

Abstract
Multi‐omics usually refers to the crossover application of multiple high‐throughput screening technologies represented by genomics, transcriptomics, single‐cell transcriptomics, proteomics and metabolomics, spatial transcriptomics, and so on, which play a great role in promoting the study of human diseases. Most of the current reviews focus on describing the development of multi‐omics technologies, data integration, and application to a particular disease; however, few of them provide a comprehensive and systematic introduction of multi‐omics. This review outlines the existing technical categories of multi‐omics, cautions for experimental design, focuses on the integrated analysis methods of multi‐omics, especially the approach of machine learning and deep learning in multi‐omics data integration and the corresponding tools, and the application of multi‐omics in medical researches (e.g., cancer, neurodegenerative diseases, aging, and drug target discovery) as well as the corresponding open‐source analysis tools and databases, and finally, discusses the challenges and future directions of multi‐omics integration and application in precision medicine. With the development of high‐throughput technologies and data integration algorithms, as important directions of multi‐omics for future disease research, single‐cell multi‐omics and spatial multi‐omics also provided a detailed introduction. This review will provide important guidance for researchers, especially who are just entering into multi‐omics medical research.
Keywords: biomarker, machine learning and deep learning, multi‐omics, neurodegenerative disease, precision medicine
Multi‐omics contains genomics, transcriptomics, proteomics, metabolomics, etc. And the experimental design considers the disease characteristics, disease model, sample size and phenotypic data. After the data integration by the approach of correlation, network and machining learning, the multi‐omics can be applied in diagnosis, biomarkers, targets of human disease and target discovery of natural compound.
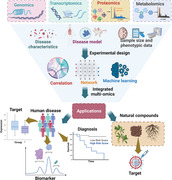
1. INTRODUCTION
As medical technology rapidly evolves, researchers need to conduct an in‐depth analysis of the pathogenesis of diseases. The omics technologies provide a high‐throughput screening method to uncover the detailed biological information of human diseases efficiently and rapidly. Usually, the omics technologies included genomics, transcriptomics, proteomics, metabolomics, single‐cell transcriptomics, single‐cell multi‐omics, spatial transcriptomics, and others. Each type of omics data provides differentially expressed disease‐associated molecules that can serve as biomarkers during disease progression and provide insight into which biological pathways or processes differ between disease and control groups. However, it should be noted that single omics technology cannot give full play to its application value in disease research. Mutations that occur in DNA will affect the expression of proteins, which may result in partial or total loss of some functions, leading to biological defects. But it is hard to tell the extent of the loss of function based on the genome alone. The level of gene expression, and ultimately how much protein is produced, both are related to the state of the disease. In addition, the occurrence of a disease may be related to a mutation in a gene, or an error in the transcription, translation, or other processes of the gene. In real research, one type of omics research can only carry out the correlation analysis with diseases, mainly reflecting the change of disease process and cannot explain the causal relationship. In Alzheimer's disease, for example, even if a biochemical molecule is statistically associated with the disease, it does not explain the complex mechanisms underlying the disease.
Integration of different types of omics data can elucidate underlying pathogenic changes of the disease, which can then be verified in further molecular researches. By integrating multi‐omics, scientists can filter out novel associations between biomolecules and disease phenotypes, identify relevant signaling pathways, and establish detailed biomarkers of disease. Therefore, the integration of various omics data will facilitate the match of associations between molecular‐disease and phenotype‐environmental factors. For instance, the molecular profiling in primary tissues through multi‐omics integration to reveal molecular mechanisms of the progress of disease, 1 estimation of the biological age of organs (liver, kidneys, etc.), and systems (immune and metabolic systems) by a multi‐omics approach for the assessment of aging status. 2 Moreover, since the occurrence and development of diseases are not only affected by the latest gene mutations, but also by the environmental factor, genetic background, gene regulation, and so on, the research of multi‐omics from different levels has also laid the foundation for the development of systems biology technology, such as multi‐omics reveals systems biology in cardiovascular disease. 3 Recently, single‐cell omics and spatial omics provide more detail information about the mechanisms of interactions between intracellular and intercellular molecules that control development, physiology, and pathology. 4 The integration analysis of single‐cell transcriptomics and spatial transcriptomics has successfully resolved the logic underlying spatially organized immune‐malignant cell networks in human colorectal cancer. 5 However, although many high‐throughput omics technologies have made good research progress in the medical field, there are few systematic introductions on how multi‐omics should be carried out in disease research, such as experimental design, data integration, analysis tools, and so on, 6 especially for researchers who are just coming or preparing to conduct multi‐omics research.
In this review, we briefly described the types of multi‐omics used in human disease research, and focused on the experimental design, data integration, application, challenges, and future development directions of multi‐omics. In particular, we provide a detailed description of the experimental design and data integration in multi‐omics research. We also have offered open‐source tools that can be used to analyze and integrate multi‐omics data, which will greatly benefit researchers who are unfamiliar with algorithms. This review will be described in sequence from omics classification, experimental design, data integration, disease application, challenges and future development directions, and conclusion, which will provide valuable guidance for researchers or newcomers in the field of multi‐omics.
2. THE CATEGORIES OF OMICS
With the development of high‐throughput technology, there are now many types of omics in the medical field, mainly including transcriptomics, proteomics, genomics and epigenomics, single‐cell omics, spatial transcriptomics, radiomics, metabolomics, and microbiomics, whose applications mainly involve cell molecular level, intestinal microbial system, and pathological imaging, and so on (Figure 1).
FIGURE 1.
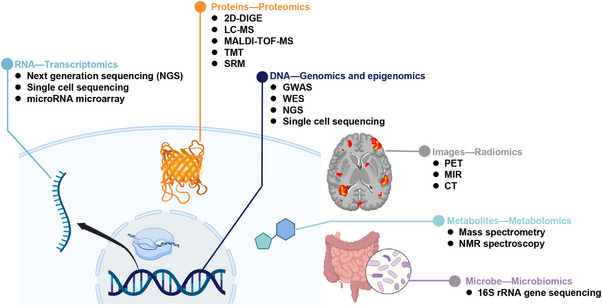
Multi‐omics approaches in disease research. Here lists the currently main available methods for each omics. RNA‐transcriptomics including next‐generation sequencing (NGS), single‐cell sequencing, and microRNA microarray. Proteins‐proteomics including 2D differential gel electrophoresis (2D‐DIGE), liquid chromatography‐mass spectrometry (LC‐MS), matrix‐assisted laser desorption ionization time‐of‐F (MALDI‐TOF‐MS), tandem mass tag (TMT), and selected reaction monitoring (SRM). DNA‐genomics and epigenomics including genome‐wide association studies (GWAS), whole‐exome sequencing (WES), next‐generation sequencing (NGS), and single‐cell sequencing. Images—radiomics including positron emission tomography (PET), magnetic resonance imaging (MRI), and computed tomography (CT). Metabolites—metabolomics including mass spectrometry and NMR spectroscopy. Microbe—microbiomics including 16S rRNA gene sequencing.
2.1. Genomics
Genomics is the application of omics in entire genomes, which aims to collect character and quantify all genes of an organism, uncover their interrelationship, and influence on the organism. Genomics is the earliest and most common application in medicine, such as The Human Genome Project. 7 Genomics usually contains the components of high‐throughput DNA sequencing, sequence assembly, and genome annotation. 8 Genome‐wide association study (GWAS) is a typical application of genomics to find out the existing sequence variation in the whole human genome, namely, single nucleotide polymorphism (SNP), and screen out disease‐related SNPs (https://www.ebi.ac.uk/gwas/). The associated technologies contained genotyping array, 9 third‐generation sequencing for whole‐genome sequencing, 10 and exome sequencing. 11 In GWAS studies, the millions of genetic variants across the genomes of multiple individuals are tested to identify genotype−phenotype associations. 12 Despite GWAS can identify novel disease‐associated susceptibility genes, biological pathways and translate these findings into clinical care, most of the acquired variants and genes have no direct biological relevance to disease. 13
2.2. Transcriptomics
Transcriptomics refers to the study of the expression of all the RNAs from a given cell population, which offer a global perspective on molecular dynamic changes induced by environment factors or pathogenic agents. The transcriptome of RNAs detection includes protein‐coding RNAs (mRNAs), long noncoding RNAs, short noncoding RNAs (microRNAs, small‐interfering RNAs, small nuclear RNAs, piwi‐interacting RNAs, and enhancer RNAs), and circular RNAs. In addition to mRNAs, noncoding RNAs also have associations with diseases, 14 such as diabetes, 15 cancer, 16 and so on, circular RNAs have a link with cardiovascular disease, 17 CNS disease, 18 and cancer. 19 Since mRNA accounts for 1%−4% of the overall transcript, the use of the transcriptome to study the impact of noncoding RNAs on disease is an important trend in the future. The usually used technology for transcriptomics is RNA‐seq, which can quality and quantify RNA transcripts from little RNA sample. 20 Currently, with the development of technology, the single‐cell transcriptome (single‐cell RNA sequencing [scRNA‐seq]) becomes hot, which can detect the transcripts of specific cell types in diseases (such as cancer, 21 Alzheimer's disease, 22 etc.).
2.3. Proteomics
Proteomics enables the maximum identification and quantification of all proteins in cells or tissues. Because the level of gene transcription is often affected by post‐transcriptional modifications, the RNA analysis usually lacks correlation with protein expression. 23 Thus, proteomics can quantify the protein expression and provide the directly associated information with the environment change or disease progression. The large‐scale study of proteins can be detected by mass spectrometry‐based method, affinity proteomics, protein chips, and reverse‐phased protein microarrays. 24 The mass‐based proteomics is widely used in modern medical research and classified as stable isotope labeling proteomics and label‐free proteomics, while labeled proteomics can shorten the detection time of mass spectrometry significantly and reduce the batch effect between samples. 25 In addition, the protein and protein interaction can also be identified by a combination of immunoprecipitation and mass spectrometry, for example, purification of target proteins by antibodies (BMPR‐1B), and followed by the detection of interacting unknown proteins by mass spectrometry, finally, acquired the interacted proteins (GDF5, GDF9, RhoD, and HSP10). 26 Noteworthy, the post‐translational modifications can be broadly found in proteins after translation, such as phosphorylation, glycosylation, ubiquitination, acetylation, and nitrosylation, 27 and those modifications are critical to intracellular signal transduction, protein transport, and enzyme activity. 28 Studying a set of modified proteins can better understand the progression of the disease, such as phosphoproteomics uncover the novel mechanism in type 2 diabetes, 29 lung adenocarcinoma, 30 nonalcoholic steatohepatitis, 31 Alzheimer's disease, 32 and so on.
2.4. Metabolomics
Metabolomics (usually containing untargeted metabolomics and targeted metabolomics) focuses on the study of a set of small molecule metabolites derived from cellular biological metabolic processes. Those metabolites include small molecule substrates, intermediates, and end products of cellular metabolism, such as carbohydrates, fatty acids, and amino acids. In general, metabolite analysis can immediately reflect dynamic changes in cell physiology, and abnormal metabolite level or ratio may induce disease. Metabolomics will help to elucidate the mechanisms of disease progression. 33 , 34 , 35 Additionally, there has a quantifiable correlation between metabolomics and other omics (genomics, transcriptomics, proteomics, etc.), such as mRNA count can predict metabolite level, 36 gut bacteria has a correlation with amino acids level in patients with fibromyalgia, 37 the expressions of creatine kinase and mitochondrial protein are consistent with acylcarnitine and acetyl‐CoA in human hypertrophic cardiomyopathy. 38 Moreover, the Human Metabolome Database (https://hmdb.ca/) is a free database containing detailed information of small molecule metabolites in the human body that can be used for metabolomics research, 39 and the corresponding metabolites analysis can be performed by MetaboAnalyst 5.0 (https://www.metaboanalyst.ca/, a free platform for metabolomics analysis). 40 Finally, the associated databases and related technologies for metabolomics include nuclear magnetic resonance (NMR) 41 and mass spectrometry (MS)‐based methods (gas chromatograph–mass spectrometer (GC‐MS), liquid chromatography tandem mass spectrometry(LC‐MS), and capillary electrophoresis–mass spectrometry (CE‐MS)). 42 , 43
2.5. Single‐cell omics
Recently, the rapid development of single‐cell sequencing technology has become popular in medical research. Single‐cell sequencing has great power in elucidating the heterogeneity of transcriptomics, genomics, and epigenomics in cellular populations, and corresponding changes in those levels. 44 To be specific, single‐cell transcriptomics aims to accurately understand the transcriptome status of heterogeneous cell populations. Single‐cell genomics reveals genetic heterogeneity from cells with or without mutation accumulation. Single‐cell epigenomics is used for detecting footprints of differentiation of individual cells. For instance, single‐cell transcriptome identified distinct transcriptional responses in lung cancer cell lines (CCLs), which were sensitive and insensitive to receptor tyrosine kinase inhibitors, and found distinct transcriptional modules that may be associated with early drug resistance. 45 Single‐cell targeted DNA sequencing obtained the information that cells that acquired RAS/MAPK mutations have a resistance to FLT3 inhibitors in acute myeloid leukemia. 46 Single‐cell ChIP‐sequencing revealed a distinct H3K27me3 pattern in resistant cells obtained from breast cancer. 47 In addition, single‐cell proteomics, another research center stage after single‐cell sequencing, 48 enables qualitative and quantitative analysis of protein composition within individual cells to reveal fundamental differences in the types and states of different individual cells, such as tumor heterogeneity, stem cell differentiation, germ cell development, and circulating tumor cells.
Moreover, single‐cell analysis has a tendency to enter the multi‐omics age. By integrating different types of molecular data (such as data on mutations, mRNAs, and proteins) from single‐cell, a comprehensive understanding of cellular processes can be achieved. Mahdessian et al. have systematically identified cell cycle‐associated heterogeneously expressed proteins at the mRNA and protein levels in single‐cell level. 49 Meanwhile, there are many assay platforms for single‐cell analysis, such as 10×Chromium Single Cell Gene Expression Solution, BD Rhapsody™ Single‐Cell Analysis System, and so on.
2.6. Spatial transcriptomics
Spatial transcriptome (ST) is a technology that preserves the spatial location of tissues and simultaneously resolves transcriptome information in tissue sections. This technique is used to analyze and describe the expression profiles of specific cell types in a spatial dimension to understand transcription differences among organs, tissues, and pathological states. ST can also be able to parse transcripts of tissues at different spatial locations. For example, ST has successfully pinpointed the type of testicular cells in a single spermatogenic tubule in mammals. 50 By detecting spatial gene expression in HER2‐positive breast tumors, ST spatially maps tumor‐associated cell types and obtains tertiary lymphoid‐like structures whose signal intensity correlates with overall survival. 51 The ST technologies include next‐generation sequencing (NGS)‐based techniques, where positional information is encoded onto transcripts prior to NGS sequencing, and imaging‐based methods include in situ sequencing, where transcripts are amplified and sequenced in tissue, and in situ hybridization (ISH)‐based methods, where imaging probes are sequentially hybridized in tissue. Moreover, spatial omics techniques have evolved to the spatial multi‐omics stage. The combination of spatial transcriptomics and proteomics has identified the multicellular mechanisms and early neurodegenerative pathways in the pathogenesis of progressive multiple sclerosis. 52
2.7. Epigenomics
Epigenomics focuses on the reversible modifications of DNA or histones that affect gene expression, and the main modification, including DNA methylation or histone modification. 53 The modifications of DNA and histone act as important roles to regulate gene expression and cellular processes (such as development or differentiation). 54 Those modifications usually can be influenced by environmental or genetic factors, and sometimes may be lasting or heritable. 55 Many researchers reported that epigenetic modifications have an association with diseases, such as type 2 diabetes, 56 cardiovascular disease, 57 cancer, 58 and so on. Epigenetic marks are often tissue‐specific, and the related research program contains Reference Epigenome Mapping Centers. 59 NGS is the common technology for epigenomics analysis.
2.8. Microbiomics
Microbiomics is to study the ecological community of microorganisms that symbiotically or pathologically live on plants and animals, including bacteria, archaea, protozoa, fungi, and viruses. 60 Due to the composition of the human microbiome being unique across individuals, the influence of the human genome on the gut microbiota is limited and generally influenced by the environment. 61 Thus, the microbiome can be used to explain clinical variations in phenotypes of interest under specific conditions better than genetic factors in humans. For those characteristics of the microbiomics, many diseases have found a relationship with the gut microbiome, such as neurological disorders, 62 renal failure, 63 and cancer. 64 The macro‐genome and macro‐transcriptome are the main technical tools for studying the microbiome, and 16S rRNA gene sequencing is the most commonly used method for microbial diversity analysis. 65
2.9. Radiomics
In the medical field, radiomics refers to extracting high‐throughput image features from the region of interest of radiographic images (computed tomography (CT), magnetic resonance (MR), positron emission tomography [PET], etc.) and the precise quantification of lesion areas, and key information (such as biomarker) through machine learning (ML) methods, 66 and ultimately aiding in the diagnosis, classification, or grading of diseases. 67 , 68 The radiomic features from PET imaging are better at predicting treatment response than conventional measures, such as tumor volume, diameter and metastases, and so on. 69 , 70 Deep learning (DL) of PET reconstruction and post‐processing of traditional reconstructed images can restore or reconstruct PET images of higher quality than traditional ordered‐subset expectation maximization through reducing image noise to obtain more details of radiation features. 71 However, class imbalances and overfitting are common defects in radiomics, such as the low prevalence of certain diseases in the cohort results in an inability to distinguish between affected and unaffected lesion areas on PET images. 71
3. EXPERIMENTAL DESIGNS AND CAUTIONS FOR MULTI‐OMICS
Due to the high cost of multi‐omics technology in experiments, different types of omics have differences in sample collection time, collection conditions, and setting groups, and samples involving human sources are more precious, careful design of experiments is often required in multi‐omics studies. And detailed design will improve the repeatability and reliability of multi‐omics results, especially for large sample sizes, multiple comparison groups, and specific data analysis experiments. 6 Moreover, the experimental design of reliable research is often inseparable from excellent statistical guidance, which can help to identify the research problem, study analysis, data interpretation, and conclusion. 72 Here, we will discuss some notable captions for multi‐omics design (Figure 2).
FIGURE 2.
