

Abstract
The combination of doxorubicin (Adriamycin) and cyclophosphamide, referred to as AC chemotherapy, is commonly used for the clinical treatment of breast and other cancers. Both agents target DNA with cyclophosphamide causing alkylation damage and doxorubicin stabilizing the topoisomerase II–DNA complex. We hypothesize a new mechanism of action whereby both agents work in concert. DNA alkylating agents, such as nitrogen mustards, increase the number of apurinic/apyrimidinic (AP) sites through deglycosylation of labile alkylated bases. Herein, we demonstrate that anthracyclines with aldehyde-reactive primary and secondary amines form covalent Schiff base adducts with AP sites in a 12-mer DNA duplex, calf thymus DNA, and MDA-MB-231 human breast cancer cells treated with nor-nitrogen mustard and the anthracycline mitoxantrone. The anthracycline–AP site conjugates are characterized and quantified by mass spectrometry after NaB(CN)H3 or NaBH4 reduction of the Schiff base. If stable, the anthracycline–AP site conjugates represent bulky adducts that may block DNA replication and contribute to the cytotoxic mechanism of therapies involving combinations of anthracyclines and DNA alkylating agents.
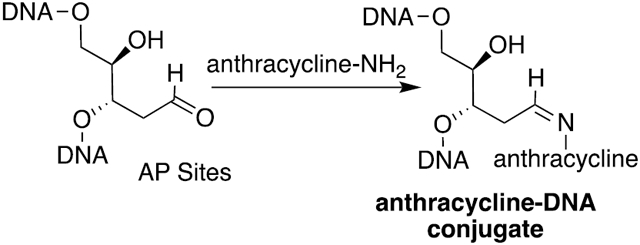
Graphical Abstract
INTRODUCTION
Anthracyclines (Figure 1) are often coadministered with nitrogen mustards (NM) to cancer patients.1 The combination of doxorubicin (Adriamycin) and cyclophosphamide, referred to as AC chemotherapy, is a common treatment for breast and ovarian cancers.1 Both agents target DNA but work by different mechanisms with doxorubicin (DOX) inhibiting topoisomerase,2 while cyclophosphamide alkylates DNA predominantly at the N7-position of 2′-deoxyguanosines (dG),3 resulting in cationic N7-dG adducts including interstrand cross-links at GNC sequences. Nitrogen mustard N7-dG adducts are chemically labile and deglycosylate to apurinic (AP) sites (Scheme 1). We have reported up to a 3.7-fold increase of AP sites in the livers of female mice following treatment with the model nitrogen mustard bis(2-chloroethyl)-ethylamine.4,5 We hypothesized a mechanism of action in which an anthracycline and NM could work in concert, where the anthracycline forms covalent Schiff base conjugates of the AP sites generated by the NM. The proposed mechanism is shown for mitoxantrone (MTX) in Scheme 1. Such bulky DNA adducts are predicted to be strong blocks to DNA replication and induce cytotoxicity.
Figure 1.

Clinically used anthracycline antitumor agents.
Scheme 1. Depurination N7-Nitrogen Mustard dG Adducts Leading to an AP Site and Subsequent Conjugation by MTXa.

aThe MTX conjugate is reduced with NaB(CN)H3 and detected by LC/MS.
Covalent anthracycline–DNA adducts have been previously proposed as an alternative mechanism of action to topoisomerase inhibition.6 One such pathway involved bioreductive activation of the anthracycline to generate an electrophilic quinone methide intermediate, which reacts with nucleophilic sites in DNA (Scheme S1).7 Support for DNA alkylation by anthracycline quinone methides in mammalian systems has not been reported.
A crystal structure of daunorubicin in double-stranded DNA revealed a methylene bridge formed between the N2-position of a dG and the daunosamine amino group (Scheme S1).8,9 The cross-linking methylene group was derived from form-aldehyde, which was a contaminant in a crystallization reagent. Subsequent studies demonstrated that formaldehyde greatly enhanced the cytotoxic potency of DOX in human breast cancer cells.10-12 One of the strongest enhancements was seen in DOX-resistant cells, suggesting that the enhanced cytotoxicity was by a topoisomerase-independent mechanism. The covalent DOX–formaldehyde–DNA cross-link is believed to be the source of the synergistic cytotoxicity. The cross-link is labile when the DNA is enzymatically digested, but 14C-labeled DOX–formaldehyde–dG adducts in intact DNA from breast cancer cells were detected by accelerator mass spectrometry.13 Other anthracyclines, such as mitoxantrone (MTX),14 pixantrone (PIX),15 and some synthetic derivatives16-18 also form covalent DNA adducts in the presence of formaldehyde in vitro. MTX is approved to treat leukemia and other cancers,19 whereas PIX is only approved for non-Hodgkin’s lymphoma treatment in Europe and remains an experimental drug in the US.20 The anthracycline–AP site conjugates might be highly cytotoxic, similar to the DOX–formaldehyde–DNA cross-link.10-12,21
Previous studies support our hypothesis that a second chemotherapeutic agent can target AP sites generated by alkylating agents and enhance cytotoxicity. The combination of temozolomide and methoxyamine (TRC102) was more cytotoxic against cultured human breast,22 ovarian,23 colon,24 and glioblastoma25 cancer cells than temozolomide alone. Temozolomide is a clinically used DNA methylating agent, and methoxyamine inhibits base excision repair by forming covalent oxime adducts with AP sites in DNA.26 The cytotoxic response of this combination was attributed to the formation of methoxyamine adducts with AP sites that result from the repair or thermal deglycosylation of methylated DNA bases. Cells that overexpress methyl purine glycosylase (MPG), which repair N7-methyl-dG or N3-methyl-2′-deoxyadenosine (dA), were further sensitized to this drug combination.22,23,27
The combination of temozolomide and methoxyamine also showed improved efficacy against colon cancer xenografts in mice over temozolomide alone and combined with O6-benzylguanine, an inhibitor of alkylguanine transferase.28 Similar results were obtained with the combination of 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) and methoxyamine in mouse xenografts.28 The combination of temozolomide and methoxyamine is the subject of three current or recently completed clinical trials.29-31
Additionally, Gates et al. hypothesized that the mutagenic side effect of hydralazine, a hydrazine-containing antihypertensive agent, was due to hydrazone formation with AP sites.32 They demonstrated that hydralazine rapidly formed a stable hydrazone conjugate with an AP site in vitro. The Gates lab also demonstrated that AP sites can form interstrand cross-links with appropriately situated dG and dA residues in the opposite strand33,34 and hypothesized that they contribute to the cytotoxicity of alkylating agents such as nitrogen mustards.35 Finally, the industrial waste semicarbazide formed a stable semicarbazone adduct with AP sites in calf thymus (CT) DNA and in exposed bacteria and rodents.36,37 These studies suggest that toxicological effects may be attributed to secondary reactions of AP sites and, at least in the case of semicarbazide, they occur in vivo.
