

Abstract
BACKGROUND
Disabling pansclerotic morphea (DPM) is a rare systemic inflammatory disorder, characterized by poor wound healing, fibrosis, cytopenias, hypogammaglobulinemia, and squamous-cell carcinoma. The cause is unknown, and mortality is high.
METHODS
We evaluated four patients from three unrelated families with an autosomal dominant pattern of inheritance of DPM. Genomic sequencing independently identified three heterozygous variants in a specific region of the gene that encodes signal transducer and activator of transcription 4 (STAT4). Primary skin fibroblast and cell-line assays were used to define the functional nature of the genetic defect. We also assayed gene expression using single-cell RNA sequencing of peripheral-blood mononuclear cells to identify inflammatory pathways that may be affected in DPM and that may respond to therapy.
RESULTS
Genome sequencing revealed three novel heterozygous missense gain-of-function variants in STAT4. In vitro, primary skin fibroblasts showed enhanced interleukin-6 secretion, with impaired wound healing, contraction of the collagen matrix, and matrix secretion. Inhibition of Janus kinase (JAK)–STAT signaling with ruxolitinib led to improvement in the hyperinflammatory fibroblast phenotype in vitro and resolution of inflammatory markers and clinical symptoms in treated patients, without adverse effects. Single-cell RNA sequencing revealed expression patterns consistent with an immunodysregulatory phenotype that were appropriately modified through JAK inhibition.
CONCLUSIONS
Gain-of-function variants in STAT4 caused DPM in the families that we studied. The JAK inhibitor ruxolitinib attenuated the dermatologic and inflammatory phenotype in vitro and in the affected family members. (Funded by the American Academy of Allergy, Asthma, and Immunology Foundation and others.)
Disabling pansclerotic morphea (DPM) is a severe systemic inflammatory disorder in the scleroderma continuum; it is characterized by poor wound healing with rapidly progressive deep fibrosis involving the mucous membranes, dermis, subcutaneous fat, fascia, muscles, and bone, leading to contractures, musculoskeletal atrophy, and articular ankylosis.1 Systemic manifestations include cytopenias and hypogammaglobulinemia, but scleroderma-associated autoantibodies are usually not present. DPM is refractory to therapy, including systemic glucocorticoids, immunosuppression, and autologous stem-cell transplantation.2 Disease pathogenesis has been attributed to abnormal collagen synthesis and deposition, vascular damage, and altered immunoregulation similar to that in other forms of scleroderma.3 Current treatment integrates multiple, broad-spectrum pharmaceutical and ancillary therapies (e.g., methotrexate, mycophenolate mofetil, and ultraviolet A light therapy),3,4 which are directed at halting disease progression but have had limited success and unacceptable side effects.5,6 DPM is associated with high morbidity and mortality due to squamous-cell carcinoma, restrictive pulmonary disease, sepsis, and gangrene, resulting in a postdiagnosis survival time of less than 10 years.1 A genetic cause has not been identified.
We describe four persons in three independent families with DPM and monoallelic gain-of-function variants in the gene encoding signal transducer and activator of transcription (STAT) 4 (STAT4). STAT proteins are recruited to activated receptors at the plasma membrane through interaction of the SH2 domain with a receptor phosphotyrosine residue generated by Janus kinase (JAK) activity. STAT proteins are also JAK substrates, and phosphorylation of a STAT leads to its dimerization, nuclear import, and activation of transcription of “downstream” genes. JAK-STAT signaling regulates cytokine responses and acts in immune responses, cell growth and differentiation, cell survival, apoptosis, and oncogenesis.7–9 STAT4 is also essential for transcriptional activation downstream of interleukin-6 receptor signaling and for transcription of interleukin-6.10 Members of the interleukin-6 family of cytokines have been proposed to coordinate immune–stroma cross-talk and have been implicated in other forms of morphea.11–13 Understanding the pathophysiology of DPM may shed light on a much larger range of disorders characterized by poor wound healing and severe, unchecked fibrosis.
METHODS
PATIENTS
Blood samples were obtained from the affected patients and immediate family members. Skin-biopsy samples were obtained from the Family 1 proband and healthy controls. All the participants (or legal guardians if the patient was a minor) provided written informed consent under their respective institutional review boards. All procedures that were performed in studies involving human participants were in accordance with the ethical standards of the respective institutions. Details of DNA isolation, library construction, and sequencing, as well as methods for site-directed mutagenesis, mammalian cell culture, transfection and expression of recombinant proteins, flow cytometry and fluorescence imaging, wound-healing assays, immunocytochemical assays, contraction assays, and enzyme-linked immunosorbent assays, are described in Supplementary Appendix 1, available with the full text of this article at NEJM.org.
STATISTICAL ANALYSIS
For in vitro experiments, statistical analyses and graphing were performed in Microsoft Excel and GraphPad Prism, version 5.03 (GraphPad Software) with the two-tailed, unpaired Student’s t-test or the Kruskal–Wallis test; significant P values were confirmed with the use of the two-tailed nonparametric Mann–Whitney test. Flow-cytometry data were analyzed with FlowJo software. Data are expressed as means and standard errors. No samples were excluded from the analyses, and no randomization of samples was performed. A P value of less than 0.05 was considered to indicate statistical significance. For 10x Genomics single-cell RNA-sequencing data analyses, see Supplementary Appendix 1.
RESULTS
PATIENT CHARACTERISTICS
We evaluated three unrelated kindreds with features of DPM (Table S1 in Supplementary Appendix 1). Family 1 was identified at the University of California, San Diego; mucosal lesions had developed in Patient 1 beginning at 3 years of age, and the first symptom that developed in Patient 2 was polyarthritis. In both patients, the condition had progressed to include inflammation and ulcerative lesions of the skin and joint swelling and contractures; both had received diagnoses of DPM by means of skin biopsy. Patient 3 had been enrolled in the National Institutes of Health (NIH) Undiagnosed Diseases Program and was admitted to the NIH Clinical Center at 25 years of age. He had been ascertained in Greece as having an autoinflammatory disease with joint swelling at 3 years of age, hand contractures at 7 years of age, and progressive skin ulcerations beginning at 8 years of age, and he had been included in a pediatric series of patients with DPM.1 Patient 4 had been referred to the University of Pittsburgh Scleroderma Center and was enrolled in tandem in the National Registry of Childhood Onset Scleroderma, after a nonhealing skin ulcer had developed at 9 months of age and progressive muscle wasting had developed by 12 months of age, leading to the clinical diagnosis of DPM. All four patients had had disease onset before 5 years of age, with signs of mucosal ulcerations and skin sclerosis (Fig. 1A through 1E, and Fig. S1 in Supplementary Appendix 1). The results of hematoxylin and eosin staining of skin-biopsy samples were consistent with morphea (Fig. 1F and 1G). Musculoskeletal imaging showed deep-tissue inflammation and sclerosis of several levels from subcutis to muscle, findings consistent with DPM.
Figure 1. Clinical Manifestations of Disabling Pansclerotic Morphea (DPM).
