

Abstract
In the over 100 years since the recognition of pulmonary hypertension (PH), immense progress and significant achievements have been made with regard to understanding the pathophysiology of the disease and its treatment. These advances have been mostly in idiopathic pulmonary arterial hypertension (IPAH), which was classified as Group 1 Pulmonary Hypertension (PH) at the Second World Symposia on PH in 1998. However, the pathobiology of PH due to chronic lung disease, classified as Group 3 PH, remains poorly understood and its treatments thus remain limited. We review the history of the classification of the five groups of PH and aim to provide a state-of-the-art review of the understanding of the pathogenesis of Group 1 PH and Group 3 PH including insights gained from novel high-throughput omics technologies that have revealed heterogeneities within these categories as well as similarities between them. Leveraging the substantial gains made in understanding the genomics, epigenomics, proteomics, and metabolomics of PAH to understand the full spectrum of the complex, heterogeneous disease of PH is needed. Multimodal omics data as well as supervised and unbiased machine learning approaches after careful consideration of the powerful advantages as well as of the limitations and pitfalls of these technologies could lead to earlier diagnosis, more precise risk stratification, better predictions of disease response, new sub-phenotype groupings within types of PH, and identification of shared pathways between PAH and other types of PH that could lead to new treatment targets.
Introduction
In the over 100 years since the recognition of pulmonary hypertension (PH), immense progress and significant achievements have been made with regard to understanding the pathophysiology of the disease (22, 234). These advances have been mostly in idiopathic pulmonary arterial hypertension (IPAH), which for almost 100 years was known as primary pulmonary hypertension (PPH) and was the paradigmatic disease in the field of PH. Beginning with the first World Health Organization (WHO) Symposium on PH in Geneva in 1973, clinicians and researchers attempted to define PH using hemodynamic parameters while delineating distinct categories of PH according to histopathology and presumed mechanism, at that time describing three patterns: plexiform pulmonary arteriopathy, pulmonary veno-occlusive disease, and chronic thromboembolic disease (71). Twenty years later at the Second World Symposia on PH, a 5-group classification of PH was established (Table 1) (58). Pulmonary arterial hypertension (PAH), termed Group 1 PH, was defined as a resting mean pulmonary artery pressure (mPAP) greater than or equal to 25 mmHg, a pulmonary capillary wedge pressure (PCWP) less than or equal to 15 mmHg, and a pulmonary vascular resistance (PVR) greater than or equal to 3 Wood units, and was characterized histopathologically by concentric laminar intimal proliferation of medium and small sized pulmonary arteries and plexiform lesions; Group 2 encompassed patients with PH due to left heart disease; Group 3 comprised PH associated with respiratory disorders or hypoxemia, Group 4 was defined as chronic thromboembolic pulmonary hypertension (CTEPH), and Group 5 PH included other diseases known to alter the pulmonary vasculature and promote PH (167). Since then, four additional World Symposia have been held, most recently in Nice in 2018, each time incorporating advances in the understanding of the clinical presentation, pathophysiology, and pathology of PH including the identification of genetic and familial causes, adding to an ever-growing list of diseases categorized as Group 1 PH.
Table 1.
World Symposia on Pulmonary Hypertension—Classification of PH
| Group 1 PH |
| Idiopathic PAH |
| Heritable PAH |
| Drug- and toxin-induced PAH |
| PAH associated with: |
| Connective tissue disease |
| HIV infection |
| Portal hypertension |
| Congenital heart disease |
| Schistosomiasis |
| PAH with long-term response to calcium channel blockers |
| PAH with overt features of venous/capillaries involvement (PVOD/PCH) |
| Persistent PH of the newborn syndrome |
| Group 2 PH due to left heart disease |
| PH due to heart failure with preserved LVEF |
| PH due to heart failure with reduced LVEF |
| Valvular heart disease |
| Congenital/acquired cardiovascular conditions leading to postcapillary PH |
| Group 3 PH due to lung disease and/or hypoxia |
| Obstructive lung disease |
| Restrictive lung disease |
| Other lung disease with mixed restrictive/obstructive pattern |
| Hypoxia without lung disease including high altitude |
| Developmental lung disorders |
| Group 4 PH due to pulmonary artery obstructions |
| Chronic thromboembolic PH (CTEPH) |
| Other pulmonary artery obstructions |
| Group 5 PH with unclear and/or multifactorial mechanisms |
| Hematologic disorders |
| Systemic and metabolic disorders |
| Others |
| Complex congenital heart disease |
Definition of abbreviations: PAH, pulmonary arterial hypertension; PVOD, pulmonary vascular occlusive disease; PCH, pulmonary capillary hemangiomatosis; LVEF, left ventricular ejection fraction.
Adapted, with permission, from Simmoneau G, et al., 2019 (58). Reproduced with permission of the ERS Copyright 2022: European Respiratory Journal.
However, there is a growing recognition of the heterogeneity of PAH (176, 204), likely paralleling the significant molecular and clinical heterogeneity of all groups of PH, especially in what is currently termed Group 3 PH. While the modification of the PH classification system is largely driven by the study of IPAH, it is unclear whether the insights gained in IPAH can be extended to other forms of PH, which are more frequent and have a larger impact on morbidity and mortality than IPAH. Shortcomings of the PH classification are further pronounced considering the discrepancies within and limited understanding of the varied entities including hypoxia, chronic obstructive pulmonary disease (COPD), and pulmonary fibrosis all clustered within Group 3 PH, implying a common nature to these disparate entities.
While the development of several pulmonary vasodilator therapies has dramatically improved the quality of life and survival of PAH patients, the mainstay of treatment of Group 3 PH has largely remained oxygen delivery and addressing the underlying cause of hypoxemia. Only one drug, inhaled treprostinil, has been approved by the Food and Drug Administration (FDA) for the treatment of PH due to interstitial lung disease (ILD), and this was derived from existing inhaled vasodilator therapy for PAH (220). There is a pressing unmet need for advances in treatment options for PH in lung disease, where prevalence is high and outcomes are poor, affecting 38% to 80% of idiopathic pulmonary fibrosis (IPF) patients (106, 126, 166) and 25% of patients with mild to moderate COPD, but up to 90% in those with severe Global Initiative for Chronic Obstructive Lung Disease (GOLD) Stage IV COPD (18, 87). PH carries a markedly higher mortality in patients with chronic lung disease compared to those without PH. Alarmingly, some epidemiologic studies have found that Group 3 PH has equally poor if not worse mortality compared to PAH patients despite a lower severity of PH and right ventricle (RV) dysfunction (Figure 1) (50, 138), highlighting the urgent need to understand the pathobiologic underpinnings of group 3 PH and to discover new therapies.
Figure 1.
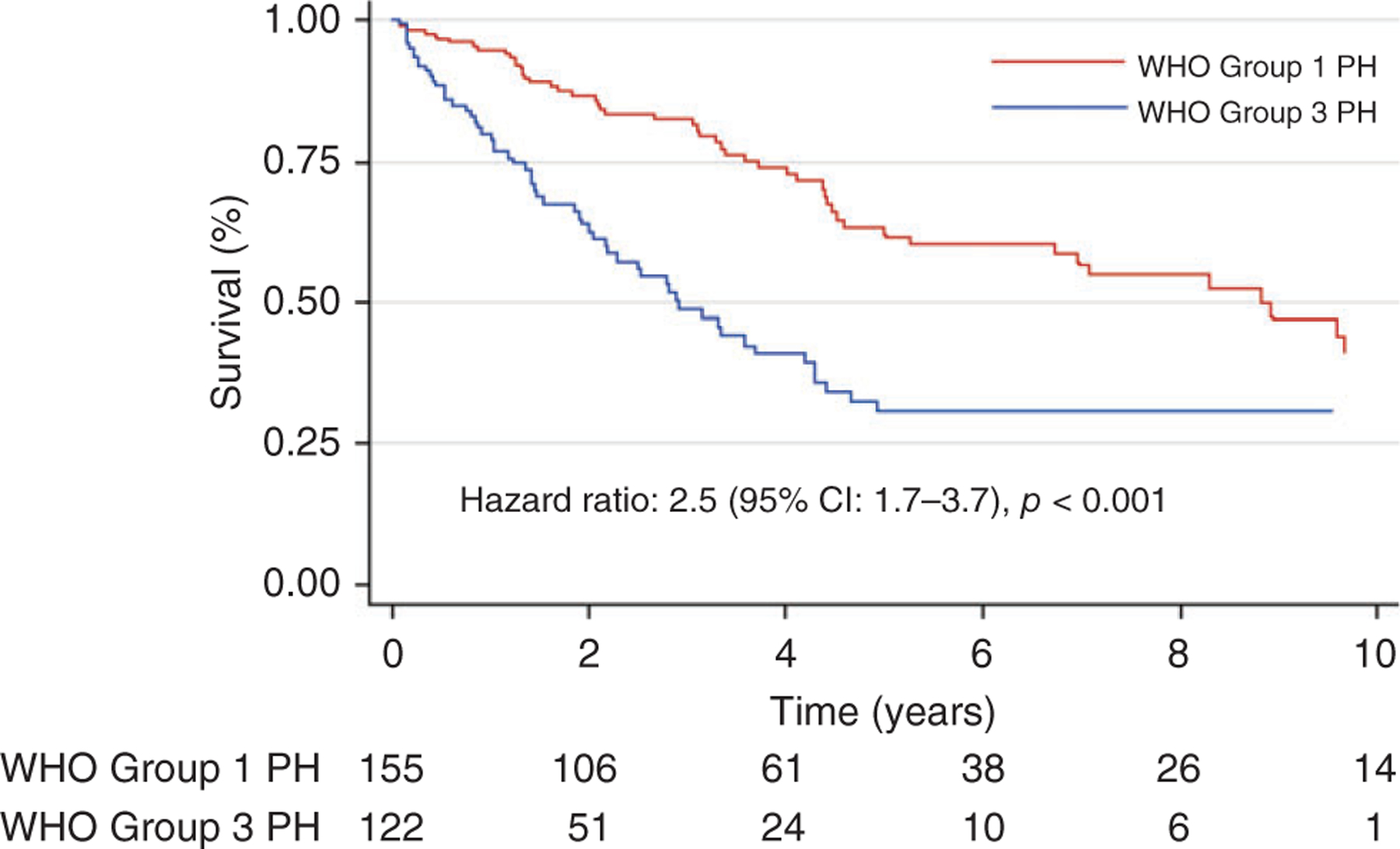
Survival in Group 1 and Group 3 PH. Reused, with permission, from Prins KW, et al., 2018 (138), American Thoracic Society.
The mechanisms contributing to the development of PH in lung diseases remain poorly understood. Hypoxic-mediated vasoconstriction has long been thought to be the primary driver of all forms of group 3 PH; however, PH can develop in both COPD and IPF in the absence of hypoxemia, and correction of hypoxemia with supplemental oxygen therapy only partially reverses PH (9, 31, 49). Other key pathobiological concepts that have been promoted include PH due to the destructive lung process itself and the resultant excessive vasoconstriction and vascular destruction leading to vessel paucity, excessive vascular growth, and inflammation.
The complexity of these paradigms and their potential interactions underlines the difficulty in finding a unifying or prevailing hypothesis in PH associated with chronic lung diseases. The limited understanding of the pathobiology of Group 3 PH may have been the basis of the taxonomy developed in the initial World Symposia, but in this new era of advanced technologies including single-cell sequencing and molecular phenotyping, this classification requires re-examination aimed at revealing how similar these conditions are to one another or to Group 1 PH. Despite the significant impact, inroads, and developments resulting from the recognition of PH as an entity and its initial classification scheme, the continued grouping of dissimilar pathobiologic processes under one umbrella may now impede the ability to study and recognize heterogeneous mechanisms implicated in not only Group 3, but also Group 1 PH, and thus contribute to the paucity of novel therapies for these diseases.
Advances in the genetic characterization of the disease as well as the advent of “omic” technologies have revealed extensive heterogeneity among the Group 1 PH diseases. Machine learning (ML) models have been applied to understand the pathobiology of PAH and have revealed heterogeneities across entities that were once considered to be etiologically homogeneous (86). These findings are consistent with clinical observations that some patients respond to certain pharmacologic therapies, while others do not (118). These approaches have formed the basis for interpreting other forms of PH in the framework of Group 1 PH, but the dangers related to inappropriate translation of these findings to highly complex diseases, such as COPD and pulmonary fibrosis, must be acknowledged.
Our goal is to provide a state-of-the-art review of the understanding of the pathogenesis of Group 1 PH and Group 3 PH, and in so doing will highlight significant knowledge gaps in the underlying molecular pathology of these highly prevalent forms of PH. We will also emphasize new scientific advances in the era of pulmonary vascular multimodal “omics,” using comprehensive high-throughput molecular analyses that can enable more precise phenotyping within the current umbrellas of PH. Given the multidimensionality of new data that can be generated in the present age of molecular medicine, continued adherence to the compartmentalized knowledge of these insights does not capture how best to understand PH across its disease spectrum. These approaches can aid in uncovering previously hidden similarities and key differences in diseases currently residing under different classifications (128). In the future, machine learning and artificial intelligence may provide one path to the discovery of novel, personalized therapies in all forms of PH.
The Normal Pulmonary Vasculature
The healthy pulmonary circulation is a low-pressure, high-compliance system, allowing the RV to accommodate high blood flow (i.e., cardiac output, Q) with minimal work and preventing fluid from escaping the pulmonary vessels into the lung tissue. PVR is related to the pressure and flow through the pulmonary circulation, summarized by the equation PVR = (mPAP – PCWP)/Q. A large meta-analysis performed by Kovacs et al. demonstrated that in healthy individuals resting mPAP typically falls below 20 mmHg (mean 14.0 ± 3.3 mmHg); in the same review, PVR in healthy individuals was 0.93 ± 0.38 WU (97). PVR may also be described by Poiseuille’s law, where PVR is proportional to the product of the volume flow rate, viscosity coefficient, and tube length, and inversely proportional to the tube radius to the fourth power (123). Increased volume flow can be accommodated through pulmonary vascular recruitment and/or distensibility (98). Thus, reduction in pulmonary arteriolar vessel caliber (the radius in Pouiselle’s equation) due to increased vascular tone or remodeling can markedly increase the resistance across the pulmonary vasculature. As will be described in this article, hypoxic pulmonary vasoconstriction and pulmonary vessel wall remodeling in the various diseases of PH increase PVR and thus increase pulmonary arterial pressure.
Pathobiology of Group 1 PH
The age of vasoconstriction
As early as in the 1950s, clinicians and investigators defined PAH using physiologic and hemodynamic descriptions and focused their efforts on understanding its pathogenic mechanisms. A period that could be termed “the age of vasoconstriction” then followed until the early 1990s, during which much work was focused on understanding the functional mechanisms governing pulmonary vascular tone that would drive vasoconstriction and the ensuing pulmonary pathological lesions of PAH. The vasoconstriction paradigm was centered upon the identification of hypoxic vasoconstriction, which led to the discovery of chemical mediators of vasoconstriction or vasodilation, and from these, the development of therapeutic agents that could counter excessive vasoconstriction or promote vasodilation: prostacyclin analogs (219), endothelin receptor blockers (155), and pharmacologic enhancers of endogenously produced nitric oxide (159), all of which remain the mainstays of the modern PAH treatment (80).
A focus on pulmonary vascular remodeling
By the late 1980s, it became increasingly clear that vasoconstriction alone could not completely explain PH pathophysiology, as in many patients there was a “fixed” component of PH due to pulmonary vascular remodeling that was unresponsive to vasodilator therapy (149, 170). Accordingly, the field shifted its focus to study the mechanisms of pulmonary vascular remodeling, initially by identifying the cell types contributing to medial and adventitial thickening, as these were the changes most evident in the hypoxic animal models of the disease which were dominant at the time (117, 180). An overabundance of adventitial fibroblasts and medial smooth muscle cells (SMCs) was consistently observed in these compartments (180). These studies also pointed out that SMCs and adventitial fibroblasts, rather than behaving uniformly, are each phenotypically heterogeneous populations with diverse functions responding in distinct ways to specific stress stimuli (39, 56, 225). However, animal models of chronic PH due to environmental hypoxia (209), or for that matter monocrotaline injection (141), fall short of fully capturing the type and extent of pulmonary vascular remodeling seen in severe forms of human PAH (179, 212). The lungs of most patients with IPAH, the prototypic form of severe PH in humans, demonstrate a complex variety of vascular lesions which cannot be reproduced by chronic environmental hypoxia or monocrotaline. While the newer model of PH involving administration of the vascular endothelial growth factor (VEGF) inhibitor Sugen followed by hypoxic exposure may perhaps model human PAH more closely, it likewise does not recapitulate the cellular complexity of the human condition (2, 188, 192). Further, the relatively short time frame of pulmonary vascular remodeling in hypoxic rodent models (i.e., up to 5–7 weeks, and usually imposed on young, growing animals) is unlikely to capture the diverse and dynamic temporal changes present in the human disease, which at presentation comprises a large age span and has widespread involvement of the pulmonary vascular bed.
The cancer hypothesis of pulmonary hypertension
In the 2000s, newly available technologies opened up possibilities of understanding the molecular, metabolic, and genetic/epigenetic basis of the disease (32, 42, 229), leading to discoveries that framed a new concept, the so-called “cancer-like hypothesis of PH.” This hypothesis, largely based on human studies, addressed mechanistically the pathogenesis of complex vascular lesions in PAH (211). Plexiform lesions in human IPAH lungs were found to be monoclonal, which strongly suggested that stem- or progenitor-like cells could acquire mutations enabling either increased cell proliferation or decreased cell death (102, 202), becoming genetically unstable (5, 51, 230), losing suppressive cell growth signals (such as the transforming growth factor-β family), and activating signaling pathways to favor their long term survival (114). The cancer-like paradigm flourished and enabled the identification of ever more complex signaling pathways involved in cellular metabolism, extracellular matrix (ECM) remodeling, and alterations of the immune system, among others (66, 140, 147, 173, 191). Importantly, work in this area led to many potentially exciting new ideas regarding therapeutic targets (Table 2) (140).
Table 2.
