

Abstract
Neuronal synchronization at gamma frequency (30-100 Hz: γ) is impaired in early-stage Alzheimer's disease (AD) patients and AD models. Oligomeric Aβ1-42 caused a concentration-dependent reduction of γ-oscillation strength and regularity while increasing its frequency. The mTOR1 inhibitor rapamycin prevented the Aβ1-42-induced suppression of γ-oscillations, whereas the mTOR activator leucine mimicked the Aβ1-42-induced suppression. Activation of the downstream kinase S6K1, but not inhibition of eIF4E, was required for the Aβ1-42-induced suppression. The involvement of the mTOR/S6K1 signaling in the Aβ1-42-induced suppression was confirmed in Aβ-overexpressing APP/PS1 mice, where inhibiting mTOR or S6K1 restored degraded γ-oscillations. To assess the network changes that may underlie the mTOR/S6K1 mediated γ-oscillation impairment in AD, we tested the effect of Aβ1-42 on IPSCs and EPSCs recorded in pyramidal neurons. Aβ1-42 reduced EPSC amplitude and frequency and IPSC frequency, which could be prevented by inhibiting mTOR or S6K1. These experiments indicate that in early AD, oligomer Aβ1-42 impairs γ-oscillations by reducing inhibitory interneuron activity by activating the mTOR/S6K1 signaling pathway, which may contribute to early cognitive decline and provides new therapeutic targets.
Keywords: β-amyloid;, mTOR, S6K1, IPSC, gamma oscillation, APP/PS1
INTRODUCTION
Alzheimer's disease (AD) is a major threat to the well-being of the aging population. Extracellular β-amyloid (Aβ) precipitation and intracellular tau accumulation are hallmark pathologies, and hippocampus-associated spatial memory impairment is one of the early clinical symptoms in AD patients [1, 2]. Increased accumulation of aberrant Aβ has been attributed to suppressed autophagy [3]. However, in mild cognitive impairment (MCI) and early stages of AD, there is a pathological heterogeneity, and a significant proportion of non-demented aged subjects show typical AD pathologies [4, 5]. Therefore, the quest for the immediate cause of early cognitive decline in AD is imperative.
Synchronization of neuronal activity at frequencies in the gamma band (30-100 Hz: γ) provides a millisecond-precision timing matrix that facilitates inter-neuronal communication and determines the dynamic coupling of activity between brain areas [6, 7]. Hippocampal γ-oscillations of the local field potential measure the synchronization of neuronal activity, which correlates with indexes of working memory [8-10] and spatial memory [11]. γ-oscillations in the hippocampus have been associated with navigation [12] and hippocampus-based memory encoding/retrieval [13, 14].
Decreased γ-oscillations have been reported in AD animal models' prefrontal, lateral entorhinal, and olfactory cortex areas [9, 15, 16] and in AD patients [17]. A reduction in the global synchronization index in the γ band, a measure used to quantify synchronization between brain areas, correlates with cognitive decline in AD [18] and is already observed in MCI [19]. In transgenic AD models that produce high levels of Aβ, γ-oscillations are reduced at very early ages [16, 20-23]. Oligomeric Aβ causes a significant reduction in spontaneous network activity in the hippocampal area CA1 [24]. Fibrillar Aβ acutely degrades mouse hippocampal γ-oscillations in a concentration- and time-dependent manner [25, 26]. Interestingly, AD has been associated with neuronal hyper-excitability and epileptiform activity, primarily during reduced γ-oscillatory activity, which has been implicated in the progression of AD [27].
These studies suggest that impairments of synchronization at γ frequencies contribute to cognitive deficits in the early stages of AD [28, 29], and treatments that restore γ-oscillations could be a promising therapeutic target [23], but the molecular mechanisms underlying the Aβ-induced change of hippocampal γ-generating circuits remain elusive.
Gamma oscillations emerge from rhythmic inhibitory postsynaptic currents (IPSCs) that synchronize neuronal firing [30]. Because γ-oscillations are very energy-demanding [31], Aβ may reduce hippocampal γ-oscillations as the result of impairment of key metabolic processes involved in energy homeostasis. One option we pursued here is that Aβ affects energy homeostasis through changes in the mechanistic target of rapamycin (mTOR), a conserved Ser/Thr kinase that forms two multi-protein complexes known as mTOR complex 1 (mTOR1) and 2 (mTOR2)[32, 33], which weakens synaptic transmission [34] and mitochondrial function [35]. Evidence from AD in both patients and mouse models shows that hyper-activation of the mTOR pathway occurs at the early stages of AD [36, 37], and an increase in two mTOR downstream targets, p70 S6 kinase polypeptide 1 (S6K1) and eukaryotic translation initiation factor 4E (eIF4E)-binding proteins (4E-BPs), occurs already in MCI [38]. Reducing S6K1 expression improves spatial memory and synaptic plasticity in a mouse model of AD [39].
In the present study, we demonstrate that oligomeric Aβ causes impairment of hippocampal γ-oscillations through activating mTOR/S6K1 signaling.
MATERIALS AND METHODS
Animals
All animal experiments followed the "Principles of laboratory animal care" (NIH publication No. 86-23, revised 1985), as well as guidelines and regulations of the Ethics Committee of Xinxiang Medical College. C57BL/6J mice of either sex (3-4-week-old, unless specifically mentioned) were obtained from Beijing HFK Bioscience Co. The APP/PS1, double transgenic mice, were obtained from cross-breeding single transgenic mice expressing human APPK670N/M671L with single transgenic mice expressing human PS1M146L [40]. The mouse colonies were kept on a 12-hour light/dark cycle in temperature- and humidity-controlled rooms. Food and water were available ad libitum.
DNA samples were isolated from the tail tip of APP/PS1 transgenic mice at 3 weeks old for genotyping, using PCR with human APP primers and human PS1 oligo primers. For APP, the forward primer was 5’- GAC TGA CCA CTC GAC CAG GTT CTG -3’and the reverse primer was 5’- CTT GTA AGT TGG ATT CTC ATA TCC G -3’. For PS1, the forward primer was 5’-GAC AAC CAC CTG AGC AAT AC-3’, and the reverse primer was 5’-CAT CTT GCT CCA CCA CCT GCC-3’. APP/PS1 double transgenic mice displayed two target bands, while wild-type mice displayed no bands. APP/PS1 mice and wild-type mice were kept under standard conditions for up to 4-6 months in age.
Slice preparation
Hippocampal slices were cut as previously described [41]. Animals were anesthetized by intraperitoneal injection of chloral hydrate (400 mg/kg). Horizontal brain slices (350 μm) containing the ventral hippocampus were cut at 4-5 °C in a cutting solution saturated with carbogen (95% O2 and 5% CO2) using a Leica VT1000S vibratome (Leica Microsystems UK, Milton Keynes, UK). The cutting solution contained (in mM): 225 sucrose, 3 KCl, 1.25 NaH2PO4, 24 NaHCO3, 6 MgSO4, 0.5 CaCl2, and 10 glucoses (pH 7.4; 305 mOsm l-1). Immediately after slicing, sections were transferred and maintained in an interface chamber, continuously perfused with artificial cerebrospinal fluid (aCSF). The aCSF contained (in mM): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 24 NaHCO3, 2 MgSO4, 2 CaCl2 and 10 glucoses (pH 7.4; 305 mOsm l-1), saturated with carbogen. The slices were allowed to equilibrate at room temperature for at least 30 minutes before being moved to the recording chamber, where they were placed at the interface between aCSF (4-5 ml/min) at 32 °C and warm moist carbogen that maintained a thin film of aCSF covering the slice, to ensure applied substances could diffuse into the area recorded.
Extracellular field potential recordings
Extracellular field potentials were recorded from the stratum pyramidal of area CA3, with aCSF-filled glass pipette recording electrodes (3-5 MΩ) [42]. Field potentials were amplified with Neurolog NL106 AC-coupled amplifiers (Digitimer, Welwyn Garden City, UK) and band-pass filtered at 2-200 Hz with Neurolog NL125 filters (Digitimer). After the mains line noise was removed with Humbug noise eliminators (Digitimer), the signal was digitized and sampled at 2 kHz using a CED-1401 Plus (Cambridge Electronic Design, Cambridge, UK) and Spike-2 software (Cambridge Electronic Design).
Ten minutes after placing the slices in the recording chamber, kainate (100 nM) was added to the aCSF to induce γ-oscillations. Gamma oscillations were recorded from area CA3c, where oscillation amplitude is normally the largest and least affected by the faster intrinsic γ-oscillations in CA1[42].
Oscillation power was calculated from the power spectrum, generated by fast Fourier transforms over 60-s epochs (1Hz bin size, Hanning window, FFT size 2048). The summated power in the γ frequency range (set at 20-60 Hz for 32 °C): γ power, was used for quantification of the γ-oscillation strength. The peak frequency was determined as the local maximum of the power spectrum in the γ frequency range. Waveform auto-correlograms were calculated over 60-s band-pass (10-200 Hz) filtered epochs.
To ensure γ power was stable before drugs and/or Aβ peptide were added, the kainate-induced oscillatory activity was left to develop for at least 60 minutes. The average over the 5 minutes before the application, was taken as a baseline control. To quantify the effect of the drug or Aβ peptide on γ-oscillations, the average measure during the last 5 minutes of the application was normalized to the baseline control. In a separate set of experiments, the slices were pretreated with drugs for 10 minutes after 60 minutes in kainate, followed by 60 minutes of Aβ peptide addition. The average measure during the last 5 minutes of the Aβ peptide addition was normalized to the baseline control, to quantify the effect of the drug plus Aβ peptide on γ-oscillations. The power of γ-oscillations varies between slices and continues to grow gradually in kainate. To control for the effect of time, the changes induced by drugs or Aβ peptides were compared to the changes with time in a control group, where slices were treated with kainate only for 120 minutes, and the average measure over the last 5 minutes was normalized to the baseline measure.
