

INTRODUCTION
Tyrosine kinase inhibitor (TKI) therapy has improved the survival of several cancers in the United States, including metastatic renal cell carcinoma (mRCC), hepatocellular carcinoma, thyroid cancer, colorectal cancer, and chronic myeloid leukemia (CML).1–5 Although TKIs have remained effective treatment options, frequent cardiac adverse effects (AEs) have been recognized (Fig. 1). Hypertension (HTN), QT prolongation, arrhythmias, left ventricular systolic dysfunction (LVSD), and heart failure (HF) are among the most common cardiac AEs reported.2,4,6 The incidence of cancer increases with age, and the cardiotoxicity of TKIs is especially relevant among patients with cancer who have preexisting cardiac dysfunction, risk factors, and/or previous treatment with cardiotoxic chemotherapy.6,7 Clinical trials often exclude patients with poor cardiac function, and a cancer clinical trial population may not accurately reflect patient comorbidities. Therefore, to decrease cardiac morbidity while increasing quality of life in cancer survivors, cardiologists and oncologists must be familiar with the cardiotoxicity profiles of these emerging cancer therapies and the mechanisms that mediate their effects.
Fig. 1.
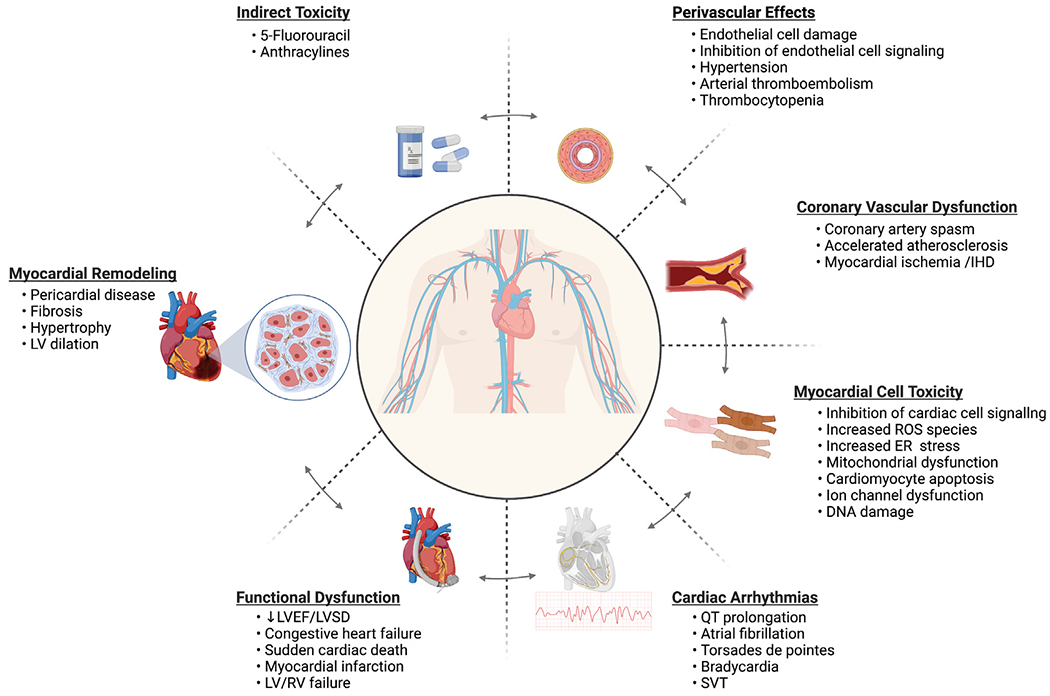
Consequences and mechanisms of direct and indirect tyrosine kinase inhibitor (TKI)-induced cardiotoxicity. ER, endoplasmic reticulum; IHD, ischemic heart disease; LV/RV, left ventricle/right ventricle; LVSD, left ventricular systolic dysfunction; ROS, reactive oxygen species; SVT, supraventricular tachycardia. (Created with BioRender.com.)
TKI-induced cardiotoxicity is mediated through the inhibition of target receptors such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and stem cell factor (c-KIT) receptors on nontarget cells.4,8 Inhibition of these highly expressed receptors on cancer cells is effective in reducing cancer growth and progression. However, the inhibition of these receptors on cell types in the cardiovascular system, from cardiomyocytes to fibroblasts, and endothelial cells (ECs) promotes cardiovascular injury.2 It has been hypothesized that VEGF/VEGF receptor (VEGFR) inhibition along with subsequent changes in the balance of endothelin-1 (ET-1) and nitric oxide (NO) is responsible for HTN, the most common cardiac AE associated with TKI use and can lead to dose reductions of therapy.2,4,9 Furthermore, current evidence suggests that the “off”-target effects of TKIs on cardiomyocyte receptors leads to dysregulation of ion channel function and turnover, contributing to increased incidence of arrhythmias in treatment recipients.4,6,10,11 Concurrent or sequential use of TKIs in patients with previous or concurrent exposure to other cancer drugs (eg, anthracyclines, 5-fluorouracil) may exacerbate cardiac injury, accelerating morbidity and the development of HF (Table 1).4,12
Table 1.
Selected tyrosine kinase inhibitors, their receptor targets, cardiotoxicity incidence and proposed cardiotoxic signaling mechanisms
| Agents | Receptor Target(s) | Cardiotoxicity | Preclinical Models | Proposed Cardiotoxic Signaling Mechanisms | Reference |
|---|---|---|---|---|---|
| Axitiniba (Inlyta) | VEGFR-1, −2, −3 PDGFR-/β c-KIT |
HTN AT QT Pericarditis LVSD |
NA | NA | 25,26,47 |
| Cabozantiniba (Cabometyx) | VEGFR-2 CDK RET |
HTN AT ↓ LVEF |
NA | Inhibition of VEGFR-2 signaling leads to decreased expression of endothelial NO synthase and diminished NO synthesis, which disrupts the balance of NO and ET-1 promoting vasoconstriction, increased peripheral resistance, and increased blood pressure.a | 26,47 |
| Dasatinibb (Sprycel) | c-KIT PDGFR-/β EphA2 ABL BRAF Src kinase |
CHF LVSD QT Thrombocytopenia HTN MI |
Rat primary cardiomyocytes | Activation of ER stress response signaling leads to cellular apoptosis. | 26,72,96 |
| Gefitinib (Iressa) | EGFR1 (ERBB1) | MI | H9c2 ventricular cardiomyocytes | Increased expression of BNP and β-MHC along with decreased the levels of α-MHC, promotes cardiac hypertrophy in vivo and in vitro due to activation of cardiac apoptosis and oxidative stress pathways (ie, increased caspase-3, p53 and HO1). | 97,99 |
| Imatinib (Gleevec) | c-KIT Bcr-Abl PDGFR-/β |
HTN QT IHD CHF LVSD |
Rat primary cardiomyocytes | Activation of ER stress pathways, mitochondrial dysfunction, and increased ROS precipitates cellular apoptosis and necrosis in cultured cells and murine hearts. Increased expression of protein kinase Cδ (PKCδ), a kinase with pro-apoptotic effects in the heart. |
26,34,95,96,100 |
| Lapatinib (Tykerb) | EGFR1 (ERBB1) ERBB2 |
HTN QT ↓ LVEF LVSD |
NA | Increased ratio of pro-apoptotic BCL-Xs to BCL-XL proteins, which may lead to ATP depletion, reduced cardiac contractility, and cardiac cell death via mitochondrial induced apoptosis. | 26,99,100 |
| Nilotinib (Tasigna) | Bcr-Abl DDR1/2 PDGFR-/β c-KIT |
HTN QT IHD SCD |
Rat primary cardiomyocytes | Activation of ER stress response pathways leading to cell death. Direct inhibition of hERG potassium channels reduce IKr promoting QT prolongation and arrhythmias. |
26,72,96 |
| Pazopaniba,b (Votrient) | VEGFR-1, −2, −3 PDGFR-/β c-KIT FGFR1/3 MCSFR-1 B-RAF |
HTN AF HF Torsades de pointes AT LVSD |
Atrial HL-1 cells; C57BL/6 Mice |
Inhibition of VEGFR on cardiomyocytes reduces PI3K/Akt signaling leading to activation of proapoptotic pathways. Inhibition of FGFR-1 and −2 results in impaired cardiac response to stress and reduced contractility. |
2,26,47 |
| Ponatiniba (Iclusig) | Bcr-Abl FLT3 c-KIT VEGFR-2 PDGFR Src kinase FGFR1-3 |
HTN QT HF MI LVSD |
hiPSC-induced cardiomyocytes; Zebrafish; NRVMs |
Increased accumulation of ROS and mitochondrial dysfunction. Inhibition of cardiac Akt and Erk pro-survival signaling pathways leads to cardiomyocyte apoptosis. |
26 |
| Sunitiniba,b (Sutent) | VEGFR-1, −2, −3 PDGFR-/β RET c-KIT FLT3 CSF-1R |
HTN HF QT ↓ LVEF LVSD |
NRVMs; Swiss-webster Mice; Rat H9c2 cardiomyocytes; C57BL/6J mice |
Inhibition of AMPK-mTOR signaling, ATP depletion, and impaired energy homeostasis promotes cardiomyocyte autophagy and death and contributes to LVSD. Inhibition of the RSK protein promotes mitochondrial dysfunction, which increases the release of cytochrome C (cyto C), and activation of caspase 9. Increased cyto C and activated caspase 9 initiates the mitochondrial apoptotic pathway in vitro and in vivo. Induction of cardiomyocyte apoptosis in presence of underlying cardiac pathology (HTN). |
26,55,100,89,95,100 |
| Sorafeniba,b. (Nexavar) | VEGFR-1, −2, −3 PDGFR-β B-RAF/C-RAF c-KIT FLT3 |
HTN HF MI QTc CHF LVEF AT |
Zebrafish; NRVMs |
Inhibition of Ras/Raf-1/Mek/Erk signaling pathway promotes mitochondrial dysfunction and apoptosis, which reduces cardiac cell survival. Increased activated CaMKII (ie, phosphorylated, and oxidized CaMKII), and ROS expression leads to pre-ventricular contractions and dysregulation in Ca+ homeostasis. |
10,24,26,47,89,100 |
| Vandetaniba. (Caprelsa) | VEGFR-1, −2, −3 EGFR PDGFR-β RET |
HTN HF AF QT Torsades de pointes SCD |
Postmortem human cardiac tissue; | Induced myocyte degeneration in the subendocardial zones and papillary muscles of the myocardium. | 26,34,47,59 |
| Vemurafenibb (Zelboraf) |
B-RAF | HTN QT CHF |
