microscopy image was traced using a MATLAB ROI selection tool, which was used to select the equivalent area on the fluorescent image (Figure S3B). The image was imported to ImageJ and linearized using the straighten selection tool (Figure S3C). The linearized image was reimported to MATLAB, where irrelevant pixels were removed and the image was compressed into a one-dimensional vector (Figure S2D). The signal data were normalized to the total signal, and distance was expressed as a percent of total distance along the GI tract (Figure S3E). Further details can be seen in Figure S3.
Statistical Analysis.
Statistical analysis was performed using the MATLAB 2020B statistics and machine learning package (Math-Works, Natick, MA). Statistically significant differences between groups were evaluated by one-way ANOVA, followed by a Tukey-Kramer correction for multiple comparisons, setting α = 0.05. For digital signal analysis, statistical significance was determined by cross correlation, followed by a Bonferroni correction for multiple comparisons setting α = 0.05.
3. RESULTS
Fluorescent Dye Conjugate Synthesis.
The fluorescent polymer conjugate was prepared using activated ester conjugation chemistries and purified from free dye via spin concentration (see Materials and Methods). The formation of a covalent linkage was evaluated via reverse phase HPLC, monitoring conjugate elution at 650 nm (Figure S1). Conjugation efficacy was determined via absorbance at 650 nm of a 1 mg/mL solution of the dye–polymer product, demonstrating the addition of approximately 3 μg of dye per mg of polymer, or the conversion of approximately 37% of the initial dye.
Device Fabrication and Characterization.
As demonstrated previously, planar, asymmetric microdevices were fabricated using a noncontact picoliter droplet liquid handling system (Figure 1A). Devices were fabricated from a 20% (w/w) blend of 100-AF to 80% (w/w) FS 30 D because it was observed to be sufficiently bright to enable in vivo imaging without compromising the mechanical integrity of the device bodies (data not presented). To adjust the final device size, we adjusted the volume of material deposited by controlling the number of 400 pL droplets dispensed per device. Device dimeters were measured to be 194.6 ± 8, 293.2 ± 7 μm, and 440.8 ± 9 μm (mean ±1 SD, n = 5 measurements per group): corresponding to 10 droplets, 40 droplets, and 120 droplets, respectively (Figure 1B). As previously shown, SEM images of discrete mechanically extracted devices demonstrate maintained structural stability (Figure 1).16
Fluorescent Conjugate Stability.
Devices were exposed to simulated gastric fluid or simulated intestinal fluid adjusted to pH 5 with hydrochloric acid to better mimic the murine intestinal environment (Figure 2A).22 Mean pixel intensity data were compared to a control unexposed group (Figure 2B). Simulated gastric fluid exposure for 30 min does not impact device fluorescence intensity, measured as maximum pixel intensity in a given device image. Similarly, devices exposed to simulated intestinal fluid (pH 5) for 60 min and 4 h maintained their fluorescent signal throughout the deration of their exposure. No significant differences (at α = 0.05) were detected in any pairwise comparison. Furthermore, there was no measurable change in device size (Figure S2A) (at α = 0.05), or noticeable change in their morphology (Figure S2B).
Figure 2.
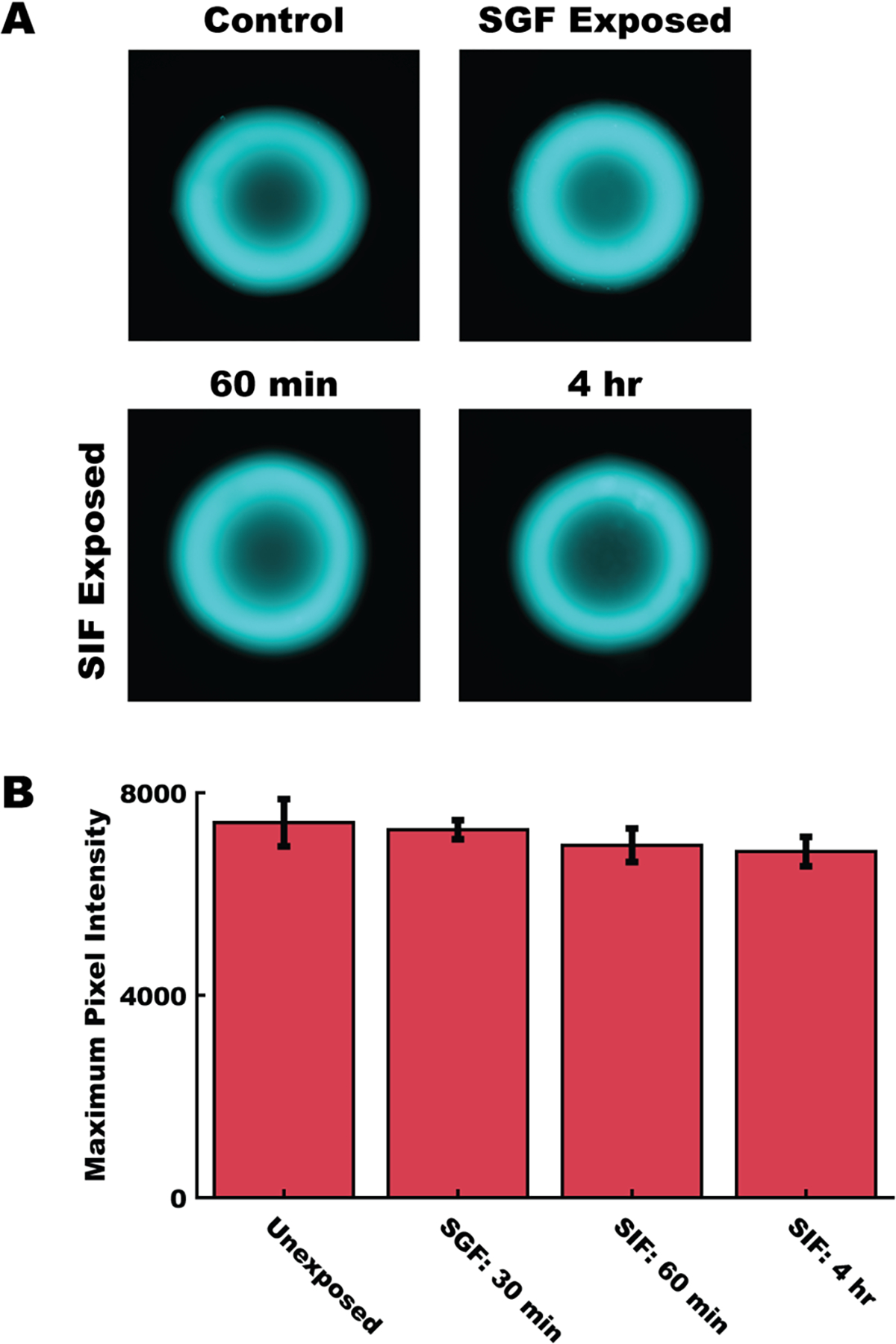
Fluorescent polymer conjugate was stable throughout the anticipated use case. (A) Representative fluorescent images of devices after exposure to simulated gastric fluid (SGF) and simulated intestinal fluid adjusted to pH 5 (SIF). (B) Comparison of maximum pixel intensity between fluorescently labeled devices exposed to SGF for 30 min, SIF adjusted to pH 5 for 60 min or 4 h. Data represents mean ±1 SD of n = 5 devices per group. No differences were detected between groups at α = 0.05, using a one-way ANOVA followed by a Tukey-Kramer correction for multiple comparisons.