Patch-clamp recordings
Whole-cell patch-clamp recordings were conducted in a submerged recording chamber perfused with aCSF (3-4 ml/minute at 30 °C). Pyramidal CA3 neurons were visually identified with a 40x water-immersion objective in an upright microscope (FN1, Nikon, Tokyo, Japan) and recorded using the whole-cell patch voltage-clamp technique, using glass pipettes (3-5 MΩ) as previously described [43]. Membrane currents were recorded with a Multiclamp 700B patch-clamp amplifier (Molecular Devices, Sunnyvale, USA), filtered at 2 kHz with a low-pass Bessel filter, and then digitized and sampled at 10 kHz with a Digidata 1440 (Molecular Devices).
All neurons included in this study had a resting membrane potential below -55 mV. The series resistance was tested before and after completion of each recording by measuring the current transient elicited by a 10-mV hyperpolarizing voltage step, and recordings were not analyzed for neurons with access resistance > 25 MΩ or if series resistance deviated >20% from the initial value.
For the recording of spontaneous excitatory postsynaptic currents (sEPSCs), electrodes were filled with (in mM): 100 Cs-methanesulfonate, 10 CsCl, 10 HEPES, 0.2 EGTA, 4 Mg-ATP, 0.3 Na-GTP and 10 Na-phosphocreatine (pH was set to 7.4 with CsOH and osmolarity was set to 290-310 mOsm l-1 with sucrose), and the membrane potential was clamped at -70 mV. 10 µM bicuculline was added to the aCSF to abolish GABAA-mediated inhibitory synaptic currents.
For the recording of spontaneous inhibitory postsynaptic currents (sIPSCs), electrodes were filled with a CsCl-based internal solution containing (in mM): 120 CsCl, 30 Hepes, 0.2 EGTA, 2 MgCl2, 1.0 CaCl2, 4.0 Mg-ATP and 5 QX-314 bromide (pH was set to 7.2 with CsOH and osmolarity was set to 290-310 mOsm l-1 with sucrose). D(-)-2-amino-5-phosphonopentanoic acid (D-AP5; 50 μM) and 2, 3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide disodium salt (NBQX; 10 μM) were added to the aCSF to block ionotropic glutamate receptor-mediated synaptic currents.
pClamp 10 software (Molecular Devices, Sunnyvale, CA, USA) was used to detect and measure synaptic events during 180 s epochs. EPSCs or IPSCs were detected on a running template (mean of ~20 events) with a well-defined baseline. Criteria for detecting spontaneous events were: exceeding a 3 pA threshold for longer than 2 ms, having a rise time shorter than 3 ms, and rising faster than the decay.
Aβ peptides or drugs were added to the incubation solution containing 100 nM kainate 30 minutes before the start of patch-clamp recordings. While incubating, slices were continuously supplied with aCSF saturated with carbogen.
Drugs
Amyloid β protein fragment 1-42 (Aβ1-42; Sigma-Aldrich, Louis, USA), rapamycin (Tocris Cookson, Bristol, UK), [2-((4-(5-ethylpyrimidin-4-yl)piperazine-1-yl) methyl)-5-(trifluoromethyl)-1H-benzo[d]imidazole] (PF4708671, Tocris Cookson), 2-[(4-(3,4-dichlorophenyl)-thiazol-2-ylhydrazono)-3-(2-nitrophenyl)]propionic acid (4EGI-1, Tocris Cookson) and NBQX (Tocris Cookson) were dissolved in dimethyl sulphoxide (DMSO). Kainate (Sigma-Aldrich), bicuculline methiodide (Tocris Cookson), and D-APV (Tocris Cookson) were dissolved in water. All drugs were stored in the freezer at 1,000-10,000 times the working solution, diluted immediately before use, and applied to the aCSF.
Aβ1-42 preparation
Aβ1-42 was prepared as described previously [44-46]. Initially, Aβ1-42 lyophilized powder was dissolved in 100% 1,1,1,3,3,3-Hexafluoro-2-Propanol (HFIP) at 1 mg/ml concentration and then incubated for 2 hours at room temperature. The Aβ1-42 solution was dried under a gentle stream of nitrogen gas and then dissolved in dimethyl sulphoxide (DMSO) under sonication for 10 minutes to obtain a stock concentration of 5 mM that was stored at -80 °C. For the Aβ1-42 monomer preparation, the Aβ1-42 stock solution was diluted in ACSF to the appropriate concentration for immediate application. For the Aβ1-42 oligomer preparation, 50 μM monomeric Aβ1-42 was incubated at 4 °C for up to 12 hours. After incubation, the Aβ1-42 solution was centrifuged at 15,000 g for 10 minutes at 4 °C, and the soluble Aβ1-42 in the supernatant was collected. For the fibrillar Aβ1-42 preparation, Aβ1-42 was diluted to 100 μM and incubated at 37 °C for 7 days. The Aβ1-42 solution was centrifuged at 5,000 g for 10 minutes at room temperature to remove the supernatant and collect the precipitation.
The biophysical and biological properties of Aβ1-42 were characterized by transmission electron microscopy (TEM) analysis [47-49]. Samples (5 μl) were diluted and deposited onto carbon-coated copper mesh grids for 5 minutes, and the liquid was absorbed with paper and negatively stained with 2% (w/v) uranyl acetate. Then, the sample grids were allowed to air dry. The samples were viewed with a JEOL JEM-1400 microscope (JEOL Ltd, Japan), and digital images were acquired with an Advanced Microscopy Techniques camera. Aβ1-42 oligomers appeared as fibril-free small globular structures that were <10 nm in diameter, and fibrillar Aβ1-42 showed long threads measuring >1 µm in length with some aggregated Aβs (Fig. 1). The morphological characteristics of these fibrils were identical to those of Aβ fibrillar structures, as has been reported [49, 50].
Figure 1.

The TEM images for monomers, oligomers, and fibrils of Aβ1-42. (A) Example of a TEM image of Aβ1-42 monomer solution. (B) Example of a TEM image of soluble Aβ1-42 oligomers taken from the supernatant after incubation of Aβ1-42 monomer solution at 4 °C for 12 h. (C) Example of a TEM image of Aβ1-42 fibrils taken from the precipitate after incubating Aβ1-42 monomer solution at 37 °C for 7 days.
Western blotting
Western blotting was performed by the methods established in our laboratory [43]. Briefly, for the preparation of total cell extracts, the dissected hippocampal area CA3 tissue was homogenized in lysis RIPA buffer containing 1% sodium dodecyl sulfate buffer in Tris-EDTA (pH7.4), 1× protease inhibitor cocktail (P8340, Sigma-Aldrich), 5mM NaF, and 1× phosphatase inhibitor cocktail (P2580, Sigma-Aldrich). The homogenate was centrifuged at 5,000 g for 10 minutes, the supernatant was collected, and total protein concentrations were measured using Bradford Assays. Proteins were electrophoretically separated in 12% SDS-PAGE gels and were transferred to a polyvinylidene fluoride membrane. The proteins were analyzed by Western blotting using antibodies against mTOR (1:500; Cell Signaling, 2972), phospho-mTOR (Ser 2448) (1:500; Cell Signaling, 2971), β-actin (1: 1,000; Abcam, ab6272). Membranes were then incubated with a horseradish peroxidenzyme-conjugatedated secondary antibody (1:2,000; Abcam, ab288151) to reveal the location of the protein bands. Immunoreactive bands were visualized by chemiluminescence using enhanced ECL reagents (BeyoECL Plus, Beyotime). Subsequently, X-ray films were exposed to the membranes and then quantitatively analyzed by Image J software, which measures the intensity of protein bands after background subtraction. Relative protein level was calculated by normalizing phosphorylated form levels and total protein levels to the β-actin levels, respectively.
Statistics
Data are expressed as mean ± standard error of the mean or medians ± min-max for non-normally distributed data. The normality of the distribution of data was assessed by the Shapiro-Wilk test. N indicates the number of hippocampal slices tested or cells recorded. Statistical comparisons between experimental conditions were made using the unpaired Student’s t-test or one-way ANOVA with Tukey’s post hoc tests for normally distributed data or the Mann-Whitney U test for non-normally distributed data. Significance was assumed when P < 0.05.
RESULTS
Oligomeric Aβ1-42 impairs γ-oscillations in hippocampal slices
We first tested the effect of Aβ1-42 oligomer on kainate-induced γ-oscillations in mouse hippocampal slices. Gamma oscillations were induced by kainate (100 nM) for 60 minutes, after which slices were perfused for 60 minutes with different concentrations (50 nM-1 μM) of Aβ1-42 oligomer. At baseline, γ power was 1925 ± 331 µV2, and the peak frequency was 33.0 ± 0.4 Hz (n= 8 slices from 8 mice). Measurements of γ power were taken at the last 5 minutes before and after 60 minutes of application of Aβ1-42 oligomer. Aβ1-42 Oligomer decreased γ power by 45% of baseline control (t16= 6.949, P< 0.001, n=10 slices from 10 mice) and increased the peak frequency by 10% (t16= 4.302, P= 0.001, example in Fig. 2A and B).
Figure 2.