HEK293 T; Isolated canine Purkinje fibers |
Inhibition of Braf increases cAMP activity with subsequent increases in PKA. PKA phosphorylation of hERG channels and reduces their ability to open during, which prolongs the repolarization period and contributes to prolonged QT intervalb and development of arrhythmias. | 2,34,71 |
Abbreviations: AF, atrial fibrillation; AMPK, AMP-activated protein kinase; AT, arterial thromboembolism; ATP, adenosine triphosphate; Bcr-Abl, breakpoint cluster region-Abelson; BNP, brain naturietic peptide; CaMKII, calcium/calmodulin-dependent protein kinase; cAMP, cyclic adenosine monophosphate; CDK, cyclin-dependent kinase; CHF, congestive heart failure; c-KIT, stem cell factor receptor; CSF-1R, colony-stimulating factor 1 receptor; DDR1/2, Discoidin domain receptor 1; 2, EGFR; epidermal growth factor receptor, EGFR; epidermal growth factor receptor, EPHA2; ephrin type-A receptor 2, ER; endoplasmic reticulum, ERK; extra-cellular-signal-regulated kinase, ET-1; endothelin-1, FGFR1/2; fibroblast growth factor receptor, FLT3; FMS-related tyrosine kinase 3, HEK293 T; human embryonic kidney cells 293 T, hERG; human ether-a-go-go-related gene, HF; heart failure, hiPSC; human induced pluripotent stem cells, HL1-HTN; hypertension, HO1; heme oxygenase 1, IHD; ischemic heart disease, IKr; potassium currents, LVEF; left ventricular ejection fraction, LVSD; left ventricular systolic dysfunction, MCSFR-1; macrophage colony-stimulating factor-1 receptor, MHC; myosin heavy chain, MI; myocardial ischemia/infarction, NO; nitric oxide, NRVMs; Neonatal rat ventricular myocytes, PDGFR; platelet derived growth factor receptors, PI3K; phosphoinositide 3-kinase, PKA; protein kinase A, QT; QT prolongation, RET; rearranged during transfection, ROS; reactive oxygen species, RSK; ribosomal S6 kinase, SCD; sudden cardiac death, Src; short for sarcoma-proto-oncogene, TKI; tyrosine kinase inhibitors, VEGFR; vascular endothelial growth factor receptors.
Note (s): All VEGFR-TKIs have the potential to cause hypertension via this molecular mechanism. Further, the mechanisms leading to VEGFR-TKIs is multifactorial and might be related to microvascular dysfunction, ATP depletion in the mitochondria, myocardial proapoptotic kinases, microvascular dysfunction, and profound vasoconstriction.
All B-RAF inhibitors have the potential to promote QTc prolongation by this mechanism.68 (NA) indicates that to the authors knowledge there are no preclinical studies, which directly evaluated these drugs on cardiomyocyte tissue.
The emerging cardiovascular side effects associated with TKIs warrant an increase in patient risk factor surveillance, further research into the mechanisms of these oncologic cardiovascular insults, and strategies to reduce TKI-induced cardiac-related morbidity. To date, concurrent treatment with clinically available drugs such as β-blockers, angiotensin-converting enzyme inhibitors (ACEIs), and angiotensin receptor blockers (ARBs) has been shown to reduce TKI-induced morbidity.2,13,14 In addition, it is hypothesized that drugs such as statins, which possess systemic pleiotropic effects, can be used with TKIs to reduce cardiotoxicity.15–18 In this review, we further assess tyrosine kinase signaling in cancer and discuss recent understandings of TKI-induced cardiotoxicity along with the intracellular signaling pathways by which these drugs disrupt cardiomyocyte function. We also broadly discuss current and possible strategies to treat and prevent cardiovascular dysfunction associated with the use of these cancer therapies.
TYROSINE KINASE INHIBITORS AND MECHANISM OF ACTION
Tyrosine kinases are crucial for extracellular signal transduction in a variety of cellular processes that regulate signaling pathways impacting cell growth, differentiation, migration, motility, and death (Fig. 2).2,19,20 The 2 major classes of tyrosine kinases are receptor tyrosine kinases (RTKs) and non-receptor tyrosine kinases (NRTKs). Tyrosine kinases are normally quiescent until activated by extracellular stimuli, growth factors ligands (eg, VEGF, PDGF, and c-KIT), or intracellular stimuli such as oxidative stress. Binding of tyrosine kinase receptors lead to the activation of tyrosine kinase in the cytoplasmic tail of the receptor, which transfers phosphate residues from adenosine triphosphate (ATP) to tyrosine residues on target protein substrates.2,21,22 Some of these protein substrates are responsible for activating the canonical Ras/Raf/Mek/Erk signaling cascade, along with possible concurrent signaling via the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) and adenosine 5′-monophosphate-activated protein kinase-mammalian target of rapamycin (AMPK-mTOR) pathways (see Fig. 2).22–24 Under normal physiologic conditions, a balance between the activity of tyrosine kinases and dephosphorylation of tyrosine residues by tyrosine phosphatases is necessary to control the timing and duration of cell signaling.19,20 As such, tight regulation of tyrosine kinase activity is critical for preserving normal cellular communication, growth, and maintenance of homeostasis.
Fig. 2.
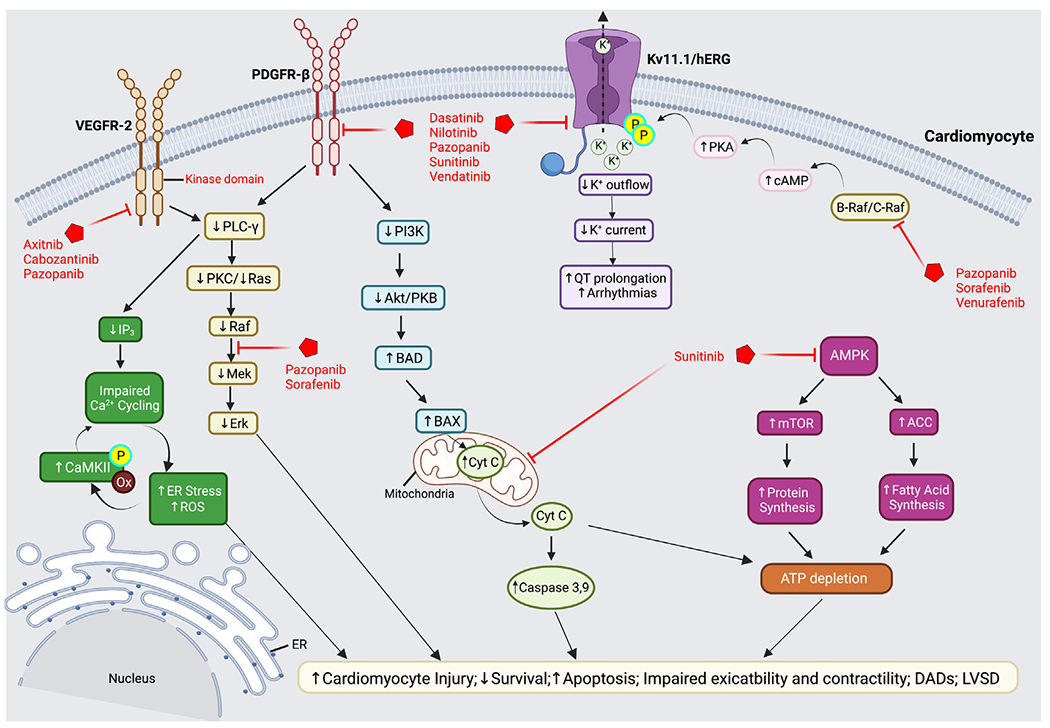
Intracellular signaling pathways mediating tyrosine kinase inhibitor cardiotoxicity. TKI-mediated inhibition of VEGFR-2 results in the downregulation of PLC-γ, which alters IP3 and Ras/Raf/Mek/Erk signal transduction cascades. Reduced IP3 contributes LVSD and reduced myocardial contractility, which results from impaired calcium cycling and increased ER stress and ROS. Increased ROS is proposed to activate and increase CaMKII phosphorylation and further dysregulation in Ca2+ homeostasis. Reduced Ras/Raf/Mek/Erk signaling is also directly affected by PDGFR-β inhibition resulting in decreased cardiomyocyte survival, increased apoptosis, and LVSD. Inhibition of PDGFR-β reduces PI3K/Akt signaling and upregulates the proapoptotic proteins, BAD and BAX, leading to mitochondrial dysfunction, release of Cyt C, activation of caspase 3 and 9, ATP depletion, and cell death. AMPK inhibition by sunitinib also depletes ATP due to increased energy sink from mTOR and ACC-mediated protein and fatty acid synthesis, respectively. Loss of ATP contributes to cardiomyocyte injury and death. TKIs also directly inhibit myocyte Kv11.1/hERG channels disrupting K+ currents. Inhibition of cytoplasmic C-RAF/B-RAF enzymes increases cAMP promoting PKA phosphorylation and inhibition of hERG channels, leading to QT prolongation and the development of arrhythmias. ACC, acetyl-coenzyme A carboxylase; Akt, protein kinase B; AMPK, adenosine 5′-monophosphate-activated protein kinase; BAD, BCL2-antagonist of cell death; BAX, BCL2-associated X protein; CaMKII, Ca2+/calmodulin-dependent protein kinase II; cAMP, cyclic adenosine monophosphate; Cyt C, cytochrome c; hERG, human ether-à-go-go; IP3, inositol-trisphosphate-3 kinase; LVSD, left ventricular systolic dysfunction; mTOR, mammalian target of rapamycin; PI3K, phosphatidyl inositol-3 kinase; PLC- γ, phospholipase C gamma; PKA/C, protein kinase A/C; PDGFR-β, platelet-derived growth factor receptor; Raf-1, rapidly accelerated fibrosarcoma-1; ROS, reactive oxygen species. (Created with BioRender.com.)