Demonstrating this putative reaction of anthracyclines and AP sites would provide new insight into the pharmacology of AC chemotherapy. We demonstrate that DOX, epirubicin (EPI), MTX, and PIX react with AP sites in a 12-mer DNA duplex. The anthracycline–AP site Schiff base intermediate is hydrolytically labile; therefore, the covalent conjugates were reduced with NaB(CN)H3 to stable secondary or tertiary amines, which is a common strategy for observing labile Schiff base intermediates. We found that MTX and PIX react with AP sites more efficiently than DOX or EPI. We quantified the reduced MTX-AP site conjugate from reaction with CT DNA and in a human breast cancer cell line (MDA-MB-231) using stable isotope dilution mass spectrometry. In this article, we describe a previously unrecognized reactivity of anthracyclines and damaged DNA that may contribute to mechanism of action of adjuvant therapies involving anthracyclines and DNA alkylating agents.
MATERIALS AND METHODS
Chemical reagents, vendors, and descriptions of experimental procedures are provided in the Supporting Information. The cell treatment with NNM and MTX, DNA isolation of AP sites, and MTX-dR formation for LC/MS measurements are reported here.
Cell Culture and Chemical Treatments and Cell Viability Assays.
MDA-MB-231 cells were obtained and authenticated by ATCC (Manassas, VA). Cells were cultured in Dulbecco’s modified Eagle medium high glucose (4.5 g/L) containing 10% fetal calf serum, penicillin (100 IU/mL), and streptomycin (100 μg/mL) at 37 °C in a humidified atmosphere of 5% CO2. For NNM and MTX cotreatment, cells (2 × 103) were seeded in 96-well plates and cultured in complete media for 72 h. The cells were washed with prewarmed phosphate-buffered saline (PBS) and pretreated with NNM for 3 h. And the cell culture media were replaced by fresh cell culture media containing NNM (0–1 mM) and MTX (0–5 μM), and cells were further incubated for 21 h. After treatment, cells were washed with prewarmed PBS and incubated with media containing 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) for cell viability. The NNM, MTX, or control solvent (cell culture for NNM and 0.1%DMSO for MTX) was used as a negative control for 24, 48, 72, and 96 h of treatment. The cells were then washed with prewarmed PBS and incubated with media containing MTT (0.5 mg/mL, 100 μL) at 37 °C for 4 h. The media were removed, and the formazan crystals were dissolved in DMSO (100 μL). The absorbance was measured at 570 nm using a SpectraMax ID3 plate reader (Molecular Devices, San Jose, CA).
MDA-MB-231 Cell Treatment with NNM and MTX and MTX-dR and AP Site Formation.
Cells (1.5 × 106) were seeded in 150 cm2 flasks and cultured in complete media for 72 h, and then washed with prewarmed PBS and treated with NNM (1 mM). After 3 h, the cells were replenished with fresh cell culture media containing NNM (1 mM) and MTX (0.6 μM) for 21 h. Then, the cells were harvested employing trypsin (0.25%) and centrifuged at 600g for 7 min. The cell pellets were then washed with cold PBS and stored at −80 °C.
MTX-dR Schiff Base Reduction in Nuclei and DNA Isolation.
Cell pellets were resuspended in lysis buffer (0.5 mL, 250 mM sucrose, 1 mM EDTA, 20 mM HEPES, pH 7.4) and passed 10 times through a 27-gauge needle to lyse the cells. The nuclei were pelleted at 3000g at 4 °C for 10 min and resuspended in reduction buffer (0.5 mL, 100 mM HEPES, 10 mM EDTA, pH 7.4). Freshly prepared 1 M NaB(CN)H3 in ethanol (50 μL) was added to the nuclei and incubated for 0,1.5, 6, 18, or 24 h at 37 °C and agitated at 900 RPM. After each incubation period, nuclear fraction was precipitated by adding 0.1 vol of NaCl (5 M) and 2 vol of ice-chilled isopropanol. After centrifugation at 21,000g for 10 min at 4 °C, the pelleted nuclear fraction was washed once with 1 mL of HEPES buffer (5 mM, pH 8.0) containing 70% ethanol and recentrifuged, and the pelleted nuclear fraction was vacuum-centrifuged to dryness.