The effect of Aβ1-42 on γ-oscillations. (A) Example of oscillatory activity recorded in stratum pyramidale of CA3 before (brown trace) and 60 minutes after perfusion with 500 nM oligomeric Aβ1-42 (yellow trace). (B) Power spectra were taken from the recordings in (A), five minutes before application (baseline control: brown line) and 55-60 minutes after oligomeric Aβ1-42 application (yellow line). (C) Example of the power spectrum before (brown) and after 500 nM monomeric Aβ1-42 application (red line). (D) Example of the power spectrum before (brown) and after 500 nM fibrillar Aβ1-42 application (orange line). (E) γ-oscillation power as % of baseline control (100 nM kainate for 120 minutes), 500 nM monomeric Aβ1-42, 500 nM oligomeric Aβ1-42 and 500 nM fibrillar Aβ1-42. n= 8 for time-only control, n= 9 for monomeric Aβ1-42, n= 10 for oligomeric Aβ1-42, n= 8 for fibrillar Aβ1-42. Significant differences with time-only control are indicated with **, P< 0.01, ***, P< 0.001. Differences between preparations are indicated with ###, P< 0.001 analyzed by unpaired t-test. (F) γ-oscillation peak frequency for the same groups, Details as in (E). (G) γ-oscillation power as % of baseline control for increasing concentrations of oligomeric Aβ1-42 and control. n=12 for time-only control, n=10 for 50 nM oligomeric Aβ1-42, n=10 for 250 nM oligomeric Aβ1-42, n=10 for 500 nM oligomeric Aβ1-42, n= 9 for 1μM oligomeric Aβ1-42. Data are presented as mean ± SEM. ***, P< 0.001, compared with time-only control, one-way ANOVA with Tukey’s post hoc test. (H) γ-oscillation peak frequency for control and increasing concentrations of oligomeric Aβ1-42. Details as in (G). (I) Auto-correlogram of the recording in A shows a 500 nM oligomeric Aβ1-42-induced reduction in oscillation regularity.
We next tested the effects of monomeric Aβ1-42 and fibrillar Aβ1-42 on kainate-induced γ-oscillations. γ power at baseline was 2325 ± 312 µV2, and the peak frequency was 32.8 ± 0.3 Hz (n= 8 slices from 8 mice). Compared with the baseline control, monomeric Aβ1-42 (500 nM) decreased γ power by 10% (t15= 2.021, P=0.061, t-test, n= 9 slices from 9 mice, Fig. 2C and E), which was significantly less than the effect of 500 nM Aβ1-42 oligomer (t16= 6.949, P< 0.001, n= 10 slices from 10 mice). Monomeric Aβ1-42 increased the peak frequency by only 4% (t15= 2.112, P= 0.052, t-test, Fig. 2C and F). Compared with the baseline control, fibrillar Aβ1-42 (500 nM) decreased γ power by 50% (t14= 6.134, P< 0.001, t-test, n= 8 slices from 8 mice, Fig. 2D and E), which was not different from the effect of 500 nM Aβ1-42 oligomer (t16= 0.508, P= 0.619). Fibrillar Aβ1-42 increased the peak frequency by 13% (t14= 6.454, P< 0.001, t-test, Fig. 2D and F). These results suggest that the oligomeric and fibrillar forms of Aβ1-42 are more potent in suppressing and accelerating γ-oscillations than monomeric Aβ1-42, as reported previously [25]. However, since it is unlikely that large fibrils can diffuse into the tissue through the extracellular space [51], the effect of the fibrillar Aβ1-42 is likely due to the formation of oligomers from fibrils [52].
To quantify the effect of different concentrations of oligomeric Aβ1-42 on γ-oscillations, comparisons were made with the effect of a time-only control: the change in γ power after application of 100 nM kainate for 120 minutes (110% of baseline). Compared with the time-only control (n= 12 slices from 12 mice), oligomeric Aβ1-42 decreased γ power by 25% at 50 nM (P< 0.001, n(Aβ)= 10 slices from 10 mice), 35% at 250 nM (P< 0.001, n= 10 slices from 10 mice), 47% at 500 nM (P< 0.001, n= 10 slices from 10 mice) and 53% at 1 μM (F4,50= 38.77, P< 0.001, one-way ANOVA with Tukey’s post hoc test, n= 9 slices from 9 mice, Fig. 2G). Oligomeric Aβ1-42 increased the γ-oscillation peak frequency by 5% (50 nM, P= 0.269), 10% (250 nM, P< 0.001), 10% (500 nM, P< 0.001) and 14% (1μM, F4,50= 11.34, P< 0.001, one-way ANOVA with Tukey’s post hoc test, Fig. 2H). γ oscillations were very regular at baseline, reflected by a peak of 0.48 ± 0.03 at 30 ± 1 ms in the autocorrelogram. After applying 500 nM oligomeric Aβ1-42, the autocorrelation peak (at 28 ± 1 ms) was reduced to 0.33 ± 0.03 (t20= 3.38, P= 0.003, n= 21 slices from 21 mice, Fig. 2I). The reduction in autocorrelation indicates that in addition to the reduced oscillation strength, the γ cycle length was less regular after oligomeric Aβ1-42 exposure.
Since 500 nM oligomeric Aβ1-42 is in the range of pathological concentrations observed in brain tissue of AD patients [53], and was not significantly different from that of 1 µM (Turkey test P= 0.841), this concentration was used for the subsequent investigations of the cellular and network mechanisms underlying the acute oligomer Aβ1-42-induced impairment of γ-oscillations. The term Aβ1-42 is used for 500 nM oligomer Aβ1-42 in the remainder of this paper.
To study whether the Aβ1-42-induced reduction of γ-oscillations is specific to kainate-induced γ-oscillations, we tested the effect of Aβ1-42 on a different in vitro model of γ-oscillations. Using the same approach as for kainate-induced γ-oscillations, Aβ1-42 reduced γ power of activity induced by carbachol (10 µM) to 47% of control power (the control power was 105% of baseline, t8= 6.80, P< 0.001, data not shown). Aβ1-42 increased the peak frequency by 7% (t8= 3.80, P= 0.005, data not shown). These data suggest that oligomeric Aβ1-42 impairs γ-oscillations in hippocampal CA3 irrespective of the in vitro model.
Activation of mTOR/S6K1 signaling mediates Aβ1-42-induced impairment of γ-oscillations
Involvement of the mechanistic target of rapamycin (mTOR) activation in AD has been shown previously [36, 54]. To explore the potential role of mTOR in modulating γ-oscillations, we tested the effect of inhibition and activation of mTOR on γ-oscillations.
Slices were pretreated with mTORC1 inhibitor rapamycin (100 nM for 10 min) and then perfused with Aβ1-42 and rapamycin for 60 minutes. Compared with the reduction of γ power induced by Aβ1-42 only (by 47% of the control, n= 10 slices from 10 mice Aβ1-42 only), pretreatment with rapamycin prevented the effect of Aβ1-42 on γ power (increased by 12% compared with baseline control, F3,42= 37.01, P= 0.369, one-way ANOVA with Tukey’s post hoc test, n= 10 slices from 10 mice, Fig. 3A-C). Rapamycin also prevented the increase in peak frequency of γ-oscillations (increased by 4% compared with the baseline control, F3,42= 13.56, P= 0.217, Fig. 3D). To control the effects of rapamycin itself, we tested the effect of rapamycin only for 60 minutes. Rapamycin-only treatment had no significant impact on the γ power (increased by 4% compared with the control, F3,42= 37.01, P= 0.158, one-way ANOVA with Tukey’s post hoc test, Fig. 3A-C) or on the peak frequency (decreased by 3% compared with the baseline control, F3,42= 13.56, P= 0.742, Fig. 3D).
Figure 3.
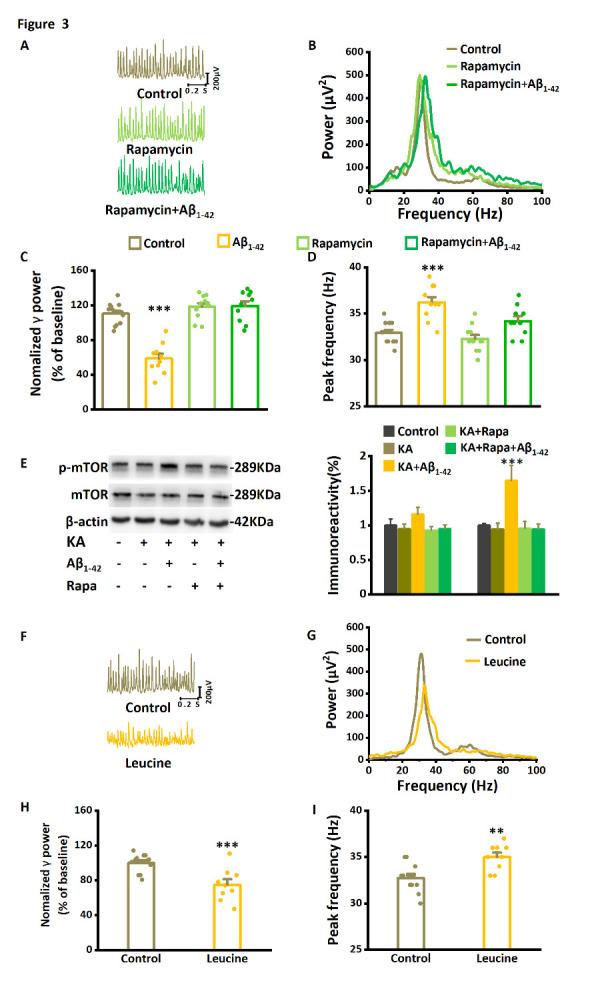
The mTOR inhibitor rapamycin prevents Aβ1-42-induced effects on γ-oscillations and suppresses Aβ1-42-induced mTOR hyperactivation. (A) Example traces of γ-oscillations for baseline control, after application of rapamycin-only, and after rapamycin plus Aβ1-42 (dark green trace). (B) Example of power spectra of γ-oscillations with the application of Aβ1-42 and rapamycin plus Aβ1-42. (C) γ-oscillation power as % of baseline control, in the presence of Aβ1-42, rapamycin only, or rapamycin with Aβ1-42. n= 12 for time-only control, n= 10 for Aβ1-42, n= 11for rapamycin only, n= 10 for rapamycin with Aβ1-42. (D) The effect of rapamycin and Aβ1-42 on the γ-oscillation peak frequency. Details as in (C). (E) Example of Western blot quantification of mTOR and the serine 2448 phosphorylated mTOR (p-mTOR). n= 9 mice for each group. (F) Example traces of γ-oscillations for baseline control and after application of leucine. (G) Example of power spectra of γ-oscillations for baseline control and after application of leucine. (H) γ-oscillation power as % of baseline control, in the presence of leucine. n= 12 for time-only control and n= 9 for leucine. The effect of leucine on the γ-oscillation peak frequency. Details as in (G). Data are presented as mean ± SEM. *, P< 0.05, **, P< 0.01, ***, P< 0.001, compared with time-only control, analyzed by one-way ANOVA with Tukey’s post hoc test in C, D, H and I. *, P< 0.05, compared with time-only control, analyzed by unpaired t test in E.
