

Summary
Analysis of brain structure, connectivity, and molecular diversity relies on effective tissue fixation. Conventional tissue fixation causes extracellular space (ECS) loss, complicating the segmentation of cellular objects from electron microscopy datasets. Previous techniques for preserving ECS in mammalian brains utilizing high-pressure perfusion can give inconsistent results owing to variations in the hydrostatic pressure within the vasculature. A more reliable fixation protocol that uniformly preserves the ECS throughout whole brains would greatly benefit a wide range of neuroscience studies. Here, we report a straightforward transcardial perfusion strategy that preserves ECS throughout the whole rodent brain. No special setup is needed besides sequential solution changes, and the protocol offers excellent reproducibility. In addition to better capturing tissue ultrastructure, preservation of ECS has many downstream advantages such as accelerating heavy-metal staining for electron microscopy, improving detergent-free immunohistochemistry for correlated light and electron microscopy, and facilitating lipid removal for tissue clearing.
Keywords: connectomics, correlated light and electron microscopy (CLEM), electron microscopy (EM), tissue fixation, tissue clearing, immunohistochemistry, extracellular space
Graphical abstract
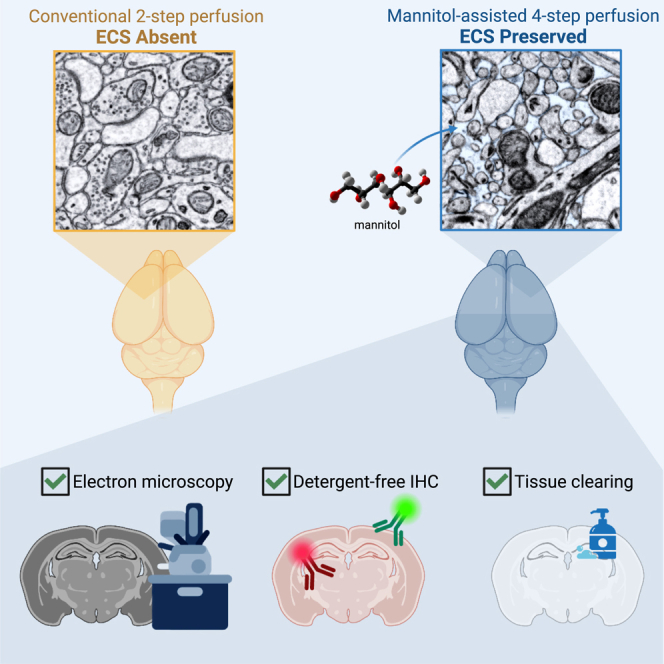
Highlights
-
•
A four-step perfusion method preserves extracellular space fixed brains
-
•
The method offers excellent scalability and reproducibility with low equipment outlay
-
•
Enhanced heavy-metal EM staining for large-scale connectomics
-
•
Improved detergent-free CLEM immunolabeling and tissue clearing
Motivation
The preservation of extracellular space (ECS) in fixed brain tissue provides a representation of structures close to its natural state, benefiting brain study in many ways. Particularly in connectomics, ECS improves the accuracy of automated segmentation and the identification of synapses. Despite the advantages, existing techniques to preserve ECS often require complex equipment or are restricted to small tissue volumes. This work presents a straightforward transcardial perfusion method to reliably preserve ECS throughout an entire rodent brain.
Lu et al. present a simple four-step perfusion fixation method that preserves the extracellular space in entire rodent brains. They demonstrate enhanced heavy-metal EM staining for connectomics, improved detergent-free immunolabeling for CLEM, and accelerated tissue clearing. This approach enables increased data quality in correlated molecular and electron microscopy brain studies.
Introduction
The brain’s dense collection of interconnected neurons underlies the animal’s behavioral repertoire. Mapping this network requires high-resolution imaging and excellent tissue quality. The critical first step in making such a map is tissue fixation, the primary goal of which is to preserve tissue structure close to its appearance in vivo. Unfortunately, this aspiration is rarely achieved because of the fixation-associated artifacts. One of these artifacts is the loss of the extracellular space (ECS) surrounding cell processes. In mammalian brains, there is about 20% ECS, which drops to less than 5% after chemical fixation with aldehydes.1 Preserving ECS in fixed brain tissues has many advantages including making the identification of neuron-to-neuron connections via synapses much easier in electron microscopy (EM),2 which is crucial for extracting connectomes from image volumes. In this report, we also show that it enables rapid diffusion of molecules into and out of the fixed brains, making post-fixation staining more efficient.
Loss of ECS is a result of the rapid influx of water into the intracellular compartment of neurons and astrocytes in response to the redistribution of ions.3 Circulatory arrest in a euthanized animal induces brain anoxia, which deactivates the Na-K pump owing to its high energy requirement. The deactivation of the pump leads to unchecked sodium ion (Na+) influx and potassium ion (K+) efflux down their electrochemical gradients.4 The entry of Na+ causes the depolarization of neurons and triggers the influx of chloride ions (Cl−).5 The accumulation of sodium chloride (NaCl) increases the intracellular osmolarity, drawing water into the neuron and causing it to swell. Additionally, the rapid accumulation of K+ in the extracellular fluid stimulates the uptake of K+ by astrocytes.6 The consequent osmotic overload promotes water inflow and swelling of the astrocytes. Tissue swelling occurs approximately 8 min after oxygen deprivation or 4 min after the Na-K pump deactivation.7 Even if significant cell swelling has not been achieved at this point, the fixative can disrupt the pumps8 and inhibit the inactivation of ion channels,9 leading to an acceleration of the swelling process. Presumably, if the fixative crosslinks cytosolic proteins sufficiently fast, ECS may be preserved to some extent. A recent fluorescence recovery after photobleaching (FRAP) experiment on cultured cells has shown that it takes paraformaldehyde more than 20 min and glutaraldehyde 4 min to fix cytosolic proteins even at 37°C.10 Therefore, it is challenging to preserve ECS using conventional fixation methods.
