

Abstract
Background
The adaptive evolution of plateau indigenous animals is a current research focus. However, phenotypic adaptation is complex and may involve the interactions between multiple genes or pathways, many of which remain unclear. As a kind of livestock with important economic value, cashmere goat has a high ability of plateau adaptation, which provides us with good materials for studying the molecular regulation mechanism of animal plateau adaptation.
Results
In this study, 32 Jiangnan (J) and 32 Tibetan (T) cashmere goats were sequenced at an average of 10. Phylogenetic, population structure, and linkage disequilibrium analyses showed that natural selection or domestication has resulted in obvious differences in genome structure between the two breeds. Subsequently, 553 J vs. T and 608 T vs. J potential selected genes (PSGs) were screened. These PSGs showed potential relationships with various phenotypes, including myocardial development and activity (LOC106502520, ATP2A2, LOC102181869, LOC106502520, MYL2, ISL1, and LOC102181869 genes), pigmentation (MITF and KITLG genes), hair follicles/hair growth (YAP1, POGLUT1, AAK1, HES1, WNT1, PRKAA1, TNKS, WNT5A, VAX2, RSPO4, CSNK1G1, PHLPP2, CHRM2, PDGFRB, PRKAA1, MAP2K1, IRS1, LPAR1, PTEN, PRLR, IBSP, CCNE2, CHAD, ITGB7, TEK, JAK2, and FGF21 genes), and carcinogenesis (UBE2R2, PIGU, DIABLO, NOL4L, STK3, MAP4, ADGRG1, CDC25A, DSG3, LEPR, PRKAA1, IKBKB, and ABCG2 genes). Phenotypic analysis showed that Tibetan cashmere goats has finer cashmere than Jiangnan cashmere goats, which may allow cashmere goats to better adapt to the cold environment in the Tibetan plateau. Meanwhile, KRTs and KAPs expression in Jiangnan cashmere goat skin was significantly lower than in Tibetan cashmere goat.
Conclusions
The mutations in these PSGs maybe closely related to the plateau adaptation ability of cashmere goats. In addition, the expression differences of KRTs and KAPs may directly determine phenotypic differences in cashmere fineness between the two breeds. In conclusion, this study provide a reference for further studying plateau adaptive mechanism in animals and goat breeding.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-023-09333-1.
Keywords: Plateau adaptability, Tibetan and jiangnan cashmere goats, Genetic, Gene expression and PSGs
Background
Changes in altitude gradients on Earth create a complex and changeable natural climate. Meanwhile, ladder-like topographical due to altitude differences is particularly evident in China; Tibet is the region with the highest altitude and the most unique climate in China. The high altitude has led to extreme climate conditions, such as high cold, hypoxia, and strong ultraviolet radiation (UVR). This extreme climate not only challenges animal’s physiological tolerance, but also adversely affects the ecosystems of the Tibet area, further leading to a decrease in the quality and quantity of livestock feed, reduced water availability, and increased disease prevalence [1, 2]. Therefore, extreme climates have limited the development of animal husbandry in Tibet area, which accounts for one-eighth of China’s total area.
To cope with extreme climate, numerous plateau indigenous animals (e.g., Bos mutus, Equus kiang, Procapra picticaudata, and Tetraogallus tibetanus, etc.) have developed a unique adaptive mechanism during evolution, specifically reflected at physiological, cellular, and molecular levels, e.g., skin tone deepening [3], a well-developed cardiopulmonary system and a high-density capillary network are formed [4, 5], and increased VEGF expression [6], etc. As such, they are excellent subjects for research on adaptive evolution. Physiological changes in plateau adaptation are often accompanied by genetic changes. Previous studies mainly focused on the former, while studies on the molecular mechanism are relatively rare, particularly in non-model organisms. Previously, research focused on known candidate genes and analyzed the differences in their sequences between animals at high and low altitudes to identify the specific variation in candidate genes of high-altitude animals. This type of research is somewhat limited because it focuses on the optimal potential adaptive phenotype genes, and makes it difficult to find new adaptive mechanisms. For example, research mainly focused on hypoxia adaptation, while ignoring cold or high radiation adaptation. Moreover, the evolution of complex adaptive physiological characteristics often results from the activity of numerous genes, and this method may ignore other important functional genes [7, 8]. Currently, advances in sequencing technologies have permitted extensive comparative genomics studies across species or populations at different altitudes. For example, Qiu et al. sequenced the genome and transcriptome of the female yak and compared them with the cattle reference genome, finding expansion of the sensory perception and energy metabolism related gene families in the yak, and metabolic enzyme and hypoxia adaptation related genes had undergone positive selection [9]. In addition, Ge et al. determined that genes associated to repair, function, angiogenesis, and hypoxia were positively selected in Tibetan antelopes, based on genome construction [10]. Further, Qu et al. performed whole-genome sequencing in ground tits and compared it to other breeds (great and yellow-cheeked tits). In ground tits, the gene family related to energy metabolism had expanded while that related to immunity and smell had contracted and the genes related to hypoxia response and bone development had undergone positive selection [11].
In contrast to the genomic research, transcriptomic analysis reveals genome-wide gene expression characteristics of animal organs, tissues, or cells at different times. If the genome sequence does not produce relevant variation to adapt to extreme climates, gene transcription can also effectively result in adaptation to extreme climates. Therefore, gene expression levels are an important molecular phenotype for identifying key genes responding to physiological challenges [12]. For example, the transcriptomic studies found accelerated evolution of genome-wide gene expression levels in Schizothorax (fish endemic to the Qinghai-Tibet Plateau), particularly in genes related to hypoxia and energy metabolism, compared with zebrafish [13, 14]. In conclusion, integrating genomics and transcriptomics can reveal the regulatory mechanisms of the adaptability of plateau indigenous animals and the development of new breeds resistant to low temperature, hypoxia, and UVR.
Cashmere goats (Capra hircus) were domesticated from wild goats (Bezoar, Capra aegagrus) in the Fertile Crescent of the Near East about 10,000 years ago, and then got spread to Europe, Africa, Asia through migration and trade, basically. At present, cashmere goat is widely found in Tibet, Xinjiang, and other regions of China. Jiangnan cashmere goat and Tibetan cashmere goat are important breeds in Xinjiang and Tibet, respectively. They were all formed by crossbreeding, and Liaoning cashmere goat as the male parent, local cashmere goat as the female parent. Cashmere goats provide inhabitants with essential living resources such as meat, milk, cashmere, and fur, and are an important income source for local herdsmen. In addition, cashmere goats have stronger adaptability to extreme climates (e.g., high cold, hypoxia, and UVR). Although within the same longitude range, the Xinjiang region is lower in altitude and has a climate more suitable for animal survival than Tibet. Thence, we used 32 Jiangnan and 32 Tibetan cashmere goats as research objects, extracted genomic DNA from their blood, and further performed whole genome resequencing of them. At the same time, we obtained a skin tissue transcriptome dataset of Tibetan and Jiangnan cashmere goats. Based on comparative genomic and transcriptomic analysis between these two breeds, we extensively screened for candidate genes associated with plateau adaptive, further investigating the plateau adaptive expression strategies of related genes (The specific experimental procedure is shown in Figure S1). In conclusion, this study provide a theoretical basis for further study the molecular regulatory mechanism of animal plateau adaptability and goat breeding.