Dried nuclear pellets containing reduced MTX-dR were resuspended in TE buffer (300 μL, 50 mM Tris-HCl, 10 mM EDTA) containing proteinase K (0.4 mg) and 2% SDS and incubated at 37 °C for 90 min at 900 rpm. Then, protein precipitation solution (200 μL) was added, and the samples were incubated on ice for 5 min. Precipitated protein was pelleted by centrifugation at 10,000g for 10 min at 4 °C. The harvested supernatant was incubated with RNAase A (150 μg) and RNAase T1 (50 U) at 37 °C for 90 min at 900 rpm, followed by enzyme removal with protein precipitation solution (100 μL). The DNA in the supernatant was precipitated by adding 0.1 vol of NaCl (5 M) and 2 vol of ice-chilled isopropanol. The precipitated DNA was desalted with 70% ethanol/30% water, followed by centrifugation. The DNA was reconstituted in LC/MS water, and its concentration was measured by UV A260 nm, assuming that one absorbance unit at 260 nm is equal to a concentration of 50 μg/mL of double-stranded DNA. The reduced MTX-Schiff base in DNA underwent nuclease digestion, followed by solid phase extraction (SPE) as reported in the Supporting Information
Monitoring AP-Site Stability during MTX-dR Reduction in Nuclei.
The isolated nuclei described above were resuspended in 0.5 mL, 100 mM HEPES, 10 mM EDTA, pH 7.4 and incubated for 90 min, 6, 18, or 24 h at 37 °C and with agitation at 900 rpm using a Thermomixer. After each incubation period, the nuclear fraction was precipitated by adding 0.1 vol of NaCl (5 M) and 2 vol of ice-chilled isopropanol as described above. The nuclear fraction was resuspended in HE (0.5 mL, 50 mM HEPES, 10 mM EDTA, pH 8.0), and the AP sites were derivatized with O-(pyridin-3-ylmethyl)hydroxylamine (PMOA), followed by quenching excess PMOA with butyraldehyde, and enzymatically digested following our published protocol (Supporting Information).5
Kinetics of the NaB(CN)H3 and NaBH4 Reduction of the MTX-dR-Schiff Base in CT DNA and Kinetics of MTX-dR Displacement with [2H8]-MTX.
CT DNA (50 μg) was dissolved in HEPES buffer (500 μL, 100 mM HEPES, 10 mM NaCl, pH 7.4). MTX (2.5 nmol) was added, and the reaction was incubated at 37 °C with mixing at 900 rpm for 2.5 h. Then, freshly prepared 1 M NaBH4 in water (25 μL) or 1 M NaB(CN)H3 in ethanol (25 or 50 μL) was added, and the samples were incubated for 0, 1, 30, 60, 90, or 180 min. At each time point, DNA was precipitated to stop the reaction by adding 0.1 vol NaCl (5 M), followed by 2 vol of ice-chilled isopropanol as described above.
The kinetics of MTX-dR displacement in CT DNA was conducted with equimolar [2H8]-MTX, which was added to the reaction mixture during the MTX-dR Schiff base reduction step with 1 M NaB(CN)H3 in ethanol (50 μL). [2H8]-MTX was added at time points 0, 30, 60, 90 or 120 min, followed by precipitation of the DNA with isopropanol as described above, and assayed for MTX-dR and [2H8]-MTX-dR by LC/MS.
LC/MSn Measurements of Reduced MTX-dR, PMOA-Modifed AP Sites, and MTX Cell Uptake.
UPLC/ESI/MSn employed a Velos Pro ion trap mass spectrometer (Thermo Scientific, San Jose, CA) using a Michrom Captive Spray source (Auburn, CA) interfaced to an UltiMate 3000 RSLCnano System (Thermo Fisher Scientific). Ion source parameters and MSn transitions are reported in the Supporting Information.
RESULTS AND DISCUSSION
Reaction of AP Sites with Anthracyclines.
DOX, EPI, MTX, and PIX (40 μM) were individually reacted with an AP site containing DNA duplex (5′-GTTGCUCGTATG-3′·5′-CATACGCGCAAC-3′, 25 μM) at 37 °C and pH 7.4 in the presence of NaB(CN)H3 (50 mM), and the reaction was monitored by HPLC (Figure 2). MTX and PIX reacted faster than DOX and EPI after 6 h; the order of reactivity was PIX > MTX > EPI > DOX. MTX and PIX reacted completely with the AP site after 24 h, while DOX and EPI reacted less efficiently (Figure S1).
Figure 2.

HPLC-UV chromatograms of the reaction of the AP-containing 12-mer duplex with DOX, EPI, MTX, and PIX reduced with NaB(CN)H3 analyzed after 6 h.
The oligodeoxynucleotide products had the correct masses for the proposed reduced AP site conjugates, and the collision-induced dissociation (CID) spectra showed a-B and W ion series consistent with the structural assignments (Figures S2-S17 and Tables S1-S4). Further, oligodeoxynucleotide products were enzymatically digested, and the hydrolysates were analyzed by liquid chromatography tandem mass spectrometry (LC/MS2). The extracted ion chromatograms (EIC) showed the presence of the reduced anthracycline–deoxyribose (dR) conjugate in addition to the four canonical nucleosides (Figures S18-S21). The CID MS2 spectra of the reduced anthracycline–dR conjugates matched those of synthetic standards (Figures 3 and S27-S29). The analysis of the reduced DOX-dR and EPI-dR conjugates were complicated by partial reduction of the C13 ketone to diastereomeric alcohols.
Figure 3.

(A) Full-scan mass spectrum of the synthetic MTX-dR reduced conjugate showing the singly protonated [M + H]+ at m/z 563.3 and the doubly protonated [M + 2H]+2 ion at m/z 282.3. (B) The MS2 scan stage mass spectrum of m/z 282.3 and (C) MS3 spectrum of the m/z 476.2 product ion are reported. (D) Proposed fragmentation mechanisms of the major ions.
Synthesis of Labeled and Unlabeled Standards and MS Characterization.