Many methods have been developed to preserve ECS in fixed brain tissues. High-pressure freezing (HPF) rapidly freezes the tissue before the anoxic cell responses begin. Tissue-damaging ice crystals are avoided by rapidly chilling samples to liquid nitrogen temperature under very high pressure. Chemical fixatives are subsequently introduced by freeze substitution. HPF preserves approximately 15% of the ECS in brain tissues1 but only in tissue samples less than 200 μm thick.11 In immersion-based fixation, a hyperosmotic buffer is often used to increase the osmolarity of extracellular fluid and counteract the influx of water.2 However, this method is limited by the diffusion rate of the buffer salts and can only be used to obtain ECS in small brain volumes, often with a decreased ECS ratio over the thickness. Preserving ECS in a whole mammalian brain via transcardial perfusion is especially challenging. Substances that are added to increase the extracellular osmolarity, such as sucrose, do not cross the blood-brain barrier (BBB).12 One way to deliver those molecules to the brain parenchyma is to disrupt the BBB by pressure.13 This method has not been widely adopted owing to the requirement of a complicated apparatus to pressurize the perfusate and inhomogeneous pressure distribution, as reflected in the inhomogeneous change of vessel shapes and diameters in the proximal and distal vasculature.14
Here, we report a transcardial perfusion strategy that preserves ECS throughout the brain. We believe that the key to preserving ECS in a large brain volume is to dissociate cell swelling and cell fixation. To achieve this, we have developed a four-step method that completely separates these two processes in which the fixative is introduced after the extracellular fluid has been replaced with a hyperosmotic milieu. This four-step perfusion protocol requires only a lab peristaltic pump and shows excellent reproducibility. We subsequently demonstrate that the preservation of ECS accelerates the heavy-metal staining of the brain tissues and improves the reproducibility of permeabilization-free immunolabeling for correlated light and EM (CLEM) to reveal the molecular identity of the cells or the location of specific molecules on EM images. Preserving ECS also enables faster and more uniform delipidation for tissue clearing.
Results
A four-step transcardial perfusion method to preserve ECS
Mannitol is a cell-impermeant carbohydrate. It has different medical applications when applied at different concentrations. At a low concentration, mannitol maintains a high extracellular osmolarity to counter the influx of water and prevent cells from swelling. As mannitol is unable to cross the BBB, mannitol is used in clinical practice to reduce elevated intracranial pressure in hydrocephalus by establishing an osmotic gradient across brain blood vessels and drawing water from the brain parenchyma to the vasculature.15 High concentrations of mannitol, which is utilized in clinical studies to improve the delivery of therapeutic agents across the BBB,16 could induce transient permeabilization of the BBB.17
By fitting different functions of mannitol to the appropriate steps, we developed a four-step strategy to preserve ECS throughout the brain (Figure 1). In the first step, fresh carbongenated artificial cerebrospinal fluid (aCSF) was transcardially perfused to remove blood from the vasculature. In the second step, a high concentration of mannitol (15 w/v%) dissolved in aCSF was perfused to induce the hyperosmotic opening of the BBB. After the BBB is open, mannitol itself can be delivered to the brain parenchyma to maintain extracellular osmolarity. Therefore, in the third step, a lower concentration of mannitol (4.5 w/v%) was applied to reestablish the ECS. Finally, in the fourth step, the fixative was perfused with 4 w/v% mannitol to fix the brain while preserving the ECS.
Figure 1.

A four-step transcardial perfusion strategy for preserving ECS in whole brains
(A) A butterfly needle connected to a peristaltic pump is inserted into the left ventricle of the heart of a deeply anesthetized animal. The solutions listed are then perfused in order. For the fixation step, the solution is different given the purpose.
(B) In the mouse brain fixed with conventional two-step perfusion, little ECS is observed.
(C) In the mouse brain fixed with the four-step perfusion protocol, the ECS is well preserved.
(D–I) EM images demonstrated the distribution of the ECS throughout a mouse brain. The ECS was painted in blue in the EM images of the (D and E) olfactory bulb, (F) motor cortex, (G) somatosensory cortex, (H) visual cortex, and (I) thalamus. The red dots in the inset schematic coronal sections indicate the locations where the images were taken. Scale bars: 1 μm.
The four-step approach is essential for preserving ECS in the brain. When a mannitol-containing fixative was perfused directly after blood clearing (i.e., a two-step perfusion), only a modest amount of ECS was preserved (Figure S1A). We also found that the third step (4.5 w/v% mannitol in aCSF) is critical. If this step was omitted (i.e., going directly from 15 w/v% mannitol in aCSF to 4 w/v% mannitol in fixative), no ECS was preserved, and the fixed brain was shrunk (Figure S1B) owing to a significant loss of ECS not related to cell swelling. This is because high concentrations of mannitol not only open the BBB but also pull water from the brain parenchyma. Decreasing the duration of the 15 w/v% mannitol or lowering the concentration of mannitol did help reduce brain shrinkage but also led to the insufficient opening of the BBB, which affected the effective delivery of mannitol in the following steps and thus decreased the amount of ECS preserved in the tissue. Therefore, we added an intermediate (third) step with only a low concentration of mannitol in aCSF. In this step, mannitol accompanying water enters the brain and reestablishes the interstitial spaces between cells, ensuring that the extracellular fluid has been completely replaced with the hyperosmolar solution to prevent cell swelling.
To identify the optimal mannitol concentration for the third step, we placed 500-μm-thick acute mouse cortex slices in aCSF containing various concentrations of mannitol for 5 min at room temperature and then transferred the tissues to cold mannitol-added fixatives. We found that 4 and 5 w/v% mannitol preserved 17.2% ± 2.4% (Figure S1C) and 18.5% ± 1.8% (Figure S1D) ESC, respectively, in the center of the tissues. 7 w/v% mannitol generated an exaggerated ECS of 26.7% ± 5.9% (Figure S1E), resulting in a more drastic shrinkage of neural processes. Therefore, we used 4.5 w/v% mannitol in the third step to establish ECS.
