

Abstract
Oropharyngeal squamous cell carcinoma (OPSCC), largely fueled by the human papillomavirus (HPV), has a complex biological and immunologic phenotype. Although HPV/p16 status can be used to stratify OPSCC patients as a function of survival, it remains unclear what drives an improved treatment response in HPV-associated OPSCC and whether targetable biomarkers exist that can inform a precision oncology approach. We analyzed OPSCC patients treated between 2000 and 2016 and correlated locoregional control (LRC), disease-free survival (DFS) and overall survival (OS) with conventional clinical parameters, risk parameters generated using deep-learning algorithms trained to quantify tumor-infiltrating lymphocytes (TILs) (OP-TIL) and multinucleated tumor cells (MuNI) and targeted transcriptomics. P16 was a dominant determinant of LRC, DFS and OS, but tobacco exposure, OP-TIL and MuNI risk features correlated with clinical outcomes independent of p16 status and the combination of p16, OP-TIL and MuNI generated a better stratification of OPSCC risk compared to individual parameters. Differential gene expression (DEG) analysis demonstrated overlap between MuNI and OP-TIL and identified genes involved in DNA repair, oxidative stress response and tumor immunity as the most prominent correlates with survival. Alteration of inflammatory/immune pathways correlated strongly with all risk features and oncologic outcomes. This suggests that development of OPSCC consists of an intersection between multiple required and permissive oncogenic and immunologic events which may be mechanistically linked. The strong relationship between tumor immunity and oncologic outcomes in OPSCC regardless of HPV status may provide opportunities for further biomarker development and precision oncology approaches incorporating immune checkpoint inhibitors for maximal anti-tumor efficacy.
INTRODUCTION
Human papillomavirus (HPV) infection with high-risk (HR) types has been linked to delayed development of oropharyngeal squamous cell carcinoma (OPSCC)1–4. This has led to an epidemic rise in incidence for OPSCC a disease formerly associated with significant tobacco exposure and relatively low prevalence in the United States5,6. Currently, HPV-associated OPSCC (HPV + OPSCC) has overtaken cervical cancer as the most common HPV-associated malignancy in the United States and incidence continues to rise. Despite increasing rates of vaccination, HPV + OPSCC is unlikely to begin to plateau for at least 2–3 decades based on disease latency post initial exposure7.
Development of precision oncology approaches requires matching tumor biology to treatment intensity in order to minimize the risk of both over- and under-treatment. This is particularly challenging in the context of OPSCC for several reasons. First, although HPV-based stratification can generally differentiate low-risk (LR) disease (treatment responsive) from HR OPSCC disease (treatment refractory)8, it remains a biologically incomplete descriptor of the disease. This is manifested by the deleterious effect of tobacco exposure on OPSCC treatment response and survival initially identified by Ang et al. and subsequently reproduced across multiple cohorts, including most recently by our group in 20208–11. Tobacco exposure has now been linked to an altered tumor immune microenvironment (TIME) by Foy et al. in oral cavity cancer (reduced CD3, CD4, CD8 and PDL1 expression via immunohistochemistry (IHC)) and our group in OPSCC (reduced CD3, CD8 expression via IHC) which may account for its effects on treatment response and oncologic outcomes12,13. Although clinically useful, stratification of OPSCC solely on the basis of HPV may fail to account for smoking-mediated immune modulation, along with other yet unclear mechanisms of aggressive tumor behavior and reduced treatment response in a subset of patients.
Second, a subset of HPV + OPSCC tumors demonstrate aggressive behavior (distant metastasis) and reduced treatment response even in the absence of tobacco exposure. The drivers of this phenomenon remain unclear, although low tumor mutational burden, reduced E7 and p16 levels14, differential transcription of E1 and E4 and associated dysregulation of PI3kinase signaling15 have all been linked to differential survival. Third, HPV itself at this time remains a non-targetable biomarker, in contrast to targetable oncogenic events such as BRAF (melanoma) mutation and EGFR (lung) mutation or amplification16,17. For these reasons we believe it important to refine the clinical and biological description of OPSCC in a manner which appropriately reflects the differential risk profile of the target patient population and the molecular profile of individual tumors. In the current manuscript, we sought to determine whether the current clinical algorithm which combines clinical datapoints (e.g. T, N-classification) with p16 IHC can be augmented by leveraging neural network algorithms focused on evaluation of the TIME and aberrant tumor cell activity in order to predict treatment response and survival in a cohort of Veterans enriched for intermediate-risk OPSCC (defined as dual exposure to HPV and tobacco). We further sought to identify transcriptional markers of reduced treatment response and survival as an important step toward a precision oncology approach to OPSCC.
MATERIALS AND METHODS
Clinical dataset
Following approval from Baylor College of Medicine and the Michael E. DeBakey Veteran’s Administration (MEDVAMC) Institutional Review Boards, we reviewed the records of Veterans with previously untreated OPSCC between January 1, 2000 and January 1, 2016. All collection and analysis of the current data was performed in a manner consistent with existing standards for clinical research (Declaration of Helsinki, US Federal Policy for the Protection of Human Subjects). Inclusion criteria included: (1) primary OPSCC, (2) tissue diagnosis at the MEDVAMC, and (3) treatment delivery at the MEDVAMC. Exclusion criteria included treatment at an outside institution and recurrent disease at presentation. Demographic information was recorded including age, gender, race, smoking history and alcohol consumption. Smoking history was collected at our institution at the time of initial diagnosis as “pack-years” consistent with current clinical practice at our institution. Clinical and pathologic features were collected including clinical stage according to the American Joint Commission on Cancer (Staging Manual 7th Edition) staging system. Results of diagnostic procedures including imaging results, biopsies, and fine needle aspirations as well as the treatments rendered and the associated dates were recorded. Note: For the current study we utilized the 7th Edition of the Staging Manual for two reasons. First, it accurately reflects the time period of patient diagnosis and treatment. Second, and more importantly, the 7th Edition is agnostic with respect to p16/HPV status. Since our goal is to evaluate a combined cohort of OPSCC enriched for dual exposure patients, and moreover to evaluate the value of p16 in the context of other parameters, it would be counterproductive to utilize the 8th Edition which itself includes HPV status in the staging of OPSCC.
p16 immunohistochemistry
All tissue analysis summarized in the current manuscript was performed using pre-treatment biopsy material from the primary tumor site (needle biopsies of nodal disease were excluded from the analysis). Formalin-fixed paraffin-embedded (FFPE) biopsy tissue blocks were retrieved from the archive maintained at the MEDVAMC Department of Pathology. Sections (5 μM) were cut from each FFPE block and mounted on positively charged slides. One section was stained with hematoxylin & eosin and reviewed by a surgical pathologist to confirm the original histopathological diagnosis and to ensure tumor adequacy. Slides were deparaffinized in Bond Dewax Solution (Leica Biosystems, Buffalo Grove, IL), and rehydrated in descending grades of 100, 90 and 70% ethanol. Endogenous peroxide activity was blocked by pretreatment with 3% hydrogen peroxide for 10 min, and antigen recovery was achieved by 20 min of heat-induced epitope retrieval. IHC stains were performed on an automated tissue-staining system using the Bond Polymer Refine Detection (Leica Biosystems, Buffalo Grove, IL) using the monoclonal antibody for p16, clone E6H4 (Ventana, Tucson, Arizona). Signal detection was performed using polymer reagent conjugated with horseradish peroxidase followed by the 3,3′-Diaminobenzidine (DAB) Chromogen Kit (Leica Biosystems, Buffalo Grove, IL). Areas of tumor were selected for analysis and confirmed by trained Pathologists (SL, WY). Representative images of tumor were captured using the Vectra3 scanner using the CRI multispectral camera using a ×20 UplanSApo objective (0.75 aperture) mounted on an Olympus microscope. (Akoya Biosciences, Menlo Park, CA). Image analysis was performed with InForm 2.4 software (Akoya) using a training algorithm defined by the operator to segment features of tissue and cells. Tissue areas were segmented to define tumor from normal epithelium, connective tissue and lymphocytes. Cell nuclei were defined by the presence of hematoxylin stain and cytoplasm was defined as the area within 6 pixels of individual nuclei. Sampled cells were defined by the operator as positive or negative and inForm was trained on these criteria. inForm counted the number of positively stained features (nuclear or cytoplasmic) in representative fields for each tissue section. The cutoff threshold was set by the operator on a subset of images and then applied to all remaining images. The percent of positively stained features was calculated as the number of positive cells divided by the total number of cells in the tumor segmented areas. These ratios were averaged across all images for each tissue section (Supplementary Fig. 1). Clinical p16 data was defined using the most recently published guidelines from Lewis Jr. et al.18 of >70% strong nuclear and cytoplasmic staining as identified by the primary pathologist involved in the individual cases. Research p16 data was determined by re-analyzing tumor specimens using the algorithm outlined above and reporting p16 values as fraction of tumor cells with strong nuclear and cytoplasmic staining and using these values as continuous variable spectrum.