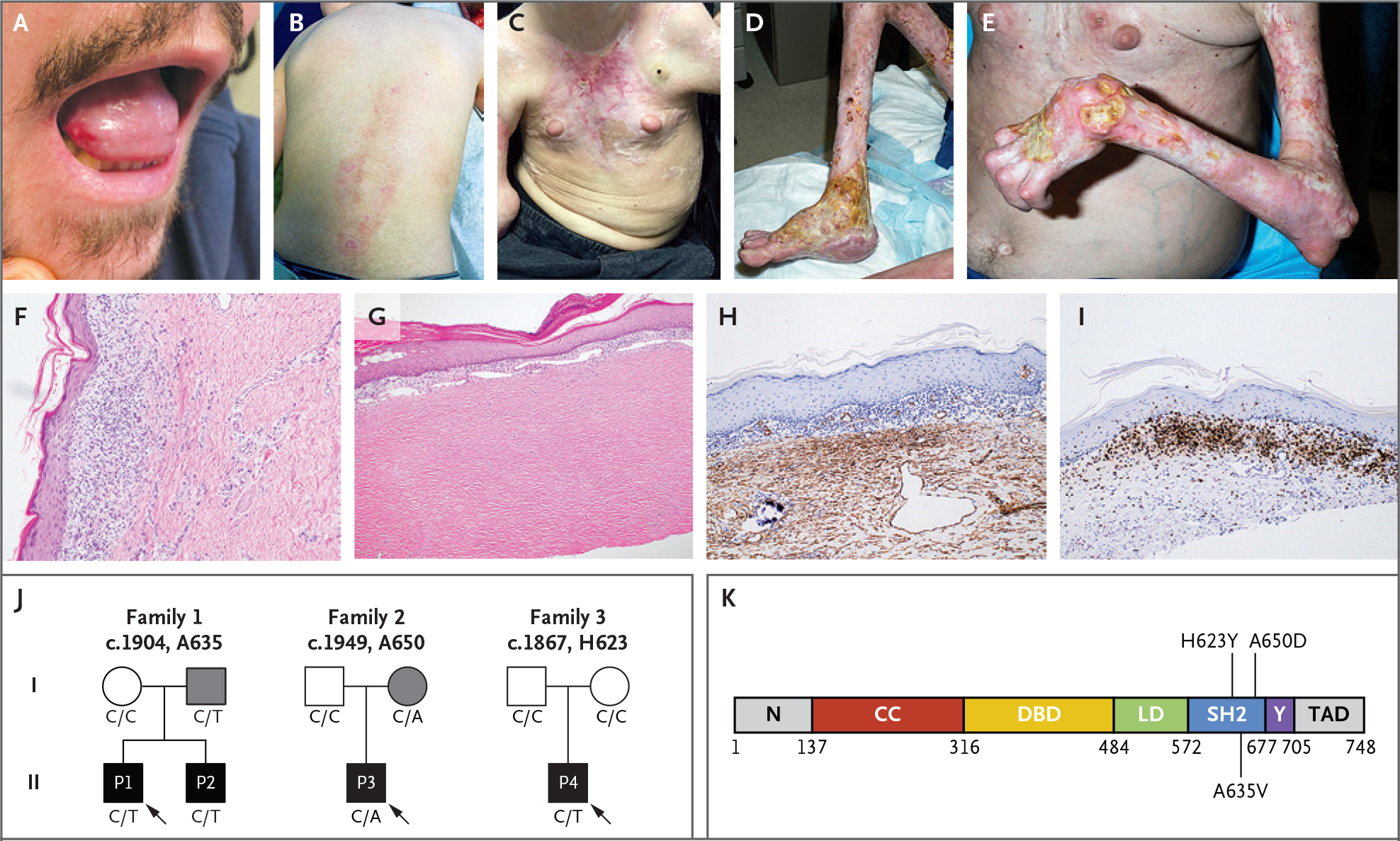
Clinical images show oral ulceration and limitation in tongue protrusion (Panel A); spreading waxy, hypopigmented lesions on the back and waxy hypopigmented “tank top” sign on the chest (Panels B and C); and ulcerations with articular ankylosis of the legs and arms (Panels D and E). Histologic sections of skin-biopsy samples show prominent inflammation (Panel F) and dermal thickening and hyalinization of morphea (Panel G). Images are shown of immunohistochemical staining for smooth-muscle action (Panel H) and CD3 (Panel I) in skin-biopsy samples before use of ruxolitinib. Family pedigrees (Panel J) are shown, with probands indicated by arrows. Circles represent female family members, squares male family members, and solid symbols persons who have received a diagnosis of DPM. Grey shading indicates persons with the STAT4 variants but with milder symptoms. The genotype at the specified locus is indicated under each person. A linear protein model (Panel K) shows the approximate locations of the identified variants in the SRC homology 2 (SH2) domain. CC denotes coiled-coil domain, DBD DNA-binding domain, LD linker domain, N the N-terminal domain, TAD transactivation domain, and Y the phosphotyrosyl-tail segment.
Laboratory evaluations showed mild neutropenia and lymphopenia, especially with respect to CD4+ T cells, and elevated serum inflammatory markers in three of the four patients. Similarly, serum levels of immunoglobulin classes IgG and IgA were reduced in three of the four patients (Fig. S2 in Supplementary Appendix 1), and no patients had detectable autoantibodies. Immunohistochemical staining of the skin (Fig. 1H and 1I) showed extensive staining for alpha smooth-muscle actin, a finding consistent with fibrosis, and CD3-positive staining in most of the subepidermal lymphocytes, which confirmed an inflammatory infiltrate composed of mostly T cells.
Despite multiple systemic immunosuppressive therapies (Figs. S3 and S4 in Supplementary Appendix 1), three of the patients had spreading of their rash and worsening ulcerations, with rapid development of contractures, muscular atrophy, and impairments in mobility. Two patients also had recurrent infections, and squamous-cell carcinoma had developed in one patient. Pulmonary nodules and infiltrates, pulmonary and portal hypertension, rapid-onset blindness (glaucoma and cataracts resulting in surgery), and sensorineural hearing loss were less frequent findings.
Two of the families had first-degree relatives with oral ulcerations, mild skin disease, or early-onset progressive hand swan-neck deformities, which suggested a possible inherited cause (Fig. 1J). For detailed clinical presentations, see Supplementary Appendix 1.
SEGREGRATION OF STAT4 VARIANTS WITH DISEASE
Independent genomic analyses identified heterozygous variants in STAT4 in all three kindreds (STAT4 c.1904 C→T, c.1949C→A, and c.1867C→T, predicting amino acid substitutions of A635V, A650D, and H623Y, respectively) (Figs. S5 and S6 in Supplementary Appendix 1). These variants were not reported in the Genome Aggregation Database, Trans-Omics for Precision Medicine database, or deCODE Allele Frequency Browser and were predicted to be damaging (Fig. S6 in Supplementary Appendix 1). Each occurs in the region of STAT4 that encodes the SH2 domain (Fig. 1K); variants affecting the SH2 domain in other STAT proteins have been implicated in other immune diseases, including chronic mucocutaneous candidiasis (STAT1) and hyper-IgE syndrome (STAT3).14–17
STAT4 VARIANTS AND GAIN-OF-FUNCTION PHENOTYPE
The clinical presentation of the patients and the inheritance pattern in the families were consistent with a gain-of-function cause. Multiple-species sequence alignment indicates that A635, A650, and H623 are evolutionarily conserved amino acids, as is the SH2 domain. In silico modeling18 predicts that they are directly involved with the phosphotyrosine peptide-binding pocket of the SH2 domain (Fig. 1K). The H623Y and A635V variants stabilize the dimer through hydrophobic interactions and thereby activate it, whereas the A650D mutation probably leads to intramonomer interactions with R705 and causes disease through a different mechanism.
To test whether the variants have a gain-of-function phenotype, we stably transfected a cell line containing a luciferase gene driven by the human IL6 promoter (a target of STAT410) with a plasmid containing either nonvariant (control) or variant STAT4. We tested each of the three variants. In the absence of stimulation, cells carrying the mutant plasmids had greater luciferase activity than those carrying the control plasmids (Fig. 2A, and Fig. S7 in Supplementary Appendix 1). To analyze phosphorylated STAT4 (pSTAT4) levels, U3A cells (deficient in STAT1 and STAT4) were stably transfected with plasmids containing control or variant STAT4. In unstimulated cells with variant STAT4, pSTAT4 levels were higher than those in cells transfected with control STAT4 (Fig. 2B). After interferon alfa stimulation, cells carrying control or variant STAT4 had similarly increased pSTAT4 levels at 30 minutes after exposure. However, the increased levels of pSTAT4 were comparatively durable in cells containing variant STAT4, showing elevated levels (as compared with cells containing control STAT4) at 240 minutes and supporting the hypothesis that variant STAT4 is resistant to mechanisms that inhibit or reduce STAT4 signaling (Fig. 2C).