We chose MTX over PIX for further studies because it has wider clinical use.19 Additionally, the ring nitrogen atom of PIX disrupts its molecular symmetry, making the nucleophilic amino groups chemically and spectroscopically distinct; this results in mixtures of conjugation products, which complicates the analyses. Finally, the symmetry of MTX makes it more amendable to the synthesis of derivatives.
MTX was reacted with dR in the presence of NaB(CN)H3, and the reduced MTX-dR standard was characterized by electrospray ionization (ESI) mass spectrometry (Figure 3A). The singly [M+H]+ and doubly [M+2H]2+ charged ions of reduced MTX-dR are observed at m/z 563.3 and 282.3, respectively, in the full-scan ESI mass spectrum. A base peak at m/z 476.2 in the CID MS2 spectrum of [M + 2H]2+ ion is attributed to loss of N-(2-hydroxyethyl)aminoethyl side chain (Figure 3B); the side chain is observed as a second prominent ion at m/z 88.1. The MS3 spectrum (Figure 3C) of the product ion at m/z 476.2 shows a principal fragment ion at m/z 206.1 attributed to 1-(2-hydroxyethyl)-1-((3S,4R)-3,4,5-trihydroxy-pentyl)-aziridin-1-ium ion. The proposed fragmentation mechanisms of the major product ions are shown in Figure 3D, which reveals the dR unit as a convenient place to incorporate isotopic labels for quantification by isotope dilution mass spectrometry.38 The MTX-[13C5]-dR standard was prepared by reductive amination with [13C5]-dR, and its CID MS2 and MS3 spectra (Figure S23) are consistent with the fragmentation mechanism shown in Figure 3D. The reduced DOX-dR, EPI-dR and PIX-dR conjugates were similarly prepared and analyzed (Figures S27-S29).
Formation of MTX-AP Conjugates in CT DNA and Kinetics of NaB(CN)H3 Reduction of the MTX-AP Schiff Base.
We observed ~50% loss of the unreduced MTX-dR Schiff base during mild ethanolic precipitation of modified CT DNA at neutral pH compared to MTX-modified DNA treated with NaB(CN)H3 before ethanol precipitation. Moreover, the unreduced MTX-dR Schiff base hydrolyzed during the ESI process at acidic pH conditions and was not detected by either infusion into the MS source or by LC/MS. We therefore characterized the kinetics of NaB(CN)H3 reduction of MTX-modified CT DNA and compared the amount of the reduced MTX-dR adduct to AP sites measured by PMOA derivatization, where the oxime-linked conjugate is stable.5
The kinetics of the Schiff base reduction of MTX-modified CT DNA was monitored over time for NaB(CN)H3 and NaBH4. The reduced MTX-dR adduct was enriched by SPE and quantified by LC/MS3, using the stable isotope dilution MS assay with the reduced MTX-[13C5]-dR internal standard. The predominant, doubly charged ion at m/z 282.3 was selected as the target ion for reduced MTX-dR measurement in DNA because it had higher ion trapping efficiency and signal of response at the MS3 scan stage than the singly charged precursor. There was a time-dependent increase in reduced MTX-dR formed, plateauing after 90 min (Figure 4A). The levels of reduced MTX-dR were comparable to the levels of AP sites derivatized with PMOA when NaB(CN)H3 was employed as the reductant (Figure 4B). PMOA reacts rapidly with AP sites in CT DNA, and the levels of PMOA-dR were constant over the 180 min incubation, indicating that the PMOA-dR oxime linkage is stable with minimal artifactual AP site formation over the incubation period. The levels of MTX-dR (1.63 ± 0.06 adducts per 105 nts) and PMOA-dR (1.82 ± 0.11 per 105 nts) are in good agreement, demonstrating that MTX reacted in high yield with AP sites in CT DNA and was efficiently reduced with NaB(CN)H3. Three independent replicate experiments on MTX-dR formation with CT DNA are reported in Figure S32. The reversibility of the MTX-Schiff base formed with the AP site in CT DNA is shown in Figure 4C. Adding equimolar [2H8]-MTX displaced approximately 35% of the MTX-dR Schiff base over the first 30 min of NaB(CN)H3 reduction (Figure 4C). These findings demonstrate the need to reduce the MTX-AP Schiff base to the stable tertiary amine for quantitative measurement of reduced MTX-dR in cells.
Figure 4.

Kinetics of MTX-dR Schiff base formation in CT DNA reduced with (A) NaB(CN)H3 or NaBH4. (B) AP sites in CT DNA derivatized with PMOA and MTX-dR Schiff base reduced with NaB(CN)H3. (C) The displacement of MTX-dR in CT DNA with [2H8]-MTX during reduction with NaB(CN)H3. (D) The displacement experiment scheme with [2H8]-MTX. Adduct levels are reported as the mean ± SD (n = 3 replicates for MTX-dR and n = 4 replicates for AP sites derivatized with PMOA). The levels of PMOA-dR and reduced MTX-dR at 180 min were not significantly different.