Computerized analysis of tissue specimens
Hematoxylin and eosin (H&E) stained slides were digitized by scanning using Ventana iScan HT at ×40 resolution (0.25 μm/pixel resolution). WSIs unsuitable for computational analysis due to blurriness, pen marking, resin bubbles were filtered out using a computational tool HistoQC19. Using a recently described algorithm20, we generated a multinucleation index (MuNI) characterizing the density of multinucleation (MN) events in epithelial (EP) regions. Briefly, two conditional generative adversarial networks (GAN) were utilized for MuNI calculation; (1) GANMN to segment MN events, and (2) GANEP to segment EP regions in the digitized images. Once the segmentation was done the MN events detected outside of EP regions were removed. MuNI was then calculated as a ratio of the total number of MN events to the total number of nuclei within EP regions. In parallel we performed a quantitative analysis of tumor-infiltrating lymphocytes (TILs) using a recently described algorithm21. First, all image nuclei were automatically segmented by employing a deep-learning-based model22. A support vector machine was then used to classify each segmented nucleus as either TIL or non-TIL based on visual features such as texture, shape, and color. Although non-TILs include cancer cells (majority), macrophages, fibroblasts, neutrophils, among other nucleated cells, in this study, they were not differentiated but treated as a single type. TIL and non-TIL clusters were built based on cell proximity (Fig. 2). From the constructed clusters of TILs and non-TILs, multiple features related to density, architecture, and co-localization were extracted21. Finally, the top features, determined by least absolute shrinkage and selection operator, were used to train a Cox regression model that assigned a risk index to each patient23. The MuNI and OP-TIL indices were generated as continuous variables and were used to stratify the patients into LR and HR categories. Averages of the MuNI and OP-TIL associated risk indices in the training cohort were defined as thresholds for risk stratification. The thresholds were then applied on the MuNI and OP-TIL indices in the current cohort to obtain binarized labels for each patient. OP-TIL and MuNI risk indices were correlated to conventional IHC-based quantification of CD3 and CD8 infiltrating lymphocytes performed on the same specimens (serial-sections) as previously described by our group13.
Fig. 2. Impact of OP-TIL risk on overall survival (OS) and locoregional control (LRC).
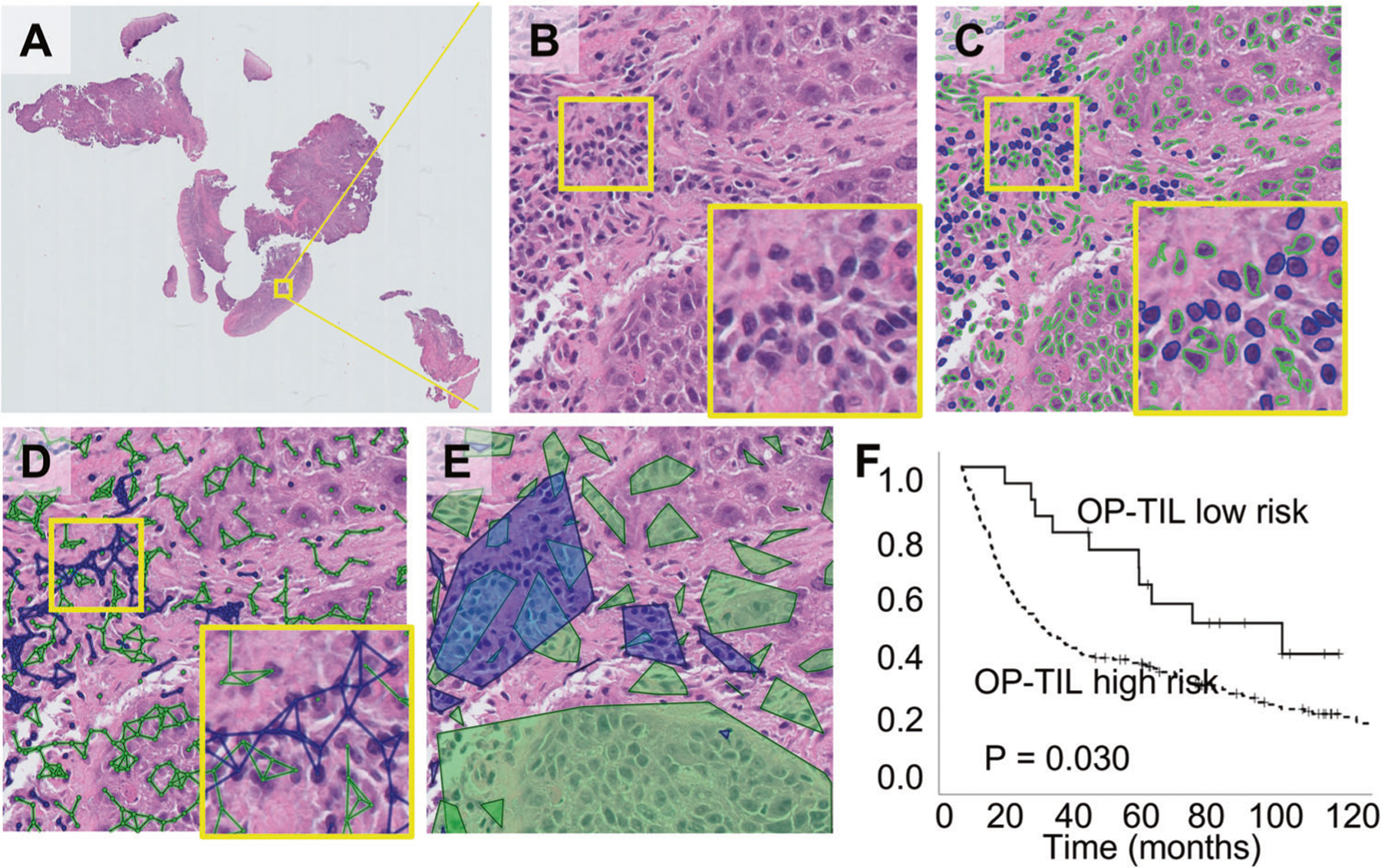
A Original H&E WSI of an OPC patient. B A representative high-power field. C Identification of cell types: TILs (blue) and non-TILs (green). D Cell subgraphs built by linking proximal nuclei of the same type. E Resulting clusters for each cell type obtained by drawing the convex hull of each subgraph. F OS analysis performed using Kaplan–Meier analysis coupled to log-rank test for the OP-TIL OS risk classifier.
Transcriptomic analysis
One hundred FFPE specimens underwent targeted transcriptomic analysis using the HTG EdgeSeq Oncology Biomarker Panel which contains 2567 probes including 4 negative and 4 positive controls alongside human universal RNA control technical replicates. Sequencing was performed using the Illumina NextSeq Sequencer. Differential gene expression analysis was performed using the DESeq2 package (version 1.14.1) available from Bioconductor. The package provides methods for estimating and testing differential expression using negative-binomial generalized linear models. Empirical Bayes methods were used to estimate dispersion and log2(fold change) with data-driven prior distributions. Correction for library size was performed using the median ratio method and dispersions were estimated with the Cox Reid-adjusted profile likelihood method. Log2 fold change was calculated using Tikhonov/ridge regularization with a zero-centered normal prior distribution with variance calculated using the observed distribution of maximum likelihood coefficients. Overlap of gene expression signatures was calculated using a Venn Diagram tool (VIB/UGent Bioinformatics & Evolutionary Genomics). Pathway analysis was performed using the Gene Ontology Resource- GO Enrichment Analysis (7/2/2021 release).
Statistical analysis
Endpoints included time to locoregional or distant recurrence and death. Imaging was used as a surrogate in the absence of a pathological report documenting recurrence. Locoregional control (LRC—date of diagnosis to date of locoregional recurrence (LRR), LRR—date of diagnosis to date of LRR), distant metastatic control (DM—date of diagnosis to date of distant metastasis), relapse-free or disease-free survival (date of primary diagnosis to date of recurrence or death; RFS/DFS) and overall survival (date of diagnosis to last documented hospital note or death note; OS) were calculated. Patients suspected of recurrence were restaged using clinical exam and imaging. Associations between clinical, biological and pathologic variables were determined by two-sided Fisher’s exact tests. Actuarial survival rates were generated using the Kaplan–Meier method, and comparisons between groups were made using log-rank statistics. Multivariate analysis was performed using Cox regression. Statistical calculations were performed with SPSS (IBM SPSS Statistics version 25). For all statistics, p-values were considered to be statistically significant if below a threshold of 0.05 (two-sided). For individual statistical tests and/or correlations, details are provided in the following section after each specific analysis.