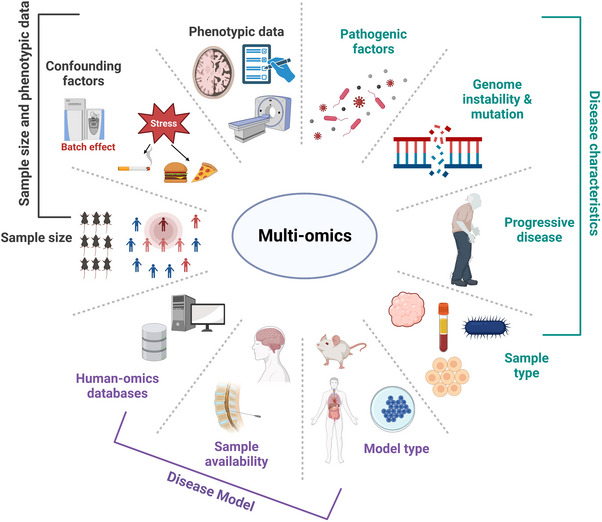
Notable recommendations for experimental design of multi‐omics. We list a lot of factors influencing the experimental results during experimental design, among them, three main factors include disease characteristics, choice of disease model, sample size, and phenotypic data. The disease characteristics contain pathogenic factors (bacteria or virus), genome instability and mutation, progressive disease (neurodegenerative disease), and sample type (tissue, cell, blood, or microbe). The disease model can be chosen according to suitable model type (human, mouse, or cell line), sample availability (unavailable samples, such as brain tissue and cerebrospinal fluid), and human‐omics databases (access to existing human disease omics data to compensate for uncommon disease models). The sample size and phenotypic data also need to be considered in multi‐omics experiment, such as the appropriate number of animal or human samples to achieve economically reliable omics results, while the appropriate number of detected samples usually according to confounding factors (batch effect, environmental stress like diet and smoking). The phenotypic data contain pathology, questionnaires, images, and so on.
3.1. Sample collection according to disease characteristics
Disease characteristics are important factors to consider in multi‐omics study design. Diseases caused by single‐gene mutations often have fewer pathogenic factors that play an important role in the development of the disease. For example, Duchenne muscular dystrophy is caused by a gene mutation of dystrophin, which helps keep muscle cells intact. 73 The mutation of phenylalanine hydroxylase gene‐induced phenylalanine accumulated in the body that leads to phenylketonuria. 74 For these types of diseases, collecting special tissue samples at specific time points and performing multi‐omics detection to deeply analyze the immediate molecular changes caused by pathogenic factors will help to enhance the understanding of the disease and find possible treatment strategies. Additionally, the pathogenic factors of some diseases also contain infectious agents, more than genetic mutations, such as Coronavirus disease 2019 (COVID‐19).
However, most diseases are caused by multiple factors rather than focusing on a single factor, and the pathogenesis is more complicated. Combinations of different factors may converge into phenotypically similar states. For these common and complex diseases, pathologic processes usually span a long time, and involve the interaction with genes, environment factors, and so on. Therefore, it is necessary to collect at multiple time points and different types of tissue samples for multi‐omics analysis, so as to reveal the mechanism of disease occurrence and development in depth. For example, in the case of Alzheimer's disease (AD), which has an uncertain etiology, multi‐omics studies often analyze AD patients or AD model mice at different ages and/or different types of samples (cerebrospinal fluid, plasma, or brain tissue). 75 , 76 , 77
3.2. Establishment of the disease model (human or animal)
The main purpose of medical research is to solve human diseases, so human‐related omics research has more translational potential than animal model‐related omics research. Several human‐omics databases have been established, including epigenome and transcriptome databases of different tissues or cell types, such as the public database of IHEC Data Portal (https://epigenomesportal.ca/ihec/) and the Human Protein Atlas (https://www.proteinatlas.org/). Moreover, there have been some established large human disease repositories, collecting medical information and samples from different patients, such as the UK biobank. 78
However, there also have some limitations in the study of human omics, and the replacement of animal disease models can solve these problems. For example, some human samples are not easy to collect, there are many confounding factors in human samples, and human‐derived cell lines cannot replicate the complex molecular network changes in vivo. While the animal model has the advantages of easy sample collection, uncontrolled sample size, clear phenotype, high reproducibility, easy environmental control, and so on. Animals play an important role in the research of diseases caused by environmental factors and can link omics data with corresponding environmental factors to reveal the pathogenesis of diseases. For instance, an animal model was used to study the effects of a high‐fat diet on nonalcoholic fatty liver disease. 79 Furthermore, comparing omics data from human and animal models will help to validate the biological relevance of the models, such as mice as useful models for functional studies of AD gene regulatory regions. 80 Nevertheless, animal model also has some application drawbacks, many transgenic models are restricted to one genetic background, some human disease manifestations cannot be reproduced in mouse models, and mouse models may not account for human biological processes in complex diseases.
3.3. Sample size and phenotypic data
The reliability of the information on omics results often depends on effect size, the sample size tested, and the magnitude of the effect of confounding factors. In terms of human studies, there have been many confounding factors, such as diet and lifestyle. Therefore, omics research on human diseases requires a large sample size to avoid misinformation and false‐positive results from individual abnormal samples. The sample size calculated from the initial efficacy is increasingly important for the reliability of the results of large‐scale studies. 81 Moreover, omics experiments often overlook the need for data analysis before and during data collection, and most fields of omics have not yet developed a gold standard for data analysis. Thus, in order to ensure that the collected data meet the requirements of subsequent analysis, the design of omics research needs to carefully consider the main objectives of the experiment and the analysis method before collecting the data. For instance, in order to acquire important molecules that are closely related to phenotype, some critical phenotypic data need to acquire in omics, such as the data of histological features, tumor stage, smoking history, and mutation status need to be collected in the study of lung adenocarcinoma. 82 The acquisition of phenotypic data of the disease is crucial for omics to reveal the molecular changes in the progression of the disease, and may directly find the key target molecules of the disease. For example, in the multi‐omics discovery of peripheral blood biomarkers of Alzheimer's disease, various cognitive impairment scale scores (MMSE and MoCA), brain imaging (PET and MRI), and other phenotype data play a decisive role in the discovery of diagnostic biomarkers. 83 , 84 , 85 , 86 Similarly, the purpose is to know the differentially expressed molecules between the disease and the control, so the experimental design should focus on the sample size 87 ; if the experiment wants to find new molecules, the omics design should focus on a higher technical coverage depth, such as long‐read transcriptome. 88 In addition, in order to avoid or minimize technically introduced errors, batch effects introduced in sample processing and data acquisition should also be considered in the design of omics experiments. 89 , 90
4. INTEGRATED ANALYSIS APPROACH FOR MULTI‐OMICS DATA
Although the detection of multi‐omics can more deeply analyze the occurrence and development of diseases, and advance the development of systems biology compared with single omics, the integration of omics data has always been a cumbersome and nonstandard difficult problem. In most cases, multiple approaches to integrating omics data at multiple levels can be employed depending on the experimental design. 91 Before the integration of multi‐omics data, we need to understand the characteristics of multi‐omics data. The multi‐omics data acquired from different technology are usually heterogeneous, 92 such as transcriptomics and proteomics have different dynamic ranges and data distribution, due to the different normalization and scaling techniques among various omics analyses. In addition, some omics may produce null values due to being below the detection line of the instrument, such as metabolomics. 93 Therefore, before integrating multi‐omics data, the imputation 94 and outlier detection 95 for each omics data should be considered separately. Next, we briefly discuss some current common methods for multi‐omics integration (Figure 3).
FIGURE 3.
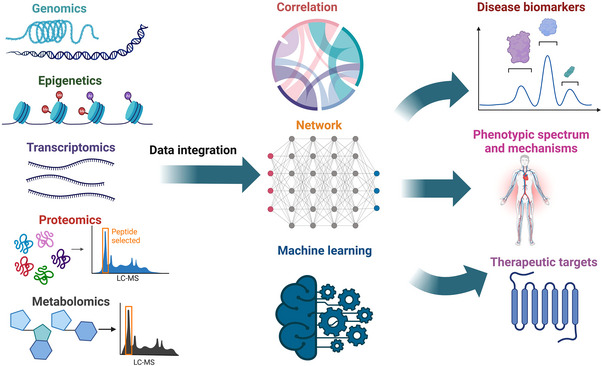
The methods for multi‐omics data integration. Here simply shows the multi‐omics data (such as genomics, epigenetics, transcriptomics, proteomics, and metabolomics) integration method based on the correlation of each omics, molecular network construction at different levels, and machine learning. The ultimate goal of data integration is to discover disease biomarkers, confirm phenotypic spectrum and mechanisms, and identify therapeutic targets.
4.1. Mining correlation and network in multi‐omics data
After acquiring multi‐omics data, the fundamental analysis contains single omics annotation, biological information derived from single omics, and biological network between two or more omics data, such as gene and metabolite or protein (enzyme) and metabolite. However, the integration of multi‐omics data is the toughest problem in dealing with multi‐omics analysis. A relatively common and simple method for multi‐omics data integration is the correlation or co‐mapping between two different omics data. For example, there is a relatively direct correspondence between transcriptome and proteome, the integration of those two omics usually contains an analysis of molecules with consistent or different trends in differentially transcribed genes and differential proteins, and a correlation of expression level between transcribed genes and proteins. 96 , 97 Sometimes, there is also a need to compare the correlation of expression level between transcriptome and proteome, and many research results have shown that the correlation between the expression of mRNA and protein is not strong, indicating that the post‐transcriptional regulation may be involved, such as in Alzheimer's disease, which has been reported that 42% of Tau‐induced transcripts were discordant with the proteome, showing the opposite direction of change. 98 Thus, the correlation between two omics will contribute to a better understanding mechanism in the process of disease occurrence and development.
In addition, the integrated multi‐omics also needs to take an effort to analyze the network between different omics data. The network serves as an established mathematical model for describing the complex interactions and regulatory mechanisms that occur in biological systems, 99 such as protein−protein interaction. 100 Network‐based approaches to the integrated analysis of multi‐omics data provide a framework to conceptualize the complex interactions of multi‐omics molecules as a collection of connected nodes (molecular features) in a network. Through these networks, it is possible to further identify associations (e.g., gene, protein, and metabolite relationships) and/or subnets (e.g., biological pathways) that can inform observed phenotypes. 101 Compared with transcriptome and proteome, metabolome records the real biochemical reactions in biological processes, and measures the metabolic level of substrate and product in chemical reactions, while transcriptome and proteome measure and quantify enzymes that catalyze biological reactions. Therefore, in multi‐omics research with metabolomics, the molecular regulatory network between each omics data and metabolites is often analyzed. However, network construction is usually applied to a single omics, and appropriate methods still need to be developed for network interactions between multi‐omics. 102 One simple and convenient approach to address multi‐omics network construction is to refer to public databases and frameworks, for example, omicsNet, 103 , 104 MetaboAnalyst, 40 and ConsensusPathDB. 105 Additionally, various algorithms (meta‐path‐based techniques, 106 random walk, 107 and module identification 108 ) of heterogeneous networks can be used to build multi‐level complex networks, such as HENA, 109 an Alzheimer's data set based on heterogeneous network, which integrated AD‐related co‐expression networks from public databases and experimental data sets to identify disease‐associated genes. Bodein et al. proposed to construct a hybrid multi‐omics network from longitudinal multi‐omics data using a random walk algorithm, highlighting key intra‐ and inter‐omics mechanisms and interactions. 110 Overall, the integrated analysis of highly complex networks in multi‐omics is not straightforward, requiring complex algorithms, and the downstream functional interpretation and validation of multi‐omics findings is not straightforward either. Therefore, for most researchers, the construction of multi‐omics networks and the interpretation of the resulting data still require open‐source tools and shared databases, such as netOmics, 110 MetaboAnalyst 5.0, 40 MergeOmics 2.0, 111 and so on (Table 1).
TABLE 1.
The online tools and resources of network‐based multi‐omics knowledge bases.
| Tool name | Biological entities | Implementation | Ref |
|---|---|---|---|
| netOmics | Genes, RNAs, proteins, metabolites | R package | 110 |
| HetioNet | Genes, SNPs, proteins, compounds, tissues, diseases | Online | 112 |
| MergeOmics 2.0 | Genes, SNPs, RNAs, proteins, metabolites | Online | 111 |
| MetaboAnalyst 5.0 | Genes, metabolites | Online | |
| MiBiOmics | Genes, RNAs, proteins, metabolites | Online | 113 |
| PathMe Viewer | Genes, proteins, metabolites | Online | 114 |
| OmicsNet 2.0 | Genes, proteins, TFs, miRNAs, metabolites | Online | 115 |
| PathwayCommons | Proteins, metabolites, drugs | Online CyPath2 a | 116 |
| Recon3D | Genes, metabolites | Online | 117 |
| BioCyc | Genes, proteins, metabolites | Online | 118 |
| PaintOmics3 | Genes, proteins, metabolites | Online | 119 |
| MetExplore | Genes, enzymes, metabolites | Online | 120 |
Applicable in Cytoscape software.
4.2. ML and DL for multi‐omics integration
With the development of high‐throughput omics technologies, multi‐omics data can also be integrated by ML‐ or DL‐based predictive algorithms to reveal the complex work of systems biology. ML is increasingly used in the development of precision medicine based on big data 121 , 122 and data mining, 123 these techniques facilitate the discovery of new omics biomarkers that can identify the molecular causes of disease. However, unlike the correlation‐ and network‐based integration approach, the approach of using ML to integrate multi‐omics data has some unique challenges. First, ML may result in class imbalance and overfitting during disease classification and subsequent model training in multi‐omics data set. 124 For example, the ML‐trained model using an imbalance data set, such as hypertension, which is only 5% of patients with endocrine hypertension, may be overfitted and lead to underperformance for unseen test data, despite high accuracy for training data. For this situation, it can be solved by collecting more data, or using weighted or normalized metrics (e.g., F1‐Score or Kappa 125 ) to measure the ML performance, oversampling, or synthetic sample generation of under‐represented class. Second, the data sets of multi‐omics usually suffer the problem of “curse of dimensionality,” which refer to inconsistency in data dimensions and features, 126 leading to redundant features in high‐dimensional space followed by misleading algorithm training. While, feature extraction and selection can be used for dimensionality reduction, such as principal component analysis (PCA), linear discriminant analysis, biofilter, and so on. 127 Third, the appropriate algorithms are essential for multi‐omics analysis, and previous reviews reported that different ML algorithms have diverse strengths and weaknesses in integrating multi‐omics data sets derived from cancer‐related research. 128 Reel et al. have proposed a flowchart that can help the interdisciplinary user to choose the appropriate algorithms from available methods. 129 Finally, ML requires high computational power and large storage for data processing and analysis, while the corresponding cost should be considered before planning an ML‐based multi‐omics workflow. Moreover, reasonable transparency and explainability are also important for multi‐omics integration by ML, and thus can be helpful for building trust in clinical decision‐making. 130 Therefore, when using ML to integrate multi‐omics data, researchers should consider all the above direct influences. Next, we will briefly introduce three ML‐based multi‐omics integration methods from the published reports, 129 , 131 which are shown in Figure 4A.
FIGURE 4.
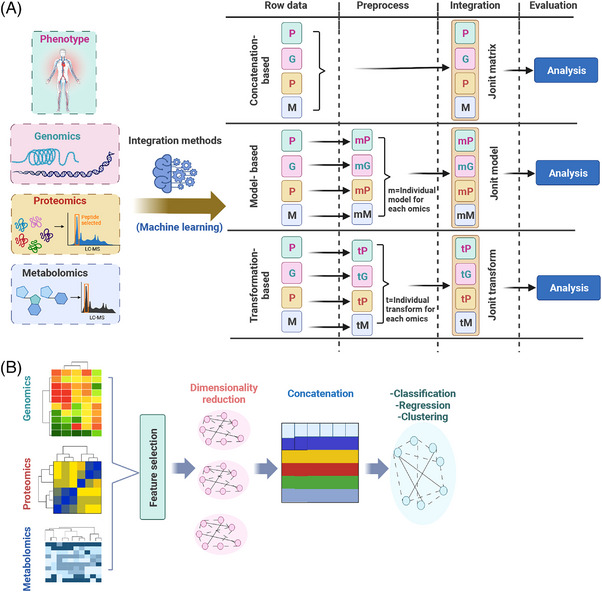
The brief process of integrating multi‐omics data with machine learning and deep learning. (A) The process of data integration by machine learning. The concatenation‐based integrated approach pipeline includes raw data from individual omics with corresponding phenotypic information, the data from the individual omics are then concatenated to form a single large matrix of multi‐omics data, and finally, supervised or unsupervised methods are used for joint matrix analysis. The model‐based integration method flow contains the establishment of the original data of various omics and the corresponding phenotypic information, develop individual models for each omics and then integrate them into a joint model, and finally, to analyze the joint model. And transformation‐based method starts with raw data of individual omics and corresponding phenotypic information, followed by developing individual transformations (in the form of graphs or kernel relations) for each omics, and then integrating it into joint transformations, and finally, analyzing it. The letters of PGPM are represented as phenotypic data (P), genomic data (G), proteomic data (P), and metabolomic data (M) in sequence. (B) The brief concept of data integration is achieved by deep learning. First, preprocess and clean the multi‐omics data, and then use conventional feature selection techniques or feature reduction methods for feature selection or dimensionality reduction to reduce the number of multi‐omics variables. Next, multiple omics variables are concatenated into one large data set for data integration. Finally, further feature selection or reduction techniques are applied to reduce the variables, and the integrated data are analyzed using classification, regression, and clustering.
4.2.1. Concatenation‐based integration methods
Concatenation‐based integrated approach pipeline includes raw data from individual omics with corresponding phenotypic information, and then, the data from the individual omics are concatenated to form a single large matrix of multi‐omics data, and finally, supervised or unsupervised methods are used for joint matrix analysis. Specifically, multiple data matrices for each sample in multi‐omics were combined into a large input matrix, and then different classical ML methods are used for data analysis. For example, data from gene expression, copy number variation, and mutation were combined into a joint matrix, and the joint was then used with classical random forest and SVM (support vector machine) to predict anticancer drug response. 132 A joint matrix of multi‐omics features (which included SNPs and mRNA gene expression) was used with the Bayesian integrative model to assess drug cytotoxicity. 133 According to the characteristics of ML, the concatenation‐based integration methods can be classed as unsupervised and supervised type. A variety of concatenation‐based unsupervised methods are available for clustering and association analysis, such as MoCluster, which can find the joint clusters among inputted multi‐omics data. 134 The Multi‐Omics Factor Analysis (MOFA) can disentangle the heterogeneity shared across different omics to discover the principal source of variability. 135 The concatenation‐based supervised learning methods that usually can be used for phenotypic prediction, such as boosted trees and SVR, have been investigated for identifying longitudinal predictors from a large multi‐omics data set of type 2 diabetes. 136 Advantages of concatenation‐based integration methods include that continuous or categorical data can be easily analyzed using ML, and the most discriminative features can be selected for a given phenotype. However, there has a challenge in concatenation‐based integration for combining multiple matrices that include data from different scales. For instance, the value of SNP data contains 0, 1, or 2, while the value of copy number data may consist of −2, −1, 0, 1, or 2, and the value of CpG loci between 0 and 1. Thus, concatenating this form of data integration can inflate high‐dimensionality during data analysis, and concatenation‐based integration is only possible applied after performing data reduction.