Targeting Signaling Pathways Involved in Cancer Biology for Controlling/Regressing Enhanced Proliferation, Survival, and Apoptosis Resistance of Lung Vascular Cells in Pulmonary Hypertension
| Molecule/Pathway | Cell Type | Status in PAH/Experimental PH | Function | Inhibitor(s)/Activator(s) Tested | Effect of Inhibitor(s)/Activator(s) |
|---|---|---|---|---|---|
| RTKs | |||||
| PDGFR | PASMCs | PASMCs: | PASMCs: | Imatinib | Inhibits proliferation of human IPAH and rat MCT-PH PASMCs (219) |
| PAECs | ↑ Human IPAH | ↑ Proliferation | Reverses MCT-PH in rats (61, 219) | ||
| ↑ Rat HPH | ↓ Apoptosis | Showed efficacy in phase II (59) and III (196) trials | |||
| Improves PVR and 6MWD in patients with severe PAH (196) | |||||
| Nilotinib | Reverses pulmonary vascular remodeling in rat MCT-PH and HPH (61) | ||||
| EGFR | PASMCs | No significant alterations in experimental/clinical PH | PASMCs: ↑ Proliferation |
PKI166 | Mediates apoptosis in PA organ culture (98) Attenuates rat MCT-PH (98) |
| ↓ Apoptosis | Gefitinib | Attenuates rat MCT-PH (127) No significant benefits in mouse HPH (127) |
|||
| Erlotinib | Attenuates rat MCT-PH (127) No significant benefits in mouse HPH (127) |
||||
| Lapatinib | No therapeutic benefit in experimental PH (127) | ||||
| FGFR | PAECs PASMCs |
↑ Human IPAH (FGF-2) ↑ Rat MCT-PH (FGF-2, FGFR1) |
PAECs: ↑ Proliferation PAECs: ↑ Proliferation |
SU5402 | Reverses rat MCT-PH (128) |
| Ras/Raf/MAPK | PAECs | ↑ Rat MCT-PH (p-Raf-1, p-ERK) ↑ Rat HPH ↑ Rat SuHx-PH (p-ERK, p-MEK1/2) |
PAECs: ↑ Proliferation Cardiomyocytes: ↓ Vasopressin-induced hypertrophy |
Sorafenib | Attenuates rat MCT-PH (81) Improves RV function in PAB rats (95) Attenuates rat HPH and SuHx-PH (56) |
| PI3K | PASMCs PAECs |
Unknown | PASMCs: ↑ Mitogen-induced proliferation and migration PAECs: ↑ Proliferation ↓ Apoptosis ↑ NO production and vasodilation |
LY294002 | Inhibits growth factor-induced PASMC proliferation and migration (159) Attenuates development of HPH in rats (17) |
| Akt | PASMCs PAAFs PAECs | PASMCs: ↑ Human IPAH ↑ Rat HPH PAAFs: ↑ Human IPAH ↑ Rat HPH PAECs: Unknown |
PASMCs: ↑ Proliferation ↓ Apoptosis PAAFs: ↑ Proliferation PAECs: ↑ Proliferation ↓ Apoptosis ↑ NO production and vasodilation |
Triciribine | Attenuates development of HPH in rats (17) |
| mTORC1 | PASMCs | ↑ Human IPAH ↑ Rat HPH ↑ Rat MCT-PH |
↑ Proliferation | Rapamycin Everolimus |
Inhibits proliferation of human IPAH PASMCs (97) Inhibits proliferation of rat MCT-PH PASMCs (226) Improves PVR and 6MWD in patients with severe PAH (23) |
| mTORC2 | PASMCs | ↑ Human IPAH ↑ Rat HPH ↑ Rat MCT-PH |
↑ Proliferation ↓ Apoptosis |
PP242 (dual) | Inhibits proliferation, induces apoptosis in human IPAH PASMCs (97) Reverses pulmonary vascular remodeling in rat HPH (97) |
| Notch3/HES | PASMCs | ↑ Human PAH ↑ Mouse HPH |
↓ Differentiation ↑ Proliferation |
DAPT | Inhibits human IPAH growth (118) Reverses mouse HPH (118) |
| FoxOs | PASMCs | ↓ Human PAH ↓ Rat MCT-PH ↓ Rat SuHx-PH |
↑ Proliferation ↓ Apoptosis |
Paclitaxel Abraxane |
Inhibits human IPAH growth (31) Reverses rat MCT-PH (31) Reverses rat SuHx-PH (31, 190) |
Definition of abbreviations: 6MWD, 6-min-walk distance; Akt, v-akt murine thymoma viral oncogene homolog; DAPT, N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester; EGFR, epidermal growth factor receptor; ERK, extracellular signal-regulated kinase; FGF-2, fibroblast growth factor 2; FGFR, fibroblast growth factor receptor; FoxO, forkhead-box class O; HES, hairy/enhancer of split; HPH, hypoxia-induced pulmonary hypertension; IPAH, idiopathic pulmonary arterial hypertension; MAPK, mitogen-activated protein kinase; MCT-PH, monocrotaline-induced PH; MEK, MAPK/ERK kinase; mTOR, mechanistic target of rapamycin; mTORC1, mTOR complex 1; mTORC2, mTOR complex 2; PA, pulmonary artery; PAB, pulmonary artery banding; PAAFs, pulmonary artery adventitial fibroblasts; PAECs, pulmonary artery endothelial cells; PAH, pulmonary arterial hypertension; PASMCs, pulmonary artery smooth muscle cells; PDGFR, platelet-derived growth factor receptor; PH, pulmonary hypertension; PI3K, phosphatidylinositol 3-kinase; PVR, pulmonary vascular resistance; RTK, receptor tyrosine kinase; RV, right ventricular; SuHx, Sugen/hypoxia.
Reused, with permission, from Pullamsetti SS, et al., 2017 (140)/with permission of American Thoracic Society.
However, as occurs with any novel disease hypothesis, contradictions and controversies have arisen, including the recognition that pulmonary vascular cells behave differently than cancer-like cells, including their low proliferation rate, sensitivity to density-dependent inhibition of cell growth, and conservation of cellular features and functions rather than de-differentiation, raising questions about the safety and efficacy of using anti-neoplastic drugs in the context of PAH (Figure 2) (66, 173). Without a critical reassessment and proper understanding of the origins, strengths, and limitations of the cancer hypothesis, there is a risk that early closure and acceptance of this as a “unifying hypothesis” for PAH can limit further advancements in the field through the exploration of new and different paradigms (34, 204).
Figure 2.
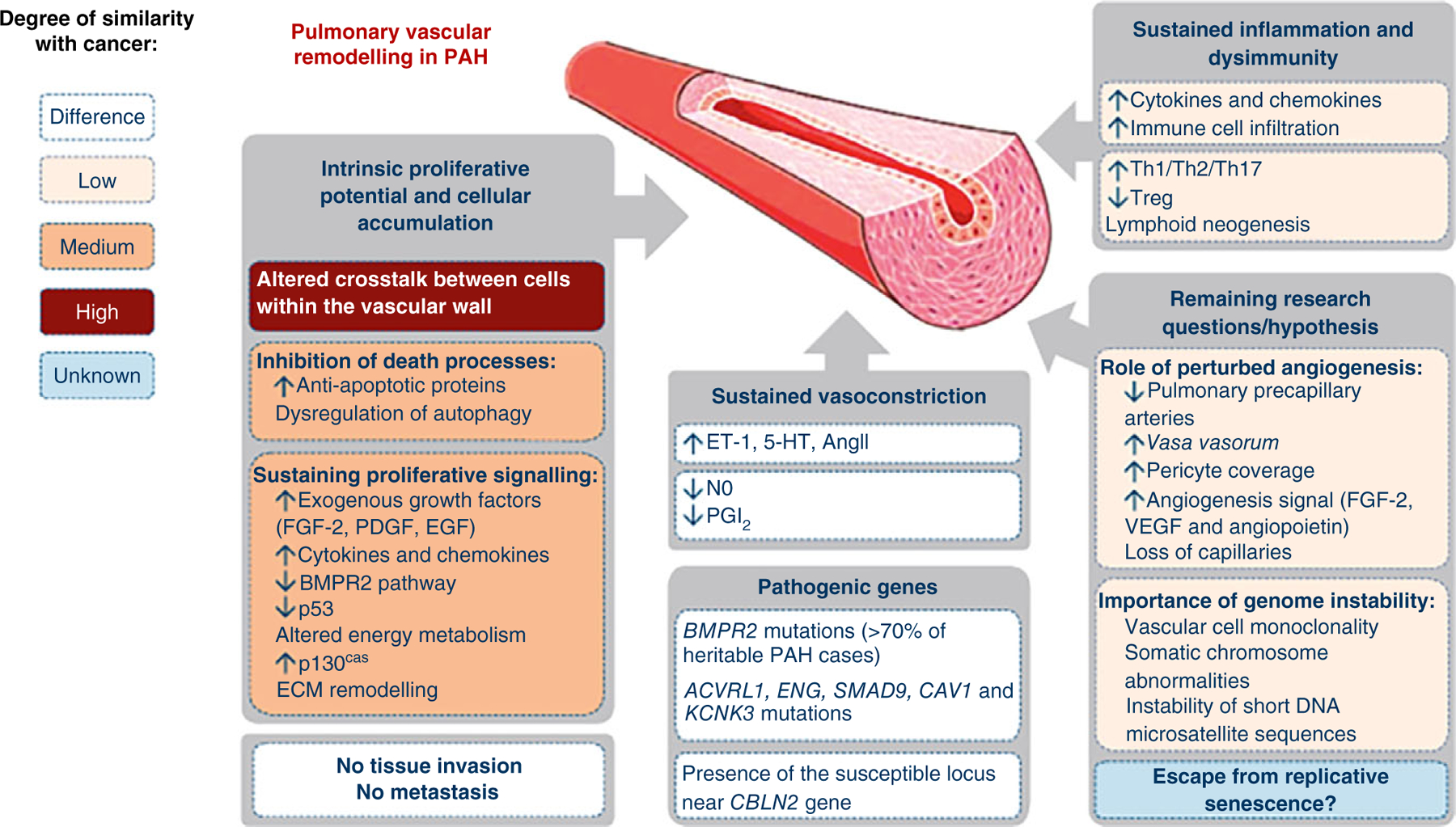
Pathogenic concepts of pulmonary arterial hypertension: Differences and similarities with cancer. FGF, fibroblast growth factor; PDGF, platelet-derived growth factor; EGF, epidermal growth factor; BMPR2, bone morphogenetic protein receptor 2; ECM, extracellular matrix; ET-1, endothelin-1; 5-HT, serotonin; NO, nitric oxide; PGI2, prostacyclin; Th, T-helper cell; Treg, regulatory T-cell; VEGF, vascular endothelial growth factor. Reused, with permission, from Guignabert C, et al., 2013 (66), European Respiratory Society.
Initiation of PAH
A significant unresolved issue in human Group 1 PH is lack of understanding of the factors involved in initiation of disease. The nature of the triggering event is unclear but has been speculated to be due to endothelial injury or death potentially due to an undefined infectious agent. This is supported by the incomplete penetrance of gene mutations found in heritable PAH, most notably bone morphogenetic protein II (BMPR2), suggesting a triggering event is needed in addition to a genetic predisposition (46, 82). It has been inferred that this hypothesis is correct based on animal studies using in particular the Sugen/hypoxia rat model, wherein a transient induction of endothelial cell (EC) death of apoptosis leads to development of complex vascular lesions (188). However, this is as of yet an unproven assumption in human PAH.
Profiling of pulmonary vascular lesions
By the time of diagnosis, patients with PAH show established, potentially irreversible, pulmonary vascular lesions. At present, there is no causal link or association between a particular type of lesion and the marked increase in PVR seen in PAH (83, 194). Several lesions are recognized either topographically as located in the intima, media, adventitia, or by the predominance of specific structural cells that compose each of the specific lesions (200): ECs likely predominate in plexiform and some concentric intima lesions (199), myofibroblasts are predominant in intima obliteration, SMCs account for media remodeling, and fibroblasts undergo activation in the adventitia. Few mapping studies have localized some obliterative lesions to branching points (35, 227), or supernumerary arteries (195), but how vascular lesions are distributed along the fractal pulmonary artery remains unclear (195). These changes of resident pulmonary vascular cells often occur in the context of perivascular inflammation, one of the most consistently reported findings in human and animal models of PH (Figure 3); importantly, perivascular inflammation is more than a bystander in the disease process, as it impacts on hemodynamic parameters (70, 160, 176, 199). None of the individual intimal or medial lesions carry a statistically significant correlation with pulmonary artery pressures or PVR (176). However, a trend is noted when hemodynamics are compared with “intima thickening.” Furthermore, the quantification of each type of lesion varies significantly in different lung regions. There is no correlation among lesions in their distribution or frequency, limiting the extent to which these data might offer clues to their potential temporal, topographical and pathogenetic relationship (176). The aspect of lesional heterogeneity is illustrated by the inability to distinguish clearly diseased lung clusters as assessed by principal component analysis based on data related to intima or medial remodeling, the number of profiles of plexiform lesions, and the degree of perivascular inflammation (176). It is also important to note that each of these elements of diversity and potentially plasticity require detailed consideration not only in PH but also in the normal pulmonary circulation. Importantly, age-related phenotypic changes in the apparently normal pulmonary circulation might differ significantly across the lifespan (176). As detailed below, the introduction of molecular profiling of these specific vascular lesions will likely offer unique insights into their molecular characteristics and the degrees that PAH lungs differ among patients with the disease.
Figure 3.
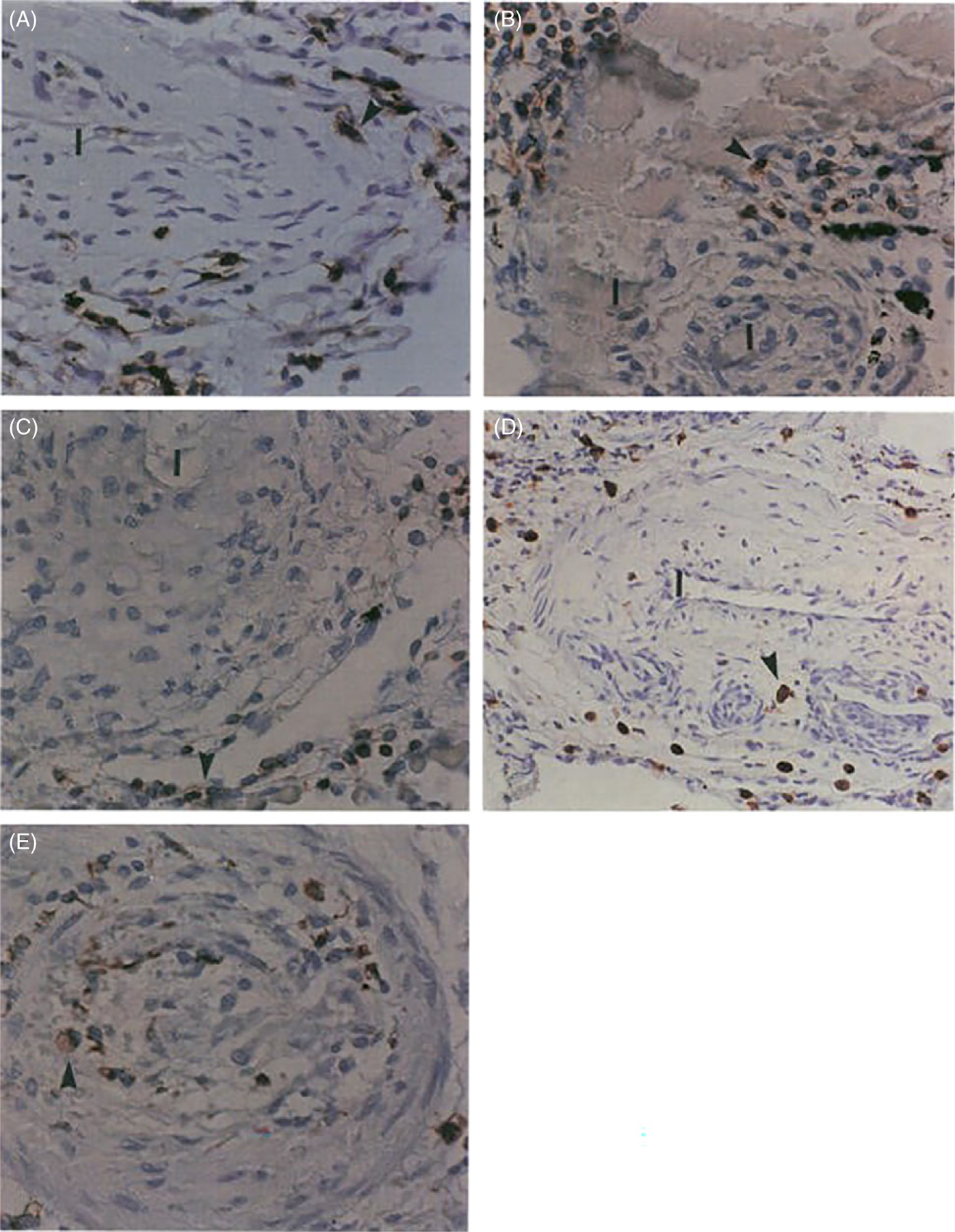
Inflammatory components in plexiform type lesions of PAH. Inflammatory cells are highlighted by the anti-CD45 immunostaining (arrowheads) (A), B-cell marker (UL-26) (B), T-cell marker (UCHSC) (C), and macrophage marker (CD68) (D and E) (arrowheads). Note infiltration of the outer portion of the plexogenic vessel (D) and vessel obliterated by concentric proliferation (E) by macrophages (immunoperoxidase, A-C, E 500X; D: 200x). I-Vascular lumen. Reused, with permission, from Tuder RM, et al., 1994 (199), from Elsevier.
The extracellular matrix in PAH
Another highly important participant in the vascular lesions in PAH is the ECM. The ECM, which is largely understudied in the PH field, is a determinant of location- and time-dependent plasticity of pulmonary vascular cells. The ECM remains in constant flux as the lung matures, ages, and responds to injury (191). ECM remodeling and pulmonary vascular stiffness occur early in the disease process and often precede increases in vascular thickness and even arterial pressure. This suggests changes in ECM and related proteins can be causal and not simply the result of changes in vascular cell phenotypes. In established IPAH, there are distinct, compartment-specific changes in the expression of genes encoding collagens, tenascin, fibronectin, and osteopontin. Additionally, there is evidence of the ongoing breakdown of elastin and collagens with increased expression of elastases and metalloproteinases (75, 78, 140). ECM remodeling and stiffness, through mechano-activation of multiple signaling pathways, can have profound effects on vascular cell phenotype and function (4, 43, 44, 190, 216). The fine proteome of pulmonary vascular lesions in human PH (notably in PAH) has not been elucidated thus far. We propose that the intra- and extracellular proteome are key to the thickening of all three vascular compartments and that there are multiple stages of matrix remodeling defined by unique macromolecular components, matrix cross-linking, and cellular signatures that need to be defined. The proteome of specific vascular lesions is predicted to contain disease driver(s), such as elements of the classic and alternative pathways of complement (57). Moreover, this lesional proteome may ultimately manifest increased pulmonary vascular stiffness and drive pathogenic enhanced cell survival/proliferation, resistance to apoptosis, and inflammation (78).
Recognizing heterogeneity within PAH
Biology and medicine have recently experienced rapid growth in groundbreaking methodologies that can be applied to interrogate fundamental cell and molecular processes. These can be used to resolve the mechanistic hierarchy of specific pathways concerning disease initiation and progression, taking into consideration the critical nature of a highly heterogeneous disease. This heterogeneity exists in clinical initiating factors, such as age at onset, clinical presentation, rate of progression, and response to therapy (184). Importantly, between- and even within-patient heterogeneity involving the types of pulmonary vascular lesions and the corresponding endotypes (i.e., underlying molecular processes driving the specific disease presentation) have been uncovered by our and other groups and underlie the almost certain high complexity of disease pathogenesis (76, 84, 94, 176). We believe that the paucity of investigations into defining these aspects of heterogeneity has resulted in a lack of recognition of subphenotypes of PAH and thereby an understanding of specific factors contributing them, hindering the development of more individualized, targeted therapies. Fortunately, there are emerging technologies that will allow us to begin to address the important issues defined above that we will address later in this article.
Pathobiology of Group 3 PH
Epidemiology of Group 3 PH
PH is a common complication of many lung diseases. The prevalence of PH associated with IPF-PH is estimated to range from 38% to 80% of patients (106, 126, 166). When compared to IPF without PH, IPF-PH patients have worse clinical outcomes, including decreased functional status, increased oxygen requirements, and, most importantly, a 5- to 8-fold increase in 1-year mortality (93, 106, 120). A significant proportion of individuals with COPD also develop PH, with the prevalence ranging from 30% to 70% (27, 88, 119, 131). Despite being associated with only mild to moderately elevated pulmonary artery pressure, PH in COPD is a major cause of morbidity and mortality in COPD, with an increased risk of exacerbations and decreased survival (28, 88, 119, 131). High altitude PH, first described in the 1950s, is observed in individuals chronically residing at altitude >2500 m (approximately 8000 ft) (103), putting approximately 140 million people around the world at risk for a disease with reported prevalence of <1% to 18% (20, 21, 61, 108, 127, 172).
Despite the recognition of the need for treatment in Group 3 PH, it remains unknown whether treatment with the pulmonary vasodilator therapies used in Group 1 PH is appropriate. Most published data using approved PAH therapies demonstrated that they are ineffective in Group 3 PH, and in some cases have shown increased mortality (36, 81, 95, 125, 145, 146). The oral and systemic pulmonary vasodilators which are used to treat PAH can worsen ventilation/perfusion mismatch in COPD (28, 119). To date, seven randomized control trials have been conducted for COPD-PH, with relatively low number of subjects, but without consistent outcomes to consistently support the use of pulmonary vasodilator use (17, 62, 148, 182, 206, 210, 213). Results from the recently completed INCREASE trial suggest benefit from inhaled treprostinil (prostacyclin) in patients with PH due to ILD (205). Still, the development of novel medical therapies for Group 3 PH is an urgent unmet need (125) but is currently limited by our poor understanding of its pathogenesis.
Comparing pathologic findings of Group 1 and Group 3 PH
Barriers to progress in understanding and finding treatments in Group 3 PH include a relative paucity of studies on the pathology of Group 3 PH compared to Group 1 PH and to differences in methodologies that make direct comparisons between these states difficult.