Figure 2. Effects of STAT4 Variants on Phosphorylation.
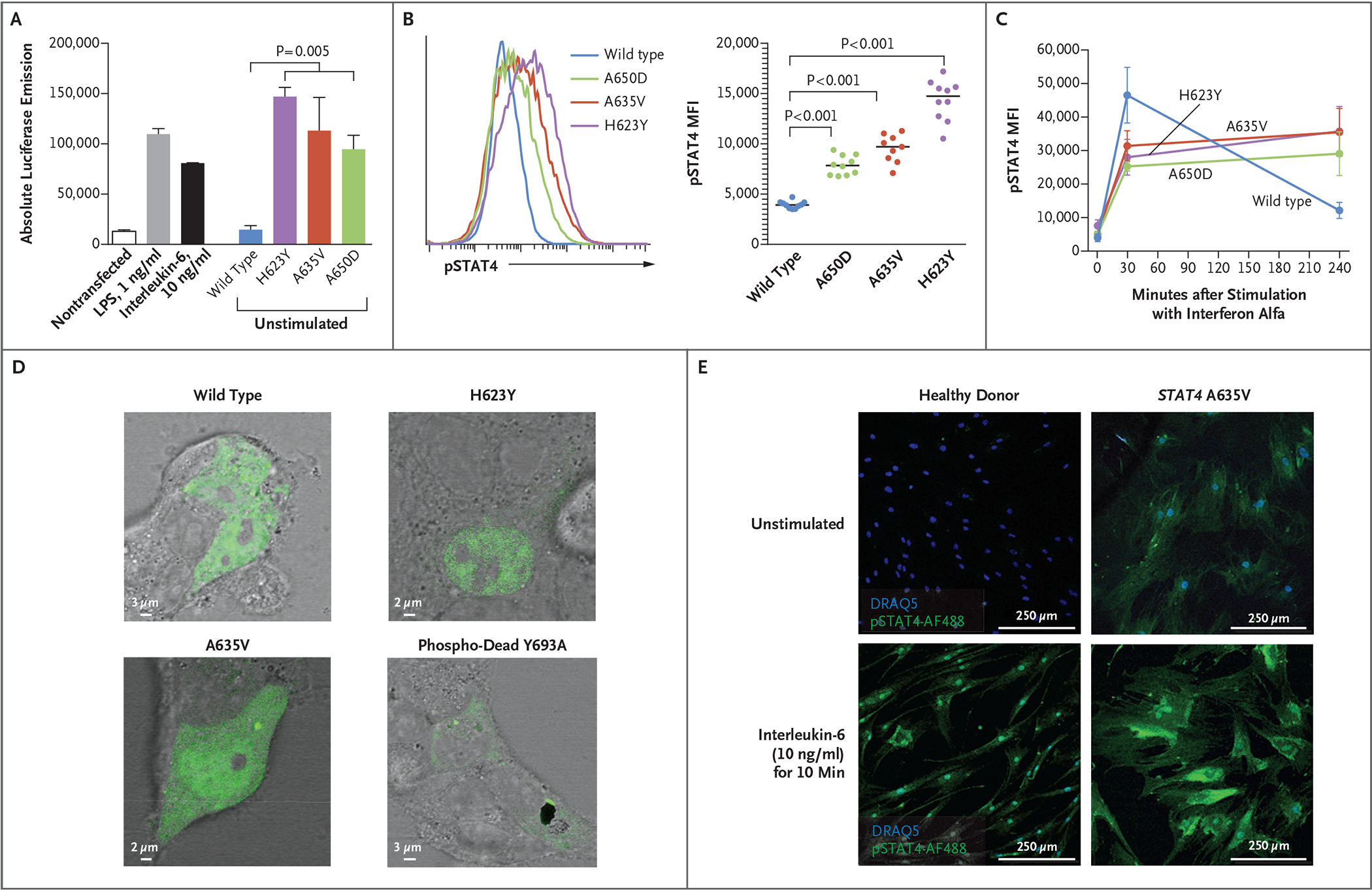
Panel A shows absolute luciferase emission from the interleukin-6 Leeporter cell line transfected with vector carrying wild-type or variant STAT4. Transcriptional activity was enhanced in the presence of STAT4 A635V, H623Y, and A650D, as compared with wild-type STAT4 and the nontransfected cell line, with or without stimulation with lipopolysaccharide (LPS) or interleukin-6. Panels B and C show STAT4 phosphorylation in U3A cells stably transfected with wild-type or variant STAT4. Flow cytometry that was used to measure the mean fluorescence intensity (MFI) of phosphorylated STAT4 (pSTAT4) showed increased pSTAT4 in unstimulated cells (Panel B) that were transfected with A635V (red), A650D (green), and H623Y (purple) variants, as compared with wild-type STAT4 (blue). In response to interferon alfa (Panel C), STAT4 phosphorylation persisted in variant cells at 240 minutes as compared with wild-type cells. In Panels A and C, error bars indicate standard errors. Panel D shows HEK293T cells transiently transfected with plasmids containing wild-type, H623Y, A635V, or phospho-dead Y693A STAT4 tagged with green fluorescent protein. Unstimulated cells that were transfected with H623Y or A635V variants had a greater accumulation of STAT4 in the nucleus than those transfected with wild-type or Y693A STAT4. Panel E shows that primary skin fibroblasts from a patient with the A635V variant had prominent pSTAT4 (green), as compared with fibroblasts from a healthy donor. Phosphorylated STAT4 staining of patient fibroblasts persisted in a perinuclear location with interleukin-6 stimulation. Nuclear staining was performed with DRAQ5 (blue).
RNA sequencing of transfected U3A cells was used to monitor global transcriptional changes caused by the variants. Variant-transfected cells that were stimulated with interferon alfa displayed a greater number and variety of differentially expressed genes than control interferon alfa–stimulated cells (Fig. S8A and S8B in Supplementary Appendix 1). Expression levels of genes encoding proteins in STAT4 pathways, including interleukin-6 and the counterregulatory suppressor of cytokine signaling (SOCS) proteins, were higher in variant-transfected cells than in control-transfected cells, even though baseline levels of total STAT4 protein were similar (Fig. S8C and S8D in Supplementary Appendix 1).
This enhanced transcriptional activity was associated with a greater accumulation of STAT4 in the nucleus of STAT4 H623Y- or A635V-transfected HEK293T cells than with control and the “phospho-dead” Y693A control variant (Fig. 2D, and Fig. S9A in Supplementary Appendix 1). Similarly, pSTAT4 was evident at baseline in primary skin fibroblasts from patients but not in fibroblasts from healthy donors despite these cells having similar levels of STAT4 expression (Fig. 2E, and Fig. S9B and S9C in Supplementary Appendix 1). Interleukin-6 stimulation enhanced nuclear and perinuclear pSTAT4 in fibroblasts (Fig. 2E), whereas interleukin-12–induced phosphorylation of STAT4 in patient peripheral-blood T cells, typical of type 1 helper T (Th1) cell skewing, was not affected, although T-cell subsets had evidence for exhaustion (Fig. S10 in Supplementary Appendix 1 and Tables S2 and S3 in Supplementary Appendix 2). Taken together, these data make a strong case that the patient-identified STAT4 variants cause a gain-of-function phenotype.