We observed ECS throughout the entire mouse brain fixed by this method, with an average ECS fraction of 16.6% ± 4.2% measured in three different mouse brain samples (Figure S1F). This value is close to the 21% measured in live rodent brains18 and 15.4% ± 5.4% in rapidly cryo-fixed brain samples1 and is much higher than the 2% ECS in conventionally fixed brains. As shown in Figures 1D–1I, this method produced a uniform distribution of ECS in the motor cortex, somatosensory cortex, and visual cortex, indicating the effectiveness of this method in preserving ECS from the anterior to the posterior of a mouse brain. We also observed variations in the distribution of ECS within individual subregions of brain areas. For example, we observed the largest ECS variation in the olfactory bulb, with only 10% in the olfactory nerve layer (Figure 1D) and 29% in the external plexiform layer (Figure 1E). We did not observe any obvious fixation artifacts (such as vacuolization, swollen mitochondria, or wrinkled cell membranes) in our samples. We have successfully performed the method in 56 mice for EM sample preparation and 23 mice for CLEM imaging (more EM images are provided in Figures S1G–S1I). The results across these experiments have shown a high degree of consistency, supporting the robustness of the method. To test the scalability of this method, we applied it to Long-Evans rats after adjusting the perfusion rate to match the cardiac output of this larger mammal. We were able to preserve a similar amount of ECS in the rat brains (Figure S2).
Preserving ECS improves heavy-metal EM staining in large volumes
Staining large brain tissues for EM imaging is challenging and time consuming. EM imaging relies on heavy-metal staining, which makes cell membranes and subcellular structures visible.19,20 Osmium tetroxide (OsO4) is the most widely used heavy-metal stain for biological tissues. It strongly reacts with unsaturated carbon–carbon bonds in lipids. This membrane-selective staining is vital for defining cell boundaries and identifying synapses. However, the membrane selectivity also makes brain tissue difficult to stain because such tissue is especially membranous, having an extremely high density of fine processes in neurons and glia. This high density of membranes means that OsO4 is depleted fast as it diffuses into a brain volume. In conventionally fixed samples where ECS is reduced, OsO4 must pass through tightly packed layers of membranes to reach the innermost part. Consequently, the staining slows dramatically for large brain tissues, and staining gradients are commonly observed.
Preserving ECS improves the efficiency of staining by increasing the diffusion efficiency of OsO4. Time-lapse X-ray micro-computed tomography (micro-CT)21 of 3-mm-thick mouse brain slabs during the application of cacodylate-buffered OsO4 showed that, in ECS-preserved mouse brain slabs, it took 9 h for osmium to penetrate the entire depth (Figure 2A). Staining time was extended by 22% to 11 h in a conventionally fixed brain slab (Figure 2B). It is possible that ECS provides an interconnected network of channels throughout the tissue so that OsO4 can be more efficiently transported to unstained regions. This fixation method provides a good starting point for developing multiple rounds of heavy-metal staining for the whole-brain connectome.
Figure 2.

Improved heavy-metal staining with ECS preservation
(A and B) Time-lapse X-ray micro-CT showed that the penetration of buffered OsO4 was faster in ECS-preserved brain samples compared with those without ECS. The brain sample turned dark with the osmium staining front moving from the surface to the center. In the ECS-preserved 3 mm brain slab (A), osmium staining took 9 h to complete, while it took 11 h in the specimen without ECS (B).
(C) Migration of buffered OsO4 in 3 mm slabs with ECS (red curve, n = 5) and without ECS (blue curve, n = 5) was measured as the shortest distances between the surface and osmication fronts from X-ray micro-CT.
(D–F) Preservation of ECS also increased the staining depth of ferrocyanide-reduced osmium in brain tissues. 1 mm biopsies were punched from the fixed brains with different fixations. Modified ROTO, the process of which is illustrated in (D), produced uniform and high-quality EM staining throughout the ECS-preserved samples (E). In conventionally fixed samples with no ECS preservation, ROTO produced an evident staining gradient, with good staining only obtained in the periphery (F-i), overstaining in the intermediate region (F-ii), and under-staining in the center (F-iii). Black scale bars in X-ray micro-CT and low-resolution EM images: 1 mm. White scale bars in high-resolution EM images: 1 μm.
Preserving ECS not only speeds staining but also improves its quality. The reduced osmium-thiocarbohydrazide-osmium (ROTO) method produces high-contrast membrane staining and has been widely used to stain small brain tissues.22 The reduced osmium (i.e., Os(VI)), made by mixing OsO4 and potassium ferrocyanide (K₄[Fe(CN)₆]), can dismutate into Os(VIII) and Os(IV). Os(IV) in the form of OsO2 has low solubility. Lipids in the tissue deplete Os(VIII) and drive the reaction to generate high concentrations of OsO2. In large tissues, this causes OsO2 precipitation, which acts as a barrier for further entry of reduced osmium into the deeper parts of the tissue.23 Therefore, ROTO can only be applied to stain small samples, thinner than ∼300 μm. Preserving ECS increased ROTO penetration to 1 mm in thickness (Figures 2D–2F). In contrast to the substantial staining gradient caused by the precipitation of OsO2 in the ECS-absent samples, there was almost no gradient in the ECS-preserved samples. High membrane contrast was achieved from the periphery to the center. We have also investigated the penetration depth of ferrocyanide-reduced osmium by applying the mixture solution to an ECS-preserved mouse brain (Figure S3). The results demonstrated that the maximum penetration depth of reduced osmium was ∼800 μm, suggesting that ROTO could be applied to dissected ECS-preserved tissues up to 1.6 mm in thickness. However, for whole-brain staining, a new method would be required.