4.2.2. Model‐based integration methods
Model‐based integration is a method of generating multiple models by using different types of data as training sets, and then generating a final model from the multiple models created in the training phase. Specifically, this integration method contains: (1) establishment of the original data of various omics and the corresponding phenotypic information; (2) development of individual models for each omics and then integrate them into a joint model; and finally, (3) analysis of the joint model. This approach can combine predictive models from different omics and facilitate the understanding of a certain phenotype among different types of data. For example, to identify genetic, genomic, and proteomic associations with ovarian cancer, model‐based integration will allow independent analysis of each of the DNA sequence data, microarray data, and proteomic data, and then integrate top‐level models from each data set to find integrated models, such as model‐based integration performed with ATHENA (Analysis Tool for Heritable and Environmental Network Associations) to create an integrative model from each data type of ovarian cancer. 137 Similarly, the model‐based integration methods also divide into unsupervised and supervised types. Various model‐based unsupervised learning methods have been achieved, such as patient‐specific data fusion, which is used for clustering of predicted cancer subtypes by combining gene expression and copy number variation data, 138 perturbation clustering for data integration and disease subtyping 139 and Bayesian consensus clustering 140 are more flexible and allow late‐stage integration of clusters. Supervised learning model‐based methods include various frameworks for developing models, such as Multi‐omics Supervised Autoencoder, which is used for pan‐cancer analysis, 141 DFNForest (hierarchical integration deep flexible neural forest) framework integrated three omics data sets to predict cancer subtype classification, 142 and so on. An advantage of model‐based integration approaches is that they can be used to combine models based on different omics types, where each model was developed from a different patient group with the same disease information. However, model‐based integration also needs specific hypothesis and analysis for each data type, and appropriate mechanisms for the resultant model combination. When integrating models constructed from different types of data, some interactions between different data types may be missed because the only variables are those detected during the data type‐specific modeling process. For instance, the methylation pattern and another protein expression pattern that are not independently associated with the results, but only through their interactions, their effects will be missed in the model‐based integration. Thus, model‐based integration is particularly suitable if each genomic data type is very heterogeneous.
4.2.3. Transformation‐based integration methods
Transformation‐based integration involves converting each omics data set into an intermediate form (such as a graph or kernel matrix), and then combining them together before elaborating any models. Briefly, this method starts with raw data of individual omics and corresponding phenotypic information, followed by developing individual transformations (in the form of graphs or kernel relations) for each omics, and then integrating it into joint transformations, and finally analyzing it. The graph‐based method provides a formal method for transforming and portraying relationships between samples of different omics. The kernel‐based method transforms the data from the original space to a higher dimensional feature space. These methods then explore linear decision functions in the feature space that are nonlinear in the original space. Various transformation‐based unsupervised methods have been introduced, such as PAMOGK, which can integrate multi‐omics data with pathways, 143 somatic mutations, transcriptomics, and proteomics data were used to identify subtypes of kidney cancer. While transformation‐based supervised learning methods are mostly kernel‐based and graph‐based algorithms, such as Multi‐Omics gRaph cOnvolutional NETworks, which uses graph convolutional networks to take advantage of both the omics features and the correlations among samples described by the similarity networks for better classification performance. 144 A major advantage of transformation‐based integration methods is that they can be used to combine broad omics studies if unique information (patient ID) is available. The disadvantage of transformation‐based integration is that each data type is transformed independently, which makes it more difficult to detect interactions between different types of data, such as an SNP and gene expression interaction. Therefore, conversion‐based integration is suitable for each data type with an associated intermediate representation, such as a kernel or a graph.
In short, the above‐described approaches for multi‐omics data sets integration are based on various supervised and unsupervised ML methods. The advantages and disadvantages of using those integrative methods, and the detailed various multi‐omics integration methods based on ML can be found in the previous review. 129
Moreover, DL has emerged as one of the most promising approaches in the integrated analysis of multi‐omics data due to its predictive performance and ability to capture nonlinear and hierarchical features. DL can integrate different omics data from clinical or health records with high specificity, sensitivity, and efficiency, 145 and has the ability to automatically capture nonlinear and hierarchical representative features with multi‐layer neural network architecture, which has an excellent performance in prediction. For example, the multi‐omics integration approach with DL can robustly predict liver cancer survival, 146 and the DL approach can predict AD on the basis of combined neuroimaging and genomics data. 147 Overall, most DL‐based data integration studies are categorized into clustering for feature selection/reduction, clinical outcome prediction, survival analysis, and subtype discovery. For these studies, the DL models for multi‐omics data integration analysis follow a general principle 148 (Figure 4B). First, preprocess and clean the multi‐omics data, and then use conventional feature selection techniques or feature reduction methods (such as PCA and autoencoder) for feature selection or dimensionality reduction to reduce the number of multi‐omics variables. Next, multiple omics variables are concatenated into one large data set for data integration. Finally, further feature selection or reduction techniques are applied to reduce the variables, and the integrated data are analyzed using classification, regression, and clustering. For example, in the survival analysis study of liver cancer reported by Chaudhary et al., 146 first, transforming features of omics data (mRNA, DNA methylation, and miRNA) using a DL framework (autoencoder) to generate new features. Second, the univariate Cox‐PH model was used for identifying survival‐associated features, which then were clustered by a K‐means clustering algorithm to identify cancer subtypes. Third, according to the cluster labels obtained from K‐means to build supervised classification model(s) using the SVM algorithm, and data samples were divided into training sets and test sets followed by fivefold cross‐validation to find the best hyperparameters of the SVM model(s). Finally, concordance index (C‐index), log‐rank p‐value of Cox‐PH regression, and brier score were used for models’ evaluation, and then, the acquired model to predict the survival risk labels of new data sets. In addition, many DL tools for integrated analysis of multi‐omics data have been developed, such as DeepOmix, DeepProg, and DeepDRK. DeepOmix can be used not only for survival prediction analysis, but also for predicting various clinical indicators, such as drug response. 149 The DeepOmix integrated different omics data as input gene layer, and connected gene layer nodes to functional layer according to the prior information of pathway or functional module defined by input. The basic idea behind the model is that the gene function module layer is introduced into the algorithm model, and the biological prior information of the gene module is fused to integrate the multi‐omics characteristic information from the sample, and then applied to the prediction of survival state. The training model can predict the survival period of patients, and obtain the low‐dimensional representation of sample data in the functional module layer. Through statistical analysis, the functional modules of genes affecting prognosis can be found. DeepProg is used to integrate multi‐omics phenotypes, such as survival to predict cancer prognosis. 150 The method can be briefly described as follows: first, the custom rank normalizations and auto‐encoders were used for dimension reduction and feature transformation, second, the univariate Cox‐PH fitting was performed on the transformed features to further select the feature subset related to survival, finally, using an unsupervised clustering approach to identify the optimal number of classes (labels) for survival subgroups followed by building a support vector machine (SVM)‐based ML model according to these classes, and then using the acquired model to predict survival groups for new patients. DeepDRK predicts cancer cell drug response by integrating pharmacogenomics data sets, providing an alternative approach to prioritizing drug repurposing in precision cancer therapy. 151 DeepDRK used a kernel‐based approach to generate integrated representations of CCL and anticancer drug interaction partners, which were then employed to train deep neural networks for drug response prediction. Briefly, various types of multi‐omics data were collected, and CCL similarity matrices based on multiple kernels were constructed, respectively, then two similarity matrices for anticancer drugs were calculated using chemical characteristics of compounds and drug−target interactions. The CCL−drug pairs were represented by concatenating multiple similarity vectors, and then training the classification model to predict drug efficacy. In addition, there also has another new method of DL for multi‐omics integration. For instance, Multi‐omics Attention Deep Learning Network focuses on the correlations between patients and omics. 152 Specifically, the authors used three fully connected layers to reduce dimensionality and extract the significant features from omics data of mRNA, DNA methylation, and miRNA, and used the self‐attention (SA) mechanism to construct the correlations between patients, respectively, for omics‐specific feature learning. The initial category labels were generated using the feature vectors learned from the SA. Then, the Multi‐Omics Correlation Discovery Network was used to learn the cross‐omics correlations in the label space for the initial label predicted of each omics data. Finally, the SoftMax classifier was used for label prediction.
However, like ML, DL also encounters challenges, such as high‐dimensional and low sample‐size data, missing data and data heterogeneity, model interpretability, and integrating clinical and environmental exposure data. In general, the method of multi‐omics data integration will become easier and more feasible for nonprofessionals to obtain more biological information through multi‐omics technology with the development of various computing frameworks and tools.
5. APPLICATIONS OF MULTI‐OMICS IN HUMAN DISEASES
In addition to the omics types, multi‐omics experimental design, and multi‐omics data integration analysis described above, we will introduce various applications of multi‐omics in human diseases, such as cancer (malignant tumor, lung cancer, liver cancer, and ovarian cancer) and neurodegeneration (Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis [ALS]). Moreover, multi‐omics have also application in drug target discovery and aging research (Figure 5). The next part will introduce some previous studies of multi‐omics in human diseases.
FIGURE 5.
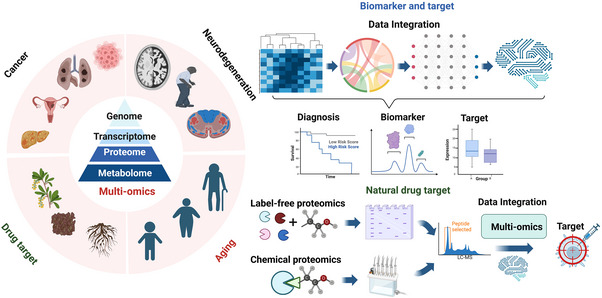
The application of multi‐omics in disease, aging, and natural drug target identification. There lists some disease‐associated applications of multi‐omics in cancer (liver cancer, lung cancer, ovarian cancer, and malignant lymphocytic tumor) and neurodegeneration (AD, PD, and ALS). Other applications, such as aging and natural drug (natural compound obtained from the flowers, seeds, or rootstock of plants) target screening, are also shown in the figure. The brief process and methods for the application of multi‐omics in their research area are listed right. For biomarker and target research, multi‐omics data are analyzed by differential expression analysis, correlation analysis, and network construction, and then, machine learning was used to obtain diagnostic biomarkers or therapeutic targets. The procedure for screening natural drug target is usually based on the integration of label‐free proteomics and chemical proteomics, and then is used for machine learning to acquire possible functional target.
5.1. Disease biomarkers and targets
Based on the outstanding advantages of multi‐omics in the discovery of disease etiological mechanisms, disease research increasingly requires the participation of multi‐omics and computational algorithms. Biomarkers can not only explore the pathogenesis at the molecular level, but also have unique advantages in accurately and sensitively evaluating early, low‐level damage, providing early warning, prognostic efficacy analysis, and accurate staging and typing of diseases. For clinical diseases, the discovery of disease biomarkers plays a guiding role in the diagnosis and prognosis of diseases. 152 While the screening of biomarkers usually requires the use of high‐throughput omics methods to measure large‐scale clinical samples, followed by screening to statistically significant differential molecules (genes, proteins, or metabolites), and finally a series of complex bioinformatics analyses to screen out the target biomarkers. Although many cancer‐related biomarkers have been identified by single omics, multi‐omics is more advantageous for cancer research. Proteomics data reveal overlapping but not identical correlations with transcriptomic and genetic data in breast cancer, 153 high‐grade serous ovarian cancer, 154 or rectal cancer, 155 and this phenomenon may account for the unique genetic and transcriptional process of proteomic alterations in the cancer process. For example, phosphoproteomic analysis of breast cancer identified G protein‐coupled receptor clusters which cannot be readily identifiable at the mRNA level. 153 Integration of various omics information is expected to be valuable in guiding targeted cancer therapy. 156 Moreover, there have been many successful examples in finding cancer biomarkers by multi‐omics, for example, diffuse large B‐cell lymphoma, 157 ovarian cancer, 158 and pancreatic cancer. 159 In addition to the application in the discovery of tumor or cancer‐related disease biomarkers, multi‐omics is also applied to find biomarkers for other diseases, such as stroke, 160 obesity, 161 cardiovascular diseases, 162 , 163 severe COVID‐19, 164 , 165 , 166 Alzheimer's disease, 167 diabetes, 168 obstructive sleep apnea, 169 and so on.
Integrated omics can also be used to investigate the influence of environmental factors on early disease formation in humans, such as sleep deficiency. Our study integrated analysis of transcriptomics, proteomics, and metabolomics in the blood of young health people suffering from transient sleep deprivation 170 (Figure 6). In this study, 32 volunteers suffered 1 day of sleep deprivation, and donated fasting blood samples prior to (Day 1) and following (Days 2 and 3) short‐term sleep deprivation. Then, the plasma was used for proteomics and metabolomics analysis, and the peripheral blood mononuclear cell (PBMC) was used for transcriptomics analysis. After integration analysis, the prominent biological pathway was immune disorders. Further detail analysis found that neutrophil‐mediated immune processes (such as neutrophil degranulation) mainly account for sleep deprivation‐induced immune disorders. And the correlation analysis showed that SOD1 and S100A8 may be served as biomarkers for immune disorder caused by sleep deprivation (Figure 6A). Noteworthily, the integrated analysis of metabolomics and proteomics showed that differentially expressed metabolites and proteins were involved in pathways of arginine and proline metabolism, tricarboxylic acid cycle (TCA) cycle has a similar trend of changes, and pyruvate and GOT1 linked these two pathways (Figure 6B). In addition, the integrated pathway analysis of proteome and transcriptome showed that differentially expressed genes and proteins shared the highest enriched score of pathways (such as neutrophil extracellular trap formation, complement and coagulation cascades, and focal adhesion) (Figure 6C).
FIGURE 6.
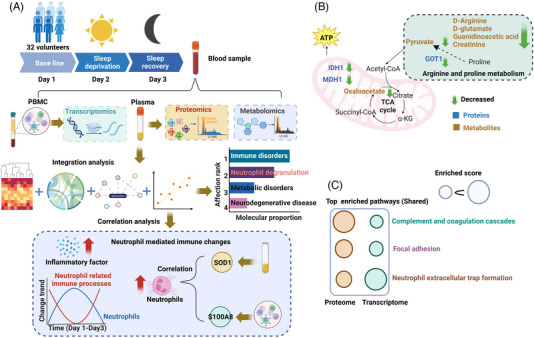
The integrated multi‐omics analysis on sleep deprivation. (A) The brief procedure and data for multi‐omics study in sleep deprivation. The blood sample obtained from 32 volunteers suffered sleep deprivation and recovery for plasma proteomics and metabolomics, PBMC transcriptomics analysis. After multi‐omics integration, the prominent biological pathway induced by sleep deprivation was immune disorders followed by metabolism disorders and neurodegenerative disease, and neutrophil degranulation was main account for the immune change. The detailed analysis showed that immune cell counts and inflammatory factor levels were elevated, and neutrophils and their mediated immune processes were highly coordinated with sleep states. The correlation analysis revealed that SOD1 and S100A8 were the most likely biomarkers of sleep deprivation‐induced immune changes. (B) The integration of proteomics and metabolomics, and the differentially expressed proteins and metabolites linked the pathway of arginine and proline metabolism, the TCA cycle. (C) The integration of proteomics and transcriptomics, and the shared top pathways enriched by differentially expressed genes and proteins are listed.
Compared with cancer or chronic diseases (diabetes), neurodegeneration acts as a major disease and lacks effective treatments, multi‐omics has important applications in the discovery of diagnostic biomarkers, pathogenic mechanisms, and therapeutic targets for neurodegenerative diseases. In view of the increasing aging of the population and the lack of effective treatments for neurodegenerative diseases, the next section will focus on the application of multi‐omics in neurodegenerative diseases, which were represented by AD, PD, and ALS, with special emphasis on the discovery of diagnostic biomarkers and therapeutic targets.