Tyrosine kinase signaling is also required for tumorigenesis, tumor growth, angiogenesis, and metastasis.19,22 In several cancers, dysregulation of tyrosine kinase signaling is responsible for the development and production of abnormal blood vessels essential to maintaining tumor growth.19,20 Notably, about 60% of human cancers overexpress VEGF, promoting tumor progression and metastasis.19 Cancer cells also express additional proangiogenic factors such as placental growth factor (PLGF), fibroblast growth factor (FGF), and PDGF, ligands of RTKs.19 As tyrosine kinase receptor signaling is a fundamental converging point for angiogenesis and tumor growth, TKIs have emerged as a pharmacologic approach to interrupt cancer growth and metastases.
Tyrosine kinases receptors can be inhibited by small molecule inhibitors that predominantly target and block the evolutionary conserved ATP-binding pocket of both RTKs and NRTKs.2,19,20 RTK inhibitors block the activity of the intracellular kinase domain, preventing trans-autophosphorylation and activation of the intracellular kinase receptor domains following ligand binding. This inhibits receptor dimerization, phosphorylation, and recruitment of downstream signaling proteins, terminating the signaling cascade.19,20 Similarly, TKIs block NRTK signaling by gaining entry to the cell and targeting intracellular kinases, blocking signal transduction. Some TKIs target multiple receptors given the conserved residues for signaling, thus inhibiting growth factors or receptors involved in angiogenesis, as well as kinases involved in tumor cell proliferation. Examples of these multitargeted receptor TKIs include axitinib, cabozantinib, pazopanib, sunitinib, and vandetanib. Targeted inhibition of VEGFR/PDGFR/c-KIT receptor signaling by TKIs is not locally restricted to the tumor environment.22,25 Owing to their lack of selectivity, TKIs also act systemically where they mediate their AEs. In this review, we focus on how dysregulation of VEGF, PDGF, and c-KIT receptor signaling by TKIs mediates cardiovascular toxicity.
RECEPTOR SIGNALING PATHWAYS AND CARDIOMYOPATHY
Several receptors targeted by TKIs, including VEGFR, PDGFR, and c-KIT, have been shown to mediate normal cardiac physiology and warrant investigation. In the following sections, we discuss the physiologic roles of these receptors and how the inhibition of these receptor signaling pathways contributes to the observed cardiac disturbances seen in patients receiving TKI therapy.
Vascular Endothelial Growth Factor/Vascular Endothelial Growth Factor Receptor Signaling
The main regulator of angiogenesis requires VEGF ligand activation of its cognate tyrosine kinase receptors, VEGFR-1, −2, and −3.19 ECs express all 3 VEGFRs, whereas VEGFR-1 and −2 are predominantly expressed within the vasculature and are activated by VEGF-A and -B. Cardiomyocytes only express VEGFR-1 and −2.4 The Ras/Raf/Mek/Erk pathway and PI3K/Akt pathway are the 2 main signaling cascade pathways activated by VEGF/VEGFR interaction (see Fig. 2).26 The activation of these signaling pathways via VEGFR-2 on ECs triggers increased vascular permeability, cell migration, proliferation, and survival.4 VEGF also plays an important role in the development, maintenance, and survival of myocardial ECs and cardiomyocytes.4 In the context of cardiovascular disease, an increase in VEGF secretion and upregulation of VEGF signaling within cardiomyocytes is essential for responding to myocardial stress and injury.4,19 In vitro and in vivo studies support this, showing that cardiomyocytes upregulate the expression of VEGFR-1 and −2, in response to hypoxia.27 In transgenic mice lacking VEGFR-1 on ECs, an increase in angiogenesis was reported along with the development of cardiomyocyte hypertrophy, suggesting a role for paracrine signaling or cross talk between ECs and cardiomyocytes.28 VEGF’s role in myocardial remodeling occurs through balanced activation of VEGFR-1, which prevents cardiomyocyte hypertrophy, and activation of VEGFR-2, which has prohypertrophic effects.29,30 Regardless of the mechanism, there are strong data suggesting that VEGF/VEGFR signaling is important for maintaining normal and adaptive function of ECs, vasculature, and cardiomyocytes.19
Platelet-Derived Growth Factor/Platelet-Derived Growth Factor Receptor Signaling
PDGF receptors (PDGFR-α/β) are ubiquitously expressed on the cell membranes of human and mouse cardiomyocytes and ECs, where they also play a role in angiogenesis and response to mechanically induced pressure overload.4,19 Overexpression of PDGFRs is found in CML and gastrointestinal stromal tumors (GIST). It is hypothesized that nonspecific effects of TKIs including the inhibition of PDGFR are responsible for the cardiac effects of these drugs. In cultured tumor cells, pazopanib is a potent inhibitor of PDGFR-β signaling,19,31 and sunitinib inhibits PDGFR-β phosphorylation in rodent tumor models (see Fig. 2).4,32–34 PDGFR-β is also upregulated in mouse models of left ventricular pressure overload via transverse aortic constriction (TAC). Although PDGFR-β is not required for normal cardiac development or function in murine models, cardiac-specific PDGFR-β knockout is associated with dysregulated left ventricular function and reduced angiogenesis following TAC when compared with TAC wild-type mice.35 In rat models, the administration of PDGF improved cardiac function following myocardial infarction (MI).36 Together, the TAC data suggest that direct cardiotoxic effects by TKIs via PGDFR signaling in the myocardium may be inconsequential and may instead be potentiated and/or unmasked by concurrent systemic disease (eg, HTN).
c-KIT Signaling
The pathogenesis of several cancers including acute myeloid leukemia, GIST, and small cell lung cancer are mediated by mutations, overexpression, or deletions of portions of the tyrosine kinase c-KIT.19,37 Although not expressed on the cardiomyocyte, a role for c-KIT in maintaining normal cardiac function was demonstrated in studies using the c-KITW/W-v transgenic model, in which one c-KIT allele is deleted and the other encodes a protein with reduced kinase activity.19,38,39 A reduction in the amount of functional c-KIT in the model led to impaired honing of bone marrow-derived proangiogenic stem/progenitor cells to regions of infarction. This impaired honing led to impaired cardiac recovery following MI and a decline in cardiac function with aging.38,39 LV structural remodeling, including chamber dilatation and hypertrophy, and LV functional deficits such as reduced left ventricular ejection fraction (LVEF) have been demonstrated in aging c-KIT mutant mice.40 Last, constitutive activation of c-KIT receptors in mice is associated with an increased cardiac myogenic and vasculogenic reparative potential after injury, with a significant improvement of survival.41 Given the totality of these findings, we postulate that the reduction of cardiomyocyte c-KIT activity may disrupt the normal cardiac response to stress or injury within human myocardium leading to increased remodeling.
Several TKIs target c-KIT in vivo8 and in vitro.42–45 Dasatinib and pazopanib are potent c-KIT inhibitors, which markedly disrupted hematopoietic progenitor cells in vivo and in vitro. On the other hand, imatinib, sunitinib, and sorafenib have shown moderate and negligible activity against c-KIT.8,42–45 However, the development of an imatinib variant lacking ABL, but retaining c-KIT inhibitor activity, did not result in cardiac dysfunction in a mouse model.19,46 The reduced potential for cardiac toxicity was attributed to a loss of ABL kinase inhibition4 and raises some questions about the role of c-KIT inhibition in the development of TKI-induced cardiotoxicity.
TYROSINE KINASE INHIBITOR-INDUCED CARDIOVASCULAR DYSFUNCTION
Hypertension
Hypertension has emerged as an important side effect common to all VEGF/VEGFR inhibitors with an incidence ranging from 16% to 80%, leading to dose reductions of therapy in clinical trials depending on the TKIs assessed.4,26 A recent network meta-analysis study assessing the cardiotoxicity risks of VEGFR-TKIs revealed that vandetanib-treated patients have the highest risk for cardiotoxicity, followed by pazopanib, axitinib, sorafenib, and sunitinib.47 In patients receiving sunitinib and axitinib, several studies suggest that HTN may serve as a predictive factor for more favorable patient response to TKIs and outcomes48,49; however, this has yet to be proved for other TKIs.2 Most of the literature has associated TKI-induced HTN with increased endothelial and cardiomyocyte dysfunction.2,50–52 One of the main mechanisms hypothesized to mediate TKI-induced HTN involves cross talk and/or paracrine signaling between ECs and cardiomyocytes.28 Presumably, this mechanism may stem from the disruption of basal VEGFR-2 signaling necessary to maintain the balance of the vasodilator, NO, and vasoconstrictor, ET-1, through the PI3K/Akt pathway.4,53 For example, pazopanib, cabozantinib, and vandetanib inhibit VEGFR-2 signaling, which normally suppresses production of ET-1, thus resulting in an increased concentration of ET-1 in blood plasma.53 ET-1 is a potent vasoconstrictor, and increased plasma levels promote sustained vasoconstriction and HTN. The PI3K pathway also plays an important role in cell survival and vasodilation from downstream NO.53 Inhibition of VEGFR-2 by pazopanib further diminishes production of NO, leading to impaired cardiomyocyte contractility.54 Recently, Ren and colleagues55 also showed that sunitinib-induced HTN is also mediated via regulation of AMPK-mTOR signaling (see Fig. 2) and warrants further investigation.