Preserving ECS also improves detergent-free immunolabeling for CLEM
One way to provide cellular context for fluorescent molecular labeling is CLEM. CLEM is a valuable tool for studying the brain, where it is important to understand both the molecular diversity of cells and their synaptic connectivity to understand function. A challenge, however, is that intracellular immunostaining with antibodies typically requires permeabilization of cell membranes with detergents, which inevitably degrades tissue integrity and makes subsequent EM segmentation unfeasible. To overcome this obstacle, we proved that permeabilization-free immunostaining could be achieved using nanobodies (∼15 kDa).24 In addition, Fulton and Briggman reported that detergent-free immunostaining with immunoglobulin G (IgG) antibodies (∼150 kDa) was possible when the ECS was preserved by drop fixation and antibodies were applied at high concentrations.25
Permeabilization-free immunohistochemistry can be further improved with the transcardial perfusion fixation approach presented here. We first confirmed that it was possible to perform permeabilization-free immunostaining on our ECS-preserved brain tissues using the anti-NeuN antibody (Figure 3A). To investigate the penetration depth of antibodies during a 5 day permeabilization-free immunostaining, we applied fluorescent anti-GFP antibodies to 400-μm-thick Thy1-YFP-H mouse brain tissues. Our results showed anti-GFP antibodies labeled ∼60 μm of the ECS-preserved tissue (Figure 3B) while remaining in the superficial layer, less than 10 μm deep, in ECS-absent tissues (Figure 3C). Using perfusion-fixed tissue with preserved ECS has several advantages compared with drop-fixed acute brain slices. Drop fixation can lead to inconsistent preservation of the ECS due to variations in the time span between circulatory arrest and the tissue being dropped into fixative. In contrast, we confirmed that four different experimenters obtained identical ECS preservation using our protocol and consistent immunohistochemical results across different preparations. Moreover, vibratoming of transcardially perfused samples helps to prevent artifacts (e.g., empty vacuoles) that may occur when sectioning unfixed tissue (Figure S4).
Figure 3.

Improved permeabilization-free immunostaining with the preservation of ECS
(A) Detergent-free immunolabeling of NeuN in mouse cerebral cortex using antibodies.
(B and C) YFP-H mouse brain tissues were stained with fluorescent anti-GFP antibodies to compare antibody penetration. Images were acquired at cross-sections to demonstrate the antibody penetration depth after 5 days of permeabilization-free immunostaining at 4°C. The results revealed that anti-GFP antibodies penetrated approximately 60 μm in ECS-preserved brain tissues (B) and less than 10 μm in ECS-absent tissues (C).
(D) PV+ interneurons in the cortex were labeled with fluorescent scFv. The light blue arrows show the PV+ interneuronal synaptic boutons (red) surrounding large pyramidal cell somata (unlabeled).
(E) Labeling of PV+ neurons with antibodies (red) and perineuronal nets (PNNs) with biotinylated wisteria floribunda agglutinin (bWFA; yellow) in the cortex. X-Z and Y-Z reslices show labeling was throughout the imaged depth of the ECS-preserved brain tissue.
(F and G) Immunolabeling of PSD-95 (magenta) in the cerebellar cortex is similar with permeabilization (F) and without permeabilization (G).
(H) Double labeling of SST+ (green) and CR+ (red) interneurons in ECS-preserved mouse cortex. EM images were taken from the yellow boxed region.
(I) The fluorescent labels were superimposed on the corresponding EM image taken from the yellow boxed region. The inset image demonstrated that the tissue ultrastructure was well preserved. The immunostaining work was carried out on 120 μm mouse brain sections except for the antibody penetration depth tests, which used 400 μm sections.
With improved ECS preservation and tissue quality, we were able to achieve permeabilization-free labeling of a variety of molecules including intracellular proteins (e.g., parvalbumin, somatostatin, calretinin stained with antibodies; Figures 3D, 3E, and 3H) and cell surface markers (e.g., perineuronal nets stained with lectin; Figure 3E). Additionally, we were able to label synaptic boutons in axon terminals of parvalbumin-positive (PV+) interneurons using single-chain variable fragment (scFv) immuno-probes (Figure 3D) and the postsynaptic density protein 95 (PSD-95) using antibodies (Figures 3F and 3G) that had previously been unstainable without detergent-based permeabilization. To demonstrate the feasibility of detergent-free CLEM, we labeled somatostatin-positive (SST+) and calbindin-positive (CR+) interneurons in the cerebral cortex of an ECS-preserved mouse brain with primary and secondary antibodies. The high-resolution EM image showed that the tissue ultrastructure was well preserved, and the immuno-labels could be easily superimposed onto the corresponding EM ultrathin sections (Figures 3H and 3I).
Efficient tissue clearing
Tissue clearing by matching the refractive index (RI) of all tissue components has widespread use in biological research.26,27 It allows for the direct imaging of large tissue volumes in their intact state without the need for time-consuming physical sectioning. To determine whether preserved ECS could facilitate clearing through more efficient and effective diffusion of lipids out of a tissue volume that allows for improved RI matching of a whole mouse brain, we performed the four-step perfusion protocol on Thy1-YFP-16 mice (n = 3). We added hydrogel precursors acrylamide (AA) and bis-acrylamide (Bis) to the PFA fixative in the fourth step of the transcardial perfusion. After post-fixation overnight, the brains were polymerized under vacuum to form polyacrylamide meshes. We then applied an electric field for 24 h to remove lipids (rather than 48 h as previously suggested28). After RI matching, the brain with preserved ECS was uniformly transparent, while the brain without ECS had a translucent core. Light-sheet microscopy verified this difference: the ECS-preserved brain allowed for imaging of the entire volume at cellular resolution, while the under-cleared core of the ECS-absent brain scattered too much light, and only the structures on the periphery could be resolved (Figure 4).
Figure 4.

Efficient tissue clearing with the preservation of ECS
Light-sheet optical sections of the hemispheres showed that (left) the brain preserved with ECS was effectively cleared and YFP could be well resolved in deep structures. (Right) The conventionally fixed brain without ECS preservation was poorly cleared after 24 h of tissue clearing. The center of the hemisphere scattered light causing a blur.
Discussion
The goal of this work was to preserve ECS in large-volume brain tissues, particularly in an entire mammalian brain. The success of the transcardial perfusion approach described here relied on dissociating cell swelling from the fixation process. After removing blood, we used varying amounts of mannitol in three different steps to achieve this goal. First, a high concentration of mannitol was used to open the BBB, which had two effects: an immediate movement of water out of the brain’s ECS into the now hypertonic vasculature, as well as allowing mannitol itself to enter the brain parenchyma. Second, a low concentration of mannitol was perfused to restore the brain’s ECS by moving water back from the vasculature and preventing cell swelling secondary to anoxia. Finally, aldehyde-based fixatives containing mannitol were perfused to preserve the brain’s ultrastructure. This multistep method resulted in ECS being preserved throughout the entire brain volume.
