

Abstract
With aging, the incidence of age-related diseases increases. Hence, age-related diseases are inevitable. However, the mechanisms by which aging leads to the onset and progression of age-related diseases remain unclear. It has been reported that inflammation is closely associated with age-related diseases and that the cGAS-STING signaling pathway, which can sense the aberrant presence of cytosolic DNA during aging and induce an inflammatory response, is an important mediator of inflammation in age-related diseases. With a better understanding of the structure and molecular biology of the cGAS-STING signaling axis, numerous selective inhibitors and agonists targeting the cGAS-STING pathway in human age-related diseases have been developed to modulate inflammatory responses. Here, we provide a narrative review of the activity of the cGAS-STING pathway in age-related diseases and discuss its general mechanisms in the onset and progression of age-related diseases. In addition, we outline treatments targeting the cGAS-STING pathway, which may constitute a potential therapeutic alternative for age-related diseases.
Keywords: cGAS-STING, age-related diseases, cardiovascular diseases, neurological disorders, neoplasms, mechanisms, therapies
Aging is an intrinsic feature of mammals associated with a gradual functional decline over time, leading to the progressive loss of physiological integrity and deterioration of multiple organ systems, a critical cause of many age-related diseases [1]. Age-related diseases are diseases that occur at quadratically increased incidence rates in the adult population with advancing age [2]. Although the biological causes of the aging process remain unknown, there are some common cellular and molecular traits known as aging hallmarks [3]. DNA integrity and stability are continuously affected by exogenous and endogenous agents throughout life, leading to the accumulation of genetic damage and genomic instability [4, 5]. Although genomic stability systems exist, the genetic damage generated during the aging process will exceed their limited repair capacity. This will eventually lead to the accumulation of excess genetic damage in cells and aging. Genomic stability systems include mechanisms that repair most of the damage to nuclear DNA, ensure the integrity of mitochondrial DNA (mtDNA), and maintain the length and functionality of telomeres [6-8]. Furthermore, the efficacy of the respiratory chain degenerates as cells and organisms age, resulting in mitochondrial dysfunction. Under normal conditions, cellular autophagy or mitophagy can eliminate aberrant cytosolic DNA via the action of nucleases in the cytosol and endolysosomal compartments. However, aging cells have autophagy or mitophagy defects, and the combination of mitochondrial dysfunction and autophagy or mitophagy defects may be responsible for several age-related diseases [9].
Genomic instability and mitochondrial dysfunction in aging are associated with the stochastic release of dsDNA from the nucleus and mitochondria into the cytoplasm [10, 11]. The aberrant presence of cytosolic dsDNA is recognized as a danger-associated molecular pattern (DAMP) by the DNA sensor cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase (cGAS) [12]. The C-terminal part of human cGAS consists of DNA-binding sites (one primary site and two additional sites) that can bind to the sugar-phosphate backbone of DNA. The catalytic pocket of the enzyme rearranges, and the conformation of cGAS changes to provide optimal interaction with the substrates when DNA binds to the primary site. DNA binding to these two additional sites is important in the formation of the minimal active enzymatic unit that acts on GTP and ATP, inducing the synthesis of cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) [13]. cGAMP functions as an endogenous second messenger and is detected by the stimulator of interferon (IFN) genes (STING) located in the endoplasmic reticulum (ER) membrane [14, 15]. When GAMP binds to STING, the STING conformation changes and STING is transferred from the ER to the Golgi apparatus through the ER-Golgi intermediate compartment (ERGIC), where it is activated [16]. Activated STING polymerizes and activates TANK-binding kinase 1 (TBK1). In addition, TBK1 recruits and phosphorylates IFN regulatory factor 3 (IRF3), resulting in its dimerization, nuclear translocation, and interaction with nuclear factor κB (NF-κB) to trigger the synthesis of IFN and inflammatory cytokines (Fig. 1) [13, 17].
Figure 1.
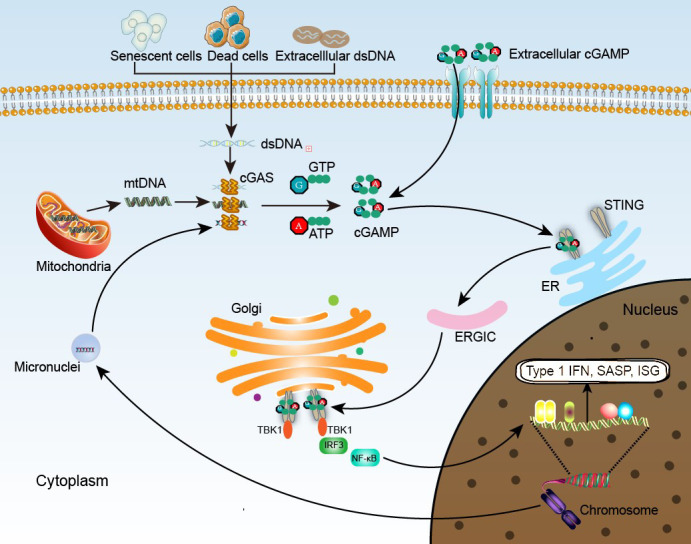
Overview of the cGAS-STING pathway in age-related diseases. cGAS is a cytosolic DNA sensor that detects dsDNA from senescent cells and dead cells, and some extracellular dsDNA, mtDNA and DNA in the micronuclei are also the main sources of cytosolic DNA. After combining with DNA, cGAS interacts with GTP and ATP, inducing the synthesis of cGAMP. In addition to intracellular synthesis, extracellular cGAMP can also enter the cell and acts as a second messenger, binding to STING located in the ER. Activated STING then transfers to the Golgi via ERGIC, where it can truly function, interacting with TBK1, IRF3, and NF-κB and mediating the production of type I IFN, SASP and ISG, promoting the inflammatory response and exerting biological effects.
Senescent cells accumulate with age. Although these cells undergo a stable cell cycle arrest and cannot duplicate, they can produce a complex secretome, including cytokines, chemokines, growth factors, and proteases. This is called the senescence-associated secretory phenotype (SASP)[18]. The functions of different SASP members differ greatly and are associated with autocrine and paracrine signaling, pro-tumorigenic and tumor-suppressive effects, and pro- and anti-inflammatory signaling [19]. The cGAS-STING pathway plays an indispensable role in cellular senescence, as cellular senescence induced by oxidative stress, DNA-damaging drugs, and oncogene activation is dependent on it [20, 21].
Senescent cells secrete SASP, such as IFN and interleukin (IL), in a cGAS-dependent manner, providing a critical paracrine signal to maintain cellular senescence. In addition, SASP can recruit immune cells to modulate the tissue immune microenvironment and monitor and eliminate tumor cells [22]. However, SASP is a double-edged sword; it can also promote tumorigenesis by inducing the proliferation, transformation, invasiveness, and metastasis of epithelial cells [23, 24].
Given the important roles of the cGAS-STING pathway in cellular senescence and age-related diseases, we critically evaluated the mechanisms of cGAS-STING in age-related diseases, as well as novel therapeutic approaches targeting this pathway.
Cardiovascular diseases (CVDs)
CVDs are the leading cause of death, accounting for almost 31% of all deaths worldwide. In 2016, it was estimated that 17.9 million people died of CVDs [25]. Although epidemiological evidence illustrates that advanced age promotes CVDs, the complete mechanisms of how aging drives CVD progression remain unclear [26]. Recent studies have indicated that the cGAS-STING pathway plays an important role in CVDs (Fig. 2) [27].
Figure 2.
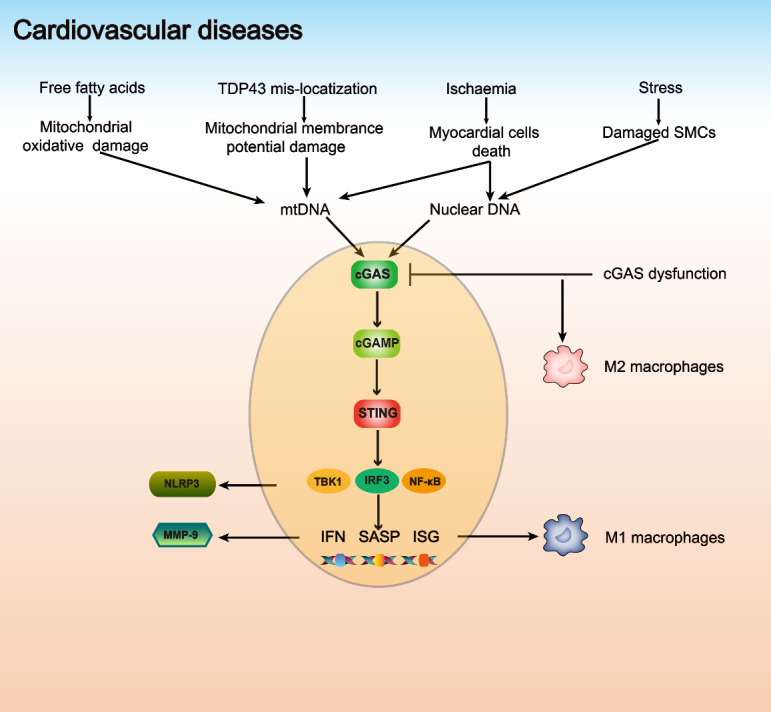
The cGAS-STING pathway in cardiovascular diseases. Free fatty acids can cause mitochondrial oxidative damage, and TDP43 mislocalization results in mitochondrial potential damage. This damage to mitochondria leads to the leakage of mtDNA into the cytoplasm. Damaged SMCs and myocardial cells release nuclear DNA; in addition, dead myocardial cells release mtDNA into the cytoplasm due to ischemia. This cytosolic dsDNA activates the cGAS-STING pathway and induces the production of NLRP3, MMP-9 and M1 macrophages (proinflammatory and antimicrobial), and inhibition of cGAS promotes the formation of M2 macrophages (healing, profibrotic, and anti-inflammatory).
Cardiac hypertrophy
Myocyte enlargement induced by prolonged and abnormal hemodynamic stress is a characteristic of pathological cardiac hypertrophy, which usually presents with hypertension. The complex responses of fibrosis, capillary thinning, increased production of proinflammatory cytokines, cellular dysfunction, and maladaptive epigenetic changes in pathological cardiac hypertrophy are major predisposing factors for maladaptive cardiac remodeling, cardiac dysfunction, and heart failure [28, 29].
TBK1/IRF3 signaling is associated with mechanical overload-induced cardiac hypertrophy [30]. The expression of STING was found to be increased both in the pathological cardiac hypertrophy heart tissues of patients and mice and correlated with the upregulated type I IFNs. STING knockout or deletion in mice remarkably alleviates cardiac hypertrophy by reducing cardiomyocyte size, ameliorating cardiac function, and decreasing hypertrophic markers, inflammatory factors, and fibrosis. Furthermore, STING knockdown abrogated the factors secreted by cardiomyocytes in vitro, which are associated with collagen accumulation in cardiac fibroblasts. These findings demonstrate that the cGAS-STING pathway plays an important regulatory role in the pathogenesis of pathological cardiac hypertrophy [31].