In studies of chronic high-altitude PH, a disease thought largely to be characterized by pure hypoxia-mediated PH, autopsies of long-term high-altitude dwellers reveal hypertrophy of the media of proximal pulmonary arteries and increased muscularization of distal arterioles with an increased ratio of distal to proximal arteries (215). Similarly, patients with COPD-PH develop hypertrophy of the muscular tunica media layer (113, 226) while the smaller arterioles develop a smooth muscle layer with a new internal elastic lamina (223). Pathology of COPD-PH has also been characterized by hyperproliferation of the intima and a thickened tunica media, both of which are primarily composed of SMCs (157, 200). COPD patients undergoing lung transplantation with severe PH had an increased remodeling score of the microvessels compared to COPD patients with moderate PH or without PH (23), which is not typical of remodeling in PAH. Animals exposed to cigarette smoke develop changes in the intimal and medial layer of small pulmonary arteries which are similar to those seen in humans (64, 113, 162). In IPF-PH, vascular remodeling is also seen prominently in the intimal and medial layers (76). In a study which compared well-characterized lungs explanted from COPD-PH and IPF-PH patients, the smaller vessels in COPD-PH lungs (<200 μm) exhibited more pronounced lumen reduction than larger vessels (200–500 μm), while both small and large-diameter vessels exhibited similar degrees of remodeling in IPF-PH (76).
In our experience and in most pathologic studies of PH in lung disease including IPF-PH, COPD-PH, PH in other forms of nonspecific interstitial pneumonitis, or hypoxic PH in the setting of high altitude or sleep apnea, the presence of plexiform lesions has not been found (29, 59). Based on the cornerstone description of the pathology of PAH (72), we define plexiform lesions as an abnormal growth of ECs with EC markers (i.e., Factor 8 related antigen, VE-cadherin, or CD31) (199) and VEGF signaling molecules (196). However, we acknowledge that a standardized definition of plexiform lesion is lacking both in PAH and in descriptions of pulmonary vascular remodeling in the setting of Group 3 PH, with some calling abnormal luminal growth representative of plexiform lesions (49). It should also be noted that recent studies demonstrate that at least certain subtypes of plexiform lesions are the result of shunt-type lesions that connect pulmonary arteries to the bronchial circulation (207, 221) which may be late manifestation of end-stage PAH rather than an inciting factor in the pathogenesis of PAH. In addition, in Group 3 PH, we have not observed concentric lesions, which would typically be composed of ECs (203). As illustrated in Figure 4, the adventitia merges with fibrotic and inflammatory processes involving alveolar septa and bridging with airways. This “fibrotic reticulum” may eventually connect with the subpleural fibrotic areas (33).
Figure 4.
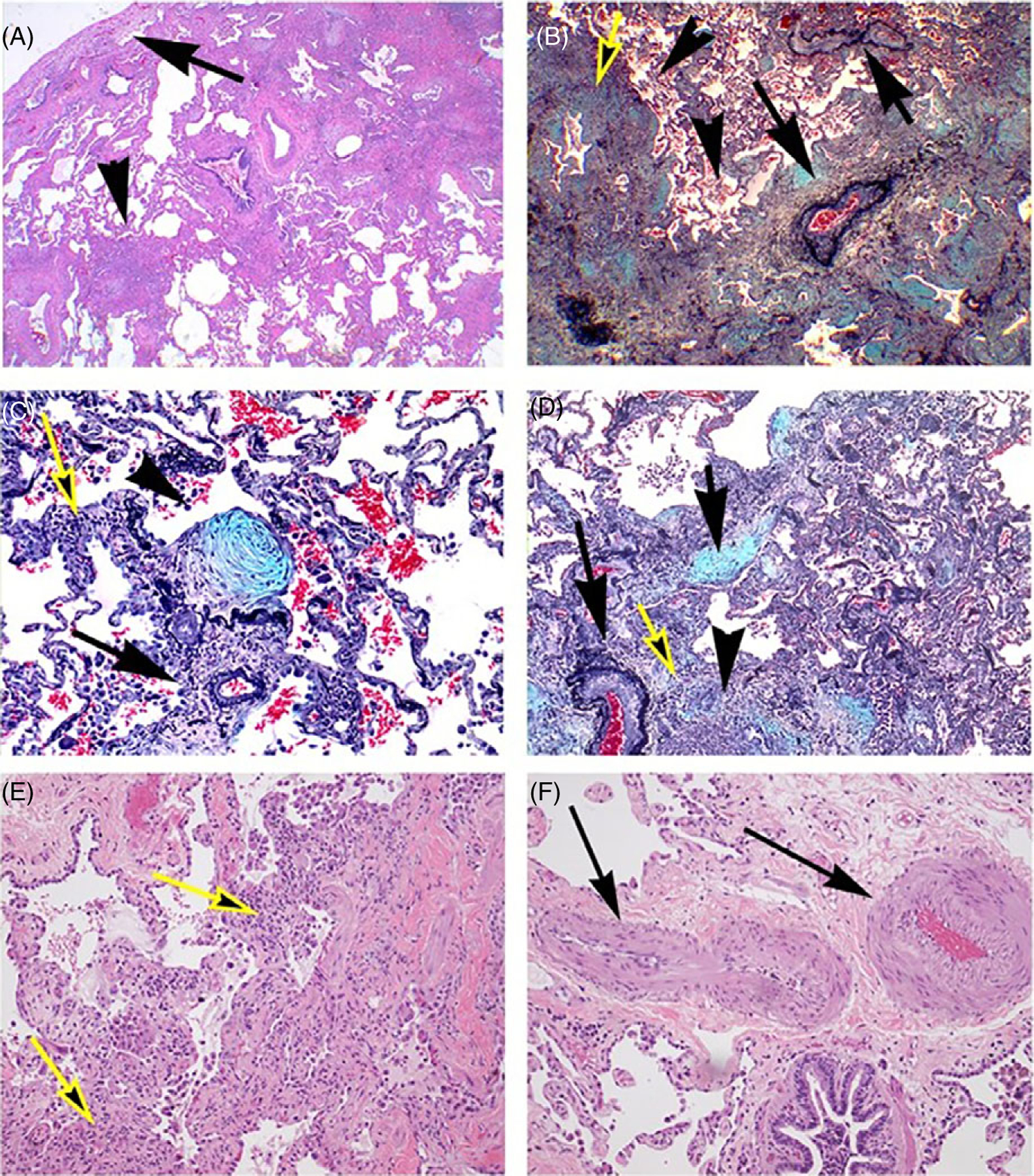
Inflammatory and fibrotic processes in usual interstitial pneumonia with marked pulmonary vascular remodeling. (A) Low magnification of subpleural region showing marked subpleural fibrosis (arrow), extending irregularly toward central regions of the lung with conglomerates of lymphoid cells (arrowhead) (Hematoxilin and Eosin). (B) Marked perivascular (arrows) and airway (yellow arrow) fibrosis and inflammation, forming a continuous process along the bronchovascular and parencymal structures. The pulmonary arteries show intima thickening, with significant obliteration of the vascular lumina (Pentachrome). (C) Exudative fibroblastic focus (arrowhead) next to small pulmonary artery (arrowhead), accompanied by scattered inflammatory cells (Pentachrome). (D) Active bridging fibrosis along alveolar and bronchovascular (yellow arrow) associated with exudative process (arrow) and inflammation (arrowhead). Note the marked obliteration of an adjacent pulmonary artery (long arrow) (Pentachrome). (E) Active inflammation and interstitial fibrosis in the advancing edge of usual interstitial pneumonia (yellow arrow) (Hematoxilin and eosin). (F) Marked media and intima thickening in muscular artery (arrows) in a region not immediately affected by UIP (arrows) (Hematoxilin and eosin).
The paradigm of vascular rarefaction that has been raised to explain the increased PVR in Group 1 PH has also been presented in IPF-PH and COPD-PH (197). Studies of COPD-PH have shown pruning of the small pulmonary vessels, causing decreased capillary density (23, 218) and correlating with severity of PH (23). Evidence for vascular rarefaction in fibrotic lung diseases has also been supported by gene expression data (37) and as well as by experimental animal studies (48). These studies, however, fall short because they did not rely on lung stereology, the gold standard for the quantification of histopathological features in the lung. It is not appropriate to compare fibrotic to nonfibrotic areas in terms of the vascular density, as these are divergent tissue types, with the single commonality that they occur in the same lung. Importantly, the studies did not consider the volume of reference tissue (i.e., the volume of fibrosis and alveolar airspace and septal tissues), which further undermines the validity of the findings.
We postulate that in IPF and perhaps in other forms of lung disease-associated PH, the process of remodeling and pathobiology begins on the “outside” (adventitial) but progresses to “inside” (luminal) injury and remodeling processes, highlighting the potential contribution of fibrotic processes and inflammation in its pathobiology, as we will highlight below. This contrasts with the marked involvement of the intima in PAH, which is suggestive of an “inside to outside” order of pathobiological events, where the adventitia then becomes a later site of remodeling in PAH and conduit of inflammation that contributes to PAH (178). In conclusion, while there are likely differences in the pathology of Group 1 and Group 3 PH, more rigorous studies are needed to confirm and establish the significance of these findings.
Hypoxic vasoconstriction and pulmonary vascular remodeling
Hypoxia is sensed via carotid body chemoreceptors (triggering the hypoxic ventilatory response), oxygen sensors in the pulmonary vasculature (triggering hypoxic vasoconstriction), and oxygen sensors in various other tissues, resulting in activation of vascular endothelial growth factor in the heart and brain and erythropoietin in the kidney and liver (74, 77). In the pulmonary vasculature, hypoxia inhibits oxygen-sensitive potassium channels. This causes depolarization of pulmonary artery SMCs and activation of voltage-gated calcium channels, which results in calcium influx and Rho kinase-mediated pulmonary artery SMC vasoconstriction. Consequently, PVR and pulmonary arterial pressure increase (67, 121). The degree of pulmonary arterial pressure elevation increases with magnitude of altitude or hypoxia and exercise (134). Sustained and exaggerated pulmonary vasoconstriction over time is thought to cause pulmonary vascular remodeling which in turn contributes to ongoing elevation in PVR (104). Remodeling may occur in part as ECs sense humoral and hemodynamic changes, which trigger vasoactive and mitogenic factor production that alter pulmonary artery SMC function, proliferation, and apoptosis (150). Additionally, there is a shift in ECs to production of endothelin, thromboxane A2, and other vasoconstrictive substances (15, 21, 53, 186).
Animal models suggest that pulmonary vascular remodeling due to pure hypoxia is rapid in onset and only partially reversed by removal from hypoxia (177). Rabinovitch and colleagues demonstrated the presence of smooth muscle-like cells in normally nonmuscular pulmonary arterioles after exposing rats to hypoxia (143, 144). Many mechanisms including recruitment of progenitor cells, distal migration of resident smooth muscle stem cells, and pericyte recruitment have been implicated in this process (40, 45, 55, 164, 177, 217, 233). The onset of neomuscularization is rapid, with changes observed within hours of hypoxic exposure and peaking at 1 week (171). The proportion of muscularized pulmonary arterioles corresponds with increasing pulmonary arterial pressure (142). With the return to normoxia, pulmonary arterial pressure decreases but does not normalize, and some structural changes persist (143, 171, 177).
As with Group 1 PH, the initiating events in the development of PH in lung disease are unknown. Although SMCs comprise most of the remodeled parts of COPD and COPD-PH pulmonary arteries, ECs are implicated in the early stages of pulmonary vascular remodeling and PH development. Cigarette smoke can directly injure ECs, and EC dysfunction contributes to emphysema development and also causes vascular stiffness due to impaired release of the endothelium-derived vasodilating agents nitric oxide (NO) and prostacyclin (189, 201). Smokers without obstructive lung disease have reduced expression of endothelial nitric oxide synthase (eNOS) (10). Hypoxia-induced EC damage can lead to over-production of the potent vasoconstrictors thromboxane A2 and endothelin-1 (ET-1) (135), which cause the neighboring SMCs to proliferate and vasospasm (187). ECs have been shown to influence pulmonary artery SMC inflammation in an autocrine manner and proliferation, hypertrophy, and collagen synthesis in a paracrine manner (110).
Gaps in understanding the pathobiology of Group 3 PH
The hypothesis that fibrosis-mediated vascular destruction and hypoxic vasoconstriction are the sole contributors to PH in IPF or severe emphysema is being increasingly challenged based on two important observations. Studies in animal models of COPD have shown that pulmonary vascular remodeling and PH precede the development of emphysema (162). Moreover, even patients with mild to moderate COPD without PH develop intimal thickening (113), as do smokers without COPD (158), caused by SMCs and deposition of elastic and collagen fibers (158). This may explain why pulmonary arterial wall thickness has not consistently been correlated with mean pulmonary arterial pressure (99, 226). Similarly, clinical data suggests that in IPF patients with similar levels of pulmonary functional impairment, some individuals develop PH while others do not (60, 122, 156). While the degree of vascular remodeling correlates with severity of regional fibrosis, studies of IPF pathology have also observed vascular remodeling in nonfibrotic regions of IPF lungs (132). These structural alterations ranged from thickening of the vascular smooth muscle layer to proliferative intimal lesions, to complete occlusion of the vessels in an appearance similar to the plexiform lesions in PAH (132), although not meeting our definition as described above. Cumulatively, this suggests that mechanisms other than fibrosis-mediated vascular destruction and hypoxic vasoconstriction occur in IPF-PH and are significant contributors to IPF-PH pathogenesis. This justifies a focus on mechanisms of pulmonary vascular remodeling in IPF-PH as well as Group 3 PH in general.
Many of the hypothesized mechanisms of pulmonary vascular remodeling in IPF or IPF-PH have been inferred from PAH pathophysiology, although as we have noted, these exact mechanisms of pulmonary vascular remodeling are unknown. Given the distinct differences in potential etiologic mechanisms that separate COPD, IPF, and PAH, it does not seem far-fetched to hypothesize that there are likely divergent mechanisms in the pathogenesis of PH in these different forms that could be detected using currently available techniques, such as with molecular profiling, but these studies have produced other surprising findings. In a study in which whole genome microarray analysis was performed on laser capture microdissected pulmonary artery profiles from COPD-PH, IPF-PH, and control donor lung explants, there was only a small overlap of differentially regulated genes between COPD-PH and IPF-PH pulmonary artery profiles compared to control lungs, while the majority of differentially expressed genes varied, with the largest observed differences seen between COPD-PH and IPF-PH in genes belonging to the retinol metabolism pathway and the ECM-receptor interaction pathway (76). The top up-regulated genes in the COPD-PH pulmonary artery profiles were then analyzed compared to donors with COPD without PH, and interestingly, no significant differences were found between these two groups. Furthermore, the severity of PH defined as increasing mPAP only correlated with minor changes in gene regulation across all samples including both COPD-PH and IPF-PH. A parallel comparison of IPF-PH with IPF lungs without PH could not be performed in this study due to the lack of access to non-PH IPF lungs; however, in an earlier study by a different group using similar laser capture microdissection techniques, the gene expression of pulmonary arteries in IPF patients with and without co-existing PH was compared to that of controls (133). The study demonstrated that factors associated with SMC and EC proliferation, Wnt signaling, complement system activation, and apoptosis were differentially expressed in the small pulmonary arteries of IPF patients compared to controls, but interestingly, unsupervised and supervised clustering analyses revealed similar gene expression in IPF-PH and IPF non-PH arterioles. Thus, pathways involved in aberrant apoptosis and vascular proliferation are already activated in IPF patients without PH. Combined, these studies highlight the likelihood that patients with COPD and IPF may develop PH through divergent mechanisms, and that much like the histopathologic changes that are seen even before the clinical development of PH, reprogramming of the transcriptome could also occur before the appearance of PH in the setting of ILD or COPD.
Inflammation in Group 3 PH
Inflammatory pathways play a central role in the development of hypoxia PH (177). Hypobaric hypoxia increases transforming growth factor β (TGF-β) secretion by primary pulmonary adventitial fibroblasts and macrophages (100). TGF-β appears to promote the Nox4 isoform of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, leading to the production of endogenous reactive oxygen species and pulmonary artery SMC proliferation (8, 183). Additionally, hypobaric hypoxia increases levels of pulmonary artery SMC IL-6, activating inflammatory responses and fibrosis. IL-6 deficient mice exposed to hypoxia demonstrate less macrophage recruitment, pulmonary vascular remodeling, and PH (115, 161). Other inflammatory pathways triggered by endoplasmic reticulum stress and mediated by GRP78, PERK, IRE1, ATF4, ATF6, CHOP, and caspase-12 may also contribute to hypoxic pulmonary vascular remodeling in subacute to chronic hypoxia (139). Finally, macrophages appear to play a role in pulmonary artery SMC proliferation (232). Hypoxia induces the production of macrophage migration inhibitor factor (MIF) leading to release of inflammatory mediators and stimulation of pulmonary artery SMC proliferation contributing to PH (168, 238).
Thus, despite a focus on ECs and pulmonary vascular smooth muscle cells (PVSMCs) in PH/PAH, many recent studies support a causative role for immune cells in PH/PAH (3, 54, 160, 199, 231). Pulmonary macrophages accumulate in PH lung tissues, and their presence is associated with worsened PH pathology (54, 100, 231). To define mechanisms underlying this observation, recent studies using PH hypoxia-induced models demonstrated that infiltrating pulmonary macrophages promote pulmonary vascular remodeling and PH (231). This data suggests that interactions between macrophages and vascular structural cells (ECs, PVSMCs, and fibroblasts) drive vascular remodeling and PH pathogenesis. Extending this observation to human IPF-PH, recent data demonstrate the presence of pulmonary macrophages around remodeled vessels. This finding suggests a causal role for pulmonary macrophages in IPF-PH development and progression, though the precise mechanisms are presently unknown.
Inflammation, which plays an important role in the pathophysiology of COPD, is also implicated in the development of pulmonary vascular remodeling and PH in COPD. Overexpression of protease-activated receptor 2 (PAR-2), whose gene expression is upregulated in ECs by cytokines, in cigarette smoke-exposed mice not only developed emphysema, but also developed muscularization of small intrapulmonary vessels; these mice also had an imbalance between vasoconstrictors (ET-1) and vasodilators (VEGF, NO synthase, and inducible iNOS) as well as increased production of growth factors involved in vascular cell proliferation (41). Myeloid-cell-specific deletion of iNOS in mice was found to ameliorate the increase in expression of CD206 on interstitial macrophages and protect against the development of PH in smoke-exposed mice (65). Furthermore, CD206-positive and iNOS-positive macrophages were seen to accumulate in the proximity of remodeled vessels in the lungs of COPD patients (65). More investigation into the crosstalk between immune cells and the other cell types in the pulmonary vasculature is required to determine whether there are targetable inflammatory pathways in the pathogenesis of Group 3 PH.
New Strategies for Defining the Molecular Pathobiology of Pulmonary Hypertension Across All Groups
While the classification of PH according to clinical characteristics has been useful in advancing the care of PH patients, it has been a precarious platform from which to discover new treatments and to build a framework to understand the complex molecular pathophysiology of numerous PH phenotypes. The vast majority of patients with PH, predominantly those with Group 2 PH due to left heart disease and Group 3 PH due to chronic lung disease, have been excluded from being able to benefit from most currently available treatments, while the search for new therapies for these groups has been largely unfruitful. Other patients currently grouped under Group 1 PH may be inappropriately included and could benefit less or even be harmed by the initiation of pulmonary vasodilator therapy, particularly in the case of patients with pulmonary veno-occlusive disease (PVOD). Big data sets generated by high-throughput molecular techniques and machine learning algorithms can be used to address the limitations of these reductionist approaches and to disentangle the complex relationships between genotype, endophenotype, and clinical phenotype of PH.
In a study of human explanted lungs from patients with PAH, PVOD, COPD, and IPF, RNA analysis of targeted sampled areas revealed that PAH and PVOD samples exhibited the greatest intergroup differences in gene expression, while pathway analysis revealed that PVOD patients shared 11-fold more similarities with patients with IPF compared to with PAH (128). Another study using laser capture microdissection of pulmonary artery profiles combined with unbiased whole genome microarrays revealed distinctly different gene expression patterns and disturbed regulatory pathways between IPF-PH and COPD-PH, with retinol metabolism and ECM receptor interaction pathways being the most affected processes (76).