Results
Genome resequencing and variation
First, the quality control was performed on the whole genome resequencing data of the 64 cashmere goats. After quality control, the clean data was mapped to the goat reference genome; the mapping rates were > 99%, 1X Coverage > 94%, and 4X Coverage > 91% of all samples. Finally, we performed SNP detection of 64 samples and obtained 15,349,261 high-quality SNPs through filtering and screening. The SNP detection statistics are shown in Table S1.
Population structure
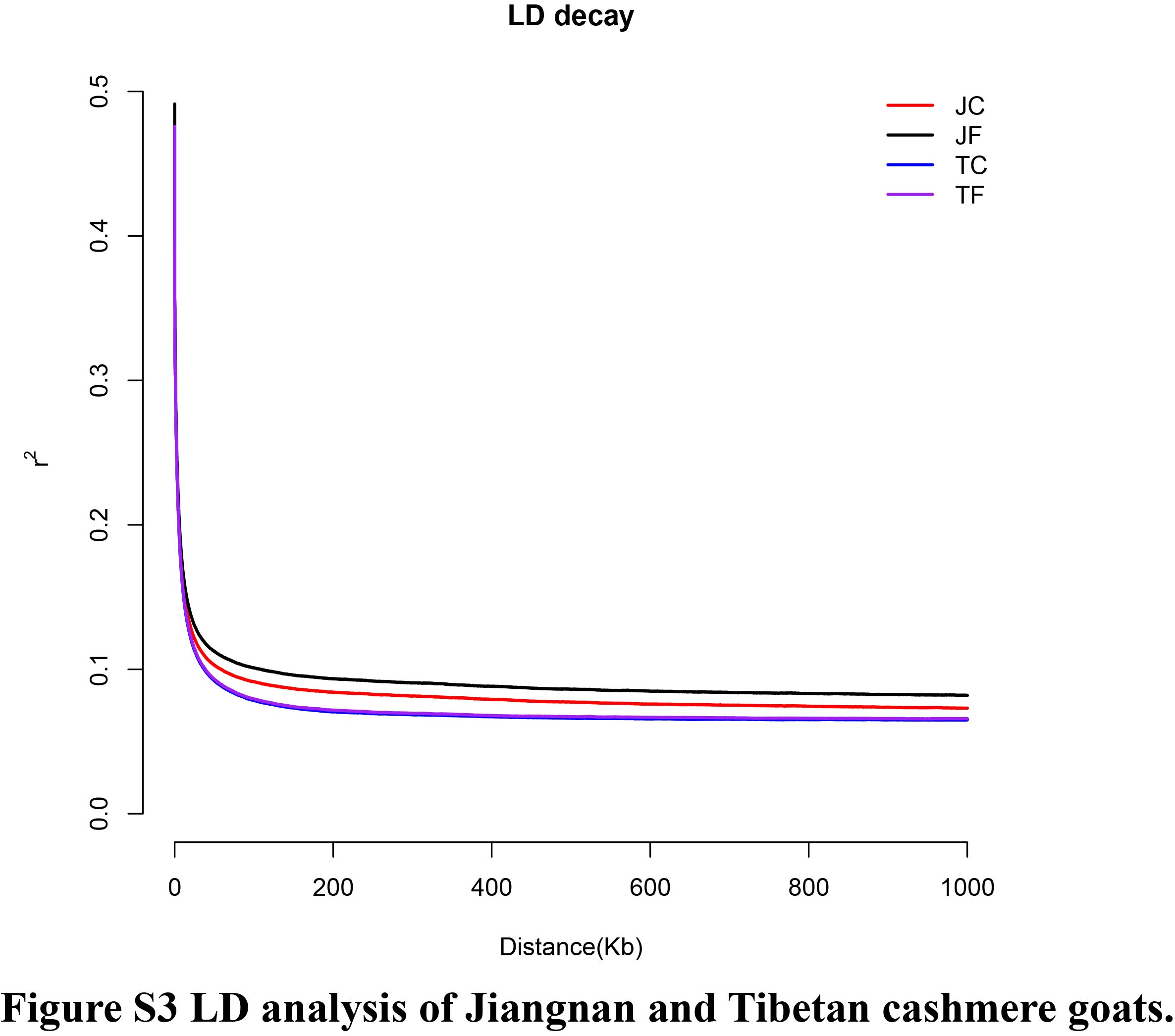
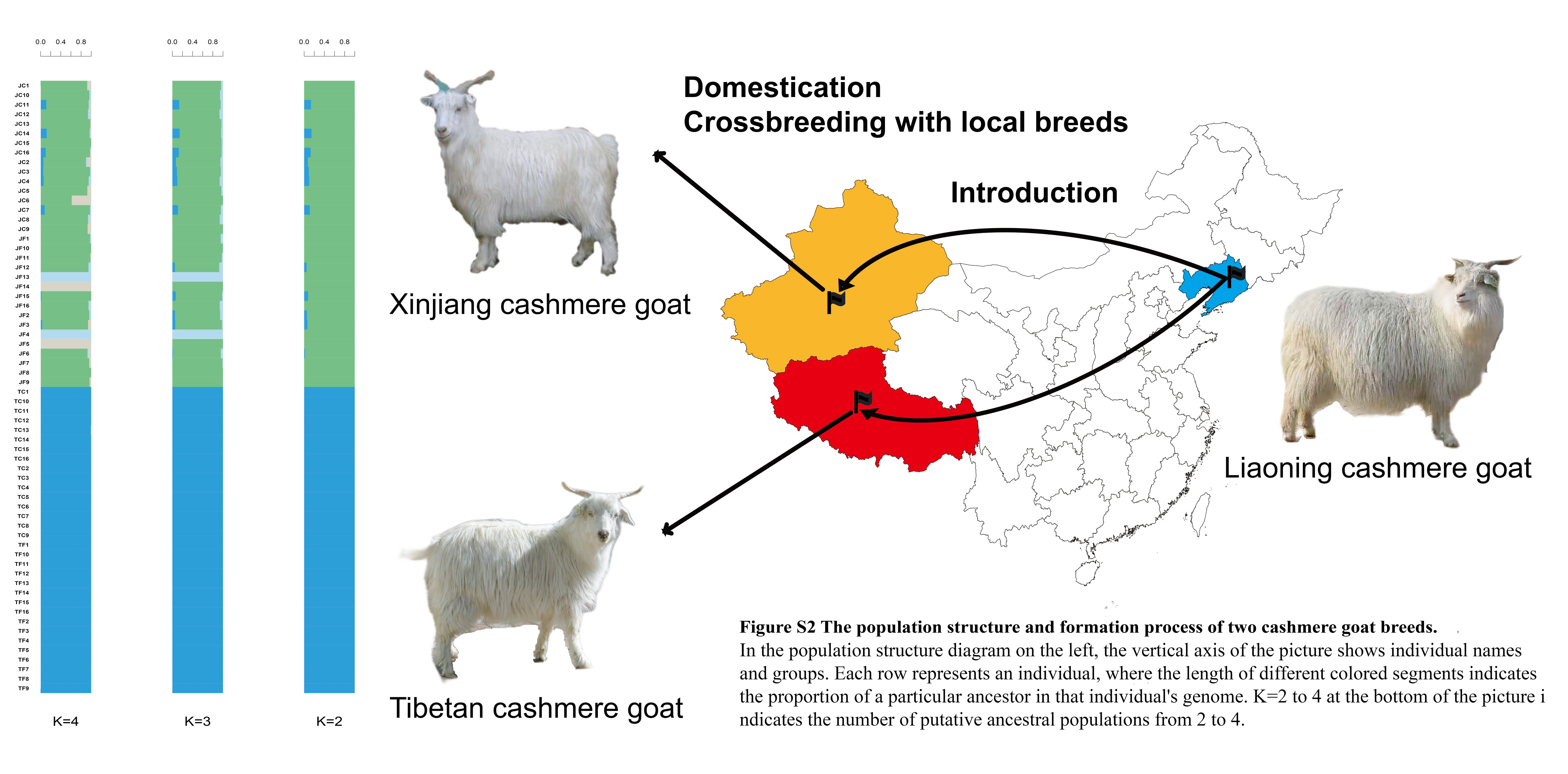
PCA divided Tibetan and Jiangnan cashmere goats into two separate clusters (Fig. 1B). There was a significant correlation between the clustering pattern of the two cashmere goat breeds and the locations where they were collected. Phylogenetic trees showed that both Tibetan and Jiangnan cashmere goats derive from a single domestication event or a common ancestor (Fig. 1C). In addition, admixture analysis (k = 2, 3, and 4) showed no obvious gene flow between Tibetan and Jiangnan cashmere goats due to geographical isolation (Figure S2). Moreover, LD (linkage disequilibrium) decayed more slowly in Tibetan cashmere goats than in Jiangnan cashmere goats (Figure S3).
