


Abstract
Machine learning (ML) has revolutionised the field of structure-based drug design (SBDD) in recent years. During the training stage, ML techniques typically analyse large amounts of experimentally determined data to create predictive models in order to inform the drug discovery process. Deep learning (DL) is a subfield of ML, that relies on multiple layers of a neural network to extract significantly more complex patterns from experimental data, and has recently become a popular choice in SBDD. This review provides a thorough summary of the recent DL trends in SBDD with a particular focus on de novo drug design, binding site prediction, and binding affinity prediction of small molecules.
Introduction
The field of drug discovery and design is one of the most vibrant areas of research, with many groups from academic and industrial settings contributing to its advancement. The identification of small molecules binding to protein or RNA targets, and the related structural and functional information is of fundamental importance to modern drug discovery. One way to identify small molecule binders is high throughput screening (HTS), where millions of compounds are tested for the desired biological activity (Martis et al., 2011). However, HTS is time-consuming and expensive (DiMasi et al., 2003; Workman, 2003; Hopkins, 2009). An alternative to HTS is computer-aided drug design (CADD) or virtual HTS (Leelananda and Lindert, 2016). CADD provides the capability of screening millions of compounds virtually. This significantly reduces the number of molecules that need to be tested biochemically. Therefore, this approach can cut cost and accelerate the preliminary stage of drug development. CADD has been successfully applied to numerous disease-related target systems advancing research in treatments for congestive heart failure (Lindert et al., 2015a; Cai et al., 2016; Aprahamian et al., 2017; Coldren et al., 2020), several types of cancer (Lee et al., 2010; Ravindranathan et al., 2010; Chuang et al., 2015; Lee et al., 2019; Fratev et al., 2021; Hantz and Lindert, 2022), and various infectious diseases (Durrant et al., 2011; Zhu et al., 2013; Sinko et al., 2014; Lindert et al., 2015b; de Sousa et al., 2020).
In practice, commonly utilised CADD approaches may vary depending on the availability and type of experimental information. The two main methods being employed are structure-based drug design (SBDD) and ligand-based drug design (LBDD). In the structure-based approach, the 3D structures of targets are known. They are generally elucidated using techniques such as X-Ray crystallography, nuclear magnetic resonance spectroscopy and cryogenic electron microscopy and can be further supplemented with molecular dynamic simulations (Salsbury, 2010), computational structure prediction (Dorn et al., 2014; Seffernick and Lindert, 2020) and design (Huang et al., 2016) methods. These structures are then used for generating or screening potential small molecule ligands by predicting interactions with the target. On the other hand, ligand-based approaches are utilised when the 3D structure of the target is unknown. However, LBDD relies on the existence of a large set of ligands whose potency against a biological target of interest is known. Using such information, a correlation between the structures and properties of known ligands and their experimentally determined biological activities can be derived. This structure–activity relationship may in turn be used to design new drugs. Over the last decade, there has been a rapid shift in the field of drug development aimed at improving CADD approaches using a variety of machine learning (ML) techniques (Cao et al., 2018). Recently, others have reviewed the general role of artificial intelligence throughout the drug discovery pipeline (Hessler and Baringhaus, 2018; Patel et al., 2020; Carracedo-Reboredo et al., 2021; Gallego et al., 2021; Paul et al., 2021; Dara et al., 2022). Notably, in this work, we provide an overview of recent applications of deep learning (DL) in CADD methods, with a specific focus on SBDD.
Structure-based drug design
SBDD is the most commonly utilised approach when the three-dimensional structure of the drug target is known. In SBDD, one first identifies the structural data of the target either experimentally or through computational modelling. Next, a docking algorithm is used to position compounds from a database into the selected region of the target. These compounds are then scored and ranked using a score function and top hits are tested experimentally. Over the past few years, SBDD methodologies have gone through significant improvements in speed and accuracy. The introduction of ML techniques to SBDD approaches has also enhanced performance (Cao et al., 2018). DL techniques, a subdivision of ML with multi-layered neural network architectures, are increasingly utilised in drug discovery to capture complex data patterns and predict outcomes (Schmidhuber, 2015). Reinforcement learning (RL) is yet another subdivision of ML, where a computer model is trained by rewarding desired outcomes and penalised for undesired ones (Botvinick et al., 2019). The combination of DL and RL techniques (DRL) in the field of drug design has also been revolutionary and has led to several breakthroughs in recent years (Olivecrona et al., 2017; Neil et al., 2018; Popova et al., 2018; Ståhl et al., 2019; Zhou et al., 2019). Applications of these techniques in SBDD can be categorised into three main areas of focus: de novo drug design, binding site prediction, and binding affinity prediction.
De novo drug design
De novo drug design refers to the exploration of a broad chemical space and creation of new chemical compounds without the need for a starting template and was first introduced by Danziger and Dean (1989). The chemical search space for identifying novel compounds is virtually infinite and sufficiently sampling this space is a primary challenge in de novo drug design. In order to resolve this issue, a set of restraints is typically incorporated. For instance, the physical and chemical properties of a target’s active site are key constraints for ligand design. Structural and chemical information plays an immense role in reducing the search space to prevent the sampling of generations of compounds that might be synthetically unobtainable. Two sampling methods, atom-based and fragment-based, are primarily used for compound generation in de novo drug design (Gillet et al., 1993; Wong et al., 2011; Schneider and Fechner, 2022). In atom-based sampling (Fig. 1a), an initial atom is used as a seed inside the active site of the target. From this seed, a diverse set of compounds are grown by varying the number of atoms and hybridization states of each atom (Nishibata and Itai, 1991). Compounds generated from atom-based sampling have high structural diversity. However, the computational cost in narrowing down the lead compound with the atom-based method can increase exponentially with the size of the compound being designed (Hartenfeller and Schneider, 2011). Alternatively, fragment-based sampling (Fig. 1b) circumvents this issue by exploring a database of fragments. These fragments are then used as a seed in the active site to build the rest of the compound. This approach significantly narrows the chemical search space while maintaining good structural diversity (Böhm, 1992; Gillet et al., 1993; Pearlman and Murcko, 1996; Clark et al., 2022).
Fig. 1.

Illustration of computational de novo drug design. (a) In atom-based drug design, the small molecule is built atom by atom by sampling additions of many different types of atoms. (b) In fragment-based drug design, the small molecule is built by sampling additions of a library of fragments.
DL and deep reinforcement learning techniques have been used in numerous cases to improve the performance of de novo drug design. Common DL architectures utilised in de novo drug design include recurrent neural network (RNN), graph neural network (GNN), and graph convolutional neural network (GCNN). RNNs make use of sequential data or time series data and are known for their internal memory which takes information from prior inputs to influence the current input and output. In contrast, traditional deep neural networks assume that inputs and outputs are independent of one another (Dupond, 2019). RNNs have been recently expanded with long short-term memory (LSTM) networks. Introduced by Hochreiter and Schmidhuber (1997), LSTM are used as the building blocks for the layers of a RNN and enable RNNs to remember inputs over a long period of time. This memory is commonly referred to as a gated cell, meaning the cell decides whether to store or delete information based on the importance or weight it assigns to the information. GNNs are a class of artificial neural networks used for processing data that can be depicted as graph structures. GNNs operate on an information diffusion mechanism, meaning a graph is processed by a set of units which are linked according to the graph connectivity. The units exchange information in a pairwise fashion until a stable equilibrium is reached (Scarselli et al., 2009). This behaviour is similar to an RNN as weights are shared in each recurrent step. In contrast to RNNs and GNNs, GCNNs do not share weights between hidden layers. GCNNs generalise classical convolutional neural networks (CNNs) to graph-structured data. In this representation, structural information can be incorporated to model connections among entities and create further insights in the data. GCNNs can utilise the graph structure and aggregate node information from the neighbourhoods in a convolutional fashion (Zhang et al., 2019). Deep reinforcement learning (DRL) combines artificial neural networks with a framework of reinforcement learning (RL) that helps agents (i.e. the algorithm making decisions/actions) learn how to reach their goals. RL considers the problem of an agent learning to make decision based on trial-and-error from rewards or punishments. DRL incorporates DL by allowing the agent to make decisions from unstructured data or domain heuristics. We will review several notable recent examples of these architectures below.
Jeon and Kim (2020) created an atom-based de novo method to design novel compounds named Molecule Optimization by Reinforcement Learning and Docking (MORLD). MORLD is a deep generative model that uses binding affinities, obtained from docking simulations, as rewards in the reinforcement learning. In MORLD, a compound is optimised by Molecule Deep Q-Networks (MolDQN) (Zhou et al., 2019), based on reinforcement learning and chemistry domain knowledge. Compounds are optimised in single action steps, meaning either an addition or removal of an atom or bond in a chemically valid manner. This is controlled by the user as atom types for modification need to be specified. The modified molecule is then evaluated with different score functions such as the synthetic accessibility (SA) (Ertl and Schuffenhauer, 2009) and quantitative estimate of drug-likeness (QED) (Bickerton et al., 2012). The SA score is a metric that is used to estimate the ease of synthesis of drug-like molecules. SA score ranges from 1 (easy to synthesise) to 10 (difficult to synthesise). QED provides a quantitative metric for assessing druglikeness of target (and/or predicted) compounds. This value ranges between 0 and 1. A compound QED score of 0 indicates that all properties of that compound are unfavourable and vice-versa. These scores are weighted and used to guide compound modification with MolDQN in the next state. This process is repeated until the compound reaches a terminal state. In the terminal state, the modified compound is docked to the target with QuickVina2 (Alhossary et al., 2015). The resulting docking score is given to MolDQN as a reward and a compound is generated. This is repeated until a specified number of training episodes are reached. MORLD was shown to significantly decrease the time needed to design a novel inhibitor for the target discoidin domain receptor 1 kinase (DDR1) compared to the deep generative model GENTRL (Zhavoronkov et al., 2019). GENTRL took approximately 46 days (with training data) to successfully design a novel inhibitor against DDR1, whereas MORLD achieved the same result in less than 2 days without any training data. MORLD was also successfully able to generate agonists for D4 dopamine receptor (D4DR) without any initial lead compound information. MORLD can be used in situations where the lead compound is available, or when the lead compound must be identified from virtual screening or from scratch. MORLD has a free web server and source code open to the public. While MORLD is a successful tool for drug design, it has a few limitations: (a) the MORLD algorithm is trained on Q-value. Q-value is the expected future rewards, that is a p-value that has been adjusted for the false discovery rate. For example, a q-value of 2% means that 2% of significant results will result in false positives. This limits the diversification of optimised compounds, (b) as an atom-based model it is unable to explore a large chemical space, and (c) the SA and QED scores are not perfect. Therefore, generated compounds are sometimes chemically inaccurate. Additionally, MORLD is also not a suitable method for drug design when the target protein is disordered or does not have a druggable binding pocket. While a relatively new program, MORLD has been cited in many studies to acknowledge the usefulness of DRL in the field of drug discovery. To date, this algorithm has been applied to only the DDR1 system as mentioned previously.