A deeper understanding of disease pathogenesis using genomics, proteomics, metabolomics, and epigenetics is needed to identify novel therapeutic targets and disease-specific biomarkers for all forms of PH. Unbiased algorithms and clustering techniques can allow for the discovery of mechanistic pathways in an agnostic fashion. The 2010 NHLBI Pulmonary Vascular Strategic Plan identified using “omics” technologies and a systems approach, combining institutional cohorts and integrating multidimensional omics, clinical, and outcome data, as top priorities in pulmonary vascular research (73). These approaches have the potential to (i) reveal molecular signatures that could be targeted with new precision methods, (ii) deeply characterize endotype-genotype-phenotype profiles to predict which patients are at greatest risk of progression and to discover signatures of treatment response, and (iii) identify using high-throughput screening molecules and repurposed drugs that could target regulatory pathways in PH. Thus far, these approaches have largely been used to understand more deeply the pathogenesis of PAH. While they have furthered the understanding of lung diseases such as COPD and IPF, PH in association with these diseases has not been studied with such precision.
Genomics
Using new sequencing techniques, several PAH-related genes have been identified in recent years with significant implications for new treatment strategies. Mutations of bone morphogenic protein receptor-2 (BMPR2), a member of the TGF-β superfamily were identified as the underlying cause of 80% of familial PAH and are involved in 25% of sporadic PAH cases (112). This was followed by discoveries of mutations in ACVRL1 (encoding activin A receptor type II-like 1) and ENG (encoding endoglin) in hereditary hemorrhagic telangiectasia, a syndrome complicated by the development of PAH, two proteins that also participate in BMPR2 signaling through dimerization (69, 193). Other mutations have been identified in transcription factors SMAD8, SMAD4, TBX4, and SOX17 which are downstream of BMPR2 signaling (124), and BMP9 (also known as growth differentiation factor 2 GDF2), the preferred ligand for the BMPR2 complex (63). The loss of BMPR2 functional signaling results in the loss of antiproliferative signaling, leading to EC dysfunction and the characteristic pulmonary vascular remodeling in PAH. The discovery of mutations in BMPR2 and genes related to BMPR2 signaling has led to the first phase 2 trials of drugs targeting this novel pathway. Sotatercept, a novel fusion protein which acts by sequestering excess ActRIIA ligands, reducing and thereby rebalancing ActRIIA-Smad2/3 pro-proliferative with BMPR2 antiproliferative signaling, was shown to reduce PVR in patients already receiving background PAH therapy after 24 weeks of treatment, and is now being tested in phase 3 clinical trials (79). A trial of the calcineurin inhibitor FK506 (tacrolimus), which upregulates BMPR2 expression, was shown to be safe and well-tolerated in PAH patients (174, 175). Preclinical trials have shown that administration of BMP9 has the potential to reverse PAH in a mouse model of PAH bearing a heterozygous knock-in of a human BMPR2 mutation, R899X, and in two severe rat models of PAH (111). Interestingly, one study has shown that BMPR2 was also significantly decreased in the lung tissue and macrophages of IPF patients with a greater decrease in IPF-PH group (30), raising the question of whether these therapies targeting BMPR2 expression and signaling could be applied in IPF-PH.
Epigenomics
The importance of epigenetic mechanisms in the development and progression of PAH has become increasingly clear and present an opportunity for new therapeutic targets, including dysregulation of noncoding RNA networks, histone acetylation and methylation, and bromodomain proteins, all of which act as epigenetic regulators of pulmonary vascular wall structure and function. Many publications have demonstrated the importance of microRNAs in PAH, with notable examples being miR-140-5p, which can upregulate the BMPR2 signaling pathway and prevent development of PAH in vivo (154), miR-126 and miR-223 of which downregulation can contribute to development of RV failure in PAH (137, 165), and the miR-130/301 family, which was shown to be a master regulator of cellular proliferation in PAH (16). Additional studies have implicated miR-124 as playing a crucial role in controlling the metabolic and proliferate state of ECs, SMCs, and adventitial fibroblasts (236, 237). DNA methylation patterns in the mitochondrial superoxide dismutase-2 (SOD2) gene, the major generator of H2O2, were found to create proliferative, apoptosis-resistant pulmonary artery smooth muscle cells (PASMCs) that led to the initiation and progression of heritable PAH in the Fawn hooded rat model of PAH, and reversal by DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine restored both SOD2 expression and the ratio of proliferation to apoptosis (6). Histone deacetylases (HDACs) and their inhibitors have been studied with great interest for their therapeutic potential, as the use of small inhibitory molecules such as valproic acid and suberoylanilide hydroxamic has been shown to successfully prevent and partially reverse PAH in rats and inhibited platelet-derived growth factor-stimulated proliferation of fibroblasts and human vascular SMCs in vitro (101, 239). Significant effects of HDAC inhibition on ventricular remodeling in PH have also been reported (25, 224). The bromodomain and extraterminal-containing (BET) protein family, which includes BRD2, BRD3, and bromodomain-containing protein 4 (BRD4), are epigenetic reader proteins that bind to specific acetylated protein residues on histone tails where they facilitate the assembly of transcription complexes and are receiving much attention in cardiovascular disease (19, 109). Upregulation of BRD4 has been found in distal pulmonary arteries and PASMC of PAH patients, and pharmacologic BRD4 inhibition has been shown to reverse experimental PAH (116). A phase 2 clinical trial using the BRD4 inhibitor apabetalone is currently in progress. Lastly, germline mutations of TET2, encoding ten-eleven translocation (TET) methylcytosine dioxygenase 2, a key enzyme in DNA methylation, were recently reported in a large cohort of patients with PAH (136).
Proteomics
Targeted, liquid chromatography and mass spectrometry-based approaches, or targeted antibody/aptamer-based approaches, can be used to generate large data sets with expression patterns of >1000 targets from a single sample. In-tandem liquid chromatography-mass spectrometry performed on whole lobe homogenates from healthy and PAH explanted lungs found differences in 25 proteins between the groups, with increased expression of chloride intracellular channel 4, a multifunctional protein involved in numerous processes implicated in PAH pathogenesis including angiogenesis, and TGF-β, endothelial growth factor, and bone morphogenetic protein signaling (1). A plasma proteomics approach using an aptamer-based assay of 1129 plasma found 20 proteins that differentiated survivors and nonsurvivors in an observational cohort of idiopathic and heritable PAH patients, with a panel of 9 proteins that provided prognostic information independent of plasma NT-proBNP concentrations and independent of the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL) risk score (Figure 5) (14, 153). This protein panel score was then validated in another independent PAH cohort, with an increasing score distinguishing low-, medium-, and high-risk groups (153). More recently, complete proteomic analysis of the plasma of patients with PAH and healthy individuals found higher levels of netrin-4, while higher thrombospondin-2 levels had a protective effect, thus identifying pathways that if inhibited may prevent development of PAH and or if potentiated could decrease the severity of established PAH (68).
Figure 5.
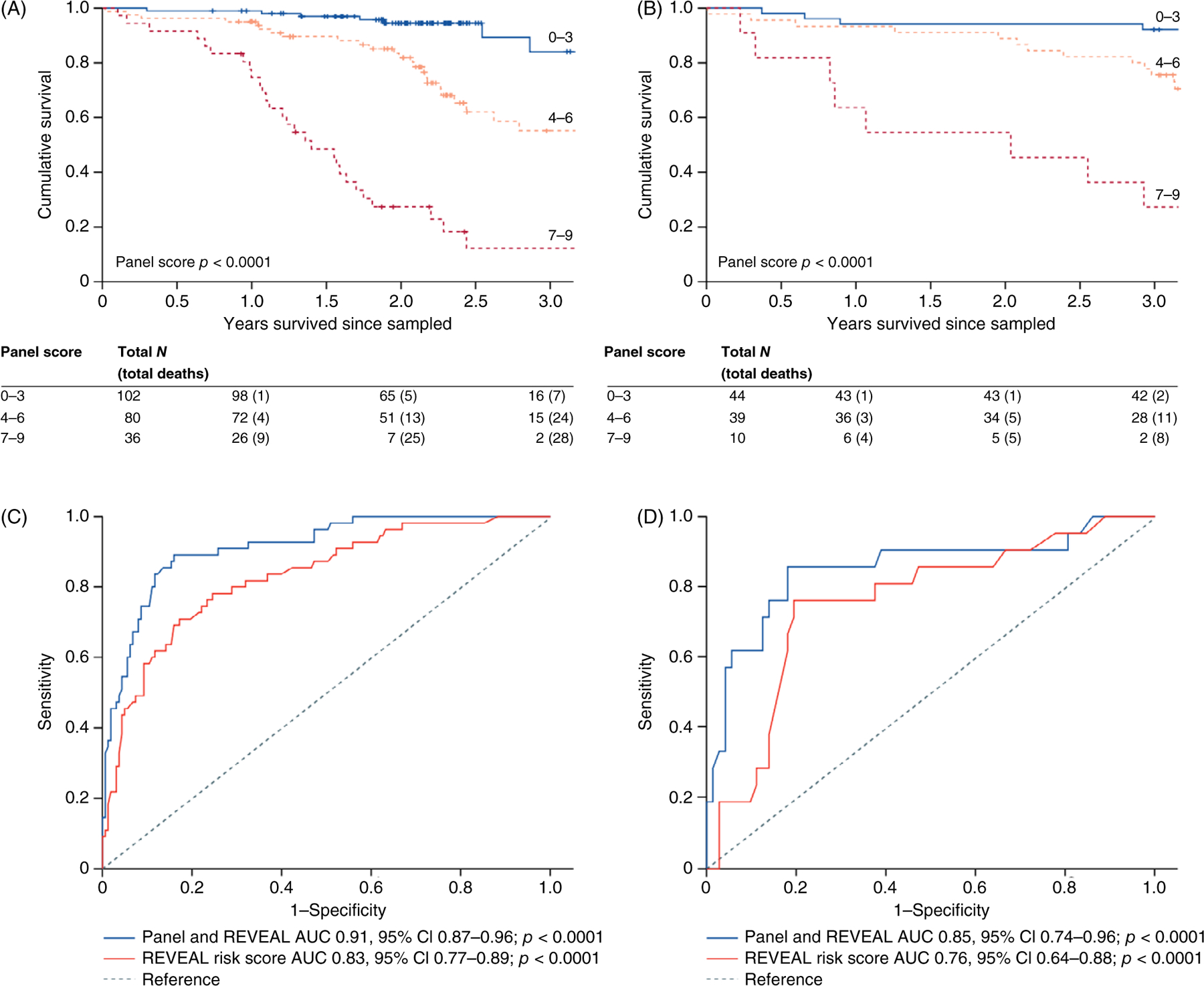
Survival analysis of based on a panel of plasma proteins and REVEAL score in an observational cohort of idiopathic and heritable PAH patients. Kaplan-Meier survival estimates in patients with different panel scores, in all patients with idiopathic pulmonary arterial hypertension from (A) cohorts 1 and 2 and (B) cohort 4. ROC analysis of Cox models before and after addition of the prognostic protein panel to the established equation, in all patients with idiopathic pulmonary arterial hypertension from (C) cohorts 1 and 2 and (D) cohort 4. ROC, receiver operating characteristic; AUC, area under the curve. Reused, with permission, from Rhodes CJ, et al., 2017 (153)/Elsevier/CC BY 4.0.
Metabolomics
One of the early insights that supported the cancer hypothesis of PAH was the observation of a switch in IPAH cells from aerobic metabolism to glycolysis (e.g., the Warburg effect), as had previously been observed in solid tumor malignancies in low oxygen environments (198). A complex picture of the interplay between cells of the pulmonary vasculature and the cells in the immediate microenvironment has since emerged, with a role for metabolic reprogramming of resident and recruited cells influencing pulmonary vascular remodeling and other hallmarks of PH including proliferation, resistance to apoptosis, inflammation, and fibrosis (38). Continued interest in energy metabolism and new metabolomics approaches including nuclear magnetic resonance (MR) and liquid chromatography-mass spectrometry have further advanced the understanding of the changes in metabolism in PH. Layered transcriptomic and metabolomic analyses of human pulmonary microvascular ECs expressing two different disease-causing mutations in BMPR2 confirmed the previously seen increase in aerobic glycolysis but also uncovered widespread metabolic changes, with significant upregulation of the pentose phosphate pathway, increases in nucleotide salvage and polyamine biosynthesis pathways, decreases in carnitine and fatty acid oxidation pathways, and major impairments of the tricarboxylic acid (TCA) cycle and failure of anaplerosis, some of which were confirmed to be present in the serum of PAH patients (52). A study using targeted mass spectrometry to perform metabolic profiling of 71 patients who also underwent right-heart catheterization and radionuclide ventriculography found an association of hemodynamic indicators of RV-pulmonary vascular dysfunction with 21 metabolites including circulating indoleamine 2,3-dioxygenase (IDO)-dependent tryptophan metabolites (TMs), TCA intermediates, purine metabolites, and arginine-nitric oxide metabolic pathway constituents (107). A comprehensive study of plasma metabolites in a large cohort of IPAH or heritable PAH patients (n = 365) found 51 metabolites that were able to distinguish PAH from healthy control subjects, with a subset of these metabolites also able to distinguish PAH from control subjects with other diseases (151). These included tRNA-specific modified nucleosides (N2,N2-dimethylguanosine, N1-methylinosine), TCA cycle intermediates (malate, fumarate), glutamate, fatty acid acylcarnitines, tryptophan, and polyamine metabolites and decreased levels of steroids, sphingomyelins, and phosphatidylcholine. Moreover, 62 metabolites were prognostic of death in PAH, with half of these independent of established prognostic markers. Interestingly, patients who responded to calcium channel blocker therapy had metabolic profiles similar to those of healthy control subjects, suggesting possible applications of these methods to improve risk stratification and identify responders to treatment. An extension of this study to patients with Group 4 PH (or CTEPH) found that untargeted analysis of plasma metabolites was able to distinguish CTEPH from control patients and that many metabolic changes were similar between CTEPH and IPAH patients, with only five metabolites able to distinguish CTEPH from IPAH (185). Another investigation into the intracellular metabolism of ECs in PH patients (which included IPAH, connective disease-associated PH, as well as CTEPH) demonstrated again a clear separation between plasma metabolites of PH patients and controls, and furthermore revealed four distinct intracellular EC metabolic clusters in patients with PH, with notably different disease severity between the clusters (24). Metabolic profiling studies thus have the potential to discover new biomarkers, pathways for further characterization and intervention, and commonalities between types of PH that could be targeted with therapeutic interventions (Figure 6).
Figure 6.
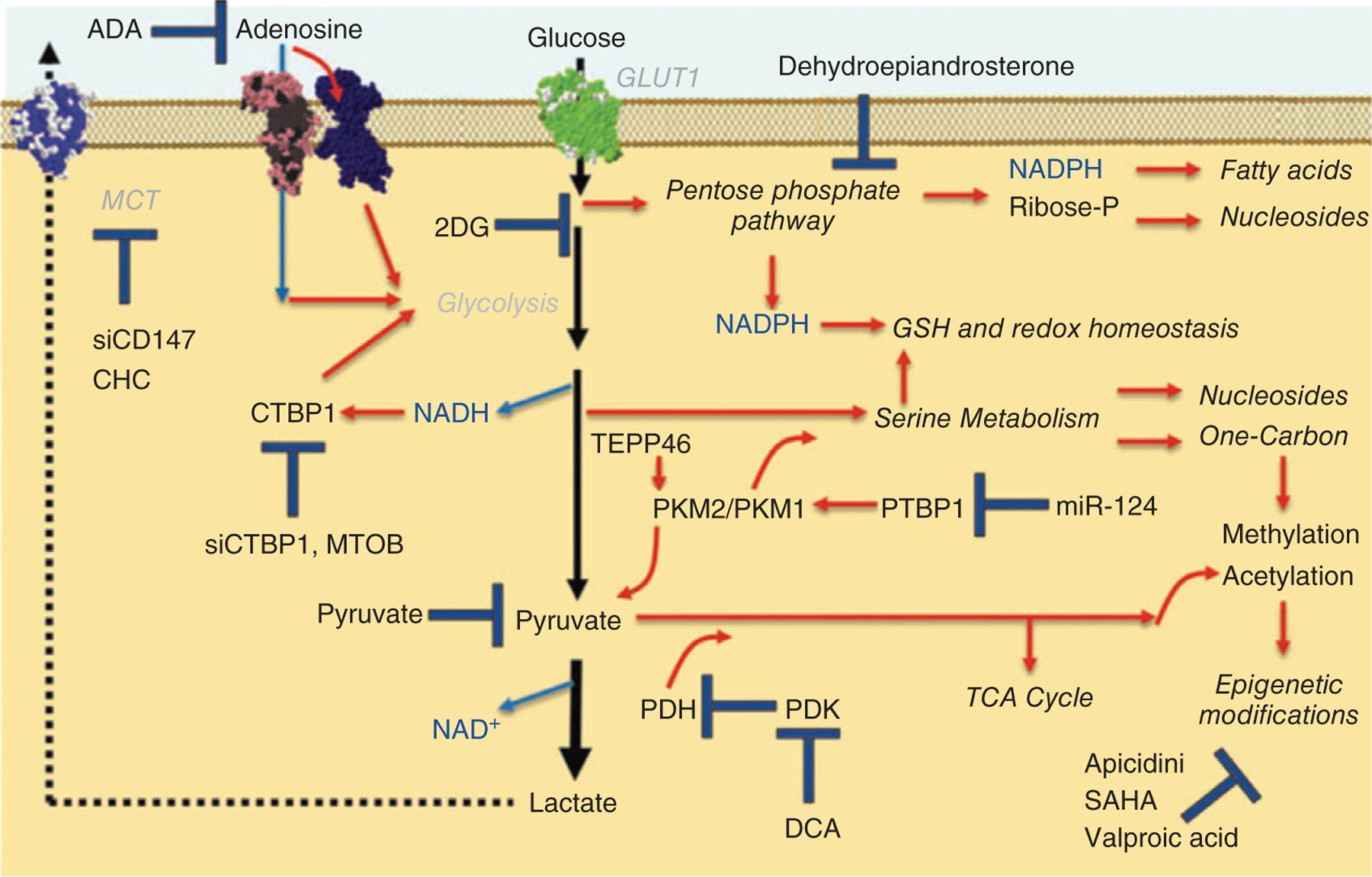
Metabolic pathways and potential therapeutic interventions in PH. CHC, a-cyano-4-hydroxycinnamic acid; DCA, dichloroacetate; PDK, pyruvate dehydrogenase kinase. Reused, with permission, from D’Alessandro A, et al., (38)/with permission of Mary Ann Liebert.
Machine learning
ML algorithms offer a computational platform to identify patterns in big data sets generated by high-throughput technologies. Although a proper understanding of their limitations and proper application of these algorithms is needed to avoid common pitfalls, recently summarized in a review by Rhodes et al. (Table 3) (152), they offer a unique opportunity to identify patterns in large omics data that can elucidate pathobiological pathways in PH (47), identify new etiological sub-populations within PH patients (129, 184, 222), reclassify types of PH using newly revealed commonalities, develop risk stratification calculators to guide clinical decision making (47, 90, 91, 105, 152), and eventually suggest novel therapeutic compounds (96).
Table 3.
Machine Learning Advantages, Challenges, and Pitfalls
| ML Advantages | |
|
| |
| Effective task-specific algorithms are available for a wide range of research applications | |
| Allows for accurate prediction and detection of complex patterns/relationships in data | |
| Permits agnostic and unbiased exploratory research, freedom from assumptions about underlying data | |
| Well-suited for high-dimensional omics data sets (where number of input variables exceeds observations) | |
| Able to accommodate several types of input variables (continuous, categorical, imaging features, etc.) | |
| Can simultaneously account for linear and nonlinear relationships between variables | |
| Models can autonomously improve while learning in real time from new data | |
|
| |
| ML Challenges and Pitfalls | Pitfall Avoidance |
|
| |
| Models trained on small data sets often have poor generalizability in other data sets | Collaborative data sharing and harmonization (particularly important in PAH, a rare disease) |
| No gold standard approaches exist for algorithm selection or hyperparameter tuning | Apply heuristic data-driven methods to objectively select algorithm and set hyperparameters |
| Algorithms can be oversensitive to noise (mislabeled data, confounding signal, assay technical artifact) | Careful attention to data collection, quality control, and preprocessing (normalization, standardization, missing value handling, batch adjustment) |
| Black box models (difficult to interpret) | Explain model decision processes (graphically); delineate which input variables drive model decisions (feature selection methods, variable importance measures) |
| Model decisions can unfairly disadvantage certain patient subgroups (algorithmic bias) | Select a cohort representative of wider disease population; include socioeconomic input variables |
| Inadequate model validation | Independent cohort validation is critical (resampling-based cross-validation on training data set is not adequate); compare model vs. existing gold standard |
| Lack of transparency in model reporting | Full disclosure of methods; share model code and anonymized data at publication; |
Definition of abbreviations: ML, machine learning; PAH, pulmonary arterial hypertension; TRIPOD, transparent reporting of a multivariable prediction model for individual prognosis or diagnosis.