WOUND HEALING, INFLAMMATION, AND FIBROSIS
STAT4 is expressed in leukocytes and skin19–21 and may perpetuate inflammation in fibroblasts derived from patients with rheumatoid arthritis.10 Given the dramatic impairment in wound healing in DPM (Fig. 1), we used primary skin fibroblasts from a patient with STAT4 A635V and unrelated healthy donors to study an in vitro model of wound healing. We first tested a scratch assay that requires all three wound-healing processes (i.e., epithelialization, contraction, and connective-tissue deposition). In the scratch assay, a fibroblast monolayer is scratched to induce a uniform gap, and the rate of closure is monitored (Fig. S11A in Supplementary Appendix 1). Skin fibroblasts from a patient with STAT4 A635V did not migrate as rapidly as fibroblasts from healthy donors and ultimately did not close the induced gap (Fig. 3A). We also found defects in individual wound-healing processes. Contraction was tested by embedding primary skin fibroblasts in a collagen matrix, followed by stimulation with transforming growth factor β (TGF-β) (Fig. S11B and S11C in Supplementary Appendix 1). Patient-derived fibroblasts had less contractility than fibroblasts from healthy donors (Fig. 3B), a finding consistent with the continued presence of inflammatory mediators.22 Immunofluorescence staining of F-actin filaments showed greater disorganization and larger cell size in patient fibroblasts than in fibroblasts from healthy donors (Fig. 3C, and Fig. S11D in Supplementary Appendix 1). Similarly, matrix secretion of procollagen α1 by patient fibroblasts was diminished relative to fibroblasts from healthy donors, whereas fibronectin secretion was unchanged, a finding consistent with poor wound healing (Fig. S11E and S11F in Supplementary Appendix 1).
Figure 3. Evaluation of the Function of Primary Skin Fibroblasts In Vitro.
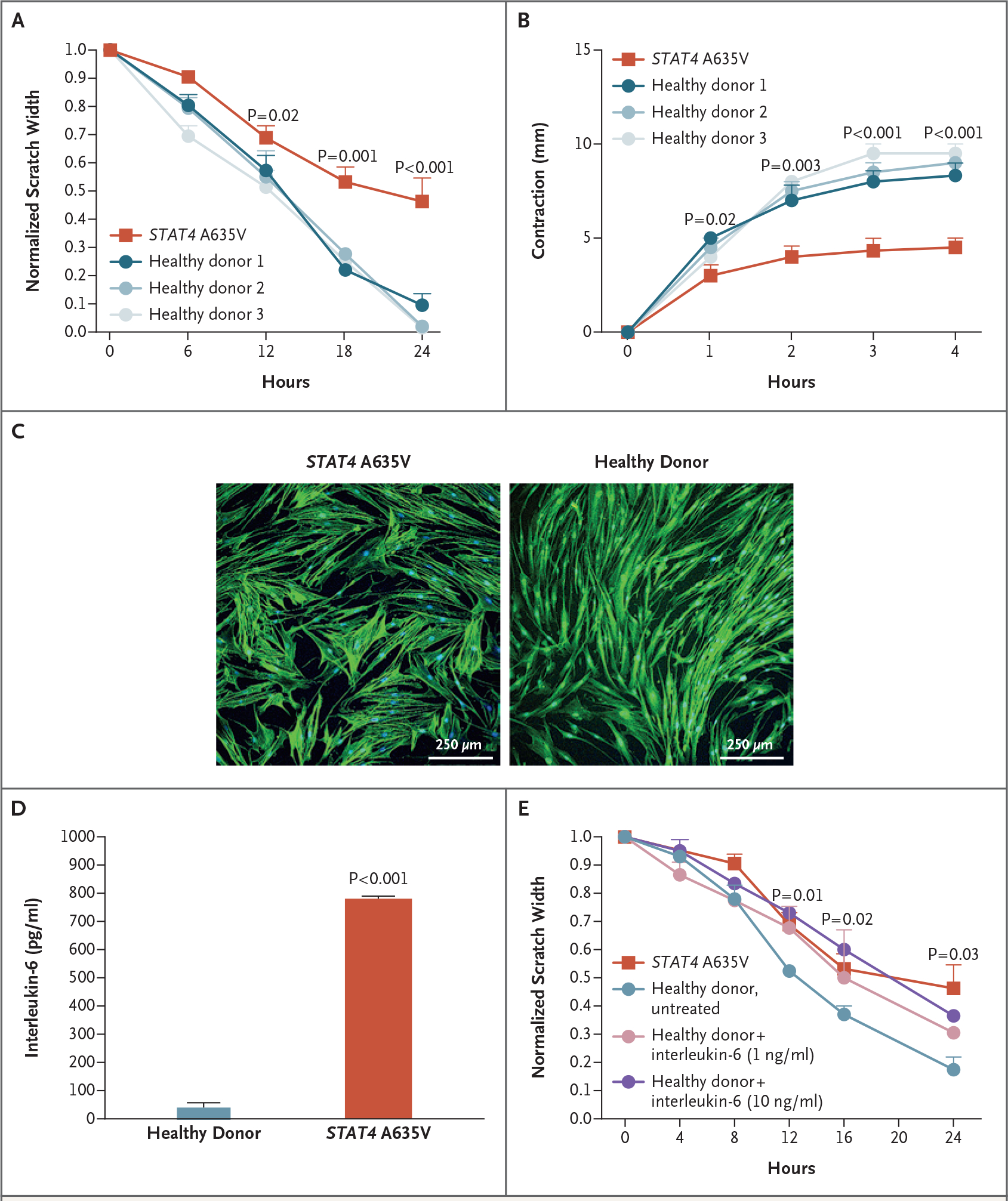
Panel A shows that wound healing as measured by scratch assay was reduced in fibroblasts from patients with STAT4 A635V (red), as compared with fibroblasts from healthy donors (blue). For the scratch assays, three experiments were performed with six scratches each. Panel B shows that transforming growth factor β–induced contraction of collagen matrix by patient-derived fibroblasts (red) was reduced relative to fibroblasts from healthy donors. Panel C shows that in F-actin immunocytochemical analysis, cell size was increased in primary skin fibroblasts from a patient with STAT4 A635V, as compared with fibroblasts from healthy donors. Panel D shows that patient fibroblasts had enhanced interleukin-6 secretion in the absence of stimulation. Panel E shows that wound healing was reduced in primary skin fibroblasts from healthy donors when treated with varying concentrations of interleukin-6, with rates approaching those of cells from patients with STAT4 A635V (three experiments with six scratches each). Throughout the figure, P values were calculated by means of two-way analysis of variance. Error bars indicate standard errors.
INTERLEUKIN-6 SECRETION BY PATIENT FIBROBLASTS
Fibroblasts secrete proinflammatory mediators, including interleukin-1, interleukin-6, TGF-β, and vascular endothelial growth factor.23 We found that unstimulated skin fibroblasts from patients secreted 12 times as much interleukin-6 as those from healthy donors (Fig. 3D). In contrast, the secretion of interleukin-1 and interferon was at the limits of detection in cultures from healthy donors and patients. To explore the potential role of interleukin-6 in the pathology of the patient fibroblasts, cells from healthy donors were pulsed with recombinant interleukin-6 every 8 hours for the duration of a scratch assay. Remarkably, interleukin-6 reduced the migration of fibroblasts derived from healthy donors, prevented scratch closure, reduced TGF-β–induced contraction, and increased the size of cells (Fig. 3E, and Fig. S12A and S12B in Supplementary Appendix 1). Together, these data suggest that the gain-of-function STAT4 A635V variant causes an autoinflammatory loop, largely mediated by interleukin-6, which drives the fibroblast phenotype. In vitro treatment with anti–interleukin-6 led to a modest improvement in fibroblast function but suggested that upstream targeting of this molecular pathway may be required to inhibit autoinflammation (Fig. S12C in Supplementary Appendix 1).