Oral Gavage and Regional Fluorescence.
For oral dosing studies, dosages were prepared by device number and adjusted to account for the difference in device mass per device between groups. Animals were dosed according to Table 1 (Figure 3A) and imaged at the specified time points, recording both ROI information and the florescent signal overlay (Figure 3B). After administration, there was a noticeable loss in device numbers in the dead volume of the syringe, amounting to between 15 and 20% of the total devices. During imaging, no pixel saturation was observed in any sample. 200 and 450 μm groups had n = 3 animals per time point, whereas the 300 μm group had n = 2 animals per time point because of the exclusion of a preliminary data set that was deemed to have insufficient fluorescent signal.
Table 1.
Dosing for In Vivo Device Transit Studies by Groupa
| droplet number (dp) | no. of devices (no. per animal) | approximate dose mass (total mg of polymer per animal) | approximate dye mass (μg per animal) | device size (μm.d) |
|---|---|---|---|---|
| 10 | 3120 | 6.2 | 18.6 | 194.6 ± 8 |
| 40 | 2340 | 18.7 | 56.1 | 293.2 ± 7 |
| 120 | 1560 | 37.4 | 112.2 | 440.8 ± 9 |
Size measurements represent the mean diameter of n = 5 devices, ± 1 SD.
Figure 3.
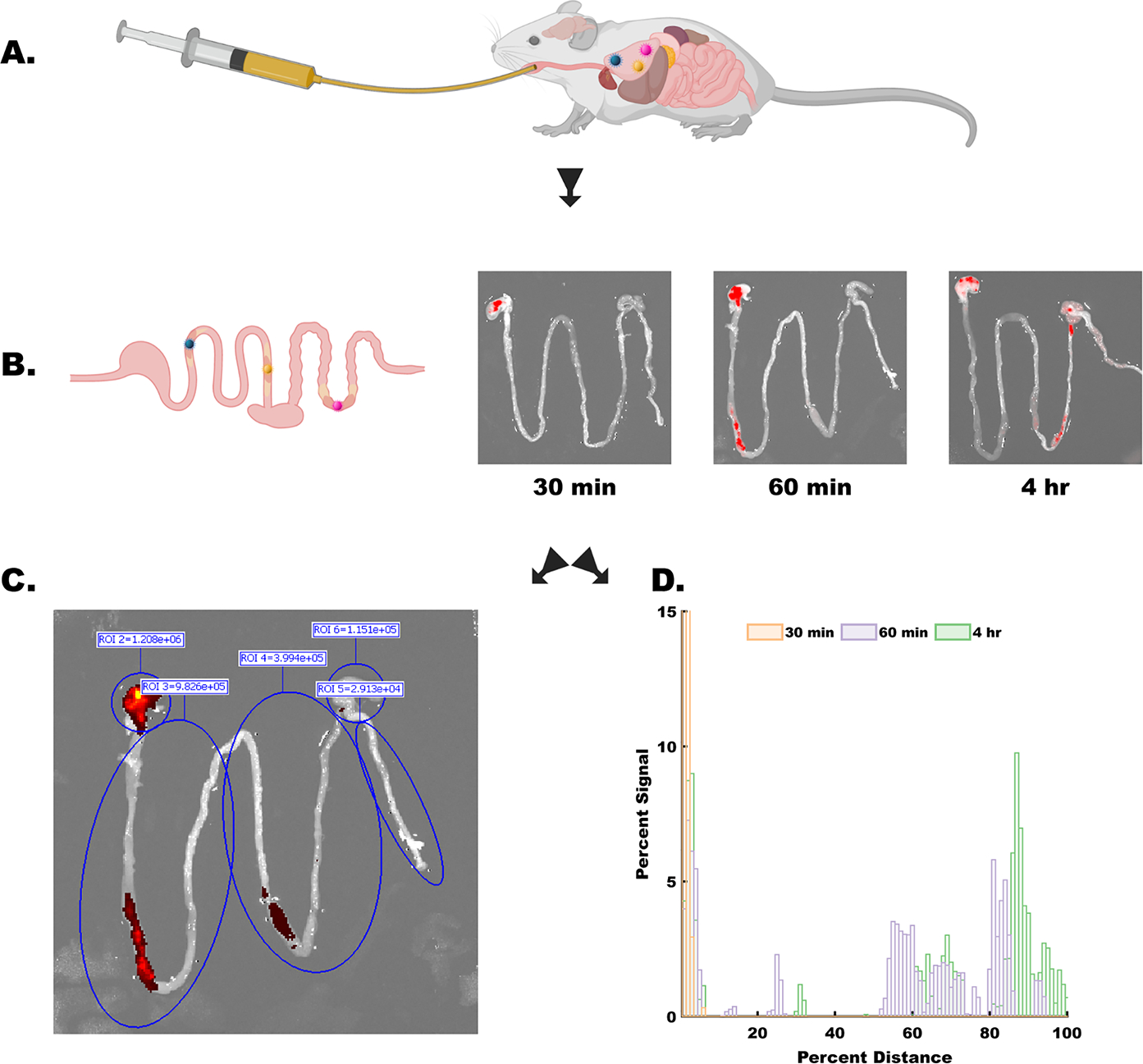
Experimental process schematic. (A) Microdevices were dosed to animals via oral gavage, directly into the stomach at time = 0. (B) At each time point (30 min, 60 min, and 4 h) animals were sacrificed, their gastrointestinal tract from the stomach to the anus was removed and imaged (representative images of 300 μm device group shown). Images and image meta-data were processed using two independent methods: (C) using preset ROIs, total fluorescent signal from major compartments was captured (in order: stomach, upper small intestine, lower small intestine, cecum, and colon). Fluorescent signal was collected as total photon counts. (D) In parallel, images were processed to extract device distribution information in a continuous manner. Data shown were extracted from images in B. Signal was normalized to the total amount of signal in each sample, and distance was normalized to the total distance of the GI tract sample. Data are presented in a semicompressed format, where each bar represents the total signal from the preceding 1% of the GI tract. More information on the image processing pipeline in Figure S3.