NaB(CN)H3 selectively reacts with the reversible MTX-AP Schiff base and likely shifts the chemical equilibrium to the Schiff base, leading to an overestimate of MTX-AP steady-state levels. We quantified the reduced MTX-AP conjugate using NaBH4 as the reductant, which will also reduce the AP-site aldehyde and prevent driving the Schiff base equilibrium. The levels of the reduced MTX-dR adduct after 1 min of reduction with NaB(CN)H3 or NaBH4 were 0.47 ± 0.03 and. 0.40 ± 0.06 adducts per 105 nts, respectively (Figure 4A). However, the reduced MTX-dR level continued to increase and plateaued at 90 min for the NaB(CN)H3 reduction, whereas the reduced MTX-dR level peaked within 5 min with NaBH4 and remained constant over the 90 min incubation. The amount of the reduced MTX-dR is ~3.3-fold lower for NaBH4-treated CT DNA than that recovered from CT DNA reduced with NaB(CN)H3 at 90 min (Figure 4A). The MTX-dR level measured after 1 min of reduction with NaBH4 may be viewed as a more realistic steady-state level of the MTX-AP Schiff base conjugate. NaBH4 is more reactive than NaB(CN)H3, but it also reacts readily with water with a half-life of seconds at pH 7–8 and is a highly basic reagent.39 We estimated that the pH of the NaBH4 reduction reaction was between 8 and 9. Intercalation probably contributes to the stability of the MTX-AP Schiff base, and alteration of the DNA structure at higher pH may affect its steady-state level.
Cytotoxicity of Bis(2-chloroethyl)amine (Nor-Nitrogen Mustard, NNM) and MTX in MDA-MB-231 Cells.
The cytotoxicity of NNM (1 μM–2.5 mM) and MTX (40 nM–5 μM) was evaluated in MDA-MB-231 cells by the MTT assay. Both compounds induced time- and concentration-dependent cytotoxicity effects (Figure 5A). However, in contrast to MTX, NNM was cytotoxic only over 100 μM (Figure 5B).
Figure 5.

Cytotoxicity of NNM and MTX in MDA-MB-231 cells. Cells were treated with (A) NNM [0.001–2.5 mM], (B) MTX [0.04–5.0 μM], or solvent control for up to 96 h. Cell viability was measured by the MTT assay every 24 h. Cytotoxicity of NNM and MTX cotreatment in MDA-MB-231 cells (C) after 24 h, and (D) after 48 h. Cells were treated with NNM [1 mM], in the presence of MTX [0.04–0.63 μM], or solvent control. Cell viability was measured by the MTT assay after 24 or 48 h. The data are compiled from three different experiments [mean ± SD]. (two-way ANOVA, Benferroni's multiple comparisons post test, NNM + MTX vs MTX, *P < 0.05; **P < 0.01, ***P < 0.005).
The cytotoxic effects of MTX and NNM cotreatment were also characterized employing varying concentrations of MTX in the presence of 1 mM NNM, which enhanced the cytotoxic effects of MTX in MDA-MB-231 human breast cancer cells (Figure 5C,D). The increase in cytotoxicity ranged from ~0.25 to 2-fold depending upon the dose of MTX employed.
NNM-Induced AP Site Formation in MDA-MB-231 Cells.
NNM forms N7-NNM-dG adducts, which depurinate to AP sites.40,41 We hypothesized that the increase in MTX cytotoxicity in MDA-MB-231 cells in combination with NNM could be attributed to the formation of cytotoxic MTX–DNA Schiff base adducts. NNM increased AP site levels in MDA-MB-231 cells in a concentration-dependent manner. Higher AP site formation occurred in dividing cells compared to confluent cells (Figure 6). The increase in AP site formation is likely attributed to the unpackaging of DNA with the removal of histones during cell proliferation, and the DNA bases are more accessible to alkylation by NNM.42
Figure 6.

Concentration-dependent increase in AP site formation in MDA-MB-231 cells. After 24 h of treatment with NNM [0.1–1 mM], AP site levels were measured following derivatization with PMOA. The data are compiled from three different experiments [mean ± SD]. Two-way ANOVA, confluence vs log phase (***P < 0.01, ***P < 0.005).
Formation of AP Sites and the MTX-AP Schiff Base in NNM and MTX Cotreated MDA-MB-231 Cells.
We measured AP-site and MTX-dR formation in MDA-MB-231 cells cotreated with NNM (1 mM) and MTX (0.6 μM) for 24 h. The isolated nuclei were lysed and treated with NaB(CN)-H3 (50 μmol) at 37 °C for up to 18 h. The DNA was isolated, enzymatically digested, and analyzed for AP sites and the reduced MTX-dR by UPLC-ESI/MSn using the Velos ion trap.4,43 Representative LC/MS2 chromatograms of AP site formation are shown in Figure S33, and LC/MS3 chromatograms of MTX-dR and its online MS3 spectra are shown in Figure S34. The kinetic data of MTX-dR and AP sites are summarized in Figure 7. The levels of reduced MTX-dR increased from 0.09 ± 0.01 adducts per 105 nts after 1.5 h to 1.50 ± 0.40 adducts per 105 nts at 18 h, after which there was no further increase even with prolonged NaB(CN)H3 reduction.
Figure 7.

MDA-MB-231 cells cotreated with 1 mM NNM and 0.6 μM MTX for 24 h. Reduced MTX-dR levels formed over 18 h in the isolated nuclei treated with NaB(CN)H3. AP site levels and stability in the nuclei of untreated and NNM-treated cells were measured at the same time points as done for MTX-dR reduction. Adduct levels are reported as the mean ± SEM (n = 4 independent replicates, ***P < 0.005, ****P < 0.0001, NS, not significant).
In a parallel experiment, we measured the persistence of AP sites in cells treated with NNM alone but under the MTX-dR reduction conditions. AP sites were derivatized with PMOA immediately and after 1.5, 6, and 18 h, as done for the MTX-dR Schiff base reduction. The AP site levels slowly decreased from 1.16 ± 0.15 AP sites per 105 nts (time t0 h after nuclei isolation) to 0.71 ± 0.10 AP sites per 105 nts at t18 h after nuclei isolation (P < 0.005, two-way ANOVA, Dunnett’s multiple comparisons test). The mean AP site level at time t18 h is also significantly lower than the mean MTX-dR level observed after 18 h of NaB(CN)H3 reduction (P < 0.0001, two-way ANOVA, Tukey’s multiple comparisons test). However, the highest mean AP site level in the NNM-treated cells immediately following nuclei isolation (time t0) is not significantly different from the mean MTX-dR level at 18 h (two-way ANOVA, Tukey’s multiple comparisons test). The decrease in AP site levels over time is likely attributed to DNA repair. Two independent replicate experiments on MTX-dR and AP sites in cells cotreated with NNM and MTX are shown in Figure S35. The detection of the reduced MTX-dR Schiff base-modified AP site in cells supports our hypothesis of a previously unrecognized reactivity of anthracycline with DNA that may contribute to cytotoxicity.