Preserving the ECS in brain tissues has been shown to be useful for identifying fine structures in EM images, such as synapses2 and cilia.29 In this study, we demonstrated that preserving the ECS improves heavy-metal staining of large brain volumes for EM. This finding could serve as the foundation for the development of whole-brain staining techniques or the improvement of existing methods.30,31,32 We also showed that preserving ECS with perfusion fixation improves permeabilization-free immunostaining, especially of fine structures such as molecules in axon terminals and postsynaptic sites. The reproducibility of immunostaining based on the results of multiple users and the better quality of the heavy-metal-stained ultrastructure make CLEM more robust. Additionally, we demonstrated that preserving ECS in the brain enabled more efficient lipid removal for this heavily membranous organ. We developed the protocol using mice and demonstrated the scalability of the approach in rats. In principle, scaling up the protocol to even larger mammals should be possible by adjusting the flow rate and time span of each step. For animals where transcardial perfusion is difficult to perform, direct carotid artery perfusion might be feasible. While those remain to be investigated, this work indicates the feasibility of using a four-step perfusion strategy for preserving ECS in a large brain volume and opens the path for many correlated molecular and EM studies.
Limitations of the study
We observed regional variation in the ECS distribution within the fixed mouse brain (Figure S1F). The local ECS ratio might not be consistent with observations in vivo or in acute slices. For researchers studying specific brain regions where less than expected ECS preservation was observed using the presented method, such as the molecular layer of the cerebellar cortex, they might consider adjusting the mannitol concentration in steps 3 and 4 to increase ECS. Moreover, this method was developed using young adult mice. For neonatal mice, we found that direct perfusion of the animal with cold fixative could preserve ECS, making the four-step perfusion process unnecessary. However, we have not tested this method on aged mice, so further investigation is required.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| NeuN | Abcam | Cat# ab190195; RRID:AB_2716282 |
| PV | Abcam | Cat# ab11427; RRID:AB_298032 |
| SST | Millipore Sigma | Cat# MAB354; RRID:AB_2255365 |
| CR | Sigma-Aldrich | Cat# C7479, RRID:AB_259024 |
| PSD-95 | Antibodies Incorporated | Cat# 75-028; RRID:AB_2292909 |
| GFP | Thermo Fisher Scientific | Cat# A-31852; RRID:AB_162553 |
| Anti-rat Fab2 A488 | Jackson ImmunoResearch | Cat# 712-546-150; RRID:AB_2340685 |
| Anti-rabbit Fab2 A594 | Jackson ImmunoResearch | Cat# 711-586-152; RRID:AB_2340622 |
| Anti-mouse IgG2a A488 | Thermo Fisher Scientific | Cat# A-21131; RRID:AB_2535771 |
| Chemicals, peptides, and recombinant proteins | ||
| NaCl | Sigma-Aldrich | Cat# S3014 |
| NaHCO3 | Sigma-Aldrich | Cat# S6014 |
| NaH2PO4 | Sigma-Aldrich | Cat# S3139 |
| KCl | Sigma-Aldrich | Cat# P3911 |
| Glucose | Sigma-Aldrich | Cat# G7528 |
| MgCl2 | Sigma-Aldrich | Cat# 63069 |
| CaCl2 | Sigma-Aldrich | Cat# 21115 |
| mannitol | Sigma-Aldrich | Cat# M4125 |
| MgSO4 | Sigma-Aldrich | Cat# 83266 |
| PBS | Sigma-Aldrich | Cat# P3813 |
| acrylamide | Sigma-Aldrich | Cat# A9099 |
| bisacrylamide | Sigma-Aldrich | Cat# M7279 |
| potassium ferrocyanide | Sigma-Aldrich | Cat# 60279 |
| NaN3 | Sigma-Aldrich | Cat# 71289 |
| Tween 20 | Sigma-Aldrich | Cat# P9416 |
| sodium cacodylate | Electron Microscopy Sciences | Cat# 12310 |
| 32% paraformaldehyde | Electron Microscopy Sciences | Cat# 15714 |
| 25% glutaraldehyde | Electron Microscopy Sciences | Cat# 16220 |
| OsO4 | Electron Microscopy Sciences | Cat# 19191 |
| thiocarbohydrazide | Electron Microscopy Sciences | Cat# 21900 |
| uranyl acetate | Electron Microscopy Sciences | Cat# 22400 |
| EMbed 812 (EPON) | Electron Microscopy Sciences | Cat# 14121 |
| acetonitrile | Electron Microscopy Sciences | Cat# 10021 |
| Hoechst 33342 solution | Thermo Fisher Scientific | Cat# 62249 |
| Streptavidin A488 | Thermo Fisher Scientific | Cat# S11223 |
| biotinylated WFA | Adipogen | Cat# VC-B-1355-M002 |
| normal goat serum | Gibco | Cat# CN5000 |
| Glycine | Invitrogen | Cat# 15527013 |
| 1N HCl | Fisher Scientific | Cat# SA48-500 |
| EasyIndex solution (RI = 1.52) | LifeCanvas | N/A |
| VA044 Thermal Polymerization Initiator | Wako Chemicals | N/A |
| Experimental models: Organisms/strains | ||
| C57BL/6J | Jackson Laboratory | strain# 000664 |
| Thy1-YFP-16 | Jackson Laboratory | strain# 003709 |
| Thy1-YFP-H | Jackson Laboratory | strain # 003782 |
| Long-Evans | Charles River Laboratories | strain# 006 |
| Software and algorithms | ||
| VAST | Berger et al.33 | https://lichtman.rc.fas.harvard.edu/vast/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Xiaotang Lu (xiaotang_lu@fas.harvard.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
C57BL/6 (strain#000664), Thy1-YFP-H (strain#003782), and Thy1-YFP-16 (strain#003709) mice were purchased from the Jackson Laboratory. Long-Evans rats were purchased from the Charles River Laboratories (stain#006). Male and Female mice were used at 16–30 weeks old. Female rats were used at 12–25 weeks, weight range 240g - 300g. All animals were housed in a 12-hour reverse dark-light cycle at 24°C and variable humidity and received water and food ad libitum. Both male and female mice were used for all experiments. Animals were chosen based on availability and age. Animal experiments were approved by the Committee on Animal Care at Harvard University.