Ma et al. (2021) created the Structure-Based de Novo Molecular Generator (SBMolGen), another DL method for de novo drug design based on Monte Carlo tree search (MCTS) (Browne et al., 2012a, 2012b) and docking simulations. Molecular generation in SBMolGen is done by ChemTS (Yang et al., 2017), and rDock (Ruiz-Carmona et al., 2014) is employed for docking generated compounds to the target. In the SBMolGen methodology, ChemTS first generates compounds with MCTS and a recurrent neural network (RNN) (Kaur and Mohta, 2019). These generated models are then docked using rDock and evaluated. Based on the top scoring model from the docking simulation, SBMolGen reweights the search tree in ChemTS. Additional molecules are then generated by ChemTS with the new weights. By repeating this process with a RNN, SBMolGen is able to generate compounds that are aware of the target it is binding to. The RNN model in SBMolGen was trained on 250,000 molecules from the ZINC database (Irwin et al., 2012). The results of SBMolGen were benchmarked with four target proteins: cyclin-dependent kinase 2 (CDK2), epidermal growth factor receptor erbB1 (EGFR), adenosine A2A receptor (AA2AR), and beta-2 adrenergic receptor (ADRB2). For each target, the generated molecules were evaluated against known actives from the Database of Useful Decoys: Enhanced (DUD-E) (Mysinger et al., 2012). Molecules generated with SBMolGen covered a larger chemical space than the ZINC data set, had an SA score of less than 3.5 and generally many of these compounds had QED scores greater than 0.8. Furthermore, SBMolGen was also able to find compounds that had a better docking score (as compared to known compounds) for all four target proteins. Fragment molecular orbital (FMO) (Kitaura et al., 1999) analysis of the target protein and generated molecules suggested that SBMolGen designed molecules with high binding affinity. Benchmark results of SBMolGen were compared against GENTRL and showed that SBMolGen was able to produce molecules that had better docking scores. While SBMolGen is faster than GENTRL, it is still limited by computation time. In addition, docking scores of SBMolGen were based on SA score, Lipinski’s rule of five (Lipinski et al., 2001) and PubChem (Kim et al., 2021) filters. While these are useful filters, comprehensive drug discovery still requires the optimization of a large number of molecular properties (Winter et al., 2019). SBMolGen has been employed as benchmark in several other method development studies.
Li et al. (2021b) created DeepLigBuilder, another atom-based DL method for de novo drug design. The deep generative model in DeepLigBuilder is able to design and optimise the 3D structures of ligands directly inside the binding pocket of the target. This method combines a graph convolutional neural network (L-Net) and MCTS, a common technique in reinforcement learning. L-Net iteratively generates and refines molecules by using a state encoder and policy network. During this stage, the state encoder analyses the state of the molecules and passes it to the policy network. The policy network then decides the type, bond order, coordinates, and the number of new atoms to be added. This combination allows the DeepLigBuilder to generate molecules with high binding affinity within the binding pocket. Benchmarking results indicated that L-Net had an overall validity of 96% compared to G-SchNet (Gebauer et al., 2022) which had an output validity of only 77%. The average RMSD of the generated compounds to optimised structures of the generated models with MMFF94 (Halgren, 1996) was only about 0.61 Å. The QED score was above 0.5 for 83.9% of the generated compounds and it was possible to correctly predict the overall distribution of molecular properties. L-Net was also able to re-create the chemical space of the training set almost identically. While L-Net’s performance was highly remarkable, it struggled to generate ring structures as observed by the high standard deviations for bond lengths and torsion angles for aromatic rings. DeepLigBuilder was also tested to generate 3D structures of compounds (as well as lead optimization of known inhibitors) inside the binding pocket of SARS-CoV-2 main protease. The MCTS (as opposed to random search) in DeepLigBuilder successfully lowered the smina scores (Koes et al., 2013) during lead optimization. Furthermore, DeepLigBuilder was able to generate compounds for SARS-CoV-2 main protease with novel chemical structures and high predicted binding affinity and had a success rate of 78.1%. DeepLigBuilder has been cited in several other studies for its use in generating lead compounds for SARS-CoV-2 as discussed above. However, to date, this algorithm has not been applied to other systems.
DeepScaffold is another DL tool for scaffold-based (can also be thought of as fragment-based) drug discovery created by Li et al. (2020). DeepScaffold is able to design compounds from cyclic skeletons, classical molecular scaffolds, and pharmacophore queries. DeepScaffold uses three different methods to enable these different inputs. When the input is a cyclic skeleton, DeepScaffold uses a graph convolutional neural network and variational autoencoder (VAE) (Kingma and Welling, 2014) to complete atom and bond types and generate a scaffold. However, if the input is a molecular scaffold, then the program uses a generative model (Li et al., 2018), from the authors’ previous study, to build the side chains. DeepScaffold can also filter out generated molecules that do not match the input pharmacophore query. DeepScaffold was trained on a dataset that contained 914,464 molecules from the ChEMBL databases with QED larger than 0.5. DeepScaffold was then evaluated for chemical validity and diversity, distribution of molecular properties and side chains. The performance of DeepScaffold to produce chemically valid and unique scaffolds was remarkable with an average of 98.9 and 69.1%, respectively. Molecular properties (molecular weight, solubility, and QED) of the test set molecules against the generated molecules indicate good correlation for scaffolds with more atoms. The model also demonstrated a diversified substitution pattern of side chains and was biased towards adding more atoms to nitrogen atoms. Finally, DeepScaffold model was tested on GPCRs. The model was able to produce chemically valid molecules, however, performed rather poorly to produce unique molecules with increasing structural complexity of scaffolds. The model was also able to reproduce known actives for the GPCRs reasonably well. DeepScaffold has been mentioned in several studies. This algorithm has not yet been applied to other target systems. However, it was referenced for successful scaffold hopping during compound design (Zhang et al., 2022). Additionally, DeepScaffold has one of the highest percentages for validity of generated compounds as compared to other scaffold-based de novo methods (Joshi et al., 2021).
MolAICal, created by Bai et al. (2021), is another fragment-based drug design algorithm. The two modules in MolAICal are trained with a DL model on fragments from the food and drug administrations (FDA) approved drugs and drug-like ligands from the ZINC database. MolAICal can directly generate 3D structures of drug-like compounds in the protein pockets. It employs a sequence-based generative model and a GNN for fragment generation and the fragment is then grown stochastically. A total of 21,064 fragments of FDA-approved drugs and 1,060,000 drug-like ligands from the ZINC database were used to train the generative model. When the grown ligand reaches a certain length, a genetic algorithm is employed to optimise the ligand into the protein pocket based on the ligand-receptor binding score. The docking score used by MolAICal is the Vinardo score (Quiroga and Villarreal, 2016) from AutoDock Vina (Trott and Olson, 2010). The Vinardo score is a summation of several energetic terms. These are Gaussian steric attractions, quadratic steric repulsions, Lennard-Jones potentials, electrostatic interactions, hydrophobic interactions, non-hydrophobic interactions, and non-directional hydrogen bonds. Vinardo scores are reported as the Gibbs free energy (ΔG) of binding in the unit of kcal mol−1. Therefore lower Vinardo scores are more favourable. However, in MolAICal, the coefficients of the Vinardo score were optimised based on a set of 2,903 protein-ligand complexes with experimental data from the PDBbind database (Wang et al., 2005). MolAICal was then tested to design drugs for glucagon receptor (GCGR) and SARS-CoV-2 main protease. For both the GCGR and the SARS-CoV-2 main protease, MolAICal was able to generate compounds that were diversified and had good theoretical binding affinity. In a recent study, MolAICal was employed to calculate the molecular synthetic accessibility score of traditional Chinese medicines in attempts to inhibit the SARS-CoV-2 nucleocapsid protein (Chen et al., 2022). In another application, MolAICal was utilised to calculate the relative binding energy of compounds from the ZINC database to inhibit carbonic anhydrase as a treatment for altitude sickness (Ali et al., 2022).
Finally, Arús-Pous and co-authors describe a fragment-based method that employs DL architecture to generate scaffolds based on SMILES string (Arús-Pous et al., 2020). In this drug-design architecture, a scaffold is generated through an RNN, that is composed of three LSTM cells (Hochreiter and Schmidhuber, 1997), from a SMILES string. Next, the scaffold is ‘decorated’ with side-chains at each attachment point in the scaffold exhaustively. The generated molecules are then filtered based on drug-like properties such as molecular weight, QED, SA, and/or user-specified properties. The decorator model was trained on a set of 4,211 Dopamine Receptor D2 active modulators. Following the training phase, the authors evaluated their generative model and the model was able to produce a diverse set of scaffolds and obtain a large amount of actives. The architecture implemented by Arús-Pous and co-authors is extremely versatile and can be combined with molecular generative architectures. This work by Arús-Pous and co-authors has been highlighted primarily in reviews of artificial intelligence in drug design.
A brief summary of all the previously mentioned methods, along with the respective links to the freely accessible code or webserver, can be found in Table 1. The methods mentioned here capture many of the key areas of how DL has influenced de novo drug design. Nonetheless, this is still an active area of research with novel applications of DL algorithms continuously being developed.
Table 1.
Summary of de novo drug design methods
| De novo drug design | |||||
|---|---|---|---|---|---|
| Method | Architecture | Sampling method | Benchmark against | Year | Code/Webserver |
| MORLD | DRL | Atom-based | GENTRL | 2020 | http://morld.kaist.ac.kr |
| SBMolGen | RNN and MCTS | Atom-based | GENTRL | 2021 | http://github.com/clinfo/SBMolGen |
| DeepLigBuilder | GCNN and MCTS | Atom-based | G-SchNet | 2021 | Not publicly available |
| DeepScaffold | GCNN and VAE | Fragment-based | None | 2020 | https://github.com/deep-scaffold |
| MolAICal | GNN and Genetic Algorithm | Fragment-based | None | 2021 | https://molaical.github.io |
| SMILES-based deep generative scaffold decorator | RNN with LSTM cells | Fragment-based | None | 2020 | https://github.com/undeadpixel/reinvent-scaffold-decorator |
Binding site prediction
Ligand binding sites in proteins (Fig. 2) encode necessary chemical and structural information for the successful execution of SBDD. Correct identification of these binding sites may help in the understanding of protein function and aid in the design of better drug-like small molecules. While these functional sites can be experimentally determined, the time and cost required to perform such experiments are quite significant. In contrast, computational prediction of protein-ligand binding sites is relatively inexpensive and significantly faster, thus expediting the drug development process. Several computational methods have been proposed over the years for structure-based binding site prediction (Huang, 2009; Le Guilloux et al., 2009; Yu et al., 2010; Tsujikawa et al., 2016; Dias et al., 2017; Wu et al., 2018). These methods can be roughly categorised as geometry-based, energy-based, and templated-based (Mylonas et al., 2021). Geometry-based methods analyse the geometry of the molecular surface of a target protein in order to locate surface cavities. Common approaches to find surface cavities within geometry-based methods include grid system scanning (Weisel et al., 2007), probe sphere filling (Yu et al., 2010) and alpha spheres (Le Guilloux et al., 2009). Energy-based binding site prediction methods involve searching for energetically favourable interactions between protein atoms and a chemical probe with the aid of a force field (Kozakov et al., 2015; Tsujikawa et al., 2016). Schrodinger’s SiteMap (Halgren, 2007, 2009) program combines geometry and energy-based properties to identify potential protein–ligand and protein–protein binding sites. Finally, template-based binding site prediction relies on the assumption that proteins that share structural similarity to the target protein also share functional similarity (Dey et al., 2013). One of the first ML-based binding site prediction algorithms was CryptoSite (Cimermancic et al., 2016). CryptoSite uses as support vector machine (SVM) to classify residues belonging to a binding site on a score range of 0 (not likely)–1 (most likely). Efforts in DL-based binding site prediction methods are fairly new and date back to as early as 2017.