Last, it is suggested that HTN seen in patients treated with TKIs is mediated by renal impairment, which involves the activation of the renin-angiotensin-aldosterone system. Because VEGF signaling is vital to the proliferation of renal glomerular ECs, it is thought that inhibition of VEGF/VEGFR signaling could contribute to capillary rarefaction in renal glomeruli, with an increased secretion of renin from the juxtaglomerular apparatus; this leads to downstream production of angiotensin II and sustained systemic vasoconstriction. However, there is currently no experimental evidence to support this hypothesis because plasma renin levels were decreased with administration of sunitinib.2,4,28 This inconsistency calls for further research into the mechanisms behind increased concentrations of ET-1 and decreased levels of NO.
Arrhythmias
The presence of arrhythmias is a primary cause of permanent discontinuation of TKIs. Patients taking TKIs that primarily inhibit VEGF/VEGFR signaling can result in QT prolongation and arrhythmias including atrial fibrillation (AF), bradycardia,56 and supraventricular tachycardia (see Fig. 1). QT prolongation is a major concern because this could lead to other serious heart effects such as torsades de pointes and sudden cardiac death.13 In clinical trials, the incidence of QT prolongation and arrhythmias is small compared to HTN.26 The incidence of QT prolongation and arrhythmias is most common with first and second generation TKIs such as sorafenib, sunitinib, and imatinib, in addition to vandetanib and nilotinib. Risk of death/serious impairment associated with arrhythmia increases greatly among patients with cancer due to older age, underlying cardiovascular disease, and use of concomitant medications.57,58 For example, in a study of 39 patients with advanced hepatocellular cancer treated with sorafenib, the incidence of AF was 5.1% when used in conjunction with chemotherapy such as 5-fluorouracil.4,12 For patients taking vandetanib, incidence of QT interval prolongation was between 8% and 11% versus 1.2% in controls59 with an increased incidence in patients taking vandetanib plus chemotherapy (QT-related events; 22%).59 Previous cardiac injury can also preclude and potentiate TKI-mediated QT prolongation and arrhythmia where underlying myocardial injury, such as LV contractile dysfunction, or HTN may provide an arrhythmogenic substrate (see Fig. 1).4 In addition, patients treated with TKIs may also experience treatment-associated hepatic dysfunction that impedes drug clearance and metabolism.2,5,60,61 Diarrhea, commonly seen in patients taking pazopanib, can contribute to electrolyte imbalances/derangements that promote QT interval prolongation.13,22,62,63
The reported incidence of AF in patients taking TKIs or other VEGF inhibitors (VEGFI) is limited to case reports, making it difficult to ascribe causation, which could be multifactorial in nature.64,65 Although the PI3K/Akt pathway has been reported as a primary mechanism for HTN, it is also a potential mechanism for AF in patients taking TKIs or other VEGFI.59,66 As previously outlined, VEGFR-2 signaling and activation of the PI3K-Akt pathway results in increased cell survival and migration. However, inhibition of PI3K-Akt signaling has been implicated in the development of AF in mouse models.4,67–70 In another preclinical study, reduced PI3K activity led to the development of AF, whereas increasing PI3K activity led to reduced atrial fibrosis and improved conduction.4,70 In support of this mechanism, reduced PI3K activation has been shown to increase the susceptibility of AF.70 Activity of PI3K in human atrial appendages isolated from patients with AF is lower than in appendages from patients in sinus rhythm.70 In another preclinical study, Lu and colleagues67 demonstrated that TKI-mediated QT interval prolongation may be mediated through a reduction in PI3K signaling and alteration of multiple ion currents. In the study, suppression of PI3K signaling in canine cardiac myocytes by TKIs and mouse hearts lacking the PI3K p110α catalytic subunit resulted in prolonged action potentials and QT intervals.67
Tyrosine kinase inhibitor-mediated potassium ion channel dysfunction
Another possible mechanism for TKI-induced arrhythmias includes changes in the electrical activity of the heart. Specifically, the dysregulation of potassium (K+) ion channels can cause preventricular contractions resulting in arrhythmias. TKI-induced arrhythmia is predominantly associated with direct ion channel inhibition of KCNH2 (Kv11.1) or the human ether-à-go-go (hERG) channel (see Fig. 2).34 The hERG channel regulates K+ efflux out of the cardiomyocyte during repolarization and comprises the “rapid” delayed rectifier K+ current (IKr) in intact heart myofibers. The inhibition of hERG channels may promote the QT interval prolongation-associated increase in the risk of arrhythmias, including the life-threatening torsades de pointes.34,71 Many of the marketed TKIs are potent direct inhibitors of the hERG channel, including dasatinib, sunitinib, and nilotinib.34,71,72 Another mechanism of QT interval prolongation in patients occurs due to cytoplasmic inhibition of the cardioprotective enzymes B-RAF and C-RAF by TKIs like vemurafenib.2,73 It has been proposed that B-RAF inhibition can lead to an increase in cyclic adenosine monophosphate (cAMP), which overactivates protein kinase A (PKA) (see Fig. 2). PKA then phosphorylates hERG channels and reduces their ability to open during action potentials, therefore decreasing IKr.2,71,73 This hyperphosphorylation of the hERG channels can also block repolarization and contribute to QT prolongation. Pazopanib and sorafenib have been reported to inhibit the hERG channel through inhibition of B-RAF signaling.2,71,73,74 This inhibition is believed to contribute to the QT prolongation reported in patients treated with these TKIs.
Tyrosine kinase inhibitor dysregulation of calcium-mediated signaling
Current research has demonstrated that TKI treatment results in dysregulation of calcium (Ca2+) through the activity of Ca2+/calmodulin-dependent protein kinase II (CaMKII). CaMKII is a multifunctional protein found in a wide array of locations throughout the body and plays an essential role in the heart (predominantly, CaMKII isoforms δ and γ). CaMKII contributes to multiple functions in the heart such as apoptosis, inflammation, and scar tissue formation.67,75,76 In disease states, overexpression of CaMKII is involved with pathologic hypertrophy,76 which promotes HF. CaMKII is vital for proper handling of Ca2+ levels and excitation-contraction coupling, and cardiomyocyte activation of CaMKII activation promotes Ca2+ waves and delayed afterdepolarizations77 primarily through phosphorylation mechanisms. Furthermore, the phosphorylation of L-type Ca2+ channels (LTCC) and ryanodine receptor (RyR2) by CaMKII facilitates Ca2+ influx, sarcoplasmic reticulum (SR) Ca2+ release in cardiomyocytes,78 and subsequent contraction.79 Increased expression of CaMKII can cause hyperphosphorylation of the LTCC, which induces Ca2+ influx,66,80 and activates RyR2 at serine 2814 to spark SR Ca2+ release events linked with HF and arrhythmias.81 Increased CaMKII expression leads to the hyperphosphorylation of RyR2, causing Ca2+ leakage from the SR and leading to arrhythmias.
CaMKII is significantly expressed in response to TKIs such as imatinib, sunitinib, and sorafenib. Ma and colleagues10 showed that rat ventricular cardiomyocytes treated with sorafenib caused a significant increase of CaMKII expression, particularly phosphorylated and oxidized CaMKII, leading to preventricular contractions and dysregulation in Ca2+ homeostasis. Fibroblasts treated with sunitinib and imatinib also showed increased expression of CaMKII.82,83 It is suggested that TKI-induced upregulation in CaMKII expression and activity is mediated by increased reactive oxygen species (ROS) production. It is proposed that ROS can oxidize and activate CaMKII at the M281/282 (methionine) position analogous to autophosphorylation. Further research demonstrated that cardiac fibroblasts treated with imatinib and sunitinib showed increased mitochondrial superoxide production, supporting a role for ROS in CaMKII activation.84 In cardiomyocytes, sorafenib treatment also caused a significant increase in cytosolic and mitochondrial ROS production,10 suggesting that CaMKII and ROS could be potential targets for modulating TKI-induced cardiotoxicity.