The reduced MTX-dR levels in cells treated with NNM and MTX were comparable to AP site levels induced by NNM treatment alone, indicating efficient reactivity of MTX with AP sites. The reduced MTX-dR adduct levels increase over the 18 h reduction period, while AP sites in cells treated with only NNM decreased over this time presumably due to repair. These results suggest that MTX may inhibit repair of the AP site, perhaps as the MTX-AP Schiff base adduct. The reduction of the Schiff base is slower in nuclear DNA than CT DNA, possibly because of other nuclear materials present and DNA packaging with histones renders the MTX-AP Schiff base less accessible for chemical reduction.
Unfortunately, NaBH4 reduction of the DNA from the MDA-MB-231 cell under the same conditions as those for CT DNA resulted in an unextractable precipitate and very poor recovery of DNA. The reduced MTX-AP adduct was not observed. Thus, we could not determine its steady-state levels directly. The reduced MTX-AP adduct levels for CT DNA and MDA-MB-231 cells were similar using NaB(CN)H3, 1.50 vs 1.63 adducts per 105 nts, although the reduction took significantly longer from the cell nuclear lysate (18 vs 3 h) (Figures 4 and 7). By analogy with the relative adduct levels between NaB(CN)H3 and NaBH4 in CT DNA, we may estimate the steady-state MTX-AP Schiff base level in MDA-MB-231 nuclear DNA at approximately 0.45 adducts per 105 nts. We acknowledge that DNA packaging with histones could alter this approximation.
Levels of Nuclear MTX in Treated MDA-MB-231 Cells.
The nuclear uptake of MTX in MDA-MB-231 cells was measured over several concentrations (0.1, 0.3, and 0.6 μM) following a 24 h treatment. The nuclear levels of MTX increased in a concentration-dependent manner and reached approximately 25% of each dose (Figure S36). The full-scan MS, MS2, and MS3 spectra for MTX are shown in Figure S37. Representative LC/MS3 chromatograms of MTX uptake in cells are shown in Figure S38. Assuming that 10 μg DNA is recovered per 106 cells and all MTX is within the DNA helix, the reduced MTX-dR formed in MDA-MB-231 cells treated with 0.6 μM MTX represents 0.016% of the nuclear uptaken dose.
CONCLUSIONS
We have demonstrated that Schiff base formation occurs between MTX and AP sites in an oligodeoxynucleotide, CT DNA, and cultured human MBA-MB-231 breast cancer cells treated with NNM and MTX. The MTX-AP Schiff base adduct was characterized and quantified by multistage ion trap mass spectrometry following NaB(CN)H3 or NaBH4 reduction of the DNA. We observed a continuous increase in the reduced MTX-dR levels over time, with the reaction reaching completion by 3 h in CT DNA and 18 h in MBA-MB-231 cells with NaB(CN)H3 as the trapping agent, whereas the MTX-dR levels peaked within 5 min with NaBH4 (CT DNA). Cotreatment of MDA-MB-231 cells with MTX and NNM enhanced cytotoxicity compared to when MDA-MB-231 cells are treated with MTX or NNM alone. These data are consistent with transient levels of nonreduced MTX-AP Schiff base contributing to the enhanced toxicity of combination therapies involving MTX with NNM. Our findings reveal a previous unknown reactivity between anthracyclines with aldehyde-reactive functional groups and AP sites in cellular DNA that could be exploited in future drug design.
Supplementary Material
ACKNOWLEDGMENTS
The National Cancer Institute funded this work through Grant P01 CA160032 (R.J.T. and C.J.R.). Cancer Center Support Grants partially support the Masonic Cancer Center, University of Minnesota (CA 077598) and Vanderbilt-Ingram Cancer Center (CA 068485). The Turesky laboratory gratefully acknowledges the support of Masonic Chair in Cancer Causation, University of Minnesota. We thank R.S. Lloyd (Oregon Health & Science University, Portland, OR) for helpful comments.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.3c00033.
Materials and experimental procedures, supplemental mass spectral analyses of reduced anthracycline-modified oligonucleotides and their enzymatic hydrolysate, MS and NMR analyses of the reduced anthracycline–deoxyribose adducts, calibration curves for MS quantification of MTX and the reduced MTX-dR adduct, and MS analyses for the formation of the reduced MTX-dR in CT DNA and MDA-MB-231 breast cancer cell (PDF)
The authors declare no competing financial interest.
Contributor Information
Medjda Bellamri, Masonic Cancer Center and Department of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
John T. Terrell, Chemistry, Vanderbilt University, Nashville, Tennessee 37235-1822, United States.
Kyle Brandt, Masonic Cancer Center and Department of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Francesca Gruppi, Chemistry, Vanderbilt University, Nashville, Tennessee 37235-1822, United States.