Fig. 2.

Illustration of computational binding site prediction. In this methodology, 3D voxels are used to identify regions of the protein as potential binding sites (shown as yellow rectangles in the figure). Next, these sites are ranked from most to least probable for a ligand to bind.
Convolutional neural networks (CNNs) are a popular DL architecture for identifying potential binding sites. CNNs are regularised versions of fully connected networks, meaning each neuron in one layer is connected to all neurons in the next layer (Albawi et al., 2017). In a 2D-CNN, the kernel or filter used to extract features from images moves in two directions (x and y) and the input/output data is three-dimensional. 2D-CNNs are typically used on image data. A region-based CNN (R-CNN), is a type of 2D-CNN that implements a selective search algorithm that generates region proposals that are then provided to the CNN architecture to compute the features (Girshick et al., 2014). Region proposals are small regions of the image that potentially contain objects of interest in the input image. The benefit of this method is that the algorithm generates approximately 2,000 region proposals which greatly reduces the computational cost compared to computing CNN features for the entire input image. In a 3D-CNN, the kernel moves in three directions (x, y, and z) and the input/output of the model are four-dimensional. These algorithms are typically used on three-dimensional image data (Hedegaard et al., 2022). A popular 3D-CNN architecture is U-Net (Ronneberger et al., 2015) which has been shown to outperform traditional sliding window convolutional networks. The model provides many advantages over patch-based segmentation approaches as it preserves the full context of the input image through an end-to-end pipeline process for the entire image and requires few training samples. One increasingly popular CNN architecture is ResNet (He et al., 2016). ResNet overcomes the vanishing gradient problem, which occurs when the neural network training algorithm attempts to find weights that bring the loss function to a minimal value. If the network has too many layers, the gradient becomes increasingly small until it disappears and optimization cannot continue. ResNet overcomes this problem by creating a deep residual learning framework. This framework consists of shortcut connections that add intermediate inputs to the output of a group of convolution blocks. The shortcut connections perform identity mapping, allowing for smoother gradient flow and ensures important features are carried until the final layers. We review several notable recent examples of these architectures below.
Jiménez et al. (2017) developed the first DL-based protein-ligand binding site prediction algorithm, DeepSite. Based on a 4-layer convolutional neural network (DCNN) architecture, DeepSite treats the target protein as an image on a 3D grid. Each atom within the voxels of the grid is defined by atom-based physicochemical properties obtained from AutoDock 4 (Morris et al., 2009). Next, the entire grid is divided into subgrids in order to defined the local properties of smaller protein areas. These subgrids are then scored by the DCNN as potential sites for ligand binding. The DCNN architecture in DeepSite was trained on 7,622 proteins from the sc-PDB (Desaphy et al., 2015) database. Pocket prediction performance was evaluated by distance and shape of the predicted binding site to that of the real binding site. To accomplish this, the authors make use of two metrics, distance to centre of binding site (DCC) and discretized volumetric overlap (DVO). These metrics were also used to benchmark DeepSite against Fpocket (Le Guilloux et al., 2009) and Concavity (Capra et al., 2009) (binding site prediction algorithms). Based on the binding site analysis metrics, DeepSite was able to provide a more accurate binding site prediction compared to Fpocket and Concavity. Further analysis of DeepSite on proteins with diverse folds in the SCOPe database showed no bias towards any specific protein fold. However, there is still room for improvement as DeepSite does not have descriptors for atom-based polarizability effects or descriptors for water molecules. Since its release, DeepSite has been a highly successful tool and has been referenced in several studies. A few notable applications are its use to study the binding process and interaction of antibiotic drugs and lysozymes (Rial et al., 2022), analysis of the potential interaction mechanism of sweeteners with aroma compounds in passion fruit juice (Xiao et al., 2022), and identifying binding pockets for drug candidates for Hepatocellular carcinoma (predominant subtype of liver cancer) (Tang et al., 2022).
FRSite, introduced by Jiang et al. (2019), is another binding site prediction method based on a region-based convolutional neural network (R-CNN), an object-detection DL architecture (Ren et al., 2015). In this methodology, the target protein is treated as a 3D image and the binding site is treated as the object within the image to be detected. Similar to DeepSite, FRSite also treats each atom in the target protein on a grid with eight descriptors from high-throughput molecular dynamics (HTMD) (Doerr et al., 2016). Next, the entire grid is fed into a region proposal network-3D (RPN-3D) module in FRSite to detect potential binding sites. Once these potential binding sites are found by RPN-3D, FRSite uses its classifier to score the binding site. FRSite was trained on the same training dataset as DeepSite. Following the training, FRSite was evaluated for DCC and DVO as well as compared to those of other prediction methods. Compared to Fpocket and DeepSite, FRSite outperformed the other algorithms in predicting more accurate binding sites of proteins. Additionally, FRSite is also able to predict the receptor grid of the binding site (which can be used to estimate the size of the pocket). The authors further showed that the DVO estimated by FRSite was more accurate than that of Fpocket. FRSite has been referenced in several review articles and as benchmark in other method development studies.
Stepniewska-Dziubinska et al. (2020) introduced another DL-based binding site prediction method called Kalasanty. Similar to DeepSite, Kalasanty also treats the target protein as a 3D image. However, Kalasanty uses a DL model that is more similar to U-Net (Ronneberger et al., 2015) to treat the problem as a 3D image segmentation problem. In this approach, each featurized segment (by atomic properties) of the protein is assigned a certain probability of being a part of a pocket where a small molecule can dock. The DL model in Kalasanty was trained on 15,860 structures with binding site data from sc-PDB dataset. Prediction results from Kalasanty were then evaluated for correct DCC and DVO as well compared to those from DeepSite. Kalasanty correctly predicted binding sites two times better than DeepSite. Furthermore, Kalasanty had DCC values lower (i.e. closer to centre of the actual binding site) than DeepSite. Additionally, the binding sites predicted by DeepSite were bigger (lower DVO) and not accurately modelled when compared to that of Kalasanty (higher DVO). While Kalasanty is a successful binding site prediction model, the dataset used to train the DL model is based on proteins with deep cavities. Therefore this model may not be suitable for cases where the binding sites are located on flat surfaces. While being a newly released method, Kalasanty has already been subjected to many benchmarks in other method development studies. In addition, relevance of Kalasanty in enzyme engineering has been discussed in this review (Singh et al., 2021).
Following the works of DeepSite (Jiménez et al., 2017), FRSite (Jiang et al., 2019), and Kalasanty (Stepniewska-Dziubinska et al., 2020), Mylonas et al. (2021) developed DeepSurf. DeepSurf uses a variant (Dimou et al., 2019) of the DL network architecture known as residual network (ResNet) (He et al., 2016). The authors propose an 18-layer ResNet to which the surface representation of the target protein is passed in the form of a 3D grid with physiochemical features. The network then predicts a binding site score for the surface presenting points. These points are then clustered and ranked to predict the binding site. The network was trained on a set of 15,182 structures from the sc-PDB database. The distance between the predicted and the real binding site centre and other metrics were used to evaluate DeepSurf. Furthermore, DeepSurf was benchmarked against DL-based methods such as DeepSite, FRSite and Kalasanty as well non-ML methods. The authors showed that compared to DeepSurf, Kalasanty was unable to predict binding sites for a large number of proteins. Interestingly, DeepSite with its shallow neural network layer performed similarly to DeepSurf. While the non-ML methods such as Fpocket (geometry-based) and AutoSite (energy-based) (Ravindranath and Sanner, 2016) performed poorly, COFACTOR (template-based) (Roy et al., 2012) performed similarly when compared to DeepSurf. However, all ML methods generally tend to predict a smaller number of binding sites that are closer to the actual binding sites compared to non-ML methods. DeepSurf has been mentioned in several other studies. In most of these cases, DeepSurf was used as benchmark for other method development and/or other reviews.
Kandel et al. (2021) developed PUResNet where the protein structure is also treated as a 3D image and the binding site as the object within the image. PUResNet employs variants of two DL architectures, ResNet and U-Net, to address this issue. PUResNet was trained on a set of 5,020 protein structures from the sc-PDB database, and evaluated with DCC, DVO and proportion of ligand inside binding pocket (PLI). The authors also benchmarked their method against Kalasanty, developed by Stepniewska-Dziubinska et al., with a k-fold cross-validation and accuracy test. Briefly, k-fold cross-validation is a method employed to estimate the predictive power of the model on new data. From the benchmarking results, it was observed that PUResNet exhibited slightly better performance during the cross-validation and accuracy test than Kalasanty, despite the fact that PUResNet was trained with only a fraction (1/3) of the dataset used to train Kalasanty. PUResNet has been referenced in several other method development studies as well as other review articles, including a review about pharmacological chaperones for rare diseases (Scafuri et al., 2022).
Along with primary binding site prediction, allosteric site prediction has also been an active area of study (Goncearenco et al., 2013; Panjkovich and Daura, 2014; Greener and Sternberg, 2015; Song et al., 2017; Tian et al., 2021). The term allosteric binding site refers to a site other than a protein’s primary (orthosteric) active site where a compound can bind, resulting in conformational and dynamic changes (Srinivasan et al., 2014). The incorporation of these dynamic changes is critical to accurately predict allosteric sites. The main difference between orthosteric and allosteric ligand binding site prediction algorithms is the inclusion of protein dynamics. Additionally, the algorithms may differ based on the dataset that the DL model was trained on, as the quality and robustness of the data significantly impacts the algorithms’ features and results. Here we review BiteNet (Kozlovskii and Popov, 2020), a DL-based method for detecting allosteric sites of target proteins from their dynamic ensembles. Similar to other site prediction methods, this work is also inspired by the applications of DL in computer vision (Islam et al., 2016). In BiteNet, the input structure of the target protein is placed on a grid and processed with a neural network composed of 10 3D convolutional layers. BiteNet then outputs the centre of the predicted allosteric binding sites, assigns scores and identifies the neighbouring residues within 6 Å radius. BiteNet captures protein dynamics through an ensemble of protein structures gathered from molecular dynamic simulations. When the input is an ensemble of protein structures, BiteNet employs three different clustering algorithms to group the predicted binding site centres and their neighbouring residues and ranks the clusters from most to least probable. The convolutional neural network in BiteNet was trained on 5,946 structures from the PDB. To test for efficiency and accuracy, the authors tested BiteNet’s predictive power on the P2X3 receptor of the ATP-gated cation channel family, the epidermal growth factor receptor of the kinase family, and the adenosine A2A receptor of the G-protein coupled receptor family. In all three cases, it was observed that BiteNet was able to detect conformation-specific allosteric binding sites. Furthermore, BiteNet was benchmarked against FPocket (Le Guilloux et al., 2009), MetaPocket (Huang, 2009), DeepSite (Jiménez et al., 2017), SiteHound (Hernandez et al., 2009) and P2Rank (Krivák and Hoksza, 2018) for precision and computational speed on an independent benchmark dataset. In terms of speed and precision, BiteNet outperformed all other methods. BiteNet has been referenced in several other method development studies.