To examine whether mTOR was activated by Aβ1-42, CA3 tissue was dissected for Western blotting after electrophysiological recordings. As shown in Figure 3E, total mTOR levels were not different between control and Aβ1-42 treatment (F4,14= 1.086, P= 0.414, one-way ANOVA with Tukey’s post hoc test, each sample collected from 3 mini slices of hippocampal CA3 region of 3 mice, n= 3 independent experiments from 9 mice). However, Aβ1-42 treatment increased serine-2448 phosphorylation of mTOR by 65% (F4,14= 6.74, P= 0.021). Interestingly, the pretreatment with rapamycin prevented the effect of Aβ1-42 on serine-2448 phosphorylation of mTOR (F4,14= 6.74, P= 0.013). The data suggest that Aβ1-42 activates mTOR without affecting total mTOR expression and that rapamycin prevents Aβ1-42-induced activation.
Because mTOR inhibition prevented Aβ1-42-induced impairment of γ-oscillations, we predicted that increasing mTOR activity would mimic the effect of Aβ1-42 on γ-oscillations. Indeed, the mTOR activator leucine (3 μM, application for 60 min) decreased γ power and increased the peak frequency (Fig. 3F, G). Leucine decreased γ power by 25% (75.0±6.3% of the baseline control, compared with the baseline control, t19= 3.868, P= 0.001, n= 12 slices from 12 mice for control, n= 9 slices from 9 mice for leucine, Fig. 3H). It increased the peak frequency by 7% (35.0±0.5 Hz, compared with 32.8±0.4 Hz in the baseline control, t19= 3.506, P= 0.002, Fig. 3I). These data suggest that rapamycin-sensitive mTOR activity is both necessary and sufficient for oligomer Aβ1-42-induced impairment of γ-oscillations.
Ribosomal protein S6 kinase beta-1(S6K1), a protein kinase downstream of mTORC1, is activated in AD [55] and MCI [38]. Pretreatment of the slices with the S6K1 inhibitor PF4708671 (20 μM for 10 min), followed by perfusion with Aβ1-42 and PF4708671 for 60 minutes, did not affect γ power (increased by 5%, compared with the baseline control, F3,39= 34.39, P= 0.935, one-way ANOVA with Tukey’s post hoc test, n= 9 slices from 9 mice for PF4708671+ Aβ1-42, Fig. 4A, B, D), or the peak frequency of γ-oscillations (increased by 1%, compared with the baseline control, F3,39= 25.95, P= 0.992, Fig. 4E). To control the effects of PF4708671 itself, we tested the impact of PF4708671 only for 60 minutes. The PF4708671-only treatment did not affect γ power (increased by 3% compared with the control, F3,39= 34.39, P= 0.836, one-way ANOVA with Tukey’s post hoc test, n= 9 slices from 9 mice for PF4708671, Fig. 4A, B, D) or the peak frequency (decreased by 5% compared with the control, F3,39= 25.95, P= 0.105, Fig. 4E). These data suggest that Aβ1-42 suppresses γ-oscillations by activating the mTOR/S6K1 pathway.
Figure 4.
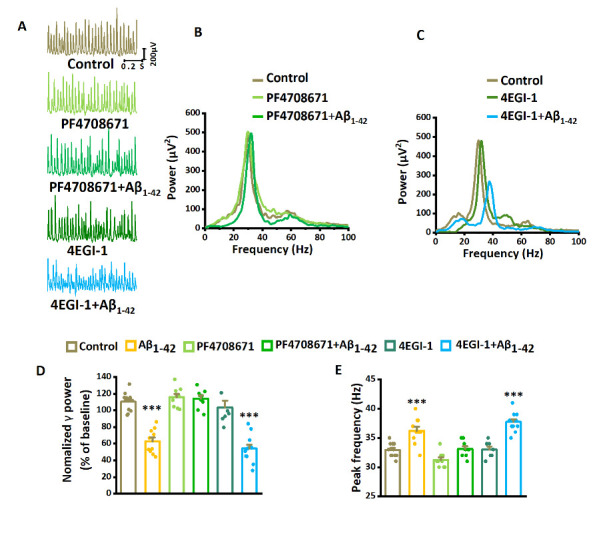
The mTOR-mediated suppression of γ-oscillations requires S6K1 activation rather than eFI4E-eIF4G interactions. (A) Example traces of γ-oscillations without and with Aβ1-42 exposure during PF4708671 application and 4EGI-1 application. (B) Example of power spectra of γ-oscillations without and with Aβ1-42 exposure during PF4708671 application. (C) Example of power spectra of γ-oscillations without and with Aβ1-42 exposure during 4EGI-1 application. (D) γ-oscillation power as % of baseline control, in the presence of Aβ1-42, PF4708671 only, PF4708671 with Aβ1-42, or 4EGI-1 only, 4EGI-1 with Aβ1-42. n= 12 for control, n= 10 for Aβ1-42, n= 9 for PF4708671 only, n= 9 for PF4708671 with Aβ1-42, n= 7 for 4EGI-1 only, n= 12 for 4EGI-1 with Aβ1-42. (E) Effect of PF4708671, 4EGI-1, and Aβ1-42 on γ-oscillation peak frequency. Details as in (D). Data are presented as mean ± SEM. ***, P< 0.001, compared with control, one-way ANOVA with Tukey’s post hoc test.
Alternatively, mTORC1 activation could affect γ-oscillations through inhibition of eIF4E, which is elevated in AD [56] and MCI [38]. Activation of mTORC1 triggers the initiation of cap-dependent mRNA translation via phosphorylation of 4E-BPs [57]. The mTOR-dependent phosphorylation of 4E-BPs causes the release of eIF4E and its subsequent binding to eIF4G and permits the formation of the eIF4F initiation complex and subsequent cap-dependent protein synthesis [58]. To determine whether eIF4E-eIF4G interactions are required for the suppression of γ-oscillations by Aβ1-42, we tested the effect of the eIF4E-eIF4G interaction inhibitor 4EGI-1. 4EGI-1 (50 μM, application for 60 min) had no significant effect on γ power (decreased by 6%, compared with the baseline control, F3, 40= 29.70, P= 0.835, n= 12 slices from 12 mice for control, n= 7 slices from 7 mice for 4EGI-1, Fig. 4A, C, D) or the peak frequency (increased by 1%, compared with the baseline control, F3, 40= 22.31, P= 0.991, Fig. 4E). Pretreatment of the slices with 4EGI-1 (50 μM for 10 min), followed by perfusion with 4EGI-1 and Aβ1-42 for 60 minutes, decreased γ power (reduced by 51%, compared with the baseline control, F3, 40= 29.70, P< 0.001, n= 12 slices from 12 mice for 4EGI-1+ Aβ1-42, Fig. 4A, C, D), and increased the peak frequency of γ-oscillations (increased by 15%, compared with the baseline control, F3, 40= 22.31, P< 0.001, Fig. 4E). The effect of Aβ1-42 on γ power in the presence of 4EGI-1 was not different from that of Aβ1-42 alone (P= 0.929). The lack of impact of eIF4E inhibition confirms the importance of the mTOR/S6K1 pathway.
Oligomeric Aβ1-42 affects EPSC and IPSC properties through the mTOR/S6K1 pathway
γ oscillations emerge from the rhythmic, mutual interactions of pyramidal neurons and interneurons in the local network [30], and extracellularly recorded γ-oscillations reflect mainly the amplitude and synchronicity of rhythmic IPSCs. To assess whether the Aβ1-42-induced impairment of γ-oscillations can result from the changes in synaptic properties and/or synchronicity, we tested the effect of Aβ1-42 on EPSCs and IPSCs, recorded in CA3 pyramidal neurons in a kainate-activated neuronal network (Fig. 5).
Figure 5.