Machine learning approach to predict disease aggressiveness based on gene expression
Patients were dichotomized into HR and LR disease if they had a distant metastasis or death before 5 years. Differentially expressed genes (DEGs) were identified by performing multiple T-test corrected by the Benjamini–Hochberg procedure with an FDR of 0.1, using JMP 13.2.1 statistical software (SAS) and correlation between survival time in months and gene expression was also determined using JMP. Matlab statistics and machine learning toolbox was used to identify features (i.e., genes) using the univariate feature ranking function based on chi-square tests, or features were manually selected based on their ranks following differential analysis of gene expression (DEG) or correlation as described above, with a fourth list comprised of genes in common following DEG and correlation. For training, 89/99 samples were randomly chosen so as to leave out 10 samples (5 aggressive and 5 non-aggressive) for subsequent validation. Three models were chosen from the Matlab menu for training and validation, which included Linear Discriminant Analysis, Logistic Regression (LR), and Support Vector Machine (quadratic SVM). For validation, sensitivity was defined as number of true aggressive tumors predicted divided by total number of true aggressive tumors, and specificity defined as true negatives predicted divided by all true negatives.
RESULTS
Patient, tumor and treatment characteristics
We analyzed data from 297 Veterans who were predominantly male, with a median age of 61, of which nearly 80% reported a tobacco use history in excess of 10 pack years (Table 1). A majority presented with T3–4 disease and nodal disease (Supplementary Table 1) and 84% of patients were slated for curative intent treatment of which concurrent chemo-radiation was the most commonly utilized (Supplementary Table 2). Among patients which received curative intent treatment 198 received chemotherapy and 51 did not. Of the patients which received chemotherapy, 153 received cisplatin monotherapy, 10 received carboplatin monotherapy, 6 received cisplatin and 5-fluorouracil and 21 received cetuximab monotherapy. The demographic characteristics of the patient population did not significantly change over the study period (Supplementary Table 3).
Table 1.
Patient characteristics.
| Mean | Median | ||
|---|---|---|---|
| Age (years) | 61.57 | 61 | |
| Income (zip code based) | $65,181 | $46,471 | |
| Distance from VA (miles) | 85.54 | 31 | |
| Number | % | ||
| Gender | Male | 295 | 99.33 |
| Female | 2 | 0.67 | |
| Race | White | 229 | 77.1 |
| Black | 65 | 21.89 | |
| Other | 3 | 1.01 | |
| Tobacco use | Yes | 256 | 86.2 |
| No | 41 | 13.8 | |
| ≥10 pack years | 232 | 79.45 | |
| <10 pack years | 60 | 20.55 | |
| Alcohol use | Yes | 247 | 84.88 |
| No | 44 | 15.12 | |
| Mean | Median | ||
| Pack years | 40.58 | 40 | |
Clinical drivers of disease control and survival
For patients treated with curative intent median overall survival (OS) was 54.7 months (mean = 75.4 months); median disease-free survival (DFS) was 30.3 months (mean = 64.4 months). T-classification impacted OS (p < 0.001) and DFS (p = 0.004) but the effect on LRC and distant metastasis (DM) did not reach statistical significance (p = 0.102, p = 0.366 respectively). The presence of nodal disease was associated with improved OS (p = 0.031) and DFS (p = 0.046) (Supplementary Fig. 2) which although apparently paradoxical is driven by the strong relationship between nodal metastasis and p16 positivity. Patients who received chemotherapy had improved OS (p = 0.014) and DFS (p = 0.010) but the effect did not reach statistical significance for LRC (p = 0.097) or DM (p = 0.103). Patients who received cisplatin monotherapy trended toward improved OS (p = 0.078; DFS (p = 0.384) compared patients who received cetuximab monotherapy. Tobacco exposure impacted OS at both a 10 (p = 0.009) and 30 pack-year (p < 0.001) threshold (Supplementary Fig. 3) as well as DFS (p = 0.002, p = 0.001 respectively) and LRC (p = 0.004, p = 0.001 respectively) but not DM. Tobacco exposure levels decreased over the study period (Table 2). P16 positivity doubled over the study period from 32% to 64% of tumors (Table 2). The most dramatic shifts were a decrease in the frequency of tumors with minimal nuclear p16 staining and a doubling of the fraction of tumors with >70% of tumor cells demonstrating strong nuclear staining. These shifts in risk factor exposure were accompanied by an improvement in OS (p = 0.009) and LRC (p = 0.035) over the study time period (Supplementary Fig. 4).
Table 2.
Risk profile changes.
| Fraction of patients | ||||
|---|---|---|---|---|
| Pack-years | 2000–2005 | 2006–2010 | 2011–2015 | |
| Tobacco exposure | <10 | 0.2 | 0.16 | 0.28 |
| ≥10 | 0.8 | 0.84 | 0.72 | |
| <30 | 0.36 | 0.28 | 0.49 | |
| ≥30 | 0.64 | 0.72 | 0.51 | |
| Mean | 44.39 | 45.46 | 32.16 | |
| Median | 40 | 41.5 | 30 | |
| p16—clinical | Neg | 0.68 | 0.48 | 0.36 |
| Pos | 0.32 | 0.52 | 0.64 | |
| p16—research | 0–25 | 0.58 | 0.41 | 0.33 |
| (% tumor cells with strong nuclear staining) | 25–50 | 0.03 | 0.02 | 0.08 |
| 50–75 | 0.12 | 0.03 | 0.06 | |
| 75–100 | 0.27 | 0.54 | 0.53 | |
P16 positivity determined using the standard clinical definition correlated strongly with OS (p < 0.001) and LRC (p < 0.001) (Fig. 1). When p16 analysis was performed using the automated imaging algorithm, the fraction of positive nuclear staining impacted LRC and OS both for the entire cohort and for patients treated with curative intent (p < 0.001 for all comparisons) (Fig. 1). We first used recursive partitioning analysis (using OS data) and identified the primary split (logworth 10.67) at 70% nuclear staining, coinciding well with the current clinical standard, and a second non-significant split (logworth 0.36) at 52%. Using LRR data we generated a significant split (logworth 13.57) at 79% and a second non-significant split (logworth 0.95) at 25%. Based on this analysis we decided to analyze the oncologic outcomes for our patients using cutoffs of 70% and 25%. Patients with intermediate nuclear staining (25–70% of nuclei) demonstrated intermediate LRC, but OS comparable with p16- OPSCC patients. When accounting for nuclear staining, the degree of cytoplasmic staining had no significant impact on OS or LRC (all p-values > 0.300). Among those patients with >70% p16 nuclear staining who received curative intent treatment tobacco exposure as a continuous variable (pack-years) had a significant effect on OS (p = 0.028, Exp (B) 1.009, 95% CI 1.001–1.017). The effect also became significant when tumors with 50–70% p16 nuclear staining were included in the analysis (p = 0.047, Exp(B) 1.007, 95% CI 1.000–1.015).
Fig. 1. Impact of p16 on overall survival (OS) and locoregional control (LRC).
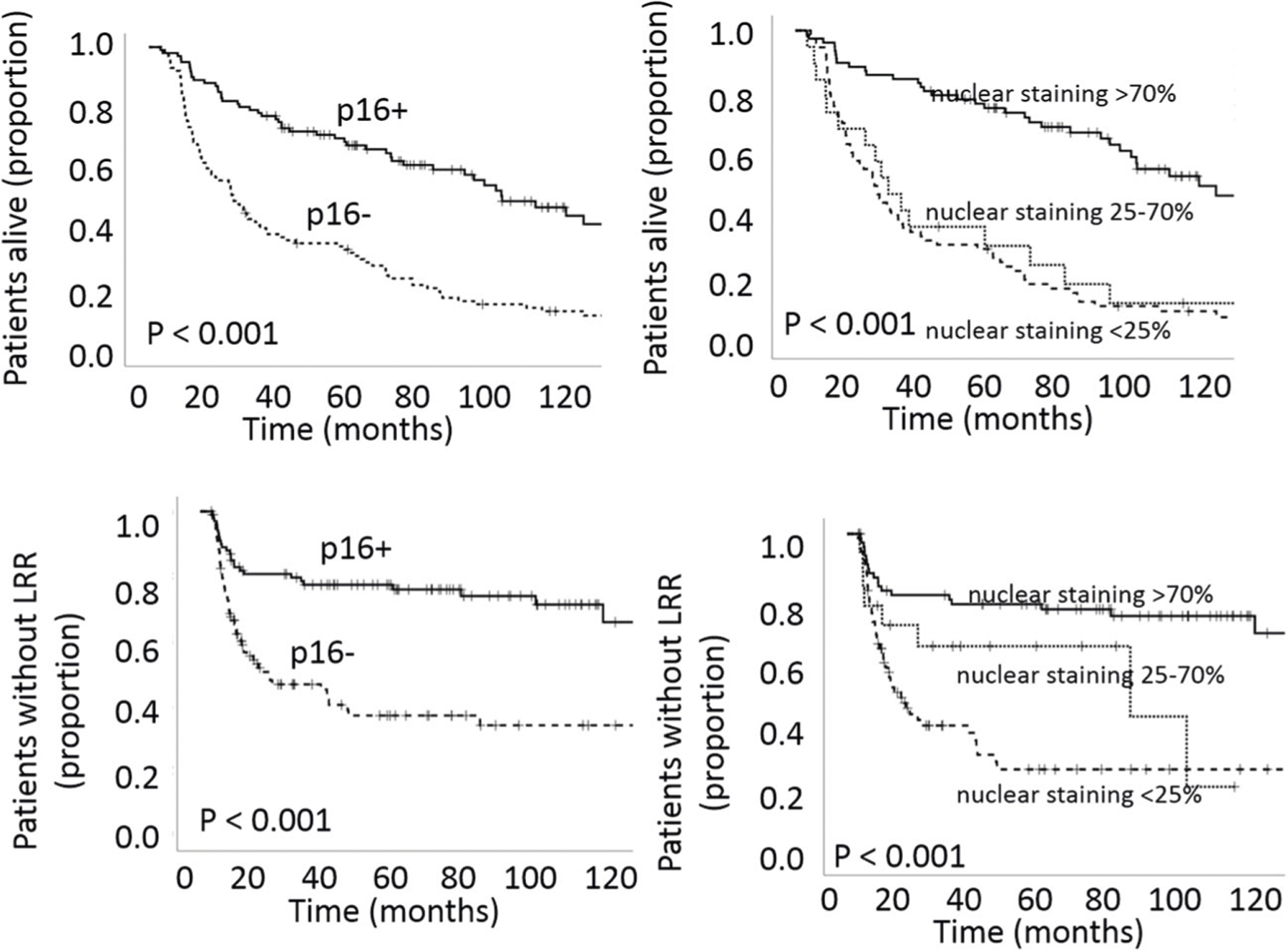
Analysis performed using Kaplan–Meier analysis coupled to log-rank test.