From this summary, it is evident that DL-based methods for binding site prediction have continuously outperformed traditional methods. A summary of the DL-based binding site prediction methods reviewed in this article is presented in Table 2. While these methods are highly successful, they are also limited. For instance, the dataset used to train most of these methods ignores noncanonical amino acids and heavy metals. Thus, these models are still not generalised enough to encompass the astronomical complexities in drug discovery and there is still a huge potential for improvement in the future.
Table 2.
Summary of binding site prediction methods
| Binding site prediction | |||||
|---|---|---|---|---|---|
| Method | Architecture | Type of site | Benchmark against | Year | Code/Webserver |
| DeepSite | 3D-CNN | Active | Fpocket and Concavity | 2017 | www.playmolecule.org/deepsite/ |
| FRSite | R-CNN | Active | Fpocket and DeepSite | 2019 | Not publicly available |
| Kalasanty | 3D-CNN (variant of U-Net) | Active | DeepSite | 2020 | https://gitlab.com/cheminfIBB/kalasanty |
| DeepSurf | 3D-CNN (variant of ResNet) | Active | DeepSite, FRSite, Kalasanty, Fpocket, AutoSite and COFACTOR | 2020 | https://github.com/stemylonas/DeepSurf |
| PUResNet | 3D-CNN (variant of ResNet and U-Net) | Active | Kalasanty | 2021 | https://github.com/jivankandel/PUResNet |
| BiteNet | 3D-CNN | Allosteric | FPocket, MetaPocket, DeepSite, SiteHound, and P2Rank | 2020 | https://github.com/i-Molecule/bitenet |
| https://sites.skoltech.ru/imolecule/tools/bitenet | |||||
Binding affinity prediction
In SBDD, an accurate prediction of protein-ligand binding affinity can facilitate the discovery of novel drug-like compounds (Fig. 3). In order to achieve such a prediction, a small molecule is typically docked to the target protein (Fig. 3a) with a molecular docking program. Then a score function (SF) is employed to predict the protein-ligand binding affinity (Fig. 3b). Such SFs (if accurate and fast) allow researchers to screen a large number of compounds to identify potential drug-like small molecules. Over the years, several SFs have been developed to approximate the interaction energy between protein and ligand (Jones et al., 1997; Jain, 2003; Friesner et al., 2004; Shivakumar et al., 2010; Trott and Olson, 2010; Wang et al., 2015). These can be classified into physics-based, empirical, and knowledge-based SFs. Physics-based methods include scoring functions based on force fields, solvation models and quantum mechanics (QM) methods. In physics-based SFs, interactions between the protein and the ligand are calculated based on terms such as van der Waals interaction, electrostatic interaction, torsion entropy, solvent effects and others. Due to the limited accuracy of these terms, QM methods hybridised with molecular mechanics (QM-MM) methods have also been utilised to address covalent interactions, polarisation and charge transfer processes (Hayik et al., 2010). While QM-MM methods are more accurate, they are also computationally expensive and impractical for screening large number of compounds. Empirical SFs estimate binding affinity by making simplistic approximations to the physics-based energy terms such as hydrogen bonds, hydrophobic effects, steric clashes and other important interactions in a protein-ligand system (Böhm, 1994). These terms are usually associated with weights that are optimised using experimental binding affinity data. Due to the simplicity of the energy terms, empirical score functions are typically less computationally expensive. Knowledge-based SFs are derived based on a statistical analysis of protein-ligand complexes from available 3D structures. In knowledge-based SFs, the frequency of different atom pairs occurring within different distances is assumed to play a key role in the interaction of the atoms. These frequencies are then converted to a distance-dependent potential of mean force (Muegge, 2006). Knowledge-based SFs are also computationally inexpensive and have similar accuracies compared to physics-based methods. However, since knowledge-based SFs do not directly account for experimental binding affinity data, discrepancies between predicted and experimental binding affinity are frequently observed (Stahl and Rarey, 2001). Recently, DL-based SFs have also been developed to predict binding affinity and have been shown to outperform traditional SF methods. These models are usually trained on a large dataset with known protein-ligand binding affinity. In DL-based SFs, feautrizing the protein-ligand system plays a key role in its ability to correctly predict the binding affinity. Here, we briefly summarise some of the DL-based methods that have been successful in predicting protein-ligand binding affinity.
Fig. 3.
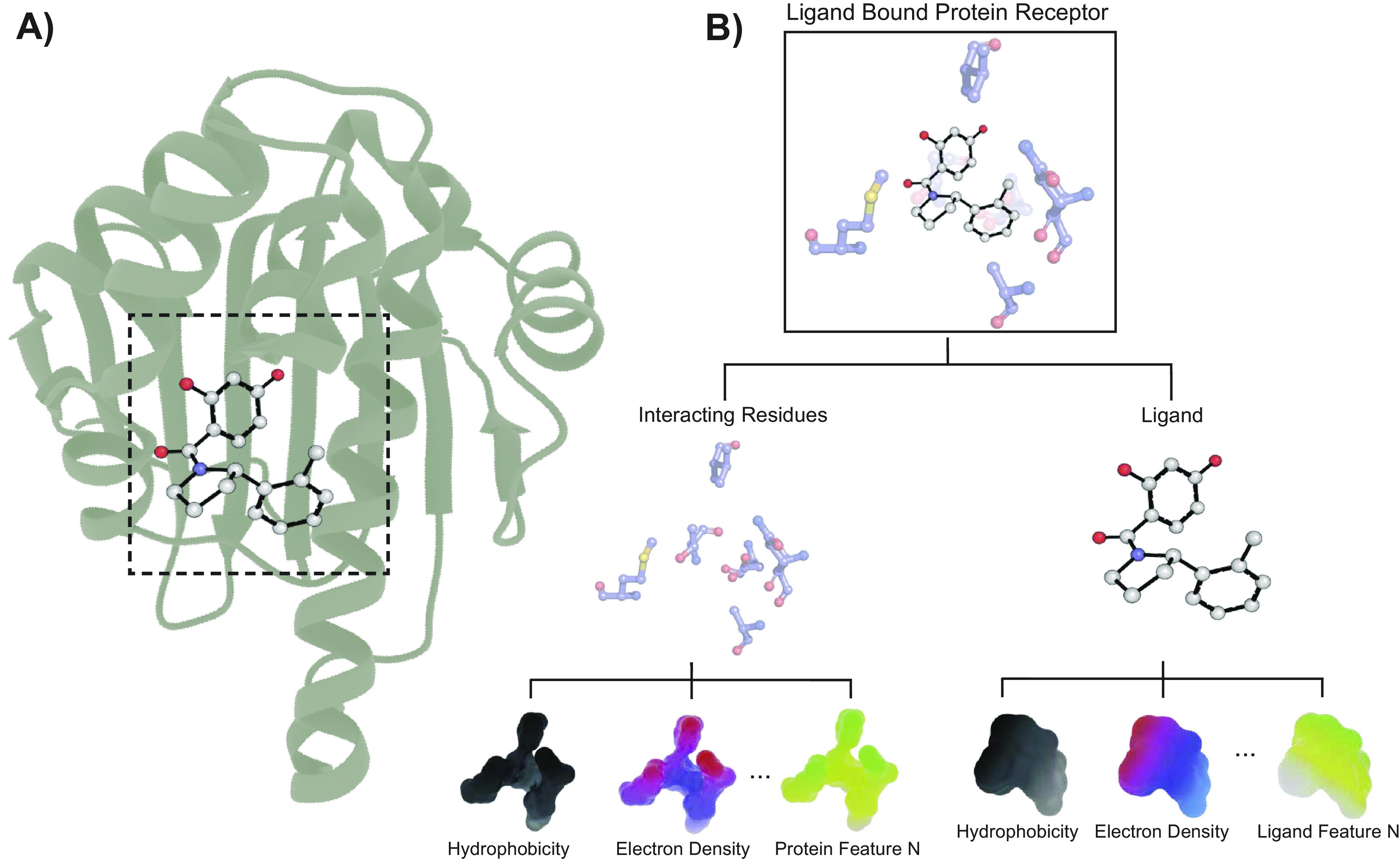
Illustration of computational binding affinity prediction. (a) Small molecule is docked into a target protein. (b) The binding site and the small molecule are then characterised with many features in order to predict the binding affinity. The atoms in the small molecule are shown in grey, blue and red for carbon, nitrogen and oxygen, respectively. The carbon, nitrogen, oxygen and sulphur of the binding site residues are shown in blue, purple, red and yellow, respectively. Bonds in the small molecule ligand are shown in black.
CNNs also serve as popular architectures in binding affinity prediction. Recent examples include variants of traditional 3D-CNNs and ResNet algorithms. Additional CNN-based architectures developed to solve binding affinity prediction are SqueezeNet (Iandola et al., 2016) and ShuffleNet (Zhang et al., 2018). SqueezeNet is a convolution neural network that utilises a design strategy aimed at reducing the number of parameters. Fewer parameters offer several advantages such as: more efficient distributed training, smaller memory storage requirements, and less communication required for over-the-air model updates. SqueezeNet accomplishes its smaller size via Fire modules which replace traditional 3 × 3 filters (kernels) with 1 × 1 filters. ShuffleNet is a CNN architecture that was designed for mobile devices with very limited computing power. In order to reduce computational cost and maintain accuracy, ShuffleNet employs pointwise group convolution and channel shuffle. Grouped convolution makes use of a multiple kernels (filters) per layer, resulting in multiple channel output per layer. This technique led to wider networks helping learn low- and high-level features (Xie et al., 2017), and it has been shown to improve classification accuracy (Xie et al., 2017). Channel shuffle is an operation that helps information flow across feature channels. We review several notable recent examples of these architectures below.