Comparisons between regional signal data were used to track the accumulation of devices in biologically relevant areas (Figure 3B and Figure 4). At the 30 min time point, the stomach signal in the 200 μm group (Figure 4) was significantly larger than the signal in the lower small intestine (p = 0.018), the cecum (p = 0.0052), and colon (p = 0.0051). By 60 min, the device population has spread such that there was no longer a statistically significant signal. Similarly, in the 450 μm group, at 30 min post gavage there was a statistical difference between the stomach and the upper and lower small intestine (p = 0.037, p = 0.037), the cecum (p = 0.0085), and the colon (p = 0.0128). At 60 min the signal in the lower small intestine of the 450 μm group was significantly higher than the upper small intestine (p = 0.029), but there were no other significant differences in any measurement at this time point. By the 4 h time point, the signal in the stomach of the 300 μm group was significantly higher than both the upper small intestine (p = 0.048) and the cecum (p = 0.049). In the 450 μm group by the 4 h time point, the particles had dispersed such that there was no longer a statistically significant difference between groups.
Figure 4.
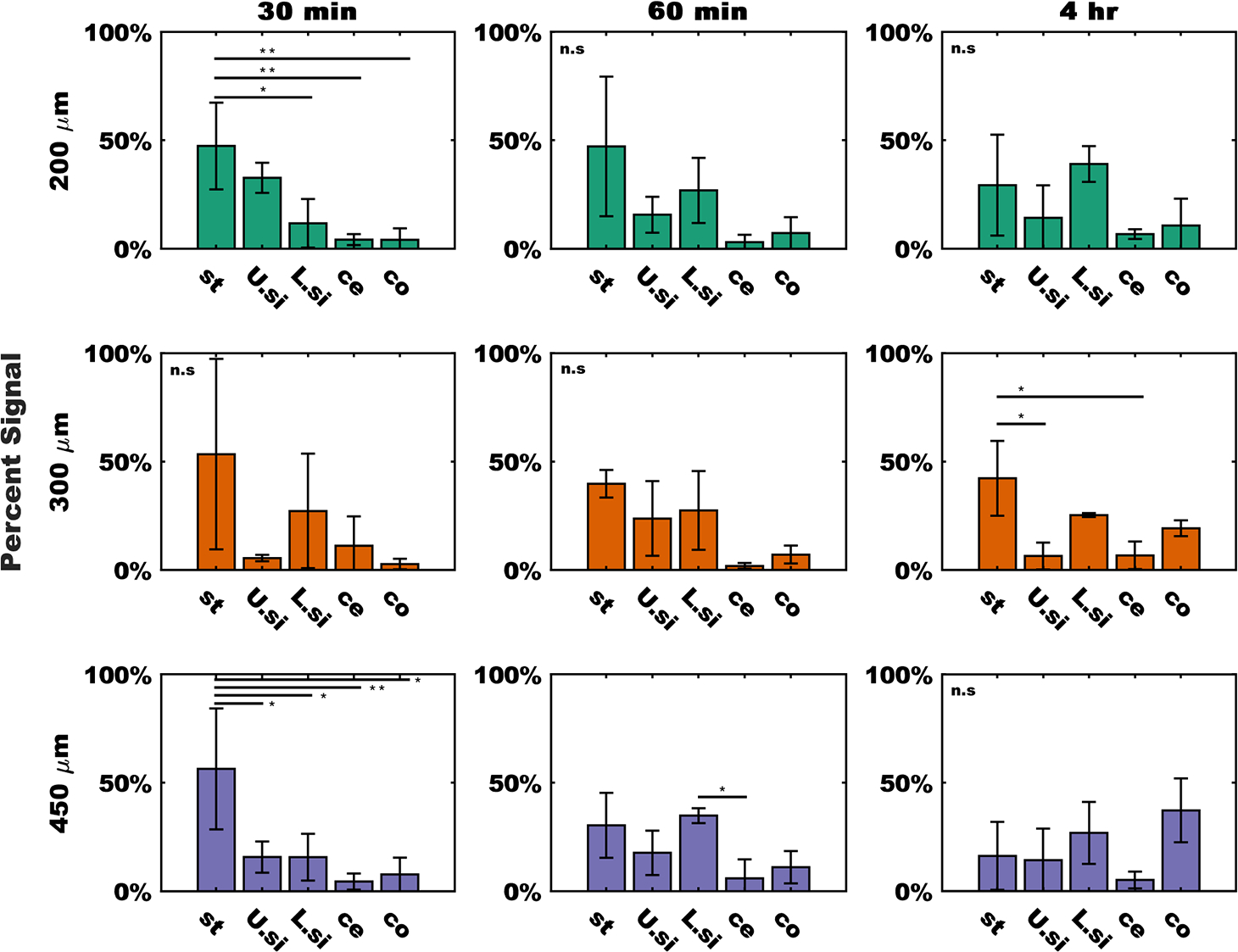
Fluorescent signal from discrete tissue regions demonstrates device transit through tissue. Signals divided by device group (rows) and time (columns). Data were normalized to the total amount of signal in each sample and expressed as a percentage. From the left to right in each plot bars represent signal from the stomach (st), upper small intestine (U.si), lower small intestine (L.si), cecum (ce), and colon (co). Data were expressed as the sample mean ±1 SD, n = 3 measurements per group for the 200 μm devices (green) and 450 μm devices (purple), n = 2 measurements per groups for 300 μm devices (orange). Statistical difference between groups was determined by one-way ANOVA followed by a Tukey-Kramer correction for multiple comparisons. * = p < 0.05, ** = p < 0.01. ns = no significant differences measured.
Visualization of Continuous In Vivo Distributions.
IVIS imaging data was processed to produce continuous signal vs distance curves for each device group at each time point (Figure S3). Figure 5 shows the results of visualizing the average distribution of each device geometry at each time point. Each bar represents the sum of the preceding 1% of the data set, in order to better represent the underlying patterns of the volatile data.
Figure 5.