TYROSINE KINASE INHIBITORSAND HEART FAILURE
Congestive heart failure (CHF) defined as a LVEF decrease of more than 10% or to less than 50% in clinical trials is among the most important heart-related functional changes that occur in response to TKI therapy (see Fig. 1).13 Among the TKIs, sunitinib, axitinib, sorafenib, and vandetanib have been associated with a reduction in LVEF and symptomatic HF.13,85 In a meta-analysis 1103 of a total of 10,647 patients from 16 randomized phase III and 5 phase II trials, the risk of CHF associated with all US Food and Drug Administration (FDA) approved VEGFR2 TKIs was evaluated.86 Ghatalia and colleagues86 observed a significant 2.69-fold increase in the risk of all grades of CHF with TKIs that target VEGFR compared with control. Given that patients with severe cardiac comorbidities are excluded from therapeutic trials, the prevalence of CHF may be higher in real-world populations. Surprisingly, third generation VEGFR2-selective TKIs such as axitinib conferred similar relative risks for CHF compared with nonspecific multitargeted TKIs (eg, sunitinib, sorafenib, and pazopanib).13,86 In a study exploring early subclinical cardiac chamber dysfunction in mRCC patients treated with TKIs, the right ventricle appeared more vulnerable to TKI therapy in the absence of pulmonary HTN, compared to the left ventricle. This observed right ventricle vulnerability is ascribed to its thinner myocardial wall.87 However, these data are inconclusive because the incidence of right-sided HF is often underreported in clinical trials.88
Systolic dysfunction and subsequent HF are hypothesized to occur because the pathways that induce the pathologic survival and abnormal proliferation of cancer cells may also regulate the survival of normal cells, including cardiomyocytes. In rat and zebrafish cardiomyocytes, sorafenib induced cardiomyocyte apoptosis through direct inhibition of the Raf/Mek/Erk signaling.89,90 VEGFR-2 signaling blockade and downstream inhibition of the PI3K/Akt pathway is also implicated in cardiomyocyte apoptosis.2,91 In mouse models, Akt activation at baseline is cardioprotective and prevents cardiomyocyte cell death through Akt-mediated inhibition of BCL-2 antagonist of cell death (BAD), a proapoptotic protein.2,91–93 In a rat model of cardiac ischemia-reperfusion injury, constitutive activation of Akt reduced cardiomyocyte apoptosis and improved cardiac function postinjury.94 In another study, the administration of exogenous VEGF prevented cardiomyocyte apoptosis and preserved cardiac function92 possibly through improved VEGF/VEGFR2/PI3K/Akt-mediated inhibition of proapoptotic proteins.2 Although it is unknown whether inhibition of the PI3K/Akt pathway by TKI-induced VEGFR-2 blockade contributes to cardiomyocyte apoptosis and HF in patients treated with TKIs, this potential pathway warrants investigation.2,19 PI3K/Akt signaling in the heart is also modulated by several other receptor pathways not affected by TKIs. Thus, further research should be conducted to determine the exact mechanism by which TKIs induce cardiomyocyte apoptosis.
Recently, it was demonstrated that the AMPK-mTOR signaling pathway may also promote TKI-mediated LVSD and cardiomyocyte death. Reduced in vivo and in vitro AMPK phosphorylation in sunitinib treated cells and mice promote cardiomyocyte cytotoxicity95 and cardiomyocyte autophagy along with impaired LVEF and LVSD in vivo.55 A selective sodium-glucose cotransporter-2 (SGLT2) inhibitor empagliflozin ameliorated this sunitinib-induced cardiac dysfunction.55 TKIs can also contribute to HF development through activation of the endoplasmic reticulum (ER) stress response96,100, and oxidative stress pathways (ie, increased caspase-3, p53),97 as well as mitochondrial dysfunction, increased ROS, and proapototic BCL-XS to BCL-XL proteins26; this may lead to ATP depletion, reduced cardiac contractility, and cardiac cell death.98–100
Cross talk between ECs and cardiomyocytes is crucial to maintain cardiac homeostasis and angiogenesis. Excessive angiogenesis may also result in cardiomyocyte hypertrophy and remodeling, which is mediated by VEGF-B and PLGF.101 VEGF binding affinity to VEGFR-2 is increased when VEGF-B and PLGF bind to VEGFR-1. Inhibition of VEGFR-2 on cardiac ECs initiates a cascade of downstream signaling changes that result in cardiomyocyte apoptosis, hypertrophy, and HTN, which can lead to cardiomyopathy and end-stage HF. In addition to VEGF signaling inhibition, the effect of TKIs on systolic function may also be caused by inhibition of FGF receptor (FGFR)-1 and −2. In the heart, normal FGFR signaling is vital for cell proliferation, differentiation, survival, and angiogenesis.2,102–104 In mice lacking FGFR-2, thrombocytosis, poor vascular function, and impaired cardiac response to ischemia102 has been reported. In a separate study, loss of FGFR-2 resulted in impaired hypertrophic response to pressure overload.105 Acute expression of FGFR-1 has also been shown to increase contractility of cardiomyocytes, whereas chronic expression leads to hypertrophy and preservation of systolic function.106 In addition, inhibition of receptors such as PDGFR and c-KIT can also disrupt coronary microvasculature through disruption of stress-induced coronary angiogenesis leading to HF.35
CURRENT AND PROPOSED TREATMENTS FOR TYROSINE KINASE INHIBITOR -INDUCED TOXICITY
Current management strategies for TKI cardiotoxicity have focused on minimizing HTN with antihypertensives, TKI dose reduction, and/or drug discontinuation.22 Among treatment options for systemic HTN are the classic agents such as ACEIs, ARBs, and non-dihydropyridine calcium channel blockers, whereas β-blockers such as carvedilol and nebivolol may be the preferred agents to reduce the risk of cardiotoxicity leading to LV dysfunction.2,13,85 Patients cotreated with metoprolol or diltiazem prevented pazopanib-mediated QT interval prolongation.2,107 As detailed earlier, there is evidence to suggest that TKI-induced HTN is mediated by reduction in VEGFR-induced NO production, disrupting ET-1 and NO balance. Exogenous NO-producing drugs such as isosorbide dinitrate or isosorbide mononitrate and ET-1 receptor blockers may have potential uses.22,108 Kruzliak and colleagues109 showed promising clinical efficacy; however, the effects of nitrates in preventing TKI-induced HTN still needs to be evaluated in larger clinical trials.22 Therefore, it is suggested that blood pressure should be normalized before treatment and monitored throughout treatment.
A growing body of evidence has demonstrated that statins and SGLT2 inhibitors also exert cardioprotective effects in several cardiovascular diseases55,110,111 and can protect the heart from chemotherapy-induced cardiac injury.85 Namely, although the mechanisms of cardiotoxicity may be different from TKIs, it was demonstrated that statins reduced HF in patients receiving anthracycline chemotherapy,112 and preserved LVEF in patients taking trastuzumab,113 suggesting a role for statins in treating drug-related cardiotoxicity.15,85 In cultured H9c2 cardiomyocytes, treatment with atorvastatin and dasatinib significantly enhanced cell survival through reduction in cell death and restoration of cardiomyocyte homeostasis.18 Hung and colleagues15 demonstrated that statins improve overall survival in patients treated with TKIs. However, a specific role for statins in reducing cardiovascular AEs was not reported and warrants further investigation.15 SGLT2 inhibitor empagliflozin was recently shown to ameliorate sunitinib-induced cardiac dysfunction both in terms of systolic blood pressure and LVEF in vivo and cardiomyocyte death and cell viability in vitro.55 These data suggest that SGLT2 inhibitor therapy could be a potential cardioprotective approach for cardiovascular complications mediated by sunitinib, but requires validation in clinical trials.55
Arrhythmias are common AEs reported in patients using TKIs (see Table 1), which are mediated by inhibition of hERG channels, leading to changes in K+ balance, dysregulation of Ca2+ and Na + homeostasis, and diarrhea. To help minimize risk of arrhythmias, patient electrolytes should be optimized and monitored before and during treatment in conjunction with monthly electrocardiography.2 Even further, diuretics and other electrolyte-depleting drugs should be avoided or used judiciously in these patients.13,17,22 In some studies, administration of β-blockers in conjunction with hydralazine was used to manage TKI-induced HTN, especially for patients with left ventricular dysfunction or arrhythmia.114,115 It is also important to address the drug interactions between antihypertensives and TKIs. For example, axitinib, cabozantinib, and pazopanib are metabolized by the CYP3A4 enzyme; hence antihypertensive drugs that inhibit CYP3A4 should be avoided to maintain the therapeutic doses and plasma clearance.2,22
Echocardiographic monitoring is an effective tool for detecting early signs of HF in patients taking TKIs.2 In a retrospective study following 23 patients with mRCC treated with TKIs, echocardiograms demonstrated early changes in left ventricular strain, which may be a precursor of TKI-induced systolic dysfunction.116,117 Moustafa and colleagues87 used velocity vector imaging to identify early subclinical cardiac chamber dysfunction secondary to TKIs in patients with mRCC. Therefore, we recommend close echocardiographic surveillance of all patients receiving TKIs starting at baseline, and at interval durations during TKI therapy, and that any observed abnormalities should result in a dose reduction or termination of treatment.2 In addition, an LVEF cutoff for TKI dose adjustment or discontinuation must be established.
Another important area of research should be the optimization of TKI drug delivery to minimize systemic AEs. The development of nanoscale drug delivery vehicles such as liposome and photoactivatable multi-inhibitor nanoliposome—which has already been developed for cabozantinib—may reduce “off”-target cardiac effects.22,118 In a pancreatic cancer model, this construct successfully reduced tumor size and metastasis following injection and near-infrared irradiation of the tumor.118 However, clinical trials are required to establish whether these delivery methods reduce cardiac adverse events.