Jiménez et al. (2018) developed K DEEP, a DL-based method that relies on 3D-convolutional neural network (3D-CNN), to predict protein-ligand binding affinity. The DL architecture in K DEEP is designed based on the image classification DL architecture SqueezeNet (Iandola et al., 2016). In K DEEP, residues of the target protein at the binding site and the ligand are placed on a grid. Next, each atom of these interacting residues and the ligand are featurized by eight pharmacophoric-like properties. These descriptors were used to train the K DEEP network on a training dataset that was composed of 3,767 protein-ligand complexes from the PDBbind (Wang et al., 2005) database. Once the network was trained, K DEEP was benchmarked against RF-Score (Ballester and Mitchell, 2010), X-Score (Wang et al., 2002), and CyScore (Cao and Li, 2014) (three other non-DL-based binding affinity prediction methods) for performance and accuracy. Briefly, RF-score is a random forest model trained on a large dataset of molecular interactions. A low RF-score generally corresponds to more favourable interactions. X-Score is an empirical score function based on terms such as van der Waals interactions, hydrogen bonding, deformation penalties, and hydrophobic effects. X-Score values are reported as the Gibbs free energy (ΔG) of binding in the unit of kcal mol−1. Therefore a lower value is indicative of more energetically favourable interactions. Cyscore is another empirical score function that has been optimised based on hydrophobic free energy, van der Waals interaction energy, hydrogen-bond energy and the ligand’s entropy. Cyscore values are comparable to the ΔG (kcal mol−1) of binding. Therefore, a lower Cyscore indicates a higher binding affinity. Compared to other binding affinity prediction methods, K DEEP exhibited the lowest root mean square error (RMSE) with respect to the predicted and experimental affinity. The Pearson’s correlation coefficient of K DEEP was higher than that of X-Score and CyScore but was very similar to that of RF-Score. Furthermore, when these methods were benchmarked against independent datasets that the network had not seen more, K DEEP underperformed compared to RF-Score. Binding affinity predictions from K DEEP are reported as the ΔG (kcal mol−1) of binding. K DEEP is a highly successful program and has been used as a tool in numerous studies. A few examples where K DEEP has been successfully utilised are: to engineer maltooligosaccharide-forming amylases and optimise the biosynthesis of maltooligosaccharides (Ding et al., 2022), to determine the inhibitory properties of antiviral drugs and their structural analogues towards coronavirus (Kalamatianos, 2022), during virtual screening (of lead compounds and analogs) to chemically inhibit processes controlled by RecA protein of Acinetobacter baumannii (Tiwari, 2022) and so forth. While K DEEP is a versatile SF for binding affinity predictions, the authors argued that additional exploration of the network architecture and a thorough featurization of the molecular descriptor could further improve the SF’s performance.
Pafnucy (Stepniewska-Dziubinska et al., 2018), developed by Stepniewska-Dzuibinska et. al, is another DL model for predicting binding affinity of protein-ligand complexes. Similar to K DEEP, Pafnucy’s 3D-CNN also treats the ligand and interacting residues of the target protein as a 3D image. This information is then used to create a 3D grid and each atom in the grid is featurized with 19 atomic properties. The network was trained on experimental dissociation constant values of 11,906 protein-ligand complexes (from the PDBbind database) and then evaluated for its performance using RMSE, mean absolute error (MAE), Pearson correlation coefficient, and standard deviation in regression (SD). Therefore, the predicted values from Pafnucy are comparable to experimental dissociation constants. Furthermore, the prediction power of Pafnucy was benchmarked against binding affinity prediction methods such as X-Score, RF-Score, ChemScore, PLP1, ChemPLP (Verdonk et al., 2003), and G-Score (SYBYL 2022). Briefly, ChemScore is an empirical score function based on simple contact terms, a simplistic model for hydrogen bonds, and a penalty function based on flexibility. PLP1 is also an empirical score function that uses terms to account for hydrogen bonds, van der Waals interactions, repulsion potentials, and piecewise linear potentials to model the steric complementarity between protein and ligand. ChemPLP is another empirical score function that is very similar to PLP1. However, in addition to the terms in PLP1, ChemPLP also considers the distance and angle-dependent hydrogen and metal bonding terms from ChemScore. G-Score is a force-field based score function. It is composed of a protein-ligand complexation term (optimised Lenard-Jones potential), a hydrogen bonding term (approximated) and an internal energy term (approximated). All score values from ChemScore, PLP1, ChemPLP, and G-score are reported as the ΔG (kcal mol−1) of binding, therefore lower scores are considered as more favourable interactions. Pafnucy has been successfully used to study the interaction of the mannose receptor of macrophages with carbohydrate ligands (Zlotnikov and Kudryashova, 2022), as well as has been used as benchmark to develop several newer methods. Pafnucy outperformed all other methods except for RF-Score v3 (Li et al., 2015). However, the observed difference in performance was negligible. Additionally, Pafnucy outperformed all 20 binding affinity prediction methods that were used in the CASF-2013 (Li et al., 2014) benchmark. Besides binding affinity prediction, the authors of Pafnucy argue that their model may also be helpful to guide ligand optimization during molecular docking.
Methods that employ DL techniques usually require a large amount of data in order for the method to be properly trained. Thus, limited data could pose a challenge. DeepAtom, a DL method for binding affinity prediction of protein-ligand complexes, developed by Li et al. (2019) addresses this concern with a novel 3D-CNN architecture. The authors of DeepAtom developed an efficient 3D-CNN architecture based on ShuffleNet (Zhang et al., 2018). Briefly, ShuffleNet is an extremely computation-efficient CNN architecture that is designed for cases with limited computational resources. Using this architecture allowed DeepAtom’s network to be designed with fewer trainable parameters, deep layers, and limited training data. Similar to other methods, DeepAtom also treats the protein-ligand complex as a 3D image. In DeepAtom the interacting residues of the target protein (defined from the centre of the ligand) as well as the ligand are voxelized in a grid. The atoms occupying the voxels are feauturized with 12 atomic properties. To evaluate DeepAtom, the authors first trained DeepAtom, Pafnucy, and RF-Score with 3,390 complexes from the PDBbind database. These models were then evaluated with Pearson correlation coefficient, MAE, RMSE and SD. DeepAtom showed significant improvement over Pafnucy and slight improvement over RF-Score in all metrics. Next, the authors re-trained DeepAtom, Pafnucy and RF-Score on a larger training dataset (9,383 complexes) and evaluated all three models for performance. DeepAtom performed significantly better than both Pafnucy and RF-Score. Since the networks in DeepAtom and Pafnucy are based on 3D-CNN, it was observed that a well-designed network architecture (DeepAtom) can yield better predictions with limited data suggesting that effective training in the deep layers results in improvements in the capacity for learning and generalisation. DeepAtom has been employed in several method development studies (as benchmark) and other review articles.
One key factor of a successful DL scoring function is being able to train the model on features that are important to protein-ligand binding affinity. While the methods discussed so far focused on features that localise to the ligand binding site, OnionNet, developed by Zheng et al. (2019), takes a different approach. The authors treat the protein-ligand complex as a 2D image with one colour channel and use a 2D-CNN architecture to train the binding affinity prediction model. OnionNet is able to capture both local and non-local interactions between the protein and ligand by grouping contacts between each atom of the ligand to atoms in the protein in a set of 60 distance groups. In this way, OnionNet uses 3,840 features to define local and non-local interaction between the target protein and ligand. The network was trained on 11,906 protein-ligand complexes from the PDBbind database. Therefore, predicted values from OnionNet are comparable to experimental dissociation constants. Following the training, the prediction model was evaluated for accuracy and also compared to other ML and non-ML binding affinity prediction methods. In terms of RMSE and Pearson correlation coefficient, OnionNet outperformed Pafnucy and RF-Score. While K DEEP performed slightly better than OnionNet, OnionNet significantly outperformed other non-ML SFs used in the CASF-2013 benchmark. The authors further showed that both the local and non-local interactions contribute significantly to the prediction capability of the model. OnionNet has been utilised as a benchmark during the development of several binding affinity prediction methods.
AK-Score developed by Kwon et al. (2020) is yet another binding affinity prediction method that was developed based on the ResNet (He et al., 2016) architecture. Similar to other methods, the protein-ligand system is represented as a 3D image on a grid, with each atom on the grid being defined with eight atomic properties. However, the network architecture uses an ensemble-based approach to perform the binding affinity prediction. AK-Score utilises 20 independently trained networks to make predictions and the average of those values is considered as the final prediction. AK-Scores are reported as the ΔG (kcal mol−1) of binding, therefore a low score corresponds to a more energetically favourable interaction. The networks were trained on 3,772 protein-ligand complexes from the PDBbind database. AK-Score was then evaluated on the CASF-2016 benchmark dataset with Pearson correlation coefficient, Spearman correlation coefficient, Kendall tau, predictive index metrics, MAE, and RSME. AK-Score with ensemble-based networks was benchmarked against AK-Score with a single network and K DEEP. These results showed that the AK-Score ensemble network model had a lower MAE and RMSE as well as higher Pearson correlation coefficient, Spearman correlation coefficient, Kendall tau and predictive index as compared to other methods. Furthermore, from feature analysis, it was shown that the excluded volume of atoms, spatial distribution of hydrophobic and aromatic atoms of the ligand and protein, as well as the distribution of hydrogen bond acceptors of the protein were particularly important for the network to accurately determine the binding affinity. Therefore, more accurate calculations of such descriptors may play an important role in improving the ensemble-based network of AK-Score. AK-Score has been reviewed in several reviews and its prediction quality has been compared in other method development studies.
Similar to AK-Score, RosENet developed by Hassan-Harrirou et al. (2020), also uses an ensemble-based 3D-CNN. However, RosENet uses a hybrid approach where it employs the 3D-CNN to combine molecular mechanics energies calculated from the Rosetta force field (Alford et al., 2017) with molecular descriptors. Briefly, the Rosetta software suite includes algorithms for computational modelling and analysis of protein structures and has enabled scientific advances in areas such as protein design, enzyme design, molecular docking and structure prediction (Leman et al., 2020). The network architecture of RosENet was developed based on the ResNet architecture. Similar to previous methods, protein-ligand complexes were represented in a 3D grid. Atoms of the protein-ligand complex were featurized with 12 molecular descriptors from HTMD (Doerr et al., 2016) and 6 molecular energies obtained from Rosetta. The network was then trained on a curated dataset of 4,463 protein-ligand complexes obtained from the PDBbind database. Therefore, the predicted values from RosENet are comparable to experimental dissociation constants. To evaluate RosENet with respect to the quality of its predictions, the network was first optimised based on a combination of multiple features and architectures, and then compared to Pafnucy and OnionNet. The authors tested two neural network architectures: ResNet (a deep residual neural network) and SqueezeNet (a smaller network architecture). RosENet showed a lower RMSE with the ResNet architecture than it did with the SqueezeNet architecture. Thus, the authors argue that the network implemented in the SqueezeNet architecture was not deep enough to generalise the molecular mechanics energies that were used to featurize the complex. RosENet was also trained with a smaller number of features, leading to a decrease in the prediction quality, however, this change was considered to be insignificant. Overall, RosENet performed better than Pafnucy and comparably to OnionNet. This suggested that the molecular energies described in RosENet may be able to represent similar local and non-local interactions described by the OnionNet. Finally, the RMSE of RosENet decreased when the average of the five best predictions was used. In future studies, the authors of RosENet propose to improve their molecular features by extracting additional dynamic properties from molecular dynamics simulations. RosENet has been featured in other method development studies and as well as review articles.