5.2. Applications of multi‐omics in neurodegenerative diseases
As the most common neurodegenerative disease, the prevalence of AD has been increasing year by year. A global public health study estimated that the number of AD patients will increase from the current 57 million in 2019 to about 150 million in 2050, 171 which leads to significant increases in medical and social costs. 172 However, there is still no effective treatment for AD, and the pathogenesis of AD is still not fully understood, although amyloid (Aβ) plaques and tau neurofibrillary tangles have revealed major pathological changes in AD. 173 Therefore, a comprehensive understanding of the molecular mechanisms underlying AD pathophysiology by multi‐omics will help to provide support for the prevention, treatment, and prognosis of AD. 174 Systematic integration of multi‐omics disciplines, including genome, transcriptome, proteome, and metabolome, will comprehensively and systematically reveal the pathophysiology of AD. 175 Genomics, the study of describing and quantifying all genes and mutations in an organism, can identify new loci that increase AD risk and is critical to understanding the underlying causes of AD, 176 such as high‐risk mutated genes of APP, PS1, Tau, APOE4, and so on. Transcriptomics, as a powerful approach to studying gene regulatory mechanisms, can map co‐expressed genomes of transcriptional programs associated with AD phenotypes. 177 Proteomics explores proteins that play a role in AD by studying protein expression, protein−protein interactions, and post‐translational modifications. 178 The changes of hippocampal proteins involved in insulin signaling and mitochondrial electron transport chain may be the crucial biological processes in AD progression. 179 In our laboratory, by integrated hippocampal proteomics and phosphoproteomics, abnormal phosphoration of GSK3β and Ppp3ca was found to induce mitochondrial dysfunction in low‐dose copper‐treated AD mice. 180 Yu et al. conducted platelet proteomics in patients with cognitive impairment in type 2 diabetes mellitus and reported that optineurin can act as a biomarker for predicting a cognitive decline, 83 and also found that PHB, UQCRH, GP1BA, and FINC may be the most promising platelet biomarkers for predicting cognitive decline in mild cognitive impairment and AD patients. 84 Metabolomics studies all the metabolites in the biological process of cells, which can reveal the persistent pathological process of AD, 181 for example, sphingolipids may act as biomarkers for early AD by metabolomics. 182
However, although signal omics can more or less provide information on the biological processes of AD progression, the regulatory and causal relationships among different levels of genes, mRNAs, regulatory factors, proteins, and metabolites are still unclear. For instance, although studies of the metabolome can be used to measure changes in biochemical pathways associated with AD pathogenesis, the relationship between systemic abnormalities of metabolism and AD pathogenesis is poorly understood, whereas integrating genetic, transcriptomic, metabolomic, and proteomic data in AD can enable the identification of AD‐specific metabolomic changes and their potential upstream genetic and transcriptional regulators. 183 Therefore, integrated multi‐omics research can explore the entire biological continuum, and may find key molecules in the progression of AD. Comprehensive and in‐depth multi‐omics research, which integrates multi‐level biological information and explains the interaction between components, is consistent with the concept of systems biology. 184 Multi‐omics analysis identified IVD, CYFIP1, and ADD2 as autoantibody biomarkers for distinguishing AD from controls, 185 biomarkers (FBP1, FBP2, RHOH, JPH2, ERAp2, and SCLT1) distinct from AD patients with APOE2 and APOE4, 186 ABCA1, CPT1A, adiponectin, and NGAL to be associated with AD pathology, 183 and 14‐3‐3 zeta/delta and clusterin associated with AD and cognitive decline. 187 Moreover, AD‐related multi‐omics data integration tools have been developed for the discovery of AD biomarkers and drug therapeutic targets, for example, Genome‐wide Positioning Systems Platform for Alzheimer's Drug Discovery (AlzGPS), which is used for the excavation of AD‐related targets and clinical‐related candidate drugs. 188 Another platform based on gene regulatory networks can integrate multi‐omics data to identify key disease pathways and driver genes. 189 Wang et al. obtained AD drug targets and biomarkers through a comprehensive analysis of multi‐omics data and animal genome‐scale metabolic model, which could eventually be validated and transformed into therapeutic or diagnostic methods. 190 Collectively, identifying the associations of AD‐related brain functional and structural changes by integrating multi‐omics studies will contribute to a more comprehensive molecular understanding of the disease pathophysiology. 191 , 192
For Parkinson's disease (PD), the second largest neurodegenerative disease in the world, its main clinical manifestations are resting tremors and movement disorders, accompanied by some mental problems, such as cognitive impairment and depression. 193 , 194 However, the early diagnosis of PD has been a difficult problem in clinical medicine, the research on the etiology and biomarkers has a very important medical value for early diagnosis. In terms of a single omics study, through GWAS analysis, genes closely related to PD have been screened, such as SCNA, MAPT, and LRRK2. 195 , 196 Zhang et al. showed that SSR1 (the signal sequence receptor subunit1) can be used as a biomarker for the early diagnosis of PD by analyzing the transcriptomics of peripheral blood of PD patients. 197 Proteomic analysis of cerebrospinal fluid of PD patients showed that OMD, CD44, VGF, PRL, and MAN2B1 were significantly correlated with PD clinical scores. 198 Metabolomic study of sebum shows that sebum can be used as a biomarker for PD diagnosis. 199 In the integration of multi‐omics, an integrated study of the PD gut microbiome and metabolome revealed that low short‐chain fatty acids were significantly associated with poorer cognition in PD, and lower butyrate levels were associated with worse postural instability gait disorder scores. 200
Finally, for ALS which is characterized by loss of upper and lower motor neurons, multi‐omics technology has also been applied to study biomarkers for its diagnosis and prognosis. 201 , 202 For instance, a multi‐omics integration approach not only identifies neurotrophic factors and histamine as potential therapeutic targets, but also provides additional guidance for the personalized medical applications for ALS. 203 GWAS have reported highly pathogenic mutations in ALS genes, such as C9ORF72, FUS, OPTN, SOD1, TARDBP, TBK1, and TDP‐43. 204 ST of SOD1‐G93A transgenic mice reveals sphingomyelin signaling as a therapeutic target for ALS. 205 UCHL1, MAP2, and GPNMB are reported to be promising biomarker candidates for ALS using proteomic studies of cerebrospinal fluid and spinal cord. 206 Patin et al. reported that arginine and proline metabolism may be used as targets for ALS therapy through muscle and brain metabolomics studies in SOD1‐G93A transgenic mice. 207 Moreover, omics has also reported many possible therapeutic targets or biological processes for neurodegenerative diseases. We reported that Mucolipin‐1 may be the target which can ameliorate mitophagy defect in 5×FAD mouse, 208 and confirmed that TrkB receptor agonist R13 promoted mitochondrial biogenesis and function in 5×FAD and SOD1‐G93A mouse by upregulated PGC‐1α, NRF1, and TFAM by mitochondriomics. 209 , 210
Overall, multi‐omics has many applications in the study of neurodegenerative biomarkers and therapeutic targets. It is believed that with the development of high‐throughput histology technologies and integration algorithms, multi‐omics technologies will reveal more in‐depth molecular network changes in neurodegenerative diseases.
5.3. Applications in aging research
In addition to the application in the basic and clinical research of disease, multi‐omics is also being explored in aging science. Aging is a time‐dependent physiological process characterized by DNA mutations, epigenetic alterations, abnormal protein aggregation and autophagy disturbances, immune impairment, mitochondrial dysfunction, telomere shortening, abnormal intracellular signaling, nutrient‐sensing obstacle, and so on. 211 These changes impair the normal function of cells and contribute to the development of age‐related diseases. And since aging is not only the greatest risk factor for many chronic diseases, but also a major cause of functional decline, there is a need to develop methods to measure the rate of aging in a given individual. The study of aging can be addressed by systematically describing the relationship between organ biomarkers, phenotypes (molecular biomarkers), and clinical presentation. 212 A major and future trend in the field of aging is the development of omics‐based biomarkers, and these biomarkers have greater potential for assessing multifactorial processes. Over the past few years, genes, gene products, epigenetic modifications, and/or metabolites during aging have been profiled by high‐throughput sequencing, mass spectrometry, and other techniques. Aging is a very complex process that is influenced by a myriad of factors in addition to genetic predisposition, and the extent to which genes influence variation in the human aging process remains controversial. 213 , 214
In particular, genomics analysis of centenarians provides insights into genetic susceptibility to exceptional longevity. 215 , 216 Human lifespan GWAS of more than 500,000 participants proved that previous research found that APOE, FOXO3, 217 and 5q33.3 218 are longevity genes, and proposed five new genes related to longevity heritability. 219 As epigenetic modifications are highly variable over the life cycle and potential biomarkers in response to aging, epigenomics shows a slow decline in total DNA methylation levels with age, and cytosine methylation containing CpG sites is hypermethylated or hypomethylation at different genomic locations with age. 220 , 221 The transcriptome is markedly altered during aging, and age‐associated transcriptome changes are highly tissue‐specific. 222 Mamoshina et al. identified tissue‐specific biomarkers of aging through transcriptomic analysis of muscle tissue using ML algorithms. 223 Plasma proteomic studies of healthy humans of various ages have found biomarkers highly correlated with age, such as growth differentiation factor 15. 224 Lehallier et al. analyzed the plasma proteomics of 4263 healthy people aged 18−95, and revealed specific changes in the proteome in the fourth, seventh, and eighty decades. 225 Compared with other omics, metabolomics has strong advantages in the sensitivity and predictability of the physiological state of the body in aging research. 226 Previous studies have reported significant changes in plasma concentrations of a large number of metabolites correlated with aging. 227 In a plasma metabolome study of a large cohort of men and women followed by up to 20 years follow‐up, higher concentrations of isocitrate and the bile acid taurocholate were associated with lower odds of longevity. 228 However, single‐omics studies do not systematically account for the deep relationships between genes, proteins, metabolites, and other molecules in the aging process. For instance, epigenomics alone cannot reveal how epigenetic changes specifically regulate transcriptional changes and gene expression, whereas integrating data from transposase‐accessible chromatin (ATAC‐seq), RNA‐seq, and ChIP‐seq can identify major regulatory elements and key genes of aging. 229 Through the integrated application of multi‐omics, molecules closely related to aging have been discovered, for example, transcription factors E2F4, TEAD1, and AP‐1 have been found to be key factors regulating aging by integrating transcriptome and epigenome. 230 Song et al. revealed that NAT1, PBX1, and RRM2 may be potential biomarkers of aging and aging‐related diseases by integrating ATAC‐seq, RNA‐seq, and ChIP‐seq in a multi‐omics analysis. 229 In order to facilitate the research of big aging‐related omics data, a multi‐omics database of aging biology has also been established, such as Aging Atlas, which provides user‐friendly functionalities to explore age‐related expression changes in genes, RNA, proteins, and so on. 231 At the same time, it has also developed a multi‐omics data analysis platform and comprehensive database that provide aging‐related data in multiple species, such as AgingBank, which provided experimentally supported multi‐omics data relevant to aging in multiple species. 232
5.4. Multi‐omics in the discovery of natural drug targets
Natural products are capable of selectively and specifically interacting with myriad molecular targets due to their complex molecular frameworks and diverse biological activities. 233 Biologically active natural products often readily penetrate cell membranes and disrupt the physiological levels of the genome, transcriptome, proteome, and metabolome. These phenomena indicate that natural products have interactive properties with specific molecular targets in organisms. The discovery of new drug targets or new pharmacological effects of drugs is of great significance for expanding the scope of drug indications. 234 Therefore, the discovery of precise drug targets is crucial to the development of new drugs. Significant progress has been made in drug target discovery through chemogenomics, chemoproteomics, label‐free proteomics, and bioinformatics approaches. 235 , 236 However, any single omics approach is insufficient to clearly define precise molecular targets in complex physiological networks, while an integrated multi‐omics approach can simultaneously elucidate, define, and validate multiple potential targets and mechanisms of candidate natural products. 237 , 238 For example, a single chemical proteomics strategy can obtain 14 possible targets of perfluorooctanoic acid (PFOA)‐induced toxicity and carcinogenic potential in humans; however, only the simultaneous combination of targeted metabolomics enabled the identification of acetyl‐CoA carboxylase 1 (ACACA, ACCa) and acetyl‐CoA carboxylase 2 (ACACB, ACCb) as bona fide binding targets of PFOA. 239 Using genomic and proteomic (DARTS, drug affinity responsive target stability, a method to identify targets of small molecules 240 ) integration, the natural product ecumicin was identified to inhibit Mycobacterium tuberculosis by targeting ClpC1. 241 The natural product baicalin improved diet‐induced obesity and hepatic steatosis through carnitine palmitoyltransferase 1 (CPT1), which was also discovered through chemical proteomics and bioinformatics (gene ontology). 242 Zhong et al. used transcriptomic and proteomic (DARTS) to discover that the natural product arctigenin played a role in renal protection through targeting PP2A. 243 Previous research applied metabolomics and chemical proteomics (ABPP) to identify acetyl‐CoA carboxylase 1 (Acaca) and acetyl‐CoA carboxylase 2 (Acacb) as binding targets of PFOA 239 (Table 2). Through these existing research reports, it can be shown that multi‐omics is a promising approach to the discovery of natural drug targets.
TABLE 2.
Published reports on multi‐omics discovery of natural drug targets.
| Methods | Compounds | Targets | Type of disease or model study | Potential applications | Ref |
|---|---|---|---|---|---|
| Transcriptomics and proteomics (DARTS) | Arctigenin | PP2A | Diabetic kidney disease | Hypertension; inflammation | 243 |
| Genomics and proteomics (DARTS) | Ecumicin | ClpC1 | Tuberculosis | Lead compounds for antituberculosis drugs | 241 |
| Chemical proteomics and bioinformatics (gene ontology) | Baicalin | CPT1 | Obesity and hepatic steatosis | Inflammation; antibacterial; obesity | 242 |
| Metabolomics and chemical proteomics (ABPP) | Perfluorooctanoic acid | Acaca, Acacb | PFOA‐induced liver toxicity | No | 239 |
| Proteomics (DARTS) and bioinformatics (gene docking) | Bithionol | NAD‐dependent dehydrogenases | Cryptococcus neoformans | Antifungal | 237 |
| ShRNA | Aurilide B | ATP1A1 | Mitochondria‐mediated apoptosis | No | 244 |
| SiRNA | QS11 | ARFGAP1 | Breast cancer | No | 245 |
| CRISPR | Ispinesib | Kinesin‐5 | Human cancer cells | Cancer | 246 |
| AI (machine learning) | β‐Lapachone | 5‐lipoxygenase | Human cancer cells | Cancer | 247 |
| AI (deep learning) | Ziprasidone | 5‐hydroxytryptamine receptor | No | No | 248 |
6. CHALLENGES AND FUTURE DIRECTIONS OF MULTI‐OMICS
Although multi‐omics plays an important role in promoting research on human diseases, aging, discovery of natural drug targets, and so on, it also faces many problems and challenges. Here, we will discuss some challenges and future directions for multi‐omics research.
6.1. Challenges and opportunities for multi‐omics integration
An early approach to multi‐omics analysis was the separate analysis of different types of data and then combined the results to obtain a comprehensive network of molecular interactions. With significant developments in this field, algorithmic meta‐analysis frameworks and methods have become the primary means of a comprehensive analysis of multi‐omics data. 92 , 249 However, multi‐omics integration analysis also faces some challenges and opportunities, such as missing value handling, heterogeneity between different omics, difficulties in interpreting multi‐omics models, and problems in data annotation, storage, and computing resources (Figure 7). 250 In terms of missing values in omics data, the treatment of missing values is a more critical issue in the integrated analysis of multi‐omics data. Missing values can lead to undetectable features in some samples, for example, in proteomic and metabolomic analysis. The complete measurements are not available for some samples in multi‐omics, which may lead to more missing values. For example, in cohort studies, not all individuals can collect all types of omics data, so the number of individuals with complete records is usually much smaller than the overall sample size. 251 And the missing values among various omics can influence the correlation analysis between different omics data, and interrupt the integration analysis between omics. While the missing values among omics can be solved by using a method of multiple imputation in a multiple factor analysis framework. 252 Missing values in some samples may be due to quality control procedures, which usually can be solved by using the k‐nearest neighbor weighted imputation method for trans‐omics block missing data, 253 such as MOFA, which can perform sample subgroup identification, data imputation, and abnormal sample detection for multi‐omics data. 135 However, missing value imputation methods can reduce the reliability of the resulting data set, 254 and generate data structures that violate the independence assumptions required by many statistical frameworks. Therefore, in integrated analyzes of multi‐omics data, sensitivity analysis was performed on missing value imputations to assess their impact on downstream analysis. In addition, for nonspecialist data processors, the researcher usually should try to avoid the intragroup or intergroup errors in samples from experimental manipulations, and minimize the testing times of omics instruments to avoid missing values due to batch benefits, especially for omics studies of large cohorts. In addition, the settings of instrument parameters need to be corrected in time to avoid data loss caused by instrument noise.
FIGURE 7.
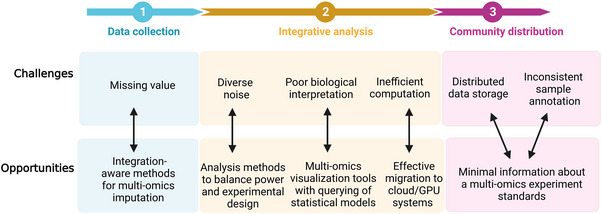
Challenges and opportunities for data integration of multi‐omics. The analysis challenges together with possible solutions are presented for three different levels, such as data collection, integrative analysis, and community distribution.
The heterogeneity among different omics is also an important factor affecting the integration of multi‐omics data. Different omics technologies have different precision levels, and the signal‐to‐noise ratio in multi‐omics measurements often affects the integration of multi‐omics data. For instance, proteomics favors the detection of abundantly expressed proteins, which is largely absent in transcriptomics. Due to this differential signature, analysis of the relationship between gene and protein expression is influenced by proteomic data. 81 Algorithms have been developed to estimate the optimal sample size required for different omics to achieve a given statistical power. 81 However, this treatment can lead to different amounts of sample data collected by different types of omics, resulting in many multi‐omics integration statistics methods not being applicable. Therefore, there is a need to find new analytical approaches that enable homogenous capabilities across multi‐omics data. In addition, the interpretability of multi‐omics models has outstanding advantages for understanding the complexity of life at the molecular level. In most cases, however, interpretability refers to the identification of biomarkers and the biological processes in which they are involved. Functional enrichment analysis is often used in the interpretation of multi‐omics data, 255 , 256 but these methods also have inherent limitations in providing mechanistic insights across molecular layers. Pathway diagrams have also been used for interpretation of multi‐omics data, 257 but this approach lacks the flexibility to reveal novel regulatory relationships. In addition, Networks (such as Cytoscape 258 ) that combine multi‐omics data, even though more general, can easily become too large, complex, and difficult to interpret. In the future, it is necessary to develop a calculation method combining the mathematical formula of multi‐layer adjustment with interactive visualization.
Data annotation and storage is also a challenge for multi‐omics data integration. Integrated annotation of multi‐omics data often requires software provided by technology suppliers; however, this software may be not publicly shared. Despite the abundance of publicly available multi‐omics studies and databases, retrieving multi‐omics data for specific biological entities remains a challenge. It is difficult to connect different types of omics data under the same research topic on various data platforms. Typically, various omics data sets are hosted in specific types of repositories, such as European Genome Archive (EGA, a sequencing database), 259 MetaboLights (a metabolite database), 260 and ProteomeXchang (a protein database). 261 Therefore, hyperlinks are the only option to connect the same study across different omics databases. For example, construct metadata summaries to concatenate multiple omics data files from the same sample and then used them on a public database file. 262 The European Bioinformatics Institute has developed a unified query system that can quickly find omics data sets responding to the same keyword across repositories. 263 In the future, metadata description frameworks that define batch, experimental conditions, preprocessing, and replication will need to enable the linking of samples across omics data files and enable efficient reuse of multiple omics data.
In the future, with the expansion of samples and features, multi‐omics data will become more complex, and the development of precision medicine requires the integration of multi‐omics data sets from large cohorts of patients. 264 The inclusion of other types of data, such as radiomics and single‐cell omics into multi‐omics research, will further increase the complexity of data processing. It is worth noting that cloud servers have been developed to help large‐scale data storage and data sharing, saving the operation and maintenance costs of computing infrastructure. 265 Moreover, the processing of large amounts of data also requires high computing power, and cloud computing infrastructure needs to use sufficiently powerful computing units to achieve fast multi‐omics data processing, such as the often‐used graphics processing unit (GPU). 266 GPUs can also meet the demand of the DL processing, which is often used in multi‐omics data analysis. 267 In summary, developing efficient computing software and processing technology based on cloud services will facilitate the integration of multi‐omics data.