Fig. 1.

Sampling and genomic landscape of the divergence of Tibetan and Jiangnan cashmere goat breeds. (A) Map of cashmere goat sampling. The orange and red areas in the figure are the sampling areas of Jiangnan and Tibetan cashmere goat, respectively. In the map, a is Aksu Prefecture; b, c, and d are Ritu County, Gaize County, and Nima County, respectively. (B) Principal components of cashmere goat samples. (C) Phylogenetic tree of 64 cashmere goats. (D) FST in 5-kb sliding windows across autosomes between Tibetan and Jiangnan cashmere goats. (E) FST Pi selection elimination analysis. The X-axis of the scatter diagram is the log2(Pi (π) ratio) value and the Y-axis is the FST value, which correspond to the frequency distribution diagram above the scatter diagram and the frequency distribution diagram on the right, respectively. The area of red scatter points is the top 5% area selected by Pi (π) ratio and FST.
Selective sweeps
We scanned the genome for regions with extreme FST divergence and the highest differences in Pi (π) ratio in 5-kb sliding windows in two compare groups (J vs. T and T vs. J) to further detect the PSGs on chromosomes (Fig. 1D F).
In total, we identified 553 J vs. T PSGs (based on π and Fst, selected genes in the top 5% region) and 608 T vs. J PSGs (based on π and Fst, selected genes in the top 5% region). GO and KEGG enrichment analysis of the PSGs in J vs. T showed that they were mainly enriched in cell adhesion (GO:0007155), positive regulation of apoptotic process (GO:0043065), extracellular region (GO:0005576), cytosol (GO:0005829), and centrosome (GO:0005813). Interestingly, PARP1, BMF, CDC25A, and ATR are associated with cellular responses to UV (GO:0034644), CD6 and ELANE are associated with acute inflammatory responses to antigenic stimulus (GO:0002438), and LTA and TNF genes are associated with positive regulation of chronic inflammatory responses to antigenic stimulus (GO:0002876) (Table S2 and S3).
GO and KEGG analysis in T vs. J further showed that PSGs were mainly enriched in the following functions: response to nuclear speck (GO:0016607), cytosol (GO:0005829), nucleoplasm (GO:0005654), calcium ion binding (GO:0005509), small GTPase binding (GO:0031267), zinc ion binding (GO:0008270), regulation of actin cytoskeleton (chx04810), and PI3K-Akt signaling pathway (chx04151). Notably, PSGs were enriched for ATP binding (GO:0005524) in J vs. T and T vs. J. Moreover, LOC106502520, ACTN3, and LOC102181869 are associated with regulation of skeletal muscle contraction force (GO:0014728); LOC106502520, MYL2, ISL1, and LOC102181869 genes are associated with ventricular cardiac muscle tissue morphogenesis (GO:0055010); LOC106502520, ATP2A2, and LOC102181869 are associated with regulation of heart contraction force (GO:0002026), cardiac muscle hypertrophy in response to stress (GO:0014898), or regulation of slow-twitch skeletal muscle fiber contraction (GO:0031449); PRKAA1, TNKS, WNT5A, VAX2, RSPO4, and CSNK1G1 are associated with Wnt signaling (GO:0016055); DUOX1, WRN, PNKP, GCLM, and DUOX2 are associated with response to oxidative stress (GO:0006979); and PRKAA1, LOC106502520, MYL2, DAG1, ATP2A2, ITGB7, and LOC102181869 are associated with hypertrophic cardiomyopathy (chx05410) (Table S2 and S3).
Gene expression profiles in skin tissues of Jiangnan and Tibetan cashmere goats
Genome-wide gene expression pattern cluster analysis showed that, eight Tibetan cashmere goats and seven Jiangnan cashmere goats were clustered into different clusters, respectively (Fig. 2A). However, Ce individuals and Fe individuals are not fully distinguished in the cluster of eight Tibetan cashmere goats or seven Jiangnan cashmere goats. WGCNA analysis further assigned genome-wide genes into modules of different colors, and the results showed that all modules showed distinct differential expression patterns in Tibetan and Jiangnan cashmere goats (Fig. 2B).
Fig. 2.

transcriptomic analysis of Jiangnan and Tibetan cashmere goat skin tissue. (A) Clustering tree of 15 Jiangnan or Tibetan cashmere goat skin transcriptomes. (B) Expression patterns of genes within different color modules in skin tissues of Tibetan and Jiangnan cashmere goats. The values in the rectangles represent the correlation between gene expression patterns in different modules and breeds. (C) Volcano plot of DEGs. (D) GO enrichment analysis of DEGs.
The analysis of DEGs showed that 1426 genes (log2FC ≥ 2 or ≤ − 2, p < 0.01) were differentially expressed in Tibetan and Jiangnan cashmere goats, of which 653 DEGs were up-regulated and 773 DEGs were down-regulated (Fig. 2C). GO and KEGG enrichment of DEGs showed that (Fig. 2 and Table S4), the up-regulated DEGs were predominantly enriched into keratin filament (GO:0045095), intermediate filament (GO:0005882), structural molecule activity (GO:0005198), and small GTPase binding (GO:0031267). Furthermore, it is also worth noting that, EDNRB and EDN2 genes are enriched for vein smooth muscle contraction (GO:0014826), FGF23 and FGF21 genes are enriched for positive regulation of MAPKKK cascade by fibroblast growth factor receptor signaling pathway (GO:0090080), LOC102170378 and AGT genes are enriched for regulation of cardiac conduction (GO:1,903,779), GPR143, DCT, and TYR genes are enriched for melanin biosynthetic process (GO:0042438) and melanosome membrane (GO:0033162), etc. Then, the down-regulated DEGs were predominantly enriched into translation (GO:0006412), extracellular space (GO:0005615), ribosome (GO:0005840), structural constituent of ribosome (GO:0003735), and calcium ion binding (GO:0005509). It is still worth noting that, WNT7A and CTHRC1 genes are enriched for Wnt signaling pathway, planar cell polarity pathway (GO:0060071), MYH7B and TNNT2 genes are enriched for cardiac myofibril (GO:0097512), etc.