OP-TIL and MuNI association with oncologic outcomes
We recently showed that TILs correlate strongly with disease stage, p16 positivity and heavy tobacco exposure using conventional IHC13. Here we applied a high throughput computational algorithm which quantifies TILs using automated imaging as well as spatial tumor cell—immunocyte interactions. First, we tested 2 OP-TIL risk stratifiers (defined using a pre-existing training cohort) for correlation with OS and LRC. The OS risk stratifier correlated with OS when analyzed in the entire patient cohort (p = 0.030); the DFS risk stratifier trended toward significant correlation with LRC (p = 0.062) (Fig. 2). Second, using data from 138 patients for whom both CD3 and CD8 IHC data and OP-TIL data were available (from the same tumor specimens, serially sectioned), we analyzed the relationship between OP-TIL risk determination, conventional TIL IHC data, p16 and tobacco exposure. OP-TIL risk determination correlated with CD3/8 risk classification as previously reported by our group13, p16 and tobacco exposure (Table 3) in a relatively linear and quantitative fashion. Pearson bivariate analysis identified correlation between: (1) OSscore and CD8 infiltration p < 0.001 (−0.441), (2) OSscore and CD3 infiltration p < 0.001 (−0.454), (3) DFSscore and CD8 infiltration p = 0.001 (−0.287) and (4) DFSscore and CD3 infiltration p = 0.003 (−0.353). MN was described in 2012 by Lewis Jr. et al. as a feature associated with OPSCC treatment response and patient survival24. MuNI was strongly correlated with LRC and OS (p < 0.001 for both parameters) (Fig. 3) and demonstrated partial correlation with p16 positivity (Pearson correlation of the binary values for each parameter (p < 0.01 (−0.458)) or continuous variables (p < 0.01 (0.519)) and tobacco exposure (p < 0.01 (0.196)).
Table 3.
Correlation between OP-TIL, IHC and risk factors.
| Mean | Mean | p-value | p-value | ||
|---|---|---|---|---|---|
| OP-TIL (OS) | OP-TIL (DFS) | OP-TIL (OS) | OP-TIL (DFS) | ||
| CD3/8 combined classifier | Cold | −0.98 | −0.14 | ||
| Hot | −1.27 | −0.2 | <0.001 | <0.001 | |
| p16 nuclear staining frequency | >70% | −1.23 | −0.18 | ||
| <70% | −0.99 | −0.15 | <0.001 | <0.001 | |
| Tobacco exposure (pack-years) | 0–10 | −1.26 | −0.19 | ||
| 10–30 | −1.1 | −0.17 | 0.022 | 0.221 | |
| >30 | −1.04 | −0.15 | <0.001 | 0.003 |
Fig. 3. Impact of MuNI on overall survival (OS) and locoregional control (LRC).
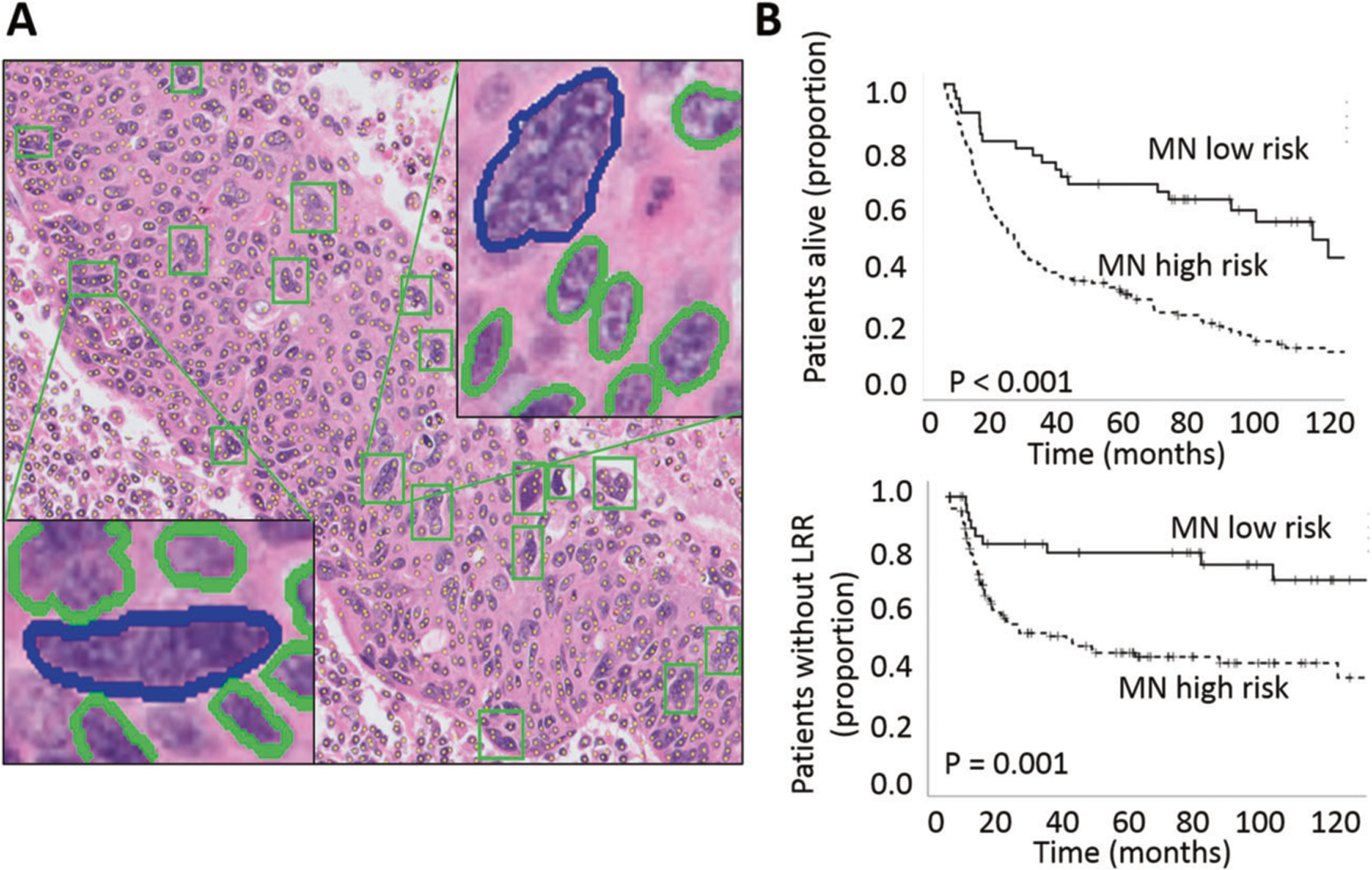
A A representative high-power field from the H&E image of an OPC patient with an overlay of automatically detected MN events by GANMN, shown by green boxes. Two of them are illustrated in enlarged views, where MN events are shown in blue and cancerous nuclei in green. B OS and LRC analysis performed using Kaplan–Meier analysis coupled to log-rank test for the MuNI risk classifier.