SUMMARY AND FUTURE DIRECTIONS
The prevalence of TKI-mediated cardiovascular complications remains high and can lead to increased comorbidity with HTN as well as life-threatening cardiac effects including arrhythmias and HF. The intracellular signaling cascades that are associated with TKI cardiotoxicity are currently not well understood. Many TKIs systemically inhibit multiple signaling pathways, which makes the investigation of the pathophysiological mechanisms underlying their cardiotoxicity challenging. Furthermore, there are no proven strategies or biomarkers that predict TKI-induced cardiac dysfunction. A multipronged approach may be required to address this issue. First, a standardized mechanism of cardiac surveillance should be established for patients on TKI therapy, which may be possible through interdisciplinary partnerships between cardiologists and oncologists. Second, an understanding of the mechanisms that promote adverse cardiac effects of TKIs is necessary for the development of cardioprotective strategies. Clinical trials must also explore the utility of administering cardioprotective drugs concurrently with TKIs or include patients with cardiovascular comorbidities to better reflect a real-world population. In addition, the investigation of TKI-mediated disruption of cardiac ion channel function can provide insight into the mechanisms of arrhythmias and HF. Collectively, the data summarized in this review suggests that further research into the general role of tyrosine kinases in cardiac biology is essential for combating TKI-induced cardiotoxicity.
KEY POINTS
Tyrosine kinase inhibitor (TKI) therapy has markedly improved survivorship of patients with cancer; however, these novel therapies also target specific signaling pathways integral to normal cardiovascular physiology, promoting cardiotoxicity.
The pathophysiology of TKI-induced cardiovascular complications involves a complex interplay of changes in the balance of endothelin-1 (ET-1) and nitric oxide (NO), inhibition of vascular endothelial growth factor receptor (VEGFR) and phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) signaling, dysregulation of cardiac ion channels, and cardiomyocyte apoptosis, all of which contribute to the development of systolic dysfunction and heart failure (HF).
TKI-induced cardiotoxicity is potentially treatable with angiotensin-converting enzyme inhibitors (ACEIs), non-dihydropyridine calcium channel blockers, and beta (β)-blockers. Notably, the β-blocker nebivolol increases NO signaling and may be of particular interest. However, clinical trials are required to assess the efficacy of these cardioprotective agents against TKI-mediated hypertension (HTN), arrhythmias, and HF.
Deeper insight into the signaling pathways underlying TKI-associated cardiotoxic sequelae may lead to recognition of new kinase pathways integral to cardiovascular biology, development of novel therapies, and effective cardioprotective treatments.
CLINICS CARE POINTS
For the pathophysiologic mechanism that promotes TKI-mediated cardiotoxicity, remember:
To evaluate and manage coexisting comorbidities, risk factors, and previous patient exposure to other chemotherapies that promote cardiac injury because their underlying pathophysiology may promote or exacerbate TKI-mediated cardiac disease.
More preclinical studies are warranted to illuminate other novel kinase pathways that are critical to cardiac physiology and the development of arrhythmia and HF.
Although rodent and cultured cardiomyocyte models have provided insights into TKI-associated cardiovascular dysfunction, human translational studies will be critical to elucidate underlying mechanisms.
FUNDING
The authors report that this work was supported by the following grants T32HL134616 and R01 NIHMS1018036.
DISCLOSURE
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the article. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants, patents received or pending, or royalties.
REFERENCES
- 1.Sasaki K, Strom SS, O’Brien S, et al. Relative survival in patients with chronic-phase chronic myeloid leukaemia in the tyrosine-kinase inhibitor era: analysis of patient data from six prospective clinical trials. Lancet Haematol 2015;2(5):e186–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Justice CN, Derbala MH, Baich TM, et al. The impact of pazopanib on the cardiovascular system. J Cardiovasc Pharmacol Ther 2018;23(5):387–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Fuchs HE, et al. Cancer statistics, 2021. CA Cancer J Clin 2021;71(1):7–33. [DOI] [PubMed] [Google Scholar]
- 4.Dobbin SJH, Petrie MC, Myles RC, et al. Cardiotoxic effects of angiogenesis inhibitors. Clin Sci 2021;135(1):71–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III Trial. J Clin Oncol 2010;28(6):1061–8. [DOI] [PubMed] [Google Scholar]
- 6.Pinkhas D, Ho T, Smith S. Assessment of pazopanib-related hypertension, cardiac dysfunction and identification of clinical risk factors for their development. Cardiooncology 2017;3(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Assuncao BMBL, Handschumacher MD, Brunner AM, et al. Acute leukemia is associated with cardiac alterations before chemotherapy. J Am Soc Echocardiogr 2017;30(11):1111–8. [DOI] [PubMed] [Google Scholar]
- 8.Galanis A, Mark L. Inhibition of c-Kit by tyrosine kinase inhibitors. Haematologica 2015;100(3):e77–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kappers MHW, van Esch JHM, Sluiter W, et al. Hypertension induced by the tyrosine kinase inhibitor sunitinib is associated with increased circulating endothelin-1 levels. Hypertension 2010;56(4):675–81. [DOI] [PubMed] [Google Scholar]
- 10.Ma W, Liu M, Liang F, et al. Cardiotoxicity of sorafenib is mediated through elevation of ROS level and CaMKII activity and dysregulation of calcium homoeostasis. Basic Clin Pharmacol Toxicol 2020;126(2):166–80. [DOI] [PubMed] [Google Scholar]
- 11.Shah DR, Shah RR, Morganroth J. Tyrosine kinase inhibitors: their on-target toxicities as potential indicators of efficacy. Drug Saf 2013;36(6):413–26. [DOI] [PubMed] [Google Scholar]
- 12.Petrini I, Lencioni M, Ricasoli M, et al. Phase II trial of sorafenib in combination with 5-fluorouracil infusion in advanced hepatocellular carcinoma. Cancer Chemother Pharmacol 2012;69(3):773–80. [DOI] [PubMed] [Google Scholar]
- 13.Zamorano JL, Lancellotti P, Rodriguez Muñoz D, et al. 2016 ESC position paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC committee for practice guidelines: the task force for cancer treatments and cardiovascular toxicity of the European society of cardiology (ESC). Eur Heart J 2016;37(36):2768–801. [DOI] [PubMed] [Google Scholar]
- 14.Kalay N, Basar E, Ozdogru I, et al. Protective effects of carvedilol against anthracycline-induced cardiomyopathy. J Am Coll Cardiol 2006;48(11):2258–62. [DOI] [PubMed] [Google Scholar]
- 15.Hung MS, Chen IC, Lee CP, et al. Statin improves survival in patients with EGFR-TKI lung cancer: a nationwide population-based study In: Souglakos J, editor. PLoS One 2017;12(2):e0171137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matusewicz L, Czogalla A, Sikorski AF. Attempts to use statins in cancer therapy: an update. Tumor Biol 2020;42(7). 10.1177/1010428320941760. [DOI] [PubMed] [Google Scholar]
- 17.Medeiros BC, Possick J, Fradley M. Cardiovascular, pulmonary, and metabolic toxicities complicating tyrosine kinase inhibitor therapy in chronic myeloid leukemia: Strategies for monitoring, detecting, and managing. Blood Rev 2018;32(4):289–99. [DOI] [PubMed] [Google Scholar]
- 18.Enoma E, Wei L, Chen H. The impact of statin on chemotherapy-induced cardiotoxicity. FASEB J 2019;33(S1). 10.1096/fasebj.2019.33.1_supplement.833.12. [DOI] [Google Scholar]
- 19.Chen MH, Kerkelä R, Force T. Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapeutics. Circulation 2008;118(1):84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du Z, Lovly CM. Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer 2018;17(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Metibemu DS, Akinloye OA, Akamo AJ, et al. Exploring receptor tyrosine kinases-inhibitors in Cancer treatments. Egypt J Med Hum Genet 2019;20(1):35. [Google Scholar]
- 22.Milling RV, Grimm D, Krüger M, et al. Pazopanib, cabozantinib, and vandetanib in the treatment of progressive medullary thyroid cancer with a special focus on the adverse effects on hypertension. Int J Mol Sci 2018;19(10):3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asnani A, Moslehi JJ, Adhikari BB, et al. Preclinical models of cancer therapy–associated cardiovascular toxicity: a scientific statement from the american heart association. Circ Res 2021;129(1):e21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strumberg D, Clark JW, Awada A, et al. Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: a review of four phase I trials in patients with advanced refractory solid tumors. Oncologist 2007;12(4):426–37. [DOI] [PubMed] [Google Scholar]
- 25.Buza V, Rajagopalan B, Curtis AB. Cancer treatment–induced arrhythmias: focus on chemotherapy and targeted therapies. Circ Arrhythm Electrophysiol 2017;10(8):e005443. [DOI] [PubMed] [Google Scholar]
- 26.Bronte E, Galvano A, Novo G, et al. Cardiotoxic effects of anti-VEGFR tyrosine kinase inhibitors. Cardio-Oncol 2017;69–89. [Google Scholar]
- 27.Zentilin L, Puligadda U, Lionetti V, et al. Cardiomyocyte VEGFR-1 activation by VEGF-B induces compensatory hypertrophy and preserves cardiac function after myocardial infarction. FASEB J 2010;24(5):1467–78. [DOI] [PubMed] [Google Scholar]
- 28.Lankhorst S, Saleh L, Danser AJ, et al. Etiology of angiogenesis inhibition-related hypertension. Curr Opin Pharmacol 2015;21:7–13. [DOI] [PubMed] [Google Scholar]