Adapted, with permission, from Rhodes CJ, et al., 2022 (152).
There are two types of ML algorithms:
Supervised algorithms (e.g., decision trees and random forests of decision trees, artificial neural networks, multivariate logistic regression) are applied when training a model to recognize a known event or estimate a specific measurement. For example, if 1000 proteins are measured in 500 patients with PAH and 500 patients without PAH, a supervised ML model can be “trained” to identify a pattern in those 1000 proteins that detects whether a patient has PAH. This is referred to as a “classification” model. If the objective was to use these 1000 proteins to estimate a continuous variable (e.g., mPAP, PVR), supervised ML models can also be trained for that purpose, which is known as a “regression” model. These kinds of models can be very useful for creating a diagnostic algorithm, estimating invasive parameters through imaging or blood markers (89, 90), or estimating prognostic risk stratification scores (12, 47, 85, 91). Supervised ML was used to train an algorithm using electronic health record healthcare utilization data from PAH patients in England to preemptively predict the development of idiopathic PAH, which was cross-validated in a subgroup of the cohort not used in the training, and was found to have excellent specificity and negative predictive value (92). Another outcome risk assessment model based on Bayesian analysis outperformed both the REVEAL (13) and REVEAL 2.0 calculators in the prediction of 1-year survival on the same cohort and external validation cohorts (85). However, assumptions about similarities and differences based on traditional PH categories and misclassification can result in bias and limit the detection of important similarities and differences in supervised ML.
Unsupervised algorithms (e.g., principal component analysis, k-means clustering) are applied to data to detect patterns when not looking for a specific event. For example, if 1000 proteins were measured in proteins in 1000 PAH patients along with their phenotypic attributes, unsupervised ML algorithms could tell if these patients cluster into sub-groups. These algorithms have been used in other diseases to reclassify and uncover subgroups, such as in a study of patients with heart failure with preserved ejection fraction (HFpEF), in which 67 phenotypic variables were analyzed and classified patients into three groups that had unique clinical characteristics, cardiac structure and function, invasive hemodynamics, and outcomes (163). Another study utilized exercise profiles using data from pulmonary function testing, cardiopulmonary exercise testing, and metabolic data to define four exercise groups with different variables that predicted hospitalization in patients with exercise intolerance (130). In studies using unsupervised ML in PAH, Sweatt et al. (184) utilized an unsupervised ML algorithm to explore if circulating markers of inflammation could identify distinct sub-populations of PAH patients and discovered four distinct immune phenotypes distinct from clinical subtypes which had differing clinical risk metrics and long-term survival. Other researchers utilizing similar techniques on whole blood transcriptomics identified that PAH patients fell into three subgroups based on up- and down-regulation of certain genes that were associated with poor, moderate, and good prognosis (86). In summary, unsupervised ML has great potential to reclassify diseases, allowing patients with shared perturbations in genomic, proteomic, or metabolic profiles to be partitioned into subgroups in an agnostic data-driven manner. However, clustering by unsupervised ML is highly sensitive to confounding sources of heterogeneity, such as comorbidities, medications, and batch effects, thus it is critical that any investigators using these techniques carefully consider their cohort selection and ensure data quality control before data processing.
ML algorithms can identify patients at greatest risk that would potentially benefit from more aggressive up-front therapy, and to identify those most likely to respond to therapy. Currently, patients with a new diagnosis of PAH undergo right heart catheterization to obtain hemodynamic measurements and to determine vasoresponsiveness to inhaled nitric oxide, which predicts a good long-term response to therapy with calcium channel blockers (169). ML offers more opportunities to identify other biomarkers indicative of potential treatment response and to measure early treatment response or failure. A recently concluded phase 2 trial evaluating the effect of rituximab B-cell depletion therapy in systemic sclerosis (SSc)-PAH utilized supervised ML models to identify baseline biomarkers that predicted a clinical response at 4 weeks (235). Training of each model began with 168 input variables including cytokines, chemokines, factors, immunoglobulin subclasses, autoantibodies, B-cell subsets, and clinical features, involved iterative algorithm resampling runs and recursive feature elimination to select variables subsets that best classified clinical responders, and identified that low concentrations of rheumatoid factor, IL-12, and IL-17 were consistently found to be favorable predictors of rituximab response (receiver operating characteristic area under the curve 0.88–0.95) (235).
While grouping and risk stratification using machine learning models requires validation in independent cohorts, future trial designs should increasingly incorporate these methods to optimize patient selection, and once validated, treatment response signatures can be used to guide therapeutic decisions.
Lessons learned from the implementation of these big data approaches combined with machine learning could allow the field of pulmonary vascular disease (PVD) research to benefit from emerging techniques which are already being successfully employed across a spectrum of disciplines. However, due to the rare nature of PAH and the relatively small size of clinical registries compared to more common diseases, laboratories studying pulmonary vascular disease have applied unsupervised (86, 184) and supervised (128) machine learning techniques to datasets with only a few hundred observations, compared to much larger data sets that have been used in other fields. As a result, while these innovative studies are exposing the PVD community to exciting new ways of analyzing high-throughput data, their findings will need to be validated across different datasets and with different techniques before seriously challenging the central dogma of PVD.
We propose that adoption of machine learning technology and advances could be made by offering the PVD research community access to data from across multiple centers added to a publicly available growing body of records, which could become the gold standard for validation. This would ensure that any laboratory generating data for an ongoing study would: (i) Be more likely to use established data acquisition methodology that could be validated with overlapping markers before publication; (ii) Offer researchers a quantitative comparison of their results with established datasets, which would serve as a simultaneous validation of the acquisition and analysis pipeline; and (iii) Prevent saturating the literature with underpowered and unvalidated results.
Discovering novel drug therapies
High-throughput screening (HTS) is a drug discovery process that allows automated testing of large numbers of chemical and/or biological compounds for a specific biological target. This process has been used successfully in discovering new treatments for rare diseases. Treatment of cystic fibrosis (CF), a disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene which previously shortened patient life expectancy by decades, has been revolutionized by the use of HTS to identify small-molecule potentiators (208, 228) resulting in the development of drugs that are now available to treat 90% of known genetic mutations in CF (11). Artificial intelligence (AI) technologies provide further promise of rapidly speeding up the process of identifying and testing candidate drugs. Traditional HTS methods begin with a screening library of up to 1 million compounds, with each costing US$50 to $100 for a total cost of several million U.S. dollars, followed by years of compound optimization and identifying preclinical drug candidates, while AI can assist in all steps of the pipeline for new drug development (26). A virtual compound library of several billion molecules can be screened within a few days, followed by a few months to a year to identify preclinical drug candidates using AI to predict the physical properties of drugs in terms of bioavailability, bioactivity, and toxicity, 3D structures of target proteins, drug-protein interactions, and to perform retrosynthesis pathway prediction as a method for designing the organic synthesis of a drug candidate (7, 214). A deep learning model was trained to predict molecules with antibacterial activity and discovered a novel antibiotic, halicin, that demonstrated bactericidal activity against a diverse spectrum of pathogens including Mycobacterium tuberculosis and multi-drug resistant Enterobacteriaceae and Acinetobacter, as well as eight other antibacterial compounds that are structurally unique compared to all known antibiotics (181). Combining insights into the molecular mechanisms underlying the pathogenesis of PH with high-throughput drug screening techniques with assistance from AI has the potential to discover new categories of treatments for this disease at a much more rapid pace than has been seen to date in the field of pulmonary vascular disease.
Conclusions
Despite significant advances in the understanding of pathogenesis of PH and its treatments, significant gaps remain in understanding the initiating factors and the role of pathologic lesions in PAH, and strides must be made to standardize the techniques and definitions applied to lung pathology in both Group 1 and Group 3 PH. Advances in scientific technologies have led to deeper, richer understandings of the genetic and pathobiologic basis of pulmonary vascular disease and have revealed similarities between as well as heterogeneity within the currently defined WHO groupings of Group 1 and Group 3 PH. Greater emphasis remains on Group 1 PH or PAH, while the use of these technologic advances to understand the pathobiology of PH in lung disease as well as other types of PH currently excluded from the Group 1 PH classification remains limited. Leveraging the substantial gains made in understanding the genomics, epigenomics, metabolomics, and proteomics of PAH to understand the full spectrum of the complex, heterogeneous disease of all forms of PH is needed. Multimodal omics data as well as supervised and unbiased machine learning approaches after careful study design and selection of patients could lead to earlier diagnosis, precise risk stratification, predictions of disease response, new sub-phenotype groupings within types of PH, and identification of similar subgroups and pathways between PAH and other types of PH that could lead to new treatment targets. These insights combined with high-throughput drug screening technologies with the assistance of artificial intelligence could offer a more rapid pathway to the development of new therapies.
Didactic Synopsis.
Major teaching points
New methodologies have demonstrated significant heterogeneity in the types of pulmonary vascular lesions observed in pulmonary arterial hypertension (PAH). They have also revealed both pathobiologic similarities as well as differences between PAH and other diseases in which pulmonary hypertension (PH) is observed, such as idiopathic pulmonary fibrosis (IPF).
Patients with pulmonary hypertension secondary to lung disease (Group 3 PH) have equally poor if not worse mortality compared to those with pulmonary arterial hypertension (Group 1 PH) despite having lower pulmonary arterial pressure and right ventricular dysfunction, highlighting the urgent need to understand its pathobiologic underpinnings, which could lead to identification of new therapeutic options.
At present, it remains unclear whether treatment of other groups of PH with the pulmonary vasodilator therapies used in Group 1 PH is appropriate or potentially harmful.
While there are likely pathologic differences between Group 1 and Group 3 PH, application of rigorous techniques including lung stereology (i.e. quantification of three-dimensional anatomic abnormalities) and precise localization of vascular lesions (for example in relationship to areas of fibrosis) will help define the significance of these differences.
Multimodal omics data as well as supervised and unbiased machine learning could lead to earlier diagnosis, more precise risk stratification, better predictions of disease response, new sub-phenotype groupings within types of PH, and identification of shared pathways between PAH and other types of PH that could lead to new treatment targets.
References
- 1.Abdul-Salam VB, Wharton J, Cupitt J, Berryman M, Edwards RJ, Wilkins MR. Proteomic analysis of lung tissues from patients with pulmonary arterial hypertension. Circulation 122: 2058–2067, 2010. [DOI] [PubMed] [Google Scholar]
- 2.Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 121: 2747–2754, 2010. [DOI] [PubMed] [Google Scholar]
- 3.Abid S, Marcos E, Parpaleix A, Amsellem V, Breau M, Houssaini A, Vienney N, Lefevre M, Derumeaux G, Evans S, Hubeau C, Delcroix M, Quarck R, Adnot S, Lipskaia L. CCR2/CCR5-mediated macrophage-smooth muscle cell crosstalk in pulmonary hypertension. Eur Respir J 54: 1802308, 2019. [DOI] [PubMed] [Google Scholar]
- 4.Acharya AP, Tang Y, Bertero T, Tai YY, Harvey LD, Woodcock CC, Sun W, Pineda R, Mitash N, Konigshoff M, Little SR, Chan SY. Simultaneous pharmacologic inhibition of yes-associated protein 1 and glutaminase 1 via inhaled poly(lactic-co-glycolic) acid-encapsulated microparticles improves pulmonary hypertension. J Am Heart Assoc 10: e019091, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aldred MA, Comhair SA, Varella-Garcia M, Asosingh K, Xu W, Noon GP, Thistlethwaite PA, Tuder RM, Erzurum SC, Geraci MW, Coldren CD. Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am J Respir Crit Care Med 182: 1153–1160, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, Dyck JR, Gomberg-Maitland M, Thebaud B, Husain AN, Cipriani N, Rehman J. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation 121: 2661–2671, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baig MH, Ahmad K, Roy S, Ashraf JM, Adil M, Siddiqui MH, Khan S, Kamal MA, Provaznik I, Choi I. Computer aided drug design: Success and limitations. Curr Pharm Des 22: 572–581, 2016. [DOI] [PubMed] [Google Scholar]
- 8.Bailey DM, Dehnert C, Luks AM, Menold E, Castell C, Schendler G, Faoro V, Gutowski M, Evans KA, Taudorf S, James PE, McEneny J, Young IS, Swenson ER, Mairbäurl H, Bärtsch P, Berger MM. High-altitude pulmonary hypertension is associated with a free radical-mediated reduction in pulmonary nitric oxide bioavailability. J Physiol 588: 4837–4847, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barberà JA, Blanco I. Pulmonary hypertension in patients with chronic obstructive pulmonary disease. Drugs 69: 1153–1171, 2009. [DOI] [PubMed] [Google Scholar]
- 10.Barbera JA, Peinado VI, Santos S, Ramirez J, Roca J, Rodriguez-Roisin R. Reduced expression of endothelial nitric oxide synthase in pulmonary arteries of smokers. Am J Respir Crit Care Med 164: 709–713, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Barry PJ, Mall MA, Alvarez A, Colombo C, de Winter-de Groot KM, Fajac I, KA MB, EF MK, Ramsey BW, Sutharsan S, Taylor-Cousar JL, Tullis E, Ahluwalia N, Jun LS, Moskowitz SM, Prieto-Centurion V, Tian S, Waltz D, Xuan F, Zhang Y, Rowe SM, Polineni D, Group VXS. Triple therapy for cystic fibrosis Phe508del-gating and -residual function genotypes. N Engl J Med 385: 815–825, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bauer Y, de Bernard S, Hickey P, Ballard K, Cruz J, Cornelisse P, Chadha-Boreham H, Distler O, Rosenberg D, Doelberg M, Roux S, Nayler O, Lawrie A. Identifying early pulmonary arterial hypertension biomarkers in systemic sclerosis: Machine learning on proteomics from the DETECT cohort. Eur Respir J 57, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benza RL, Gomberg-Maitland M, Elliott CG, Farber HW, Foreman AJ, Frost AE, McGoon MD, Pasta DJ, Selej M, Burger CD, Frantz RP. Predicting survival in patients with pulmonary arterial hypertension: The REVEAL risk score calculator 2.0 and comparison with ESC/ERS-based risk assessment strategies. Chest 156: 323–337, 2019. [DOI] [PubMed] [Google Scholar]
- 14.Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hyper-tension: Insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 122: 164–172, 2010. [DOI] [PubMed] [Google Scholar]
- 15.Berger MM, Dehnert C, Bailey DM, Luks AM, Menold E, Castell C, Schendler G, Faoro V, Mairbäurl H, Bärtsch P, Swenson ER. Transpulmonary plasma ET-1 and nitrite differences in high altitude pulmonary hypertension. High Alt Med Biol 10: 17–24, 2009. [DOI] [PubMed] [Google Scholar]
- 16.Bertero T, Lu Y, Annis S, Hale A, Bhat B, Saggar R, Saggar R, Wallace WD, Ross DJ, Vargas SO, Graham BB, Kumar R, Black SM, Fratz S, Fineman JR, West JD, Haley KJ, Waxman AB, Chau BN, Cottrill KA, Chan SY. Systems-level regulation of microRNA networks by miR-130/301 promotes pulmonary hypertension. J Clin Invest 124: 3514–3528, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blanco I, Santos S, Gea J, Guell R, Torres F, Gimeno-Santos E, Rodriguez DA, Vilaro J, Gomez B, Roca J, Barbera JA. Sildenafil to improve respiratory rehabilitation outcomes in COPD: A controlled trial. Eur RespirJ 42: 982–992, 2013. [DOI] [PubMed] [Google Scholar]
- 18.Blanco I, Tura-Ceide O, Peinado VI, Barbera JA. Updated perspectives on pulmonary hypertension in COPD. Int J Chron Obstruct Pulmon Dis 15: 1315–1324, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borck PC, Guo LW, Plutzky J. BET epigenetic reader proteins in cardiovascular transcriptional programs. Circ Res 126: 1190–1208, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brito J, Siques P, López R, Romero R, León-Velarde F, Flores K, Lüneburg N, Hannemann J, Böger RH. Long-term intermittent work at high altitude: Right heart functional and morphological status and associated cardiometabolic factors. Front Physiol 9: 248, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brito J, Siques P, Pena E. Long-term chronic intermittent hypoxia: A particular form of chronic high-altitude pulmonary hypertension. Pulm Circ 10: 5–12, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brittain EL, Hemnes AR. Introduction to review series on pulmonary vascular disease and right ventricular heart failure. Circ Res 130: 1362–1364, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bunel V, Guyard A, Dauriat G, Danel C, Montani D, Gauvain C, Thabut G, Humbert M, Castier Y, Dorfmüller P, Mal H. Pulmonary arterial histologic lesions in patients with COPD with severe pulmonary hypertension. Chest 156: 33–44, 2019. [DOI] [PubMed] [Google Scholar]