Free fatty acids generated by injections of streptozotocin and a high-fat diet (HFD) in diabetes models can impair mitochondrial function. In addition, the leakage of mtDNA into the cytosol activates the cGAS-STING pathway in mice. Its activation subsequently promotes the formation of the nucleotide-binding oligomerization domain-like receptor pyrin domain containing 3 (NLRP3) inflammasome in the hearts of diabetic mice and the secretion of proinflammatory cytokines related to cardiac pyroptosis and chronic inflammation, ultimately promoting pathological cardiac hypertrophy [32]. HFD can induce cardiac dysfunction and promote maladaptive cardiac remodeling via chronic activation of the cGAS-STING signaling axis. Protein kinase B (Akt) and AMP-dependent protein kinase (AMPK) are closely related to cardiac energy metabolism and the maintenance of cardiac homeostasis. Double knockout of cardiac Akt isoform (Akt2) and AMPK accentuated the HFD-induced increases in heart weight, cardiac hypertrophy, myocardial apoptosis, cardiomyocyte contractile, and oxidative stress. Akt2-AMPK double ablation enhanced the HFD-induced activation of the cGAS-STING signaling pathway and promoted the production of proinflammatory cytokines. Cardiac function was improved, and myocardial hypertrophy was reduced after inhibiting cGAS or STING [33, 34]. The remarkable mitochondrial oxidative damage to cardiomyocytes in patients with chronic kidney disease (CKD) causes mtDNA leakage, which activates the cGAS-STING-NFκB pathway and NFκB-transactivated ornithine decarboxylase (ODC1)-putrescine metabolic flux, inducing cardiac hypertrophy. Inhibition of the myocardial mitochondria-STING-NFκB-ODC1 axis using genetic or pharmacological approaches significantly alleviated CKD-associated cardiac hypertrophy [35].
Atherosclerosis (AS)
AS is a chronic inflammatory disease that affects the large- and medium-sized arteries. Early in AS, scavenging macrophages clear atherogenic lipoproteins and lead to the accumulation of intracellular lipids and the formation of foam cells. The residual lipid from the dead foam cells is deposited in the arteries along with the proliferation of the fibrous matrix and smooth muscle cells, forming the characteristic plaques of AS. Plaque rupture is the most common cause of coronary artery thrombosis and can result in heart attack and stroke [36-38]. Inflammation has been recognized as an important mediator of the onset and progression of AS [39].
The mislocalization of DNA/RNA binding protein transactive response DNA-binding protein 43 kDa (TDP43) in the cytoplasm impairs the mitochondrial membrane potential, causes oxidative stress, and induces the release of mtDNA into the cytoplasm. Increased TDP43 expression in both oxidized LDL (oxLDL)-treated macrophages and peripheral blood mononuclear cells from patients with coronary artery disease (CAD) can activate inflammation via NF-κB signaling in a cGAS-dependent manner; TDP43 knockout in macrophages significantly alleviated AS progression['\ 40]. Furthermore, it was recently demonstrated that stromal interacting molecule 1 (STIM1) regulates the cytosolic Ca2+ concentration and activates the cGAS-STING pathway, promoting vascular smooth muscle cell (VSMC) proliferation and apoptosis, resulting in fragile plaque formation and AS progression [41]. Mitochondrial damage in the VSMCs of CKD patients activates the cGAS-STING pathway and promotes the expression of type I IFN, leading to loss and thinning of the fibrous cap of VSMCs. This is responsible for the vulnerability of atherosclerotic plaques, accelerated atherosclerosis development, and the high morbidity rate of CVDs in patients with CKD [42].
Myocardial infarction (MI)
MI is characterized by myocardial cell death caused by prolonged ischemia, which is a minor event in a lifelong chronic disease that could be the first manifestation of CAD, occurring during unstable periods of AS or recurring constantly in established MI patients. MI is a leading cause of global disability and death [43].
Massive myocardial cell death induced by ischemia during MI increases the release of nuclear DNA and mtDNA. The accumulated cytosolic DNA then stimulates the cGAS-STING pathway, inducing the production of IRF3 and its target prototypical cytokine type I IFNs (IFNα and IFNβ) transcripts. The IRF3-IFN axis was activated in different populations of IFN-inducible cells in MI mice. cGAS deficiency, genetic disruption of IRF3-dependent signaling, or antibody blockade of the type I IFN receptor in mice results in the downregulation of inflammatory cytokines and chemokines, alleviated inflammatory cell infiltration, and improved cardiac function and survival after MI [44]. Furthermore, selective small-molecule STING inhibitors, C178 and H-151, could reduce pathological remodeling, improve cardiac function, and alleviate heart failure after MI by disturbing the palmitoylation of STING. Cao et al. ligated the LAD coronary artery of mice to induce MI and found that cGAS-STING consistently and significantly increased the mRNA expression of IFN-stimulated genes (ISGs), including IRF7, IFN-induced protein with tetratricopeptide repeat 1 (IFIT1), IFIT3, chemokine (C-X-C motif) ligand 10 (CXCL10), and cluster of differentiation 14 (CD14). Importantly, the expression of STING increased without influencing the expression of major proinflammatory cytokines, such as IL1β, IL6, IL6, and tumor necrosis factor α (TNFα), by the inactivation of the cGAS-STING pathway at the level of transcript abundance [45]. M1-subtype macrophages (pro-inflammatory and antimicrobial) and M2-subtype macrophages (healing, profibrotic, and anti-inflammatory) are the major cell types in early MI [46, 47]. Cytosolic DNA induced the expression of the M1 markers inducible nitric oxide synthase (iNOS) and CXCL10 via the cGAS-STING pathway. However, cGAS signaling deficiency promotes the abundance of M2 macrophages, which can promote myofibroblast angiogenesis and transformation to enhance myocardial wound repair, alleviate pathological ventricular remodeling, and improve survival. cGAS-STING pathway activation in human heart failure samples was confirmed by the dynamic changes in cGAS and CXCL10, which were consistent with those observed in mice [45].
Aortic aneurysm (AA) and dissection (AAD)
AA is the second most common aortic disease, characterized by permanent dilation of the thoracic and abdominal aortic regions compared to normal individuals[48]. Aortic dissection is characterized by true and false lumens of the aortic wall due to tears in the inner layer. Advanced age and vascular inflammation are two common risk factors for AA and dissection (AAD) [49]. The most important features of AAD are extracellular matrix (ECM) depletion and progressive loss of smooth muscle cells (SMCs) [50].
Significant DNA damage and leakage were observed in the aortic media and adventitia cells of patients with AAD, which resulted in the accumulation of cytosolic DNA and activation of the STING-TBK1-IRF3 pathway. This was confirmed by the upregulated expression and phosphorylation of STING, TBK1, and IRF3 in aortic tissues from patients with AAD. The STING-TBK1-IRF3 signaling pathway is critical in SMC necroptosis and apoptosis induced by ROS and cytosolic DNA. The DNA from damaged SMCs can activate the STING-TBK1-IRF3 signaling pathway in macrophages, inducing the production of matrix metalloproteinase (MMP)-9, and ultimately contributing to aortic degeneration and AAD formation. In the Sting gt/gt mouse model of sporadic AAD model, aortic enlargement and fiber disruption were alleviated, and the incidence of AAD was decreased in both the thoracic and abdominal aortic regions compared with the sporadic AAD wild-type mouse model. The TBK1 inhibitor amlexanox can significantly preserve the aortic structure and reduce aortic dilatation and the AAD incidence, improving survival and preventing the progression of AAD [51].
Neurological disorders
Aging is associated with several neurodegenerative diseases, and age-related brain inflammation, known as “inflammaging,” can lead to cognitive decline and deficits. The SASP phenotype induced by the cGAS-STING pathway plays an important role in neuroinflammation and age-related neurological disorders (Fig. 3) [52].
Figure 3.

The cGAS-STING pathway in neurological disorders. mHTT has a high affinity for interacting with TIM23, the interaction of which damages the function of mitochondria. Furthermore, DRP1 and LRRK2 mutations and Parkin and PINK1 mutations also cause mitochondrial dysfunction, resulting in mtDNA leakage. Melatonin stabilizes mitochondria and inhibits mtDNA release. ATM mutation, TREX1 mutation and DNA oxidative damage contribute to the increase in nuclear DNA. Activation of the cGAS-STING pathway promotes Aβ-induced neurotoxicity. TOLLIP, steroids, IFNAR1 blockade (ablation, deficiency and antibody) and IRF3 knockdown inhibit the cGAS-STING pathway.
Alzheimer’s disease (AD)
AD is the most common cause of dementia, affecting the daily activities of patients with AD. It is a major cause of dependence, disability, and mortality, and is recognized as a global public health priority. AD usually occurs in elderly individuals, resulting in insidious progressive problems in episodic memory. The representative pathological features of AD are extracellular accumulation of senile plaques containing amyloid-beta (Aβ), amyloid plaques, and neurofibrillary tangles (NFTs) containing hyperphosphorylated tau, accompanied by neuropil threads, dystrophic neurites, associated astrogliosis, microglial activation, and cerebral amyloid angiopathy [53].
Advanced age is the greatest risk factor for AD, mainly occurring in people over 65 years of age [54]. Neuroinflammation contributes to AD pathology, as confirmed by the enhanced microglial and astroglial activation, upregulated proinflammatory cytokine levels in the brains of patients with AD, and the decreased incidence of AD in individuals treated with nonsteroidal anti-inflammatory drugs. Type I IFN aggravates neuroinflammation and promotes AD development. Inhibition of IFN signaling within the aged brain partially rescues hippocampal neurogenesis and cognitive function and re-establishes IFN1-dependent choroid plexus activity during aging [55, 56]. The choroid plexus transcriptome demonstrated an overall upregulated expression of type I IFN response genes at all ages in AD mouse models [57]. Modulation of neuroinflammatory responses by inhibiting type I IFN signaling may provide new potential therapeutic targets in AD. Type I IFN receptor-1 (IFNAR1) is a highly specific transducer of type I IFN. Upon binding to type I IFN, IFNAR1 can trigger tyrosine phosphorylation of many proteins, including JAK, TYK2, and STAT, further regulating immune cell recruitment and inflammatory progression. Current research suggests that the removal of type 1 IFN signaling can reduce neuroinflammation and delay phenotypic progression. IFNAR1-deficient mice displayed a decrease in type I IFN and proinflammatory cytokines. Although astrocyte reactivity was enhanced, microglial proliferation around amyloid plaque deposition was attenuated. In addition, conditioned media from IFNAR1-/- primary glia treated with Aβ1-42 were less toxic to primary cultured neurons [58]. The expression of IFNα in the brains of the APP/PS1 transgenic mouse model of AD was two-fold higher than that in control brains, and ablation of IFNAR1 and IRF3 or IRF7 knockdown protected cells against Aβ-induced neurotoxicity [59, 60]. Taken together, these findings indicate that the cGAS-STING-IFN pathway plays a critical role in the onset and development of AD, and that treatments targeting this pathway can alleviate IFN-related neuroinflammation and delay the progression of AD to some extent [61].