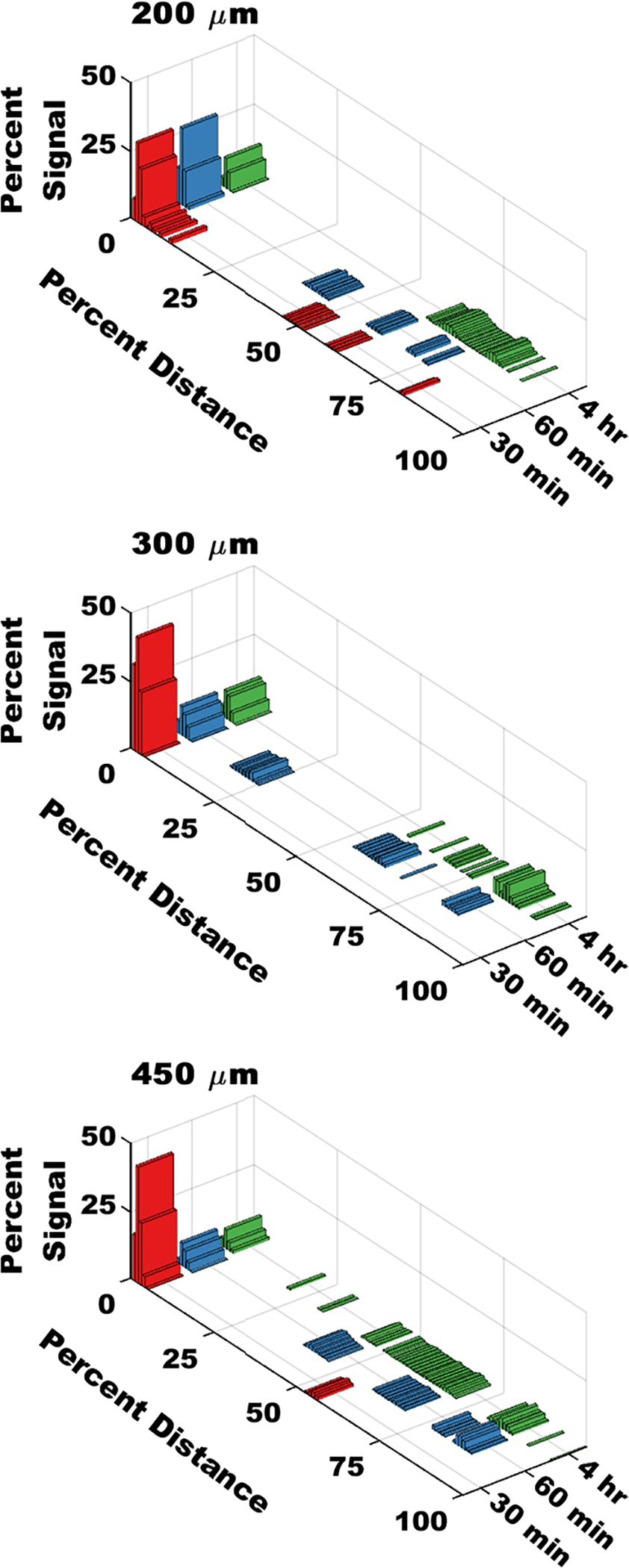
Continuous distributions show device translocation through the GI tract over time. Signal data were normalized to the total amount of signal in each sample, and expressed as a percentage. Distance was expressed as a percentage of the total distance along the GI tract. Each bar represents signal summed over the preceding 1% of the length of the GI tract. Data represent the mean of n = 3 separate measurements for the 200 and 450 μm groups, and n = 2 separate measurements for the 300 μm group.
To link these distribution results to a more rigorous statistical method, we calculated the cross-correlation coefficients pairwise between each group at each time point, as well as a p-value associated with that comparison (Figure 6). In this context, the cross-correlation coefficients for two vectors can be interpreted as a metric of similarity or goodness-of-fit. Cross-correlation coefficients can also be used for hypothesis testing against the null-result of no correlation between the compared signals. When comparing overlaid fluorescence vs distance signals at each time point between device groups (Figure 6A–C), a p-value of less than α = 0.05 indicates significant correlation between the groups, whereas a larger p-value fails to reject the null-hypothesis of no correlation (Figure 6D–F). For the purpose of this analysis, we excluded signal from the stomach compartment (~0–15% of total distance) and instead focused on signal from motile devices in the small intestine, cecum, and colon (Figure S2E). At 30 min, we observed no correlation between the signal from the 150 and 450 μm groups, but because of limited signal in this region from the 300 μm group, we were unable to draw any comparisons to that signal. At 60 min, we observed no statistical correlation between any groups, suggesting that all groups present statistically distinct distribution profiles. At 4 h post gavage, there was a statistically significant correlation between the signals from all device groups, suggesting all groups were similar at α = 0.05. The family-wise error rate was limited to α = 0.05 using a Bonferroni correction for multiple comparisons. These results were further supported by confirming the intragroup correlation between replicates of the same device group and time point (Figure S4).
Figure 6.
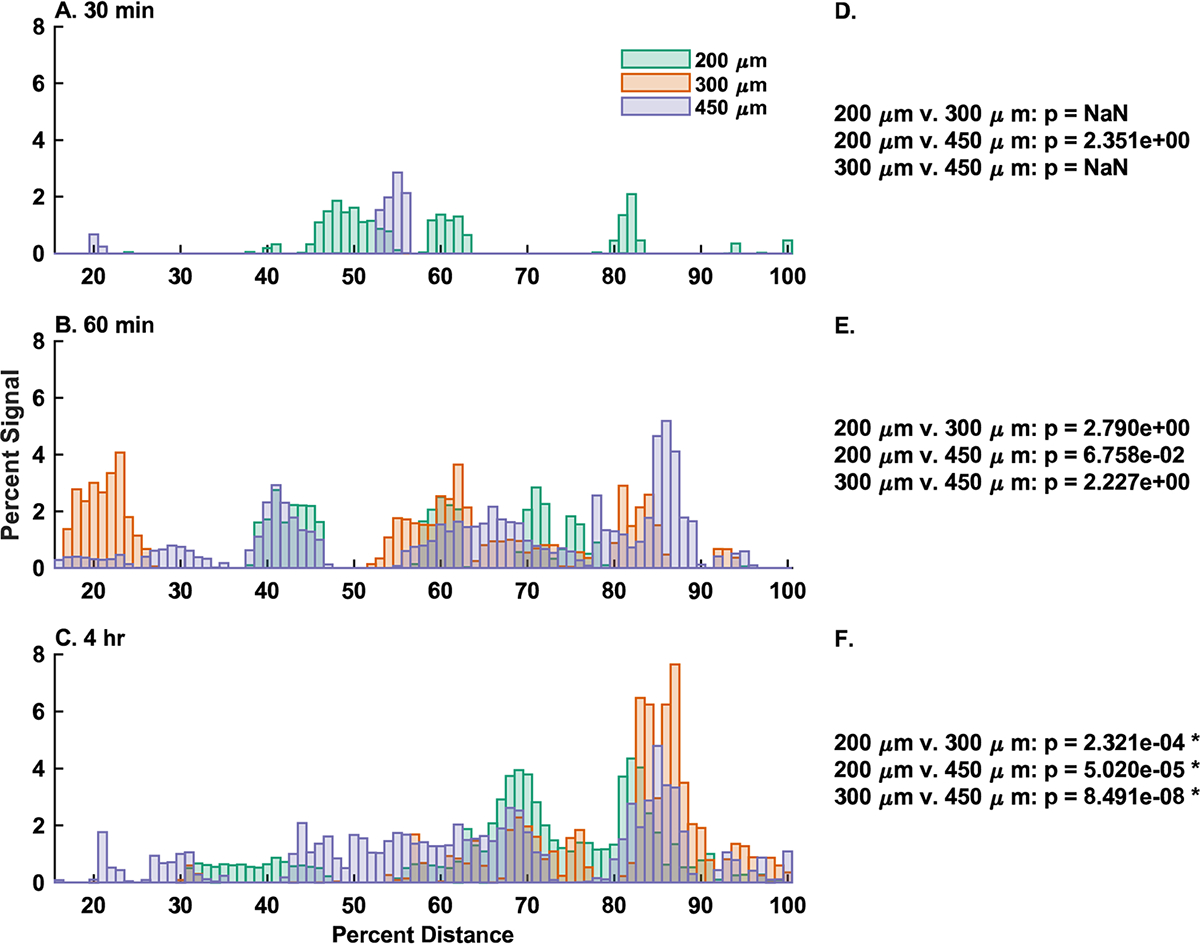
Comparison of distribution signals by-time indicate group stratification. (A–C) Overlays of each device group at (A, D) 30 min, (B, E) 60 min, and (C, F) 4 h presented after gastric emptying (15–100% total distance). Signal data are normalized to the total signal present in each sample, and distance is normalized to the total distance along the GI tract. Each bar represents the arithmetic mean of the cumulative signal from the preceding 1% of total distance from n = 3 animals for the 200 and 450 μm groups, and n = 2 animals for the 300 μm group. (D–F) Cross correlation results for pairwise comparisons between groups. p-values less than p = 0.05 represent a statistically significant correlation between the indicated signals, whereas larger p values fail to reject the null-hypothesis of no correlation. In the 30 min group (D), there was insufficient signal from the 300 μm group for comparison. Family-wise error rate was limited to α = 0.05 using a Bonferroni correction for multiple comparisons. * = p < 0.05 and indicates a statistically significant correlation.