- 24.Carlsen J, Henriksen HH, Marin de Mas I, Johansson PI. An explorative metabolomic analysis of the endothelium in pulmonary hypertension. Sci Rep 12: 13284, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cavasin MA, Stenmark KR, McKinsey TA. Emerging roles for histone deacetylases in pulmonary hypertension and right ventricular remodeling (2013 Grover Conference series). Pulm Circ 5: 63–72, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan HCS, Shan H, Dahoun T, Vogel H, Yuan S. Advancing drug discovery via artificial intelligence. Trends Pharmacol Sci 40: 592–604, 2019. [DOI] [PubMed] [Google Scholar]
- 27.Chaouat A, Bugnet AS, Kadaoui N, Schott R, Enache I, Ducoloné A, Ehrhart M, Kessler R, Weitzenblum E. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 172: 189–194, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Chaouat A, Naeije R, Weitzenblum E. Pulmonary hypertension in COPD. Eur RespirJ 32: 1371–1385, 2008. [DOI] [PubMed] [Google Scholar]
- 29.Chatterjee K, Tarawneh AR, Alam S. Out of proportion pulmonary hypertension in obstructive lung diseases. Curr Opin Pulm Med 24: 161–172, 2018. [DOI] [PubMed] [Google Scholar]
- 30.Chen NY, Collum SD, Luo F, Weng T, Le TT AMH, Philip K, Molina JG, Garcia-Morales LJ, Cao Y, Ko TC, Amione-Guerra J, Al-Jabbari O, Bunge RR, Youker K, Bruckner BA, Hamid R, Davies J, Sinha N, Karmouty-Quintana H. Macrophage bone morphogenic protein receptor 2 depletion in idiopathic pulmonary fibrosis and Group III pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 311: L238–L254, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collum SD, Amione-Guerra J, Cruz-Solbes AS, DiFrancesco A, Hernandez AM, Hanmandlu A, Youker K, Guha A, Karmouty-Quintana H. Pulmonary hypertension associated with idiopathic pulmonary fibrosis: Current and future perspectives. Can Respir J 2017: 1430350, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Consortium TIP, Lane KB, Machado RD, Pauciulo MW, Thompson JR, Philips JA III, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2 encoding a TGF-B receptor cause familiar pulmonary hypertension. Nat Genet 26: 81–84, 2000. [DOI] [PubMed] [Google Scholar]
- 33.Cool CD, Groshong SD, Rai PR, Henson PM, Stewart JS, Brown KK. Fibroblast foci are not discrete sites of lung injury or repair: The fibroblast reticulum. Am J Respir Crit Care Med 174: 654–658, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cool CD, Kuebler WM, Bogaard HJ, Spiekerkoetter E, Nicolls MR, Voelkel NF. The hallmarks of severe pulmonary arterial hypertension: The cancer hypothesis - ten years later. Am J Physiol Lung Cell Mol Physiol 318 (6): L1115–L1130, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cool CD, Stewart JS, Werahera P, Miller GJ, Williams RL, Voelkel NF, Tuder RM. Three-dimensional reconstruction of pulmonary arteries in plexiform pulmonary hypertension using cell specific markers: Evidence for a dynamic and heterogeneous process of pulmonary endothelial cell growth. Am J Pathol 155: 411–419, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Corte TJ, Keir GJ, Dimopoulos K, Howard L, Corris PA, Parfitt L, Foley C, Yanez-Lopez M, Babalis D, Marino P, Maher TM, Renzoni EA, Spencer L, Elliot CA, Birring SS, O’Reilly K, Gatzoulis MA, Wells AU, Wort SJ, Group BS. Bosentan in pulmonary hypertension associated with fibrotic idiopathic interstitial pneumonia. Am J Respir Crit Care Med 190: 208–217, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cosgrove GP, Brown KK, Schiemann WP, Serls AE, Parr JE, Geraci MW, Schwarz MI, Cool CD, Worthen GS. Pigment epithelium-derived factor in idiopathic pulmonary fibrosis: A role in aberrant angiogenesis. Am J Respir Crit Care Med 170: 242–251, 2004. [DOI] [PubMed] [Google Scholar]
- 38.D’Alessandro A, El Kasmi KC, Plecita-Hlavata L, Jezek P, Li M, Zhang H, Gupte SA, Stenmark KR. Hallmarks of pulmonary hypertension: Mesenchymal and inflammatory cell metabolic reprogramming. Antioxid Redox Signal 28: 230–250, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Das M, Dempsey EC, Reeves JT, Stenmark KR. Selective expansion of fibroblast subpopulations from pulmonary artery adventitia in response to hypoxia. Am J Physiol Lung Cell Mol Physiol 282: L976–L986, 2002. [DOI] [PubMed] [Google Scholar]
- 40.Davie NJ, Crossno JT Jr, Frid MG, Hofmeister SE, Reeves JT, Hyde DM, Carpenter TC, Brunetti JA, McNiece IK, Stenmark KR. Hypoxia-induced pulmonary artery adventitial remodeling and neovascularization: Contribution of progenitor cells. Am J Physiol Lung Cell Mol Physiol 286: L668–L678, 2004. [DOI] [PubMed] [Google Scholar]
- 41.De Cunto G, Cardini S, Cirino G, Geppetti P, Lungarella G, Lucattelli M. Pulmonary hypertension in smoking mice over-expressing protease-activated receptor-2. Eur RespirJ 37: 823–834, 2011. [DOI] [PubMed] [Google Scholar]
- 42.Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) Is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 67: 737–744, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dieffenbach PB, Haeger CM, Coronata AMF, Choi KM, Varelas X, Tschumperlin DJ, Fredenburgh LE. Arterial stiffness induces remodeling phenotypes in pulmonary artery smooth muscle cells via YAP/TAZ-mediated repression of cyclooxygenase-2. Am J Physiol Lung Cell Mol Physiol 313: L628–L647, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dieffenbach PB, Mallarino Haeger C, Rehman R, Corcoran AM, Coronata AMF, Vellarikkal SK, Chrobak I, Waxman AB, Vitali SH, Sholl LM, Padera RF, Lagares D, Polverino F, Owen CA, Fredenburgh LE. A novel protective role for matrix metalloproteinase-8 in the pulmonary vasculature. Am J Respir Crit Care Med 204: 1433–1451, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dierick F, Solinc J, Bignard J, Soubrier F, Nadaud S. Progenitor/stem cells in vascular remodeling during pulmonary arterial hypertension. Cell 10: 1338, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dunmore BJ, Jones RJ, Toshner MR, Upton PD, Morrell NW. Approaches to treat pulmonary arterial hypertension by targeting BMPR2: From cell membrane to nucleus. Cardiovasc Res 117: 2309–2325, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Errington N, Iremonger J, Pickworth JA, Kariotis S, Rhodes CJ, Rothman AM, Condliffe R, Elliot CA, Kiely DG, Howard LS, Wharton J, Thompson AAR, Morrell NW, Wilkins MR, Wang D, Lawrie A. A diagnostic miRNA signature for pulmonary arterial hypertension using a consensus machine learning approach. EBioMedicine 69: 103444, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Farkas L, Farkas D, Ask K, Moller A, Gauldie J, Margetts P, Inman M, Kolb M. VEGF ameliorates pulmonary hypertension through inhibition of endothelial apoptosis in experimental lung fibrosis in rats. J Clin Invest 119: 1298–1311, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Farkas L, Gauldie J, Voelkel NF, Kolb M. Pulmonary hypertension and idiopathic pulmonary fibrosis: A tale of angiogenesis, apoptosis, and growth factors. Am J Respir Cell Mol Biol 45: 1–15, 2011. [DOI] [PubMed] [Google Scholar]
- 50.Fauvel C, Raitière O, Belkacem NS, Dominique S, Artaud-Macari E, Viacroze C, Schleifer D, Bauer F. Prognostic importance of Kidney, Heart and Interstitial lung diseases (KHI triad) in PH: A machine learning study. Arch Cardiovasc Dis 113: 630–641, 2020. [DOI] [PubMed] [Google Scholar]
- 51.Federici C, Drake KM, Rigelsky CM, McNelly LN, Meade SL, Comhair SA, Erzurum SC, Aldred MA. Increased mutagen sensitivity and DNA damage in pulmonary arterial hypertension. Am J Respir Crit Care Med 192: 219–228, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fessel JP, Hamid R, Wittmann BM, Robinson LJ, Blackwell T, Tada Y, Tanabe N, Tatsumi K, Hemnes AR, West JD. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm Circ 2: 201–213, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Firth AL, Remillard CV, Platoshyn O, Fantozzi I, Ko EA, Yuan JX. Functional ion channels in human pulmonary artery smooth muscle cells: Voltage-dependent cation channels. Pulm Circ 1: 48–71, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Florentin J, Coppin E, Vasamsetti SB, Zhao J, Tai YY, Tang Y, Zhang Y, Watson A, Sembrat J, Rojas M, Vargas SO, Chan SY, Dutta P. Inflammatory macrophage expansion in pulmonary hypertension depends upon mobilization of blood-borne monocytes. J Immunol 200: 3612–3625, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, Roedersheimer MT, van Rooijen N, Stenmark KR. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol 168: 659–669, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frid MG, Dempsey EC, Durmowicz AG, Stenmark KR. Smooth muscle cell heterogeneity in pulmonary and systemic vessels. Importance in vascular disease. Arterioscler Thromb Vasc Biol 17: 1203–1209, 1997. [DOI] [PubMed] [Google Scholar]
- 57.Frid MG, McKoen BA, Thurman JM, Maron BA, Li M, Zhang H, Kumar S, Sullivan T, Laskowsky J, Fini MA, Hu S, Tuder RM, Gandjeva A, Wilkins MR, Rhodes CJ, Ghataorphe P, Leopold JA, Wang R-S, Holers VM, Stenmark KR. Immunoglobulin-driven complement activation regulates pro-inflammatory remodeling in pulmonary hypertension. Am J Respir Crit Care Med 201 (2): 224–239, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur RespirJ 53: 1801913, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ghigna M-R, Dorfmüller P Pulmonary vascular disease and pulmonary hypertension. Diagn Histopathol 25: 304–312, 2019. [Google Scholar]
- 60.Ghigna MR, Mooi WJ, Grunberg K. Pulmonary hypertensive vasculopathy in parenchymal lung diseases and/or hypoxia: Number 1 in the Series “Pathology for the clinician” Edited by Peter Dorfmuller and Alberto Cavazza. Eur Respir Rev 26: 170003, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gou Q, Shi R, Zhang X, Meng Q, Li X, Rong X, Gawa Z, Zhuoma N, Chen X. The prevalence and risk factors of high-altitude pulmonary hypertension among native Tibetans in Sichuan Province China. High Alt Med Biol 21: 327–335, 2020. [DOI] [PubMed] [Google Scholar]
- 62.Goudie AR, Lipworth BJ, Hopkinson PJ, Wei L, Struthers AD. Tadalafil in patients with chronic obstructive pulmonary disease: A randomised, double-blind, parallel-group, placebo-controlled trial. Lancet Respir Med 2: 293–300, 2014. [DOI] [PubMed] [Google Scholar]
- 63.Graf S, Haimel M, Bleda M, Hadinnapola C, Southgate L, Li W, Hodgson J, Liu B, Salmon RM, Southwood M, Machado RD, Martin JM, Treacy CM, Yates K, Daugherty LC, Shamardina O, Whitehorn D, Holden S, Aldred M, Bogaard HJ, Church C, Coghlan G, Condliffe R, Corris PA, Danesino C, Eyries M, Gall H, Ghio S, Ghofrani HA, Gibbs JSR, Girerd B, Houweling AC, Howard L, Humbert M, Kiely DG, Kovacs G, MacKenzie Ross RV, Moledina S, Montani D, Newnham M, Olschewski A, Olschewski H, Peacock AJ, Pepke-Zaba J, Prokopenko I, Rhodes CJ, Scelsi L, Seeger W, Soubrier F, Stein DF, Suntharalingam J, Swietlik EM, Toshner MR, van Heel DA, Vonk Noordegraaf A, Waisfisz Q, Wharton J, Wort SJ, Ouwehand WH, Soranzo N, Lawrie A, Upton PD, Wilkins MR, Trembath RC, Morrell NW. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat Commun 9: 1416, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gredic M, Blanco I, Kovacs G, Helyes Z, Ferdinandy P, Olschewski H, Barbera JA, Weissmann N. Pulmonary hypertension in chronic obstructive pulmonary disease. Br J Pharmacol 178: 132–151, 2021. [DOI] [PubMed] [Google Scholar]
- 65.Gredic M, Wu C-Y, Hadzic S, Pak O, Savai R, Kojonazarov B, Doswada S, Weiss A, Weigert A, Guenther A, Brandes RP, Schermuly RT, Grimminger F, Seeger W, Sommer N, Kraut S, Weissmann N. Myeloid-cell-specific deletion of inducible nitric oxide synthase protects against smoke-induced pulmonary hypertension in mice. Eur Respir J 59: 2101153, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guignabert C, Tu L, Le Hiress M, Ricard N, Sattler C, Seferian A, Huertas A, Humbert M, Montani D. Pathogenesis of pulmonary arterial hypertension: Lessons from cancer. Eur Respir Rev 22: 543–551, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hainsworth R, Drinkhill MJ. Cardiovascular adjustments for life at high altitude. Respir Physiol Neurobiol 158: 204–211, 2007. [DOI] [PubMed] [Google Scholar]
- 68.Harbaum L, Rhodes CJ, Wharton J, Lawrie A, Karnes JH, Desai AA, Nichols WC, Humbert M, Montani D, Girerd B, Sitbon O, Boehm M, Novoyatleva T, Schermuly RT, Ghofrani HA, Toshner M, Kiely DG, Howard LS, Swietlik EM, Graf S, Pietzner M, Morrell NW, Wilkins MR, U.K. National Institute for Health Research BioResource Rare Diseases Consortium UKPAHCSC, Consortium USPAHB. Mining the plasma proteome for insights into the molecular pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med 205: 1449–1460, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harrison RE, Berger R, Haworth SG, Tulloh R, Mache CJ, Morrell NW, Aldred MA, Trembath RC. Transforming growth factor-beta receptor mutations and pulmonary arterial hypertension in childhood. Circulation 111: 435–441, 2005. [DOI] [PubMed] [Google Scholar]
- 70.Hassoun PM, Mouthon L, Barbera JA, Eddahibi S, Flores SC, Grimminger F, Jones PL, Maitland ML, Michelakis ED, Morrell NW, Newman JH, Rabinovitch M, Schermuly R, Stenmark KR, Voelkel NF, Yuan JX, Humbert M. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol 54: S10–S19, 2009. [DOI] [PubMed] [Google Scholar]
- 71.Hatano S, Strasser T, and World Health Organization. Primary pulmonary hypertension: report on a WHO meeting, Geneva, 15–17 October 1973/edited by Shuichi Hatano and Toma Strasser Geneva: World Health Organization, 1975. [Google Scholar]
- 72.Heath D, Edwards JE. The pathology of hypertensive pulmonary vascular disease; a description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac septal defects. Circulation 18: 533–547, 1958. [DOI] [PubMed] [Google Scholar]
- 73.Hemnes AR, Beck GJ, Newman JH, Abidov A, Aldred MA, Barnard J, Berman Rosenzweig E, Borlaug BA, Chung WK, SAA C, Erzurum SC, Frantz RP, Gray MP, Grunig G, Hassoun PM, Hill NS, Horn EM, Hu B, Lempel JK, Maron BA, Mathai SC, Olman MA, Rischard FP, Systrom DM, WHW T, Waxman AB, Xiao L, Yuan JX, Leopold JA, Group PS. PVDOMICS: A multi-center study to improve understanding of pulmonary vascular disease through phenomics. Circ Res 121: 1136–1139, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hochachka PW. Defense strategies against hypoxia and hypothermia. Science 231: 234–241, 1986. [DOI] [PubMed] [Google Scholar]
- 75.Hoffmann J, Marsh LM, Pieper M, Stacher E, Ghanim B, Kovacs G, Konig P, Wilkens H, Haitchi HM, Hoefler G, Klepetko W, Olschewski H, Olschewski A, Kwapiszewska G. Compartment-specific expression of collagens and their processing enzymes in intrapulmonary arteries of IPAH patients. Am J Physiol Lung Cell Mol Physiol 308: L1002–L1013, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hoffmann J, Wilhelm J, Marsh LM, Ghanim B, Klepetko W, Kovacs G, Olschewski H, Olschewski A, Kwapiszewska G. Distinct differences in gene expression patterns in pulmonary arteries of patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis with pulmonary hypertension. Am J Respir Crit Care Med 190: 98–111, 2014. [DOI] [PubMed] [Google Scholar]
- 77.Huicho L Postnatal cardiopulmonary adaptations to high altitude. Respir Physiol Neurobiol 158: 190–203, 2007. [DOI] [PubMed] [Google Scholar]
- 78.Humbert M, Guignabert C, Bonnet S, Dorfmuller P, Klinger JR, Nicolls MR, Olschewski AJ, Pullamsetti SS, Schermuly RT, Stenmark KR, Rabinovitch M. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur Respir J 53: 1801887, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Humbert M, McLaughlin V, Gibbs JSR, Gomberg-Maitland M, Hoeper MM, Preston IR, Souza R, Waxman A, Escribano Subias P, Feldman J, Meyer G, Montani D, Olsson KM, Manimaran S, Barnes J, Linde PG, de Oliveira PJ, Badesch DB, Investigators PT. Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med 384: 1204–1215, 2021. [DOI] [PubMed] [Google Scholar]
- 80.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med 351: 1425–1436, 2004. [DOI] [PubMed] [Google Scholar]
- 81.Idiopathic Pulmonary Fibrosis Clinical Research Network, Zisman DA, Schwarz M, Anstrom KJ, Collard HR, Flaherty KR, Hunninghake GW. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med 363: 620–628, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.International PPHC Lane KB, Machado RD Pauciulo MW, Thomson JR Phillips JA 3rd, Loyd JE Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet 26: 81–84, 2000. [DOI] [PubMed] [Google Scholar]
- 83.Jonigk D, Golpon H, Bockmeyer CL, Maegel L, Hoeper MM, Gottlieb J, Nickel N, Hussein K, Maus U, Lehmann U, Janciauskiene S, Welte T, Haverich A, Rische J, Kreipe H, Laenger F. Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am J Pathol 179: 167–179, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jonigk D, Stark H, Braubach P, Neubert L, Shin H-o, Izykowski N, Welte T, Janciauskiene S, Warnecke G, Haverich A, Kuehnel M, Laenger F. Morphological and molecular motifs of fibrosing pulmonary injury patterns. J Pathol Clin Res 5: 256–271, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kanwar MK, Gomberg-Maitland M, Hoeper M, Pausch C, Pittow D, Strange G, Anderson JJ, Zhao C, Scott JV, Druzdzel MJ, Kraisangka J, Lohmueller L, Antaki J, Benza RL. Risk stratification in pulmonary arterial hypertension using Bayesian analysis. Eur Respir J 56: 2000008, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kariotis S, Jammeh E, Swietlik EM, Pickworth JA, Rhodes CJ, Otero P, Wharton J, Iremonger J, Dunning MJ, Pandya D, Mascarenhas TS, Errington N, AAR T, Romanoski CE, Rischard F, JGN G, JXJ Y, THS A, Desai AA, Coghlan G, Lordan J, Corris PA, Howard LS, Condliffe R, Kiely DG, Church C, Pepke-Zaba J, Toshner M, Wort S, Gräf S, Morrell NW, Wilkins MR, Lawrie A, Wang D, Bleda M, Hadinnapola C, Haimel M, Auckland K, Tilly T, Martin JM, Yates K, Treacy CM, Day M, Greenhalgh A, Shipley D, Peacock AJ, Irvine V, Kennedy F, Moledina S, Macdonald L, Tamvaki E, Barnes A, Cookson V, Chentouf L, Ali S, Othman S, Ranganathan L, JSR G, Dacosta R, Pinguel J, Dormand N, Parker A, Stokes D, Ghedia D, Tan Y, Ngcozana T, Wanjiku I, Polwarth G, Mackenzie Ross RV, Suntharalingam J, Grover M, Kirby A, Grove A, White K, Seatter A, Creaser-Myers A, Walker S, Roney S, Elliot CA, Charalampopoulos A, Sabroe I, Hameed A, Armstrong I, Hamilton N, AMK R, Swift AJ, Wild JM, Soubrier F, Eyries M, Humbert M, Montani D, Girerd B, Scelsi L, Ghio S, Gall H, Ghofrani A, Bogaard HJ, Noordegraaf AV, Houweling AC, AHIT V, Schotte G. Biological heterogeneity in idiopathic pulmonary arterial hypertension identified through unsupervised transcriptomic profiling of whole blood. Nat Commun 12: 7104, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kessler R, Faller M, Weitzenblum E, Chaouat A, Aykut A, Ducoloné A, Ehrhart M, Oswald-Mammosser M. “Natural History” of pulmonary hypertension in a series of 131 patients with chronic obstructive lung disease. Am J Respir Crit Care Med 162: 219–224, 2001. [DOI] [PubMed] [Google Scholar]
- 88.Kessler RFM, Fourgaut G, Mennecier B, Weitzenblum E. Predictive factors of hospitalization for acute exacerbation in a series of 64 patietns with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 159: 158–164, 1999. [DOI] [PubMed] [Google Scholar]