The effect of Aβ1-42 on spontaneous IPSCs and EPSCs. (A) Examples of IPSCs recorded in kainate aCSF only (control) after addition of Aβ1-42 exposure, in the presence of rapamycin or PF4708671, and after leucine application. (B) Waveform averages of the recordings in (A). (C) sIPSC amplitude of control (n= 14 from 5 mice), in the presence of Aβ1-42 (n= 20 from 7 mice), rapamycin only (n= 13 from 7 mice), rapamycin with Aβ1-42 (n= 14 from 5 mice), or PF4708671 only (n= 14 from 6 mice), PF4708671 with Aβ1-42 (n= 13 from 5 mice), or leucine only (n= 11 from 5 mice). Data are presented as mean ± SEM. *, P< 0.05, compared with control, unpaired Student’s t-tests. (D) sIPSC frequency for the same conditions and with same details as in (C). ***, P< 0.001, compared with control. (E) sIPSC decay time for the same conditions and with same details as in (C). **, P< 0.01, compared with control. (F) Examples of EPSCs recorded in kainate aCSF only (control) after addition of Aβ1-42 exposure, in the presence of rapamycin or PF4708671, and after leucine application. (G) Waveform averages of the recordings in (F). (H) sEPSC amplitude of control (n= 12 from 5 mice), in the presence of Aβ1-42 (n= 14 from 6 mice), rapamycin only (n= 14 from 5 mice), rapamycin with Aβ1-42 (n= 13 from 6 mice), or PF4708671 only (n= 12 from 5 mice), PF4708671 with Aβ1-42(n= 14 from 6 mice), or leucine only (n= 14 from 7 mice). Data are presented as mean ± SEM. *, P< 0.05; **, P< 0.01, compared with control, unpaired Student’s t-tests. (I) sEPSC frequency for the same conditions and with same details as in (H). ***, P< 0.001, compared with control. (J) sEPSC decay time for the same conditions and with same details as in (H).
Hippocampal slices were treated with kainate (100 nM) or kainate plus Aβ1-42 for at least 30 minutes before pyramidal cells were recorded in whole-cell patch voltage-clamp mode. In submerged slices, kainate caused increased activity, but did not generate γ-oscillations. Spontaneous IPSCs were pharmacologically isolated and were inward at -70 mV (Fig. 5A). Aβ1-42 reduced the IPSC frequency by 45% (compared with a control frequency of 11.8±1.1 Hz, t32= 3.62, P= 0.001, n= 14 neurons from 5 mice for control group, n= 20 neurons from 7 mice for Aβ1-42 only, Fig. 5D, Table 1) but did not affect the IPSC amplitude (the control amplitude was 34.7±2.7 pA, Fig. 5C, Table 1) or the IPSC decay time (5.9±0.4 ms in control, Fig. 5E, Table 1). Aβ1-42 reduced the EPSC amplitude by 31% (compared with a controll amplitude of 18.5±1.4 pA, t24= 2.92, P= 0.007, n= 12 neurons from 5 mice for control group, n= 14 neurons from 6 mice for Aβ1-42 only, Fig. 5F and H, Table 2), and the EPSC frequency by 40% (the control frequency was 9.5±0.9 Hz, t24= 3.53, P= 0.002, Fig. 5F and I, Table 2), but did not affect the EPSC decay time (8.1±0.6 ms in control, Fig. 5G and J, Table 2).
Table 1.
Effect of mTOR/S6K1 on spontaneous IPSC in hippocampal CA3.
| Drug | N | Rm (GΩ) |
Amplitude (pA) |
P(t-test with control) |
Frequency (Hz) |
P(t-test with control) |
Decay time (ms) |
P(t-test with control) |
|---|---|---|---|---|---|---|---|---|
| Control | 14(5) | 193±17 | 34.7±2.7 | – | 11.8±1.1 | – | 5.9±0.4 | – |
| Aβ1-42 | 20(7) | 201±15 | 30.5±2.9 | 0.32 | 6.6±0.9 | 0.001*** | 6.5±0.6 | 0.408 |
| leucine | 11(5) | 194±16 | 25.8±2.4 | 0.025* | 6.3±0.7 | <0.001*** | 8.2±0.7 | 0.007** |
| rapamycin | 13(5) | 198±15 | 33.0±3.0 | 0.678 | 12.4±1.4 | 0.756 | 5.7±0.4 | 0.747 |
| rapamycin + Aβ1-42 | 14(5) | 196±18 | 30.5±2.8 | 0.292 | 11.9±1.9 | 0.946 | 5.2±0.4 | 0.244 |
| PF4708671 | 14(6) | 208±19 | 37.9±3.6 | 0.49 | 13.7±1.2 | 0.4 | 6.3±0.6 | 0.552 |
| PF4708671 + Aβ1-42 | 13(5) | 207±16 | 31.4±1.3 | 0.288 | 12.2±1.7 | 0.85 | 6.1±0.1 | 0.684 |
Table 2.
Effect of mTOR/S6K1 on spontaneous EPSC in hippocampal CA3.
| Drug | N | Rm (GΩ) |
Amplitude (pA) |
P(t-test with control) |
Frequency (Hz) |
P(t-test with control) |
Decay time (ms) |
P(t-test with control) |
|---|---|---|---|---|---|---|---|---|
| Control | 12(5) | 187±10 | 18.5±1.4 | – | 9.5±0.9 | – | 8.2±0.6 | – |
| Aβ1-42 | 14(6) | 218±20 | 12.8±1.3 | 0.007** | 5.7±0.7 | 0.002** | 8.8±0.4 | 0.38 |
| leucine | 14(7) | 193±10 | 14.8±0.9 | 0.037* | 5.3±0.5 | <0.001*** | 7.9±0.4 | 0.741 |
| rapamycin | 14(5) | 215±24 | 19.9±1.1 | 0.422 | 10.9±1.0 | 0.257 | 6.5±0.3 | 0.04* |
| rapamycin + Aβ1-42 | 13(6) | 201±17 | 21.4±1.7 | 0.471 | 10.4±1.2 | 0.777 | 6.3±0.5 | 0.013* |
| PF4708671 | 12(5) | 215±17 | 20.1±1.2 | 0.457 | 11.0±0.9 | 0.321 | 6.6±0.3 | 0.021* |
| PF4708671 + Aβ1-42 | 14(6) | 209±8 | 19.8±1.0 | 0.225 | 9.8±0.6 | 0.569 | 6.4±0.2 | 0.035* |
Because the Aβ1-42-induced impairment of γ-oscillations was involved in mTOR activation, we predicted that rapamycin would prevent the Aβ1-42-induced changes in IPSC and EPSC properties. Hippocampal slices were pretreated with 100 nM rapamycin or rapamycin plus Aβ1-42 in the present with 100 nM kainate for at least 30 minutes. Rapamycin alone did not affect IPSC or EPSC properties (Fig. 5, Table 1, 2). After pretreatment with rapamycin for 10 min, adding Aβ1-42 had no effect on the amplitude or frequency of IPSCs and EPSCs (Fig. 5, Table 1, 2), but decreased the decay time of EPSCs by 20% (t24= 2.69, P= 0.013, Fig. 5, Table 1, 2).
Because leucine could mimic the effect of Aβ1-42 on γ-oscillations, we predicted that leucine would reduce both IPSC and EPSC frequency and EPSC amplitude. Using the same approach as with the application of Aβ1-42, leucine (3 μM) was applied with 100 nM kainate to slices for 30 minutes before the whole-cell recordings were started. Compared with the control, leucine decreased the IPSC frequency by 47% (t23= 3.96, P< 0.001, n=11 neurons from 5 mice, Fig. 5A and D, Table 1) and increased the IPSC decay time by 40% (t23= 2.99, P= 0.007, Fig. 5B and E, Table 1). The IPSCs amplitude was reduced by 26% (t23= 2.40, P= 0.025, Fig. 5A and C). Leucine decreased the EPSC frequency by 45% (t24= 4.36, P< 0.001, n=14 neurons from 7 mice, Fig. 5F and I, Table 2) and reduced the EPSC amplitude by 20% (t24= 2.20, P= 0.037, Fig. 5F and H). Leucine did not affect the EPSC decay time (Fig. 5G and J, Table 2).
Since PF4708671 could prevent the effect of Aβ1-42 on γ-oscillations, we tested whether S6K1 activation was involved in the effect of Aβ1-42 on IPSC and EPSC properties. As described for rapamycin, hippocampal slices were pretreated with PF4708671 (20 μM). PF4708671 alone had no significant effect on the amplitude or frequency of IPSCs and EPSCs (Fig. 5, Tables 1 and 2) but slightly reduced the EPSC decay time (by 20%, t24= 2.46, P= 0.021, n=12 neurons from 5 mice, Fig. 5, Table 2). In the presence of PF4708671, Aβ1-42 did not affect IPSC amplitude or the frequency of IPSCs or EPSCs (Tables 1 and 2), but slightly reduced the EPSC decay time (by 22%, t23= 2.24, P= 0.035, n=14 neurons from 6 mice, Table 2). These data suggest that the Aβ1-42-induced changes to IPSC and EPSC properties are mediated through mTOR/S6K1 activation.
Degeneration of hippocampal γ-oscillations in APP/PS1 mice and rescue by inhibiting mTOR/S6K1 signaling
Gamma oscillations are markedly reduced in patients diagnosed with AD [59]. To test whether γ-oscillations are affected in an AD animal model that over-expresses Aβ, we compared γ-oscillations induced by 100 nM kainate in slices from APP/PS1 mice (4-6-month-old) with those from age-matched wild-type mice (Fig. 6A, B). The γ power of the oscillations recorded in slices from APP/PS1 mice was 64% lower than that recorded in slices from wildtype mice (Mann-Whitney U test P= 0.008, n= 18 slices from 18 mice for WT, n= 15 slices from 15 mice for APP/PS1 mice, Fig. 6A-C) and the peak frequency was higher (by 3%, Mann-Whitney U test P< 0.05, Fig. 6D). Whereas the γ-oscillation in slices of wildtype mice was very regular, reflected by peaks of 0.42±0.05 at 32±1 ms in the auto-correlogram, the autocorrelation peak of APP/PS1 mice was smaller (0.29±0.03, t31= 2.141, P= 0.040) at 29±1 ms (example in Fig. 6E), indicating that the oscillation was less regular.