6.2. Single‐cell analysis as the future direction of multi‐omics
Single‐cell multi‐omics is the most promising development direction in the field of multi‐omics, which can infer regulatory models between multilayer molecules with better accuracy. Accounting for cell type heterogeneity will facilitate the modeling of complex biological processes (tumor 268 ) and understanding of function in highly differentiated organs (brain 4 ). Multiple omics studies on the same cell have been successful, such as combining RNA‐sequencing, DNA methylation, and/or protein abundance at the single‐cell level. 269 , 270 In view of the multiple omics data collected on the same cell, as they have the same cell information, the characteristics of different modalities can be matched, 271 which is conducive to the subsequent integrated analysis, thereby improving the level of multi‐omics research. The main components of single‐cell multi‐omics analysis include single‐cell isolation, barcoding, and sequencing techniques for measuring multiple types of molecules from the same cell, as well as a comprehensive analysis of molecules measured at the single‐cell level to identify cell types and their functions related to pathophysiological processes based on molecular characteristics. The single‐cell omics usually focus on the data of mRNA‐genome, mRNA‐DNA methylation, mRNA‐chromatin accessibility, and mRNA‐protein. Here, we briefly introduce some detection technologies of the above single‐cell multi‐omics from the previous review 270 (Table 3).
TABLE 3.
Some technologies for single‐cell multi‐omics.
| Category | Technologies | Separation methods | Feature | Ref |
|---|---|---|---|---|
| Genome‐transcriptome | G&T‐seq | Flow cytometry; bead‐based separation | Medium cell throughput; automation | 272 |
| DR‐seq | Cell picking by pipette; preamplification and tagging of DNA and RNA followed by splitting | Low cell throughput | 273 | |
| Transcriptome‐DNA methylome | scM&T‐seq | Flow cytometry; bead‐based separation followed by bisulfite treatment | Medium cell throughput; automation | 277 |
| scMT‐seq | Micropipetting for isolation of single nuclei | Low cell throughput; partial automation | 281 | |
| Transcriptome‐chromatin accessibility | sci‐CAR | Combinatorial indexing; lysate splitting followed by library preparation | High cell throughput | 282 |
| SNARE‐seq | Microfluidic channels; open chromatin tagmentation followed by dual‐omics capture | High cell throughput | 283 | |
| Transcriptome‐proteome | PEA/STA | Microfluidic channels; reverse transcription of PEA probe and RNA followed by targeted amplification | Medium cell throughput; automation | 279 |
| PLAYR | Flow or mass cytometry; detection of amplified product of PLAYR probe pair and antibody staining | High cell throughput | 280 |
For integrated analysis of single‐cell genome and transcriptome data, there have several approaches to achieve this analysis, such as genome and transcriptome sequencing (G&T‐seq), 272 gDNA‐mRNA sequencing (DR‐seq), 273 and so on. The approaches for single‐cell whole‐genome sequencing (scWGS) contain multiple displacement amplification, 274 PicoPLEX (Rubicon Genomics PicoPLEX Kit), and so on, while the methods for scRNA‐seq include switching mechanism at the 5′end of the RNA transcript (Smart‐seq), 275 and cell expression by linear amplification and sequencing (CEL‐seq). 276 G&T‐seq uses the oligo‐dT‐coated magnetic beads to separate poly‐A‐tailed mRNAs from gDNA, and then sequencing by Smart‐seq2 and scWGS protocols, respectively. With this technology, Macaulay et al. reported that genomic imbalance on the chromosomes of a subpopulation of HCC38‐BL cells was consistent with changes in gene expression in unbalanced regions. 272 The technology for integrating the signal‐cell data of transcriptome and epigenome is that the gDNA and RNA in the same single cell were separated and amplified using the G&T‐seq program, and then the single‐cell methylome and transcriptome sequencing (scBS‐seq) program was applied to the amplified gDNA to generate DNA methylome data. 277 Using this technique, previous research found a link between epigenetic and transcriptional signatures associated with tissue‐specific mouse stem cell aging. 278 The integration analysis of transcriptome and proteome data can be conducted by proximity extension assay/specific RNA target amplification (PEA/STA), 279 proximity ligation assay for RNA (PLAYR), 280 and so on. For PEA/STA method, adjacent‐dependent hybridization of DNA oligonucleotides attached to antibody pairs was performed using adjacent extension assay (PEA) labeled antibody pairs to convert proteins into DNA oligonucleotides, and RT mRNA using random RT primers to generate cDNA. Then, the DNA oligonucleotides and cDNA were amplified by PCR and quantified using quantitative PCR or sequencing. Using the PEA approach, Genshaft et al. found that a subpopulation of glioblastoma cells showed significant changes in mRNA and protein abundance following BMP4 treatment, and the proteins were able to define the response more accurately to BMP4. 279
In addition, the analysis for single‐cell multi‐omics data is also complex. The general approaches can be divided as follows: correlation analysis between single‐cell mono‐omics data, combined analysis of all types of single‐cell omics data to generate an overall single‐cell atlas, analyze one type of single‐cell data and then integrate another single‐cell data type. For example, REAP‐seq was used to analyze PBMCs and calculated the Pearson correlation between single‐cell mRNA and protein expression of immune cell markers, and found that protein quantification was more sensitive than mRNA quantification for markers with low mRNA expression. 284 Stoeckius et al. integrated cellular protein and transcriptome measurements into an efficient, single‐cell readout, and found that the protein expression level can be used to further subdivide the cell population identified by RNA‐seq with subtle mRNA expression differences, such as the (NK cell population. 285 In addition, there are other methods for single‐cell multi‐omics integration, such as bi‐order canonical correlation analysis (bi‐CCA), 286 UnpairReg (regression analysis on unpaired observations), 287 PyLiger, 288 nonnegative matrix factorization algorithm (UINMF), 289 and so on.
However, although single‐cell multi‐omics data provide a wealth of biological information, their integrated analysis faces the same types of challenges described above. In addition, there is a lack of technology capable of generating multimodal data, resulting in comprehensive studies that often rely on the integration of modalities from different data sets. Notably, cross‐omics matching of cell types by using publicly available data from different modalities can greatly increase the number of single‐cell multi‐omics studies, such as MATCHER, an ensemble method dedicated to single‐cell‐specific data. 290 Integration of single‐cell multi‐omics data also hinders the use of batch data processing methods due to specific technical limitations, such as cell‐level noise. Therefore, in the future, it also needs to develop relevant methods to interpret the biological significance of the data generated from a single type of cell.
In a word, the future field of single‐cell multi‐omics will not only address existing technical limitations, but also is a significant breakthrough in single‐cell ChIP‐sep, proteomics, metabolomics, and single‐cell omics combined with cellular‐level imaging and morphological analysis. Furthermore, future single‐cell multi‐omics will require the development of methods to decipher the biological interpretation of the complex signals linked between the different modalities. Finally, incorporating patient clinical data into single‐cell omics studies will help explain molecular regulatory models of health and disease at the cellular level.
6.3. Application of multi‐omics in precision medicine
The concept of precision medicine refers to an emerging approach to provide disease treatment options and prevention strategies based on individualized genetic, environmental, and lifestyle factors, in other words, the shift from one‐size‐fits‐all medicine to a broader paradigm of precision medicine (right drug, right patient, right dose, right time). 291 Clinical data can be categorized into phenotypic (physiological assessment, disease scoring, imaging, health questionnaires, etc.) or multi‐omics molecular (genomics, transcriptomics, proteomics and metabolomics, etc.). Genetics research combined with clinical data provides the basis for precision medicine, such as translational genomics. 292 And the study of blood proteomics can provide pathogenic target proteins for personalized therapy. 293 Moreover, based on multi‐omics, there are significant advantages in explaining the molecular patterns of complex diseases, and identifying key nodes of disease at multiple levels of convergence, thus maximizing the precision of identifying new drug targets, endotypes, or biomarkers. However, the development of biomarkers based on multi‐omics is a key step in the realization of precision medicine. 294 For example, the development of biomarker‐based companion diagnostics achieves precision medicine in the field of oncology. 295 , 296 While multi‐omics has difficulty in explaining the complex interactions between genetics, gene regulation, and proteins at multiple biological levels, networks can use their network topology to identify key nodes that are critical for screening drug targets and biomarkers for complex diseases. 297 , 298 In addition, based on the ML and DL methods discussed above, it is also helpful for multi‐omics to play an important role in the identification of biomarkers, and ultimately realize precision medicine. 129 At present, there are also many review articles clarifying the wide application of multi‐omics in tumor and immune‐related precision medicine. 299 , 300 Similarly, in the field of neurodegenerative diseases, there are also research initiatives based on multi‐omics technology to realize precision medicine, such as the multi‐omics study of the gut microbial ecosystem in Parkinson's disease, 200 brain tissue in Alzheimer's disease, 75 induced pluripotent stem cell lines from ALS patients. 301 In addition, multi‐omics has also been applied to precision medicine research on metabolic‐related diseases, such as type 2 diabetes. 302
In general, multi‐omics plays a pivotal role in the development of precision medicine. With the development of high‐throughput technology and artificial intelligence algorithms, precision medicine based on multi‐omics will become the main trend of disease diagnosis, treatment, and prognosis in the future.
7. CONCLUSION
The study of biological multi‐omics data systematically reveals the physiological or pathological molecular map in health or disease state, and the development of multi‐omics technology is mainly to achieve precision medicine in the future. It is undeniable that multi‐omics is becoming increasingly important in medical research, and further development of its technology will likely explain the pathogenesis of major diseases (such as cancer and neurodegenerative diseases) and provide an important molecular theoretical basis for the clinical diagnosis, treatment, and prognosis. However, the current development of multi‐omics is also facing many challenges, such as the design of multi‐omics experiments mentioned above, especially the integrated analysis of multi‐omics data.
With the development of medical treatment and high‐throughput technology, various omics technologies are also integrated into disease research. In addition to the application of advanced omics technology, high‐level research reports often show their innovation and novelty in very sophisticated experimental design or analysis methods. Therefore, at the beginning of the experiment, special attention should be paid to whether the design of the experiment corresponds to the expected research purpose, after all, omics analysis is often both time‐consuming and costly. When conducting multi‐omics research, we must consider whether the type of omics, analysis techniques, and collected data indicators can achieve the set research goals or solve existing research problems. In other words, only a multi‐omics design approach suitable for explaining the purpose of the experiment is needed here, rather than just forcing the use of as many types of omics in the study as possible. It is believed that some suggestions on the design of multi‐omics experiments in this review may be helpful to guide researchers to carry out multi‐omics experiments in the future.
In addition, the large amount of data sets generated by multi‐omics also brings great difficulties to the data analysis. In this review, we also discussed some of the more commonly used multi‐omics data analysis and integration methods. For researchers with relatively weak computing algorithms, the way of multi‐omics data integration is often based on the correlation between various omics data and the discovery of related upper‐lower‐level regulatory networks, so as to further explain the biological significance of the data. However, researchers who are more proficient in bioinformatics and algorithms usually develop ML or DL‐related algorithms to integrate multi‐omics data, since ML and DL have significant advantages in multi‐omics data integration. For instance, ML and DL have excellent data processing capabilities for both linear and nonlinear data, and use supervised or unsupervised learning capabilities for disease prediction or biomarker discovery. Fortunately, open‐source platforms and software are also being developed to help noncomputational medical researchers integrate and analyze multi‐omics data, such as Hiplot and the “Wu Kong” platform (Table 4). Therefore, as more and more open‐source tools will be developed and accessible for the integrated analysis of multi‐omics data in the future, the application of multi‐omics will become more and more easy for most researchers.
TABLE 4.
Open‐source tools for the integrated analysis of multi‐omics data.
| Tool name | Omics type | Website | Advantages | Ref |
|---|---|---|---|---|
| OmicsAnalyst | Transcriptomics, proteomics, metabolomics, microbiome | https://www.omicsanalyst.ca | Correlation network analysis, cluster heat map analysis, and dimension reduction analysis all have comprehensive options for parameter customization, view customization, and target analysis. | 303 |
| OmicsNet 2.0 | Genomics, transcriptomics, proteomics, metabolomics, microbiome | https://www.omicsnet.ca/ |
Web‐based multi‐omics analysis platform supporting 2D and 3D network visualization exploration. The Random Walk with Restart algorithm can be used to search for candidate disease markers. |
115 |
| Hiplot | Genomics, transcriptomics, proteomics | https://hiplot.com.cn/ | Have a variety of omics analysis, bioinformation analysis modules, especially in the possession of high‐quality visual mapping tools. | 304 |
| Wu Kong |
Genomics, transcriptomics, proteomics, phosphoproteomics Metabolomics |
https://www.omicsolution.com/ | Complete one‐click omics result generation, as well as a variety of statistical analysis, functional analysis, visualization analysis, clinical data analysis, multi‐omics cross‐association, and other modules. | 305 |
| OmicStudio | Genomics, transcriptomics, proteomics, metabolomics, microbiome | https://www.omicstudio.cn/ | A variety of modules for single‐cell omics analysis, easy data entry types, and high‐quality visual mapping tools are available. | 306 |
| Majorbio Cloud | Genomics, transcriptomics, proteomics, metabolomics, microbiome | https://cloud.majorbio.com/ | Provides interactive analysis reports that produce analysis results from bioinformation workflows into Web‐based interactive analysis reports. | 307 |
In conclusion, with the development of high‐throughput technology, multi‐omics technology will become inseparable in the research of diseases, and finally realize precision medicine.
AUTHOR CONTRIBUTIONS
C.C. and J.W. drafted the manuscript and Figures. D.P., X.W., Y.X., J.Y., and L.W. helped Figure modification. X.Y., M.Y., and G.P.L. revised the manuscript. All authors have read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
Not appllicable.
ACKNOWLEDGMENTS
All Figures were made using Biorender and acquired publishing license (agreement number: MQ25BZH9EN, NL25BZJVTK, XH25BZHUUX, RN25BZINSJ, RE25BZISBC, NB25EO2PZS, MG25BZK4TS, YQ25BZHMQT). The publishing license of all the figures in this review are available upon reasonable request.
Chen C, Wang J, Pan D, et al. Applications of multi‐omics analysis in human diseases. MedComm. 2023;4:e315. 10.1002/mco2.315
Contributor Information
Xifei Yang, Email: xifeiyang@gmail.com.
Min Yang, Email: yangmin@jsinm.org.
Gong‐Ping Liu, Email: liugp111@mail.hust.edu.cn.
DATA AVAILABILITY STATEMENT
Data in this study are available upon reasonable request.