- 89.Kheyfets VO, Schafer M, Podgorski CA, Schroeder JD, Browning J, Hertzberg J, Buckner JK, Hunter KS, Shandas R, Fenster BE. 4D Magnetic resonance flow imaging for estimating pulmonary vascular resistance in pulmonary hypertension. J Magn Reson Imaging 44: 914–922, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kheyfets VO, Sucharov CC, Truong U, Dunning J, Hunter K, Ivy D, Miyamoto S, Shandas R. Circulating miRNAs in pediatric pulmonary hypertension show promise as biomarkers of vascular function. Oxidative Med Cell Longev 2017: 4957147, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kheyfets VO, Sweatt A, Gomberg-Maitland M, Ivy D, Kiely D, Lawrie A, Zamanian R, Stemark K. Computational platform for doctor-AI cooperation in AI prognostication: A pilot study. ERJ Open Research Submitted Sept 24 2022. In Press. [DOI] [PMC free article] [PubMed]
- 92.Kiely DG, Doyle O, Drage E, Jenner H, Salvatelli V, Daniels FA, Rigg J, Schmitt C, Samyshkin Y, Lawrie A, Bergemann R. Utilising artificial intelligence to determine patients at risk of a rare disease: Idiopathic pulmonary arterial hypertension. Pulm Circ 9: 2045894019890549, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kimura M, Taniguchi H, Kondoh Y, Kimura T, Kataoka K, Nishiyama O, Aso H, Sakamoto K, Hasegawa Y. Pulmonary hypertension as a prognostic indicator at the initial evaluation in idiopathic pulmonary fibrosis. Respiration 85: 456–463, 2013. [DOI] [PubMed] [Google Scholar]
- 94.King CS, Shlobin OA. The trouble with group 3 pulmonary hypertension in interstitial lung disease: Dilemmas in diagnosis and the conundrum of treatment. Chest 158: 1651–1664, 2020. [DOI] [PubMed] [Google Scholar]
- 95.King TE Jr, Behr J, Brown KK, du Bois RM, Lancaster L, de Andrade JA, Stahler G, Leconte I, Roux S, Raghu G. BUILD-1: A randomized placebo-controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 177: 75–81, 2008. [DOI] [PubMed] [Google Scholar]
- 96.Kissinger H, Schmidt E, Huttenlocher DP, Schouten S. The Age of AI: And our Human Future New York: Little Brown and Company, 2021, p. ix, 254 pages. [Google Scholar]
- 97.Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: A systematic review. Eur Respir J 34: 888–894, 2009. [DOI] [PubMed] [Google Scholar]
- 98.Krenz GS, Dawson CA. Flow and pressure distributions in vascular networks consisting of distensible vessels. Am J Physiol Heart Circ Physiol 284: H2192–H2203, 2003. [DOI] [PubMed] [Google Scholar]
- 99.Kubo K, Ge RL, Koizumi T, Fujimoto K, Yamanda T, Haniuda M, Honda T. Pulmonary artery remodeling modifies pulmonary hypertension during exercise in severe emphysema. Respir Physiol 120: 71–79, 2000. [DOI] [PubMed] [Google Scholar]
- 100.Kumar R, Mickael C, Kassa B, Sanders L, Hernandez-Saavedra D, Koyanagi DE, Kumar S, Pugliese SC, Thomas S, McClendon J, Maloney JP, Janssen WJ, Stenmark KR, Tuder RM, Graham BB. Interstitial macrophage-derived thrombospondin-1 contributes to hypoxia-induced pulmonary hypertension. Cardiovasc Res 116: 2021–2030, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lan B, Hayama E, Kawaguchi N, Furutani Y, Nakanishi T. Therapeutic efficacy of valproic acid in a combined monocrotaline and chronic hypoxia rat model of severe pulmonary hypertension. PLoS One 10: e0117211, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee SD, Shroyer KR, Markham NE, Cool CD, Voelkel NF, Tuder RM. Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J Clin Investig 101: 927–934, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.León-Velarde F, Maggiorini M, Reeves JT, Aldashev A, Asmus I, Bernardi L, Ge RL, Hackett P, Kobayashi T, Moore LG, Penaloza D, Richalet JP, Roach R, Wu T, Vargas E, Zubieta-Castillo G, Zubieta-Calleja G. Consensus statement on chronic and subacute high altitude diseases. High Alt Med Biol 6: 147–157, 2005. [DOI] [PubMed] [Google Scholar]
- 104.León-Velarde F, Villafuerte FC, Richalet JP. Chronic mountain sickness and the heart. Prog Cardiovasc Dis 52: 540–549, 2010. [DOI] [PubMed] [Google Scholar]
- 105.Leopold JA, Maron BA, Loscalzo J. The application of big data to cardiovascular disease: Paths to precision medicine. J Clin Invest 130: 29–38, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lettieri CJ, Nathan SD, Barnett SD, Ahmad S, Shorr AF. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest 129: 746–752, 2006. [DOI] [PubMed] [Google Scholar]
- 107.Lewis GD, Ngo D, Hemnes AR, Farrell L, Domos C, Pappagianopoulos PP, Dhakal BP, Souza A, Shi X, Pugh ME, Beloiartsev A, Sinha S, Clish CB, Gerszten RE. Metabolic profiling of right ventricular-pulmonary vascular function reveals circulating biomarkers of pulmonary hypertension. J Am Coll Cardiol 67: 174–189, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lichtblau M, Saxer S, Furian M, Mayer L, Bader PR, Scheiwiller PM, Mademilov M, Sheraliev U, Tanner FC, Sooronbaev TM, Bloch KE, Ulrich S. Cardiac function and pulmonary hypertension in Central Asian highlanders at 3250 m. Eur Respir J 56: 1902474, 2020. [DOI] [PubMed] [Google Scholar]
- 109.Lin S, Du L. The therapeutic potential of BRD4 in cardiovascular disease. Hypertens Res 43: 1006–1014, 2020. [DOI] [PubMed] [Google Scholar]
- 110.Liu X, Zhang S, Wang X, Wang Y, Song J, Sun C, Chen G, Yang G, Tao Y, Hu Y, Bu D, Huang Y, Du J, Jin H. Endothelial cell-derived SO(2) controls endothelial cell inflammation, smooth muscle cell proliferation, and collagen synthesis to inhibit hypoxic pulmonary vascular remodelling. Oxidative Med Cell Longev 2021: 5577634, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Long L, Ormiston ML, Yang X, Southwood M, Graf S, Machado RD, Mueller M, Kinzel B, Yung LM, Wilkinson JM, Moore SD, Drake KM, Aldred MA, Yu PB, Upton PD, Morrell NW. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat Med 21: 777–785, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Machado RD, Southgate L, Eichstaedt CA, Aldred MA, Austin ED, Best DH, Chung WK, Benjamin N, Elliott CG, Eyries M, Fischer C, Graf S, Hinderhofer K, Humbert M, Keiles SB, Loyd JE, Morrell NW, Newman JH, Soubrier F, Trembath RC, Viales RR, Grunig E. Pulmonary arterial hypertension: A current perspective on established and emerging molecular genetic defects. Hum Mutat 36: 1113–1127, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Magee F, Wright JL, Wiggs BR, Pare PD, Hogg JC. Pulmonary vascular structure and function in chronic obstructive pulmonary disease. Thorax 43: 183–189, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Masri FA, Xu W, Comhair SA, Asosingh K, Koo M, Vasanji A, Drazba J, nand-Apte B, and Erzurum SC. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 293: L548–L554, 2007. [DOI] [PubMed] [Google Scholar]
- 115.Maston LD, Jones DT, Giermakowska W, Resta TC, Ramiro-Diaz J, Howard TA, Jernigan NL, Herbert L, Maurice AA, Gonzalez Bosc LV. Interleukin-6 trans-signaling contributes to chronic hypoxia-induced pulmonary hypertension. Pulm Circ 8: 2045894018780734, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Meloche J, Potus F, Vaillancourt M, Bourgeois A, Johnson I, Deschamps L, Chabot S, Ruffenach G, Henry S, Breuils-Bonnet S, Tremblay E, Nadeau V, Lambert C, Paradis R, Provencher S, Bonnet S. Bromodomain-containing protein 4: The epigenetic origin of pulmonary arterial hypertension. Circ Res 117: 525–535, 2015. [DOI] [PubMed] [Google Scholar]
- 117.Meyrick B, Reid L. Hypoxia-induced structural changes in the media and adventitia of the rat hilar pulmonary artery and their regression. Am J Pathol 100: 151–178, 1980. [PMC free article] [PubMed] [Google Scholar]
- 118.Michelakis ED, Gurtu V, Webster L, Barnes G, Watson G, Howard L, Cupitt J, Paterson I, Thompson RB, Chow K, O’Regan DP, Zhao L, Wharton J, Kiely DG, Kinnaird A, Boukouris AE, White C, Nagendran J, Freed DH, Wort SJ, Gibbs JSR, Wilkins MR. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci Transl Med 9: eaao4583, 2017. [DOI] [PubMed] [Google Scholar]
- 119.Minai OA, Chaouat A, Adnot S. Pulmonary hypertension in COPD: Epidemiology, significance, and management: Pulmonary vascular disease: The global perspective. Chest 137: 39S–51S, 2010. [DOI] [PubMed] [Google Scholar]
- 120.Minai OA, Santacruz JF, Alster JM, Budev MM, McCarthy K. Impact of pulmonary hemodynamics on 6-min walk test in idiopathic pulmonary fibrosis. Respir Med 106: 1613–1621, 2012. [DOI] [PubMed] [Google Scholar]
- 121.Moudgil R, Michelakis ED, Archer SL. Hypoxic pulmonary vasoconstriction. J Appl Physiol (1985) 98: 390–403, 2005. [DOI] [PubMed] [Google Scholar]
- 122.Nadrous HF, Pellikka PA, Krowka MJ, Swanson KL, Chaowalit N, Decker PA, Ryu JH. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Chest 128: 2393–2399, 2005. [DOI] [PubMed] [Google Scholar]
- 123.Naeije R, Chesler N. Pulmonary circulation at exercise. Compr Physiol 2: 711–741, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nasim MT, Ogo T, Ahmed M, Randall R, Chowdhury HM, Snape KM, Bradshaw TY, Southgate L, Lee GJ, Jackson I, Lord GM, Gibbs JS, Wilkins MR, Ohta-Ogo K, Nakamura K, Girerd B, Coulet F, Soubrier F, Humbert M, Morrell NW, Trembath RC, Machado RD. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum Mutat 32: 1385–1389, 2011. [DOI] [PubMed] [Google Scholar]
- 125.Nathan SD, Behr J, Collard HR, Cottin V, Hoeper MM, Martinez FJ, Corte TJ, Keogh AM, Leuchte H, Mogulkoc N, Ulrich S, Wuyts WA, Yao Z, Boateng F, Wells AU. Riociguat for idiopathic interstitial pneumonia-associated pulmonary hypertension (RISE-IIP): A randomised, placebo-controlled phase 2b study. Lancet Respir Med 7: 780–790, 2019. [DOI] [PubMed] [Google Scholar]
- 126.Nathan SD, Shlobin OA, Ahmad S, Koch J, Barnett SD, Ad N, Burton N, Leslie K. Serial development of pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respiration 76: 288–294, 2008. [DOI] [PubMed] [Google Scholar]
- 127.Negi PC, Marwaha R, Asotra S, Kandoria A, Ganju N, Sharma R, Kumar RV, Bhardwaj R. Prevalence of high altitude pulmonary hypertension among the natives of Spiti Valley—a high altitude region in Himachal Pradesh, India. High Alt Med Biol 15: 504–510, 2014. [DOI] [PubMed] [Google Scholar]
- 128.Neubert L, Borchert P, Stark H, Hoefer A, Vogel-Claussen J, Warnecke G, Eubel H, Kuenzler P, Kreipe HH, Hoeper MM, Kuehnel M, Jonigk D. Molecular Profiling of Vascular Remodeling in Chronic Pulmonary Disease. Am J Pathol 190: 1382–1396, 2020. [DOI] [PubMed] [Google Scholar]
- 129.Oldham WM, Hess E, Waldo SW, Humbert M, Choudhary G, Maron BA. Integrating haemodynamics identifies an extreme pulmonary hypertension phenotype. Eur Respir J 58: 2004625, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Oldham WM, Oliveira RKF, Wang RS, Opotowsky AR, Rubins DM, Hainer J, Wertheim BM, Alba GA, Choudhary G, Tornyos A, MacRae CA, Loscalzo J, Leopold JA, Waxman AB, Olschewski H, Kovacs G, Systrom DM, Maron BA. Network analysis to risk stratify patients with exercise intolerance. Circ Res 122: 864–876, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Oswald-Mammosser M, Weitzenblum E, Quoix E, Moser G, Chaouat A, Charpentier C, Kessler R. Prognostic factors in COPD patients receiving long-term oxygen therapy. Importance of pulmonary artery pressure. Chest 107: 1193–1198, 1995. [DOI] [PubMed] [Google Scholar]
- 132.Parra ER, David YR, da Costa LR, Ab’Saber A, Sousa R, Kairalla RA, de Carvalho CR, Filho MT, Capelozzi VL. Heterogeneous remodeling of lung vessels in idiopathic pulmonary fibrosis. Lung 183: 291–300, 2005. [DOI] [PubMed] [Google Scholar]
- 133.Patel NM, Kawut SM, Jelic S, Arcasoy SM, Lederer DJ, Borczuk AC. Pulmonary arteriole gene expression signature in idiopathic pulmonary fibrosis. Eur Respir J 41: 1324–1330, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Penaloza D, Arias-Stella J. The heart and pulmonary circulation at high altitudes: Healthy highlanders and chronic mountain sickness. Circulation 115: 1132–1146, 2007. [DOI] [PubMed] [Google Scholar]
- 135.Polverino F, Laucho-Contreras ME, Petersen H, Bijol V, Sholl LM, Choi ME, Divo M, Pinto-Plata V, Chetta A, Tesfaigzi Y, Celli BR, Owen CA. A pilot study linking endothelial injury in lungs and kidneys in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 195: 1464–1476, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Potus F, Pauciulo MW, Cook EK, Zhu N, Hsieh A, Welch CL, Shen Y, Tian L, Lima P, Mewburn J, D’Arsigny CL, Lutz KA, Coleman AW, Damico R, Snetsinger B, Martin AY, Hassoun PM, Nichols WC, Chung WK, Rauh MJ, Archer SL. Novel mutations and decreased expression of the epigenetic regulator TET2 in pulmonary arterial hypertension. Circulation 141: 1986–2000, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Potus F, Ruffenach G, Dahou A, Thebault C, Breuils-Bonnet S, Tremblay E, Nadeau V, Paradis R, Graydon C, Wong R, Johnson I, Paulin R, Lajoie AC, Perron J, Charbonneau E, Joubert P, Pibarot P, Michelakis ED, Provencher S, Bonnet S. Downregulation of MicroRNA-126 contributes to the failing right ventricle in pulmonary arterial hypertension. Circulation 132: 932–943, 2015. [DOI] [PubMed] [Google Scholar]
- 138.Prins KW, Rose L, Archer SL, Pritzker M, Weir EK, Kazmirczak F, Misialek JR, Thenappan T. Disproportionate Right Ventricular Dysfunction and Poor Survival in Group 3 Pulmonary Hypertension. Am J Respir Crit Care Med 197: 1496–1499, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pu X, Lin X, Duan X, Wang J, Shang J, Yun H, Chen Z. Oxidative and endoplasmic reticulum stress responses to chronic high-altitude exposure during the development of high-altitude pulmonary hypertension. High Alt Med Biol 21: 378–387, 2020. [DOI] [PubMed] [Google Scholar]
- 140.Pullamsetti SS, Savai R, Seeger W, Goncharova EA. Translational advances in the field of pulmonary hypertension. From cancer biology to new pulmonary arterial hypertension therapeutics. Targeting cell growth and proliferation signaling hubs. Am J Respir Crit Care Med 195: 425–437, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rabinovitch M Investigational approaches to pulmonary hypertension. Toxicol Pathol 19: 458–469, 1991. [DOI] [PubMed] [Google Scholar]
- 142.Rabinovitch M, Gamble W, Nadas AS, Miettinen OS, Reid L. Rat pulmonary circulation after chronic hypoxia: Hemodynamic and structural features. Am J Phys 236: H818–H827, 1979. [DOI] [PubMed] [Google Scholar]
- 143.Rabinovitch M, Gamble WJ, Miettinen OS, Reid L. Age and sex influence on pulmonary hypertension of chronic hypoxia and on recovery. Am J Phys 240: H62–H72, 1981. [DOI] [PubMed] [Google Scholar]
- 144.Rabinovitch M, Konstam MA, Gamble WJ, Papanicolaou N, Aronovitz MJ, Treves S, Reid L. Changes in pulmonary blood flow affect vascular response to chronic hypoxia in rats. Circ Res 52: 432–441, 1983. [DOI] [PubMed] [Google Scholar]
- 145.Raghu G, Behr J, Brown KK, Egan JJ, Kawut SM, Flaherty KR, Martinez FJ, Nathan SD, Wells AU, Collard HR, Costabel U, Richeldi L, de Andrade J, Khalil N, Morrison LD, Lederer DJ, Shao L, Li X, Pedersen PS, Montgomery AB, Chien JW, O’Riordan TG, ARTEMIS-IPF Investigators. Treatment of idiopathic pulmonary fibrosis with ambrisentan: A parallel, randomized trial. Ann Intern Med 158: 641–649, 2013. [DOI] [PubMed] [Google Scholar]
- 146.Raghu G, Million-Rousseau R, Morganti A, Perchenet L, Behr J, Group MS. Macitentan for the treatment of idiopathic pulmonary fibrosis: The randomised controlled MUSIC trial. Eur Respir J 42: 1622–1632, 2013. [DOI] [PubMed] [Google Scholar]
- 147.Rai PR, Cool CD, King JAC, Stevens T, Burns N, Winn RA, Kasper M, Voelkel NF. The cancer paradigm of severe pulmonary arterial hypertension. Am J Respir Crit Care Med 178: 558–564, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rao RS, Singh S, Sharma BB, Agarwal VV, Singh V. Sildenafil improves six-minute walk distance in chronic obstructive pulmonary disease: A randomised, double-blind, placebo-controlled trial. Indian J Chest Dis Allied Sci 53: 81–85, 2011. [PubMed] [Google Scholar]
- 149.Reeves JT, Rubin LJ. The pulmonary circulation: Snapshots of progress. Am J Respir Crit Care Med 157: S101–S108, 1998. [DOI] [PubMed] [Google Scholar]
- 150.Remillard CV, Yuan JX. High altitude pulmonary hypertension: Role of K+ and Ca2+ channels. High Alt Med Biol 6: 133–146, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rhodes CJ, Ghataorhe P, Wharton J, Rue-Albrecht KC, Hadinnapola C, Watson G, Bleda M, Haimel M, Coghlan G, Corris PA, Howard LS, Kiely DG, Peacock AJ, Pepke-Zaba J, Toshner MR, Wort SJ, Gibbs JS, Lawrie A, Graf S, Morrell NW, Wilkins MR. Plasma metabolomics implicates modified transfer RNAs and altered bioenergetics in the outcomes of pulmonary arterial hypertension. Circulation 135: 460–475, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rhodes CJ, Sweatt AJ, Maron BA. Harnessing big data to advance treatment and understanding of pulmonary hypertension. Circ Res 130: 1423–1444, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rhodes CJ, Wharton J, Ghataorhe P, Watson G, Girerd B, Howard LS, Gibbs JSR, Condliffe R, Elliot CA, Kiely DG, Simonneau G, Montani D, Sitbon O, Gall H, Schermuly RT, Ghofrani HA, Lawrie A, Humbert M, Wilkins MR. Plasma proteome analysis in patients with pulmonary arterial hypertension: An observational cohort study. Lancet Respir Med 5: 717–726, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rothman AM, Arnold ND, Pickworth JA, Iremonger J, Ciuclan L, Allen RM, Guth-Gundel S, Southwood M, Morrell NW, Thomas M, Francis SE, Rowlands DJ, Lawrie A. MicroRNA-140-5p and SMURF1 regulate pulmonary arterial hypertension. J Clin Invest 126: 2495–2508, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, Pulido T, Frost A, Roux S, Leconte I, Landzberg M, Simonneau G. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 346: 896–903, 2002. [DOI] [PubMed] [Google Scholar]
- 156.Ryu JH, Krowka MJ, Pellikka PA, Swanson KL, McGoon MD. Pulmonary hypertension in patients with interstitial lung diseases. Mayo Clin Proc 82: 342–350, 2007. [DOI] [PubMed] [Google Scholar]
- 157.Sakao S, Voelkel NF, Tatsumi K. The vascular bed in COPD: Pulmonary hypertension and pulmonary vascular alterations. Eur Respir Rev 23: 350–355, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Santos S, Peinado VI, Ramirez J, Melgosa T, Roca J, Rodriguez-Roisin R, Barbera JA. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur Respir J 19: 632–638, 2002. [DOI] [PubMed] [Google Scholar]
- 159.Sastry BK, Narasimhan C, Reddy NK, Raju BS. Clinical efficacy of sildenafil in primary pulmonary hypertension: A randomized, placebo-controlled, double-blind, crossover study. J Am Coll Cardiol 43: 1149–1153, 2004. [DOI] [PubMed] [Google Scholar]
- 160.Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, Fink L, Scheed A, Ritter C, Dahal BK, Vater A. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 186: 897–908, 2012. [DOI] [PubMed] [Google Scholar]
- 161.Savale L, Tu L, Rideau D, Izziki M, Maitre B, Adnot S, Eddahibi S. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir Res 10: 6, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Seimetz M, Parajuli N, Pichl A, Veit F, Kwapiszewska G, Weisel FC, Milger K, Egemnazarov B, Turowska A, Fuchs B, Nikam S, Roth M, Sydykov A, Medebach T, Klepetko W, Jaksch P, Dumitrascu R, Garn H, Voswinckel R, Kostin S, Seeger W, Schermuly RT, Grimminger F, Ghofrani HA, Weissmann N. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell 147: 293–305, 2011. [DOI] [PubMed] [Google Scholar]