THERAPEUTIC TARGETING WITH JAK INHIBITORS
To determine whether the hyperinflammatory fibroblast phenotype could be ameliorated in vitro by disrupting STAT4 signaling, we treated cultures of primary skin fibroblasts from patients with the JAK inhibitor ruxolitinib. Ruxolitinib significantly reduced interleukin-6 secretion at concentrations achievable in serum (Fig. 4A). Similarly, ruxolitinib enhanced the rate of scratch closure of patient fibroblasts to one nearly identical to that of fibroblasts from healthy donors (Fig. 4B) but did not affect the rate of scratch closure of fibroblasts from healthy donors (data not shown). The results of other assays of individual steps in wound healing showed less response to ruxolitinib treatment (Fig. S13 in Supplementary Appendix 1). Ruxolitinib treatment also reduced the total amount of pSTAT4 and its nuclear localization in unstimulated U3A cells and patient fibroblasts (Fig. 4C and 4D). However, the persistent levels of pSTAT4, even in the absence of new phosphorylation (Fig. 4E and 4F), suggest that the mutant STAT4 dimers are more stable than dimers of wild-type STAT4, which would be consistent with in silico predictions.
Figure 4. Janus Kinase Inhibition, Reduction of STAT4 Phosphorylation, and Wound Healing.
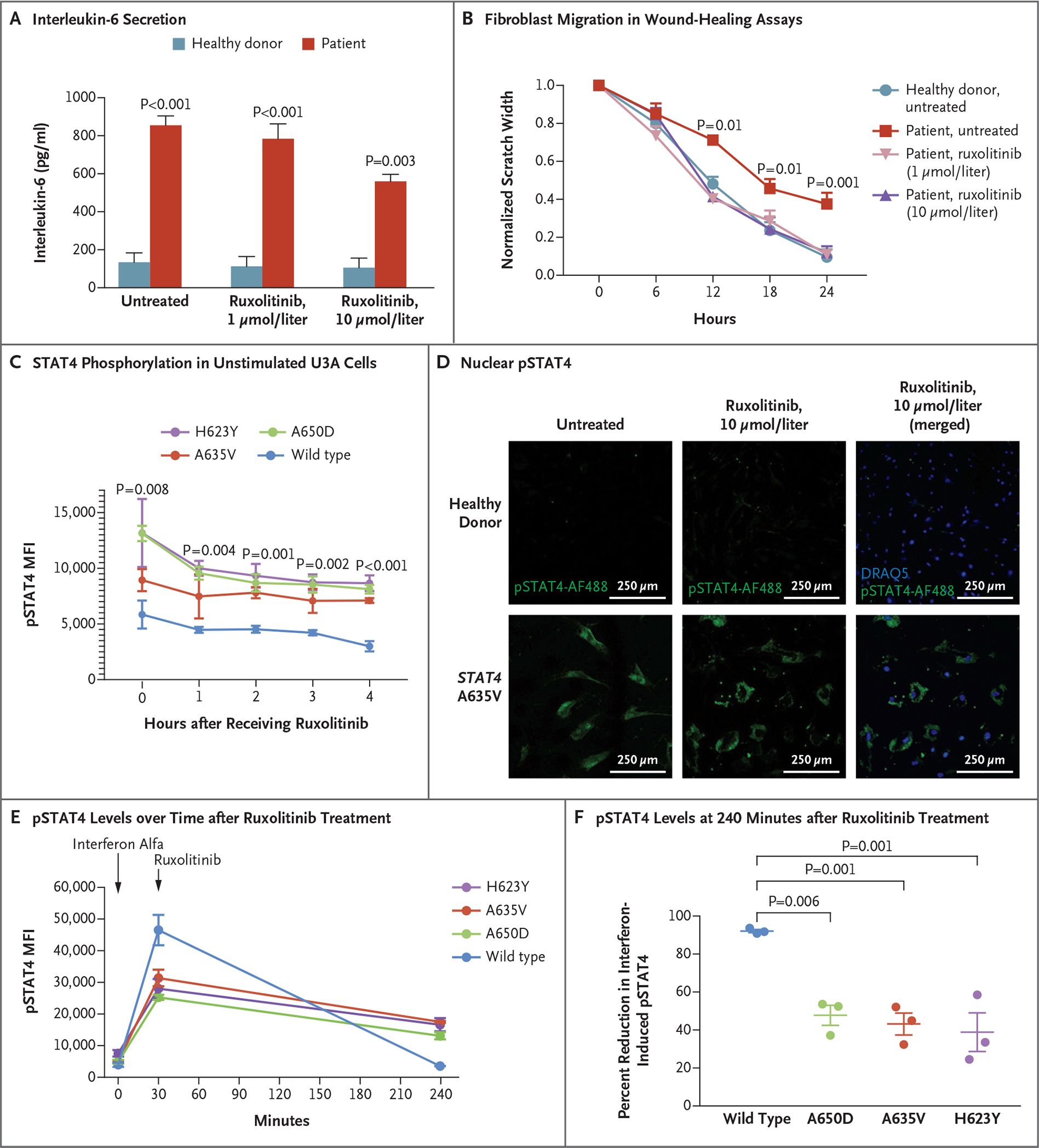
Panel A shows that patient fibroblasts had enhanced interleukin-6 secretion that was responsive to ruxolitinib. Data for healthy donors are a summary of three such donors. The P value for patient cells treated with ruxolitinib as compared with untreated patient cells is 0.02. Panel B shows that pretreatment with ruxolitinib led to enhanced fibroblast migration in wound-healing assays, with closure at 24 hours that was similar to that in unaffected fibroblasts (three experiments with six scratches each). Panel C shows that STAT4 phosphorylation in unstimulated U3A cells was reduced after treatment with ruxolitinib. Panel D shows that nuclear pSTAT4 was reduced in response to ruxolitinib treatment of patient fibroblasts. Panels E and F show that interferon alfa–stimulated U3A cells expressing variant STAT4 had higher levels of pSTAT4 than cells expressing wild-type STAT4 at 240 minutes. Ruxolitinib treatment decreased phosphorylation in cell lines expressing variant STAT4 and in those expressing wild-type STAT4. In Panel F, mean decreases and standard errors are shown. Throughout the figure, P values were calculated by means of two-way analysis of variance. Error bars indicate standard errors.
The identification of gain-of-function STAT4 variants led us to initiate oral ruxolitinib treatment in Patients 1 and 2. Peripheral-blood samples, collected from Patient 1 while he was receiving treatment and from Patient 2 while he was not receiving treatment, were analyzed by means of single-cell RNA sequencing to identify cell types (Fig. S14 in Supplementary Appendix 1) and to analyze differential gene expression in specific cell types such as natural killer cells and T cells, which are known to highly express STAT4 (Supplementary Appendix 2). In these cell types, pathway analysis based on differentially expressed genes showed that ruxolitinib treatment decreased the activity of genes such as IFNG, IFNA, TNF, IL6 and STAT1, which control multiple inflammatory pathways. These cell types were also associated with increased expression of genes in pathways controlled by upstream regulators (Fig. 5A, and Fig. S15 in Supplementary Appendix 1).
Figure 5. Ruxolitinib Treatment and Resolution of the Inflammatory Phenotype.