Huntington’s disease (HD)
HD is a progressive neurodegenerative disease accompanied by motor, cognitive, and psychiatric disorders. A CAG repeat expansion with a polyglutamine strand of variable length confers direct toxicity to the mutant huntingtin (mHTT) protein, the main cause of HD[62]. HD can manifest throughout life, and the typical symptoms are chorea and dystonia, incoordination, cognitive decline, and behavioral difficulties, usually occurring in middle-aged individuals [63]. CAG repeats, but not the polyglutamine strand length, determine the age at HD onset[64]. Correia et al. conducted genome-wide association studies to discover genetic modifiers of HD and demonstrated that the age of HD onset was associated with oxidative DNA damage, DNA repair, and mitochondrial function pathways [65].
mHTT has a high affinity for translocase of the inner mitochondrial membrane 23 (TIM23), and their interaction can reduce the transportation of mitochondrial matrix proteins, altering the mitochondrial proteome and leading to the profound mitochondrial dysfunction documented in HD. mHtt can enhance the activity of mitochondrial fission GTPase dynamin-related protein-1 (DRP1) by binding to DRP1, resulting in excessive fragmentation and abnormal mitochondrial distribution. Subsequently, the increase in ROS causes mitochondrial damage and massive release of mtDNA, which triggers cGAS signaling and leads to the production of proinflammatory cytokines. Exploring new drugs that block the increase in ROS and the release of mtDNA may provide novel treatment options for aging and neurodegenerative diseases such as HD. Melatonin protects neurons from mHTT-mediated neurotoxicity. However, melatonin progressively decreases with age and in HD, and is a main cause of HD onset [66, 67]. mtDNA damage of the striatum and cerebral cortex in HD mice progressively increased and was eight times more frequent than nuclear DNA damage. An age-dependent increase in mtDNA damage contributes to mitochondrial dysfunction in HD and is an early biomarker of HD-associated neurodegeneration [68]. Cytosolic mtDNA in HD mice and mHTT-expressing cells stimulates the cGAS-STING-IRF3 pathway and subsequent production of proinflammatory cytokines. Evidence of cGAS-STING-IRF3 signaling pathway activation has also been reported in the early stage of human HD striatum, indicating that mtDNA release into the cytoplasm plays an important role in HD inflammation as an initial agent to activate the cGAS-STING-IRF3 signaling pathway [67]. Toll-interacting protein (TOLLIP) negatively regulates the Toll-like receptor (TLR) signaling pathway, which is associated with a variety of inflammatory diseases. TOLLIP is a stabilizer of STING and mediates the clearance of HD-linked polyQ protein aggregates, which can inhibit the STING-induced immune response by sequestering TOLLIP away from STING. The expression of TOLLIP decreases in the striatum of HD mice [69, 70].
Parkinson’s disease (PD)
Selective neuronal loss in the substantia nigra and other brain areas is a characteristic of PD, the most common serious movement disorder and second most common neurodegenerative disease worldwide. Age is the strongest risk factor for PD. The typical syndromes of PD are rigidity, rest tremor, and bradykinesia [71, 72]. The pathological features of PD include damage to dopaminergic projections from the substantia nigra pars compacta to the caudate nucleus and putamen (striatum), and its pathological hallmarks are intraneuronal Lewy bodies and Lewy neurites [73]. Neuroinflammation is a critical cause of progressive PD. Activated microglia and upregulated proinflammatory cytokine expression were detected in both PD patients and animal models, and increased neuroinflammation damaged dopaminergic neuronal cells, resulting in PD [74, 75].
Mutations in the leucine-rich-repeat kinase 2 (LRRK2) gene in familial PD are recognized as the most common cause. Asymptomatic PD subjects with LRRK2 mutations have higher proinflammatory cytokine levels, which in turn increase LRRK2 kinase activity [76, 77]. Furthermore, LRRK2 maintains mitochondrial homeostasis, and LRRK2 deficiency in macrophages improves the basal levels of IFN. Oxidative stress mediated by purine metabolites and mitochondrial fission induced by DRP1 in LRRK2 knockout macrophages contributes to mtDNA leakage into the cytosol, which chronically activates the cGAS-STING pathway, and reduces DRP1 expression. Antioxidant treatment alleviates mitochondrial damage and decreases type I IFN expression in LRRK2 knockout macrophages [78]. Type I IFN signaling is upregulated both in the postmortem brains of PD patients and in a PD mouse model induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). IFNAR1-deficient mice displayed decreased type I IFN signaling, proinflammatory response, and dopaminergic neuron loss. Monoclonal IFNAR1 antibody blockade treatment significantly upregulated striatal dopamine levels and improved behavior. This indicated that blocking IFNAR1 reduces MPTP-induced neuroinflammatory responses and dopaminergic cell death [79]. Parkin and PINK1 remove damaged mitochondria via mitophagy and mitigate STING-induced inflammation. Mutations in Parkin and PINK1 result in the accumulation of mtDNA mutations with age, the main cause of early onset PD. Blocking monoclonal IFNAR1 antibody treatment or STING deficiency rescued the inflammation induced by exhaustive exercise and mtDNA mutation, motor defects, and loss of dopaminergic neurons in aged Parkin-deficient mice [80].
Aicardi-Goutières syndrome (AGS)
AGS is a recessive and progressive neurological disease associated with inflammation. The constitutive upregulation of type I IFN expression mediated by AGS-associated gene mutations is considered a major cause of AGS pathogenesis. Thus, ‘type I interferonopathy’ is also suggested to define the wider spectrum of diseases induced by mutations of AGS-associated genes [81, 82].
Aberrant activation of the cGAS-STING signal by self-DNA can lead to an abnormal innate immune response, which is the primary pathogenic factor in AGS and causes other severe autoimmune diseases. Three-prime repair exonuclease 1 (TREX1) dysfunction has been suggested to be associated with autoimmune diseases and inflammation. Mutations in TREX1, a 3’-DNA exonuclease that degrades cytoplasmic DNA, result in DNA degradation disorders. Furthermore, cytoplasmic chromatin DNA fragments contain DNA damage, and oxidative damage to DNA can inhibit the exonuclease activities of TREX1, collectively leading to the accumulation of cytosolic DNA [83, 84]. The increased cytosolic DNA subsequently provokes the cGAS-STING pathway and induces the production of type I IFN[85]. The upregulated expression of cGAMP in the inflamed hearts of TREX1-deficient mice confirmed the activation of cGAS in vivo. The mortality and pathology of TREX1-deficient mice with IFNAR deficiency significantly improved, indicating the important role of the cGAS-STING-IFN signaling axis in TREX1 mutation. Given the close relationship between AGS and TREX1 mutations, cGAS inhibitors may be a potential therapy for patients with AGS [86-88].
Ataxia-telangiectasia (A-T)
A-T is a progressive neurodegenerative disorder characterized by immune dysfunction, predisposition to cancer, cerebellar degeneration, and telangiectasia. The underlying cause of A-T is mutations in the ataxia telangiectasia mutated (ATM) gene, which are important for dealing with DNA damage and maintaining genomic stability [89, 90]. More evidence indicates that chronic inflammation plays a critical role in the onset and development of A-T [91].
Increased numbers of micronuclei and nuclear shape abnormalities are observed in the olfactory neurosphere-derived cells (ONS) of A-T patients, and SASP genes, including IL1α, IL-6, and IL-8, are upregulated. Interfering with the expression of cGAS and STING with siRNAs decreased the abundance of SASP genes. In addition, A-T brain organoid senescence is reduced, and A-T neuropathology is improved by cGAS or STING inhibition [92]. Quek et al. constructed an ATM knockout rat model with paralysis and spinal cord atrophy, which was similar to older patients and milder forms instead of cerebellar atrophy. They found that the levels of cytosolic DNA dramatically increased in the neurons and glia of ATM-deficient rats. The accumulated DNA in the cytoplasm consequently activated the cGAS-STING signaling axis, accompanied by cytokine upregulation, formation of an inflammatory microenvironment, and constitutive activation of microglia, contributing to neuronal dysfunction and death. Short-term treatment with steroids, such as betamethasone, could significantly reduce the loss of motor neurons in the spine, presumably via its anti-inflammatory function [93].
Neoplasms
Inflammation induced by the cGAS-STING signaling pathway is an essential component of innate immunity that can defend against microbial infections. However, aberrant, and chronic inflammatory stimulation is responsible for some autoimmune diseases, including lethal anemia, polyarthritis, and systemic lupus erythematosus. Recent studies indicated that inflammation plays a crucial role in all phases of tumorigenesis, including onset, progression, malignant invasion, metastasis, morbidity, and mortality [94-96]. The downstream effector TBK1 of the activated cGAS-STING signaling pathway has been recognized as a critical mediator of persistent inflammatory responses that contribute to carcinogenesis (Fig. 4) [97, 98].
Figure 4.

The cGAS-STING pathway in neoplasms. MUS81, docetaxel, paclitaxel, arginine starvation, and P. acnes contribute to the increase in dsDNA in the cytoplasm. Carboplatin upregulates the expression of TREX1, which increases cytosolic dsDNA. SHP2 phosphorylates PARP1 and inhibits DNA repair, resulting in the accumulation of dsDNA. Cytosolic dsDNA is recognized by cGAS, and downstream signaling pathways are activated. The activated cGAS-STING pathway induces apoptosis and antitumor immunity, and these effects together lead to tumor cell death. JAK2/STAT3 and ENPP1 can inhibit the cGAS-STING pathway.
Colorectal cancer (CRC)
CRC is the fourth leading cause of cancer-related deaths worldwide, accounting for approximately 10% of all annually diagnosed cancers and 9.2% of global deaths. Advanced age is a high-risk factor of CRC [99, 100]. Growing evidence suggests that chemotherapy for CRC induces multiple forms of DNA damage, of which DNA double-strand breaks (DSB) are the most severe. If left unrepaired, DSB can lead to mutations that promote an antitumor immune response. Therefore, targeted therapy provides a new strategy for CRC to improve therapeutic efficacy and reduce the recurrence rate. Downregulation of cGAS and STING and decreased transcription of TBK1 and IFN in CRC tissues indicate that the cGAS-STING pathway is inhibited in CRC. The cGAS-STING pathway is essential for the antitumor immunity of CRC, the disruption of which may be a critical mechanism for immune escape of tumor cells [101, 102].
Exogenous cGAMP injection into tumors can provoke the cGAS-STING pathway and promote the expression of IFN, initiating antitumor immunity [103]. Higher STING expression in patients with CRC is associated with longer overall and recurrence-free survival[104]. Zhu et al. generated a colitis-associated CRC mouse model and found that STING-deficient mice exhibited increased colonic inflammation, hyperplasia, tumor burden, and severe intestinal pathology, suggesting that STING plays an essential role in suppressing colorectal tumorigenesis[105]. Poly ADP-ribose polymerase 1 (PARP1) is dephosphorylated by src homology-2 domain-containing protein tyrosine phosphatase-2 (SHP2) when DNA is damaged. This compromises DNA repair and contributes to the accumulation of cytosolic dsDNA, resulting in activation of the cGAS-STING signaling pathway. The agonist lovastatin can enhance the activity of SHP2 and promote the chemotherapeutic effect in colon cancer by activating the cGAS-STING signaling pathway, which could be a better alternative for colon cancer treatment [106]. The cGAS-STING signaling axis may be a potential therapeutic target for CRC [107].
Melanoma
Melanoma is a potentially fatal malignancy with an increasing incidence and mortality; it usually occurs in young and middle-aged people, and the incidence increases linearly from the age of 25 to 50 years [108-110].