REFERENCES
- 1. Kreitmaier P, Katsoula G, Zeggini E. Insights from multi‐omics integration in complex disease primary tissues. Trends Genet. 2023;39(1):46‐58. [DOI] [PubMed] [Google Scholar]
- 2. Nie C, Li Y, Li R, et al. Distinct biological ages of organs and systems identified from a multi‐omics study. Cell Rep. 2022;38(10):110459. [DOI] [PubMed] [Google Scholar]
- 3. Joshi A, Rienks M, Theofilatos K, Mayr M. Systems biology in cardiovascular disease: a multiomics approach. Nat Rev Cardiol. 2021;18(5):313‐330. [DOI] [PubMed] [Google Scholar]
- 4. Armand EJ, Li J, Xie F, Luo C, Mukamel EA. Single‐cell sequencing of brain cell transcriptomes and epigenomes. Neuron. 2021;109(1):11‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pelka K, Hofree M, Chen JH, et al. Spatially organized multicellular immune hubs in human colorectal cancer. Cell. 2021;184(18):4734‐4752. e4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hasin Y, Seldin M, Lusis A. Multi‐omics approaches to disease. Genome Biol. 2017;18(1):83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Collins FS, Morgan M, Patrinos A. The Human Genome Project: lessons from large‐scale biology. Science. 2003;300(5617):286‐290. [DOI] [PubMed] [Google Scholar]
- 8. Horner DS, Pavesi G, Castrignano T, et al. Bioinformatics approaches for genomics and post genomics applications of next‐generation sequencing. Brief Bioinform. 2010;11(2):181‐197. [DOI] [PubMed] [Google Scholar]
- 9. Ragoussis J. Genotyping technologies for genetic research. Annu Rev Genomics Hum Genet. 2009;10:117‐133. [DOI] [PubMed] [Google Scholar]
- 10. Zhao L, Shi Y, Lau HC, et al. Uncovering 1058 novel human enteric DNA viruses through deep long‐read third‐generation sequencing and their clinical impact. Gastroenterology. 2022;163(3):699‐711. [DOI] [PubMed] [Google Scholar]
- 11. Flannick J, Mercader JM, Fuchsberger C, et al. Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature. 2019;570(7759):71‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Uffelmann E, Huang QQ, Munung NS, et al. Genome‐wide association studies. Nat Rev Methods Primers. 2021;1(1):59. [Google Scholar]
- 13. Tam V, Patel N, Turcotte M, Bosse Y, Pare G, Meyre D. Benefits and limitations of genome‐wide association studies. Nat Rev Genet. 2019;20(8):467‐484. [DOI] [PubMed] [Google Scholar]
- 14. Schmitz SU, Grote P, Herrmann BG. Mechanisms of long noncoding RNA function in development and disease. Cell Mol Life Sci. 2016;73(13):2491‐2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arnes L, Akerman I, Balderes DA, Ferrer J, Sussel L. Betalinc1 encodes a long noncoding RNA that regulates islet beta‐cell formation and function. Genes Dev. 2016;30(5):502‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gupta RA, Shah N, Wang KC, et al. Long non‐coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464(7291):1071‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rai AK, Lee B, Hebbard C, Uchida S, Garikipati VNS. Decoding the complexity of circular RNAs in cardiovascular disease. Pharmacol Res. 2021;171:105766. [DOI] [PubMed] [Google Scholar]
- 18. Mehta SL, Dempsey RJ, Vemuganti R. Role of circular RNAs in brain development and CNS diseases. Prog Neurobiol. 2020;186:101746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li J, Sun D, Pu W, Wang J, Peng Y. Circular RNAs in cancer: biogenesis, function, and clinical significance. Trends Cancer. 2020;6(4):319‐336. [DOI] [PubMed] [Google Scholar]
- 20. Wang Z, Gerstein M, Snyder M. RNA‐Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y, Wang D, Peng M, et al. Single‐cell RNA sequencing in cancer research. J Exp Clin Cancer Res. 2021;40(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen Y, Colonna M. Microglia in Alzheimer's disease at single‐cell level. Are there common patterns in humans and mice? J Exp Med. 2021;218(9):e20202717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rogers S, Girolami M, Kolch W, et al. Investigating the correspondence between transcriptomic and proteomic expression profiles using coupled cluster models. Bioinformatics. 2008;24(24):2894‐2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weston AD, Hood L. Systems biology, proteomics, and the future of health care: toward predictive, preventative, and personalized medicine. J Proteome Res. 2004;3(2):179‐196. [DOI] [PubMed] [Google Scholar]
- 25. Chen X, Sun Y, Zhang T, Shu L, Roepstorff P, Yang F. Quantitative proteomics using isobaric labeling: a practical guide. Genomics Proteomics Bioinformatics. 2021;19(5):689‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jia J, Jin J, Chen Q, et al. Eukaryotic expression, Co‐IP and MS identify BMPR‐1B protein–protein interaction network. Biol Res. 2020;53(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mann M, Jensen ON. Proteomic analysis of post‐translational modifications. Nat Biotechnol. 2003;21(3):255‐261. [DOI] [PubMed] [Google Scholar]
- 28. Wu R, Haas W, Dephoure N, et al. A large‐scale method to measure absolute protein phosphorylation stoichiometries. Nat Methods. 2011;8(8):677‐683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Batista TM, Jayavelu AK, Wewer Albrechtsen NJ, et al. A cell‐autonomous signature of dysregulated protein phosphorylation underlies muscle insulin resistance in type 2 diabetes. Cell Metab. 2020;32(5):844‐859. e845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu JY, Zhang C, Wang X, et al. Integrative proteomic characterization of human lung adenocarcinoma. Cell. 2020;182(1):245‐261. e217. [DOI] [PubMed] [Google Scholar]
- 31. Li Y, Xu J, Lu Y, et al. DRAK2 aggravates nonalcoholic fatty liver disease progression through SRSF6‐associated RNA alternative splicing. Cell Metab. 2021;33(10):2004‐2020. e2009. [DOI] [PubMed] [Google Scholar]
- 32. Morshed N, Ralvenius WT, Nott A, et al. Phosphoproteomics identifies microglial Siglec‐F inflammatory response during neurodegeneration. Mol Syst Biol. 2020;16(12):e9819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang R, Li B, Lam SM, Shui G. Integration of lipidomics and metabolomics for in‐depth understanding of cellular mechanism and disease progression. J Genet Genomics. 2020;47(2):69‐83. [DOI] [PubMed] [Google Scholar]
- 34. Newgard CB. Metabolomics and metabolic diseases: where do we stand? Cell Metab. 2017;25(1):43‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cui L, Lu H, Lee YH. Challenges and emergent solutions for LC‐MS/MS based untargeted metabolomics in diseases. Mass Spectrom Rev. 2018;37(6):772‐792. [DOI] [PubMed] [Google Scholar]
- 36. Cavicchioli MV, Santorsola M, Balboni N, Mercatelli D, Giorgi FM. Prediction of metabolic profiles from transcriptomics data in human cancer cell lines. Int J Mol Sci. 2022;23(7):e20202717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clos‐Garcia M, Andres‐Marin N, Fernandez‐Eulate G, et al. Gut microbiome and serum metabolome analyses identify molecular biomarkers and altered glutamate metabolism in fibromyalgia. EBioMedicine. 2019;46:499‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Previs MJ, O'Leary TS, Morley MP, et al. Defects in the proteome and metabolome in human hypertrophic cardiomyopathy. Circ Heart Fail. 2022;15(6):e009521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wishart DS, Guo A, Oler E, et al. HMDB 5.0: the Human Metabolome Database for 2022. Nucleic Acids Res. 2022;50(D1):D622‐D631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pang Z, Zhou G, Ewald J, et al. Using MetaboAnalyst 5.0 for LC‐HRMS spectra processing, multi‐omics integration and covariate adjustment of global metabolomics data. Nat Protoc. 2022;17(8):1735‐1761. [DOI] [PubMed] [Google Scholar]
- 41. Viant MR, Rosenblum ES, Tieerdema RS. NMR‐based metabolomics: a powerful approach for characterizing the effects of environmental stressors on organism health. Environ Sci Technol. 2003;37(21):4982‐4989. [DOI] [PubMed] [Google Scholar]
- 42. Halket JM, Waterman D, Przyborowska AM, Patel RK, Fraser PD, Bramley PM. Chemical derivatization and mass spectral libraries in metabolic profiling by GC/MS and LC/MS/MS. J Exp Bot. 2005;56(410):219‐243. [DOI] [PubMed] [Google Scholar]
- 43. Soga T, Imaizumi M. Capillary electrophoresis method for the analysis of inorganic anions, organic acids, amino acids, nucleotides, carbohydrates and other anionic compounds. Electrophoresis. 2001;22(16):3418‐3425. [DOI] [PubMed] [Google Scholar]
- 44. Kashima Y, Sakamoto Y, Kaneko K, Seki M, Suzuki Y, Suzuki A. Single‐cell sequencing techniques from individual to multiomics analyses. Exp Mol Med. 2020;52(9):1419‐1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Suzuki A, Matsushima K, Makinoshima H, et al. Single‐cell analysis of lung adenocarcinoma cell lines reveals diverse expression patterns of individual cells invoked by a molecular target drug treatment. Genome Biol. 2015;16(1):66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McMahon CM, Ferng T, Canaani J, et al. Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 2019;9(8):1050‐1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Grosselin K, Durand A, Marsolier J, et al. High‐throughput single‐cell ChIP‐seq identifies heterogeneity of chromatin states in breast cancer. Nat Genet. 2019;51(6):1060‐1066. [DOI] [PubMed] [Google Scholar]
- 48. Perkel JM. Single‐cell proteomics takes centre stage. Nature. 2021;597(7877):580‐582. [DOI] [PubMed] [Google Scholar]
- 49. Mahdessian D, Cesnik AJ, Gnann C, et al. Spatiotemporal dissection of the cell cycle with single‐cell proteogenomics. Nature. 2021;590(7847):649‐654. [DOI] [PubMed] [Google Scholar]
- 50. Chen H, Murray E, Sinha A, et al. Dissecting mammalian spermatogenesis using spatial transcriptomics. Cell Rep. 2021;37(5):109915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Andersson A, Larsson L, Stenbeck L, et al. Spatial deconvolution of HER2‐positive breast cancer delineates tumor‐associated cell type interactions. Nat Commun. 2021;12(1):6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kaufmann M, Schaupp AL, Sun R, et al. Identification of early neurodegenerative pathways in progressive multiple sclerosis. Nat Neurosci. 2022;25(7):944‐955. [DOI] [PubMed] [Google Scholar]
- 53. Stricker SH, Koferle A, Beck S. From profiles to function in epigenomics. Nat Rev Genet. 2017;18(1):51‐66. [DOI] [PubMed] [Google Scholar]
- 54. Zhu J, Adli M, Zou JY, et al. Genome‐wide chromatin state transitions associated with developmental and environmental cues. Cell. 2013;152(3):642‐654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Taudt A, Colome‐Tatche M, Johannes F. Genetic sources of population epigenomic variation. Nat Rev Genet. 2016;17(6):319‐332. [DOI] [PubMed] [Google Scholar]
- 56. Ling C, Ronn T. Epigenetics in human obesity and type 2 diabetes. Cell Metab. 2019;29(5):1028‐1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. van der Harst P, de Windt LJ, Chambers JC. Translational perspective on epigenetics in cardiovascular disease. J Am Coll Cardiol. 2017;70(5):590‐606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12‐27. [DOI] [PubMed] [Google Scholar]
- 59. Bernstein BE, Stamatoyannopoulos JA, Costello JF, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28(10):1045‐1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Berg G, Rybakova D, Fischer D, et al. Microbiome definition re‐visited: old concepts and new challenges. Microbiome. 2020;8(1):103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lax S, Smith DP, Hampton‐Marcell J, et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science. 2014;345(6200):1048‐1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cryan JF, O'Riordan KJ, Sandhu K, Peterson V, Dinan TG. The gut microbiome in neurological disorders. Lancet Neurol. 2020;19(2):179‐194. [DOI] [PubMed] [Google Scholar]
- 63. Wang X, Yang S, Li S, et al. Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut. 2020;69(12):2131‐2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cheng WY, Wu CY, Yu J. The role of gut microbiota in cancer treatment: friend or foe? Gut. 2020;69(10):1867‐1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Claesson MJ, Wang Q, O'Sullivan O, et al. Comparison of two next‐generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 2010;38(22):e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Uribe CF, Mathotaarachchi S, Gaudet V, et al. Machine learning in nuclear medicine: Part 1‐Introduction. J Nucl Med. 2019;60(4):451‐458. [DOI] [PubMed] [Google Scholar]
- 67. Lambin P, Rios‐Velazquez E, Leijenaar R, et al. Radiomics: extracting more information from medical images using advanced feature analysis. Eur J Cancer. 2012;48(4):441‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kumar V, Gu Y, Basu S, et al. Radiomics: the process and the challenges. Magn Reson Imaging. 2012;30(9):1234‐1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. van Rossum PS, Fried DV, Zhang L, et al. The incremental value of subjective and quantitative assessment of 18F‐FDG PET for the prediction of pathologic complete response to preoperative chemoradiotherapy in esophageal cancer. J Nucl Med. 2016;57(5):691‐700. [DOI] [PubMed] [Google Scholar]
- 70. van Helden EJ, Vacher YJL, van Wieringen WN, et al. Radiomics analysis of pre‐treatment [(18)F]FDG PET/CT for patients with metastatic colorectal cancer undergoing palliative systemic treatment. Eur J Nucl Med Mol Imaging. 2018;45(13):2307‐2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mayerhoefer ME, Materka A, Langs G, et al. Introduction to radiomics. J Nucl Med. 2020;61(4):488‐495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nakagawa S, Cuthill IC. Effect size, confidence interval and statistical significance: a practical guide for biologists. Biol Rev Camb Philos Soc. 2007;82(4):591‐605. [DOI] [PubMed] [Google Scholar]
- 73. Duan D, Goemans N, Takeda S, Mercuri E, Aartsma‐Rus A. Duchenne muscular dystrophy. Nat Rev Dis Primers. 2021;7(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. van Spronsen FJ, Blau N, Harding C, Burlina A, Longo N, Bosch AM. Phenylketonuria. Nat Rev Dis Primers. 2021;7(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bai B, Wang X, Li Y, et al. Deep multilayer brain proteomics identifies molecular networks in Alzheimer's disease progression. Neuron. 2020;105(6):975‐991. e977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Barisano G, Kisler K, Wilkinson B, et al. A “multi‐omics” analysis of blood–brain barrier and synaptic dysfunction in APOE4 mice. J Exp Med. 2022;219(11):e20221137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bundy JL, Vied C, Badger C, Nowakowski RS. Sex‐biased hippocampal pathology in the 5XFAD mouse model of Alzheimer's disease: a multi‐omic analysis. J Comp Neurol. 2019;527(2):462‐475. [DOI] [PubMed] [Google Scholar]
- 78. Sudlow C, Gallacher J, Allen N, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12(3):e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. HM AE, Veidal SS, Feigh M, et al. Multi‐omics characterization of a diet‐induced obese model of non‐alcoholic steatohepatitis. Sci Rep. 2020;10(1):1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gjoneska E, Pfenning AR, Mathys H, et al. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer's disease. Nature. 2015;518(7539):365‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tarazona S, Balzano‐Nogueira L, Gomez‐Cabrero D, et al. Harmonization of quality metrics and power calculation in multi‐omic studies. Nat Commun. 2020;11(1):3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gillette MA, Satpathy S, Cao S, et al. Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell. 2020;182(1):200‐225. e235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yu H, Liu Y, He T, et al. Platelet biomarkers identifying mild cognitive impairment in type 2 diabetes patients. Aging Cell. 2021;20(10):e13469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Yu H, Liu Y, He B, et al. Platelet biomarkers for a descending cognitive function: a proteomic approach. Aging Cell. 2021;20(5):e13358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yang Y, Knol MJ, Wang R, et al. Epigenetic and integrative cross‐omics analyses of cerebral white matter hyperintensities on MRI. Brain. 2023;146(2):492–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Jiang J, Wang M, Alberts I, et al. Using radiomics‐based modelling to predict individual progression from mild cognitive impairment to Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2022;49(7):2163‐2173. [DOI] [PubMed] [Google Scholar]
- 87. Schurch NJ, Schofield P, Gierlinski M, et al. How many biological replicates are needed in an RNA‐seq experiment and which differential expression tool should you use? RNA. 2016;22(6):839‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Huang KK, Huang J, Wu JKL, et al. Long‐read transcriptome sequencing reveals abundant promoter diversity in distinct molecular subtypes of gastric cancer. Genome Biol. 2021;22(1):44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Goh WWB, Wang W, Wong L. Why batch effects matter in omics data, and how to avoid them. Trends Biotechnol. 2017;35(6):498‐507. [DOI] [PubMed] [Google Scholar]
- 90. Moossavi S, Fehr K, Khafipour E, Azad MB. Repeatability and reproducibility assessment in a large‐scale population‐based microbiota study: case study on human milk microbiota. Microbiome. 2021;9(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Palsson B, Zengler K. The challenges of integrating multi‐omic data sets. Nat Chem Biol. 2010;6(11):787‐789. [DOI] [PubMed] [Google Scholar]
- 92. Bersanelli M, Mosca E, Remondini D, et al. Methods for the integration of multi‐omics data: mathematical aspects. BMC Bioinformatics. 2016;17(2):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Antonelli J, Claggett BL, Henglin M, et al. Statistical workflow for feature selection in human metabolomics data. Metabolites. 2019;9(7):143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Liew AW, Law NF, Yan H. Missing value imputation for gene expression data: computational techniques to recover missing data from available information. Brief Bioinform. 2011;12(5):498‐513. [DOI] [PubMed] [Google Scholar]
- 95. Vivian J, Eizenga JM, Beale HC, Vaske OM, Paten B. Bayesian framework for detecting gene expression outliers in individual samples. JCO Clin Cancer Inform. 2020;4:160‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wang D, Eraslan B, Wieland T, et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol Syst Biol. 2019;15(2):e8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kumar D, Bansal G, Narang A, Basak T, Abbas T, Dash D. Integrating transcriptome and proteome profiling: strategies and applications. Proteomics. 2016;16(19):2533‐2544. [DOI] [PubMed] [Google Scholar]
- 98. Mangleburg CG, Wu T, Yalamanchili HK, et al. Integrated analysis of the aging brain transcriptome and proteome in tauopathy. Mol Neurodegener. 2020;15(1):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Barabasi AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5(2):101‐113. [DOI] [PubMed] [Google Scholar]
- 100. Stelzl U, Worm U, Lalowski M, et al. A human protein–protein interaction network: a resource for annotating the proteome. Cell. 2005;122(6):957‐968. [DOI] [PubMed] [Google Scholar]
- 101. Chakravorty D, Banerjee K, Saha S. Integrative omics for interactomes. In: Singh S, ed. Synthetic Biology: Omics Tools and Their Applications. Springer Singapore; 2018:39‐49. [Google Scholar]
- 102. Yugi K, Kubota H, Hatano A, Kuroda S. Trans‐omics: how to reconstruct biochemical networks across multiple ‘omic’ layers. Trends Biotechnol. 2016;34(4):276‐290. [DOI] [PubMed] [Google Scholar]
- 103. Zhou G, Xia J. Using OmicsNet for network integration and 3D visualization. Curr Protoc Bioinformatics. 2019;65(1):e69. [DOI] [PubMed] [Google Scholar]
- 104. Zhou G, Xia J. OmicsNet: a web‐based tool for creation and visual analysis of biological networks in 3D space. Nucleic Acids Res. 2018;46(W1):W514‐W522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Herwig R, Hardt C, Lienhard M, Kamburov A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat Protoc. 2016;11(10):1889‐1907. [DOI] [PubMed] [Google Scholar]
- 106. Himmelstein DS, Baranzini SE. Heterogeneous network edge prediction: a data integration approach to prioritize disease‐associated genes. PLoS Comput Biol. 2015;11(7):e1004259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Valdeolivas A, Tichit L, Navarro C, et al. Random walk with restart on multiplex and heterogeneous biological networks. Bioinformatics. 2019;35(3):497‐505. [DOI] [PubMed] [Google Scholar]