We utilized the clinical and biological characteristics described above to determine whether patients with OPSCC can be stratified with respect to LRC and OS to a greater or equivalent degree than using p16 positivity. Using Cox regression for patients who received curative intent treatment (Supplementary Table 4) we found that T-classification and p16 status correlated significantly with OS and N-classification and p16 status correlated significantly with LRC. Given the previously identified partial correlations between tobacco exposure, p16 status, MuNI and OP-TIL risk stratifiers we then conducted a secondary analysis. Median survival for patients with tobacco exposure <10 pack-years and >70% p16 staining was 85.95 months. Among these LR patients, a high MuNI score reduced survival to 72.86 months. Patients with a high MuNI score and a low OP-TIL score demonstrated an even lower median survival of 55.77 months. The combination of dichotomized MuNI, OP-TIL and p16 scores generated a profound differentiation in survival between LR and HR patients for patients treated with curative intent (Fig. 4). At 5 years post treatment, ~90% of LR patients were without disease compared to ~20% of HR patients, a greater differentiation than for any of the classifiers alone (p = 0.001 for OS, p = 0.005 for LRC). Mean time to LRR for the LR patients was 102 months, whereas for the HR patients it was 36 months. The hazard ratio for p16-defined HR patients was 4.2 (95% CI 2.5–7.1) for LRC and 3.9 (95% CI 2.6–5.8) for OS. The hazard ratio for combined classifier-defined HR patients was 10.2 (95% CI 1.4–74.0) for LRC and 6.0 (95% CI 1.9–19.4) for OS.
Fig. 4. Impact of combined p16, OP-TIL OS and MN on overall survival (OS) and locoregional control (LRC).
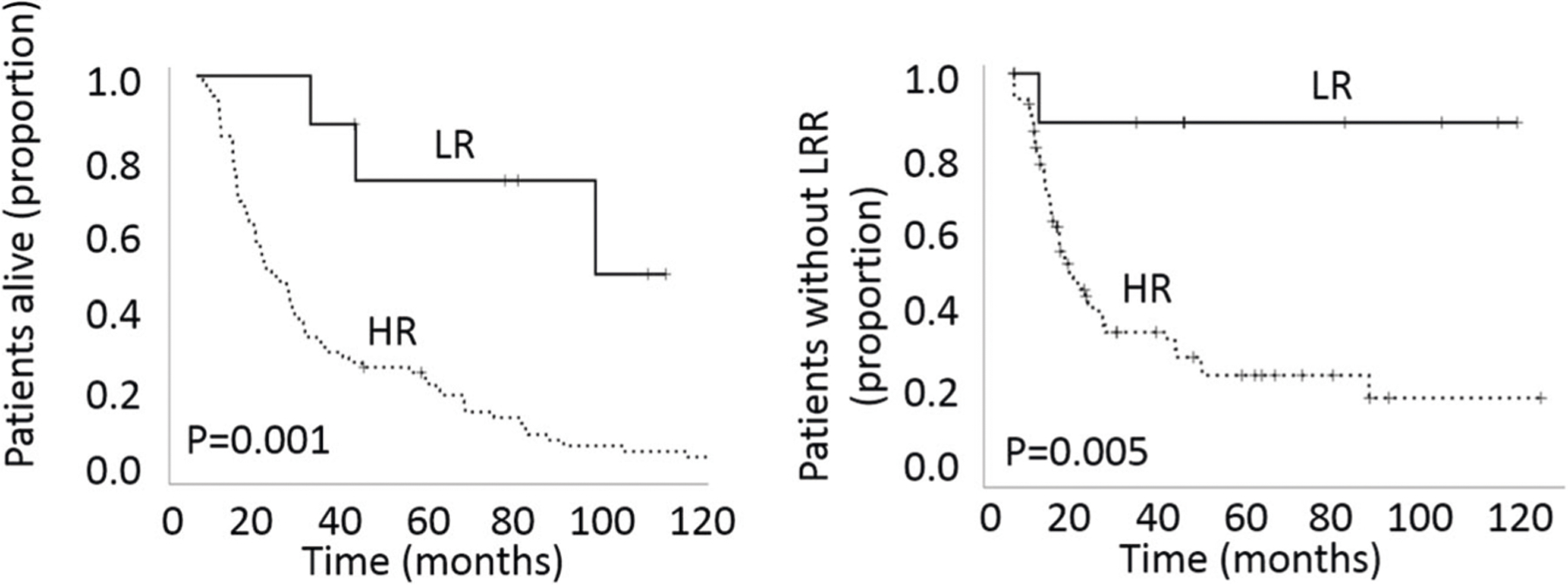
Analysis performed using Kaplan–Meier analysis coupled to log-rank test. LR = patients with low-risk MuNI and OP-TIL scores and p16 positive tumors; HR = patients with high-risk MuNI and OP-TIL scores and p16 negative tumors.
Transcriptomic signature of OPSCC as a function of risk factors
A targeted transcriptomic analysis was performed on 100 specimens from patients treated with curative intent. To confirm that these specimens accurately reflected the larger cohort (Fig. 5A) and previously published clinical data we evaluated the interaction between tobacco exposure and p16 status with respect to OS and demonstrated a clear correlation with our previous publications and existing literature (Fig. 5B)13,25,26. We first dichotomized these specimens into LR and HR based on the 4 individual features outlined above: tobacco exposure (using a 10 pack year cutoff), p16 status, MuNI and OP-TIL. For each individual feature, we identified DEGs using an adjusted p-value <0.05 and a 1.5-fold change as the cutoffs for the analysis. For p16, 75 genes were downregulated in p16 LR tumors compared to p16 HR tumors, and 58 genes were upregulated (Supplementary Table 5). Of note, SLC7A11 (cysteine-glutamate transport), SLC7A8 (amino acid transport), PYGL (glycogen metabolism), ANPEP (peptide metabolism), SLC5A1 (glucose transport), ODC1 (polyamine metabolism) were all significantly upregulated in p16 HR tumors. For tobacco exposure, 16 genes were downregulated in LR tumors compared to HR tumors, and 15 genes were upregulated (Supplementary Table 6). Thirteen of sixteen genes downregulated in non-smokers were shared with p16 positive tumors and 8/15 genes upregulated in non-smokers were shared with p16 positive tumors including CDKN2A. HR MuNI tumors demonstrated 530 downregulated DEGs and 98 upregulated DEGs compared to LR MuNI tumors (Supplementary Table 7). LR MuNI and LR p16 tumors shared 49 downregulated DEGs and 20 upregulated DEGs, a minority in both cases. A total of 15 DEGs were shared between all 3 features with 12 downregulated in LR tumors and 3 upregulated in LR tumors. GO analysis of these genes identified 6 pathways with FDR < 0.05 including regulation of immune system process (enriched 6-fold, FDR 0.037) and regulation of lymphocyte migration (enriched 63-fold, FDR 0.041). This suggests that p16, tobacco exposure and MN biologically converge at least partially on an immune OPSCC signature (Supplementary Fig. 5).
Fig. 5. Identification of genes associated with aggressive OPSCC.