The candidate genes associated with cashmere growth in Jiangnan and Tibetan cashmere goats
We measured the cashmere MFD of the 64 goats (Tibetan and Jiangnan cashmere goats) in this study, and further calculated the CVFD and FDSD (Fig. 3A). Meanwhile, we found that the MFD of Jiangnan cashmere goats is higher than that of Tibetan cashmere goats. All phenotypic data were used for GWAS.
Fig. 3.

SNPs or candidate genes associated with cashmere fineness. (A) Statistics of three cashmere fineness phenotypes in Tibetan and Jiangnan cashmere goats. (B) GWAS of cashmere fineness, and selective sweeps analysis of trait-associated candidate genes
GWAS further showed that some SNPs were associated with the MFD, FDSD, and CVFD of Tibetan or Jiangnan cashmere goats. These SNPs were annotated to 11 (MFD), 16 (FDSD), and 13 (CVFD) genes, respectively (Fig. 3B). Combined with selective sweeps, these genes were found in regions with low genetic diversity (Fig. 3B). This may indicate that cashmere fineness has a similar genetic basis in different goat breeds. We further focused on the expression patterns of candidate genes in Tibetan and Jiangnan cashmere goats, including SLC35F3 (Down), PAMR1 (Down), DCP1B (Up), HS3ST4 (Down), AKAP13 (Down), MYO3B (Down), GABRA2 (Up), RALYL (Up), QRFPR (Down), TRPM3 (Down), PTPRT (Up), MAP6 (Up), and MACROD2 (Up); they all had significant differential expression (log2FC ≥ 1 or ≤ − 1, P < 0.05;Figure S4). Simultaneously, down-regulated DEG HS3ST4 was associated with both FDSD and CVFD. However, there is no evidence showing that these candidate genes regulate wool growth and follicle development in animals. Hence, the effect of these genes on cashmere fineness remains to be verified.
In addition, keratin (KRTs) and keratin-associated proteins (KAPs) are the main structural proteins of animal hair and play a decisive role in its physical properties [15, 16]. In transcriptomic analysis, 24 differentially expressed KRT or KAP genes (including LOC102178483, KRT82, LOC102179595, LOC102185436, LOC10218376, LOC102184223, LOC102183211, LOC108638297, LOC102184693, LOC102185150, LOC102170546, LOC102177517, KRT84, LOC100861184, LOC108638292, LOC102172755, LOC102171368, LOC102170264, LOC108638299, LOC108638288, LOC102176522, LOC108634945, LOC102174594, and LOC102182538) were enriched in keratin filaments (GO:0045095), and 14 differentially expressed KRT or KAP genes (Including LOC102179881, KRTAP11-1, KRTAP15-1, KRT35, LOC100861175, LOC100861181, LOC100861381, LOC102176726, LOC100860930, LOC102168573, KRT39, KRT26, LOC102176457, and KAP8) were enriched in intermediate filaments (GO:0005882). The expression of the above genes in the skin tissue of Tibetan cashmere goats was significantly lower than that of Jiangnan cashmere goats (log2FC of the above genes >2). This may explain the finer texture of Tibetan cashmere.
The PSGs associated with pigmentation in Jiangnan and Tibetan cashmere goats
In T vs. J, we found two PSGs–KITLG and microphthalmia-associated transcription factor (MITF) genes on chromosome 5 and 22(Figs. 1D and 4A), respectively. MITF and KITLG genes play a crucial role in the melanin biosynthesis and pigmentation [17, 18]. Among them, MITF plays an important role in mammalian skin and hair melanin deposition, mainly regulating the transcription of three pigmentation enzymes, TYR, TYRP-1, and TYRP-2/DCT (Fig. 4B) [18–21].
Fig. 4.

Functional genomic basis of differences in melanin synthesis between Tibetan and Jiangnan cashmere goats. (A) FST of all SNPs in chromosome 22 between Tibetan and Jiangnan cashmere goats. (B) Schematic overview of the melanogenesis pathway. The schematic was drew based on KEGG imagery [62–64]. (C) FST and Pi (π) ratio of MITF gene on chromosome 22
In the goat reference genome, we found seven transcripts of MITF genes: X1, X2, X3, X4, and X5-1–3. transcriptomic analysis showed high MITF expression in the skin of both cashmere goat breads, but the average MITF expression level in the skin of Tibetan cashmere goats was higher than in Jiangnan cashmere goats. We further analyzed the expression characteristics of the seven MITF transcripts in Jiangnan and Tibetan cashmere goat skin tissue; the expression levels of X2 (XM_005695740.3), X5-1 (XM_018067161.1) and X4 (XM_018067158.1) in Tibetan cashmere goat skin tissue were significantly higher than those in Jiangnan cashmere goats (The log2FC of these 3 transcripts > 2)(Fig. 4A). However, the expression levels of the three downstream genes (TYR, TYRP1, and DCT) regulated by MITF gene were very low (FPKMs of all three genes: 100 ~ 0). Based on selective sweeps analysis, the sequence segment of MITF gene where exons 2, 3, 4, and 5 are located was most obvious selected for environmental adaptation (Fig. 4C). However, TYR, TYRP1 and DCT genes were not selected, and their sequence structures may be relatively conserved among different breeds of cashmere goat.
Discussion
Extreme environments (low temperature, low oxygen, and UVR) have adverse effects on the survival of plateau organisms, limiting the development of animal husbandry in plateau areas. The adaptation of indigenous animals like cashmere goats to the extreme plateau environment can help us understand the molecular regulatory mechanisms underlying their independent adaptation to the plateau environment.
During long-term domestication and genetic improvement of Tibetan and Jiangnan cashmere goats, the blood of Liaoning cashmere goat was introduced to improve their reproductive performance. The Tibetan and Jiangnan cashmere goat populations in this study shared a common ancestor. Phylogenetic and PCA analysis of 64 cashmere goats showed that, even if these two breeds may have originated from the same ancestor, long-term environmental adaptation and selection separated them. LD analysis further revealed that the two breeds were subject to varying degrees of natural or artificial selection. Based on population structure analysis and genome-wide gene expression pattern analysis, the environmental differences (particularly regional differences in altitude) and geographic isolation led to differences in genome sequence structure and genome-wide expression patterns between these two breeds. The differences between the two groups indicate that cashmere goats adapted to the extreme environment of the plateau by changing their genome structure and gene expression levels. Selective sweep analysis further identified a total of 553 J vs. T and 608 T vs. J PSGs associated with environmental adaptation; these PSGs may function in different adaptive mechanisms in response to numerous adverse environment factors.