- 29.Hiratsuka S, Minowa O, Kuno J, et al. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci 1998;95(16):9349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou Y, Bourcy K, Kang YJ. Copper-induced regression of cardiomyocyte hypertrophy is associated with enhanced vascular endothelial growth factor receptor-1 signalling pathway. Cardiovasc Res 2009;84(1):54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar R, Knick VB, Rudolph SK, et al. Pharmacokinetic-pharmacodynamic correlation from mouse to human with pazopanib, a multikinase angiogenesis inhibitor with potent antitumor and antiangiogenic activity. Mol Cancer Ther 2007;6(7):2012–21. [DOI] [PubMed] [Google Scholar]
- 32.Abrams TJ, Lee LB, Murray LJ, et al. SU11248 inhibits KIT and platelet-derived growth factor receptor NL in preclinical models of human small cell lung cancer. Mol Cancer Ther 2003;2:471–8. [PubMed] [Google Scholar]
- 33.Mendel DB, Laird AD, Xin X, et al. In Vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 2003;9:327–37. [PubMed] [Google Scholar]
- 34.Lamore SD, Kohnken RA, Peters MF, et al. Cardiovascular toxicity induced by kinase inhibitors: mechanisms and preclinical approaches. Chem Res Toxicol 2020;33(1):125–36. [DOI] [PubMed] [Google Scholar]
- 35.Chintalgattu V, Ai D, Langley RR, et al. Cardiomyocyte PDGFR-β signaling is an essential component of the mouse cardiac response to load-induced stress. J Clin Invest 2010;120(2):472–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsieh PCH, MacGillivray C, Gannon J, et al. Local controlled intramyocardial delivery of platelet-derived growth factor improves postinfarction ventricular function without pulmonary toxicity. Circulation 2006;114(7):637–44. [DOI] [PubMed] [Google Scholar]
- 37.Abbaspour Babaei M, Kamalidehghan B, Saleem M, et al. Receptor tyrosine kinase (c-Kit) inhibitors: a potential therapeutic target in cancer cells. Drug Des Devel Ther 2016;10:2443–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ayach BB, Yoshimitsu M, Dawood F, et al. Stem cell factor receptor induces progenitor and natural killer cell-mediated cardiac survival and repair after myocardial infarction. Proc Natl Acad Sci 2006;103(7):2304–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fazel S Cardioprotective c-kit+ cells are from the bone marrow and regulate the myocardial balance of angiogenic cytokines. J Clin Invest 2006;116(7):1865–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ye L, Zhang EY, Xiong Q, et al. Aging kit mutant mice develop cardiomyopathy. In: Qin G, editor. PLoS One 2012;7(3):e33407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Di Siena S, Gimmelli R, Nori SL, et al. Activated c-Kit receptor in the heart promotes cardiac repair and regeneration after injury. Cell Death Dis 2016;7(7):e2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Talpaz M, Paquette R, Blackwood-Chirchir MA, et al. Dasatinib in imatinib-resistant philadelphia chromosome–positive leukemias. N Engl J Med 2006;354(24):2531–41. [DOI] [PubMed] [Google Scholar]
- 43.Demetri GD, Lo Russo P, MacPherson IRJ, et al. Phase I dose-escalation and pharmacokinetic study of dasatinib in patients with advanced solid tumors. Clin Cancer Res 2009;15(19):6232–40. [DOI] [PubMed] [Google Scholar]
- 44.Brazzelli V, Grasso V, Barbaccia V, et al. Hair depigmentation and vitiligo-like lesions in a leukaemic paediatric patient during chemotherapy with dasatinib. Acta Derm Venereol 2012;92(2):218–9. [DOI] [PubMed] [Google Scholar]
- 45.Hurwitz HI, Dowlati A, Saini S, et al. Phase I trial of pazopanib in patients with advanced cancer. Clin Cancer Res 2009;15(12):4220–7. [DOI] [PubMed] [Google Scholar]
- 46.Fernández A, Sanguino A, Peng Z, et al. An anticancer C-Kit kinase inhibitor is reengineered to make it more active and less cardiotoxic. J Clin Invest 2007;117(12):4044–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hou W, Ding M, Li X, et al. Comparative evaluation of cardiovascular risks among nine FDA-approved VEGFR-TKIs in patients with solid tumors: a Bayesian network analysis of randomized controlled trials. J Cancer Res Clin Oncol 2021;147(8):2407–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rini BI, Cohen DP, Lu DR, et al. Hypertension as a biomarker of efficacy in patients with metastatic renal cell carcinoma treated with sunitinib. J Natl Cancer Inst 2011;103(9):763–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rixe O, Billemont B, Izzedine H. Hypertension as a predictive factor of Sunitinib activity. Ann Oncol 2007;18(6):1117. [DOI] [PubMed] [Google Scholar]
- 50.Cameron AC, Touyz RM, Lang NN. Vascular complications of cancer chemotherapy. Can J Cardiol 2016;32(7):852–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Touyz RM, Lang NN, Herrmann J, et al. Recent advances in hypertension and cardiovascular toxicities with vascular endothelial growth factor inhibition. Hypertension 2017;70(2):220–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Touyz RM, Herrmann SMS, Herrmann J. Vascular toxicities with VEGF inhibitor therapies–focus on hypertension and arterial thrombotic events. J Am Soc Hypertens 2018;12(6):409–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horowitz JR, Rivard A, van der Zee R, et al. Vascular endothelial growth factor/vascular permeability factor produces nitric oxide–dependent hypotension: evidence for a maintenance role in quiescent adult endothelium. Arterioscler Thromb Vasc Biol 1997;17(11):2793–800. [DOI] [PubMed] [Google Scholar]
- 54.Colliva A, Braga L, Giacca M, et al. Endothelial cell–cardiomyocyte crosstalk in heart development and disease. J Physiol 2020;598(14):2923–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ren C, Sun K, Zhang Y, et al. Sodium–glucose cotransporter-2 inhibitor empagliflozin ameliorates sunitinib-induced cardiac dysfunction via regulation of AMPK–mTOR signaling pathway–mediated autophagy. Front Pharmacol 2021;12:664181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shioyama W, Oka T, Kamada R, et al. Symptomatic sinus bradycardia in a patient with solitary fibrous tumor/hemangiopericytoma treated with pazopanib. Intern Med 2021;60(18):2973–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lenihan DJ, Oliva S, Chow EJ, et al. Cardiac toxicity in cancer survivors: cardiac toxicity in cancer survivors. Cancer 2013;119:2131–42. [DOI] [PubMed] [Google Scholar]
- 58.Leiva O, AbdelHameid D, Connors JM, et al. Common pathophysiology in cancer, atrial fibrillation, atherosclerosis, and thrombosis. JACC Cardiooncol 2021;3(5):619–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morabito A, Piccirillo MC, Falasconi F, et al. Vandetanib (ZD6474), a dual inhibitor of vascular endothelial growth factor receptor (VEGFR) and epidermal growth factor receptor (EGFR) tyrosine kinases: current status and future directions. Oncologist 2009;14(4):378–90. [DOI] [PubMed] [Google Scholar]
- 60.Brell JM. Prolonged QTc interval in cancer therapeutic drug development: defining arrhythmic risk in malignancy. Prog Cardiovasc Dis 2010;53(2):164–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ahmad K, Dorian P. Drug-induced QT prolongation and proarrhythmia: an inevitable link? Europace 2007;9(Supplement 4):iv16–22. [DOI] [PubMed] [Google Scholar]
- 62.Dy GK, Adjei AA. Understanding, recognizing, and managing toxicities of targeted anticancer therapies: toxicities of targeted anticancer therapies. CA Cancer J Clin 2013;63(4):249–79. [DOI] [PubMed] [Google Scholar]
- 63.Hutson TE, Davis ID, Machiels JPH, et al. Efficacy and safety of pazopanib in patients with metastatic renal cell carcinoma. J Clin Oncol 2010;28(3):475–80. [DOI] [PubMed] [Google Scholar]
- 64.Mego M, Reckova M, Obertova J, et al. Increased cardiotoxicity of sorafenib in sunitinib-pretreated patients with metastatic renal cell carcinoma. Ann Oncol 2007;18(11):1906–7. [DOI] [PubMed] [Google Scholar]
- 65.O’Neal WT, Lakoski SG, Qureshi W, et al. Relation between cancer and atrial fibrillation (from the REasons for geographic and racial differences in stroke study). Am J Cardiol 2015;115(8):1090–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Holden SN, Eckhardt SG, Basser R, et al. Clinical evaluation of ZD6474, an orally active inhibitor of VEGF and EGF receptor signaling, in patients with solid, malignant tumors. Ann Oncol 2005;16(8):1391–7. [DOI] [PubMed] [Google Scholar]
- 67.Lu Z, Wu CYC, Jiang YP, et al. Suppression of phosphoinositide 3-kinase signaling and alteration of multiple ion currents in drug-induced long QT syndrome. Sci Transl Med 2012;4(131):131ra50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abedi H, Zachary I. Vascular endothelial growth factor stimulates tyrosine phosphorylation and recruitment to new focal adhesions of focal adhesion kinase and paxillin in endothelial cells. J Biol Chem 1997;272(24):15442–51. [DOI] [PubMed] [Google Scholar]
- 69.Gerber HP, McMurtrey A, Kowalski J, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. J Biol Chem 1998;273(46):30336–43. [DOI] [PubMed] [Google Scholar]
- 70.Pretorius L, Du XJ, Woodcock EA, et al. Reduced phosphoinositide 3-kinase (p110α) activation increases the susceptibility to atrial Fibrillation. Am J Pathol 2009;175(3):998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bronte E, Bronte G, Novo G, et al. What links BRAF to the heart function? new insights from the cardiotoxicity of BRAF inhibitors in cancer treatment. Oncotarget 2015;6(34):35589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu Z, Cang S, Yang T, et al. Cardiotoxicity of tyrosine kinase inhibitors in chronic myelogenous leukemia therapy. Hematol Rep 2009;1(1):4. [Google Scholar]