Takashima et al. established a melanoma mouse model by injecting B16D8 melanoma cells into mice, which were sensitive to the cytotoxicity of natural killer (NK) cells. STING-knockout mice and STING-disrupted B16D8 cells were used to probe the critical role of STING in NK cell sensitivity in a melanoma mouse model. The proliferation and survival of STING-disrupted B16D8 cells were unaffected, and the expression of IFNβ mRNA did not increase when stimulated with cGAMP. The growth of STING-disrupted B16D8 cells was much faster than that of normal B16D8 cells in STING knockout mice, indicating that STING is involved in natural regulation and inhibits tumor proliferation[111].
As a primary epigenetic regulator, protein arginine methyltransferase 5 (PRMT5) is involved in various cell growth processes and has been reported to be associated with the development and progression of melanoma. PRMT5 methylation of IFN gamma inducible protein 16 (IFI16) and its mouse homolog IFI204, which plays a vital role in the cGAS-STING pathway, attenuates the production of IFN and chemokines induced by solute DNA, further inhibiting the activity of the cGAS-STING pathway. NLR family CARD domain containing 5 (NLRC5) is a member of a class of genes that regulate MHCI antigen presentation. The elevated NLRC5 expression enhanced MHCI cell surface expression and inhibited melanoma growth more effectively. Furthermore, upregulation of IFI16, IFI204, and NLRC5 could limit melanoma growth in mice and improve the survival of patients with melanoma. PRMT5 function is negatively associated with antitumor immunity. PRMT5 methylates IFI16/IFI204 and inhibits NLRC5 transcription, suppresses inflammation and antigen presentation, and promotes melanoma growth. PRMT5 inhibition antagonizes melanoma growth in immunocompetent mice [112]. TREX1 is essential for DNA replication and repair, but its expression is downregulated in human melanoma cell lines. Carboplatin activates the cGAS-STING pathway by upregulating TREX1 expression to inhibit proliferation and induce apoptosis in melanoma cells. Targeting the TREX1/cGAS-STING signaling axis could be a potential therapy for human melanoma [113].
Breast tumors
Breast tumors are the most common female malignancy worldwide, accounting for 30% of all female cancers in 2021 [114]. Parkes et al. reported a molecular subtype of breast cancer with a defective DNA damage response (DDR), which demonstrated increased DNA in the cytoplasm and constitutive activation of the cGAS-STING-TBK1-IRF3 pathway [115]. The activated cGAS-STING axis promoted the proliferation of triple-negative breast cancer (TNBC) cells [116].
Chromosomal instability (CIN), a tumor hallmark, is caused by persistent errors in chromosomal segregation during mitosis. It is considered a major driver of metastasis in tumors, such as breast tumors. CIN induced by continuous errors in chromosome segregation contributes to the generation of micronuclei, which are one of the main sources of cytosolic DNA after rupture. Chronic stimulation of cGAS-STING and its downstream effector NF-κB due to CIN is responsible for breast tumor metastasis [117]. Ectonucleotide pyrophosphatase/ phosphodiesterase 1 (ENPP1), which selectively degrades extracellular cGAMP and interferes with immune cell infiltration, is upregulated in TNBC cells with CIN, contributing to resistance to immunotherapy and tumor metastasis [118]. ENPP1 inhibitors may restore innate immune signaling and potentiate antitumor immune responses [119]. Metastatic breast tumor cells in the brain can transfer the second messenger cGAMP to adjacent astrocytes as paracrine signals, which activate STING and mediate the generation of inflammatory cytokines to promote metastatic tumor growth and chemoresistance [120]. Hong et al. found that drug-induced CIN TNBC cells could trigger IL-6 and signal transducer and activator of transcription 3 (STAT3)-mediated signaling to prevent tumor cell death, depending on the activation of the cGAS-STING pathway. Tocilizumab targets the IL-6 receptor and disturbs IL-6-STAT3 signaling, inhibiting the proliferation of TNBC cells with CIN. It has been reported that the application of immune checkpoint inhibitors, such as paclitaxel, in patients with TNBC could induce CIN and the production of micronuclei, which consequently provoked the cGAS-STING pathway. The combined use of tocilizumab with drugs mediating CIN could be a promising therapeutic target for activating IL-6 signaling in cancers, such as TNBC [121, 122].
Prostate cancer (PCa)
PCa is the second most frequently diagnosed malignancy and the fifth leading cause of cancer-related deaths in men[123]. PCa is recognized as a heterogeneous age-related disorder; the risk of PCa increases significantly with age, and over 85% of newly diagnosed cases are older than 60 years [2, 124]. Recent bioinformatics research has also demonstrated that senescence-related gene prognostic indices can predict PCa prognosis [125].
Cytosolic dsDNA has been detected in various human PCa cells, and dsDNA continually increases after bromodeoxyuridine labeling, demonstrating that genomic DNA is the source of cytosolic DNA in PCa cells[126]. However, STING signaling is defective in human DU145 PCa cells due to the Janus kinase 2 (JAK2)/STAT3 pathway, and STING agonists fail to provoke the cGAS-STING pathway and promote the production of type I IFN. Blocking the IL-6 or JAK2/STAT3 pathway can restore the responsiveness of DU145 cells to STING agonists [127]. DNA structure-specific endonuclease ultraviolet-sensitive 81 (MUS81) is associated with the amount of DNA in the cytoplasm of both human and mouse PCa cells. MUS81 induces the expression of type I IFNs and chemokines, promotes the immune responses of T cells, and enhances phagocytosis of PCa cells in a STING-dependent manner [126]. Propionibacterium acnes (P. acnes) is the most common microorganism in normal prostate tissues and is abundant in PCa tissues. It can activate the cGAS-STING pathway and induce the expression of IFN, which subsequently promotes PCa growth [128, 129].
Arginine synthesis deficiency, a common characteristic of cancers, causes depletion of α-ketoglutarate and epigenetic silencing of metabolic genes related to oxidative phosphorylation and DNA repair via inactivation of histone demethylases. Cancer cells with arginine deficiency exhibit mitochondrial dysfunction, transcriptional reprogramming, and eventual cell death, resulting in chromatin leakage and DNA damage. This leads to cytosolic DNA accumulation and activation of the cGAS-STING pathway. Moreover, the resulting cGAS-STING activation may further enhance its killing effects. Consequently, PCa growth was inhibited in response to the upregulation of type I IFN and recruitment of immune cells. Arginine starvation can be an effective therapy for PCa by activating the cGAS-STING pathway [130]. Ma et al. combined chemotherapy with checkpoint blockade immunotherapy in PCa to investigate the therapeutic effect and found that docetaxel therapy promoted the expression of IFN, and T cell infiltration mediated by the cGAS-STING pathway in PCa patient samples. Correspondingly, docetaxel therapy enhanced intratumor-infiltrating T cells in a PCa xenograft mouse model and promoted the expression of programmed cell death-1 (PD1) and programmed cell death-ligand 1 (PD-L1), potentiating the anti-PD1 blockade therapeutic effect in PCa xenograft mice. Furthermore, prostate-specific antigen progression-free survival of the combination of docetaxel and anti-PD1 blockade was much better than that of anti-PD1 blockade alone in a metastatic castration-resistant prostate cancer (CRPC) cohort [131].
Conclusions and perspectives
During human life, genomic stability is challenged by various endogenous and environmental factors. Many mechanisms, collectively named DDR, maintain genomic stability, by recognizing and repairing DNA damage. However, genomic damage still accumulates when genomic damage is excessive and DDR is deficient or disrupted, leading to genomic instability [132]. Genomic instability is a hallmark of aging and a leading cause of age-related diseases, which are closely related to the aberrant presence of cytosolic DNA and activation of the cGAS-STING pathway [3, 133].
Inflammation induced by the cGAS-STING signaling pathway is important for the onset of many age-related diseases. However, the cGAS-STING pathway plays different roles in different age-related diseases (Table 1). Its aberrant activation can lead to pathological cell proliferation, cellular dysfunction, and death, ultimately resulting in a detrimental disease state. Activated cGAS-STING pathway-mediated inflammation increases immune cell infiltration, promotes antitumor immunity, and reduces the risk of tumor metastasis in some cancers. Downregulation or deletion of the cGAS-STING pathway may be an important cause of tumorigenesis and progression. In contrast, the cGAS-STING pathway can also promote tumor proliferation and metastasis [117, 134].
Table 1.
List of age-related diseases and supporting evidence associated with the cGAS-STING pathway.