Among the various physiological activities of animals, respiration is the most important for animal survival. However, hypoxia is one of the most significant features of the Tibetan plateau, which will inevitably affect animal breathing. Based on previous studies, the physiological and morphological characteristics of different animals’ adaptation to hypoxia are different. Some plateau native animals have strong cardiopulmonary functions and dense capillaries, such as yak, plateau bar-headed geese, and Tibetan antelope. Studies have also shown that the plateau pika responds to the cold and hypoxic plateau environment by maintaining a high resting metabolic rate, non-shivering heat production, and improving oxygen utilization [22]. The adaptation of deer mice to the plateau is achieved by increasing the blood oxygen affinity. The studies have shown that increased oxygen affinity of deer mice with the altitude increase and is related to the distribution frequency of alleles at different altitudes [23]. In our study, three PSGs were related to regulation of the heart contraction force (including LOC106502520, ATP2A2, and LOC102181869) and four PSGs related to ventricular cardiac muscle tissue morphogenesis (Including LOC106502520, MYL2, ISL1, and LOC102181869) were selected in Tibetan cashmere goats; the mutations in the above genes may enhance the myocardial function of Tibetan cashmere goats.
Another environmental characteristic of the plateau is the cold; the thick hair on the animal’s body helps resisting the cold. Our study found that, compared with Jiangnan cashmere goats, Tibetan cashmere goats have finer cashmere. The mathematical theory of reaction diffusion proposed by Nagorcka et al. [24] considered hair follicle size as ultimate determinant of the volume and fiber diameter of the dermal papilla. Moore’s progenitor cell theory also supports this view [25]. Moreover, Adelson et al. found that fiber diameter and hair follicle density are inversely proportional [26]. Based on the above results, we consider that Tibetan cashmere goats may have smaller and denser hair follicles, which play a positive role in resisting the cold and UVR. In follow-up GWAS, a few SNPs associated with cashmere fineness traits (MFD, CVFD, and FDSD) were identified; the candidate genes obtained by SNP annotation were not directly associated with hair follicle or hair growth. In selective sweep analysis, we found PSGs enriched in positive regulation of Notch signaling pathway (involving the PSGs YAP1, POGLUT1, AAK1, HES1, and WNT1), Wnt signaling pathway (involving the PSGs PRKAA1, TNKS, WNT5A, VAX2, RSPO4, and CSNK1G1), and PI3K-Akt signaling pathway (Involving the PSGs PHLPP2, CHRM2, PDGFRB, PRKAA1, MAP2K1, IRS1, LPAR1, PTEN, PRLR, IBSP, CCNE2, CHAD, ITGB7, TEK, JAK2, and FGF21) were affected by environmental adaptive selection. Demehri et al. [27] found that ligand binding to Notch receptors will activate the hair follicle stem cells, so that the hair follicles enter the growth phase. Andl et al. [28] found that WNT signals are required for the initiation of hair follicle development. Lu et al. [29] found that amphiregulin promotes hair regeneration of skin-derived precursors via PI3K pathways. Therefore, we speculate that a mutation in the above PSGs may regulate differences in hair follicle development between the two cashmere goat breeds, thereby affecting cashmere fineness traits. Meanwhile, we found that the expression levels of KRT and KAP genes in the skin tissue of Tibetan cashmere goats were significantly lower than those of Jiangnan cashmere goats, which may be another key factor determining cashmere fineness.
The extreme climate increases the risk of cancer in plateau animals, such as gastric or skin cancer [30–32]. Among them, the increase of animal skin cancer caused by the increased solar UVR has attracted global attention; this also poses a serious threat to natural ecology, especially reproduction. The skin is the largest protective organ, and the melanin in it is the main component shielding UVR [33]. Melanin is secreted, generated, and stored by melanocytes, mainly distributed in the epidermis and dermis and subcutis [34, 35], and its quantity is related to biological species, location, age, etc. Melanin prevents skin and organ damage by absorbing UV photons and free radicals [36] to protect DNA from UV damage [37]. In this study, MITF and KITLG were environmental adaptive and selected; these two genes are closely related to melanin biosynthesis and transport. The expression level of three MITF transcripts in the skin tissue of Tibetan cashmere goat was higher than in Jiangnan cashmere goats. Hence, we speculate that sequence mutation due to environmental adaptive selection may be an important factor affecting MITF gene expression. However, we noticed that TYR, TYRP1, and DCT, which are directly involved in melanin synthesis had low expression levels, directly regulated by MITF. In addition, these three genes were not subjected to strong environmental adaptive selection. Therefore, we speculate that the maturation period (Sampling period) may not be critical for melanin synthesis and deposition in the skin tissues of the two cashmere goat breeds. At the same time, we have no direct evidence of a significant difference in melanin deposition in the skin tissues between the two breeds. Nonetheless, we do not exclude environmental adaptation mutations in the MITF and KITLG at different growth stages between the two cashmere goat breeds.
In addition to the above-mentioned regulatory mechanisms in response to cancer, we found that PSGs (J vs. T PSGs: KITLG, UBE2R2, PIGU, NOL4L, STK3, DIABLO, ADGRG1, MAP4, CDC25A, DSG3, and MITF) with Pi (π) ratio > 0.5 and FST > 0.4 were more or less associated with cancer (For example: UBE2R2 gene–cervical cancer [38]; PIGU and DIABLO genes–colorectal cancer [39, 40]; NOL4L, STK3 and MAP4 genes–ovarian cancer [41–43]; ADGRG1 gene–tumorigenesis [44]; CDC25A gene–human tumors [45]; DSG3 gene–head and neck cancer [46]; LEPR, PRKAA1, and IKBKB genes–gastric cancer [47–49]; ABCG2 gene–breast cancer [50]). GO enrichment analysis also found that five PSGs (including AREL1, TNIP1, DUOXA1, DUOXA2, and JAK2) associated with inflammatory response conditions were also selected for environmental adaptation. In summary, we speculate that, the environmental adaptive selection in PSGs related to diseases such as cancer is the most obvious.
Conclusion
In this study, we identified the 553 J vs. T and 608 T vs. J PSGs in cashmere goats, which are mainly associated with carcinogenesis, pigmentation, myocardial tissue development, and hair growth. Meanwhile, the specific functions of these PSGs still deserve further study. In addition, we found that the plateau adaptation of cashmere goats was also closely related to the genome-wide gene expression pattern. For example, compared with the Jiangnan cashmere goat, the cashmere of the Tibetan cashmere goat is significantly finer, which may be because the expression levels of KRT and KAP gene family members in the skin tissue of the Jiangnan cashmere goat were significantly lower than that of the Tibetan cashmere goat.
Materials and methods
Experimental animals
A total of 32 1-year-old female Jiangnan cashmere goats were selected from the breeding center of Wenshu County, Aksu Prefecture, Xinjiang Province. They belonged from the Aerken group (Numbers(n) = 8), Samusak group (n = 8), Tuniazi group (n = 8), and Yiming group (n = 8). A total of 32 1-year-old female Tibetan cashmere goats were selected from the original breed of cashmere goats in Ngari Prefecture (Ritu County, n = 9; Gaize County, n = 11) and Nagqu Prefecture (Nima County, n = 12)(Fig. 1A). Before the experiment, all cashmere goats were healthy and raised by grazing.