Robert J. Turesky, Masonic Cancer Center and Department of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States
Carmelo J. Rizzo, Chemistry, Biochemistry, and Vanderbilt-Ingram Cancer Center, Vanderbilt University, Nashville, Tennessee 37235-1822, United States
REFERENCES
- (1).Jones SE; Durie BG; Salmon SE Combination chemotherapy with adriamycin and cyclophosphamide for advanced breast cancer. Cancer 1975, 36, 90–97. [DOI] [PubMed] [Google Scholar]
- (2).Martins-Teixeira MB; Carvalho I Antitumour anthracyclines: Progress and perspectives. ChemMedChem 2020, 15, 933–948. [DOI] [PubMed] [Google Scholar]
- (3).Povirk LF; Shuker DE DNA damage and mutagenesis induced by nitrogen mustards. Mutat. Res 1994, 318, 205–226. [DOI] [PubMed] [Google Scholar]
- (4).Chen H; Cui Z; Hejazi L; Yao L; Walmsley SJ; Rizzo CJ; Turesky RJ Kinetics of DNA adducts and abasic site formation in tissues of mice treated with a nitrogen mustard. Chem. Res. Toxicol 2020, 33, 988–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chen H; Yao L; Brown C; Rizzo CJ; Turesky RJ Quantitation of apurinic/apyrimidinic sites in isolated DNA and in mammalian tissue with a reduced level of artifacts. Anal. Chem 2019, 91, 7403–7410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gates K. Covalent modification of DNA by natural products. In Comprehensive Natural Products Chemistry; Barton D; Nakanishi K; Meth-Cohn O; Kool E, Eds.; Elsevier Science, 1999; Vol. 7, pp 491–552. [Google Scholar]
- (7).Wang P; Song Y; Zhang L; He H; Zhou X Quinone methide derivatives: Important intermediates to DNA alkylating and DNA cross-linking actions. Curr. Med. Chem 2005, 12, 2893–2913. [DOI] [PubMed] [Google Scholar]
- (8).Wang AHJ; Gao YG; Liaw YC; Li YK Formaldehyde cross-links daunorubicin and DNA efficiently: HPLC and X-ray diffraction studies. Biochemistry 1991, 30, 3812–3815. [DOI] [PubMed] [Google Scholar]
- (9).Gao YG; Liaw YC; Li YK; van der Marel GA; van Boom JH; Wang AH Facile formation of a crosslinked adduct between DNA and the daunorubicin derivative MAR70 mediated by formaldehyde: molecular structure of the MAR70-d(CGTnACG) covalent adduct. Proc. Natl. Acad. Sci. U.S.A 1991, 88, 4845–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Cutts SM; Nudelman A; Rephaeli A; Phillips DR The power and potential of doxorubicin-DNA adducts. IUBMB Life 2005, 57, 73–81. [DOI] [PubMed] [Google Scholar]
- (11).Cutts S; Rephaeli A; Nudelman A; Ugarenko M; Phillips D Potential therapeutic advantages of doxorubicin when activated by formaldehyde to function as a DNA adduct-forming agent. Curr. Top. Med. Chem 2015, 15, 1409–1422. [DOI] [PubMed] [Google Scholar]
- (12).Koch TH; Barthel BL; Kalet BT; Rudnicki DL; Post GC; Burkhart DJ Anthracycline-formaldehyde conjugates and their targeted prodrugs. Top. Curr. Chem 2008, 283, 141–170. [DOI] [PubMed] [Google Scholar]
- (13).Coldwell KE; Cutts SM; Ognibene TJ; Henderson PT; Phillips DR Detection of Adriamycin-DNA adducts by accelerator mass spectrometry at clinically relevant Adriamycin concentrations. Nucleic Acids Res. 2008, 36, No. e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Parker BS; Cullinane C; Phillips DR Formation of DNA adducts by formaldehyde-activated mitoxantrone. Nucleic Acids Res. 1999, 27, 2918–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Evison BJ; Mansour OC; Menta E; Phillips DR; Cutts SM Pixantrone can be activated by formaldehyde to generate a potent DNA adduct forming agent. Nucleic Acids Res. 2007, 35, 3581–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ankers EA; Evison BJ; Phillips DR; Brownlee RTC; Cutts SM Design, synthesis, and DNA sequence selectivity of formaldehyde-mediated DNA-adducts of the novel N-(4-aminobutyl) acridine-4-carboxamide. Bioorg. Med. Chem. Lett 2014, 24, 5710–5715. [DOI] [PubMed] [Google Scholar]
- (17).Mansour OC; Evison BJ; Sleebs BE; Watson KG; Nudelman A; Rephaeli A; Buck DP; Collins JG; Bilardi RA; Phillips DR; Cutts SM New anthracenedione derivatives with improved biological activity by virtue of stable drug-DNA adduct formation. J. Med. Chem 2010, 53, 6851–6866. [DOI] [PubMed] [Google Scholar]
- (18).Pumuye PP; Evison BJ; Konda SK; Collins JG; Kelso C; Medan J; Sleebs BE; Watson K; Phillips DR; Cutts SM Formaldehyde-activated WEHI-150 induces DNA interstrand cross-links with unique structural features. Bioorg. Med. Chem 2020, 28, No. 115260. [DOI] [PubMed] [Google Scholar]
- (19).Evison BJ; Sleebs BE; Watson KG; Phillips DR; Cutts SM Mitoxantrone, more than just another topoisomerase II poison. Med. Res. Rev 2016, 36, 248–299. [DOI] [PubMed] [Google Scholar]
- (20).Pean E; Flores B; Hudson I; Sjoberg J; Dunder K; Salmonson T; Gisselbrecht C; Laane E; Pignatti F The European Medicines Agency review of pixantrone for the treatment of adult patients with multiply relapsed or refractory aggressive non-Hodgkin’s B-cell lymphomas: summary of the scientific assessment of the committee for medicinal products for human use. Oncologist 2013, 18, 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Taatjes DJ; Koch TH Nuclear targeting and retention of anthracycline antitumor drugs in sensitive and resistant tumor cells. Curr. Med. Chem 2001, 8, 15–29. [DOI] [PubMed] [Google Scholar]