| Age-related disease | cGAS-STING activity | Pathogenesis | Human sample | Animal model | Therapy | Therapeutic effect | Ref | |
|---|---|---|---|---|---|---|---|---|
| Cardiovascular diseases | Cardiac hypertrophy | In human: upregulated STING and type I IFNs In animal: upregulated STING and type I IFNs |
- | DCM and HCM patients | Cardiac hypertrophy mice induced by AB and Ang II | STING knockout | Alleviating inflammation, fibrosis, and attenuating cardiac hypertrophy | [31] |
| In animal: upregulated cGAS, cGAMP, TBK1, IRF3, type I IFN, TNF-α, IL-1β and IL-18 | Free fatty acids | - | DCM mice induced by streptozotocin and HFD | cGAS and STING knockdown and inhibitor (Ru.521 and C176-1) | Alleviating myocardial pyroptosis, inflammation, and attenuating cardiac hypertrophy | [32] | ||
| In animal: upregulated cGAS, STING, TNFαand IL-1β | Akt2-AMPKα2 double ablation | - | Akt2-/- and AMPKα2-/- mice; HFD-induced cardiac hypertrophy mice | cGAS and STING inhibitors (PF-06928215 and Astin C) | Ameliorating HFD-induced contractile dysfunction | [33] | ||
| Atherosclerosis | In human: upregulated of NF-κB, IL-6 and TNF-α In animal: downregulated IL-6 and TNF-α after TDP43 knockout |
TDP43 mis-localization | CAD patients peripheral blood sample | TDP43-/- mice | NF-κB inhibitors (PDTC and SC75741) | Alleviating AS in TDP43-/- mice | [40] | |
| Myocardial infarction | In animal: downregulated type I IFN in IRF3 knockout mice, upregulated ISGs in WT mice | Dead myocardial cells | - | cGAS and STING deficient mice | Genetic disrupton or antibody blockade of IFNAR1 | Improving heart function and survival | [44] | |
| In human: upregulated cGAS and CXCL10 In animal: upregulated STING, CXCL10,IFIT1,IFIT3, IRF7 and CD14 |
Dead myocardial cells | Failing and nonfailing hearts tissues | LAD coronary artery ligation-induced MI mice; cGAS-null mice | cGAS knockout | Improving myocardial repair after infarction | [45] | ||
| Aortic aneurysm and dissection | In human: upregulated STING, TBK1 and IRF3 In animal: upregulated STING |
DNA from damaged SMCs | AAD patient aortic tissues | HFD and Ang II-induced AAD model; Sting deficient mice | TBK1 Inhibitor (amlexanox) | Partially preventing AAD development | [51] | |
| Neurological disorders | Alzheimer’s disease | In human: upregulated type I IFN, TNFα, IL-1β, and IL-6 In animal: upregulated type I IFN |
- | AD patient prefrontal cortex | AD mouse model; IFNAR1-/- mice | IFNAR1 ablation | Protection against Aβ-induced toxicity | [57] [59] |
| Huntington’s disease | In human: upregulated cGAS, STING and IRF3 In animal: upregulated cGAS, STING, IRF3, type I IFN, IL-1β, IL-6 and IL-18 |
Melatonin decrease | HD patient striatum samples | HD mice model | cGAS inhibitor (Ru.521); melatonin | Inhibition of cGAS-STING pathway | [67] | |
| In animal: upregulated cGAS, STING and IRF3 | - | - | TOLLIP-/- mice | TOLLIP knockout | Impairing STING signaling and ameliorating HD | [70] | ||
| Parkinson’s disease | In LRRK2 KO macrophages: cGAS, STING, IRF3, IFN and ISG | LRRK2 mutation | - | LRRK2 knockout mice | reducing DRP1 and antioxidants | Alleviating mitochondrial damage and decreasing type I IFN | [78] | |
| In human: upregulated IL-6, IL-1β, CCL2, and CCL4 In animal: upregulated cGAMP, IL-6, IFNβ1, IL-12, IL-13, CXCL1, CCL2, and CCL4 |
Parkin and PINK1 mutations | - | Parkin-/-, PINK1-/- and STING-/- mice | Blocking IFNAR1 or STING knockout | Alleviating inflammation and PD | [80] | ||
| Aicardi-Goutières syndrome | In animal: upregulated cGAMP and IFN | TREX1 mutation | - | TREX1 deficient mice | IFNAR1 knockout | The mortality and pathology significantly improved | [86-87] | |
| Ataxia-telangiectasia | In brain organoid: downregulated cGAS, STING and SASP (IL-6, IL-8, IL1α and CCL20) via RNA interference | ATM mutation | A-T patient nasal biopsies | - | cGAS and STING inhibitors (aspirin and H-151) | Reduction of senescence phenotypes and inflammation | [92] | |
| In animal: upregulated STING, TBK1, NF-κB, IFNβ, IL-1β | ATM mutation | - | ATM knockout mice | Steroids (betamethasone) | Reducing the loss of motor neurons | [93] | ||
| Neoplasms | Colorectal cancer | In cells: downregulated cGAS, STING, IRF3 and IFN | - | - | STING knockout mice | Viral oncolytic therapy | Tumor cells growth reduction | [101] |
| In cells: upregulated STING and IFN after cGAMP stimulation | - | - | shSHP2 mice | SHP2 agonist (lovastatin) | Delayed DNA damage repair and enhanced antitumor immunity | [106] | ||
| Melanoma | In cell: upregulated cGAS, STING, TBK1, IRF3 and TNF after TREX1 overexpression | TREX1 decrease | Human sinonasal mucosal melanoma | - | Carboplatin | Inducing apoptosis and inhibiting proliferation | [113] | |
| Breast tumors | In human: upregulated cGAS, STING, TBK1, IRF3 | CIN | Human breast cancer samples | - | Tocilizumab | Inducing tumor cells death | [121] | |
| Prostate cancer | In cell: upregulated IRF3 and type I IFN | Nuclear MUS81 decrease | Human PCa samples | - | - | - | [126] | |
| In cells: upregulated cGAS, STING, TBK1 and type I IFN | Arginine | - | Arginine-restriction xenograft mouse model | Arginine starvation | Increased immune cells infiltration and PCa growth inhibition | [130] | ||
| In human: upregulated cGAS, STING and IFN | - | PCa patient tumor samples | CRPC mice | Docetaxel | Immune microenvironment remodeling and enhancing immunotherapy | [131] | ||
For each age-related disease described in the text, the evidence for causality or correlation with the cGAS-STING pathway, human sample(s), animal model(s), therapy targeting this pathway and its effect are listed. cGAS, cyclic guanosine monophosphate (GMP)- adenosine monophosphate (AMP) synthase; cGAMP, cyclic guanosine monophosphate-adenosine monophosphate; STING, stimulator of interferon genes; IFN, interferon; TBK1, TANK-binding kinase 1; IRF3, interferon regulatory factor 3; IL, interleukin; TNF, tumor necrosis factor; DCM, diabetic cardiomyopathy; HCM, hypertrophic cardiomyopathy; AB, aortic banding; Ang II, angiotensin II; NLRP3, nucleotide-binding oligomerization domain-like receptor pyrin domain containing 3; Akt2, protein kinase B 2; AMPK, AMP-dependent protein kinase; TDP43, Transactive response DNA-binding protein ~43 kDa; CAD, coronary artery disease; ISG, interferon stimulated gene; CXCL10, chemokine (C-X-C motif) Ligand 10; IFIT, interferon-induced protein with tetratricopeptide repeat; CD14, cluster of differentiation 14; AAD, aortic aneurysm and dissection; HFD, high-fat diet; TOLLIP, toll-interacting protein; MI, myocardial infarction; SMCs, smooth muscle cells; MMP-9, matrix metalloproteinase (MMP)-9; AD, Alzheimer’s disease; IFNAR1, type I interferon-α receptor 1; HD,Huntington’s disease; LRRK2, leucine-rich repeat kinase 2; DRP1, dynamin-related protein 1; SHP2, Src homology-2 domain-containing protein tyrosine phosphatase-2; TREX1, three-prime repair exonuclease 1; CIN, chromosomal instability; MUS81, DNA structure-specific endonuclease ultraviolet-sensitive 81; CRPC, castration-resistant prostate cancer.
In addition to malignant age-related diseases, inflammation also plays an important role in the development and progression of benign age-related diseases such as benign prostatic hyperplasia (BPH), which mainly develops in the transition zone and enlarges with age. BPH is a hyperplastic process with an increased number of epithelial and stromal cells, but the stromal-to-epithelial ratios in BPH samples are significantly heterogeneous [135]. SASP plays a crucial role in tumorigenesis and age-related diseases. Recent studies have suggested SASP can be triggered by cGAS-STING signaling pathway via accumulation of cytoplasmic DNA in senescent cells. Given the critical role of the cGAS-STING pathway in inflammation and aging, it may also be involved in the development and progression of BPH. Choi et al. used senescent biomarker senescent-associated beta-galactosidase to detect BPH specimens and demonstrated that senescence was only presented in epithelial cells, but not in stromal cells [136]. Although senescent epithelial cells cannot proliferate anymore in BPH, we propose that senescent epithelial cells may secrete SASP via the cGAS-STING pathway to promote the proliferation of stromal cells, which could be a potential mechanism by which aging results in BPH [137, 138]. However, given that there are no studies have been reported on the cGAS-STING pathway and BPH, further studies are needed to confirm the hypothesis.
Treatment with inhibitors or agonists targeting the cGAS-STING pathway in age-related diseases should be prudent depending on the function of this pathway in these diseases. Some drugs targeting the cGAS-STING pathway used for the treatment of diseases are listed in Table 2 [87, 103, 139-159]. However, as most of these drugs are still in the experimental/clinical trial stage, they cannot be used in humans to address aging and age-related diseases [160]. Further studies are needed to identify ideal inhibitors or agonists targeting the cGAS-STING pathway that can be used in humans. These agents may provide new insights into and therapeutic alternatives for age-related diseases.
Table 2.
The inhibitors and agonists of the cGAS-STING pathway.
| Target | Agents | Function or property | Species specificity | Advantage | Disadvantage | Application model | Treatment effect | Ref |
|---|---|---|---|---|---|---|---|---|
| cGAS inhibitors | AMDs | Blocking dsDNA/cGAS binding | Human and mouse cGAS | Widely used with a strong safety profile | - | Trex1-/- mouse model | Effective for the treatment of AGS mice | [87] |
| EGCG | Disrupting cGAS activation | Human and mouse cGAS | Specifically inhibit inflammation caused by cGAS activation | Effect of EGCG on cGAS activity is clearly dependent on G3BP1 | AGS patient cells and AGS mouse model | Effective in treating cGAS-mediated autoinflammation | [139] | |
| Aspirin | Inhibiting cGAS by acetylation | Human and mouse cGAS | Widely used with a strong safety profile | - | AGS patient cells and AGS mouse model | Effective in treating AGS and potentially other DNA-mediated autoimmune diseases | [140] | |
| RU.521 | Occupying the catalytic site of cGAS | Mouse cGAS | Selective inhibition of cGAS dsDNA-dependent enzymatic activity in vitro and in cells | Poor inhibitor of recombinant human cGAS | AGS mouse model | Suppressing type I IFN expression | [141] | |
| G140, G150 | Inhibiting cGAS activity | Human cGAS | Specific and potent small-molecule inhibitors for human cGAS | The utility for therapeutic treatment of cGAS-related human diseases is uncertain | Human monocytic THP1 cells and mouse macrophage RAW 264.7 cells | Inhibiting dsDNA-induced cGAS activity | [142] | |
| PAH | Inhibiting cGAS activity | Human and mouse cGAS | No side effects and toxicity with biological safety in vivo | Inhibitory mechanisms are uncertain | AGS mouse model | Ameliorating self-DNA-induced autoinflammatory responses | [143] | |
| Compound 25 | Inhibiting cGAS activity | Human and mouse cGAS | Superior in vivo anti-inflammatory effects | Structural optimization is needed to achieve higher potency | Lipopolysaccharide-induced mouse model | Ihibiting the dsDNA-induced phosphorylation of STING/TBK1/IRF3 signaling and the mRNA expression of ISGs | [144] | |
| A151 | Competing with DNA | Mouse cGAS | Into the damaged brain parenchyma from the blood | - | Ischemic mouse brains model | Protecting against brain damage and improving neurodeficits | [145] | |
| STING inhibitors | Compound 18 | Competing with cGAMP | Human STING | Good oral exposure and slow binding kinetics | - | Human monocytic THP1 cells | Inhibiting cGAMP-induced IFNβ production | [146] |
| Astin C | Blocking IRF3 recruitment onto the STING | Human and mouse STING | Well-tolerated compound with minimal cytotoxic side effects | The in vivo effectiveness is uncertain | Trex1-/- BMDMs cells and Trex1-/- mouse model | Inhibiting the expression of type I IFN and pro-inflammatory cytokines and alleviating the autoinflammatory responses | [147] | |
| NO2-FA | Inhibiting STING palmitoylation | Human and mouse STING | Natural antiinflammatory mediators | Not yet used in clinical practice | Fibroblasts from SAVI patients | Inhibiting production of type I IFN | [148] | |
| C-178, C-176 | Inhibiting STING palmitoylation | Mouse STING | In vivo inhibitory capacity is not limited by the short serum half-life | Low affinity to human STING | Trex1-/- mouse model | Amelioration of various signs of systemic inflammation | [149] | |
| C-170, C-171, H-151 | Blocking palmitoylation-induced clustering of STING | Human and mouse STING | Highly potent and selective small-molecule inhibitor | - | Trex1-/- mouse model | Reducing systemic cytokine responses | [149] | |
| SN-011 | Blocking CDN binding and STING activation | Human and mouse STING | Potent and selective inhibitor with high affinity and specificity | - | Trex1-/- mouse model | Ameliorating autoimmune pathology and preventing death | [150] | |
| STING agonists | DMXAA | Non-CDNs | Mouse STING | Potent and specific therapeutic effect on mice | Lacking the ability to activate human STING | Mouse xenotransplantation models | Induction of cytokines and disrupting tumor vascularization | [151] |
| CMA | Non-CDNs | Mouse STING | Potent and specific therapeutic effect on mice | Lacking the ability to activate human STING | Mouse macrophages | Inducing IRF3 phosphorylation and Ifnb mRNA translation | [152] | |
| c(di-GMP) | CDNs | Mouse STING | Mobilizing abscopal immunity when combined with checkpoint modulation | Low affinity to human STING | Bilaterally-implanted TRAMP-C2 tumors mouse model | Mediating regression of injected tumors | [153] | |
| 3'3'-cGAMP | CDNs | Mouse STING | More efficient than DMXAA in activating STING | - | Chronic lymphocytic leukemia and multiple myeloma mouse model | Inducing apoptosis and tumor regression | [154] | |
| ML RR-S2 CDA | CDNs | Human and mouse STING | Activating all human and mouse STING alleles | Intratumor injection is necessary to achieve maximal therapeutic effect | 4T-1 colon or CT26 mammary carcinomas mouse model | Inducing tumor regression and resistant to the same tumor cell line | [103] | |
| ABZI | Non-CDNs | Human and mouse STING | Intravenous administration can induce adaptive CD8 T cell response in vivo | - | Syngeneic colon tumours mouse model | Strong anti-tumour activity with complete and lasting regression of tumours | [155] | |
| STING-LNP | Non-CDNs | Mouse STING | Efficiently delivered the STING agonist to the cytoplasm | - | B16-F10-luc2 lung metastatic mouse model | Increasing the expression of CD3, CD4, PD-1 and IFN in lung metastases | [156] | |
| MSA-2 | Non-CDNs | Human and mouse STING | Favorable activity and tolerability profiles | - | MC38 syngeneic tumors mouse model | Inducing tumor regressions | [157] | |
| PC7A | Non-CDNs | Human and mouse STING | Polyvalent STING agonist with prolonged cytokine expression | - | MC38 and TC-1 tumour models | Notably extended the survival | [158] | |
| SR-717 | Non-CDNs | Human and mouse STING | Substantial efficacy without considerable toxicity | - | B16.F10 melanomas mouse model | Reduction in tumor growth and the increased survival | [159] |
cGAS, cyclic guanosine monophosphate (GMP)- adenosine monophosphate (AMP) synthase; cGAMP, cyclic guanosine monophosphate-adenosine monophosphate; STING, stimulator of interferon genes; IFN, interferon; TBK1, TANK-binding kinase 1; IRF3, interferon regulatory factor 3; ISG, interferon stimulated gene; AMDs: antimalarial drugs; EGCG: epigallocatechin gallate; PAH: perillaldehyde; BMDMs: bone marrow-derived macrophages; SAVI: STING-associated vasculopathy with onset in infancy; CDN: cyclic dinucleotide; ABZI: amidobenzimidazole.