- 108. Zachariou M, Minadakis G, Oulas A, Afxenti S, Spyrou GM. Integrating multi‐source information on a single network to detect disease‐related clusters of molecular mechanisms. J Proteomics. 2018;188:15‐29. [DOI] [PubMed] [Google Scholar]
- 109. Sugis E, Dauvillier J, Leontjeva A, et al. HENA, heterogeneous network‐based data set for Alzheimer's disease. Sci Data. 2019;6(1):151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Bodein A, Scott‐Boyer MP, Perin O, Le Cao KA, Droit A. Interpretation of network‐based integration from multi‐omics longitudinal data. Nucleic Acids Res. 2022;50(5):e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ding J, Blencowe M, Nghiem T, et al. Mergeomics 2.0: a web server for multi‐omics data integration to elucidate disease networks and predict therapeutics. Nucleic Acids Res. 2021;49(W1):W375‐W387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Himmelstein DS, Lizee A, Hessler C, et al. Systematic integration of biomedical knowledge prioritizes drugs for repurposing. eLife. 2017;6:e26726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Zoppi J, Guillaume JF, Neunlist M, Chaffron S. MiBiOmics: an interactive web application for multi‐omics data exploration and integration. BMC Bioinformatics. 2021;22(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Domingo‐Fernandez D, Mubeen S, Marin‐Llao J, Hoyt CT, Hofmann‐Apitius M. PathMe: merging and exploring mechanistic pathway knowledge. BMC Bioinformatics. 2019;20(1):243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Zhou G, Pang Z, Lu Y, Ewald J, Xia J. OmicsNet 2.0: a web‐based platform for multi‐omics integration and network visual analytics. Nucleic Acids Res. 2022;50(W1):W527‐533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Rodchenkov I, Babur O, Luna A, et al. Pathway Commons 2019 Update: integration, analysis and exploration of pathway data. Nucleic Acids Res. 2020;48(D1):D489‐D497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Brunk E, Sahoo S, Zielinski DC, et al. Recon3D enables a three‐dimensional view of gene variation in human metabolism. Nat Biotechnol. 2018;36(3):272‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Caspi R, Billington R, Ferrer L, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016;44(D1):D471‐D480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Hernandez‐de‐Diego R, Tarazona S, Martinez‐Mira C, et al. PaintOmics 3: a web resource for the pathway analysis and visualization of multi‐omics data. Nucleic Acids Res. 2018;46(W1):W503‐W509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Cottret L, Frainay C, Chazalviel M, et al. MetExplore: collaborative edition and exploration of metabolic networks. Nucleic Acids Res. 2018;46(W1):W495‐W502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Delavan B, Roberts R, Huang R, Bao W, Tong W, Liu Z. Computational drug repositioning for rare diseases in the era of precision medicine. Drug Discov Today. 2018;23(2):382‐394. [DOI] [PubMed] [Google Scholar]
- 122. Zou Q, Chen L, Huang T, Zhang Z, Xu Y. Machine learning and graph analytics in computational biomedicine. Artif Intell Med. 2017;83:1. [DOI] [PubMed] [Google Scholar]
- 123. Cheng PF, Dummer R, Levesque MP. Data mining The Cancer Genome Atlas in the era of precision cancer medicine. Swiss Med Wkly. 2015;145:w14183. [DOI] [PubMed] [Google Scholar]
- 124. Haas R, Zelezniak A, Iacovacci J, Kamrad S, Townsend S, Ralser M. Designing and interpreting ‘multi‐omic’ experiments that may change our understanding of biology. Curr Opin Syst Biol. 2017;6:37‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Jeni LA, Cohn JF, De La Torre F. Facing imbalanced data recommendations for the use of performance metrics. Int Conf Affect Comput Intell Interact Workshops. 2013;2013:245‐251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Misra BB, Langefeld CD, Olivier M, Cox LA. Integrated omics: tools, advances, and future approaches. J Mol Endocrinol. 2018;JME–18–0055. [DOI] [PubMed] [Google Scholar]
- 127. Meng C, Zeleznik OA, Thallinger GG, Kuster B, Gholami AM, Culhane AC. Dimension reduction techniques for the integrative analysis of multi‐omics data. Brief Bioinform. 2016;17(4):628‐641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Sathyanarayanan A, Gupta R, Thompson EW, Nyholt DR, Bauer DC, Nagaraj SH. A comparative study of multi‐omics integration tools for cancer driver gene identification and tumour subtyping. Brief Bioinform. 2020;21(6):1920‐1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Reel PS, Reel S, Pearson E, Trucco E, Jefferson E. Using machine learning approaches for multi‐omics data analysis: a review. Biotechnol Adv. 2021;49:107739. [DOI] [PubMed] [Google Scholar]
- 130. Gunning D, Stefik M, Choi J, Miller T, Stumpf S, Yang G‐Z. XAI—explainable artificial intelligence. Science Robotics. 2019;4(37):eaay7120. [DOI] [PubMed] [Google Scholar]
- 131. Ritchie MD, Holzinger ER, Li R, Pendergrass SA, Kim D. Methods of integrating data to uncover genotype–phenotype interactions. Nat Rev Genet. 2015;16(2):85‐97. [DOI] [PubMed] [Google Scholar]
- 132. Stetson LC, Pearl T, Chen Y, Barnholtz‐Sloan JS. Computational identification of multi‐omic correlates of anticancer therapeutic response. BMC Genomics. 2014;15(7):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Fridley BL, Lund S, Jenkins GD, Wang L. A Bayesian integrative genomic model for pathway analysis of complex traits. Genet Epidemiol. 2012;36(4):352‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Meng C, Helm D, Frejno M, Kuster B. moCluster: identifying joint patterns across multiple omics data sets. J Proteome Res. 2016;15(3):755‐765. [DOI] [PubMed] [Google Scholar]
- 135. Argelaguet R, Velten B, Arnol D, et al. Multi‐omics factor analysis—a framework for unsupervised integration of multi‐omics data sets. Mol Syst Biol. 2018;14(6):e8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Prélot L, Draisma H, Anasanti MD, et al. Machine learning in multi‐omics data to assess longitudinal predictors of glycaemic health. bioRxiv; 2018. PPR: PPR8021. doi: 10.1101/358390 [DOI]
- 137. Kim D, Li R, Dudek SM, Ritchie MD. ATHENA: identifying interactions between different levels of genomic data associated with cancer clinical outcomes using grammatical evolution neural network. BioData Min. 2013;6(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Yuan Y, Savage RS, Markowetz F. Patient‐specific data fusion defines prognostic cancer subtypes. PLoS Comput Biol. 2011;7(10):e1002227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Nguyen T, Tagett R, Diaz D, Draghici S. A novel approach for data integration and disease subtyping. Genome Res. 2017;27(12):2025‐2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Lock EF, Dunson DB. Bayesian consensus clustering. Bioinformatics. 2013;29(20):2610‐2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Tan K, Huang W, Hu J, Dong S. A multi‐omics supervised autoencoder for pan‐cancer clinical outcome endpoints prediction. BMC Med Inform Decis Mak. 2020;20(3):129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Xu J, Wu P, Chen Y, Meng Q, Dawood H, Dawood H. A hierarchical integration deep flexible neural forest framework for cancer subtype classification by integrating multi‐omics data. BMC Bioinformatics. 2019;20(1):527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Tepeli YI, Unal AB, Akdemir FM, Tastan O. PAMOGK: a pathway graph kernel‐based multiomics approach for patient clustering. Bioinformatics. 2021;36(21):5237‐5246. [DOI] [PubMed] [Google Scholar]
- 144. Wang T, Shao W, Huang Z, et al. MOGONET integrates multi‐omics data using graph convolutional networks allowing patient classification and biomarker identification. Nat Commun. 2021;12(1):3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Min S, Lee B, Yoon S. Deep learning in bioinformatics. Brief Bioinform. 2017;18(5):851‐869. [DOI] [PubMed] [Google Scholar]
- 146. Chaudhary K, Poirion OB, Lu L, Garmire LX. Deep learning‐based multi‐omics integration robustly predicts survival in liver cancer. Clin Cancer Res. 2018;24(6):1248‐1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Lin E, Lin CH, Lane HY. Deep learning with neuroimaging and genomics in Alzheimer's disease. Int J Mol Sci. 2021;22(15):7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Kang M, Ko E, Mersha TB. A roadmap for multi‐omics data integration using deep learning. Brief Bioinform. 2022;23(1):bbab454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Zhao L, Dong Q, Luo C, et al. DeepOmix: a scalable and interpretable multi‐omics deep learning framework and application in cancer survival analysis. Comput Struct Biotechnol J. 2021;19:2719‐2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Poirion OB, Jing Z, Chaudhary K, Huang S, Garmire LX. DeepProg: an ensemble of deep‐learning and machine‐learning models for prognosis prediction using multi‐omics data. Genome Med. 2021;13(1):112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Wang Y, Yang Y, Chen S, Wang J. DeepDRK: a deep learning framework for drug repurposing through kernel‐based multi‐omics integration. Brief Bioinform. 2021;22(5):bbab048. [DOI] [PubMed] [Google Scholar]
- 152. Biomarkers Definitions Working Group . Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69(3):89‐95. [DOI] [PubMed] [Google Scholar]
- 153. Mertins P, Mani DR, Ruggles KV, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534(7605):55‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Zhang H, Liu T, Zhang Z, et al. Integrated proteogenomic characterization of human high‐grade serous ovarian cancer. Cell. 2016;166(3):755‐765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Zhang B, Wang J, Wang X, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513(7518):382‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Migliozzi S, Oh YT, Hasanain M, et al. Integrative multi‐omics networks identify PKCdelta and DNA‐PK as master kinases of glioblastoma subtypes and guide targeted cancer therapy. Nat Cancer. 2023;4:181‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Liang XJ, Song XY, Wu JL, et al. Advances in multi‐omics study of prognostic biomarkers of diffuse large B‐cell lymphoma. Int J Biol Sci. 2022;18(4):1313‐1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Xiao Y, Bi M, Guo H, Li M. Multi‐omics approaches for biomarker discovery in early ovarian cancer diagnosis. EBioMedicine. 2022;79:104001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Mayerle J, Kalthoff H, Reszka R, et al. Metabolic biomarker signature to differentiate pancreatic ductal adenocarcinoma from chronic pancreatitis. Gut. 2018;67(1):128‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Montaner J, Ramiro L, Simats A, et al. Multilevel omics for the discovery of biomarkers and therapeutic targets for stroke. Nat Rev Neurol. 2020;16(5):247‐264. [DOI] [PubMed] [Google Scholar]
- 161. Aleksandrova K, Egea Rodrigues C, Floegel A, Ahrens W. Omics biomarkers in obesity: novel etiological insights and targets for precision prevention. Curr Obes Rep. 2020;9(3):219‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Tang WH. Biomarkers in cardiovascular diseases: how can the “‐omics” revolution be applicable at the bedside. Introduction. Prog Cardiovasc Dis. 2012;55(1):1–2. [DOI] [PubMed] [Google Scholar]
- 163. Doran S, Arif M, Lam S, et al. Multi‐omics approaches for revealing the complexity of cardiovascular disease. Brief Bioinform. 2021;22(5):bbab061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Bernardes JP, Mishra N, Tran F, et al. Longitudinal multi‐omics analyses identify responses of megakaryocytes, erythroid cells, and plasmablasts as hallmarks of severe COVID‐19. Immunity. 2020;53(6):1296‐1314. e1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. COvid‐19 Multi‐omics Blood ATlas (COMBAT) Consortium . A blood atlas of COVID‐19 defines hallmarks of disease severity and specificity. Cell. 2022;185(5):916‐938 e958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Byeon SK, Madugundu AK, Garapati K, et al. Development of a multiomics model for identification of predictive biomarkers for COVID‐19 severity: a retrospective cohort study. Lancet Digit Health. 2022;4(9):e632‐e645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Aerqin Q, Wang ZT, Wu KM, He XY, Dong Q, Yu JT. Omics‐based biomarkers discovery for Alzheimer's disease. Cell Mol Life Sci. 2022;79(12):585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Zhou W, Sailani MR, Contrepois K, et al. Longitudinal multi‐omics of host‐microbe dynamics in prediabetes. Nature. 2019;569(7758):663‐671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Tripathi A, Melnik AV, Xue J, et al. Intermittent hypoxia and hypercapnia, a hallmark of obstructive sleep apnea, alters the gut microbiome and metabolome. mSystems. 2018;3(3):e00020–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Chen C, Wang J, Yang C, et al. Multiomics analysis of human peripheral blood reveals marked molecular profiling changes caused by one night of sleep deprivation. MedComm. 2023;4(3):e252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. GBD 2019 Dementia Forecasting Collaborators . Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health. 2022;7(2):e105‐e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Jia J, Wei C, Chen S, et al. The cost of Alzheimer's disease in China and re‐estimation of costs worldwide. Alzheimers Dement. 2018;14(4):483‐491. [DOI] [PubMed] [Google Scholar]
- 173. Scheltens P, De Strooper B, Kivipelto M, et al. Alzheimer's disease. Lancet. 2021;397(10284):1577‐1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Pena‐Bautista C, Baquero M, Vento M, Chafer‐Pericas C. Omics‐based biomarkers for the early Alzheimer disease diagnosis and reliable therapeutic targets development. Curr Neuropharmacol. 2019;17(7):630‐647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Hampel H, Nistico R, Seyfried NT, et al. Omics sciences for systems biology in Alzheimer's disease: state‐of‐the‐art of the evidence. Ageing Res Rev. 2021;69:101346. [DOI] [PubMed] [Google Scholar]
- 176. Pimenova AA, Raj T, Goate AM. Untangling genetic risk for Alzheimer's disease. Biol Psychiatry. 2018;83(4):300‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Mostafavi S, Gaiteri C, Sullivan SE, et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer's disease. Nat Neurosci. 2018;21(6):811‐819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Wesseling H, Mair W, Kumar M, et al. Tau PTM profiles identify patient heterogeneity and stages of Alzheimer's disease. Cell. 2020;183(6):1699‐1713. e1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. He K, Nie L, Zhou Q, et al. Proteomic profiles of the early mitochondrial changes in APP/PS1 and ApoE4 transgenic mice models of Alzheimer's disease. J Proteome Res. 2019;18(6):2632‐2642. [DOI] [PubMed] [Google Scholar]
- 180. Chen C, Jiang X, Li Y, et al. Low‐dose oral copper treatment changes the hippocampal phosphoproteomic profile and perturbs mitochondrial function in a mouse model of Alzheimer's disease. Free Radic Biol Med. 2019;135:144‐156. [DOI] [PubMed] [Google Scholar]
- 181. Mahajan UV, Varma VR, Griswold ME, et al. Dysregulation of multiple metabolic networks related to brain transmethylation and polyamine pathways in Alzheimer disease: a targeted metabolomic and transcriptomic study. PLoS Med. 2020;17(1):e1003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Varma VR, Oommen AM, Varma S, et al. Brain and blood metabolite signatures of pathology and progression in Alzheimer disease: a targeted metabolomics study. PLoS Med. 2018;15(1):e1002482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Horgusluoglu E, Neff R, Song WM, et al. Integrative metabolomics‐genomics approach reveals key metabolic pathways and regulators of Alzheimer's disease. Alzheimers Dement. 2022;18(6):1260‐1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Zupanic A, Bernstein HC, Heiland I. Systems biology: current status and challenges. Cell Mol Life Sci. 2020;77(3):379‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. San Segundo‐Acosta P, Montero‐Calle A, Jernbom‐Falk A, et al. Multiomics profiling of Alzheimer's disease serum for the identification of autoantibody biomarkers. J Proteome Res. 2021;20(11):5115‐5130. [DOI] [PubMed] [Google Scholar]
- 186. Madrid L, Moreno‐Grau S, Ahmad S, et al. Multiomics integrative analysis identifies APOE allele‐specific blood biomarkers associated to Alzheimer's disease etiopathogenesis. Aging (Albany, NY). 2021;13(7):9277‐9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Clark C, Dayon L, Masoodi M, Bowman GL, Popp J. An integrative multi‐omics approach reveals new central nervous system pathway alterations in Alzheimer's disease. Alzheimers Res Ther. 2021;13(1):71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Zhou Y, Fang J, Bekris LM, et al. AlzGPS: a genome‐wide positioning systems platform to catalyze multi‐omics for Alzheimer's drug discovery. Alzheimers Res Ther. 2021;13(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Wang M, Li A, Sekiya M, et al. Transformative network modeling of multi‐omics data reveals detailed circuits, key regulators, and potential therapeutics for Alzheimer's disease. Neuron. 2021;109(2):257‐272. e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Wang H, Robinson JL, Kocabas P, et al. Genome‐scale metabolic network reconstruction of model animals as a platform for translational research. Proc Natl Acad Sci U S A. 2021;118(30):e2102344118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Xie L, He B, Varathan P, et al. Integrative‐omics for discovery of network‐level disease biomarkers: a case study in Alzheimer's disease. Brief Bioinform. 2021;22(6):bbab121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Vialle RA, de Paiva Lopes K, Bennett DA, Crary JF, Raj T. Integrating whole‐genome sequencing with multi‐omic data reveals the impact of structural variants on gene regulation in the human brain. Nat Neurosci. 2022;25(4):504‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Ge WH, Lin Y, Li S, Zong X, Ge ZC. Identification of biomarkers for early diagnosis of acute myocardial infarction. J Cell Biochem. 2018;119(1):650‐658. [DOI] [PubMed] [Google Scholar]
- 194. Hatano T, Saiki S, Okuzumi A, Mohney RP, Hattori N. Identification of novel biomarkers for Parkinson's disease by metabolomic technologies. J Neurol Neurosurg Psychiatry. 2016;87(3):295‐301. [DOI] [PubMed] [Google Scholar]
- 195. Hardy J. Genetic analysis of pathways to Parkinson disease. Neuron. 2010;68(2):201‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Redensek S, Trost M, Dolzan V. Genetic determinants of Parkinson's disease: can they help to stratify the patients based on the underlying molecular defect? Front Aging Neurosci. 2017;9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Zhang W, Shen J, Wang Y, Cai K, Zhang Q, Cao M. Blood SSR1: a possible biomarker for early prediction of Parkinson's disease. Front Mol Neurosci. 2022;15:762544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Karayel O, Virreira Winter S, Padmanabhan S, et al. Proteome profiling of cerebrospinal fluid reveals biomarker candidates for Parkinson's disease. Cell Rep Med. 2022;3(6):100661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Sinclair E, Trivedi DK, Sarkar D, et al. Metabolomics of sebum reveals lipid dysregulation in Parkinson's disease. Nat Commun. 2021;12(1):1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Tan AH, Chong CW, Lim SY, et al. Gut microbial ecosystem in Parkinson disease: new clinicobiological insights from multi‐omics. Ann Neurol. 2021;89(3):546‐559. [DOI] [PubMed] [Google Scholar]
- 201. Morello G, Salomone S, D'Agata V, Conforti FL, Cavallaro S. From multi‐omics approaches to precision medicine in amyotrophic lateral sclerosis. Front Neurosci. 2020;14:577755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Mitropoulos K, Katsila T, Patrinos GP, Pampalakis G. Multi‐omics for biomarker discovery and target validation in biofluids for amyotrophic lateral sclerosis diagnosis. OMICS. 2018;22(1):52‐64. [DOI] [PubMed] [Google Scholar]
- 203. Volonte C, Morello G, Spampinato AG, et al. Omics‐based exploration and functional validation of neurotrophic factors and histamine as therapeutic targets in ALS. Ageing Res Rev. 2020;62:101121. [DOI] [PubMed] [Google Scholar]
- 204. Kim G, Gautier O, Tassoni‐Tsuchida E, Ma XR, Gitler AD. ALS genetics: gains, losses, and implications for future therapies. Neuron. 2020;108(5):822‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Maniatis S, Aijo T, Vickovic S, et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science. 2019;364(6435):89‐93. [DOI] [PubMed] [Google Scholar]