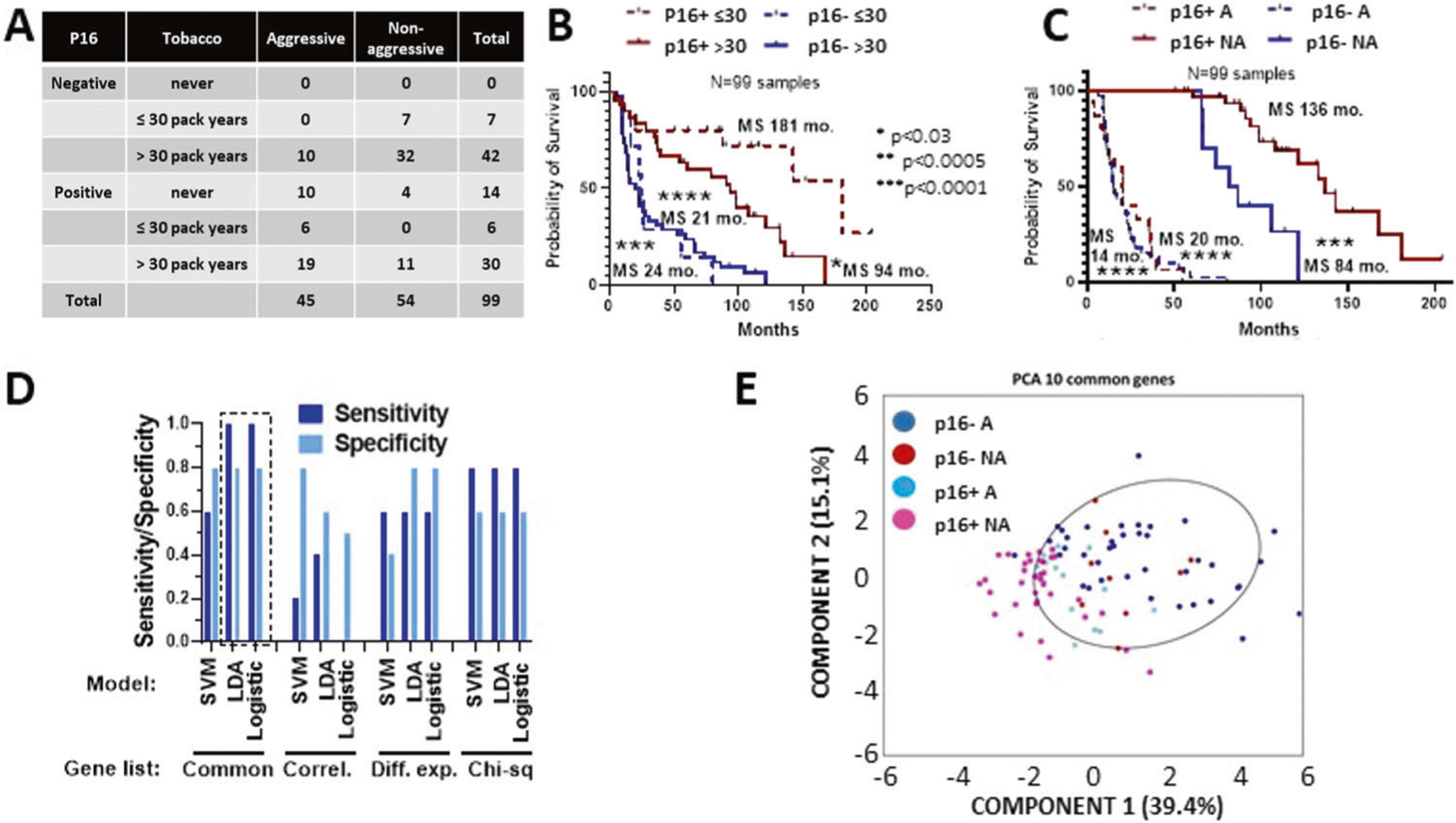
A Relevant clinical characteristics of cohort chosen for focused transcriptional gene expression analysis. Aggressive (A) disease was defined as associated with death or distant metastasis within 5 years; non-aggressive (NA) disease was defined as no death or distant metastasis within 5 years. B Survival of the RNA cohort stratified by tobacco exposure and p16 status. C Survival of RNA cohort stratified by aggressive (A) vs non-aggressive (NA) classification and p16 status. D Validation results from machine learning models using different gene selection lists: Common= 10 genes in common between top 20 ranking differentially expressed and most correlated genes; Correl = top 10 most correlated genes with survival time; Diff. exp. = top 10 most significantly different genes between aggressive and non-aggressive tumors; Chi-sq = top 10 genes using univariate ranking function in Matlab. E Principal component analysis (PCA) using RNA expression from the 10 genes on the common list demonstrating the genes could separate A p16+ and p16-neg tumors from NA p16+ OPSCC tumors.
Since the OP-TIL algorithm relies primarily on immune infiltration and spatial interactions between immunocytes and tumor cells to predict treatment response and survival, we sought to measure the biological overlap between this feature and intrinsic OPSCC biological features. LR OP-TIL tumors demonstrated downregulation of 495 DEGs compared to HR OP-TIL tumors with upregulation of 47 DEGs (Supplementary Table 8). Thirty-eight DEGs were shared between p16 LR and OP-TIL LR tumors; 33 were downregulated, 5 were upregulated. GO analysis revealed the following pathways enriched more than 100 fold with a concomitant FDR < 0.01: positive regulation of immature T cell proliferation, positive regulation of dendritic cell antigen processing and presentation, positive regulation of prostaglandin biosynthetic process, fever generation and regulation of macrophage migration inhibitory factor signaling pathway. MuNI and OP-TIL features shared 447 DEGs which were downregulated in LR tumors. GO analysis revealed enrichment in multiple immune/inflammatory pathways: positive regulation of metabolic process(enriched 2.6-fold, FDR 10−40), positive regulation of macromolecule metabolic process (enriched 2.6-fold; FDR 10−39), positive regulation of chronic inflammatory response (enriched 44.6-fold, FDR 0.03) and positive regulation of chronic inflammatory response to antigenic stimulus (enriched 44.6-fold, FDR 0.04). We subsequently restricted the analysis to p16 positive tumors and identified 12 genes which were upregulated in LR MuNI tumors: IL16 (chemotaxis), IRF8 (interferon signaling, CD3D (TIL activity), RIPK3 (TNF signaling), CCR7 (chemotaxis), CXXC5 (DNA damage), DSC2 (migration), IL2RB (chemotaxis), MEF2C (lymphopoiesis), PIK3CD (immune response), SPINK5 (inflammation), SUSD3 (migration). GO analysis of these genes demonstrated: lymphocyte activation (enriched 26-fold, FDR < 0.001), lymphocyte differentiation (enriched 33.8-fold, FDR < 0.01) and mononuclear cell differentiation (enriched 29.4-fold, FDR < 0.01).
Transcriptomic signature of OPSCC as a function of oncologic outcomes
We then correlated gene expression levels (Supplementary Table 9) with 2 clinical outcomes most closely related to treatment failure, namely LRR and DM. Twenty two genes correlated with LRR with an FDR ≤ 0.1 (CES1 (xenobiotic processing), FGF19 (metastasis), BAP1 (DNA damage), IL6 (chemotaxis), TYK2 (interferon signaling), FANCL (DNA repair), NUP62 (replication), TCF7L1 (senescence), TCF3 (DNA binding), IRF3 (interferon signaling), PDK2 (metabolism), SMARCA4 (transcription), PNKP (DNA repair), FAF1 (apoptosis), ALDH2 (oxidative stress), PRKX (differentiation), SMAD7 (TGF signaling), CD40 (inflammation), CDKN1B (proliferation), ABCD3 (fatty acid metabolism), ARID1B (proliferation), ITPKB (signaling)) and 1 gene correlated with DM with an FDR ≤ 0.1 (CXCL5 (chemotaxis)). GO analysis of genes associated with LRR identified enrichment in pathways both intrinsic to tumor cells and pathways regulating the interaction between tumor cells and immunocytes including: regulation of DNA binding transcription factor activity (enriched 13.3-fold, FDR < 0.01), cytokine mediated signaling pathway (enriched 8.4-fold, FDR < 0.05) and cellular response to cytokine stimulus (enriched 6.5-fold, FDR < 0.05).
Finally, we dichotomized tumors into aggressive (A) OPSCC and non-aggressive (NA) OPSCC as a function of survival and/or distant metastasis at the 5-year mark which would be considered by most clinicians to be adequate to differentiate recurrent disease for a new primary or define an oncologic “cure” (Fig. 5C). Next, we compared DEGs between A and NA tumors while ignoring p16 status (Supplementary Tables 10–12). We then utilized machine learning tools to determine whether expression levels for genes of interest could be used to predict whether a patient’s tumor was aggressive or non-aggressive; selected genes were limited to the top 10 differentially expressed or highly correlated ones or the intersection of these two lists. The RNA expression cohort was split to utilize 89 randomly chosen samples for training using 3 different models, leaving 10 samples out with a 50% split between aggressive and non-aggressive classification for validation. Principal component analysis using expression of the 10 common genes (AHNAK2 (metastasis), ANPEP (metastasis), BCL2 (apoptosis), CES1 (xenobiotic processing), CXCL8 (IL-8; chemotaxis), FADD (apoptosis), FANCG (DNA repair), FGF19 (metastasis), FOSL1 (proliferation) and IL1A (IL-1alpha; inflammation)) (Fig. 5) revealed that they were reasonably good at separating both p16+ and p16− aggressive tumors from non-aggressive p16+ tumors, but less robust distinguishing aggressive and non-aggressive tumors within p16-neg disease.
DISCUSSION
Although dichotomized into “low-risk” HPV-associated disease and “high-risk” non-HPV-associated disease by the 8th Edition of the AJCC Staging Manual, there is now ample evidence that OPSCC biology occasionally defies this binary risk prediction. Our transcriptomic data link genes involved in 3 distinct processes with LRR: (1) DNA repair/proliferation, (2) inflammation and (3) metabolic processing of oxidative stress. Although there is evidence linking HPV to differential DNA repair and activation of cell death cascades, in OPSCC the link to inflammation/immunity and metabolism remains relatively weak27. MN, a morphologic feature of tumor cells associated with reduced treatment response and aggressive behavior has now been shown to be prognostic of OPSCC treatment response independent of HPV status20,24. In the current study we confirmed that although MN and p16 status do correlate, this correlation is incomplete suggesting that HPV status alone does not fully capture the biology intrinsic to OPSCC tumor cells. This is consistent with recent studies which demonstrate substantial transcriptomic variability among HPV + OPSCC15 and is particularly relevant for genes involved in metabolic processing of exogenous or oxidative stress such as CES1, a target of Nrf2, a master regulator of oxidative stress response linked to cisplatin and radiation resistance in multiple solid tumor types28. Whether differential activation of metabolic and oxidative stress genes drives the development of MN irrespective of HPV status will require combining molecular manipulation of individual genes using preclinical OPSCC models with spatial genomic and transcriptomic analysis of patient tumors.