Acknowledgement
This program was supported by the National Natural Science Foundation of China (Grant Nos. 81974099, 82170785, 81974098, 82170784).
Footnotes
Conflict of interest
All authors declare that no competing interest associated with the manuscript needs to be disclosed.
References
- [1].McHugh D, Gil J (2018). Senescence and aging: Causes, consequences, and therapeutic avenues. J Cell Biol, 217:65-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chang AY, Skirbekk VF, Tyrovolas S, Kassebaum NJ, Dieleman JL (2019). Measuring population ageing: an analysis of the Global Burden of Disease Study 2017. Lancet Public Health, 4: e159-e167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013). The hallmarks of aging. Cell, 153:1194-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hoeijmakers JH (2009). DNA damage, aging, and cancer. N Engl J Med, 361:1475-1485. [DOI] [PubMed] [Google Scholar]
- [5].Moskalev AA, Shaposhnikov MV, Plyusnina EN, Zhavoronkov A, Budovsky A, Yanai H, et al. (2013). The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res Rev, 12:661-684. [DOI] [PubMed] [Google Scholar]
- [6].Lord CJ, Ashworth A (2012). The DNA damage response and cancer therapy. Nature, 481:287-294. [DOI] [PubMed] [Google Scholar]
- [7].Blackburn EH, Greider CW, Szostak JW (2006). Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med, 12:1133-1138. [DOI] [PubMed] [Google Scholar]
- [8].Kazak L, Reyes A, Holt IJ (2012). Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol, 13:659-671. [DOI] [PubMed] [Google Scholar]
- [9].Green DR, Galluzzi L, Kroemer G (2011). Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science, 333:1109-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Miller KN, Victorelli SG, Salmonowicz H, Dasgupta N, Liu T, Passos JF, et al. (2021). Cytoplasmic DNA: sources, sensing, and role in aging and disease. Cell, 184:5506-5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Akbari M, Shanley DP, Bohr VA, Rasmussen LJ (2021). Cytosolic Self-DNA-A Potential Source of Chronic Inflammation in Aging. Cells, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sun L, Wu J, Du F, Chen X, Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science, 339:786-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Decout A, Katz JD, Venkatraman S, Ablasser A (2021). The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol, 21:548-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. (2013). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science, 339:826-830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ishikawa H, Barber GN (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature, 455:674-678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fang R, Jiang Q, Guan Y, Gao P, Zhang R, Zhao Z, et al. (2021). Golgi apparatus-synthesized sulfated glycosaminoglycans mediate polymerization and activation of the cGAMP sensor STING. Immunity, 54:962-975.e968. [DOI] [PubMed] [Google Scholar]
- [17].Zhang X, Bai XC, Chen ZJ (2020). Structures and Mechanisms in the cGAS-STING Innate Immunity Pathway. Immunity, 53:43-53. [DOI] [PubMed] [Google Scholar]
- [18].Birch J, Gil J (2020). Senescence and the SASP: many therapeutic avenues. Genes Dev, 34:1565-1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kuilman T, Peeper DS (2009). Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer, 9:81-94. [DOI] [PubMed] [Google Scholar]
- [20].Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. (2017). Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature, 550:402-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Glück S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, et al. (2017). Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol, 19:1061-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. (2007). Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature, 445:656-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J (2001). Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A, 98:12072-12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol, 6:2853-2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Evans MA, Sano S, Walsh K (2020). Cardiovascular Disease, Aging, and Clonal Hematopoiesis. Annu Rev Pathol, 15:419-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Donato AJ, Machin DR, Lesniewski LA (2018). Mechanisms of Dysfunction in the Aging Vasculature and Role in Age-Related Disease. Circ Res, 123:825-848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Oduro PK, Zheng X, Wei J, Yang Y, Wang Y, Zhang H, et al. (2022). The cGAS-STING signaling in cardiovascular and metabolic diseases: Future novel target option for pharmacotherapy. Acta Pharm Sin B, 12:50-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shimizu I, Minamino T (2016). Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol, 97:245-262. [DOI] [PubMed] [Google Scholar]
- [29].Hu D, Cui YX, Wu MY, Li L, Su LN, Lian Z, et al. (2020). Cytosolic DNA sensor cGAS plays an essential pathogenetic role in pressure overload-induced heart failure. Am J Physiol Heart Circ Physiol, 318:H1525-h1537. [DOI] [PubMed] [Google Scholar]
- [30].Sun H, Wang Y (2014). Interferon regulatory factors in heart: stress response beyond inflammation. Hypertension, 63:663-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhang Y, Chen W, Wang Y (2020). STING is an essential regulator of heart inflammation and fibrosis in mice with pathological cardiac hypertrophy via endoplasmic reticulum (ER) stress. Biomed Pharmacother, 125:110022. [DOI] [PubMed] [Google Scholar]
- [32].Yan M, Li Y, Luo Q, Zeng W, Shao X, Li L, et al. (2022). Mitochondrial damage and activation of the cytosolic DNA sensor cGAS-STING pathway lead to cardiac pyroptosis and hypertrophy in diabetic cardiomyopathy mice. Cell Death Discov, 8:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gong Y, Li G, Tao J, Wu NN, Kandadi MR, Bi Y, et al. (2020). Double knockout of Akt2 and AMPK accentuates high fat diet-induced cardiac anomalies through a cGAS-STING-mediated mechanism. Biochim Biophys Acta Mol Basis Dis, 1866:165855. [DOI] [PubMed] [Google Scholar]
- [34].Rech L, Abdellatif M, Pöttler M, Stangl V, Mabotuwana N, Hardy S, et al. (2022). Small molecule STING inhibition improves myocardial infarction remodeling. Life Sci, 291:120263. [DOI] [PubMed] [Google Scholar]
- [35].Han W, Du C, Zhu Y, Ran L, Wang Y, Xiong J, et al. (2022). Targeting Myocardial Mitochondria-STING-Polyamine Axis Prevents Cardiac Hypertrophy in Chronic Kidney Disease. JACC Basic Transl Sci, 7:820-840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Falk E (2006). Pathogenesis of atherosclerosis. J Am Coll Cardiol, 47:C7-12. [DOI] [PubMed] [Google Scholar]
- [37].Tabares-Guevara JH, Villa-Pulgarin JA, Hernandez JC (2021). Atherosclerosis: immunopathogenesis and strategies for immunotherapy. Immunotherapy, 13:1231-1244. [DOI] [PubMed] [Google Scholar]
- [38].Zhu Y, Xian X, Wang Z, Bi Y, Chen Q, Han X, et al. (2018). Research Progress on the Relationship between Atherosclerosis and Inflammation. Biomolecules, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Libby P, Ridker PM, Hansson GK (2011). Progress and challenges in translating the biology of atherosclerosis. Nature, 473:317-325. [DOI] [PubMed] [Google Scholar]
- [40].Huangfu N, Wang Y, Xu Z, Zheng W, Tao C, Li Z, et al. (2021). TDP43 Exacerbates Atherosclerosis Progression by Promoting Inflammation and Lipid Uptake of Macrophages. Front Cell Dev Biol, 9:687169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wan X, Tian J, Hao P, Zhou K, Zhang J, Zhou Y, et al. (2022). cGAS-STING Pathway Performance in the Vulnerable Atherosclerotic Plaque. Aging Dis, 13:1606-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bi X, Du C, Wang X, Wang XY, Han W, Wang Y, et al. (2021). Mitochondrial Damage-Induced Innate Immune Activation in Vascular Smooth Muscle Cells Promotes Chronic Kidney Disease-Associated Plaque Vulnerability. Adv Sci (Weinh), 8:2002738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Thygesen K, Alpert JS, White HD (2007). Universal definition of myocardial infarction. J Am Coll Cardiol, 50:2173-2195. [DOI] [PubMed] [Google Scholar]
- [44].King KR, Aguirre AD, Ye YX, Sun Y, Roh JD, Ng RP Jr., et al. (2017). IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med, 23:1481-1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cao DJ, Schiattarella GG, Villalobos E, Jiang N, May HI, Li T, et al. (2018). Cytosolic DNA Sensing Promotes Macrophage Transformation and Governs Myocardial Ischemic Injury. Circulation, 137:2613-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, et al. (2013). Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol, 62:24-35. [DOI] [PubMed] [Google Scholar]
- [47].Lawrence T, Natoli G (2011). Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol, 11:750-761. [DOI] [PubMed] [Google Scholar]
- [48].Skotsimara G, Antonopoulos A, Oikonomou E, Papastamos C, Siasos G, Tousoulis D (2022). Aortic Wall Inflammation in the Pathogenesis, Diagnosis and Treatment of Aortic Aneurysms. Inflammation, 45:965-976. [DOI] [PubMed] [Google Scholar]
- [49].Chen MT, Chung CH, Ke HY, Peng CK, Chien WC, Shen CH (2021). Risk of Aortic Aneurysm and Dissection in Patients with Tuberculosis: A Nationwide Population-Based Cohort Study. Int J Environ Res Public Health, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Guo DC, Papke CL, He R, Milewicz DM (2006). Pathogenesis of thoracic and abdominal aortic aneurysms. Ann N Y Acad Sci, 1085:339-352. [DOI] [PubMed] [Google Scholar]
- [51].Luo W, Wang Y, Zhang L, Ren P, Zhang C, Li Y, et al. (2020). Critical Role of Cytosolic DNA and Its Sensing Adaptor STING in Aortic Degeneration, Dissection, and Rupture. Circulation, 141:42-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Paul BD, Snyder SH, Bohr VA (2021). Signaling by cGAS-STING in Neurodegeneration, Neuroinflammation, and Aging. Trends Neurosci, 44:83-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lane CA, Hardy J, Schott JM (2018). Alzheimer's disease. Eur J Neurol, 25:59-70. [DOI] [PubMed] [Google Scholar]
- [54].Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, et al. (2021). Alzheimer's disease. Lancet, 397:1577-1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Taylor JM, Moore Z, Minter MR, Crack PJ (2018). Type-I interferon pathway in neuroinflammation and neurodegeneration: focus on Alzheimer's disease. J Neural Transm (Vienna), 125:797-807. [DOI] [PubMed] [Google Scholar]