- 206. Oeckl P, Weydt P, Thal DR, Weishaupt JH, Ludolph AC, Otto M. Proteomics in cerebrospinal fluid and spinal cord suggests UCHL1, MAP2 and GPNMB as biomarkers and underpins importance of transcriptional pathways in amyotrophic lateral sclerosis. Acta Neuropathol. 2020;139(1):119‐134. [DOI] [PubMed] [Google Scholar]
- 207. Patin F, Corcia P, Vourc'h P, et al. Omics to explore amyotrophic lateral sclerosis evolution: the central role of arginine and proline metabolism. Mol Neurobiol. 2017;54(7):5361‐5374. [DOI] [PubMed] [Google Scholar]
- 208. Chen C, Yang C, Wang J, et al. Melatonin ameliorates cognitive deficits through improving mitophagy in a mouse model of Alzheimer's disease. J Pineal Res. 2021;71(4):e12774. [DOI] [PubMed] [Google Scholar]
- 209. Li T, Li X, Huang X, et al. Mitochondriomics reveals the underlying neuroprotective mechanism of TrkB receptor agonist R13 in the 5xFAD mice. Neuropharmacology. 2022;204:108899. [DOI] [PubMed] [Google Scholar]
- 210. Li X, Chen C, Zhan X, et al. R13 preserves motor performance in SOD1(G93A) mice by improving mitochondrial function. Theranostics. 2021;11(15):7294‐7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Johnson AA, Stolzing A. The role of lipid metabolism in aging, lifespan regulation, and age‐related disease. Aging Cell. 2019;18(6):e13048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Gross AL, Carlson MC, Chu NM, et al. Derivation of a measure of physiological multisystem dysregulation: results from WHAS and health ABC. Mech Ageing Dev. 2020;188:111258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Kaplanis J, Gordon A, Shor T, et al. Quantitative analysis of population‐scale family trees with millions of relatives. Science. 2018;360(6385):171‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Gogele M, Pattaro C, Fuchsberger C, Minelli C, Pramstaller PP, Wjst M. Heritability analysis of life span in a semi‐isolated population followed across four centuries reveals the presence of pleiotropy between life span and reproduction. J Gerontol A Biol Sci Med Sci. 2011;66(1):26‐37. [DOI] [PubMed] [Google Scholar]
- 215. Zenin A, Tsepilov Y, Sharapov S, et al. Identification of 12 genetic loci associated with human healthspan. Commun Biol. 2019;2:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Fortney K, Dobriban E, Garagnani P, et al. Genome‐wide scan informed by age‐related disease identifies loci for exceptional human longevity. PLoS Genet. 2015;11(12):e1005728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Broer L, Buchman AS, Deelen J, et al. GWAS of longevity in CHARGE consortium confirms APOE and FOXO3 candidacy. J Gerontol A Biol Sci Med Sci. 2015;70(1):110‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Nygaard M, Thinggaard M, Christensen K, Christiansen L. Investigation of the 5q33.3 longevity locus and age‐related phenotypes. Aging (Albany, NY). 2017;9(1):247‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Timmers PR, Mounier N, Lall K, et al. Genomics of 1 million parent lifespans implicates novel pathways and common diseases and distinguishes survival chances. eLife. 2019;8:e39856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Horvath S, Raj K. DNA methylation‐based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018;19(6):371‐384. [DOI] [PubMed] [Google Scholar]
- 221. Unnikrishnan A, Freeman WM, Jackson J, Wren JD, Porter H, Richardson A. The role of DNA methylation in epigenetics of aging. Pharmacol Ther. 2019;195:172‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Palmer D, Fabris F, Doherty A, Freitas AA, de Magalhaes JP. Ageing transcriptome meta‐analysis reveals similarities and differences between key mammalian tissues. Aging (Albany, NY). 2021;13(3):3313‐3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Mamoshina P, Volosnikova M, Ozerov IV, et al. Machine learning on human muscle transcriptomic data for biomarker discovery and tissue‐specific drug target identification. Front Genet. 2018;9:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Tanaka T, Biancotto A, Moaddel R, et al. Plasma proteomic signature of age in healthy humans. Aging Cell. 2018;17(5):e12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Lehallier B, Gate D, Schaum N, et al. Undulating changes in human plasma proteome profiles across the lifespan. Nat Med. 2019;25(12):1843‐1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Hoffman JM, Lyu Y, Pletcher SD, Promislow DEL. Proteomics and metabolomics in ageing research: from biomarkers to systems biology. Essays Biochem. 2017;61(3):379‐388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Yu Z, Zhai G, Singmann P, et al. Human serum metabolic profiles are age dependent. Aging Cell. 2012;11(6):960‐967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Cheng S, Larson MG, McCabe EL, et al. Distinct metabolomic signatures are associated with longevity in humans. Nat Commun. 2015;6:6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Song Q, Hou Y, Zhang Y, et al. Integrated multi‐omics approach revealed cellular senescence landscape. Nucleic Acids Res. 2022;50(19):10947‐10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Wang Y, Liu L, Song Y, Yu X, Deng H. Unveiling E2F4, TEAD1 and AP‐1 as regulatory transcription factors of the replicative senescence program by multi‐omics analysis. Protein Cell. 2022;13(10):742‐759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Aging Atlas Consortium . Aging Atlas: a multi‐omics database for aging biology. Nucleic Acids Res. 2021;49(D1):D825‐D830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Gao Y, Shang S, Guo S, et al. AgingBank: a manually curated knowledgebase and high‐throughput analysis platform that provides experimentally supported multi‐omics data relevant to aging in multiple species. Brief Bioinform. 2022;23(6):bbac438. [DOI] [PubMed] [Google Scholar]
- 233. Carlson EE. Natural products as chemical probes. ACS Chem Biol. 2010;5(7):639‐653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Nandi T, Pradyuth S, Singh AK, Chitkara D, Mittal A. Therapeutic agents for targeting desmoplasia: current status and emerging trends. Drug Discov Today. 2020;S1359–6446(20)30365–2. [DOI] [PubMed] [Google Scholar]
- 235. Dai L, Li Z, Chen D, et al. Target identification and validation of natural products with label‐free methodology: a critical review from 2005 to 2020. Pharmacol Ther. 2020;216:107690. [DOI] [PubMed] [Google Scholar]
- 236. Chang J, Kim Y, Kwon HJ. Advances in identification and validation of protein targets of natural products without chemical modification. Nat Prod Rep. 2016;33(5):719‐730. [DOI] [PubMed] [Google Scholar]
- 237. Park YD, Sun W, Salas A, et al. Identification of multiple cryptococcal fungicidal drug targets by combined gene dosing and drug affinity responsive target stability screening. mBio. 2016;7(4):e01073–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Dos Santos BS, da Silva LC, da Silva TD, et al. Application of omics technologies for evaluation of antibacterial mechanisms of action of plant‐derived products. Front Microbiol. 2016;7:1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Shao X, Ji F, Wang Y, et al. Integrative chemical proteomics‐metabolomics approach reveals Acaca/Acacb as direct molecular targets of PFOA. Anal Chem. 2018;90(18):11092‐11098. [DOI] [PubMed] [Google Scholar]
- 240. Lomenick B, Hao R, Jonai N, et al. Target identification using drug affinity responsive target stability (DARTS). Proc Natl Acad Sci U S A. 2009;106(51):21984‐21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Chang J, Kwon HJ. Discovery of novel drug targets and their functions using phenotypic screening of natural products. J Ind Microbiol Biotechnol. 2016;43(2‐3):221‐231. [DOI] [PubMed] [Google Scholar]
- 242. Dai J, Liang K, Zhao S, et al. Chemoproteomics reveals baicalin activates hepatic CPT1 to ameliorate diet‐induced obesity and hepatic steatosis. Proc Natl Acad Sci U S A. 2018;115(26):E5896‐E5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Zhong Y, Lee K, Deng Y, et al. Arctigenin attenuates diabetic kidney disease through the activation of PP2A in podocytes. Nat Commun. 2019;10(1):4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Takase S, Kurokawa R, Arai D, et al. A quantitative shRNA screen identifies ATP1A1 as a gene that regulates cytotoxicity by aurilide B. Sci Rep. 2017;7(1):2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Zhang Q, Major MB, Takanashi S, et al. Small‐molecule synergist of the Wnt/beta‐catenin signaling pathway. Proc Natl Acad Sci U S A. 2007;104(18):7444‐7448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Kasap C, Elemento O, Kapoor TM. DrugTargetSeqR: a genomics‐ and CRISPR‐Cas9‐based method to analyze drug targets. Nat Chem Biol. 2014;10(8):626‐628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Rodrigues T, Werner M, Roth J, et al. Machine intelligence decrypts beta‐lapachone as an allosteric 5‐lipoxygenase inhibitor. Chem Sci. 2018;9(34):6899‐6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Wen M, Zhang Z, Niu S, et al. Deep‐learning‐based drug–target interaction prediction. J Proteome Res. 2017;16(4):1401‐1409. [DOI] [PubMed] [Google Scholar]
- 249. Pierre‐Jean M, Deleuze JF, Le Floch E, Mauger F. Clustering and variable selection evaluation of 13 unsupervised methods for multi‐omics data integration. Brief Bioinform. 2020;21(6):2011‐2030. [DOI] [PubMed] [Google Scholar]
- 250. Tarazona S, Arzalluz‐Luque A, Conesa A. Undisclosed, unmet and neglected challenges in multi‐omics studies. Nat Comput Sci. 2021;1(6):395‐402. [DOI] [PubMed] [Google Scholar]
- 251. Conesa A, Beck S. Making multi‐omics data accessible to researchers. Sci Data. 2019;6(1):251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Voillet V, Besse P, Liaubet L, San Cristobal M, Gonzalez I. Handling missing rows in multi‐omics data integration: multiple imputation in multiple factor analysis framework. BMC Bioinformatics. 2016;17(1):402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Dong X, Lin L, Zhang R, et al. TOBMI: trans‐omics block missing data imputation using a k‐nearest neighbor weighted approach. Bioinformatics. 2019;35(8):1278‐1283. [DOI] [PubMed] [Google Scholar]
- 254. Messer K, Vaida F, Hogan C. Robust analysis of biomarker data with informative missingness using a two‐stage hypothesis test in an HIV treatment interruption trial: AIEDRP AIN503/ACTG A5217. Contemp Clin Trials. 2006;27(6):506‐517. [DOI] [PubMed] [Google Scholar]
- 255. Canzler S, Hackermuller J. multiGSEA: a GSEA‐based pathway enrichment analysis for multi‐omics data. BMC Bioinformatics. 2020;21(1):561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Arneson D, Bhattacharya A, Shu L, Makinen VP, Yang X. Mergeomics: a web server for identifying pathological pathways, networks, and key regulators via multidimensional data integration. BMC Genomics. 2016;17(1):722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Long Y, Lu M, Cheng T, Zhan X, Zhan X. Multiomics‐based signaling pathway network alterations in human non‐functional pituitary adenomas. Front Endocrinol (Lausanne). 2019;10:835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Su G, Morris JH, Demchak B, Bader GD. Biological network exploration with Cytoscape 3. Curr Protoc Bioinformatics. 2014;47:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Lappalainen I, Almeida‐King J, Kumanduri V, et al. The European Genome‐phenome Archive of human data consented for biomedical research. Nat Genet. 2015;47(7):692‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Haug K, Cochrane K, Nainala VC, et al. MetaboLights: a resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2020;48(D1):D440‐D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Deutsch EW, Csordas A, Sun Z, et al. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res;45(D1):D1100‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Byrd JB, Greene AC, Prasad DV, Jiang X, Greene CS. Responsible, practical genomic data sharing that accelerates research. Nat Rev Genet. 2020;21(10):615‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Perez‐Riverol Y, Bai M, da Veiga Leprevost F, et al. Discovering and linking public omics data sets using the Omics Discovery Index. Nat Biotechnol. 2017;35(5):406‐409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Angione C. Human systems biology and metabolic modelling: a review‐from disease metabolism to precision medicine. Biomed Res Int. 2019;2019:8304260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Oh M, Park S, Kim S, Chae H. Machine learning‐based analysis of multi‐omics data on the cloud for investigating gene regulations. Brief Bioinform. 2021;22(1):66‐76. [DOI] [PubMed] [Google Scholar]
- 266. Alyass A, Turcotte M, Meyre D. From big data analysis to personalized medicine for all: challenges and opportunities. BMC Med Genomics. 2015;8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Dias R, Torkamani A. Artificial intelligence in clinical and genomic diagnostics. Genome Med. 2019;11(1):70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Fan J, Slowikowski K, Zhang F. Single‐cell transcriptomics in cancer: computational challenges and opportunities. Exp Mol Med. 2020;52(9):1452‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Zhu C, Preissl S, Ren B. Single‐cell multimodal omics: the power of many. Nat Methods. 2020;17(1):11‐14. [DOI] [PubMed] [Google Scholar]
- 270. Lee J, Hyeon DY, Hwang D. Single‐cell multiomics: technologies and data analysis methods. Exp Mol Med. 2020;52(9):1428‐1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Forcato M, Romano O, Bicciato S. Computational methods for the integrative analysis of single‐cell data. Brief Bioinform. 2021;22(1):20‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Macaulay IC, Haerty W, Kumar P, et al. G&T‐seq: parallel sequencing of single‐cell genomes and transcriptomes. Nat Methods. 2015;12(6):519‐522. [DOI] [PubMed] [Google Scholar]
- 273. Dey SS, Kester L, Spanjaard B, Bienko M, van Oudenaarden A. Integrated genome and transcriptome sequencing of the same cell. Nat Biotechnol. 2015;33(3):285‐289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Dean FB, Hosono S, Fang L, et al. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci U S A. 2002;99(8):5261‐5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Ramskold D, Luo S, Wang YC, et al. Full‐length mRNA‐seq from single‐cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 2012;30(8):777‐782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Hashimshony T, Wagner F, Sher N, Yanai I. CEL‐Seq: single‐cell RNA‐seq by multiplexed linear amplification. Cell Rep. 2012;2(3):666‐673. [DOI] [PubMed] [Google Scholar]
- 277. Angermueller C, Clark SJ, Lee HJ, et al. Parallel single‐cell sequencing links transcriptional and epigenetic heterogeneity. Nat Methods. 2016;13(3):229‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Hernando‐Herraez I, Evano B, Stubbs T, et al. Ageing affects DNA methylation drift and transcriptional cell‐to‐cell variability in mouse muscle stem cells. Nat Commun. 2019;10(1):4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Genshaft AS, Li S, Gallant CJ, et al. Multiplexed, targeted profiling of single‐cell proteomes and transcriptomes in a single reaction. Genome Biol. 2016;17(1):188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Frei AP, Bava FA, Zunder ER, et al. Highly multiplexed simultaneous detection of RNAs and proteins in single cells. Nat Methods. 2016;13(3):269‐275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Hu Y, Huang K, An Q, et al. Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol. 2016;17:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Cao J, Cusanovich DA, Ramani V, et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science. 2018;361(6409):1380‐1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Chen S, Lake BB, Zhang K. High‐throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat Biotechnol. 2019;37(12):1452‐1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Peterson VM, Zhang KX, Kumar N, et al. Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol. 2017;35(10):936‐939. [DOI] [PubMed] [Google Scholar]
- 285. Stoeckius M, Hafemeister C, Stephenson W, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14(9):865‐868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Dou J, Liang S, Mohanty V, et al. Bi‐order multimodal integration of single‐cell data. Genome Biol. 2022;23(1):112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Yuan Q, Duren Z. Integration of single‐cell multi‐omics data by regression analysis on unpaired observations. Genome Biol. 2022;23(1):160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Lu L, Welch JD. PyLiger: scalable single‐cell multi‐omic data integration in Python. Bioinformatics. 2022;38(10):2946‐2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Kriebel AR, Welch JD. UINMF performs mosaic integration of single‐cell multi‐omic datasets using nonnegative matrix factorization. Nat Commun. 2022;13(1):780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Welch JD, Hartemink AJ, Prins JF. MATCHER: manifold alignment reveals correspondence between single cell transcriptome and epigenome dynamics. Genome Biol. 2017;18(1):138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Seyhan AA, Carini C. Are innovation and new technologies in precision medicine paving a new era in patients centric care? J Transl Med. 2019;17(1):114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Zeggini E, Gloyn AL, Barton AC, Wain LV. Translational genomics and precision medicine: moving from the lab to the clinic. Science. 2019;365(6460):1409‐1413. [DOI] [PubMed] [Google Scholar]
- 293. Lamb JR, Jennings LL, Gudmundsdottir V, Gudnason V, Emilsson V. It's in our blood: a glimpse of personalized medicine. Trends Mol Med. 2021;27(1):20‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Olivier M, Asmis R, Hawkins GA, Howard TD, Cox LA. The need for multi‐omics biomarker signatures in precision medicine. Int J Mol Sci. 2019;20(19):4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Mankoff DA, Edmonds CE, Farwell MD, Pryma DA. Development of companion diagnostics. Semin Nucl Med. 2016;46(1):47‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Jorgensen JT. Companion and complementary diagnostics: clinical and regulatory perspectives. Trends Cancer. 2016;2(12):706‐712. [DOI] [PubMed] [Google Scholar]
- 297. Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network‐based approach to human disease. Nat Rev Genet. 2011;12(1):56‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Cowen L, Ideker T, Raphael BJ, Sharan R. Network propagation: a universal amplifier of genetic associations. Nat Rev Genet. 2017;18(9):551‐562. [DOI] [PubMed] [Google Scholar]
- 299. Raufaste‐Cazavieille V, Santiago R, Droit A. Multi‐omics analysis: paving the path toward achieving precision medicine in cancer treatment and immuno‐oncology. Front Mol Biosci. 2022;9:962743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Valenti F, Falcone I, Ungania S, et al. Precision medicine and melanoma: multi‐omics approaches to monitoring the immunotherapy response. Int J Mol Sci. 2021;22(8):3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Baxi EG, Thompson T, Li J, et al. Answer ALS, a large‐scale resource for sporadic and familial ALS combining clinical and multi‐omics data from induced pluripotent cell lines. Nat Neurosci. 2022;25(2):226‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Hu C, Jia W. Multi‐omics profiling: the way towards precision medicine in metabolic diseases. J Mol Cell Biol. 2021;13(8):576‐593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Zhou G, Ewald J, Xia J. OmicsAnalyst: a comprehensive web‐based platform for visual analytics of multi‐omics data. Nucleic Acids Res. 2021;49(W1):W476‐W482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Li J, Miao B, Wang S, et al. Hiplot: a comprehensive and easy‐to‐use web service for boosting publication‐ready biomedical data visualization. Brief Bioinform. 2022;23(4):bbac261. [DOI] [PubMed] [Google Scholar]
- 305. Wang S, Li W, Hu L, Cheng J, Yang H, Liu Y. NAguideR: performing and prioritizing missing value imputations for consistent bottom‐up proteomic analyses. Nucleic Acids Res. 2020;48(14):e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Lyu F, Han F, Ge C, et al. OmicStudio: a composable bioinformatics cloud platform with real‐time feedback that can generate high‐quality graphs for publication. iMeta. 2023;2(1):e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Ren Y, Yu G, Shi C, et al. Majorbio Cloud: a one‐stop, comprehensive bioinformatic platform for multiomics analyses. iMeta. 2022;1(2):e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data in this study are available upon reasonable request.