Sample collection
Cashmere samples were collected from Jiangnan and Tibetan cashmere goats from the 10 cm posterior margin of the scapula above the body’s left midline. Cashmere is naturally dried after washing according to a conventional process. A fiber diameter optical analyzer (OFDA2000) was used to determine the cashmere mean fiber diameter (MFD)(Table S5), fiber diameter standard deviation (FDSD), and coefficient of variation of fiber diameter (CVFD) under a constant temperature of 20℃ ± 2℃ and humidity of 65% ± 4%.
Together with the cashmere sample, we collected 5 mL of blood in an anticoagulation tube and stored it in a refrigerator at − 20℃. The cashmere mean fiber diametkit (TIANGENG, USA) was used to extract DNA from cashmere goats. DNA quality and concentration were determined using 1.0% agarose gel electrophoresis and the Qubit 2.0 (Thermo, USA). The results of agarose gel electrophoresis of 64 DNA samples are showed in Figure S5.
Whole-genome resequencing
A total of 64 DNA samples, including 32 Jiangnan cashmere goats and 32 Tibetan cashmere goats, were analyzed. Sequencing library construction was performed as follows: 1 µg of genomic DNA was randomly cut with Covaris; then, the fragments were screened with Agent AMPure XP-Medium Kit with an average size of 200–400 bp. The 64 single strand circle DNAs were formatted as final qualified libraries, which were sequenced at 10× average depth by BGISEQ-500.
Variant discovery and genotyping
High-quality sequencing data of 64 cashmere goats were mapped to the goat reference genome (ARS1.2, GCA_001704415.2) by using BWA software (Parameter: mem-t 4-M) [51]; the mapped results were deduplicated using the SAMTOOLS software (Parameter: rmdup) [52]. GATK software [53] was used to detect SNPs in multiple samples; high-quality SNPs were obtained by filtering and screening under the following conditions:
1) Supporting number (depth of coverage) of SNPs ≥ 2.
2) The proportion of MIS (missing) < 10%.
3) Minimum allele frequency > 5%.
Principal component and phylogenetic analysis
Principal component analysis (PCA) was based on all SNPs using GCTA software [54]. Subsequently, the Treebest-1.9.2 software (http://treesoft.sourceforge.net/index.shtml) was used to calculate the distance matrix also based on the SNPs of individuals. The distance between two individuals i and j was calculated by the following formula:
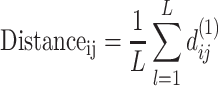 |
L in the formula is the length of the region of high quality SNPs, assuming that the allele at position 1 is A/C, then:
If the genotypes of both individuals are AA:
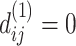
If the genotypes of two individuals are AA and AC respectively:
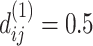
If the genotypes of both individuals are AC:
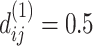
If the genotypes of two individuals are AA and CC respectively:
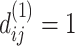
And on this basis a phylogenetic tree was constructed using the neighbor-joining method. Bootstrap values were obtained after up to 1000 calculations.
Structural analysis
We estimated the ancestry of each individual using the genome-wide unlinked SNP dataset and the model-based assignment software program ADMIXTURE [55] to quantify genome-wide admixture between Jiangnan and Tibetan cashmere goats. ADMIXTURE was run for each possible group number (K = 2, 3, and 4) with 200 bootstrap replicates to estimate the parameter standard errors used to determine the optimal group number (K).
Linkage disequilibrium (LD) analysis
We separated the 64 samples by breed and cashmere fineness, and used Haploview software [56] to calculate the r2 for each of the four populations (Fine cashmere (Fe) group: JF and TF; Coarse cashmere (Ce) group: JC and TC). It is generally accepted that there is a linkage between SNPs in the range of r2≥r2MAX/2. An r2 value of 0.175 was used for subsequent analysis. When the r2 value is 0.175, the distance is 20 Kb.
Genome scanning for potential selected genes
To identify the potential selected genes (PSGs) that have undergone environmental adaptive selection and investigate the differences between cashmere goat breeds, we measured the allele frequency (FST) and the genetic diversity (Pi (π)) ratio was compared using a sliding window (5 kb window) by using VCFtools. Meanwhile, 5% of windows with the highest FST and Pi (π) ratio were considered candidate divergent windows, thereby becoming the potential selected region. The Pi (π) ratio between Jiangnan and Tibetan cashmere goat or between Tibetan and Jiangnan cashmere goatswas calculated as log2(Pi ratio(PiJ/PiT)) or log2(Pi ratio(PiT/PiJ)), reflecting the loss of nucleotide diversity in Jiangnan cashmere goat relative to Tibetan cashmere goat or Tibetan cashmere goat relative to Jiangnan cashmere goat.
Genome-wide association analysis
Genome-wide association analysis (GWAS) of cashmere fineness traits (including MFD, FDSD and CVFD) in the goat population (including 32 Jiangnan and 32 Tibetan cashmere goats) was performed using GEMMA [57], using the significance (P-value) of the association to screen out potential candidate SNPs. In this study SNPs with -log10(P)>4 were considered significant SNPs. During GWAS, individual relationship and population stratification were the main factors causing false-positive associations. Therefore, a mixed linear model was used for GWAS, using a population genetic structure as a fixed effect and individual relationships as a random effect to correct for the effects of population structure and individual relationships:
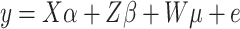 |
where y is the phenotypic trait, X is the indicator matrix of the fixed effect, α is the estimated parameter of the fixed effect; Z is the indicator matrix of the SNP, β is the effect of the SNP; W is the only matrix of the random effect, µ is the predicted random individual, and e is the random residual, obeying e~(0, δe2).
Transcriptomic analysis
The 16 goat transcriptomes were obtained from the NCBI of SRA (https://www.ncbi.nlm.nih.gov/sra/?term=), including eight mature female Jiangnan (BioProject: PRJNA778726) and eight mature female Tibetan (BioProject: PRJNA643003) cashmere goat skin tissue transcriptomes. SRA Toolkit was used to download these data. The clean reads of the 16 transcriptomes were mapped to the goat reference genome (ARS1.2, GCA_001704415.2) using HISAT 2.0 software [58]; the mapping rates of all samples were > 95%. Then, transcript assembly and quantification were performed using StringTie software [59], and fragments per kilobase million (FPKM) was used as a measure of transcript or gene expression levels. The genome-wide gene expression pattern analysis of the 16 samples was performed using WGCNA software [60]. Finally, differentially expressed gene (DEG) analysis was performed using DESeq 2.0 [61].
Functional enrichment analysis of candidate genes
GO and KEGG annotation of candidate genes was performed using the online tool DAVID (https://david.ncifcrf.gov/) with default parameters. The GO annotation is divided into three levels: Biological process, Cellular component, and Molecular function.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
Not applicable.
Author Contribution
C. W., S. M., and X. F.: Methodology, Writing-Original Draft. C.W., S.M., B.Z., and C.Q.: Formal analysis, investigation. L.S., S.M., and J.D.: Validation, Data Curation. C.W., S.M., B.Z., and Y.W.: Writing-Review Editing, Conceptualization. X.F. and C.W.: Supervision, Project administration. All authors have read and approved the final version of the manuscript.