- [56].Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, et al. (2014). Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science, 346:89-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Mesquita SD, Ferreira AC, Gao F, Coppola G, Geschwind DH, Sousa JC, et al. (2015). The choroid plexus transcriptome reveals changes in type I and II interferon responses in a mouse model of Alzheimer's disease. Brain Behav Immun, 49:280-292. [DOI] [PubMed] [Google Scholar]
- [58].Minter MR, Moore Z, Zhang M, Brody KM, Jones NC, Shultz SR, et al. (2016). Deletion of the type-1 interferon receptor in APPSWE/PS1ΔE9 mice preserves cognitive function and alters glial phenotype. Acta Neuropathol Commun, 4:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Taylor JM, Minter MR, Newman AG, Zhang M, Adlard PA, Crack PJ (2014). Type-1 interferon signaling mediates neuro-inflammatory events in models of Alzheimer's disease. Neurobiol Aging, 35:1012-1023. [DOI] [PubMed] [Google Scholar]
- [60].Minter MR, Main BS, Brody KM, Zhang M, Taylor JM, Crack PJ (2015). Soluble amyloid triggers a myeloid differentiation factor 88 and interferon regulatory factor 7 dependent neuronal type-1 interferon response in vitro. J Neuroinflammation, 12:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Chen K, Lai C, Su Y, Bao WD, Yang LN, Xu PP, et al. (2022). cGAS-STING-mediated IFN-I Response in Host Defense and Neuroinflammatory Diseases. Curr Neuropharmacol, 20:362-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].McColgan P, Tabrizi SJ (2018). Huntington's disease: a clinical review. Eur J Neurol, 25:24-34. [DOI] [PubMed] [Google Scholar]
- [63].Walker FO (2007). Huntington's disease. Lancet, 369:218-228. [DOI] [PubMed] [Google Scholar]
- [64].CAG Repeat Not Polyglutamine Length Determines Timing of Huntington's Disease Onset (2019). Cell, 178:887-900.e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Correia K, Harold D, Kim KH, Holmans P, Jones L, Orth M, et al. (2015). The Genetic Modifiers of Motor OnsetAge (GeM MOA) Website: Genome-wide Association Analysis for Genetic Modifiers of Huntington's Disease. J Huntingtons Dis, 4:279-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Yablonska S, Ganesan V, Ferrando LM, Kim J, Pyzel A, Baranova OV, et al. (2019). Mutant huntingtin disrupts mitochondrial proteostasis by interacting with TIM23. Proc Natl Acad Sci U S A, 116:16593-16602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Jauhari A, Baranov SV, Suofu Y, Kim J, Singh T, Yablonska S, et al. (2020). Melatonin inhibits cytosolic mitochondrial DNA-induced neuroinflammatory signaling in accelerated aging and neurodegeneration. J Clin Invest, 130:3124-3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Acevedo-Torres K, Berríos L, Rosario N, Dufault V, Skatchkov S, Eaton MJ, et al. (2009). Mitochondrial DNA damage is a hallmark of chemically induced and the R6/2 transgenic model of Huntington's disease. DNA Repair (Amst), 8:126-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Li M, Feng B, Wang L, Guo S, Zhang P, Gong J, et al. (2015). Tollip is a critical mediator of cerebral ischaemia-reperfusion injury. J Pathol, 237:249-262. [DOI] [PubMed] [Google Scholar]
- [70].Pokatayev V, Yang K, Tu X, Dobbs N, Wu J, Kalb RG, et al. (2020). Homeostatic regulation of STING protein at the resting state by stabilizer TOLLIP. Nat Immunol, 21:158-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Samii A, Nutt JG, Ransom BR (2004). Parkinson's disease. Lancet, 363:1783-1793. [DOI] [PubMed] [Google Scholar]
- [72].Tolosa E, Garrido A, Scholz SW, Poewe W (2021). Challenges in the diagnosis of Parkinson's disease. Lancet Neurol, 20:385-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Fearnley JM, Lees AJ (1991). Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain, 114(Pt 5):2283-2301. [DOI] [PubMed] [Google Scholar]
- [74].Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. (2016). Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson's Disease. Cell, 167:1469-1480.e1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Gelders G, Baekelandt V, Van der Perren A (2018). Linking Neuroinflammation and Neurodegeneration in Parkinson's Disease. J Immunol Res, 2018:4784268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Singleton AB, Farrer MJ, Bonifati V (2013). The genetics of Parkinson's disease: progress and therapeutic implications. Mov Disord, 28:14-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Wallings RL, Tansey MG (2019). LRRK2 regulation of immune-pathways and inflammatory disease. Biochem Soc Trans, 47:1581-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Weindel CG, Bell SL, Vail KJ, West KO, Patrick KL, Watson RO (2020). LRRK2 maintains mitochondrial homeostasis and regulates innate immune responses to Mycobacterium tuberculosis. Elife, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Main BS, Zhang M, Brody KM, Ayton S, Frugier T, Steer D, et al. (2016). Type-1 interferons contribute to the neuroinflammatory response and disease progression of the MPTP mouse model of Parkinson's disease. Glia, 64:1590-1604. [DOI] [PubMed] [Google Scholar]
- [80].Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. (2018). Parkin and PINK1 mitigate STING-induced inflammation. Nature, 561:258-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, et al. (2015). Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A, 167a:296-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Crow YJ, Shetty J, Livingston JH (2020). Treatments in Aicardi-Goutières syndrome. Dev Med Child Neurol, 62:42-47. [DOI] [PubMed] [Google Scholar]
- [83].Vizioli MG, Liu T, Miller KN, Robertson NA, Gilroy K, Lagnado AB, et al. (2020). Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev, 34:428-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Gehrke N, Mertens C, Zillinger T, Wenzel J, Bald T, Zahn S, et al. (2013). Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity, 39:482-495. [DOI] [PubMed] [Google Scholar]
- [85].Hemphill WO, Simpson SR, Liu M, Salsbury FR Jr., Hollis T, Grayson JM, et al. (2021). TREX1 as a Novel Immunotherapeutic Target. Front Immunol, 12:660184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Stetson DB, Ko JS, Heidmann T, Medzhitov R (2008). Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell, 134:587-598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Gray EE, Treuting PM, Woodward JJ, Stetson DB (2015). Cutting Edge: cGAS Is Required for Lethal Autoimmune Disease in the Trex1-Deficient Mouse Model of Aicardi-Goutières Syndrome. J Immunol, 195:1939-1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, et al. (2013). Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol, 12:1159-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, Lederman HM (2016). Ataxia telangiectasia: a review. Orphanet J Rare Dis, 11:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Quek H, Luff J, Cheung K, Kozlov S, Gatei M, Lee CS, et al. (2017). Rats with a missense mutation in Atm display neuroinflammation and neurodegeneration subsequent to accumulation of cytosolic DNA following unrepaired DNA damage. J Leukoc Biol, 101:927-947. [DOI] [PubMed] [Google Scholar]
- [91].Zaki-Dizaji M, Akrami SM, Azizi G, Abolhassani H, Aghamohammadi A (2018). Inflammation, a significant player of Ataxia-Telangiectasia pathogenesis? Inflamm Res, 67:559-570. [DOI] [PubMed] [Google Scholar]
- [92].Aguado J, Chaggar HK, Gómez-Inclán C, Shaker MR, Leeson HC, Mackay-Sim A, et al. (2021). Inhibition of the cGAS-STING pathway ameliorates the premature senescence hallmarks of Ataxia-Telangiectasia brain organoids. Aging Cell, 20: e13468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Quek H, Luff J, Cheung K, Kozlov S, Gatei M, Lee CS, et al. (2017). A rat model of ataxia-telangiectasia: evidence for a neurodegenerative phenotype. Hum Mol Genet, 26:109-123. [DOI] [PubMed] [Google Scholar]
- [94].Grivennikov SI, Greten FR, Karin M (2010). Immunity, inflammation, and cancer. Cell, 140:883-899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Nagata S, Hanayama R, Kawane K (2010). Autoimmunity and the clearance of dead cells. Cell, 140:619-630. [DOI] [PubMed] [Google Scholar]
- [96].Trinchieri G (2012). Cancer and inflammation: an old intuition with rapidly evolving new concepts. Annu Rev Immunol, 30:677-706. [DOI] [PubMed] [Google Scholar]
- [97].Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN (2014). Inflammation-driven carcinogenesis is mediated through STING. Nat Commun, 5:5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature, 462:108-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, Wallace MB (2019). Colorectal cancer. Lancet, 394:1467-1480. [DOI] [PubMed] [Google Scholar]
- [100].Li J, Ma X, Chakravarti D, Shalapour S, DePinho RA (2021). Genetic and biological hallmarks of colorectal cancer. Genes Dev, 35:787-820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Xia T, Konno H, Ahn J, Barber GN (2016). Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep, 14:282-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Zhong G, Peng C, Chen Y, Li J, Yang R, Wu M, et al. (2018). Expression of STING and PD-L1 in colorectal cancer and their correlation with clinical prognosis. Int J Clin Exp Pathol, 11:1256-1264. [PMC free article] [PubMed] [Google Scholar]
- [103].Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. (2015). Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep, 11:1018-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Chon HJ, Kim H, Noh JH, Yang H, Lee WS, Kong SJ, et al. (2019). STING signaling is a potential immunotherapeutic target in colorectal cancer. J Cancer, 10:4932-4938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Zhu Q, Man SM, Gurung P, Liu Z, Vogel P, Lamkanfi M, et al. (2014). Cutting edge: STING mediates protection against colorectal tumorigenesis by governing the magnitude of intestinal inflammation. J Immunol, 193:4779-4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Wei B, Xu L, Guo W, Wang Y, Wu J, Li X, et al. (2021). SHP2-Mediated Inhibition of DNA Repair Contributes to cGAS-STING Activation and Chemotherapeutic Sensitivity in Colon Cancer. Cancer Res, 81:3215-3228. [DOI] [PubMed] [Google Scholar]
- [107].Reisländer T, Groelly FJ, Tarsounas M (2020). DNA Damage and Cancer Immunotherapy: A STING in the Tale. Mol Cell, 80:21-28. [DOI] [PubMed] [Google Scholar]
- [108].Rastrelli M, Tropea S, Rossi CR, Alaibac M (2014). Melanoma: epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo, 28:1005-1011. [PubMed] [Google Scholar]