Accurate prediction of binding affinity is a key step during the drug discovery process. Despite several limitations, recently developed DL-based methods for binding affinity prediction have generally significantly outperformed traditional methods. A summary of such DL-based methods as reviewed in this article can be found in Table 3. These studies indicate the availability of successful in silico tools for rapid and accurate prediction of protein-ligand binding affinity. Additionally, a few ML-based methods (Aggarwal and Koes, 2020; Bao et al., 2021) have also been trained to predict ‘the root mean square deviation (RMSD)’ of a docked structure with reference to the native bound structure. Deep Binding Structure RMSD Prediction model (DeepBSP) (Bao et al., 2021) is one such method that employs a 3D-CNN (same model architecture as K DEEP) to achieve this purpose. DeepBSP exhibited accuracy on predicting RMSD. Furthermore, the docking power of DeepBSP (through model selection with lowest predicted RMSD), outperformed all other scoring functions benchmarked in CASF-2016. While these methods have been successful, this field continues to grow and evolve with new advancements in ML.
Table 3.
Summary of binding affinity prediction methods
| Binding affinity prediction | |||||
|---|---|---|---|---|---|
| Method | Architecture | Features used | Benchmark against | Year | Code/Webserver |
| K DEEP | 3D-CNN (variant of SqueezeNet) | Atomic descriptors for local interactions | RF-Score, X-Score, and CyScore | 2018 | www.playmolecule.org/kdeep/ |
| Pafnucy | 3D-CNN | Atomic descriptors for local interactions | X-Score, RF-Score, ChemScore, ChemPLP, PLP1, and G-Score | 2018 | https://gitlab.com/cheminfIBB/pafnucy |
| DeepAtom | 3D-CNN (variant of ShuffleNet) | Atomic descriptors for local interactions | Pafnucy and RF-Score | 2019 | Not publicly available |
| OnionNet | 2D-CNN | Atomic descriptors for local and non-local interactions | Pafnucy, RF-Score, and K DEEP | 2019 | http://github.com/zhenglz/onionnet/ |
| AK-Score | 3D-CNN ensemble-based (variant of ResNet) | Atomic descriptors for local interactions | AK-Score single network and K DEEP | 2020 | Not publicly available |
| RosENet | 3D-CNN (variant of ResNet) | Molecular energies from Rosetta and atomic descriptors | K DEEP, Pafnucy, and OnionNet | 2020 | https://github.com/DS3Lab/RosENet/tree/master/models |
Conclusion
Over recent years, ML has become an increasingly popular field of study. Application of these methods to biological problems will only continue to grow as academic and industry users create better tools to predict biomolecular structure, treat disease, and improve public health. In this work, we summarised recent DL applications in three structure-based drug discovery categories: de novo drug design, binding site prediction and binding affinity prediction. In the interest of the reader, we also provide links to the various methods’ publicly available source code or webservers. The use of ML (more recently DL) methods is not limited solely to SBDD, and they have also been applied to all other areas of the drug discovery pipeline such as LBDD (Bahi and Batouche, 2018), lead optimization (de Souza Neto et al., 2020), and assessment of absorption (Shin et al., 2018), metabolism (Wang et al., 2020; Litsa et al., 2021), binding kinetics (De Benedetti and Fanelli, 2018; Mardt et al., 2018; Gao et al., 2019; Nunes-Alves et al., 2020; Feizpour et al., 2021; Obeid et al., 2021; Hoffmann et al., 2022), efficacy (Lin et al., 2018; Benning et al., 2020; Li et al., 2021a; Zhu et al., 2021) and toxicity properties (Ferreira and Andricopulo, 2019; Liu et al., 2019; Shi et al., 2019; Cáceres et al., 2020; Feinberg et al., 2020). While these studies are not covered in this review, interested readers are encouraged to review these recent works as well.
With the expansion of large datasets, DL algorithms will only become more accurate, thereby increasing cost-effectiveness and time efficiency. However, a major critique of DL methods is their inherit ‘black-box’ nature. In order for the use of large datasets to be truly successful, we must begin to understand the nature of DL algorithms and the connections they make. Another common pitfall of any ML algorithm is the quality of its training dataset(s). In any biological application, an algorithm may easily become hyper-specific to a certain motif if it is not exposed to an unbiased training set. This lack of generalisation prevents the algorithm from solving real-world problems.
Recently the company DeepMind and the Baker research group have released AlphaFold (Jumper et al., 2021) and RoseTTAFold (Baek et al., 2021), DL algorithms for protein structure prediction. These algorithms have been credited with ‘solving the protein folding problem’. Given the impressive prediction capabilities of these tools, it is natural to investigate how relevant such DL-based protein conformations are for drug design. For example, recently the crystal structure of the σ2 receptor has been reported (Alon et al., 2021). In this study, the authors also screened a large library of compounds to the X-ray structure (through molecular docking), which resulted in the identification of around 130 active compounds. However, the molecular docking of these same hits scored relatively poorly against the AlphaFold model. This suggests that additional efforts are needed to employ predicted structures successfully in drug discovery. Within the last year, the CEO of DeepMind announced the plan of applying their knowledge of artificial intelligence to drug discovery with the formation of a new company called Isomorphic Labs (https://www.isomorphiclabs.com). This year another company, CHARM Therapeutics (https://charmtx.com), in the UK have launched DragonFold. DragonFold is the first DL algorithm to rapidly predict protein-ligand co-folding. While we wait on more details of these developments, they however foreshadow a very promising future for the field of computer-aided drug discovery, as the brightest minds from industry and academia continue to collaborate and improve upon ML applications. While the future of the field appears very promising, we must also keep in mind the ethics of good science. As demonstrated by Urbina et al. (2022), artificial intelligence was utilised to predict the toxicity of small molecules. The algorithm developed by the Collaborations team predicted many compounds with higher toxicity as compared to the nerve agent VX. The team calls for a strengthening of ethical training of students, as well as for artificial intelligence drug discovery companies to create a code of conduct to properly train employees and establish protection measures for their technology.
Open Peer Review
To view the open peer review materials for this article, please visit http://doi.org/10.1017/qrd.2022.12.
References
- Aggarwal R and Koes DR (2020) Learning RMSD to improve protein–ligand scoring and pose selection.
- Albawi S, Mohammed TA and Al-Zawi S (2017) Understanding of a convolutional neural network. In 2017 International Conference on Engineering and Technology (ICET). Antalya, Turkey: IEEE, pp. 1–6. [Google Scholar]
- Alford RF, Leaver-Fay A, Jeliazkov JR, O’Meara MJ, Dimaio FP, Park H, Shapovalov MV, Renfrew PD, Mulligan VK, Kappel K, Labonte JW, Pacella MS, Bonneau R, Bradley P, Dunbrack RL, Das R, Baker D, Kuhlman B, Kortemme T and Gray JJ (2017) The Rosetta all-atom energy function for macromolecular modeling and design. Journal of Chemical Theory and Computation 13(6), 3031–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhossary A, Handoko SD, Mu Y and Kwoh CK (2015) Fast, accurate, and reliable molecular docking with QuickVina 2. Bioinformatics 31(13), 2214–2216. [DOI] [PubMed] [Google Scholar]
- Ali A, Warsi MH, Rahman MA, Ahsan MJ and Azam F (2022) Toward the discovery of a novel class of leads for high altitude disorders by virtual screening and molecular dynamics approaches targeting carbonic anhydrase. International Journal of Molecular Sciences 23(9), 5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon A, Lyu J, Braz JM, Tummino TA, Craik V, O’Meara MJ, Webb CM, Radchenko DS, Moroz YS, Huang X-P, Liu Y, Roth BL, Irwin JJ, Basbaum AI, Shoichet BK and Kruse AC (2021) Structures of the σ2 receptor enable docking for bioactive ligand discovery. Nature 600(7890), 759–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprahamian ML, Tikunova SB, Price MV, Cuesta AF, Davis JP and Lindert S (2017) Successful identification of cardiac troponin calcium sensitizers using a combination of virtual screening and ROC analysis of known troponin C binders. Journal of Chemical Information and Modeling 57(12), 3056–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arús-Pous J, Patronov A, Bjerrum EJ, Tyrchan C, Reymond JL, Chen H and Engkvist O (2020) SMILES-based deep generative scaffold decorator for de-novo drug design. Journal of Cheminformatics 12(1), 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek M, Dimaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD, Millán C, Park H, Adams C, Glassman CR, Degiovanni A, Pereira JH, Rodrigues AV, Dijk AAV, Ebrecht AC, Opperman DJ, Sagmeister T, Buhlheller C, Pavkov-Keller T, Rathinaswamy MK, Dalwadi U, Yip CK, Burke JE, Garcia KC, Grishin NV, Adams PD, Read RJ and Baker D (2021) Accurate prediction of protein structures and interactions using a three-track neural network. Science 373(6557), 871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahi M and Batouche M (2018) Deep learning for ligand-based virtual screening in drug discovery. In 2018 3rd International Conference on Pattern Analysis and Intelligent Systems (PAIS). Tebessa, Algeria: IEEE, pp. 1–5. [Google Scholar]
- Bai Q, Tan S, Xu T, Liu H, Huang J and Yao X (2021) MolAICal: A soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Briefings in Bioinformatics 22(3), bbaa161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballester PJ and Mitchell JB (2010) A machine learning approach to predicting protein–ligand binding affinity with applications to molecular docking. Bioinformatics 26(9), 1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao J, He X and Zhang JZH (2021) DeepBSP—A machine learning method for accurate prediction of protein–ligand docking structures. Journal of Chemical Information and Modeling 61(5), 2231–2240. [DOI] [PubMed] [Google Scholar]
- Benning L, Peintner A, Finkenzeller G and Peintner L (2020) Automated spheroid generation, drug application and efficacy screening using a deep learning classification: A feasibility study. Scientific Reports 10(1), 11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickerton GR, Paolini GV, Besnard J, Muresan S and Hopkins AL (2012) Quantifying the chemical beauty of drugs. Nature Chemistry 4(2), 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhm HJ (1992) The computer program LUDI: A new method for the de novo design of enzyme inhibitors. Journal of Computer-Aided Molecular Design 6(1), 61–78. [DOI] [PubMed] [Google Scholar]
- Böhm HJ (1994) The development of a simple empirical scoring function to estimate the binding constant for a protein–ligand complex of known three-dimensional structure. Journal of Computer-Aided Molecular Design 8(3), 243–256. [DOI] [PubMed] [Google Scholar]
- Botvinick M, Ritter S, Wang JX, Kurth-Nelson Z, Blundell C and Hassabis D (2019) Reinforcement learning, fast and slow. Trends in Cognitive Sciences 23(5), 408–422. [DOI] [PubMed] [Google Scholar]
- Browne CB, Powley E, Whitehouse D, Lucas SM, Cowling PI, Rohlfshagen P, Tavener S, Perez D, Samothrakis S and Colton S (2012a) A survey of Monte Carlo tree search methods. IEEE Transactions on Computational Intelligence and AI in Games 4(1), 1–43. [Google Scholar]
- Browne CB, Powley E, Whitehouse D, Lucas SM, Cowling PI, Rohlfshagen P, Tavener S, Perez D, Samothrakis S and Colton S (2012b) A survey of Monte Carlo tree search methods.