Because inhibition of the mTOR/S6K1 pathway could prevent the Aβ1-42-induced reduction of γ-oscillations, we predicted that inhibition of the mTOR/S6K1 pathway would restore γ-oscillations in APP/PS1 mice. To test this, 200 nM rapamycin or 20 μM PF4708671 were applied after the kainate-induced γ-oscillations were established for 60 minutes (Fig. 7A). Compared with the APP/PS1 control, the application of rapamycin increased γ power (example in Fig. 7A and B) by 25% (the control γ power was 105% of baseline, P< 0.05, Mann-Whitney U test, n= 15 slices from 15 mice for control, n= 14 slices from 14 mice for rapamycin, Fig. 7C) and reduced the peak frequency by 3% (P< 0.05, Mann-Whitney U test, Fig. 7D). Compared with the APP/PS1 control, the application of PF4708671 increased the γ power (example in Fig. 7A and B) by 19% (P< 0.05, Mann-Whitney U test, n=15 slices from 15 mice for PF4708671, Fig. 7C) and reduced the peak frequency by 3% (P< 0.05, Mann-Whitney U test, Fig. 7D). To determine the role of eIF4E-eIF4G interactions in the suppression of γ-oscillations in APP/PS1 mice, we also tested the effect of 4EGI-1 on γ-oscillations. 4EGI-1 (50 μM, application for 60 min) had no significant effect on γ power (increased by 1% compared with the control, P< 0.05, Mann-Whitney U test, n= 14 slices from 14 mice, Fig. 7A, B and C) or the peak frequency (increased by 6% compared with the control, P< 0.05, Mann-Whitney U test, Fig. 7D). This suggests that the degradation of γ oscillations in APP/PS1 mice is partly caused by an over-activity of the mTOR/S6K1 pathway.
Figure 7.

The suppression of γ-oscillations in APP/PS1 mice depends on mTOR/S6K1 pathway hyper-activation. Example of traces. (A) and power spectra (B) of γ-oscillations recorded in slices from APP/PS1 mouse without and with the application of rapamycin, PF4708671, or 4EGI-1. (C) γ-oscillation power in APP/PS1 mice as % of baseline control, rapamycin, PF4708671 or 4EGI-1. n=15 for control, n=14 for rapamycin, n=15 for PF4708671 and n=14 for 4EGI-1. (D) γ-oscillation peak frequency in APP/PS1 mice in control and after rapamycin, PF4708671, or 4EGI-1. Details as in (C). Data are presented as median±min-max, *, P< 0.05, analyzed by Mann-Whitney U test. (E) Representative Western blot quantification of mTOR and the serine 2448 phosphorylated mTOR (p-mTOR). Relative protein level was calculated by normalizing phosphorylated form levels to their corresponding total protein levels and total protein levels to the β-actin levels, respectively. Data are presented as mean ± SEM. *, P< 0.05, compared with WT, t-test, n=12 mice for each group.
To examine whether mTOR was over-activated in APP/PS1 mice, hippocampal CA3 tissue was dissected for Western blotting. Total mTOR levels were not different between APP/PS1 and wild-type mice (t4= 0.551, P= 0.601, each sample collected from 3 mini slices of hippocampal CA3 region of 3 mice, n= 4 independent experiments from 12 mice, Fig. 7E). However, compared to that in wildtype, mTOR serine 2448 phosphorylation in CA3 of APP/PS1 mice was increased by 38% (t4= 3.216, P= 0.018, Fig. 7E). The increased mTOR serine 2448 phosphorylation was strikingly similar to the increase induced by Aβ1-42 in wildtype slices (Fig. 3E) and suggests that the mTOR/S6K1 pathway mediates the disruption of γ-oscillations in AD.
DISCUSSION
In this study, we investigated the role of oligomeric Aβ1-42 in modulating hippocampal γ-oscillations in vitro. Aβ1-42 strongly suppressed γ-oscillations while increasing the peak frequency. Aβ1-42-induced hyper-activation of mTOR was both necessary and sufficient to suppress γ-oscillations through activation of S6K1 rather than through inhibition of eIF4E. Activation of the mTOR/S6K1 pathway decreased spontaneous EPSC amplitude and frequency and IPSC frequency. Inhibition of the mTOR/S6K1 pathway rescued the impairment of hippocampal γ-oscillations in APP/PS1 mice.
Oligomeric Aβ1-42 is increased in the brain tissue of AD patients and animal models [60]. Because increased levels of oligomer Aβ1-42 are observed early in the development of AD, it has been the target of AD therapies [61, 62].
The oligomeric Aβ1-42 concentrations that were effective in suppressing γ-oscillations are within the concentration range of oligomeric Aβ, found in wet brain tissue of AD patients [53]. Still, no information is available about the oligomeric Aβ1-42 concentration in the extracellular space. Aβ1-42 was more potent in its oligomeric or fibrillar form than as monomers, which confirms a study by Kurudenkandy et al.[25] and is in line with the increased toxicity of the oligomeric form [63, 64]. Although the fibrillar Aβ1-42 was equally potent as the oligomeric form, it is unlikely that large fibrils can affect γ-oscillation-generating networks directly [25] because the average extracellular space is only ~50 nm [51]. Most likely, fibrils are turned into oligomers in the tissue through a fibril-catalyzed secondary nucleation reaction [52]. The < 10 nm diameter oligomers will probably diffuse into the tissue and affect the γ-generating networks.
A similar impairment of γ-oscillations was observed in the 4-month-old APP/PS1 mouse [16] and has been described at very early stages in other AD animal models [20, 21, 23]. Early-stage AD is associated with the hyper-activation of mTOR in patients and animal models [36, 37]. Oligomeric Aβ1-42 caused a hyper-activation of mTOR that was necessary for the effect of Aβ1-42 on γ-oscillations, as shown by the effect of the mTORC1 inhibitor rapamycin. Hyperactivation of mTOR was also demonstrated in the hippocampus of 4-month-old APP/PS1 mice. Rescuing impaired γ-oscillations in slices from APP/PS1 mice by rapamycin confirms that mTOR1 hyper-activation is necessary for the Aβ1-42 effect on γ-oscillations. Hyperactivation of mTORC1 was also sufficient for the Aβ1-42-induced impairment of γ-oscillations, since activating the mTOR1 pathway in neurons by leucine [65], could mimic the Aβ1-42 effect. The rapamycin-sensitive mTORC1 suppresses autophagy and promotes protein synthesis by phosphorylating S6K1 and 4E-BPs [66]. Whether the acute impact of Aβ1-42 on γ-oscillations is caused by suppression of autophagy remains to be determined. Our results identify the mTOR/S6K1 pathway as responsible for the suppression of γ-oscillations. Because eIF4E phosphorylation levels are elevated significantly in the later stages of AD, where they are associated with tau hyper-phosphorylation [56], the mTOR/S6K1-induced suppression of γ-oscillations by oligomeric Aβ1-42 may be instrumental in the cognitive deficits, especially in the early stages of AD. In addition, a lack of γ-oscillations was proposed to contribute to the development of AD-associated pathology [23].
The suppression of γ-oscillations by mTOR hyperactivation, whether by Aβ1-42, leucine or in the APP/PS1 AD model, was invariably accompanied by an increase in peak frequency, which was reduced by rapamycin that restored γ power as well. This inverse relation between γ power and peak frequency is expected since the IPSC amplitude determines the cycle length [67]. It suggests that the Aβ1-42-induced reduction in power and increase in the frequency of γ-oscillations, results from a reduced amplitude of the combined IPSCs of interneurons contributing to the synchronization. However, mTOR hyper-activation did not affect the amplitude of spontaneous (not rhythmic) IPSCs, indicating normal GABAergic synaptic transmission. The mTOR/S6K1-induced reduction of IPSC frequency points to a reduced presynaptic GABA release probability. This can be due to a reduction of AMPA receptor-mediated fast EPSCs on interneurons. Fuchs et al. [10] showed that a reduced AMPA receptor-mediated recruitment of parvalbumin-expressing interneurons caused a reduction of γ power, an increase in peak frequency, and a reduction in regularity, a pattern identical to the Aβ1-42 effect on γ-oscillations. The Aβ1-42-induced reduced amplitude and frequency of spontaneous EPSCs in CA3 pyramidal neurons can explain a reduced interneuron activation. Our results are in line with the observations of Ramirez et al.[68], who showed that rapamycin increases the frequency of miniature EPSCs of rat hippocampal primary neurons by modulating neurotransmitter release [68]. Interestingly, a decreased glutamate release probability and a reduced spontaneous EPSC frequency were also observed in the CA3 region of 4-month-old APP/PS1 mice [69, 70]. Our observations differ from those of Kurudenkandy et al. [25], who report increased EPSC amplitude and frequency and decreased IPSC amplitude and frequency. However, they tested a high concentration (1 µM) of the fibrillar form Aβ1-42, producing an unknown concentration of oligomer Aβ1-42 [52]. This advocates using a controlled oligomer preparation [44, 45].