There is now clear evidence that intra-tumor processes including mechanism of cell death generate shifts in the TIME29–34. Both necrosis and apoptosis can release sufficient signals for immune activation and surveillance of solid tumors, and hyperactivation of programmed cell death cascades may in fact generate a sufficiently favorable TIME to impact treatment response and survival. There is reason to evaluate the DEG analysis summarized above using a holistic approach which considers shifts in DNA damage/repair and shifts in TIME to represent a mechanistically linked driver of treatment response and survival particularly in light of recently published data indicating aggressive disease behavior in HPV + OPSCC with defective DNA repair and genomic instability35. This concept is supported by the dramatic overlap of DEGs in tumors with high levels of multinucleation (MuNI) and unfavorable interactions between TILs and tumor cells (OP-TIL) and the fact that 9/12 genes upregulated in tumors with low levels of MN are genes involved in inflammation/immunity. Multiple analyses of patient specimens and preclinical studies have linked both endogenous and exogenous shifts in tumor metabolism with alterations in TIME in multiple solid tumors including HNSCC36–39. Furthermore, there is now increasing evidence that oxidative stress response mechanisms such as Nrf2 can play a critical role in regulating tumor immunity suggesting that the transcriptomic shifts described here may be mechanistically related40,41. Finally, we and others have linked metabolic shifts in metabolite transport, especially amino acids with resistance to cisplatin and ferroptosis agonists which may generate secondary immunologically relevant signals42.
It is clear that p16 as a surrogate marker of HPV oncogenic infection remains a potent risk stratifier even in OPSCC patient cohorts with extensive tobacco exposure such as the one evaluated here6,8,9,25. Its clinical impact dominates all other clinical and biological stratifiers on multivariate analysis. It is therefore highly unlikely that another specific biomarker will replace p16 in the near future for clinical purposes. However, clinical and biological evidence from other investigators and our group indicates that additional information will be needed to separate “low-risk” and “intermediate-risk” OPSCC cohorts sufficiently in order to support safe, yet effective treatment de-escalation for OPSCC patients with favorable tumor biology. Previously reported institutional trials aimed at de-escalation relied heavily on p16/HPV testing to broadly define the study cohorts, but generally included other features of LR disease in the final inclusion criteria (e.g. low T-classification; lack of tobacco exposure)43,44. ECOG3311 also utilized additional risk features following surgical resection of HPV + OPSCC to stratify patients between arms of variable treatment intensity. Our data indicate that MuNI and OP-TIL may improve upon p16-based stratification. That p16 alone is an incomplete measure of favorable tumor immunity is borne out by multiple studies including our previous work which demonstrated that tobacco exposure is associated with ~40% reduction in CD8 + TILs even in p16 positive OPSCC tumors13. By combining intrinsic tumor cell features (e.g. p16 overexpression, MuNI) with direct measures of TIME (e.g. OP-TIL, or alternatively the newly developed inflamed T-cell gene expression profile (TGEP)45,46) we may be able to not only consistently identify the most appropriate OPSCC candidates for de-escalation, but also begin to define the patient population most likely to respond to ICIs. We believe that our data support a concept recently discussed by Fialova et al., namely the potential of tumor immunity as a prognostic tool either as standalone or combined with HPV status47.
Limitations of the current study include its size and built-in selection bias driven by its single institutional nature. An additional limitation is the potential for tissue sampling bias as previous studies have demonstrated variability in TIME between the core and periphery of solid tumors48, along with a limited validation cohort for the machine learning-based analysis of DEGs. Future studies have been designed to combine bulk transcriptomic measurements with newer techniques which allow us to generate transcriptomic data with spatial resolution directly from FFPE slides and correlate it closely with MuNI, OP-TIL and conventional IHC data in order to reduce some of the inherent limitations of measuring gene expression across multiple cell types present in the same solid tumor. Strengths include a comprehensive clinical annotation of the chosen specimens which substantially surpasses what can be extracted from existing public databases and the combination of high throughput imaging-based risk stratification, with conventional clinical stratification strategies and a transcriptional analysis. The MuNI risk stratification algorithm has now already been validated across multiple HPV + OPSCC cohorts and thus represents a robust and consistent prognostic marker of OPSCC treatment response and survival20. The OP-TIL algorithm has also been validated in a multi-institutional HPV + OPSCC cohort and correlates well with our previously published CD3/CD8 classifier for intermediate-risk OPSCC13,21. Unlike multiplexed IHC/IF, both OP-TIL and MuNI can be rapidly applied to existing hematoxylin & eosin slides from diagnostic biopsies and thus be rapidly deployed in a manner minimally intrusive to existing clinical pathways for cancer diagnosis. By combining these algorithms with a more detailed transcriptomic analysis of OPSCC tumors we suggest 2 translational strategies deserve additional investigation. First, that response to conventional chemo-radiation strategies may reflect at least in part, an interaction between tumor cells and immunocytes. Second, TIME may be a prognostic biomarker of chemo-radiation response and ICI effectiveness regardless of HPV status. Between the newly defined inflamed T-cell gene expression profile (TGEP)45,46 that can be captured using targeted transcriptomics, quantitative IHC for cytotoxic CD8 + T cells as recently described by our group and the newly developed OP-TIL algorithm, it may be possible to define a sensitive and specific biomarker of treatment response in OPSCC which could more faithfully replicate the complex biological and immunologic reality and thus serve to develop a precision oncology approach to this complex disease.
Supplementary Material
Funding
This material is the result of work supported with resources and the use of facilities of the Michael E. DeBakey VA Medical Center and was specifically supported by an institutional pilot grant (V.C.S). A.G.S. has received support from the Department of Veterans Affairs Million Veteran Program (BX004183-01A1) and the National Institutes of Health (U01DE028233). This work was supported by a Career Development Award to V.C.S. from the Veterans Administration Clinical Science Research and Development division (1IK2CX001953). A.M.’s work is supported from grants supported by the National Cancer Institute under award numbers 1U24CA199374-01, R01CA249992-01A1, R01CA202752-01A1, R01CA208236-01A1, R01CA216579-01A1, R01CA220581-01A1, R01CA257612-01A1, 1U01CA239055-01, 1U01CA248226-01, 1U54CA254566-01, National Heart, Lung and Blood Institute 1R01HL15127701A1, R01HL15807101A1, National Institute of Biomedical Imaging and Bioengineering 1R43EB028736-01, National Center for Research Resources under award number 1 C06 RR12463-01 VA Merit Review Award IBX004121A from the United States Department of Veterans Affairs Biomedical Laboratory Research and Development Service, the Office of the Assistant Secretary of Defense for Health Affairs, through the Breast Cancer Research Program (W81XWH-19-1-0668), the Prostate Cancer Research Program (W81XWH-15-1-0558, W81XWH-20-1-0851), the Lung Cancer Research Program (W81XWH-18-1-0440, W81XWH-20-1-0595), the Peer Reviewed Cancer Research Program (W81XWH-18-1-0404, W81XWH-21-1-0345), the Kidney Precision Medicine Project (KPMP) Glue Grant, the Ohio Third Frontier Technology Validation Fund, the Clinical and Translational Science Collaborative of Cleveland (UL1TR0002548) from the National Center for Advancing Translational Sciences (NCATS) component of the National Institutes of Health and NIH roadmap for Medical Research, The Wallace H. Coulter Foundation Program in the Department of Biomedical Engineering at Case Western Reserve University. Sponsored research agreements from Bristol Myers-Squibb, Boehringer-Ingelheim, and Astrazeneca. H.D.S. is supported by R01CA168485-06.
Footnotes
COMPETING INTERESTS
There are no competing interests or conflicts of interest directly related to this work. A.M. is an equity holder in Elucid Bioimaging and in Inspirata Inc. In addition he has served as a scientific advisory board member for Inspirata Inc, Astrazeneca, Bristol Meyers-Squibb and Merck. Currently he serves on the advisory board of Aiforia Inc and currently consults for Caris, Roche and Aiforia. He also has sponsored research agreements with Philips, AstraZeneca, Boehringer-Ingelheim and Bristol Meyers-Squibb. His technology has been licensed to Elucid Bioimaging. He is also involved in a NIH U24 grant with PathCore Inc, and 3 different R01 grants with Inspirata Inc. J.P.Z. is founder, equity shareholder, and chair of the board for Droplet Biosciences (Cambridge, MA), equity shareholder in Summit Biolabs (Aurora, CO), and a consultant for Merck.