Method details
Solutions
The aCSF solution was composed of 125 mM NaCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 2.5 mM KCl, 26 mM glucose, 1 mM MgCl2, and 2 mM CaCl2. MgCl2 and CaCl2 were added after the solution was bubbled with carbogen gas for 20 min. The aCSF solution was made fresh before each use and was used at room temperature. The mannitol-added aCSF was prepared by mixing mannitol at a certain weight percentage with the fresh aCSF. For example, to make 10 mL of 15 w/v% mannitol aCSF, 1.5 g of mannitol was added to a conical tube, the fresh aCSF was added to a volume of 10 mL, and the mixture was stirred using an ultrasonic bath until no solid particles were visible.
The stock cacodylate buffer (2x) was composed of 300 mM sodium cacodylate, 8 mM MgSO4, 4 mM CaCl2, and was titrated with 1N HCl to a pH of 7.4. The EM fixatives were made by mixing 4 w/v% mannitol, 2 w/v% PFA, 2.5 w/v% glutaraldehyde, and 150 mM cacodylate solution. The fixative for permeabilization-free immunostaining was made by mixing 4% w/v mannitol, 4% w/v PFA, and 1x PBS. The hydrogel fixative for tissue clearing was made by mixing 4 w/v% mannitol, 3 w/v% PFA, 3% w/v acrylamide, 0.02 w/v% bisacrylamide, 2.5 mg/mL VA044 Thermal Polymerization Initiator, and 1x PBS. The fixatives were chilled on ice before use.
Transcardial perfusion to preserve ECS in the mouse brain
The mouse was anesthetized with an overdose of isoflurane and placed on the dissection tray. The chest was then opened, and a 21-gauge butterfly needle was inserted through the left ventricle, and a small cut was made on the right atrium. The mouse was perfused transcardially at a flow rate of 10 mL/min using a Masterflex Peristaltic pump with fresh aCSF for 2-3 minutes to remove the blood. The liver should change color from dark red to pale brown after this step. Pause the pump briefly and transfer the rubber tubing from the Falcon tube containing aCSF to the tube containing mannitol-added aCSF. Be careful not to introduce any air bubbles into the tubing during the operation. Then, turn the pump back on and perfuse a 15 w/v% mannitol aCSF solution for 1 minute to increase the BBB permeability, followed by perfusing a 4.5 w/v% mannitol aCSF solution for 5 minutes. Finally, perfuse an ice-cold fixative for 5 minutes. For different applications, the different fixative described above was used.
After perfusion, the brain was carefully removed from the skull and transferred to a vial containing the same fixative solution for further fixing for at least 24 hours with gentle agitation at 4°C. The fixed brain can be used as is or, if thin slices are desired, they can be cut using a Leica VT1000 S vibrating blade microtome in the cold fixative solution.
Transcardial perfusion for rats
The procedure for perfusing the rat was similar to the mouse perfusion, except that a 16-gauge blunt needle was used, and the flow rate was adjusted to 30 mL/min. After opening the chest, the needle was inserted through the left ventricle into the ascending aorta with the tip of the needle visible in the beginning section of the ascending aorta. The needle was then clamped with a hemostat and aCSF was perfused for 3-5 minutes to remove the blood. This was followed by perfusion of a 15 w/v% mannitol aCSF solution for 1 minute, a 4 w/v% mannitol aCSF solution for 5 minutes, and cold fixative for 5 minutes.
Modified reduced osmium-thiocarbohydrazide-osmium (ROTO) staining for EM imaging
A modified ROTO protocol was used to stain the brain samples for EM imaging. All procedures were conducted at room temperature unless specified otherwise. For 1 mm-thick brain slices or 1 mm biopsy punches, the samples were rinsed 3 x 20 min in a 150 mM sodium cacodylate buffer (pH 7.4) and stained in a solution containing 2 w/v% OsO4, 1.5% w/v potassium ferrocyanide and 0.15M cacodylate buffer for 3 hours on a rotator. After washing 3 x 20 min in ddH2O, the samples were stained in filtered 1 w/v% thiocarbohydrazide aqueous solution for 1.5 hours. The samples were then washed 3 x 20 min in ddH2O and stained again with 2 w/v% OsO4 aqueous solution for 3 hours. The samples were then washed 3 x 20 min and transferred to 1 w/v% uranyl acetate aqueous solution, wrapped in foil to protect from light, for overnight staining (∼12 hours). On the next day, the samples were washed 3 x 20 min in water.
The stained sample was dehydrated through a graded (25, 50, 75, 100, 100%, 20 min each) acetonitrile series. After dehydration, samples were infiltrated at room temperature with 25% EPON:acetonitrile for 6 hours, 50% EPON:acetonitrile for 12 hours, 75% EPON:acetonitrile for 12 hours, 100% EPON for 12 hours, fresh 100% EPON for another 12 hours on a rotator. The fully infiltrated samples were then placed in a flat embedding mold and incubated in a 60°C oven for two days.
X-Ray MicroCT imaging
The microCT of the mouse brain was acquired using a Zeiss Xradia 520 Versa X-ray microscope. For time-lapse imaging, the brain was wrapped in a biopsy nylon mesh bag and placed in a 20 mL scintillation vial. The vial was then filled with a 2 w/v% OsO4 solution buffered in 150 mM cacodylate and tightly sealed. The vial was then transferred to a rotator or nutating mixer for staining. At different reaction time points, the vial was removed from the mixer, attached to the microCT sample holder using double-sided tape, and moved to the X-ray imaging chamber for imaging. The sample was imaged using a 0.4× objective lens at a tube voltage of 60 kV and output power of 5 W, with an exposure time of 2 seconds for each projection. Each tomographic scan involved rotating the sample 360° and acquiring 401 projection images, which took approximately 30 minutes. After the scan, the vial was returned to the mixer to continue staining. For resin-embedded brain samples, the block was directly attached to the microCT sample holder for imaging.