Alternatively, the mTOR hyper-activity-induced suppression of γ-oscillations may be caused by reduced intrinsic excitability of interneurons. Supporting this hypothesis, Verret et al. [22] reported a reduction in γ-oscillations in human APP mice and AD patients, linked to decreased levels of the interneuron-specific voltage-gated sodium channel subunit Nav1.1 in parvalbumin-expressing interneuron. Reduced intrinsic excitability of interneurons can also result from a metabolic impairment of fast-firing interneurons that are very energy-demanding during γ-oscillations. Gamma oscillations are exquisitely sensitive to metabolic stress [71] and more vulnerable in early AD [72]. mTOR hyper-activation affects metabolic processes involved in energy homeostasis [32, 33] and impairs mitochondrial function [35], which reduces interneuron activity [31, 34] and leads to reduced hippocampal γ-oscillations in aged mice [31].
The mTOR hyper-activation-induced impairment of γ-oscillations is likely to contribute to the cognitive impairment observed in early-stage AD and MCI since γ activity serves as an elementary operator of brain function and communication, as reviewed by Basar [73]. Impairment of γ activity has been related to the impairment of working memory [10], spatial reference memory [11], and context-dependent memory deficits [22], cognitive deficits typical for early stages of AD.
Our study does not exclude other ways by which mTOR hyperactivity can contribute to cognitive deficits [74], but restoring γ-oscillations is a potential target for early-stage AD therapy. For instance, restoring the intrinsic excitability of parvalbumin-expressing interneurons, increased γ-oscillations, and reversed cognitive impairment in the human APP AD model [22]. Interestingly, restoring degenerated γ-oscillations in an AD model by optogenetically driving parvalbumin-expressing interneurons reduced Aβ accumulation [23], suggesting that restoration of γ-oscillations may have not only short-term cognitive benefits but can even attenuate AD-associated pathology.
Acknowledgments
This work was supported by the Natural Science Foundation of China (grants numbers 81701070, 81771517, 91632305, 81261120570), the science and technology planning project of Henan province (212102310824). This study was initiated and designed by C-B L, M V, J-Z W and Y-L W; Y-L W, J-G W, S-L G, F-L G, and X Y performed brain slice electrophysiology recordings and analyzed the data; S-L G, E-J L, and B-Y F performed Western blotting; C-B L, M V and Y-L W wrote and revised the manuscript. All authors contributed to the article and approved the submitted version. The authors declare that the research was conducted without any commercial or financial relationships that could be construed as a potential conflict of interest. The datasets generated for this study are available on request from the corresponding author.
Footnotes
Declaration of competing interest
The authors declare no potential conflict of interest.
References
- [1].Selkoe DJ, Hardy J (2016). The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med, 8:595-608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ferrari C, Sorbi S (2021). The complexity of Alzheimer's disease: an evolving puzzle. Physiol Rev, 101:1047-1081. [DOI] [PubMed] [Google Scholar]
- [3].Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, et al. (2017). Mitophagy and Alzheimer's Disease: Cellular and Molecular Mechanisms. Trends Neurosci, 40:151-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Price JL, McKeel DW Jr., Buckles VD, Roe CM, Xiong C, Grundman M, et al. (2009). Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging, 30:1026-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT (2011). Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med, 1:a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Buzsaki G, Draguhn A (2004). Neuronal oscillations in cortical networks. Science, 304:1926-1929. [DOI] [PubMed] [Google Scholar]
- [7].Womelsdorf T, Fries P (2007). The role of neuronal synchronization in selective attention. Curr Opin Neurobiol, 17:154-160. [DOI] [PubMed] [Google Scholar]
- [8].Csicsvari J, Jamieson B, Wise KD, Buzsaki G (2003). Mechanisms of gamma oscillations in the hippocampus of the behaving rat. Neuron, 37:311-322. [DOI] [PubMed] [Google Scholar]
- [9].Liu T, Bai W, Wang J, Tian X (2015). An aberrant link between gamma oscillation and functional connectivity in Abeta-mediated memory deficits in rats. Behav Brain Res, 297:51-58. [DOI] [PubMed] [Google Scholar]
- [10].Fuchs EC, Zivkovic AR, Cunningham MO, Middleton S, Lebeau FE, Bannerman DM, et al. (2007). Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior. Neuron, 53:591-604. [DOI] [PubMed] [Google Scholar]
- [11].Lu CB, Jefferys JG, Toescu EC, Vreugdenhil M (2011). In vitro hippocampal gamma oscillation power as an index of in vivo CA3 gamma oscillation strength and spatial reference memory. Neurobiol Learn Mem, 95:221-230. [DOI] [PubMed] [Google Scholar]
- [12].Jensen O, Lisman JE (2005). Hippocampal sequence-encoding driven by a cortical multi-item working memory buffer. Trends Neurosci, 28:67-72. [DOI] [PubMed] [Google Scholar]
- [13].Montgomery SM, Buzsaki G (2007). Gamma oscillations dynamically couple hippocampal CA3 and CA1 regions during memory task performance. Proc Natl Acad Sci U S A, 104:14495-14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Carr MF, Karlsson MP, Frank LM (2012). Transient slow gamma synchrony underlies hippocampal memory replay. Neuron, 75:700-713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].AlvaradoMartínez R, SalgadoPuga K, PeñaOrtega F (2013). Amyloid beta inhibits olfactory bulb activity and the ability to smell. PLoS One, 8:e75745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Klein AS, Donoso JR, Kempter R, Schmitz D, Beed P (2016). Early Cortical Changes in Gamma Oscillations in Alzheimer's Disease. Front Syst Neurosci, 10:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Stam CJ, van Cappellen van Walsum AM, Pijnenburg YA, Berendse HW, de Munck JC, Scheltens P, et al. (2002). Generalized synchronization of MEG recordings in Alzheimer's Disease: evidence for involvement of the gamma band. J Clin Neurophysiol, 19:562-574. [DOI] [PubMed] [Google Scholar]
- [18].Lee SH, Park YM, Kim DW, Im CH (2010). Global synchronization index as a biological correlate of cognitive decline in Alzheimer's disease. Neurosci Res, 66:333-339. [DOI] [PubMed] [Google Scholar]
- [19].Koenig T, Prichep L, Dierks T, Hubl D, Wahlund LO, John ER, et al. (2005). Decreased EEG synchronization in Alzheimer's disease and mild cognitive impairment. Neurobiol Aging, 26:165-171. [DOI] [PubMed] [Google Scholar]
- [20].Driver JE, Racca C, Cunningham MO, Towers SK, Davies CH, Whittington MA, et al. (2007). Impairment of hippocampal gamma-frequency oscillations in vitro in mice overexpressing human amyloid precursor protein (APP). Eur J Neurosci, 26:1280-1288. [DOI] [PubMed] [Google Scholar]
- [21].Goutagny R, Gu N, Cavanagh C, Jackson J, Chabot JG, Quirion R, et al. (2013). Alterations in hippocampal network oscillations and theta-gamma coupling arise before Abeta overproduction in a mouse model of Alzheimer's disease. Eur J Neurosci, 37:1896-1902. [DOI] [PubMed] [Google Scholar]
- [22].Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, et al. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell, 149:708-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Iaccarino HF, Singer AC, Martorell AJ, Rudenko A, Gao F, Gillingham TZ, et al. (2016). Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature, 540:230-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Balleza-Tapia H, Huanosta-Gutierrez A, Marquez-Ramos A, Arias N, Pena F (2010). Amyloid beta oligomers decrease hippocampal spontaneous network activity in an age-dependent manner. Curr Alzheimer Res, 7:453-462. [DOI] [PubMed] [Google Scholar]
- [25].Kurudenkandy FR, Zilberter M, Biverstal H, Presto J, Honcharenko D, Stromberg R, et al. (2014). Amyloid-beta-induced action potential desynchronization and degradation of hippocampal gamma oscillations is prevented by interference with peptide conformation change and aggregation. J Neurosci, 34:11416-11425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nerelius C, Sandegren A, Sargsyan H, Raunak R, Leijonmarck H, Chatterjee U, et al. (2009). Alpha-helix targeting reduces amyloid-beta peptide toxicity. Proc Natl Acad Sci U S A, 106:9191-9196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].HA B (2015). Neuroscience porefront review seizures in alzheimers disease. Neuronscience, 286:251-263. [Google Scholar]
- [28].Palop JJ, Mucke L (2010). Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci, 13:812-818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang S, Li K, Zhao S, Zhang X, Yang Z, Zhang J, et al. (2020). Early-stage dysfunction of hippocampal theta and gamma oscillations and its modulation of neural network in a transgenic 5xFAD mouse model. Neurobiol Aging, 94:121-129. [DOI] [PubMed] [Google Scholar]
- [30].Fisahn A, Pike FG, Buhl EH, Paulsen O (1998). Cholinergic induction of network oscillations at 40 Hz in the hippocampus in vitro. Nature, 394:186-189. [DOI] [PubMed] [Google Scholar]
- [31].Lu CB, Vreugdenhil M, Toescu EC (2012). The effect of aging-associated impaired mitochondrial status on kainate-evoked hippocampal gamma oscillations. Neurobiol Aging, 33:2692-2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pedros I, Patraca I, Martinez N, Petrov D, Sureda FX, Auladell C, et al. (2016). Molecular links between early energy metabolism alterations and Alzheimer's disease. Front Biosci (Landmark Ed), 21:8-19. [DOI] [PubMed] [Google Scholar]