Finally, we analyzed the correlation lag between each device group at each time point (Figure 7). Correlation lag measures the number of discrete steps along the x-axis; a signal must be shifted to align with a given fixed signal. In this analysis, each lag step equates to shifting the signal backward (negative) or forward (positive) 1% of the total distance along the GI tract. In groups in which we were unable to reject the null hypothesis of no correlation, we observed unity peaks at nonzero lags, which indicated that these signals differ in distance along the GI tract. Although in groups that show significant correlation with one another, we observed unity peaks at 0 lag, showing that the fluorescence vs distance signals were superimposable to a scalar factor. All fluorescent images used in this study can be seen in Figure S5.
Figure 7.
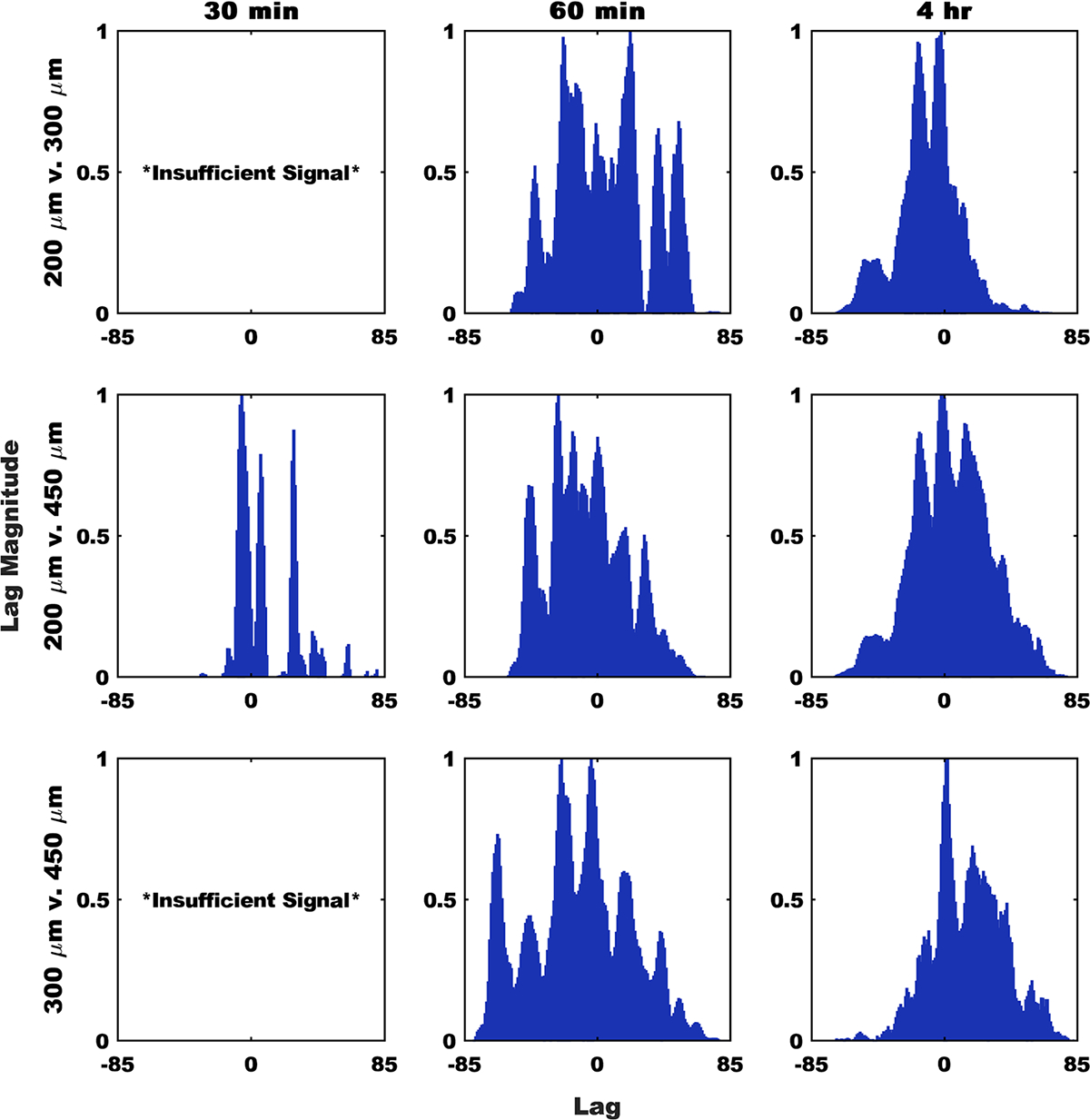
Signal lag shows devices accumulate differentially along the GI tract over time. The presented biased correlation lag analysis results account for shifts along the entirety of the analyzed portion of the GI tract (excluding the stomach). Signals were normalized to the maximum in each group. Peaks at positive lag values represent the second term needing to shift in the positive direction to match the first term, whereas negative lag values indicate the second term moving in the negative direction. Because of an insufficient signal, no comparisons to the 300 μm group were possible at the 30 min time point.
4. DISCUSSION
Fluorescent Device Fabrication.