EM imaging
The sample block was cut into 30 nm ultrathin sections using a Leica ultramicrotome and collected on a home-built automated tape collecting system on carbon-coated Kapton tape.34 The tape was then cut into appropriate lengths and mounted on a 4-inch silicon wafer. The sections were imaged with a Zeiss Sigma SEM using a backscattered electron detector at a working distance of approximately 7.3 mm, with an incident electron energy of 8 keV and a dwell time of 2 μs.
Detergent-free immunostaining
Information on the primary and secondary antibodies and bWFA used in this study, along with dilution ratios, was provided in Table S1. After perfusion fixation, the 120 μm sections were cut and collected in cold PBS using Leica VT1000 S vibrating blade microtome. The region of interest was dissected from the brain slice and placed into a 3.7mL glass vial with a pair of paintbrushes. The sample was rinsed in cold PBS for 3 x 10 min and blocked in the solution containing 10% normal goat serum, 100 mM glycine, 0.05% NaN3, and PBS for 3 hours. During waiting, the staining solution was made by mixing the antibody stock with the dilution solution containing 3% normal goat serum, 100 mM glycine, 0.05% NaN3, and PBS. After blocking was done, add the primary antibody solution to the vial and incubate for 3-5 days. After this, the sample was washed with PBS for 3 x 10 min and then left in PBS to rinse overnight. On the next day, the secondary antibody solution was added to the sample and incubated for 3 days. After staining, the sample was washed with PBS for 3 x 10 min and then stained with Hoechst (diluted 1:5000 in PBS) for 1 hour at room temperature or overnight at 4°C. All the steps, unless otherwise stated, were carried out on a rotator in a refrigerator (4°C).
For CLEM, after fluorescent imaging, the tissue was post-fixed in a fixative containing 4 w/v% mannitol, 2 w/v% PFA, 2.5 w/v% glutaraldehyde, and 150 mM cacodylate solution for at least one day at 4°C. It was then processed for EM imaging as described above.
Whole brain clearing
After perfusion fixation, the Thy1-YFP-16 mouse brain was post-fixed at 4°C for one day. The fixed brain was then placed into a 50 mL conical tube filled with fresh hydrogel fixative and transferred to the X-CLARITY polymerization device (Logos Biosystems) to polymerize for three hours. The vacuum was set at -90 kPa, temperature of 37°C. After polymerization, the brain was transferred immediately to a new conical tube filled with cold PBS to prevent further cross-linking. The brain was kept in PBS at 4°C for overnight rinsing. The next day, the brain was warmed up to room temperature and then transferred to the X-CLARITY ETC chamber for electrophoretic lipid extraction. The reservoir was filled with the electrophoretic tissue-clearing solution, and the system was run at 1A and 37°C for 24 hours. After delipidation, the brain was placed in a conical tube filled with 50 mL PBST (PBS with 0.1% Tween 20) and wrapped in foil to protect it from light. The tube was placed on a nutating mixer for overnight agitation. The brain was then transferred to the EasyIndex solution (RI = 1.52, LifeCanvas) to match the refractive index. The cleared brain was imaged with a Zeiss Lightsheet microscope.
Acknowledgments
We thank Wenjie Yin and Jinglin Zhao for ECS tracing and proofreading. X.L. is supported by the NIH BRAIN Initiative award K99MH128891. This research was also supported by NIH grant U19NS104653.
Author contributions
X.L. developed the method. X.H. and E.S. helped with detergent-free immunostaining and data collection. Y.M. conducted ECS auto-segmentation. R.L.S. helped with EM imaging. X.L. and J.W.L. wrote the paper.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: July 5, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2023.100520.
Contributor Information
Xiaotang Lu, Email: xiaotang_lu@fas.harvard.edu.
Jeff W. Lichtman, Email: jeff@mcb.harvard.edu.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Korogod N., Petersen C.C.H., Knott G.W. Ultrastructural analysis of adult mouse neocortex comparing aldehyde perfusion with cryo fixation. Elife. 2015;4:e05793. doi: 10.7554/eLife.05793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pallotto M., Watkins P.V., Fubara B., Singer J.H., Briggman K.L. Extracellular space preservation aids the connectomic analysis of neural circuits. Elife. 2015;4:e08206. doi: 10.7554/eLife.08206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raichle M.E. The pathophysiology of brain ischemia. Ann. Neurol. 1983;13:2–10. doi: 10.1002/ana.410130103. [DOI] [PubMed] [Google Scholar]
- 4.Liang D., Bhatta S., Gerzanich V., Simard J.M. Cytotoxic edema: mechanisms of pathological cell swelling. Neurosurg. Focus. 2007;22:E2. doi: 10.3171/foc.2007.22.5.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rungta R.L., Choi H.B., Tyson J.R., Malik A., Dissing-Olesen L., Lin P.J.C., Cain S.M., Cullis P.R., Snutch T.P., MacVicar B.A. The cellular mechanisms of neuronal swelling underlying cytotoxic edema. Cell. 2015;161:610–621. doi: 10.1016/j.cell.2015.03.029. [DOI] [PubMed] [Google Scholar]
- 6.Leis J.A., Bekar L.K., Walz W. Potassium homeostasis in the ischemic brain. Glia. 2005;50:407–416. doi: 10.1002/glia.20145. [DOI] [PubMed] [Google Scholar]
- 7.Joshi I., Andrew R.D. Imaging anoxic depolarization during ischemia-like conditions in the mouse hemi-brain slice. J. Neurophysiol. 2001;85:414–424. doi: 10.1152/jn.2001.85.1.414. [DOI] [PubMed] [Google Scholar]
- 8.Mărgineanu D.G., Rucăreanu C., Flonta M.L., Finichiu D. Glutaraldehyde inhibits the active transport of sodium and the oxygen consumption, while increasing the water diffusional permeability in frog skin. Arch. Int. Physiol. Biochim. 1984;92:305–312. doi: 10.3109/13813458409071171. [DOI] [PubMed] [Google Scholar]
- 9.Ulbricht W. Sodium channel inactivation: molecular determinants and modulation. Physiol. Rev. 2005;85:1271–1301. doi: 10.1152/physrev.00024.2004. [DOI] [PubMed] [Google Scholar]