- [109].MacKie RM, Hauschild A, Eggermont AM (2009). Epidemiology of invasive cutaneous melanoma. Ann Oncol, 20 Suppl 6:vi1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Markovic SN, Erickson LA, Rao RD, Weenig RH, Pockaj BA, Bardia A, et al. (2007). Malignant melanoma in the 21st century, part 1: epidemiology, risk factors, screening, prevention, and diagnosis. Mayo Clin Proc, 82:364-380. [DOI] [PubMed] [Google Scholar]
- [111].Takashima K, Takeda Y, Oshiumi H, Shime H, Okabe M, Ikawa M, et al. (2016). STING in tumor and host cells cooperatively work for NK cell-mediated tumor growth retardation. Biochem Biophys Res Commun, 478:1764-1771. [DOI] [PubMed] [Google Scholar]
- [112].Kim H, Kim H, Feng Y, Li Y, Tamiya H, Tocci S, et al. (2020). PRMT5 control of cGAS/STING and NLRC5 pathways defines melanoma response to antitumor immunity. Sci Transl Med, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Ma Z, Xiong Q, Xia H, Liu W, Dai S, Cai S, et al. (2021). Carboplatin activates the cGAS-STING pathway by upregulating the TREX-1 (three prime repair exonuclease 1) expression in human melanoma. Bioengineered, 12:6448-6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Siegel RL, Miller KD, Fuchs HE, Jemal A (2021). Cancer Statistics, 2021. CA Cancer J Clin, 71:7-33. [DOI] [PubMed] [Google Scholar]
- [115].Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, et al. (2017). Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J Natl Cancer Inst, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Liu LC, Shen YC, Wang YL, Wu WR, Chang LC, Chen YH, et al. (2022). Growth-promoting function of the cGAS-STING pathway in triple-negative breast cancer cells. Front Oncol, 12:851795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, et al. (2018). Chromosomal instability drives metastasis through a cytosolic DNA response. Nature, 553:467-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Li J, Duran MA, Dhanota N, Chatila WK, Bettigole SE, Kwon J, et al. (2021). Metastasis and Immune Evasion from Extracellular cGAMP Hydrolysis. Cancer Discov, 11:1212-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Cogan D, Bakhoum SF (2020). Re-awakening Innate Immune Signaling in Cancer: The Development of Highly Potent ENPP1 Inhibitors. Cell Chem Biol, 27:1327-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. (2016). Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature, 533:493-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Hong C, Schubert M, Tijhuis AE, Requesens M, Roorda M, van den Brink A, et al. (2022). cGAS-STING drives the IL-6-dependent survival of chromosomally instable cancers. Nature, 607:366-373. [DOI] [PubMed] [Google Scholar]
- [122].Hu Y, Manasrah BK, McGregor SM, Lera RF, Norman RX, Tucker JB, et al. (2021). Paclitaxel Induces Micronucleation and Activates Pro-Inflammatory cGAS-STING Signaling in Triple-Negative Breast Cancer. Mol Cancer Ther, 20:2553-2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. (2021). Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin, 71:209-249. [DOI] [PubMed] [Google Scholar]
- [124].Rebello RJ, Oing C, Knudsen KE, Loeb S, Johnson DC, Reiter RE, et al. (2021). Prostate cancer. Nat Rev Dis Primers, 7:9. [DOI] [PubMed] [Google Scholar]
- [125].Feng D, Shi X, You J, Xiong Q, Zhu W, Wei Q, et al. (2022). A cellular senescence-related gene prognostic index for biochemical recurrence and drug resistance in patients with prostate cancer. Am J Cancer Res, 12:3811-3828. [PMC free article] [PubMed] [Google Scholar]
- [126].Ho SS, Zhang WY, Tan NY, Khatoo M, Suter MA, Tripathi S, et al. (2016). The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity, 44:1177-1189. [DOI] [PubMed] [Google Scholar]
- [127].Suter MA, Tan NY, Thiam CH, Khatoo M, MacAry PA, Angeli V, et al. (2021). cGAS-STING cytosolic DNA sensing pathway is suppressed by JAK2-STAT3 in tumor cells. Sci Rep, 11:7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Radej S, Szewc M, Maciejewski R (2022). Prostate Infiltration by Treg and Th17 Cells as an Immune Response to Propionibacterium acnes Infection in the Course of Benign Prostatic Hyperplasia and Prostate Cancer. Int J Mol Sci, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Fischer K, Tschismarov R, Pilz A, Straubinger S, Carotta S, McDowell A, et al. (2020). Cutibacterium acnes Infection Induces Type I Interferon Synthesis Through the cGAS-STING Pathway. Front Immunol, 11:571334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Hsu SC, Chen CL, Cheng ML, Chu CY, Changou CA, Yu YL, et al. (2021). Arginine starvation elicits chromatin leakage and cGAS-STING activation via epigenetic silencing of metabolic and DNA-repair genes. Theranostics, 11:7527-7545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Ma Z, Zhang W, Dong B, Xin Z, Ji Y, Su R, et al. (2022). Docetaxel remodels prostate cancer immune microenvironment and enhances checkpoint inhibitor-based immunotherapy. Theranostics, 12:4965-4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Jackson SP, Bartek J (2009). The DNA-damage response in human biology and disease. Nature, 461:1071-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Li T, Chen ZJ (2018). The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med, 215:1287-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, et al. (2018). Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature, 563:131-136. [DOI] [PubMed] [Google Scholar]
- [135].Roehrborn CG (2008). Pathology of benign prostatic hyperplasia. Int J Impot Res, 20 Suppl 3: S11-18. [DOI] [PubMed] [Google Scholar]
- [136].Choi J, Shendrik I, Peacocke M, Peehl D, Buttyan R, Ikeguchi EF, et al. (2000). Expression of senescence-associated beta-galactosidase in enlarged prostates from men with benign prostatic hyperplasia. Urology, 56:160-166. [DOI] [PubMed] [Google Scholar]
- [137].He S, Sharpless NE (2017). Senescence in Health and Disease. Cell, 169:1000-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Loo TM, Miyata K, Tanaka Y, Takahashi A (2020). Cellular senescence and senescence-associated secretory phenotype via the cGAS-STING signaling pathway in cancer. Cancer Sci, 111:304-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Liu ZS, Cai H, Xue W, Wang M, Xia T, Li WJ, et al. (2019). G3BP1 promotes DNA binding and activation of cGAS. Nat Immunol, 20:18-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Dai J, Huang YJ, He X, Zhao M, Wang X, Liu ZS, et al. (2019). Acetylation Blocks cGAS Activity and Inhibits Self-DNA-Induced Autoimmunity. Cell, 176:1447-1460.e1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Vincent J, Adura C, Gao P, Luz A, Lama L, Asano Y, et al. (2017). Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun, 8:750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Lama L, Adura C, Xie W, Tomita D, Kamei T, Kuryavyi V, et al. (2019). Development of human cGAS-specific small-molecule inhibitors for repression of dsDNA-triggered interferon expression. Nat Commun, 10:2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Chu L, Li C, Li Y, Yu Q, Yu H, Li C, et al. (2021). Perillaldehyde Inhibition of cGAS Reduces dsDNA-Induced Interferon Response. Front Immunol, 12:655637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Tan J, Wu B, Chen T, Fan C, Zhao J, Xiong C, et al. (2021). Synthesis and Pharmacological Evaluation of Tetrahydro-γ-carboline Derivatives as Potent Anti-inflammatory Agents Targeting Cyclic GMP-AMP Synthase. J Med Chem, 64:7667-7690. [DOI] [PubMed] [Google Scholar]
- [145].Li Q, Cao Y, Dang C, Han B, Han R, Ma H, et al. (2020). Inhibition of double-strand DNA-sensing cGAS ameliorates brain injury after ischemic stroke. EMBO Mol Med, 12: e11002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Siu T, Altman MD, Baltus GA, Childers M, Ellis JM, Gunaydin H, et al. (2019). Discovery of a Novel cGAMP Competitive Ligand of the Inactive Form of STING. ACS Med Chem Lett, 10:92-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Li S, Hong Z, Wang Z, Li F, Mei J, Huang L, et al. (2018). The Cyclopeptide Astin C Specifically Inhibits the Innate Immune CDN Sensor STING. Cell Rep, 25:3405-3421.e3407. [DOI] [PubMed] [Google Scholar]
- [148].Hansen AL, Buchan GJ, Rühl M, Mukai K, Salvatore SR, Ogawa E, et al. (2018). Nitro-fatty acids are formed in response to virus infection and are potent inhibitors of STING palmitoylation and signaling. Proc Natl Acad Sci U S A, 115: E7768-e7775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, et al. (2018). Targeting STING with covalent small-molecule inhibitors. Nature, 559:269-273. [DOI] [PubMed] [Google Scholar]
- [150].Hong Z, Mei J, Li C, Bai G, Maimaiti M, Hu H, et al. (2021). STING inhibitors target the cyclic dinucleotide binding pocket. Proc Natl Acad Sci U S A, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, et al. (2013). Structure-function analysis of STING activation by c[G(2',5')pA(3',5')p] and targeting by antiviral DMXAA. Cell, 154:748-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Cavlar T, Deimling T, Ablasser A, Hopfner KP, Hornung V (2013). Species-specific detection of the antiviral small-molecule compound CMA by STING. Embo j, 32:1440-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, Curran MA (2017). Intratumoral STING Activation with T-cell Checkpoint Modulation Generates Systemic Antitumor Immunity. Cancer Immunol Res, 5:676-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Tang CH, Zundell JA, Ranatunga S, Lin C, Nefedova Y, Del Valle JR, et al. (2016). Agonist-Mediated Activation of STING Induces Apoptosis in Malignant B Cells. Cancer Res, 76:2137-2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang SY, et al. (2018). Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature, 564:439-443. [DOI] [PubMed] [Google Scholar]
- [156].Nakamura T, Sato T, Endo R, Sasaki S, Takahashi N, Sato Y, et al. (2021). STING agonist loaded lipid nanoparticles overcome anti-PD-1 resistance in melanoma lung metastasis via NK cell activation. J Immunother Cancer, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Pan BS, Perera SA, Piesvaux JA, Presland JP, Schroeder GK, Cumming JN, et al. (2020). An orally available non-nucleotide STING agonist with antitumor activity. Science, 369. [DOI] [PubMed] [Google Scholar]
- [158].Li S, Luo M, Wang Z, Feng Q, Wilhelm J, Wang X, et al. (2021). Prolonged activation of innate immune pathways by a polyvalent STING agonist. Nat Biomed Eng, 5:455-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].C hin EN, Yu C, Vartabedian VF, Jia Y, Kumar M, Gamo AM, et al. (2020). Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic. Science, 369:993-999. [DOI] [PubMed] [Google Scholar]
- [160].Wang Y, Luo J, Alu A, Han X, Wei Y, Wei X (2020). cGAS-STING pathway in cancer biotherapy. Mol Cancer, 19:136. [DOI] [PMC free article] [PubMed] [Google Scholar]