The goal of this study was to better understand the impact of microdevice diameter on their in vivo distribution and transit kinetics and the potential use of device diameter as an engineering handle for future applications. Previously, we reported the use of additive manufacturing to fabricate microdevices of tunable sizes, entirely using materials present in FDA-approved oral dosage forms.16 Here, we build upon that work by leveraging the flexibility and consistency of the printed microdevice approach in order to fabricate materials that can be used to better understand the transit of microscale drug delivery systems through the GI tract. With this information, we can more rationally design materials to target specific regions of the GI tract or a desired transit time depending on the desired therapeutic profile. Despite the breadth of efforts to develop a microscale device-like system for oral delivery applications, many approaches utilize particles on a similar size scale, between 200 and 500 μm in diameter.15,23 The reasons for this clustering are manyfold, but are most likely tied to the ease of manufacturing particles at this size scale using photolithography and replica molding.24 Because of the ease of tunability in our device printing approach, we were not limited in the same manner. This allowed us the opportunity to better understand the transit implications of device size, by producing microdevices that maintained a consistent material profile but ranged in size by nearly 3-fold. Other groups have developed devices for oral delivery using additive manufacturing. These strategies produce materials on the millimeter scale using micro stereolithography printing, which can achieve more complex designs but are limited by the resolution of the instrument, the inflexibility of the CAD design process, and the necessity of photo-cross-linkable materials or specific printer resin filament.6,25
Fluorescence tracking has previously been applied to understand the location of materials in the gastrointestinal tract and associated tissues.5,7,17 By conjugating our enteric materials directly to a near IR fluorophore, we reduced the need for disruptive processes, such as microsurgery or tissue digestion, for detection, which may have otherwise disturbed device placement. As shown in Figure 2, the fluorescently labeled microdevices retain their fluorescent signal throughout a simulated use case, suggesting that there was limited loss of fluorescence intensity through chemical means or via erosion of the labeled S 100. Eudragit S 100 is a material designed to erode above pH 7. However, because the murine GI tract never exceeds pH 5, the devices remain in an intact state.12,22 The observed stability of the fluorescent microdevices confirms that all signal observed in the GI tract is related directly to intact devices, and not to eroded polymer or disassociated free-dye, underlining the utility of these materials in tracking applications in the murine GI tract.
Regional Device Distributions.
To understand the impact of microdevice size on transit, we first examined how the accumulation of particles in each tissue compartment varied over time. As shown in Figure 4, In both the 200 and 450 μm groups at the 30 min time point, the signal in the stomach was significantly larger than the signal from the more distal compartments. This suggests that devices were still largely retained in the stomach. In the 200 μm group, there was no significant difference between the upper small intestine and stomach groups at 30 min, which suggests that some of the devices may have already emptied into the duodenum. In most samples, there were few differences between groups at the 60 min and 4 h time points, which is indicative of device dispersion throughout the GI tract. In the 450 μm group, we observed that there was a difference between the signal in the lower small intestine and the cecum, which suggests that the devices were still largely retained in the small intestine and had not yet permeated to the colon. In the 300 μm group at 4 h, we observed a significant difference between the signal in the stomach and signal from the upper small intestine and cecum, which also indicates that even after 4 h there was still a significant population of devices that were retained in the stomach. These data as a whole demonstrate that once dosed via oral gavage, devices distribute throughout the GI tract over the course of hours. These results also suggest that smaller devices seem to empty from the stomach faster and distribute more quickly, whereas larger devices were more likely to be retained in the upper portion of the GI tract and stomach for longer periods of time. These findings are similar to those by Tuleu et al., who saw that in the rat GI tract, larger particles tended to transit and exit slower than smaller particles fabricated from the same material.26 These results have implications in the potential use cases for planar microdevices as delivery platforms in murine models. For example, if larger devices are retained for extended periods in the stomach, they might have applications in extended-release or long-acting treatments.
Continuous Device Distributions and Quantitative Methods.
Most studies investigating particle transit in the GI tract discuss material accumulation and distribution regionally, by summing the signal within a given area, as we have done above. However, utilization of high-resolution fluorescence imaging systems allows us to extract detailed information from images, using image analysis tools, allowing for the correlation of fluorescent signals to their specific location in the GI tract. This level of resolution has implications in further understanding the interaction of materials with local environments in the GI tract and providing insight into the in vivo behavior of microdevices. To accomplish this, we developed an image analysis workflow that extracts the fluorescent data from our tissue imaging samples, and ties it to position data in the GI tract (Figure S3). To account for differences between animals, and for differences in dosing due to particle loss, we expressed our results as a percentage of the total signal observed at a given percent distance along the GI tract. As shown in Figure 5, we observed devices across the entirety of the GI tract in all device groups. As expected, a larger signal was seen at the proximal side of the GI tract (red bars), that then distributes down the GI tract in later time points (blue and green bars). Given that the average adult murine gastrointestinal tract is 50 cm in length, and given that on average our samples measured 2000 pixels long, we can conclude that a pixel in these images could correspond to as little as 250 μm, or about one device diameter.27 The bars in Figures 5 and 6 correspond to a sum of the signal derived from 1% of the total length of the GI tract, or around 20 pixels, meaning that each bar with a nonzero signal likely represents tens of devices.
To more rigorously understand these patterns, we applied cross correlation analysis, as derived from its use in digital signal processing.28 Cross correlation can be applied to uncover patterns between signals in the same dimensional space. Cross correlation can also be a tool for hypothesis testing, similar to the Pearson product moment correlation. Specifically, cross-correlation can be used to determine if two signals corelate in a statistically significant manner, or if they fail to reject the null-hypothesis of no correlation. In Figure 6, we observed that, after exiting the stomach, the device signals adopt unique distributions. We confirm this using cross correlation to demonstrate that at 30 min and 1 h post gavage, different device sizes produce different distributions in the GI tract. By the 4 h time point, we can no longer distinguish between device groups via cross correlation, which may be due to a concentrated signal near the cecum and colon washing out other signals, or signal loss due to device elimination. This result was further validated by analysis of the intragroup variation (Figure S4). We show that signals from the same time point and group tend to be well correlated, suggesting that the differences we observed in the cross-correlation analysis in Figures 6 and 7 are due to differences in the behavior of different device sizes and not signal variability. To better understand how our device signals differed, we looked at the lag from our cross-correlation analysis. Correlation lag quantifies a time or space delay between two signals in units of the minimum sampling distance.29 In the case of our systems, this equates to 1% of the total distance along the GI tract. Correlation lag analysis further confirms that, in the case of planar microdevices, the GI transit rate is inversely corelated with device diameters, as shown by the dominance of positive lag peaks between small and large device groups (Figure 7). We hypothesize that this is likely due to the increased surface area of the larger devices, leading to increased adhesion at the mucosal epithelium. Previously, it was shown that the increase in microdevice surface area through the addition of nanotopographical features increased residence time, so it follows that a similar increase in device surface area would replicate those results.8 The one exception we observed is the dominance of negative lag peaks in the 200 μm vs 450 μm comparison at 60 min post gavage. We speculate that this is the result of the 200 μm devices beginning to be eliminated at this point, effectively reducing the residual signal and shifting the first moment of the signal in the negative direction. In total, these results seem to suggest that until at least 60 min post gavage, the rate of stomach emptying and the rate of device transit through the GI tract, was inversely correlated with device size. In summary, our continuous distribution analysis show that it is reasonable to assume that microdevices are likely to begin to interact with any specific target location along the murine GI tract within one hour after administration, but as seen in Figure 4, the extent of this interaction may change from 1 to 4 h. It is also clear from the presence of multiple discrete peaks in signal vs distance curve from each group/time point that devices do not transit in unison but instead transit as independent particles. These results suggest that in the case of a specific acute local target, such as in the case of acute inflammatory disease, or in the case of specific microbial environments, discrete local areas are likely to encounter multiple devices nonsynchronously over the course of many hours. We were unable to capture full device elimination by the 4 h time point, so it is also reasonable to assume that the residence time of these particles was greater than 4 h. Residence time is likely also impacted by device size, and so depending on the needs of the specific application, a microdevice diameter can be selected to achieve the desired transit kinetics and distribution profile.