ETHICS APPROVAL/CONSENT TO PARTICIPATE
All procedures and care were reviewed and approved by the Baylor College of Medicine Institutional Review Board.
ADDITIONAL INFORMATION
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41379-022-01024-8.
DATA AVAILABILITY
All data not explicitly included in the accompanying tables and figures will be made available upon reasonable request.
REFERENCES
- 1.Gravitt PE & Winer RL Natural History of HPV Infection across the Lifespan: Role of Viral Latency. Viruses 9, 267 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gupta SM & Mania-Pramanik J Molecular mechanisms in progression of HPV-associated cervical carcinogenesis. J. Biomed. Sci 26, 28 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3.Kreimer AR et al. The epidemiology of oral HPV infection among a multinational sample of healthy men. Cancer Epidemiol. Biomark. Prev 20, 172–182 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castellsague X Natural history and epidemiology of HPV infection and cervical cancer. Gynecol. Oncol 110, S4–S7 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Dahlstrom KR et al. An evolution in demographics, treatment, and outcomes of oropharyngeal cancer at a major cancer center: a staging system in need of repair. Cancer 119, 81–89 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sandulache VC et al. Oropharyngeal squamous cell carcinoma in the veteran population: association with traditional carcinogen exposure and poor clinical outcomes. Head Neck 37, 1246–1253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Fakhry C & D’Souza G Projected association of human papillomavirus vaccination with oropharynx cancer incidence in the US, 2020–2045. JAMA Oncol 7, e212907 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ang KK et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med 363, 24–35 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fakhry C et al. Validation of NRG oncology/RTOG-0129 risk groups for HPV-positive and HPV-negative oropharyngeal squamous cell cancer: Implications for risk-based therapeutic intensity trials. Cancer 125, 2027–2038 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fakhry C et al. Human papillomavirus and overall survival after progression of oropharyngeal squamous cell carcinoma. J. Clin. Oncol 32, 3365–3373 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gillison ML et al. Tobacco smoking and increased risk of death and progression for patients with p16-positive and p16-negative oropharyngeal cancer. J. Clin. Oncol 30, 2102–2111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foy JP et al. The immune microenvironment of HPV-negative oral squamous cell carcinoma from never-smokers and never-drinkers patients suggests higher clinical benefit of IDO1 and PD1/PD-L1 blockade. Ann. Oncol 28, 1934–1941 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Kemnade JO et al. CD8 infiltration is associated with disease control and tobacco exposure in intermediate-risk oropharyngeal cancer. Sci. Rep 10, 243 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Facompre ND et al. Identifying predictors of HPV-related head and neck squamous cell carcinoma progression and survival through patient-derived models. Int. J. Cancer 147, 3236–3249 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gleber-Netto FO, et al. Variations in HPV function are associated with survival in squamous cell carcinoma. JCI Insight 4, e124762 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giugliano F et al. First line treatment of BRAF mutated advanced melanoma: Does one size fit all? Cancer Treat Rev 99, 102253 (2021). [DOI] [PubMed] [Google Scholar]
- 17.Bonomi PD et al. Predictive biomarkers for response to EGFR-directed monoclonal antibodies for advanced squamous cell lung cancer. Ann. Oncol 29, 1701–1709 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewis JS Jr. et al. Human Papillomavirus Testing in Head and Neck Carcinomas: Guideline From the College of American Pathologists. Arch. Pathol. Lab. Med. 142, 559–597 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Janowczyk A, Zuo R, Gilmore H, Feldman M & Madabhushi A HistoQC: An Open-Source Quality Control Tool for Digital Pathology Slides. JCO Clin. Cancer Inf 3, 1–7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koyuncu CF, et al. Computerized tumor multinucleation index (MuNI) is prognostic in p16+ oropharyngeal carcinoma: a multi-site validation study. J. Clin. Investig 131, e145488 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corredor G, et al. An Imaging Biomarker of Tumor-Infiltrating Lymphocytes to Risk-Stratify Patients with HPV-Associated Oropharyngeal Cancer. J. Natl Cancer Inst djab215 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahmood F et al. Deep Adversarial Training for Multi-Organ Nuclei Segmentation in Histopathology Images. IEEE Trans. Med. Imaging 39, 3257–3267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corredor G, et al. A watershed and feature-based approach for automated detection of lymphocytes on lung cancer images. Med. Imaging 105810R, 2018 (2018). [Google Scholar]
- 24.Lewis JS Jr., Scantlebury JB, Luo J & Thorstad WL Tumor cell anaplasia and multinucleation are predictors of disease recurrence in oropharyngeal squamous cell carcinoma, including among just the human papillomavirus-related cancers. Am. J. Surg. Pathol 36, 1036–1046 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elhalawani H et al. Tobacco exposure as a major modifier of oncologic outcomes in human papillomavirus (HPV) associated oropharyngeal squamous cell carcinoma. BMC Cancer 20, 912 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vawda N, Banerjee RN & Debenham BJ Impact of Smoking on Outcomes of HPV-related Oropharyngeal Cancer Treated with Primary Radiation or Surgery. Int. J. Radiat. Oncol. Biol. Phys 103, 1125–1131 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Rampias T, Sasaki C & Psyrri A Molecular mechanisms of HPV induced carcinogenesis in head and neck. Oral Oncol 50, 356–363 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Ge PX et al. Short-term standard alcohol consumption enhances platelet response to clopidogrel through inhibition of Nrf2/Ces1 pathway and induction of Cyp2c in mice. Life Sci 279, 119268 (2021). [DOI] [PubMed] [Google Scholar]
- 29.Dudek AM, Garg AD, Krysko DV, De Ruysscher D & Agostinis P Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev 24, 319–333 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Green DR, Ferguson T, Zitvogel L & Kroemer G Immunogenic and tolerogenic cell death. Nat. Rev. Immunol 9, 353–363 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson TS, McGaha T & Munn DH Chemo-Immunotherapy: Role of Indoleamine 2,3-Dioxygenase in Defining Immunogenic Versus Tolerogenic Cell Death in the Tumor Microenvironment. Adv. Exp. Med. Biol 1036, 91–104 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Obeid M et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med 13, 54–61 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Tesniere A et al. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ 15, 3–12 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Zhou J et al. Immunogenic cell death in cancer therapy: present and emerging inducers. J. Cell. Mol. Med 23, 4854–4865 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schrank TP et al. Genomic heterogeneity and copy number variant burden are associated with poor recurrence-free survival and 11q loss in human papillomavirus-positive squamous cell carcinoma of the oropharynx. Cancer 127, 2788–2800 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krupar R et al. Immunometabolic determinants of chemoradiotherapy response and survival in head and neck squamous cell carcinoma. Am. J. Pathol 188, 72–83 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Veeramachaneni R, et al. Metformin generates profound alterations in systemic and tumor immunity with associated antitumor effects. J. Immunother. Cancer 9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng RN et al. Histidine supplementation improves insulin resistance through suppressed inflammation in obese women with the metabolic syndrome: a randomised controlled trial. Diabetologia 56, 985–994 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Ippolito L, Morandi A, Giannoni E & Chiarugi P Lactate: A Metabolic Driver in the Tumour Landscape. Trends Biochem. Sci 44, 153–166 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Friedmann Angeli JP & Meierjohann S NRF2-dependent stress defense in tumor antioxidant control and immune evasion. Pigment Cell Melanoma Res 34, 268–279 (2021). [DOI] [PubMed] [Google Scholar]
- 41.Rojo de la Vega M, Chapman E & Zhang DD NRF2 and the Hallmarks of Cancer. Cancer Cell 34, 21–43 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu W et al. Acquisition of Cisplatin Resistance Shifts Head and Neck Squamous Cell Carcinoma Metabolism toward Neutralization of Oxidative Stress. Cancers (Basel) 2020; 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carnevale C, Ortiz-Gonzalez I, Ortiz-Gonzalez A, Bodi-Blanes L & Til-Perez G Early T1-T2 stage p16+ oropharyngeal tumours. Role of upfront transoral robotic surgery in de-escalation treatment strategies. A review of the current literature. Oral Oncol 113, 105111 (2021). [DOI] [PubMed] [Google Scholar]
- 44.Ma DJ et al. Phase II Evaluation of Aggressive Dose De-Escalation for Adjuvant Chemoradiotherapy in Human Papillomavirus-Associated Oropharynx Squamous Cell Carcinoma. J. Clin. Oncol 37, 1909–1918 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cristescu R et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018; 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ayers M et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig 127, 2930–2940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fialova A, Koucky V, Hajduskova M, Hladikova K & Spisek R Immunological Network in Head and Neck Squamous Cell Carcinoma-A Prognostic Tool Beyond HPV Status. Front. Oncol 10, 1701 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galon J & Lanzi A Immunoscore and its introduction in clinical practice. Q J. Nucl. Med. Mol. Imaging 64, 152–161 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data not explicitly included in the accompanying tables and figures will be made available upon reasonable request.