- 10.Huebinger J., Spindler J., Holl K.J., Koos B. Quantification of protein mobility and associated reshuffling of cytoplasm during chemical fixation. Sci. Rep. 2018;8:17756. doi: 10.1038/s41598-018-36112-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Studer D., Humbel B.M., Chiquet M. Electron microscopy of high pressure frozen samples: bridging the gap between cellular ultrastructure and atomic resolution. Histochem. Cell Biol. 2008;130:877–889. doi: 10.1007/s00418-008-0500-1. [DOI] [PubMed] [Google Scholar]
- 12.Banks W.A. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol. 2009;9(Suppl 1):S3. doi: 10.1186/1471-2377-9-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cragg B. Preservation of extracellular space during fixation of the brain for electron microscopy. Tissue Cell. 1980;12:63–72. doi: 10.1016/0040-8166(80)90052-x. [DOI] [PubMed] [Google Scholar]
- 14.Schwarzmaier S.M., Knarr M.R.O., Hu S., Ertürk A., Hellal F., Plesnila N. Perfusion pressure determines vascular integrity and histomorphological quality following perfusion fixation of the brain. J. Neurosci. Methods. 2022;372:109493. doi: 10.1016/j.jneumeth.2022.109493. [DOI] [PubMed] [Google Scholar]
- 15.Del Bigio M.R., Di Curzio D.L. Nonsurgical therapy for hydrocephalus: a comprehensive and critical review. Fluids Barriers CNS. 2016;13:3. doi: 10.1186/s12987-016-0025-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rapoport S.I. Osmotic opening of the blood-brain barrier: principles, mechanism, and therapeutic applications. Cell. Mol. Neurobiol. 2000;20:217–230. doi: 10.1023/a:1007049806660. [DOI] [PubMed] [Google Scholar]
- 17.Brightman M.W., Hori M., Rapoport S.I., Reese T.S., Westergaard E. Osmotic opening of tight junctions in cerebral endothelium. J. Comp. Neurol. 1973;152:317–325. doi: 10.1002/cne.901520402. [DOI] [PubMed] [Google Scholar]
- 18.Nicholson C., Hrabětová S. Brain extracellular space: the final frontier of neuroscience. Biophys. J. 2017;113:2133–2142. doi: 10.1016/j.bpj.2017.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palade G.E. A study of fixation for electron microscopy. J. Exp. Med. 1952;95:285–298. doi: 10.1084/jem.95.3.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayat . CRC PressI Llc; 1989. Principles & Techniques of Electron Microscopy. [Google Scholar]
- 21.Ströh S., Hammerschmith E.W., Tank D.W., Seung H.S., Wanner A.A. In situ X-ray-assisted electron microscopy staining for large biological samples. Elife. 2022;11:e72147. doi: 10.7554/eLife.72147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holcomb P.S., Hoffpauir B.K., Hoyson M.C., Jackson D.R., Deerinck T.J., Marrs G.S., Dehoff M., Wu J., Ellisman M.H., Spirou G.A. Synaptic inputs compete during rapid formation of the calyx of Held: a new model system for neural development. J. Neurosci. 2013;33:12954–12969. doi: 10.1523/JNEUROSCI.1087-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hua Y., Laserstein P., Helmstaedter M. Large-volume en-bloc staining for electron microscopy-based connectomics. Nat. Commun. 2015;6:7923. doi: 10.1038/ncomms8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fang T., Lu X., Berger D., Gmeiner C., Cho J., Schalek R., Ploegh H., Lichtman J. Nanobody immunostaining for correlated light and electron microscopy with preservation of ultrastructure. Nat. Methods. 2018;15:1029–1032. doi: 10.1038/s41592-018-0177-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fulton K.A., Briggman K.L. Permeabilization-free immunohistochemistry for correlative microscopy. Elife. 2021;10:e63392. doi: 10.7554/eLife.63392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richardson D.S., Guan W., Matsumoto K., Pan C., Chung K., Ertürk A., Ueda H.R., Lichtman J.W. Tissue clearing. Nat. Rev. Methods Primers. 2021;1:84. doi: 10.1038/s43586-021-00080-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richardson D.S., Lichtman J.W. Clarifying tissue clearing. Cell. 2015;162:246–257. doi: 10.1016/j.cell.2015.06.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung K., Wallace J., Kim S.-Y., Kalyanasundaram S., Andalman A.S., Davidson T.J., Mirzabekov J.J., Zalocusky K.A., Mattis J., Denisin A.K., et al. Structural and molecular interrogation of intact biological systems. Nature. 2013;497:332–337. doi: 10.1038/nature12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheu S.-H., Upadhyayula S., Dupuy V., Pang S., Deng F., Wan J., Walpita D., Pasolli H.A., Houser J., Sanchez-Martinez S., et al. A serotonergic axon-cilium synapse drives nuclear signaling to alter chromatin accessibility. Cell. 2022;185:3390–3407.e18. doi: 10.1016/j.cell.2022.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mikula S., Binding J., Denk W. Staining and embedding the whole mouse brain for electron microscopy. Nat. Methods. 2012;9:1198–1201. doi: 10.1038/nmeth.2213. [DOI] [PubMed] [Google Scholar]
- 31.Mikula S., Denk W. High-resolution whole-brain staining for electron microscopic circuit reconstruction. Nat. Methods. 2015;12:541–546. doi: 10.1038/nmeth.3361. [DOI] [PubMed] [Google Scholar]
- 32.Song K., Feng Z., Helmstaedter M. High-contrast en-bloc staining of mouse whole-brain samples for EM-based connectomics. bioRxiv. 2022 doi: 10.1101/2022.03.30.486341. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berger D.R., Seung H.S., Lichtman J.W. VAST. (Volume Annotation and Segmentation Tool): Efficient Manual and Semi-Automatic Labeling of Large 3D Image Stacks. Front. Neural Circuits. 2018;12:88. doi: 10.3389/fncir.2018.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hayworth K.J., Morgan J.L., Schalek R., Berger D.R., Hildebrand D.G.C., Lichtman J.W. Imaging ATUM ultrathin section libraries with WaferMapper: a multi-scale approach to EM reconstruction of neural circuits. Front. Neural Circuits. 2014;8:68. doi: 10.3389/fncir.2014.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.