- Cáceres EL, Tudor M and Cheng AC (2020) Deep learning approaches in predicting ADMET properties. Future Medicinal Chemistry 12(22), 1995–1999. [DOI] [PubMed] [Google Scholar]
- Cai F, Li MX, Pineda-Sanabria SE, Gelozia S, Lindert S, West F, Sykes BD and Hwang PM (2016) Structures reveal details of small molecule binding to cardiac troponin. Journal of Molecular and Cellular Cardiology 101, 134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y and Li L (2014) Improved protein–ligand binding affinity prediction by using a curvature-dependent surface-area model. Bioinformatics 30(12), 1674–1680. [DOI] [PubMed] [Google Scholar]
- Cao C, Liu F, Tan H, Song D, Shu W, Li W, Zhou Y, Bo X and Xie Z (2018) Deep learning and its applications in biomedicine. Genomics Proteomics Bioinformatics 16(1), 17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra JA, Laskowski RA, Thornton JM, Singh M and Funkhouser TA (2009) Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure. PLOS Computational Biology 5(12), e1000585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo-Reboredo P, Liñares-Blanco J, Rodríguez-Fernández N, Cedrón F, Novoa FJ, Carballal A, Maojo V, Pazos A and Fernandez-Lozano C (2021) A review on machine learning approaches and trends in drug discovery. Computational and Structural Biotechnology Journal 19, 4538–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YM, Wei JL, Qin RS, Hou JP, Zang GC, Zhang GY and Chen TT (2022) Folic acid: A potential inhibitor against SARS-CoV-2 nucleocapsid protein. Pharmaceutical Biology 60(1), 862–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang CH, Cheng TC, Leu YL, Chuang KH, Tzou SC and Chen CS (2015) Discovery of Akt kinase inhibitors through structure-based virtual screening and their evaluation as potential anticancer agents. International Journal of Molecular Sciences 16(2), 3202–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimermancic P, Weinkam P, Rettenmaier TJ, Bichmann L, Keedy DA, Woldeyes RA, Schneidman-Duhovny D, Demerdash ON, Mitchell JC, Wells JA, Fraser JS and Sali A (2016) CryptoSite: Expanding the druggable proteome by characterization and prediction of cryptic binding sites. Journal of Molecular Biology 428(4), 709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark DE, Frenkel D, Levy SA, Li J, Murray CW, Robson B, Waszkowycz B and Westhead DR (2022) PRO_LIGAND: An approach to de novo molecular design. 1. Application to the design of organic molecules. Journal of Computer-Aided Molecular Design 9(1), 13–32. [DOI] [PubMed] [Google Scholar]
- Coldren WH, Tikunova SB, Davis JP and Lindert S (2020) Discovery of novel small-molecule calcium sensitizers for cardiac troponin C: A combined virtual and experimental screening approach. Journal of Chemical Information and Modeling 60(7), 3648–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danziger DJ and Dean PM (1989) Automated site-directed drug design: A general algorithm for knowledge acquisition about hydrogen-bonding regions at protein surfaces. Proceedings of the Royal Society of London. Series B, Biological Sciences 236(1283), 101–113. [DOI] [PubMed] [Google Scholar]
- Dara S, Dhamercherla S, Jadav SS, Babu CHM and Ahsan MJ (2022) Machine learning in drug discovery: A review. Artificial Intelligence Review 55(3), 1947–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Benedetti PG and Fanelli F (2018) Computational modeling approaches to quantitative structure–binding kinetics relationships in drug discovery. Drug Discovery Today 23(7), 1396–1406. [DOI] [PubMed] [Google Scholar]
- De Sousa ACC, Combrinck JM, Maepa K and Egan TJ (2020) Virtual screening as a tool to discover new β-haematin inhibitors with activity against malaria parasites. Scientific Reports 10(1), 3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza Neto LR, Moreira-Filho JT, Neves BJ, Maidana RLBR, Guimarães ACR, Furnham N, Andrade CH and Silva FP (2020) In silico strategies to support fragment-to-lead optimization in drug discovery. Frontiers in Chemistry 8, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desaphy J, Bret G, Rognan D and Kellenberger E (2015) sc-PDB: A 3D-database of ligandable binding sites – 10 years on. Nucleic Acids Research 43, D399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey F, Cliff Zhang Q, Petrey D and Honig B (2013) Toward a “structural BLAST”: Using structural relationships to infer function. Protein Science 22(4), 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias SED, Nguyen QT, Jorge JA and Gomes AJP (2017) Multi-GPU-based detection of protein cavities using critical points. Future Generation Computer Systems 67, 430–440. [Google Scholar]
- Dimasi JA, Hansen RW and Grabowski HG (2003) The price of innovation: New estimates of drug development costs. Journal of Health Economics 22(2), 151–185. [DOI] [PubMed] [Google Scholar]
- Dimou A, Ataloglou D, Dimitropoulos K, Álvarez F and Daras P (2019) LDS-inspired residual networks. IEEE Transactions on Circuits and Systems for Video Technology 29, 2363–2375. [Google Scholar]
- Ding N, Zhao B, Han X, Li C, Gu Z and Li Z (2022) Starch-binding domain modulates the specificity of maltopentaose production at moderate temperatures. Journal of Agricultural and Food Chemistry 70, 9057–9065. [DOI] [PubMed] [Google Scholar]
- Doerr S, Harvey MJ, Noé F and De Fabritiis G (2016) HTMD: High-throughput molecular dynamics for molecular discovery. Journal of Chemical Theory and Computation 12(4), 1845–1852. [DOI] [PubMed] [Google Scholar]
- Dorn M, Silva MBE, Buriol LS and Lamb LC (2014) Three-dimensional protein structure prediction: Methods and computational strategies. Computational Biology and Chemistry 53, 251–276. [DOI] [PubMed] [Google Scholar]
- Dupond S (2019) A thorough review on the current advance of neural network structures. Annual Reviews in Control 14, 200–230. [Google Scholar]
- Durrant JD, Cao R, Gorfe AA, Zhu W, Li J, Sankovsky A, Oldfield E and Mccammon JA (2011) Non-bisphosphonate inhibitors of isoprenoid biosynthesis identified via computer-aided drug design. Chemical Biology & Drug Design 78(3), 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertl P and Schuffenhauer A (2009) Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. Journal of Cheminformatics 1(1), 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg EN, Joshi E, Pande VS and Cheng AC (2020) Improvement in ADMET prediction with multitask deep featurization. Journal of Medicinal Chemistry 63(16), 8835–8848. [DOI] [PubMed] [Google Scholar]
- Feizpour A, Marstrand T, Bastholm L, Eirefelt S and Evans CL (2021) Label-free quantification of pharmacokinetics in skin with stimulated raman scattering microscopy and deep learning. Journal of Investigative Dermatology 141(2), 395–403. [DOI] [PubMed] [Google Scholar]
- Ferreira LLG and Andricopulo AD (2019) ADMET modeling approaches in drug discovery. Drug Discovery Today 24(5), 1157–1165. [DOI] [PubMed] [Google Scholar]
- Fratev F, Gutierrez DA, Aguilera RJ, Tyagi A, Damodaran C and Sirimulla S (2021) Discovery of new AKT1 inhibitors by combination of in silico structure based virtual screening approaches and biological evaluations. Journal of Biomolecular Structure and Dynamics 39(1), 368–377. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P and Shenkin PS (2004) Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. Journal of Medicinal Chemistry 47(7), 1739–1749. [DOI] [PubMed] [Google Scholar]
- Gallego V, Naveiro R, Roca C, Ríos Insua D and Campillo NE (2021) AI in drug development: A multidisciplinary perspective. Molecular Diversity 25(3), 1461–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Jiang H, Fu K and He W (2019) On understanding degradation kinetics of pharmaceutic gelatin matrices for precision medicine: A deep learning approach. In 2019 IEEE International Conference on Big Data (Big Data). Los Angeles, CA, USA: IEEE, pp. 6060–6062. [Google Scholar]
- Gebauer NWA, Gastegger M, Hessmann SSP, Müller KR and Schütt KT (2022) Inverse design of 3d molecular structures with conditional generative neural networks. Nature Communications 13(1), 973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet V, Johnson AP, Mata P, Sike S and Williams P (1993) SPROUT: A program for structure generation. Journal of Computer-Aided Molecular Design 7(2), 127–153. [DOI] [PubMed] [Google Scholar]
- Girshick R, Donahue J, Darrell T and Malik J (2014) Rich feature hierarchies for accurate object detection and semantic segmentation. In 2014 IEEE Conference on Computer Vision and Pattern Recognition. Columbus, OH, USA: IEEE, pp. 580–587. [Google Scholar]
- Goncearenco A, Mitternacht S, Yong T, Eisenhaber B, Eisenhaber F and Berezovsky IN (2013) SPACER: Server for predicting allosteric communication and effects of regulation. Nucleic Acids Research 41, W266–W272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greener JG and Sternberg MJ (2015) AlloPred: Prediction of allosteric pockets on proteins using normal mode perturbation analysis. BMC Bioinformatics 16, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren TA (1996) Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. Journal of Computational Chemistry 17(5–6), 490–519. [Google Scholar]
- Halgren T (2007) New method for fast and accurate binding-site identification and analysis. Chemical Biology & Drug Design 69(2), 146–148. [DOI] [PubMed] [Google Scholar]
- Halgren TA (2009) Identifying and characterizing binding sites and assessing druggability. Journal of Chemical Information and Modeling 49(2), 377–389. [DOI] [PubMed] [Google Scholar]
- Hantz ER and Lindert S (2022) Actives-based receptor selection strongly increases success rate in structure-based drug design and leads to identification of 22 unique potent cancer inhibitors. ChemRxiv. Cambridge: Cambridge Open Engage. This content is a preprint and has not been peer-reviewed. [DOI] [PubMed] [Google Scholar]
- Hartenfeller M and Schneider G (2011) De novo drug design. Methods in Molecular Biology 672, 299–323. [DOI] [PubMed] [Google Scholar]
- Hassan-Harrirou H, Zhang C and Lemmin T (2020) RosENet: Improving binding affinity prediction by leveraging molecular mechanics energies with an ensemble of 3D convolutional neural networks. Journal of Chemical Information and Modeling 60(6), 2791–2802. [DOI] [PubMed] [Google Scholar]
- Hayik SA, Dunbrack R Jr and Merz KM Jr (2010) A mixed QM/MM scoring function to predict protein–ligand binding affinity. Journal of Chemical Theory and Computation 6(10), 3079–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K, Zhang X, Ren S and Sun J (2016) Deep residual learning for image recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition. Las Vegas, NV, USA: IEEE, pp. 770–778. [Google Scholar]
- Hedegaard L, Heidari N and Iosifidis A (2022) Chapter 14 - Human activity recognition. In Iosifidis A and Tefas A (eds), Deep Learning for Robot Perception and Cognition. Cambridge, MA, USA: Academic Press, pp. 341–370. [Google Scholar]
- Hernandez M, Ghersi D and Sanchez R (2009) SITEHOUND-web: A server for ligand binding site identification in protein structures. Nucleic Acids Research 37, W413–W416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessler G and Baringhaus KH (2018) Artificial intelligence in drug design. Molecules 23(10), 2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochreiter S and Schmidhuber J (1997) Long short-term memory. Neural Computation 9(8), 1735–1780. [DOI] [PubMed] [Google Scholar]
- Hoffmann B, Gerst R, Cseresnyés Z, Foo W, Sommerfeld O, Press AT, Bauer M and Figge MT (2022) Spatial quantification of clinical biomarker pharmacokinetics through deep learning-based segmentation and signal-oriented analysis of MSOT data. Photoacoustics 26, 100361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins AL (2009) Drug discovery: Predicting promiscuity. Nature 462, 167–168. [DOI] [PubMed] [Google Scholar]
- Huang B (2009) MetaPocket: A meta approach to improve protein ligand binding site prediction. Omics 13(4), 325–330. [DOI] [PubMed] [Google Scholar]
- Huang P-S, Boyken SE and Baker D (2016) The coming of age of de novo protein design. Nature 537(7620), 320–327. [DOI] [PubMed] [Google Scholar]
- Iandola FN, Han S, Moskewicz MW, Ashraf K, Dally WJ and Keutzer K (2016) SqueezeNet: AlexNet-level accuracy with 50x fewer parameters and <0.5 MB model size. arXiv preprint. arXiv:1602.07360.