Compared to regionalized quantification, our continuous signal approach provides more insight into the specific distribution profile of each microdevice population. The regionalized approach benefits some statistical tests, but can mask underlying patterns in the device distribution. The continuous signal method also functions in an unbiased manner, removing the need to classify or demarcate specific regions of the GI tract, and does not depend on a prior understanding of the interaction between the devices and the tissue target in question. We anticipate that used alongside regional quantification, our continuous signal method will allow researchers to better capture particle level interactions with tissue and unanticipated distribution patterns in an unbiased manner, identifying patterns that may be washed out in ROI-based analysis. We also anticipate that the continuous signal approach will allow researchers to better understand how their microdevices transit as a population: either in a homogeneous group or as distinct populations, which has implications in application selection as described above.
5. CONCLUSIONS
We have demonstrated the fabrication of fluorescently labeled planar microdevices and demonstrated how their size affects transit through the murine GI tract. We produced a covalently labeled polymer product using a set of enteric materials found in many FDA-approved products and showed that it remained intact in simulated murine intestinal conditions. Our study of device accumulation in tissue compartments over time shows that particles 300 μm in diameter and larger can remain at least partially in the stomach for hours, whereas devices 200 μm in diameter are emptied faster. Our results suggest that device size is inversely correlated with distribution and transit rate in vivo. Additionally, we observed that devices seem to transit independently of one another as opposed to moving in a uniform mass, meaning that instead of reaching a target site simultaneously, devices are likely to reach a site of action over the course of hours. Next, we presented the use of a method to quantify the continuous distribution of particles along the GI tract, using image analysis and digital signal processing tools. These results support our conclusions from examination of specific tissue regions and suggest that that the majority of device distribution occurs within the first 60 min after dosing, but the kinetics and extent of that distribution are a function of device size. We also see that devices seem to transit individually as opposed to as a homogeneous population, which has implications in the design of future microdevice-based therapeutics. Our continuous signal analysis also highlights that our devices are retained in the GI tract for longer than 4 h, but devices 200 μm in diameter potentially begin elimination by 60 min post dosing. These results demonstrate that microdevice diameter can be applied as an orthogonal method to control the rate of transit and distribution in the GI tract, independent of the base material or the loaded cargo. We expect that these results will inform the parameter space of microdevice-based products and enable data-driven decision making in the design of future delivery systems.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Sun Pharma Advanced Research Company (SPARC) (Vadodara, India). W.R.L. and E.H. supported by NSF GRFP grant 1650113. J.A.F. was supported by the UCSF HIVE postdoctoral fellowship. T.D. and E.H. are both supported by NIH T32 grant GM 8155-34. A.K.J. and Y.Z. are employees and minor stake holders of SPARC. IVIS data was obtained through the UCSF Helen Diller Family Comprehensive Cancer Center Preclinical Core. We gratefully acknowledge the UCSF Center for Advanced Technology for use of the sciFLEXARRAYER S3. Fluorescence microscopy data for this study were acquired at the Nikon Imaging Center at UCSF/QB3. Figure 3, the TOC graphic, and Figure S3 were created with BioRender.com.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsbiomaterials.0c01606.
Chromatographic validation of fluorescent polymer conjugate, quantification of device size stability, image collection and analysis pipeline schematic, intragroup cross correlation coefficients, collected fluorescent overlay images (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsbiomaterials.0c01606
Contributor Information
William R. Lykins, University of California, Berkeley–University of California San Franciso Graduate, Program in Bioengineering, San Francisco, California 94118, United States; Department of Bioengineering and Therapeutic Sciences, University of California San Francisco, San Francisco, California 94143, United States.
M. Eva Hansen, University of California, Berkeley–University of California San Franciso Graduate Program in Bioengineering, San Francisco, California 94118, United States; Department of Bioengineering and Therapeutic Sciences, University of California San Francisco, San Francisco, California 94143, United States.
Xiaofei Sun, Department of Medicine, University of California San Francisco, San Francisco, California 94143, United States.
Rommel Advincula, Department of Medicine, University of California San Francisco, San Francisco, California 94143, United States.
Joel A. Finbloom, Department of Bioengineering and Therapeutic Sciences, University of California San Francisco, San Francisco, California 94143, United States
Abhishek Kumar Jain, Sun Pharma Advanced Research Company, Vadodara 390010, India.
Yashoraj Zala, Sun Pharma Advanced Research Company, Vadodara 390010, India.
Averil Ma, Department of Medicine, University of California San Francisco, San Francisco, California 94143, United States.
Tejal A. Desai, University of California Berkeley–University of California San Franciso Graduate Program in Bioengineering, San Francisco, California 94118, United States Department of Bioengineering and Therapeutic Sciences, University of California San Francisco, San Francisco, California 94143, United States.