Funding
This work was supported by the National Key R&D Program of China (Grant No. 2021YFD1200902), the Xinjiang Uygur Autonomous Region innovation environment (talent, base) construction project (2021D04008, 2020Q035).
Data Availability
All the genome sequencing data have been deposited in the SRA of NCBI. PRJNA897713: Genome resequencing data of Tibetan cashmere goats.
PRJNA898437: Genome resequencing data of Jiangnan cashmere goats.
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was carried out in compliance with the ARRIVE guidelines. Sample collection was carried out under license in accordance with the Guidelines for Care and Use of Laboratory Animals of China and all study was approved by the Animal Care and Use Committee of Xinjiang Academy of Animal Science (Approval number 2020008 and 2019009).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Cuiling Wu and Shengchao Ma contributed equally to this work and should be considered co-first authors.
Contributor Information
Langda Suo, Email: suolangda163@163.com.
Xuefeng Fu, Email: fuxuefeng@xjaas.net.
References
- 1.Budke CM, Jiamin QIU, Qian W, et al. Economic effects of echinococcosis in a disease-endemic region of the Tibetan Plateau. Am J Trop Med Hyg. 2005;73(1):2–10. doi: 10.4269/ajtmh.2005.73.2. [DOI] [PubMed] [Google Scholar]
- 2.Li W, Cao Y, Fu S, et al. Tahyna virus infection, a neglected arboviral disease in the Qinghai-Tibet Plateau of China. Vector-Borne and Zoonotic Diseases. 2014;14(5):353–7. doi: 10.1089/vbz.2013.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hong W, Hong W, Weida L, et al. Skin reflectance in the Han and Tibetan nationality in China. Chin J Dermatology. 2000;33(4):257–8. [Google Scholar]
- 4.Mitra SS, Ghosh SK, Chatterjea JB. Activity of pyruvate kinase (PK) and stability of ATP in heterozygous and homozygous states for Hb-E. Bull Calcutta School Trop Med. 1968;16(4):103–4. [PubMed] [Google Scholar]
- 5.Scott GR, Egginton S, Richards JG, et al. Evolution of muscle phenotype for extreme high altitude flight in the bar-headed goose. Proc Royal Soc B: Biol Sci. 2009;276(1673):3645–53. doi: 10.1098/rspb.2009.0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3(5):418–26. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 7.Storz JF, Moriyama H. Mechanisms of hemoglobin adaptation to high altitude hypoxia. High Alt Med Biol. 2008;9(2):148–57. doi: 10.1089/ham.2007.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flowers JM, Sezgin E, Kumagai S, et al. Adaptive evolution of metabolic pathways in Drosophila. Mol Biol Evol. 2007;24(6):1347–54. doi: 10.1093/molbev/msm057. [DOI] [PubMed] [Google Scholar]
- 9.Qiu Q, Zhang G, Ma T, et al. The yak genome and adaptation to life at high altitude. Nat Genet. 2012;44(8):946–9. doi: 10.1038/ng.2343. [DOI] [PubMed] [Google Scholar]
- 10.Ge RL, Cai Q, Shen YY, et al. Draft genome sequence of the tibetan antelope. Nat Commun. 2013;4(1):1–7. doi: 10.1038/ncomms2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qu Y, Zhao H, Han N, et al. Ground tit genome reveals avian adaptation to living at high altitudes in the tibetan plateau. Nat Commun. 2013;4(1):1–9. doi: 10.1038/ncomms3071. [DOI] [PubMed] [Google Scholar]
- 12.Somero GN. Linking biogeography to physiology: evolutionary and acclimatory adjustments of thermal limits. Front Zool. 2005;2(1):1–9. doi: 10.1186/1742-9994-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang L, Wang Y, Zhang Z, et al. Comprehensive transcriptomic analysis reveals accelerated genic evolution in a Tibet fish, Gymnodiptychus pachycheilus. Genome Biol Evol. 2015;7(1):251–61. doi: 10.1093/gbe/evu279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tong C, Lin Y, Zhang C, et al. Transcriptome-wide identification, molecular evolution and expression analysis of toll-like receptor family in a Tibet fish, Gymnocypris przewalskii. Fish Shellfish Immunol. 2015;46(2):334–45. doi: 10.1016/j.fsi.2015.06.023. [DOI] [PubMed] [Google Scholar]
- 15.Gong H, Zhou H, McKenzie GW, et al. An updated nomenclature for keratin-associated proteins (KAPs) Int J Biol Sci. 2012;8(2):258. doi: 10.7150/ijbs.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou H, Gong H, Yan W, et al. Identification and sequence analysis of the keratin-associated protein 24-1 (KAP24-1) gene homologue in sheep. Gene. 2012;511(1):62–5. doi: 10.1016/j.gene.2012.08.049. [DOI] [PubMed] [Google Scholar]
- 17.Picardo M, Cardinali G. The genetic determination of skin pigmentation: KITLG and the KITLG/c-Kit pathway as key players in the onset of human familial pigmentary diseases. J Invest Dermatology. 2011;131(6):1182–5. doi: 10.1038/jid.2011.67. [DOI] [PubMed] [Google Scholar]
- 18.Beleza S, Santos AM, McEvoy B, et al. The timing of pigmentation lightening in Europeans. Mol Biol Evol. 2013;30(1):24–35. doi: 10.1093/molbev/mss207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hemesath TJ, Steingrímsson E, McGill G, et al. Microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 1994;8(22):2770–80. doi: 10.1101/gad.8.22.2770. [DOI] [PubMed] [Google Scholar]
- 20.Bentley NJ, Eisen T, Goding CR. Melanocyte-specific expression of the human tyrosinase promoter: activation by the microphthalmia gene product and role of the initiator. Mol Cell Biol. 1994;14(12):7996–8006. doi: 10.1128/mcb.14.12.7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yasumoto K, Yokoyama K, Shibata K, et al. Microphthalmia-associated transcription factor as a regulator for melanocyte-specific transcription of the human tyrosinase gene. Mol Cell Biol. 1994;14(12):8058–70. doi: 10.1128/mcb.14.12.8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ge RL, Kubo K, Kobayashi T, et al. Blunted hypoxic pulmonary vasoconstrictive response in the rodent Ochotona curzoniae (pika) at high altitude. Am J Physiol Heart Circ Physiol. 1998;274(5):H1792–9. doi: 10.1152/ajpheart.1998.274.5.H1792. [DOI] [PubMed] [Google Scholar]
- 23.Storz JF, Sabatino SJ, Hoffmann FG, et al. The molecular basis of high-altitude adaptation in deer mice. PLoS Genet. 2007;3(3):e45. doi: 10.1371/journal.pgen.0030045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagorcka BN. The reaction-diffusion (RD) theory of wool (hair) follicle initiation and development. II. Original secondary follicles. Aust J Agric Res. 1995;46(2):357–78. doi: 10.1071/AR9950357. [DOI] [Google Scholar]
- 25.Moore GPM, Jackson N, Isaacs K, et al. Pattern and morphogenesis in skin. J Theor Biol. 1998;191(1):87–94. doi: 10.1006/jtbi.1997.0567. [DOI] [PubMed] [Google Scholar]
- 26.Adelson DL, Hollis DE, Brown GH. Wool fibre diameter and follicle density are not specified simultaneously during wool follicle initiation. Aust J Agric Res. 2002;53(9):1003–9. doi: 10.1071/AR01200. [DOI] [Google Scholar]