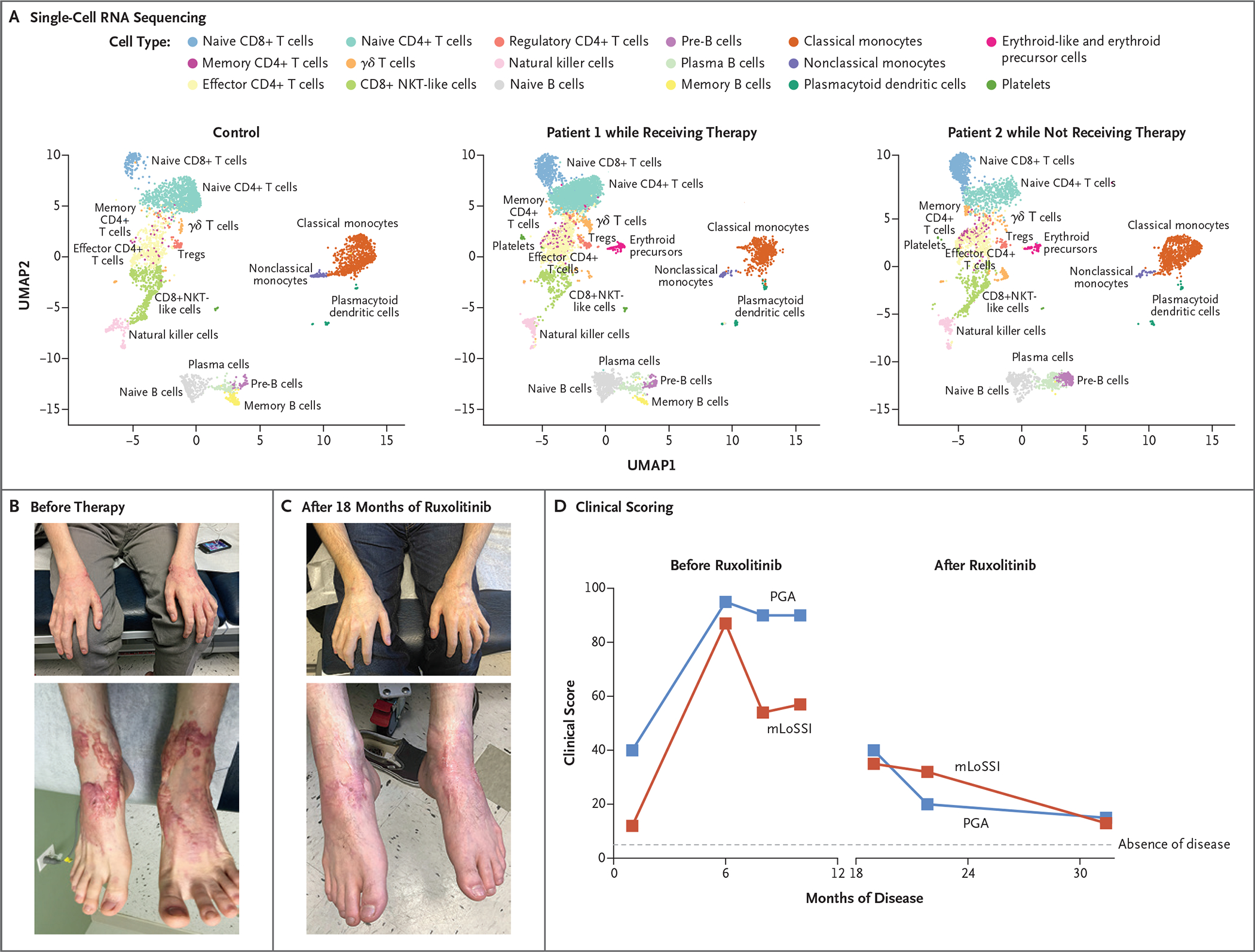
Shown are peripheral-blood mononuclear cells analyzed by means of single-cell RNA sequencing (Panel A), plotted in uniform manifold approximation and projection (UMAP) space and clustered for each of the control and patient samples. NKT cells denote natural killer T cells, and Tregs regulatory T cells. Photographs show waxy, erythematous nodular lesions on bilateral hands and feet before (Panel B) and after (Panel C) initiation of ruxolitinib therapy. Shown are clinical scores (Panel D) on the modified Localized Scleroderma Skin Severity Index (mLoSSI, red) and the Physician Global Assessment (PGA, blue) of disease activity. Scores on both scales range from 0 to 100, with higher scores indicating greater disease activity.
Initiation of oral ruxolitinib therapy yielded clinical success. Patient 1 had notable improvement in his nodular rash, without development of new lesions. After 11 months of therapy, the rash and oral ulcers had largely resolved. The most recent laboratory evaluation was notable for stable leukocyte counts, normal levels of inflammatory markers, and normal IgG and IgM levels, with persistent IgA deficiency. No adverse events have been reported during ruxolitinib treatment. At 18 months after initiation of ruxolitinib, he had discontinued all other medications, with complete resolution of his chest rash, substantial clearing on the arms and legs, and global clinical improvement (Fig. 5B, 5C, and 5D). Patient 2 has since begun therapy with ruxolitinib, with improvement in his pulmonary hypertension. He continues to receive intravenous immunoglobulin (2 g per kilogram of body weight) for IgG levels below 1000 mg per deciliter but has been able to avoid infusions for up to 2 months at a time. His neutropenia resolved, inflammatory markers normalized, anemia decreased, and thrombocytopenia stabilized (see Supplementary Appendix 1).
DISCUSSION
DPM, the most severe subtype of deep morphea within the spectrum of juvenile localized scleroderma, is characterized by rapid sclerosis in all layers of the skin, fascia, muscle, and bone.24 The cause of DPM has been elusive since the first description in 1923.24 Despite the general belief that a genetic component underlies disease development, a genetic basis for DPM has not been previously identified. Our identification of novel, autosomal dominantly inherited or de novo variants in STAT4 appears to be the first description of a gain-of-function variant in this gene and the first genetic link for DPM.
The roles played by STAT4 that could drive the multiple clinical facets of DPM10 include its involvement in Th1 cell development and function25 and in regulation of interleukin-6 by stromal cells.13,23 Genomewide association studies have also implicated single-nucleotide polymorphisms in STAT4 with multiple autoimmune diseases (Fig. S16 in Supplementary Appendix 1).26,27 Given the number of pathways in which STAT4 is a member, additional genetic modifiers of disease severity could exist, which may account for the presence of a parent with milder disease in two of the three families.
Increasing evidence points to fibroblasts as inflammatory mediators in sites of inflammation.13,23 Interleukin-6 family cytokines specifically have been proposed to coordinate immune–stroma cross-talk.11–13 Interleukin-6 has also been implicated in negatively regulating Th1 cell differentiation,28 which may explain the lack of Th1 cell skewing and reduced phosphorylation to interleukin-12, as well as in generating a chronic inflammatory disease state, which is consistent with the T-cell exhaustion29,30 we observed before treatment. The prominent interleukin-6 signature observed in our fibroblast cultures also suggests that anti–interleukin-6 monoclonal antibodies — such as tocilizumab, which is currently approved for interstitial lung disease in systemic sclerosis31 — may be an alternative therapy or may be useful in combination with JAK inhibitors in patients with DPM.
We speculated that the STAT4 gain-of-function mutations were dependent on JAK activity and explored the use of the clinically available JAK inhibitor ruxolitinib for patients in the most severely affected family. In the patient receiving consistent therapy, we observed normalization of most immunologic variables and resolution of systemic symptoms, without adverse effects. Given the multiple systems and body-surface area affected, we expect that oral systemic therapy, rather than topical JAK inhibitor therapy, would be appropriate in patients with DPM. We propose that this immunomodulatory approach may be promising for patients with refractory disease.