- 73.Gril B, Palmieri D, Qian Y, et al. Pazopanib reveals a role for tumor cell B-Raf in the prevention of HER2 1 breast cancer brain metastasis. Clin Cancer Res 2011;17(1):142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pakladok T, Hosseinzadeh Z, Almilaji A, et al. Up-Regulation of hERG K+ Channels by B-RAF. In: Barnes S, editor. PLoS One 2014;9(1):e87457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhu WZ, Wang SQ, Chakir K, et al. Linkage of β1-adrenergic stimulation to apoptotic heart cell death through protein kinase A–independent activation of Ca21/calmodulin kinase II. J Clin Invest 2003;111(5):617–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol 2011;51(4):468–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pandey V, Xie LH, Qu Z, et al. Mitochondrial contributions in the genesis of delayed afterdepolarizations in ventricular myocytes. Front Physiol 2021;12:744023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wehrens XHT, Lehnart SE, Reiken SR, et al. Ca 2+/ Calmodulin-dependent protein kinase ii phosphorylation regulates the cardiac ryanodine receptor. Circ Res 2004;94(6):e61–70. [DOI] [PubMed] [Google Scholar]
- 79.Beckendorf J, van den Hoogenhof MMG, Backs J. Physiological and unappreciated roles of CaMKII in the heart. Basic Res Cardiol 2018;113(4):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hudmon A, Schulman H. Structure–function of the multifunctional Ca21/calmodulin-dependent protein kinase II. Biochem J 2002;364(3):593–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van Oort RJ, McCauley MD, Dixit SS, et al. Ryanodine receptor phosphorylation by calcium/ calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation 2010;122(25):2669–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 2005;105(7):2640–53. [DOI] [PubMed] [Google Scholar]
- 83.Lee WS, Kim J. Cardiotoxicity associated with tyrosine kinase-targeted anticancer therapy. Mol Cell Toxicol 2018;14(3):247–54. [Google Scholar]
- 84.McMullen CJ, Chalmers S, Wood R, et al. Sunitinib and imatinib display differential cardiotoxicity in adult rat cardiac fibroblasts that involves a role for calcium/calmodulin dependent protein kinase II. Front Cardiovasc Med 2021;7:630480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Curigliano G, Lenihan D, Fradley M, et al. Management of cardiac disease in cancer patients throughout oncological treatment: ESMO consensus recommendations. Ann Oncol 2020;31(2):171–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ghatalia P, Morgan CJ, Je Y, et al. Congestive heart failure with vascular endothelial growth factor receptor tyrosine kinase inhibitors. Crit Rev Oncol Hematol 2015;94(2):228–37. [DOI] [PubMed] [Google Scholar]
- 87.Moustafa S, Ho TH, Shah P, et al. Predictors of incipient dysfunction of all cardiac chambers after treatment of metastatic renal cell carcinoma by tyrosine kinase inhibitors: cardiac chambers’ dysfunction with TKIs. J Clin Ultrasound 2016;44(4):221–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Groarke JD, Choueiri TK, Slosky D, et al. Recognizing and managing left ventricular dysfunction associated with therapeutic inhibition of the vascular endothelial growth factor signaling pathway. Curr Treat Options Cardiovasc Med 2014;16(9):335. [DOI] [PubMed] [Google Scholar]
- 89.Cheng H, Kari G, Dicker AP, et al. A novel preclinical strategy for identifying cardiotoxic kinase inhibitors and mechanisms of cardiotoxicity. Circ Res 2011;109(12):1401–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Grabowska ME, Chun B, Moya R, et al. Computational model of cardiomyocyte apoptosis identifies mechanisms of tyrosine kinase inhibitor-induced cardiotoxicity. J Mol Cell Cardiol 2021;155:66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Datta SR, Dudek H, Tao X, et al. Akt Phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997;91(2):231–41. [DOI] [PubMed] [Google Scholar]
- 92.Friehs I, Barillas R, Vasilyev NV. Vascular endothelial growth factor prevents apoptosis and preserves contractile function in hypertrophied infant heart. Circulation 2006;114(1_suppl). I-290–I-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.del Peso L, Gonzalez-Garcia M, Page C, et al. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 1997;278(5338):687–9. [DOI] [PubMed] [Google Scholar]
- 94.Matsui T, Tao J, del Monte F, et al. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation 2001;104(3):330–5. [DOI] [PubMed] [Google Scholar]
- 95.Kerkela R, Woulfe KC, Durand JB, et al. Sunitinib-induced cardiotoxicity is mediated by off-target inhibition of AMP-activated protein kinase. Clin Transl Sci 2009;2(1):15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Freebern W, Fang H, Slade M, et al. In vitro cardiotoxicity potential comparative assessments of chronic myelogenous leukemia tyrosine kinase inhibitor therapies: dasatinib, imatinib and nilotinib. Blood 2007;110(11):4582. [Google Scholar]
- 97.Korashy HM, Attafi IM, Ansari MA, et al. Molecular mechanisms of cardiotoxicity of gefitinib in vivo and in vitro rat cardiomyocyte: Role of apoptosis and oxidative stress. Toxicol Lett 2016;252:50–61. [DOI] [PubMed] [Google Scholar]
- 98.Kerkelä R, Grazette L, Yacobi R, et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med 2006;12(8):908–16. [DOI] [PubMed] [Google Scholar]
- 99.Chaar M, Kamta J, Ait-Oudhia S. Mechanisms, monitoring, and management of tyrosine kinase inhibitors-associated cardiovascular toxicities. Oncotargets Ther 2018;11:6227–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Force T, Krause DS, Van Etten RA. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer 2007;7(5):332–44. [DOI] [PubMed] [Google Scholar]
- 101.Kivelä R, Hemanthakumar KA, Vaparanta K, et al. Endothelial cells regulate physiological cardiomyocyte growth via VEGFR2-mediated paracrine signaling. Circulation 2019;139(22):2570–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Virag JAI, Rolle ML, Reece J, et al. Fibroblast growth factor-2 regulates myocardial infarct repair. Am J Pathol 2007;171(5):1431–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lieu C, Heymach J, Overman M, et al. Beyond VEGF: inhibition of the fibroblast growth factor pathway and antiangiogenesis. Clin Cancer Res 2011;17(19):6130–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Baird A, Esch F, Mormede P, et al. Molecular characterization of fibroblast growth factor: distribution and biological activities in various tissues. Recent Prog Horm Res 1986;42:143–205. [DOI] [PubMed] [Google Scholar]
- 105.Schultz JEJ, Witt SA, Nieman ML, et al. Fibroblast growth factor-2 mediates pressure-induced hypertrophic response. J Clin Invest 1999;104(6):709–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cilvik SN, Wang JI, Lavine KJ, et al. Fibroblast growth factor receptor 1 signaling in adult cardiomyocytes increases contractility and results in a hypertrophic cardiomyopathy. In: Lionetti V, editor. PLoS One 2013;8(12):e82979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Akman T, Erbas O, Akman L, et al. Prevention of pazopanib-induced prolonged cardiac repolarization and proarrhythmic effects. Arq Bras Cardiol 2014;103(5):403–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kappers MHW, de Beer VJ, Zhou Z, et al. Sunitinib-induced systemic vasoconstriction in swine is endothelin mediated and does not involve nitric oxide or oxidative stress. Hypertension 2012;59(1):151–7. [DOI] [PubMed] [Google Scholar]
- 109.Kruzliak P, Novak J, Novak M. Vascular endothelial growth factor inhibitor–induced hypertension: from pathophysiology to prevention and treatment based on long-acting nitric oxide donors. Am J Hypertens 2014;27(1):3–13. [DOI] [PubMed] [Google Scholar]
- 110.Cholesterol Treatment Trialists’ (CTT) Collaborators. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet 2012;380(9841):581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mills EJ, Rachlis B, Wu P, et al. Primary prevention of cardiovascular mortality and events with statin treatments. J Am Coll Cardiol 2008;52(22):1769–81. [DOI] [PubMed] [Google Scholar]
- 112.Seicean S, Seicean A, Plana JC, et al. Effect of statin therapy on the risk for incident heart failure in patients with breast cancer receiving anthracycline chemotherapy. J Am Coll Cardiol 2012;60(23):2384–90. [DOI] [PubMed] [Google Scholar]
- 113.Calvillo-Argüelles O, Abdel-Qadir H, Michalowska M, et al. Cardioprotective effect of statins in patients with her2-positive breast cancer receiving trastuzumab therapy. Can J Cardiol 2019;35(2):153–9. [DOI] [PubMed] [Google Scholar]
- 114.Maitland ML, Bakris GL, Black HR, et al. Initial assessment, surveillance, and management of blood pressure in patients receiving vascular endothelial growth factor signaling pathway inhibitors. J Natl Cancer Inst 2010;102(9):596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wasserstrum Y, Kornowski R, Raanani P, et al. Hypertension in cancer patients treated with anti-angiogenic based regimens. Cardio-Oncol 2015;1(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Thavendiranathan P, Poulin F, Lim KD, et al. Use of myocardial strain imaging by echocardiography for the early detection of cardiotoxicity in patients during and after cancer chemotherapy. J Am Coll Cardiol 2014;63(25):2751–68. [DOI] [PubMed] [Google Scholar]
- 117.Geyer H, Caracciolo G, Abe H, et al. Assessment of myocardial mechanics using speckle tracking echocardiography: fundamentals and clinical applications. J Am Soc Echocardiogr 2010;23(4):351–69. [DOI] [PubMed] [Google Scholar]
- 118.Spring BQ, Bryan Sears R, Zheng LZ, et al. A photoactivable multi-inhibitor nanoliposome for tumour control and simultaneous inhibition of treatment escape pathways. Nat Nanotechnol 2016;11(4):378–87. [DOI] [PMC free article] [PubMed] [Google Scholar]