- 163.Shah SJ, Katz DH, Selvaraj S, Burke MA, Yancy CW, Gheorghiade M, Bonow RO, Huang CC, Deo RC. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation 131: 269–279, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sheikh AQ, Saddouk FZ, Ntokou A, Mazurek R, Greif DM. Cell autonomous and non-cell autonomous regulation of SMC progenitors in pulmonary hypertension. Cell Rep 23: 1152–1165, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Shi L, Kojonazarov B, Elgheznawy A, Popp R, Dahal BK, Bohm M, Pullamsetti SS, Ghofrani HA, Godecke A, Jungmann A, Katus HA, Muller OJ, Schermuly RT, Fisslthaler B, Seeger W, Fleming I. miR-223-IGF-IR signalling in hypoxia- and load-induced right-ventricular failure: A novel therapeutic approach. Cardiovasc Res 111: 184–193, 2016. [DOI] [PubMed] [Google Scholar]
- 166.Shorr AF, Wainright JL, Cors CS, Lettieri CJ, Nathan SD. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur Respir J 30: 715–721, 2007. [DOI] [PubMed] [Google Scholar]
- 167.Simonneau G, Galie N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, Gibbs S, Lebrec D, Speich R, Beghetti M, Rich S, Fishman A. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 43: 5S–12S, 2004. [DOI] [PubMed] [Google Scholar]
- 168.Siques P, Pena E, Brito J, El Alam S. Oxidative stress, kinase activation, and inflammatory pathways involved in effects on smooth muscle cells during pulmonary artery hypertension under hypobaric hypoxia exposure. Front Physiol 12: 690341, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Sitbon O, Humbert M, Jagot JL, Taravella O, Fartoukh M, Parent F, Herve P, Simonneau G. Inhaled nitric oxide as a screening agent for safely identifying responders to oral calcium-channel blockers in primary pulmonary hypertension. Eur Respir J 12: 265–270, 1998. [DOI] [PubMed] [Google Scholar]
- 170.Sitbon O, Humbert M, Jais X, Ioos V, Hamid AM, Provencher S, Garcia G, Parent F, Herve P, Simonneau G. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 111: 3105–3111, 2005. [DOI] [PubMed] [Google Scholar]
- 171.Sobin SS, Tremer HM, Hardy JD, Chiodi HP. Changes in arteriole in acute and chronic hypoxic pulmonary hypertension and recovery in rat. J Appl Physiol Respir Environ Exerc Physiol 55: 1445–1455, 1983. [DOI] [PubMed] [Google Scholar]
- 172.Soria R, Egger M, Scherrer U, Bender N, Rimoldi SF. Pulmonary artery pressure and arterial oxygen saturation in people living at high or low altitude: Systematic review and meta-analysis. J Appl Physiol (1985) 121: 1151–1159, 2016. [DOI] [PubMed] [Google Scholar]
- 173.Spiekerkoetter E, Goncharova EA, Guignabert C, Stenmark K, Kwapiszewska G, Rabinovitch M, Voelkel N, Bogaard HJ, Graham B, Pullamsetti SS, Kuebler WM. Hot topics in the mechanisms of pulmonary arterial hypertension disease: Cancer-like pathobiology, the role of the adventitia, systemic involvement, and right ventricular failure. Pulm Circ 9: 2045894019889775, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Spiekerkoetter E, Sung YK, Sudheendra D, Scott V, Del Rosario P, Bill M, Haddad F, Long-Boyle J, Hedlin H, Zamanian RT. Randomised placebo-controlled safety and tolerability trial of FK506 (tacrolimus) for pulmonary arterial hypertension. Eur Respir J 50, 2017. [DOI] [PubMed] [Google Scholar]
- 175.Spiekerkoetter E, Tian X, Cai J, Hopper RK, Sudheendra D, Li CG, El-Bizri N, Sawada H, Haghighat R, Chan R, Haghighat L, de Jesus PV, Wang L, Reddy S, Zhao M, Bernstein D, Solow-Cordero DE, Beachy PA, Wandless TJ, Ten Dijke P, Rabinovitch M. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest 123: 3600–3613, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, Jessup M, Grizzle WE, Aldred MA, Cool CD, Tuder RM. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med 186: 261–272, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: Cellular and molecular mechanisms. Circ Res 99: 675–691, 2006. [DOI] [PubMed] [Google Scholar]
- 178.Stenmark KR, Frid MG, Graham BB, Tuder RM. Dynamic and diverse changes in the functional properties of vascular smooth muscle cells in pulmonary hypertension. Cardiovasc Res 114: 551–564, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Stenmark KR, McMurtry IF. Vascular remodeling versus vasoconstriction in chronic hypoxic pulmonary hypertension: A time for reappraisal? Circ Res 97: 95–98, 2005. [DOI] [PubMed] [Google Scholar]
- 180.Stenmark KR, Mecham RP. Cellular and molecular mechanisms of pulmonary vascular remodeling. Annu Rev Physiol 59: 89–144, 1997. [DOI] [PubMed] [Google Scholar]
- 181.Stokes JM, Yang K, Swanson K, Jin W, Cubillos-Ruiz A, Donghia NM, MacNair CR, French S, Carfrae LA, Bloom-Ackermann Z, Tran VM, Chiappino-Pepe A, Badran AH, Andrews IW, Chory EJ, Church GM, Brown ED, Jaakkola TS, Barzilay R, Collins JJ. A deep learning approach to antibiotic discovery. Cell 180: 688–702 e613, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Stolz D, Rasch H, Linka A, Di Valentino M, Meyer A, Brutsche M, Tamm M. A randomised, controlled trial of bosentan in severe COPD. Eur Respir J 32: 619–628, 2008. [DOI] [PubMed] [Google Scholar]
- 183.Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, Karwande SV, Stringham JC, Bull DA, Gleich M, Kennedy TP, Hoidal JR. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 290: L661–L673, 2006. [DOI] [PubMed] [Google Scholar]
- 184.Sweatt AJ, Hedlin HK, Balasubramanian V, Hsi A, Blum LK, Robinson WH, Haddad F, Hickey PM, Condliffe R, Lawrie A, Nicolls MR, Rabinovitch M, Khatri P, Zamanian RT. Discovery of distinct immune phenotypes using machine learning in pulmonary arterial hypertension. Circ Res 124: 904–919, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Swietlik EM, Ghataorhe P, Zalewska KI, Wharton J, Howard LS, Taboada D, Cannon JE, UKNCSo PAH, Morrell NW, Wilkins MR, Toshner M, Pepke-Zaba J, Rhodes CJ. Plasma metabolomics exhibit response to therapy in chronic thromboembolic pulmonary hypertension. Eur Respir J 57: 2003201, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev 92: 367–520, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Sysol JR, Machado RF. Classification and pathophysiology of pulmonary hypertension. Contin Cardiol Educ 4: 2–12, 2018. [Google Scholar]
- 188.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mahon GM, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J 15: 427–438, 2001. [DOI] [PubMed] [Google Scholar]
- 189.Taraseviciene-Stewart L, Voelkel NF. Molecular pathogenesis of emphysema. J Clin Invest 118: 394–402, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Thenappan T, Chan SY, Weir EK. Role of extracellular matrix in the pathogenesis of pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 315: H1322–H1331, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Thenappan T, Ormiston ML, Ryan JJ, Archer SL. Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ 360: j5492, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Toba M, Alzoubi A, O’Neill KD, Gairhe S, Matsumoto Y, Oshima K, Abe K, Oka M, McMurtry IF. Temporal hemodynamic and histological progression in Sugen5416/hypoxia/normoxia-exposed pulmonary arterial hypertensive rats. Am J Physiol Heart Circ Physiol 306: H243–H250, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I, Simonneau G, Galie N, Loyd JE, Humbert M, Nichols WC, Morrell NW, Berg J, Manes A, McGaughran J, Pauciulo M, Wheeler L. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 345: 325–334, 2001. [DOI] [PubMed] [Google Scholar]
- 194.Tuder RM. How do we measure pathology in PAH (lung and RV) and what does it tell us about the disease. Drug Discov Today 19: 1257–1263, 2014. [DOI] [PubMed] [Google Scholar]
- 195.Tuder RM. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res 367: 643–649, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Tuder RM, Chacon M, Alger LA, Wang J, Taraseviciene-Stewart L, Kasahara Y, Cool CD, Bishop AE, Geraci MW, Semenza GL, Yacoub M, Polak JM, Voelkel NF. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. J Pathol 195: 367–374, 2001. [DOI] [PubMed] [Google Scholar]
- 197.Tuder RM, Cool CD. Pulmonary arteries and microcirculation in COPD with pulmonary hypertension: Bystander or culprit? Chest 156: 4–6, 2019. [DOI] [PubMed] [Google Scholar]
- 198.Tuder RM, Davis LA, Graham BB. Targeting energetic Metabolism. Am J Respir Crit Care Med 185: 260–266, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Tuder RM, Groves BM, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol 144: 275–285, 1994. [PMC free article] [PubMed] [Google Scholar]
- 200.Tuder RM, Marecki JC, Richter A, Fijalkowska I, Flores S. Pathology of pulmonary hypertension. Clin Chest Med 28: 23–42, vii, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Tuder RM, Petrache I. Pathogenesis of chronic obstructive pulmonary disease. J Clin Invest 122: 2749–2755, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Tuder RM, Radisavljevic Z, Shroyer KR, Polak JM, Voelkel NF. Monoclonal endothelial cells in appetite suppressant-associated pulmonary hypertension. Am J Respir Crit Care Med 158: 1999–2001, 1998. [DOI] [PubMed] [Google Scholar]
- 203.Tuder RM, Stacher E, Robinson J, Kumar R, Graham BB. Pathology of pulmonary hypertension. Clin Chest Med 34: 639–650, 2013. [DOI] [PubMed] [Google Scholar]
- 204.Tuder RM, Stenmark KR. Perspective: Pathobiological paradigms in pulmonary hypertension, time for reappraisal. Am J Physiol Lung Cell Mol Physiol 318: L1131–L1137, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.United Therapeutics Corporation. United Therapeutics announces INCREASE study of Tyvaso ® meets primary and all secondary endpoints https://www.prnewswire.com/news-releases/united-therapeutics-announces-increase-study-of-tyvaso-meets-primary- and-all-secondary-endpoints-301009562.html. [2/22/2022], 2022.
- 206.Valerio G, Bracciale P, Grazia D’AA. Effect of bosentan upon pulmonary hypertension in chronic obstructive pulmonary disease. Ther Adv Respir Dis 3: 15–21, 2009. [DOI] [PubMed] [Google Scholar]
- 207.van der Have O, Westöö C, Ahrné F, Tian X, Ichimura K, Dreier T, Norvik C, Kumar ME, Spiekerkoetter E, Tran-Lundmark K. Shunt-type plexiform lesions identified in the Sugen5416/hypoxia rat model of pulmonary arterial hypertension using synchrotron-based phase-contrast micro-CT. Eur RespirJ 59: 2102802, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Van Goor F, Straley KS, Cao D, Gonzalez J, Hadida S, Hazlewood A, Joubran J, Knapp T, Makings LR, Miller M, Neuberger T, Olson E, Panchenko V, Rader J, Singh A, Stack JH, Tung R, Grootenhuis PD, Negulescu P. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol 290: L1117–L1130, 2006. [DOI] [PubMed] [Google Scholar]
- 209.van Suylen RJ, Smits JF, Daemen MJ. Pulmonary artery remodeling differs in hypoxia- and monocrotaline-induced pulmonary hypertension. Am J Respir Crit Care Med 157: 1423–1428, 1998. [DOI] [PubMed] [Google Scholar]
- 210.Vitulo P, Stanziola A, Confalonieri M, Libertucci D, Oggionni T, Rottoli P, Paciocco G, Tuzzolino F, Martino L, Beretta M, Callari A, Amaducci A, Badagliacca R, Poscia R, Meloni F, Refini RM, Geri P, Baldi S, Ghio S, D’Alto M, Argiento P, Sofia M, Guardamagna M, Pezzuto B, Vizza CD. Sildenafil in severe pulmonary hypertension associated with chronic obstructive pulmonary disease: A randomized controlled multicenter clinical trial. J Heart Lung Transplant 36: 166–174, 2017. [DOI] [PubMed] [Google Scholar]
- 211.Voelkel NF, Cool CD, Lee SD, Wright L, Geraci MW, Tuder RM. Primary pulmonary hypertension between inflammation and cancer. Chest 114: 225S–230S, 1999. [DOI] [PubMed] [Google Scholar]
- 212.Voelkel NF, Tuder RM. Hypoxia-induced pulmonary vascular remodeling—A model for what human disease? J Clin Investig 106: 733–738, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Vonbank K, Ziesche R, Higenbottam TW, Stiebellehner L, Petkov V, Schenk P, Germann P, Block LH. Controlled prospective randomised trial on the effects on pulmonary haemodynamics of the ambulatory long term use of nitric oxide and oxygen in patients with severe COPD. Thorax 58: 289–293, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Voosen P The AI detectives. Science 357: 22–27, 2017. [DOI] [PubMed] [Google Scholar]
- 215.Wagenvoort CA, Wagenvoort N. Pulmonary veins in high-altitude residents: A morphometric study. Thorax 37: 931–935, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wang A, Valdez-Jasso D. Cellular mechanosignaling in pulmonary arterial hypertension. Biophys Rev 13: 747–756, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Wang X, Guo Z, Ding Z, Mehta JL. Inflammation, autophagy, and apoptosis after myocardial infarction. J Am Heart Assoc 7: e008024, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Washko GR, Nardelli P, Ash SY, Vegas Sanchez-Ferrero G, Rahaghi FN, Come CE, Dransfield MT, Kalhan R, Han MK, Bhatt SP, Wells JM, Aaron CP, Diaz AA, Ross JC, Cuttica MJ, Labaki WW, Querejeta Roca G, Shah AM, Young K, Kinney GL, Hokanson JE, Agusti A, San Jose Estepar R. Arterial vascular pruning, right ventricular size, and clinical outcomes in chronic obstructive pulmonary disease. A longitudinal observational study. Am J Respir Crit Care Med 200: 454–461, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Watkins WD, Peterson MB, Crone RK, Shannon DC, Levine L. Prostacyclin and prostaglandin E1 for severe idiopathic pulmonary artery hypertension. Lancet 1: 1083, 1980. [DOI] [PubMed] [Google Scholar]
- 220.Waxman A, Restrepo-Jaramillo R, Thenappan T, Ravichandran A, Engel P, Bajwa A, Allen R, Feldman J, Argula R, Smith P, Rollins K, Deng C, Peterson L, Bell H, Tapson V, Nathan SD. Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med 384: 325–334, 2021. [DOI] [PubMed] [Google Scholar]
- 221.Westöö C, Norvik C, Peruzzi N, van der Have O, Lovric G, Jeremiasen I, Tran PK, Mokso R, de Jesus PV, Brunnström H, Bech M, Galambos C, Tran-Lundmark K. Distinct types of plexiform lesions identified by synchrotron-based phase-contrast micro-CT. Am J Physiol Lung Cell Mol Physiol 321: L17–l28, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Wilkins MR. Personalized medicine for pulmonary hypertension: The future management of pulmonary hypertension requires a new taxonomy. Clin Chest Med 42: 207–216, 2021. [DOI] [PubMed] [Google Scholar]
- 223.Wilkinson M, Langhorne CA, Heath D, Barer GR, Howard P. A pathophysiological study of 10 cases of hypoxic cor pulmonale. QJ Med 66: 65–85, 1988. [PubMed] [Google Scholar]
- 224.Williams SM, Golden-Mason L, Ferguson BS, Schuetze KB, Cavasin MA, Demos-Davies K, Yeager ME, Stenmark KR, McKinsey TA. Class I HDACs regulate angiotensin II-dependent cardiac fibrosis via fibroblasts and circulating fibrocytes. J Mol Cell Cardiol 67: 112–125, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Wohrley JD, Frid MG, Moiseeva EP, Orton EC, Belknap JK, Stenmark KR. Hypoxia selectively induces proliferation in a specific subpopulation of smooth muscle cells in the bovine neonatal pulmonary arterial media. J Clin Invest 96: 273–281, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Wright JL, Petty T, Thurlbeck WM. Analysis of the structure of the muscular pulmonary arteries in patients with pulmonary hypertension and COPD: National Institutes of Health nocturnal oxygen therapy trial. Lung 170: 109–124, 1992. [DOI] [PubMed] [Google Scholar]
- 227.Yaginuma G, Mohri H, Takahashi T. Distribution of arterial lesions and collateral pathways in the pulmonary hypertension of congenital heart disease: A computer aided reconstruction study. Thorax 45: 586–590, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Yang H, Shelat AA, Guy RK, Gopinath VS, Ma T, Du K, Lukacs GL, Taddei A, Folli C, Pedemonte N, Galietta LJ, Verkman AS. Nanomolar affinity small molecule correctors of defective Delta F508-CFTR chloride channel gating. J Biol Chem 278: 35079–35085, 2003. [DOI] [PubMed] [Google Scholar]
- 229.Yeager ME, Halley GR, Golpon HA, Voelkel NF, Tuder RM. Microsatellite instability of endothelial cell growth and apoptosis genes within plexiform lesions in primary pulmonary hypertension. Circ Res 88: e8–e11, 2001. [DOI] [PubMed] [Google Scholar]
- 230.Yeager ME, Voelkel NF, Tuder RM. Mutational analysis of endothelial cell TGF-β receptor type II in plexiform lesions of patients with primary pulmonary hypertension. Circulation 100: I–587, 1999. [Google Scholar]
- 231.Yu YA, Malakhau Y, Yu CA, Phelan SJ, Cumming RI, Kan MJ, Mao L, Rajagopal S, Piantadosi CA, Gunn MD. Nonclassical monocytes sense hypoxia, regulate pulmonary vascular remodeling, and promote pulmonary hypertension. J Immunol 204: 1474–1485, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Yu YR, Mao L, Piantadosi CA, Gunn MD. CCR2 deficiency, dysregulation of Notch signaling, and spontaneous pulmonary arterial hypertension. Am J Respir Cell Mol Biol 48: 647–654, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Yuan K, Liu Y, Zhang Y, Nathan A, Tian W, Yu J, Sweatt AJ, Shamshou EA, Condon D, Chakraborty A, Agarwal S, Auer N, Zhang S, Wu JC, Zamanian RT, Nicolls MR, de Jesus Perez VA. Mural cell SDF1 signaling is associated with the pathogenesis of pulmonary arterial hypertension. Am J Respir Cell Mol Biol 62: 747–759, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Zaiman A, Fijalkowska I, Hassoun PM, Tuder RM. One hundred years of research in the pathogenesis of pulmonary hypertension. Am J Respir Cell Mol Biol 33: 425–431, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Zamanian RT, Badesch D, Chung L, Domsic RT, Medsger T, Pinckney A, Keyes-Elstein L, D’Aveta C, Spychala M, White RJ, Hassoun PM, Torres F, Sweatt AJ, Molitor JA, Khanna D, Maecker H, Welch B, Goldmuntz E, Nicolls MR. Safety and efficacy of B-cell depletion with rituximab for the treatment of systemic sclerosis-associated pulmonary arterial hypertension: A multicenter, double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 204: 209–221, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Zhang H, Laux A, Stenmark KR, Hu CJ. Mechanisms contributing to the dysregulation of miRNA-124 in pulmonary hypertension. IntJ Mol Sci 22: 3852, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Zhang H, Wang D, Li M, Plecita-Hlavata L, D’Alessandro A, Tauber J, Riddle S, Kumar S, Flockton A, McKeon BA, Frid MG, Reisz JA, Caruso P, El Kasmi KC, Jezek P, Morrell NW, Hu CJ, Stenmark KR. Metabolic and proliferative state of vascular adventitial fibroblasts in pulmonary hypertension is regulated through a microRNA-124/PTBP1 (polypyrimidine tract binding protein 1)/pyruvate kinase muscle axis. Circulation 136: 2468–2485, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Zhang Y, Talwar A, Tsang D, Bruchfeld A, Sadoughi A, Hu M, Omonuwa K, Cheng KF, Al-Abed Y, Miller EJ. Macrophage migration inhibitory factor mediates hypoxia-induced pulmonary hypertension. Mol Med 18: 215–223, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Zhao L, Chen CN, Hajji N, Oliver E, Cotroneo E, Wharton J, Wang D, Li M, McKinsey TA, Stenmark KR, Wilkins MR. Histone deacetylation inhibition in pulmonary hypertension: Therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation 126: 455–467, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]