- 27.Demehri S, Kopan R. Notch signaling in bulge stem cells is not required for selection of hair follicle fate. Development. 2009;136(6):891–6. doi: 10.1242/dev.030700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andl T, Reddy ST, Gaddapara T, et al. WNT signals are required for the initiation of hair follicle development. Dev Cell. 2002;2(5):643–53. doi: 10.1016/S1534-5807(02)00167-3. [DOI] [PubMed] [Google Scholar]
- 29.Lu Q, Gao Y, Fan Z, et al. Amphiregulin promotes hair regeneration of skin-derived precursors via the PI3K and MAPK pathways. Cell Prolif. 2021;54(9):e13106. doi: 10.1111/cpr.13106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lerebours A, Chapman EC, Sweet MJ, et al. Molecular changes in skin pigmented lesions of the coral trout Plectropomus leopardus. Mar Environ Res. 2016;120:130–5. doi: 10.1016/j.marenvres.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 31.Work TM, Aeby GS. Skin pathology in H awaiian goldring surgeonfish, C tenochaetus strigosus (B ennett) J Fish Dis. 2014;37(4):357–62. doi: 10.1111/jfd.12112. [DOI] [PubMed] [Google Scholar]
- 32.Jiang J, Zhao JH, Wang XL, et al. Analysis of mitochondrial DNA in tibetan gastric cancer patients at high altitude. Mol Clin Oncol. 2015;3(4):875–9. doi: 10.3892/mco.2015.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitchell DL, Paniker L, Douki T. DNA damage, repair and photoadaptation in a Xiphophorus fish hybrid. Photochem Photobiol. 2009;85(6):1384–90. doi: 10.1111/j.1751-1097.2009.00591.x. [DOI] [PubMed] [Google Scholar]
- 34.Rawls JF, Mellgren EM, Johnson SL. How the zebrafish gets its stripes. Dev Biol. 2001;240(2):301–14. doi: 10.1006/dbio.2001.0418. [DOI] [PubMed] [Google Scholar]
- 35.Zarnescu O. Ultrastructure of the skin melanophores and iridophores in paddlefish, Polyodon spathula. Micron. 2007;38(1):81–4. doi: 10.1016/j.micron.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 36.Wu GM. Studying on Genes Related with Melanin in Black Chicken.PhD Thesis.Hunan Agricultural University. China; 2003.
- 37.Cesarini JP. Melanins and their possible roles through biological evolution. Adv Space Res. 1996;18(12):35–40. doi: 10.1016/0273-1177(96)00025-7. [DOI] [Google Scholar]
- 38.Liu C, Li H, Yin Q. The lncRNA UBE2R2-AS1 suppresses cervical cancer cell growth in vitro. Open Med. 2020;15(1):1184–92. doi: 10.1515/med-2020-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang M, Wang H, Li H, et al. Identification of PIGU as the hub gene associated with KRAS mutation in colorectal cancer by coexpression analysis. DNA Cell Biol. 2020;39(9):1639–48. doi: 10.1089/dna.2020.5574. [DOI] [PubMed] [Google Scholar]
- 40.Endo K, Kohnoe S, Watanabe A, et al. Clinical significance of Smac/DIABLO expression in colorectal cancer. Oncol Rep. 2009;21(2):351–5. [PubMed] [Google Scholar]
- 41.Lin F, Zhou J, Li X, et al. NOL4L, a novel nuclear protein, promotes cell proliferation and metastasis by enhancing the PI3K/AKT pathway in ovarian cancer. Biochem Biophys Res Commun. 2021;559:121–8. doi: 10.1016/j.bbrc.2021.04.055. [DOI] [PubMed] [Google Scholar]
- 42.Wang X, Wang F, Zhang ZG et al. STK3 suppresses ovarian cancer progression by activating NF-κB signaling to recruit CD8 + T-cells. Journal of Immunology Research, 2020; 2020. [DOI] [PMC free article] [PubMed]
- 43.Yang H, Mao W, Rodriguez-Aguayo C, et al. Paclitaxel Sensitivity of Ovarian Cancer can be enhanced by knocking down pairs of Kinases that regulate MAP4 phosphorylation and Microtubule StabilityCombinatorial siRNA Therapy enhances Paclitaxel Sensitivity. Clin Cancer Res. 2018;24(20):5072–84. doi: 10.1158/1078-0432.CCR-18-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhat R, Abdulkareem NM, Yasser H, et al. ADGRG1 promotes tumorigenesis, invasion/migration, and cell-cell adhesion in triple-negative breast cancer cells. Cancer Res. 2020;80(16Supplement):5148–8. doi: 10.1158/1538-7445.AM2020-5148. [DOI] [Google Scholar]
- 45.Kang T, Wei Y, Honaker Y, et al. GSK-3β targets Cdc25A for ubiquitin-mediated proteolysis, and GSK-3β inactivation correlates with Cdc25A overproduction in human cancers. Cancer Cell. 2008;13(1):36–47. doi: 10.1016/j.ccr.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen YJ, Chang JT, Lee L, et al. DSG3 is overexpressed in head neck cancer and is a potential molecular target for inhibition of oncogenesis. Oncogene. 2007;26(3):467–76. doi: 10.1038/sj.onc.1209802. [DOI] [PubMed] [Google Scholar]
- 47.Yu H, Pan R, Qi Y, et al. LEPR hypomethylation is significantly associated with gastric cancer in males. Exp Mol Pathol. 2020;116:104493. doi: 10.1016/j.yexmp.2020.104493. [DOI] [PubMed] [Google Scholar]
- 48.Dargiene G, Streleckiene G, Skieceviciene J et al. TLR1 and PRKAA1 gene polymorphisms in the development of atrophic gastritis and gastric cancer. J Gastrointest Liver Dis, 2018; 27(4). [DOI] [PubMed]
- 49.Gong Y, Zhao W, Jia Q, et al. IKBKB rs2272736 is associated with gastric cancer survival. Pharmacogenomics and Personalized Medicine. 2020;13:345. doi: 10.2147/PGPM.S258761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Staud F, Pavek P. Breast cancer resistance protein (BCRP/ABCG2) Int J Biochem Cell Biol. 2005;37(4):720–5. doi: 10.1016/j.biocel.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 51.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McKenna A, Hanna M, Banks E, et al. The genome analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang J, Lee SH, Goddard ME, et al. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–64. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barrett JC, Fry B, Maller J, et al. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 57.Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44(7):821–4. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim D, Paggi JM, Park C, et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37(8):907–15. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pertea M, Pertea GM, Antonescu CM, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9(1):1–13. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):1–21. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28(11):1947–51. doi: 10.1002/pro.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kanehisa M, Furumichi M, Sato Y, et al. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49(D1):D545–51. doi: 10.1093/nar/gkaa970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the genome sequencing data have been deposited in the SRA of NCBI. PRJNA897713: Genome resequencing data of Tibetan cashmere goats.
PRJNA898437: Genome resequencing data of Jiangnan cashmere goats.