- [33].Tramutola A, Lanzillotta C, Di Domenico F (2017). Targeting mTOR to reduce Alzheimer-related cognitive decline: from current hits to future therapies. Expert Rev Neurother, 17:33-45. [DOI] [PubMed] [Google Scholar]
- [34].Bateup HS, Johnson CA, Denefrio CL, Saulnier JL, Kornacker K, Sabatini BL (2013). Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron, 78:510-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Miwa S, Czapiewski R, Wan T, Bell A, Hill KN, von Zglinicki T, et al. (2016). Decreased mTOR signalling reduces mitochondrial ROS in brain via accumulation of the telomerase protein TERT within mitochondria. Aging (Albany NY), 8:2551-2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Caccamo A, Maldonado MA, Majumder S, Medina DX, Holbein W, Magri A, et al. (2011). Naturally secreted amyloid-beta increases mammalian target of rapamycin (mTOR) activity via a PRAS40-mediated mechanism. J Biol Chem, 286:8924-8932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhou XW, Tanila H, Pei JJ (2008). Parallel increase in p70 kinase activation and tau phosphorylation (S262) with Abeta overproduction. FEBS Lett, 582:159-164. [DOI] [PubMed] [Google Scholar]
- [38].Tramutola A, Triplett JC, Di Domenico F, Niedowicz DM, Murphy MP, Coccia R, et al. (2015). Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. [J] Neurochem. [DOI] [PubMed] [Google Scholar]
- [39].Caccamo Antonella, Branca C, Talboom Joshua S, Shaw Darren M, Turner Dharshaun, Ma Luyao, Messina Angela, Huang Zebing, Wu Jie and Oddo Salvatore (2015). Reducing Ribosomal Protein S6 Kinase 1 Expression Improves Spatial Memory and Synaptic Plasticity in a Mouse Model of Alzheimer's Disease. The Journal of Neuroscience, 35:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, et al. (1998). Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med, 4:97-100. [DOI] [PubMed] [Google Scholar]
- [41].Wang J, Zhao J, Liu Z, Guo F, Wang Y, Wang X, et al. (2016). Acute Ethanol Inhibition of gamma Oscillations Is Mediated by Akt and GSK3beta. Front Cell Neurosci, 10:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pietersen AN, Ward PD, Hagger-Vaughan N, Wiggins J, Jefferys JG, Vreugdenhil M (2014). Transition between fast and slow gamma modes in rat hippocampus area CA1 in vitro is modulated by slow CA3 gamma oscillations. J Physiol, 592:605-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yaling Yin, Di Gao, Yali Wang, Zhi-Hao Wang, Xin Wang, Jinwang Ye, et al. (2016). Tau accumulation induces synaptic impairment and memory deficit by calcineurin-mediated nactivation of nuclear CaMKIV/CREB signaling. PNAS, 113:E3773-3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fa M, Orozco IJ, Francis YI, Saeed F, Gong Y, Arancio O (2010). Preparation of oligomeric beta-amyloid 1-42 and induction of synaptic plasticity impairment on hippocampal slices. J Vis Exp:1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Whitcomb DJ, Hogg EL, Regan P, Piers T, Narayan P, Whitehead G, et al. (2015). Intracellular oligomeric amyloid-beta rapidly regulates GluA1 subunit of AMPA receptor in the hippocampus. Sci Rep, 5:10934-10946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hyeon-Jin Kim S-CC, Chul Park, William L Klein, Dae-Kwon Lee, Grant A Krafft, Brett Chromy, Sam Cheol Lee, Seong-Tshool Hong (2002). Selective neuronal degeneration induced by soluble oligomeric amyloid beta-protein. The FASEB Journal, 17:118-120. [DOI] [PubMed] [Google Scholar]
- [47].Broersen K, Jonckheere W, Rozenski J, Vandersteen A, Pauwels K, Pastore A, et al. (2011). A standardized and biocompatible preparation of aggregate-free amyloid beta peptide for biophysical and biological studies of Alzheimer's disease. Protein Eng Des Sel, 24:743-750. [DOI] [PubMed] [Google Scholar]
- [48].Faucher P, Mons N, Micheau J, Louis C, Beracochea DJ (2015). Hippocampal Injections of Oligomeric Amyloid beta-peptide (1-42) Induce Selective Working Memory Deficits and Long-lasting Alterations of ERK Signaling Pathway. Front Aging Neurosci, 7:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, et al. (2010). Structural conversion of neurotoxic amyloid-beta(1-42) oligomers to fibrils. Nat Struct Mol Biol, 17:561-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kim HJ, Chae SC, Lee DK, Chromy B, Lee SC, Park YC, et al. (2003). Selective neuronal degeneration induced by soluble oligomeric amyloid beta protein. FASEB J, 17:118-120. [DOI] [PubMed] [Google Scholar]
- [51].Thorne RG, Nicholson C (2006). In vivo diffusion analysis with quantum dots and dextrans predicts the width of brain extracellular space. Proc Natl Acad Sci U S A, 103:5567-5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, et al. (2013). Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci U S A, 110:9758-9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yang T, Hong S, O'Malley T, Sperling RA, Walsh DM, Selkoe DJ (2013). New ELISAs with high specificity for soluble oligomers of amyloid beta-protein detect natural Abeta oligomers in human brain but not CSF. Alzheimers Dement, 9:99-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].An WL, Cowburn RF, Li L, Braak H, Alafuzoff I, Iqbal K, et al. (2003). Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer's disease. Am J Pathol, 163:591-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Antonella C, Caterina B, S TJ, M SD, Dharshaun T, Luyao M, et al. (2015). Reducing ribosomal protein S6 Kinase 1 expression improves spatial memory and synaptic plasticity in a mouse model of alzheimer's disease. The Journal of Neuroscience, 35:14042-14056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Li X, An WL, Alafuzoff I, Soininen H, Winblad B, Pei JJ (2004). Phosphorylated eukaryotic translation factor 4E is elevated in Alzheimer brain. Neuroreport, 15:2237-2240. [DOI] [PubMed] [Google Scholar]
- [57].Richter JD, Klann E (2009). Making synaptic plasticity and memory last: mechanisms of translational regulation. Genes Dev, 23:1-11. [DOI] [PubMed] [Google Scholar]
- [58].Gingras AC, Raught B, Sonenberg N (2001). Regulation of translation initiation by FRAP/mTOR. Genes Dev, 15:807-826. [DOI] [PubMed] [Google Scholar]
- [59].Uhlhaas PJ, Singer W (2006). Neural synchrony in brain disorders: relevance for cognitive dysfunctions and pathophysiology. Neuron, 52:155-168. [DOI] [PubMed] [Google Scholar]
- [60].Panza F, Lozupone M, Logroscino G, Imbimbo BP (2019). A critical appraisal of amyloid-beta-targeting therapies for Alzheimer disease. Nat Rev Neurol, 15:73-88. [DOI] [PubMed] [Google Scholar]
- [61].Izzo NJ, Xu J, Zeng C, Kirk MJ, Mozzoni K, Silky C, et al. (2014). Alzheimer's therapeutics targeting amyloid beta 1-42 oligomers II: Sigma-2/PGRMC1 receptors mediate Abeta 42 oligomer binding and synaptotoxicity. PLoS One, 9:e111899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Jeremic D, Jimenez-Diaz L, Navarro-Lopez JD (2021). Past, present and future of therapeutic strategies against amyloid-beta peptides in Alzheimer's disease: a systematic review. Ageing Res Rev, 72:101496. [DOI] [PubMed] [Google Scholar]
- [63].Hardy J, Selkoe DJ (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science, 297:353-356. [DOI] [PubMed] [Google Scholar]
- [64].Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med, 14:837-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Shih YT, Hsueh YP (2016). VCP and ATL1 regulate endoplasmic reticulum and protein synthesis for dendritic spine formation. Nat Commun, 7:11020-11036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Laplante M, Sabatini DM (2012). mTOR signaling in growth control and disease. Cell, 149:274-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Atallah BV, Scanziani M (2009). Instantaneous modulation of gamma oscillation frequency by balancing excitation with inhibition. Neuron, 62:566-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ramirez AE, Pacheco CR, Aguayo LG, Opazo CM (2014). Rapamycin protects against Abeta-induced synaptotoxicity by increasing presynaptic activity in hippocampal neurons. Biochim Biophys Acta, 1842:1495-1501. [DOI] [PubMed] [Google Scholar]
- [69].Cummings DM, Liu W, Portelius E, Bayram S, Yasvoina M, Ho SH, et al. (2015). First effects of rising amyloid-beta in transgenic mouse brain: synaptic transmission and gene expression. Brain, 138:1992-2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Richetin K, Leclerc C, Toni N, Gallopin T, Pech S, Roybon L, et al. (2015). Genetic manipulation of adult-born hippocampal neurons rescues memory in a mouse model of Alzheimer's disease. Brain, 138:440-455. [DOI] [PubMed] [Google Scholar]
- [71].Kann O, Hollnagel JO, Elzoheiry S, Schneider J (2016). Energy and potassium ion homeostasis during gamma oscillations. Front Mol Neurosci, 9:47-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Fang D, Zhang Z, Li H, Yu Q, Douglas JT, Bratasz A, et al. (2016). Increased electron paramagnetic resonance signal correlates with mitochondrial dysfunction and oxidative stress in an alzheimer's disease mouse brain. J Alzheimers Dis, 51:571-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Basar E (2013). A review of gamma oscillations in healthy subjects and in cognitive impairment. Int J Psychophysiol, 90:99-117. [DOI] [PubMed] [Google Scholar]
- [74].Izzo NJ, Staniszewski A, To L, Fa M, Teich AF, Saeed F, et al. (2014). Alzheimer's therapeutics targeting amyloid beta 1-42 oligomers I: Abeta 42 oligomer binding to specific neuronal receptors is displaced by drug candidates that improve cognitive deficits. PLoS One, 9:e111898. [DOI] [PMC free article] [PubMed] [Google Scholar]