- (22).Rinne M; Caldwell D; Kelley MR Transient adenoviral N-methylpurine DNA glycosylase overexpression imparts chemotherapeutic sensitivity to human breast cancer cells. Mol. Cancer Ther 2004, 3, 955–967. [PubMed] [Google Scholar]
- (23).Fishel ML; Kelley MR The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Mol. Aspects Med 2007, 28, 375–395. [DOI] [PubMed] [Google Scholar]
- (24).Liu LL; Nakatsuru Y; Gerson SL Base excision repair as a therapeutic target in colon cancer. Clin. Cancer Res 2002, 8, 2985–2991. [PubMed] [Google Scholar]
- (25).Montaldi AP; Sakamoto-Hojo ET Methoxyamine sensitizes the resistant glioblastoma T98G cell line to the alkylating agent temozolomide. Clin. Exp. Med 2013, 13, 279–288. [DOI] [PubMed] [Google Scholar]
- (26).Liu L; Gerson SL Therapeutic impact of methoxyamine: Blocking repair of abasic sites in the base excision repair pathway. Curr. Opin. Invest. Drugs 2004, 5, 623–627. [PubMed] [Google Scholar]
- (27).Rinne ML; He Y; Pachkowski BF; Nakamura J; Kelley MR N-Methylpurine DNA glycosylase overexpression increases alkylation sensitivity by rapidly removing non-toxic 7-methylguanine adducts. Nucleic Acids Res. 2005, 33, 2859–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Liu L; Yan L; Donze JR; Gerson SL Blockage of abasic site repair enhances antitumor efficacy of 1,3-bis-(2-chloroethyl)-1-nitrosourea in colon tumor xenografts. Mol. Cancer Ther 2003, 2, 1061–1066. [PubMed] [Google Scholar]
- (29).TRC102 and Temozolomide for Relapsed Solid Tumors and Lymphomas; National Library of Medicine: Bethesda (MD). https://clinicaltrials.gov/ct2/show/NCT01851369/. [Google Scholar]
- (30).Methoxyamine and Temozolomide in Treating Patients with Recurrent Glioblastoma; National Library of Medicine: Bethesda (MD). https://clinicaltrials.gov/ct2/show/NCT02395692/. [Google Scholar]
- (31).Methoxyamine and Temozolomide in Treating Patients with Advanced Solid Tumors; National Library of Medicine: Bethesda (MD). https://clinicaltrials.gov/ct2/show/NCT00892385/. [Google Scholar]
- (32).Melton D; Lewis CD; Price NE; Gates KS Covalent adduct formation between the antihypertensive drug hydralazine and abasic sites in double- and single-stranded DNA. Chem. Res. Toxicol 2014, 27, 2113–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Johnson KM; Price NE; Wang J; Fekry MI; Dutta S; Seiner DR; Wang Y; Gates KS On the formation and properties of interstrand DNA-DNA cross-links forged by reaction of an abasic site with the opposing guanine residue of 5′-CAp sequences in duplex DNA. J. Am. Chem. Soc 2013, 135, 1015–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Price NE; Johnson KM; Wang J; Fekry MI; Wang Y; Gates KS Interstrand DNA-DNA cross-link formation between adenine residues and abasic sites in duplex DNA. J. Am. Chem. Soc 2014, 136, 3483–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Nejad MI; Johnson KM; Price NE; Gates KS A new cross-link for an old cross-linking drug: the nitrogen mustard anticancer agent mechlorethamine generates cross-links derived from abasic sites in addition to the expected drug-bridged cross-links. Biochemistry 2016, 55, 7033–7041. [DOI] [PubMed] [Google Scholar]
- (36).Wang Y; Chan HW; Chan W Facile formation of a DNA adduct of semicarbazide on reaction with apurinic/apyrimidinic sites in DNA. Chem. Res. Toxicol 2016, 29, 834–840. [DOI] [PubMed] [Google Scholar]
- (37).Wang Y; Wong TY; Chan W Quantitation of the DNA adduct of semicarbazide in organs of semicarbazide-treated rats by isotope-dilution liquid chromatography-tandem mass spectrometry: A comparative study with the RNA adduct. Chem. Res. Toxicol 2016, 29, 1560–1564. [DOI] [PubMed] [Google Scholar]
- (38).Tretyakova N; Goggin M; Sangaraju D; Janis G Quantitation of DNA adducts by stable isotope dilution mass spectrometry. Chem. Res. Toxicol 2012, 25, 2007–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Yamamoto J, Ed. Rohm and Haas: The Sodium Borohydride Digest, 2003. [Google Scholar]
- (40).Gates KS; Nooner T; Dutta S Biologically relevant chemical reactions of N7-alkylguanine residues in DNA. Chem. Res. Toxicol 2004, 17, 839–856. [DOI] [PubMed] [Google Scholar]
- (41).Hemminki K. DNA-binding products of nornitrogen mustard, a metabolite of cyclophosphamide. Chem.-Biol. Interact 1987, 61, 75–88. [DOI] [PubMed] [Google Scholar]
- (42).Antonin W; Neumann H Chromosome condensation and decondensation during mitosis. Curr. Opin. Cell Biol 2016, 40, 15–22. [DOI] [PubMed] [Google Scholar]
- (43).Gruppi F; Hejazi L; Christov PP; Krishnamachari S; Turesky RJ; Rizzo CJ Characterization of nitrogen mustard formamidopyrimidine adduct formation of bis(2-chloroethyl)-ethylamine with calf thymus DNA and a human mammary cancer cell line. Chem. Res. Toxicol 2015, 28, 1850–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.