Supplementary Material
Acknowledgments
Supported by grants from the American Academy of Allergy, Asthma, and Immunology Foundation (to Dr. Broderick), the Ludwig Institute for Cancer Research (to Dr. Putnam), the University of California, San Diego, Department of Pediatrics (to Dr. Broderick), and the Novo Nordisk Foundation (to Dr. Lewis). The project was supported in part by a grant from Deutsche Forschungsgemeinschaft (to Dr. Oda), extramural funding from the NIH (including UH2 AI153029 and R35 GM119850, to Dr. Lewis), and a National Institute of General Medical Sciences Postdoctoral Research Associate Fellowship (to Dr. Philips). Additional grant support came from the NIH (T32GM8806, to Mr. Baghdassarian), the California Institute for Regenerative Medicine (CL1-00511, to Ms. McVicar and Dr. Liu), the Hydrocephalus Association (HA-60832, to Ms. McVicar and Dr. Liu), the NIH Shared Instrumentation Grant Program (S10 OD026929), and the NIH Clinical and Translational Science Awards Program (UL1TR001442). Additional support for this project was provided by the Scleroderma Research Foundation and the NIH Intramural Research Programs, including the Intramural Research Programs of the National Human Genome Research Institute, the National Institute of Arthritis and Musculoskeletal and Skin Diseases, the National Institute of Allergy and Infectious Diseases, the Biowulf High-Performance Computing Cluster of the Center for Information Technology, the Undiagnosed Diseases Program of the Common Fund of the Office of the Director of the NIH, and the NIH Clinical Center.
We thank Drs. Hal M. Hoffman, Manish Butte, and Seema S. Aceves for helpful discussion and the patients and families who participated.
APPENDIX
The authors’ affiliations are as follows: the Bioinformatics and Systems Biology Program (H.B.), the Department of Pediatrics (H.B., O.S.C., V.K.H., N.E.L.), the Center for Computational Biology and Bioinformatics, Department of Medicine (A.M., R.S., K.M.F.), the Institute for Genomic Medicine (K.J.), the Department of Bioengineering (N.E.L.), the Division of Allergy, Immunology, and Rheumatology, Department of Pediatrics (J.C., L.B.), and the Department of Medicine (C.D.P.), University of California, San Diego, Sanford Burnham Prebys Medical Discovery Institute (R.M., Y.L.), and the San Diego Branch, Ludwig Institute for Cancer Research (C.D.P.), La Jolla, and the Department of Pathology (S.M.T.), Rady Children’s Institute for Genomic Medicine (S.C., D.D.), and Rady Children’s Hospital Foundation (J.C., L.B.), Rady Children’s Hospital, San Diego — all in California; the Inflammatory Disease Section (S.A.B., B.M., M.N., S.R., P.P.C., N.H., E.F.R., D.L.K., H.O.), the Oncogenesis and Development Section (N.D.), and the Undiagnosed Diseases Program, Medical Genetics Branch (D.R.M., W.A.G.), National Human Genome Research Institute, the Molecular Immunology and Inflammation Branch (R.P., J.J.O.), the Light Imaging Section (D.R.) and the Translational Immunology Section (M.G.), Office of Science and Technology, and the Protein Expression Laboratory (E.E., N.R.W.), National Institute of Arthritis and Musculoskeletal and Skin Diseases, and the Genetics and Pathogenesis of Allergy Section, Laboratory of Allergic Diseases (C.A.M.), and the Translational Autoinflammatory Disease Section (R.G.-M.), National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, and the Department of Cell Biology and Molecular Genetics, University of Maryland, College Park (B.M.) — all in Maryland; Sanford School of Medicine, University of South Dakota, Sioux Falls (S.A.B.); the Division of Rheumatology and Clinical Immunology, University of Pittsburgh (D.M.S.), University of Pittsburgh Medical Center, Children’s Hospital of Pittsburgh (A.S., G.W., K.T.), and the University of Pittsburgh Scleroderma Center (A.S., G.W., K.T.) — all in Pittsburgh; the Division of Pediatric Allergy, Immunology, and Rheumatology, Columbia University, New York (J.D.M.); and Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases and the Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany (H.O.).
Footnotes
Disclosure forms as provided by the authors are available with the full text of this article at NEJM.org.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health (NIH).
The authors’ affiliations are listed in the Appendix.
Contributor Information
Hratch Baghdassarian, Bioinformatics and Systems Biology Program, University of California, San Diego, California Department of Pediatrics, University of California, San Diego, California.
Sarah A. Blackstone, Inflammatory Disease Section, National Human Genome Research Institute, Maryland Sanford School of Medicine, University of South Dakota, Sioux Falls, Pittsburgh.
Owen S. Clay, Department of Pediatrics, University of California, San Diego, California
Rachael Philips, Molecular Immunology and Inflammation Branch, Office of Science and Technology, Maryland
Brynja Matthiasardottir, Inflammatory Disease Section, National Human Genome Research Institute, Maryland Department of Cell Biology and Molecular Genetics, University of Maryland, College Park, Maryland.
Michele Nehrebecky, Inflammatory Disease Section, National Human Genome Research Institute, Maryland
Vivian K. Hua, Department of Pediatrics, University of California, San Diego, California
Rachael McVicar, Sanford Burnham Prebys Medical Discovery Institute, La Jolla, California
Yang Liu, Sanford Burnham Prebys Medical Discovery Institute, La Jolla, California
Suzanne M. Tucker, Department of Pathology, Rady Children’s Hospital, San Diego, California
Davide Randazzo, Light Imaging Section, Office of Science and Technology, Maryland
Natalie Deuitch, Oncogenesis and Development Section, National Human Genome Research Institute, Maryland
Sofia Rosenzweig, Inflammatory Disease Section, National Human Genome Research Institute, Maryland
Adam Mark, Center for Computational Biology and Bioinformatics, Department of Medicine, University of California, San Diego, California
Roman Sasik, Center for Computational Biology and Bioinformatics, Department of Medicine, University of California, San Diego, California
Kathleen M. Fisch, Center for Computational Biology and Bioinformatics, Department of Medicine, University of California, San Diego, California
Pallavi Pimpale Chavan, Inflammatory Disease Section, National Human Genome Research Institute, Maryland
Elif Eren, Protein Expression Laboratory, National Institute of Arthritis and Musculoskeletal and Skin Diseases, Maryland
Norman R. Watts, Protein Expression Laboratory, National Institute of Arthritis and Musculoskeletal and Skin Diseases, Maryland
Chi A. Ma, Genetics and Pathogenesis of Allergy Section, Laboratory of Allergic Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland
Massimo Gadina, Translational Immunology Section, Office of Science and Technology, Maryland
Daniella M. Schwartz, Division of Rheumatology and Clinical Immunology, University of Pittsburgh, Pittsburgh
Anwesha Sanyal, University of Pittsburgh Medical Center, Children’s Hospital of Pittsburgh, Pittsburgh University of Pittsburgh Scleroderma Center, Pittsburgh.
Giffin Werner, University of Pittsburgh Medical Center, Children’s Hospital of Pittsburgh, Pittsburgh University of Pittsburgh Scleroderma Center, Pittsburgh.
David R. Murdock, Undiagnosed Diseases Program, Medical Genetics Branch, National Human Genome Research Institute, Maryland
Nobuyuki Horita, Inflammatory Disease Section, National Human Genome Research Institute, Maryland
Shimul Chowdhury, Rady Children’s Institute for Genomic Medicine, Rady Children’s Hospital, San Diego, California
David Dimmock, Rady Children’s Institute for Genomic Medicine, Rady Children’s Hospital, San Diego, California
Kristen Jepsen, Institute for Genomic Medicine, University of California, San Diego, California
Elaine F. Remmers, Inflammatory Disease Section, National Human Genome Research Institute, Maryland
Raphaela Goldbach-Mansky, Translational Autoinflammatory Disease Section, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland
William A. Gahl, Undiagnosed Diseases Program, Medical Genetics Branch, National Human Genome Research Institute, Maryland
John J. O’Shea, Molecular Immunology and Inflammation Branch, Office of Science and Technology, Maryland
Joshua D. Milner, Division of Pediatric Allergy, Immunology, and Rheumatology, Columbia University, New York
Nathan E. Lewis, Department of Pediatrics, University of California, San Diego, California Department of Bioengineering, University of California, San Diego, California.