REFERENCES
- (1).Homayun B; Lin X; Choi HJ Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals. Pharmaceutics 2019, 11 (3), 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Brown TD; Whitehead KA; Mitragotri S Materials for Oral Delivery of Proteins and Peptides. Nat. Rev. Mater. 2020, 5, 127. [Google Scholar]
- (3).Ahadian S; Finbloom JA; Mofidfar M; Diltemiz SE; Nasrollahi F; Davoodi E; Hosseini V; Mylonaki I; Sangabathuni S; Montazerian H Micro and Nanoscale Technologies in Oral Drug Delivery. Adv. Drug Delivery Rev. 2020, 157, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Renukuntla J; Vadlapudi AD; Patel A; Boddu SHS; Mitra AK Approaches for Enhancing Oral Bioavailability of Peptides and Proteins. Int. J. Pharm. 2013, 447 (1–2), 75–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Li J; Thamphiwatana S; Liu W; Esteban-Fernández De Ávila B; Angsantikul P; Sandraz E; Wang J; Xu T; Soto F; Ramez V; Wang X; Gao W; Zhang L; Wang J Enteric Micromotor Can Selectively Position and Spontaneously Propel in the Gastrointestinal Tract. ACS Nano 2016, 10 (10), 9536–9542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Vaut L; Juszczyk JJ; Kamguyan K; Jensen KE; Tosello G; Boisen A 3D Printing of Reservoir Devices for Oral Drug Delivery: From Concept to Functionality through Design Improvement for Enhanced Mucoadhesion. ACS Biomater. Sci. Eng. 2020, 6, 2478. [DOI] [PubMed] [Google Scholar]
- (7).Li D; Zhuang J; He H; Jiang S; Banerjee A; Lu Y; Wu W; Mitragotri S; Gan L; Qi J Influence of Particle Geometry on Gastrointestinal Transit and Absorption Following Oral Administration. ACS Appl. Mater. Interfaces 2017, 9 (49), 42492–42502. [DOI] [PubMed] [Google Scholar]
- (8).Chirra HD; Shao L; Ciaccio N; Fox CB; Wade JM; Ma A; Desai TA Planar Microdevices for Enhanced In Vivo Retention and Oral Bioavailability of Poorly Permeable Drugs. Adv. Healthcare Mater. 2014, 3 (10), 1648–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Schmidt C; Lautenschlaeger C; Collnot EM; Schumann M; Bojarski C; Schulzke JD; Lehr CM; Stallmach A Nano- and Microscaled Particles for Drug Targeting to Inflamed Intestinal Mucosa - A First in Vivo Study in Human Patients. J. Controlled Release 2013, 165 (2), 139–145. [DOI] [PubMed] [Google Scholar]
- (10).Fox C; Chirra H; Desai T Planar Bioadhesive Microdevices: A New Technology for Oral Drug Delivery. Curr. Pharm. Biotechnol. 2014, 15 (7), 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Jørgensen J; Jepsen M; Nielsen L; Dufva M; Nielsen H; Rades T; Boisen A; Müllertz A Microcontainers for Oral Insulin Delivery - in Vitro Studies of Permeation Enhancement. Eur. J. Pharm. Biopharm. 2019, 143, 98. [DOI] [PubMed] [Google Scholar]
- (12).Nollenberger K; Albers J Poly(Meth)Acrylate-Based Coatings. Int. J. Pharm. 2013, 457 (2), 461–469. [DOI] [PubMed] [Google Scholar]
- (13).Thakral S; Thakral NK; Majumdar DK Eudragit®: A Technology Evaluation. Expert Opin. Drug Delivery 2013, 10 (1), 131–149. [DOI] [PubMed] [Google Scholar]
- (14).Christfort JF; Guillot AJ; Melero A; Thamdrup LHE; Garrigues TM; Boisen A; Zór K; Nielsen LH Cubic Microcontainers Improve In Situ Colonic Mucoadhesion and Absorption of Amoxicillin in Rats. Pharmaceutics 2020, 12 (4), 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Nielsen LH; Keller SS; Boisen A Microfabricated Devices for Oral Drug Delivery. Lab Chip 2018, 18 (16), 2348–2358. [DOI] [PubMed] [Google Scholar]
- (16).Nemeth CL; Lykins WR; Tran H; ElSayed MEH; Desai TA Bottom-Up Fabrication of Multilayer Enteric Devices for the Oral Delivery of Peptides. Pharm. Res. 2019, 36 (6), 89. [DOI] [PubMed] [Google Scholar]
- (17).Ma Y; Fuchs AV; Boase NRB; Rolfe BE; Coombes AGA; Thurecht KJ The in Vivo Fate of Nanoparticles and Nanoparticle-Loaded Microcapsules after Oral Administration in Mice: Evaluation of Their Potential for Colon-Specific Delivery. Eur. J. Pharm. Biopharm. 2015, 94, 393–403. [DOI] [PubMed] [Google Scholar]
- (18).Shen C; Yang Y; Shen B; Xie Y; Qi J; Dong X; Zhao W; Zhu W; Wu W; Yuan H; Lu Y Self-Discriminating Fluorescent Hybrid Nanocrystals: Efficient and Accurate Tracking of Translocation via Oral Delivery. Nanoscale 2018, 10 (1), 436–450. [DOI] [PubMed] [Google Scholar]
- (19).Bhutiani N; Samykutty A; McMasters KM; Egilmez NK; McNally LR In Vivo Tracking of Orally-Administered Particles within the Gastrointestinal Tract of Murine Models Using Multi-spectral Optoacoustic Tomography. Photoacoustics 2019, 13, 46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Oosegi T; Onishi H; Machida Y Gastrointestinal Distribution and Absorption Behavior of Eudragit-Coated Chitosan-Prednisolone Conjugate Microspheres in Rats with TNBS-Induced Colitis. Int. J. Pharm. 2008, 348 (1–2), 80–88. [DOI] [PubMed] [Google Scholar]
- (21).Panthani MG; Khan TA; Reid DK; Hellebusch DJ; Rasch MR; Maynard JA; Korgel BA In Vivo Whole Animal Fluorescence Imaging of a Microparticle-Based Oral Vaccine Containing (CuInSexS2- x)/ZnS Core/Shell Quantum Dots. Nano Lett. 2013, 13 (9), 4294–4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).McConnell EL; Basit AW; Murdan S Measurements of Rat and Mouse Gastrointestinal PH, Fluid and Lymphoid Tissue, and Implications for in-Vivo Experiments. J. Pharm. Pharmacol. 2010, 60 (1), 63–70. [DOI] [PubMed] [Google Scholar]
- (23).Fox CB; Kim J; Le LV; Nemeth CL; Chirra HD; Desai TA Micro/Nanofabricated Platforms for Oral Drug Delivery. J. Controlled Release 2015, 219, 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Sant S; Tao SL; Fisher OZ; Xu Q; Peppas NA; Khademhosseini A Microfabrication Technologies for Oral Drug Delivery. Adv. Drug Delivery Rev. 2012, 64 (6), 496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Vaut L; Jensen KE; Tosello G; Khosla A; Furukawa H; Boisen A Additive Manufacturing of Microreservoir Devices for Oral Drug Delivery Using an Acculas BA-30 Micro-Stereolithography Instrument: A Feasibility Study. J. Electrochem. Soc. 2019, 166 (9), B3257–B3263. [Google Scholar]
- (26).Tuleu C; Andrieux C; Boy P; Chaumeil JC Gastrointestinal Transit of Pellets in Rats: Effect of Size and Density. Int. J. Pharm. 1999, 180 (1), 123–131. [DOI] [PubMed] [Google Scholar]
- (27).Ogiolda L; Wanke R; Rottmann O; Hermanns W; Wolf E Intestinal Dimensions of Mice Divergently Selected for Body Weight. Anat. Rec. 1998, 250 (3), 292–299. [DOI] [PubMed] [Google Scholar]
- (28).Derrick TR; Thomas JM Time Series Analysis: The Cross-Correlation Function. Innov. Anal. Hum. Mov. 2004, 46. [Google Scholar]
- (29).Azaria M; Hertz D Time Delay Estimation by Generalized Cross Correlation Methods. IEEE Trans. Acoust., Speech, Signal Process. 1984, 32 (2), 280–285. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.