Johanna Chang, Division of Allergy, Immunology, and Rheumatology, Department of Pediatrics, University of California, San Diego, California Rady Children’s Hospital Foundation, Rady Children’s Hospital, San Diego, California.
Daniel L. Kastner, Inflammatory Disease Section, National Human Genome Research Institute, Maryland
Kathryn Torok, University of Pittsburgh Medical Center, Children’s Hospital of Pittsburgh, Pittsburgh University of Pittsburgh Scleroderma Center, Pittsburgh.
Hirotsugu Oda, Inflammatory Disease Section, National Human Genome Research Institute, Maryland Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases and the Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany.
Christopher D. Putnam, Department of Medicine, University of California, San Diego, California San Diego Branch, Ludwig Institute for Cancer Research, La Jolla, California.
Lori Broderick, Division of Allergy, Immunology, and Rheumatology, Department of Pediatrics, University of California, San Diego, California Rady Children’s Hospital Foundation, Rady Children’s Hospital, San Diego, California.
References
- 1.Wollina U, Looks A, Uhlemann C, Wollina K. Pansclerotic morphea of childhood-follow-up over 6 years. Pediatr Dermatol 1999;16:2 45–7. [DOI] [PubMed] [Google Scholar]
- 2.Moll M, Holzer U, Zimmer C, Rieber N, Kuemmerle-Deschner JB. Autologous stem cell transplantation in two children with disabling pansclerotic morphea. Pediatr Rheumatol Online J 2011; 9: Suppl 1:77. abstract. [Google Scholar]
- 3.Gruss C, Stücker M, Kobyletzki G, Schreiber D, Altmeyer P, Kerscher M. Low dose UVA1 phototherapy in disabling pansclerotic morphoea of childhood. Br J Dermatol 1997;136:293–4. [DOI] [PubMed] [Google Scholar]
- 4.Kowal-Bielecka O, Distler O. Use of methotrexate in patients with scleroderma and mixed connective tissue disease. Clin Exp Rheumatol 2010;2 8: Suppl 61:S160–S163. [PubMed] [Google Scholar]
- 5.Forsea A-M, Cretu A-N, Ionescu R, Giurcaneanu C. Disabling pansclerotic morphea of childhood — unusual case and management challenges. J Med Life 2008;1:348–54. [PMC free article] [PubMed] [Google Scholar]
- 6.Soh HJ, Samuel C, Heaton V, Renton WD, Cox A, Munro J. Challenges in the diagnosis and treatment of disabling pansclerotic morphea of childhood: case-based review. Rheumatol Int 2019;39:933–41. [DOI] [PubMed] [Google Scholar]
- 7.Philips RL, Wang Y, Cheon H, et al. The JAK-STAT pathway at 30: much learned, much more to do. Cell 2022;185:3857–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shuai K Modulation of STAT signaling by STAT-interacting proteins. Oncogene 2000;19:2638–44. [DOI] [PubMed] [Google Scholar]
- 9.Xin P, Xu X, Deng C, et al. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int Immunopharmacol 2020;80:106210. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen HN, Noss EH, Mizoguchi F, et al. Autocrine loop involving IL-6 family member LIF, LIF receptor, and STAT4 drives sustained fibroblast production of inflammatory mediators. Immunity 2017;46:2 20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurzinski K, Torok KS. Cytokine profiles in localized scleroderma and relationship to clinical features. Cytokine 2011;55:157–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Torok KS, Kurzinski K, Kelsey C, et al. Peripheral blood cytokine and chemokine profiles in juvenile localized scleroderma: T-helper cell-associated cytokine profiles. Semin Arthritis Rheum 2015;45:284–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.West NR. Coordination of immune-stroma crosstalk by IL-6 family cytokines. Front Immunol 2019;10:1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chakraborty A, Dyer KF, Cascio M, Mietzner TA, Tweardy DJ. Identification of a novel Stat3 recruitment and activation motif within the granulocyte colony-stimulating factor receptor. Blood 1999;93:15–24. [PubMed] [Google Scholar]
- 15.Holland SM, DeLeo FR, Elloumi HZ, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 2007;357:1608–19. [DOI] [PubMed] [Google Scholar]
- 16.Jägle S, Heeg M, Grün S, et al. Distinct molecular response patterns of activating STAT3 mutations associate with penetrance of lymphoproliferation and autoimmunity. Clin Immunol 2020;210:108316. [DOI] [PubMed] [Google Scholar]
- 17.Okada S, Asano T, Moriya K, et al. Human STAT1 gain-of-function heterozygous mutations: chronic mucocutaneous candidiasis and type I interferonopathy. J Clin Immunol 2020;40:1065–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mao X, Ren Z, Parker GN, et al. Structural bases of unphosphorylated STAT1 association and receptor binding. Mol Cell 2005;17:761–71. [DOI] [PubMed] [Google Scholar]
- 19.Nishio H, Matsui K, Tsuji H, Tamura A, Suzuki K. Immunolocalisation of the janus kinases (JAK) — signal transducers and activators of transcription (STAT) pathway in human epidermis. J Anat 2001;198:581–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamamoto K, Kobayashi H, Arai A, Miura O, Hirosawa S, Miyasaka N. cDNA cloning, expression and chromosome mapping of the human STAT4 gene: both STAT4 and STAT1 genes are mapped to 2q32.2–>q32.3. Cytogenet Cell Genet 1997;77:207–10. [DOI] [PubMed] [Google Scholar]
- 21.Zhong Z, Wen Z, Darnell JE Jr. Stat3 and Stat4: members of the family of signal transducers and activators of transcription. Proc Natl Acad Sci U S A 1994;91:4806–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ehrlich HP, Wyler DJ. Fibroblast contraction of collagen lattices in vitro: inhibition by chronic inflammatory cell mediators. J Cell Physiol 1983;116:345–51. [DOI] [PubMed] [Google Scholar]
- 23.Kendall RT, Feghali-Bostwick CA. Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol 2014;5:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diaz-Perez JL, Connolly SM, Winkelmann RK. Disabling pansclerotic morphea of children. Arch Dermatol 1980;116:169–73. [PubMed] [Google Scholar]
- 25.Wurster AL, Tanaka T, Grusby MJ. The biology of Stat4 and Stat6. Oncogene 2000;19:2577–84. [DOI] [PubMed] [Google Scholar]
- 26.Lessard CJ, Li H, Adrianto I, et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren’s syndrome. Nat Genet 2013;45:1284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Remmers EF, Plenge RM, Lee AT, et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med 2007;357:977–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diehl S, Anguita J, Hoffmeyer A, et al. Inhibition of Th1 differentiation by IL-6 is mediated by SOCS1. Immunity 2000;13:805–15. [DOI] [PubMed] [Google Scholar]
- 29.Mognol GP, Spreafico R, Wong V, et al. Exhaustion-associated regulatory regions in CD8+ tumor-infiltrating T cells. Proc Natl Acad Sci U S A 2017; 114(13):E 2776–E2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kurachi M CD8+ T cell exhaustion. Semin Immunopathol 2019;41:3 27–37. [DOI] [PubMed] [Google Scholar]
- 31.Khanna D, Lin CJF, Furst DE, et al. Long-term safety and efficacy of tocilizumab in early systemic sclerosis-interstitial lung disease: open-label extension of a phase 3 randomized controlled trial. Am J Respir Crit Care Med 2022;205:674–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.