- Irwin JJ, Sterling T, Mysinger MM, Bolstad ES and Coleman RG (2012) ZINC: A free tool to discover chemistry for biology. Journal of Chemical Information and Modeling 52(7), 1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam SMS, Rahman S, Rahman MM, Dey EK and Shoyaib M (2016) Application of deep learning to computer vision: A comprehensive study. In 2016 5th International Conference on Informatics, Electronics and Vision (ICIEV). Dhaka, Bangladesh: IEEE, pp. 592–597. [Google Scholar]
- Jain AN (2003) Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. Journal of Medicinal Chemistry 46(4), 499–511. [DOI] [PubMed] [Google Scholar]
- Jeon W and Kim D (2020) Autonomous molecule generation using reinforcement learning and docking to develop potential novel inhibitors. Scientific Reports 10(1), 22104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Wei Z, Zhang S, Wang S, Wang X and Li Z (2019) FRSite: Protein drug binding site prediction based on faster R-CNN. Journal of Molecular Graphics and Modelling 93, 107454. [DOI] [PubMed] [Google Scholar]
- Jiménez J, Doerr S, Martínez-Rosell G, Rose AS and De Fabritiis G (2017) DeepSite: Protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 33(19), 3036–3042. [DOI] [PubMed] [Google Scholar]
- Jiménez J, Škalič M, Martínez-Rosell G and De Fabritiis G (2018) KDEEP: Protein–ligand absolute binding affinity prediction via 3D-convolutional neural networks. Journal of Chemical Information and Modeling 58(2), 287–296. [DOI] [PubMed] [Google Scholar]
- Jones G, Willett P, Glen RC, Leach AR and Taylor R (1997) Development and validation of a genetic algorithm for flexible docking. Journal of Molecular Biology 267(3), 727–748. [DOI] [PubMed] [Google Scholar]
- Joshi RP, Gebauer NWA, Bontha M, Khazaieli M, James RM, Brown JB and Kumar N (2021) 3D-scaffold: A deep learning framework to generate 3D coordinates of drug-like molecules with desired scaffolds. The Journal of Physical Chemistry B 125(44), 12166–12176. [DOI] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P and Hassabis D (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596(7873), 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalamatianos KG (2022) In silico drug repurposing for coronavirus (COVID-19): Screening known HCV drugs against the SARS-CoV-2 spike protein bound to angiotensin-converting enzyme 2 (ACE2) (6M0J). Molecular Diversity 23, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel J, Tayara H and Chong KT (2021) PUResNet: Prediction of protein–ligand binding sites using deep residual neural network. Journal of Cheminformatics 13(1), 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur M and Mohta A (2019) A review of deep learning with recurrent neural network. In 2019 International Conference on Smart Systems and Inventive Technology (ICSSIT). Tirunelveli, India: IEEE, pp. 460–465. [Google Scholar]
- Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, Li Q, Shoemaker BA, Thiessen PA, Yu B, Zaslavsky L, Zhang J and Bolton EE (2021) PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Research 49(D1), D1388–D1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingma DP and Welling M (2014) Auto-encoding variational bayes. In International Conference for Learning Representations.
- Kitaura K, Ikeo E, Asada T, Nakano T and Uebayasi M (1999) Fragment molecular orbital method: An approximate computational method for large molecules. Chemical Physics Letters 313(3), 701–706. [Google Scholar]
- Koes DR, Baumgartner MP and Camacho CJ (2013) Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. Journal of Chemical Information and Modeling 53(8), 1893–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakov D, Grove LE, Hall DR, Bohnuud T, Mottarella SE, Luo L, Xia B, Beglov D and Vajda S (2015) The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nature Protocols 10(5), 733–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlovskii I and Popov P (2020) Spatiotemporal identification of druggable binding sites using deep learning. Communications Biology 3(1), 618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivák R and Hoksza D (2018) P2Rank: Machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. Journal of Cheminformatics 10(1), 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y, Shin WH, Ko J and Lee J (2020) AK-score: Accurate protein-ligand binding affinity prediction using an ensemble of 3D-convolutional neural networks. International Journal of Molecular Sciences 21(22), 8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guilloux V, Schmidtke P and Tuffery P (2009) Fpocket: An open source platform for ligand pocket detection. BMC Bioinformatics 10, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Jeong KW, Lee Y, Song JY, Kim MS, Lee GS and Kim Y (2010) Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors. European Journal of Medicinal Chemistry 45(11), 5420–5427. [DOI] [PubMed] [Google Scholar]
- Lee JH, Lin WC, Wen TK, Wang C and Lin YT (2019) Inhibiting two cellular mutant epidermal growth factor receptor tyrosine kinases by addressing computationally assessed crystal ligand pockets. Future Medicinal Chemistry 11(8), 833–846. [DOI] [PubMed] [Google Scholar]
- Leelananda SP and Lindert S (2016) Computational methods in drug discovery. Beilstein Journal of Organic Chemistry 12, 2694–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leman JK, Weitzner BD, Lewis SM, Adolf-Bryfogle J, Alam N, Alford RF, Aprahamian M, Baker D, Barlow KA, Barth P, Basanta B, Bender BJ, Blacklock K, Bonet J, Boyken SE, Bradley P, Bystroff C, Conway P, Cooper S, Correia BE, Coventry B, Das R, De Jong RM, Dimaio F, Dsilva L, Dunbrack R, Ford AS, Frenz B, Fu DY, Geniesse C, Goldschmidt L, Gowthaman R, Gray JJ, Gront D, Guffy S, Horowitz S, Huang PS, Huber T, Jacobs TM, Jeliazkov JR, Johnson DK, Kappel K, Karanicolas J, Khakzad H, Khar KR, Khare SD, Khatib F, Khramushin A, King IC, Kleffner R, Koepnick B, Kortemme T, Kuenze G, Kuhlman B, Kuroda D, Labonte JW, Lai JK, Lapidoth G, Leaver-Fay A, Lindert S, Linsky T, London N, Lubin JH, Lyskov S, Maguire J, Malmström L, Marcos E, Marcu O, Marze NA, Meiler J, Moretti R, Mulligan VK, Nerli S, Norn C, Ó’Conchúir S, Ollikainen N, Ovchinnikov S, Pacella MS, Pan X, Park H, Pavlovicz RE, Pethe M, Pierce BG, Pilla KB, Raveh B, Renfrew PD, Burman SSR, Rubenstein A, Sauer MF, Scheck A, Schief W, Schueler-Furman O, Sedan Y, Sevy AM, Sgourakis NG, Shi L, Siegel JB, Silva DA, Smith S, Song Y, Stein A, Szegedy M, Teets FD, Thyme SB, Wang RY, Watkins A, Zimmerman L and Bonneau R (2020) Macromolecular modeling and design in Rosetta: Recent methods and frameworks. Nature Methods 17(7), 665–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Han L, Liu Z and Wang R (2014) Comparative assessment of scoring functions on an updated benchmark: 2. Evaluation methods and general results. Journal of Chemical Information and Modeling 54(6), 1717–1736. [DOI] [PubMed] [Google Scholar]
- Li Y, Hu J, Wang Y, Zhou J, Zhang L and Liu Z (2020) DeepScaffold: A comprehensive tool for scaffold-based de novo drug discovery using deep learning. Journal of Chemical Information and Modeling 60(1), 77–91. [DOI] [PubMed] [Google Scholar]
- Li H, Leung KS, Wong MH and Ballester PJ (2015) Improving AutoDock vina using random forest: The growing accuracy of binding affinity prediction by the effective exploitation of larger data sets. Molecular Informatics 34(2–3), 115–126. [DOI] [PubMed] [Google Scholar]
- Li Y, Pei J and Lai L (2021b) Structure-based de novo drug design using 3D deep generative models. Chemical Science 12(41), 13664–13675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Rezaei MA, Li C and Li X (2019) DeepAtom: A framework for protein–ligand binding affinity prediction. In 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). San Diego, CA, USA: IEEE, pp. 303–310. [Google Scholar]
- Li J, Wu J, Zhao Z, Zhang Q, Shao J, Wang C, Qiu Z and Li W (2021a) Artificial intelligence-assisted decision making for prognosis and drug efficacy prediction in lung cancer patients: A narrative review. Journal of Thoracic Disease 13(12), 7021–7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang L and Liu Z (2018) Multi-objective de novo drug design with conditional graph generative model. Journal of Cheminformatics 10(1), 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin E, Kuo P-H, Liu Y-L, Yu YW-Y, Yang AC and Tsai S-J (2018) A deep learning approach for predicting antidepressant response in major depression using clinical and genetic biomarkers. Frontiers in Psychiatry 9, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindert S, Li MX, Sykes BD and Mccammon JA (2015a) Computer-aided drug discovery approach finds calcium sensitizer of cardiac troponin. Chemical Biology & Drug Design 85(2), 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindert S, Tallorin L, Nguyen QG, Burkart MD and Mccammon JA (2015b) In silico screening for Plasmodium falciparum enoyl-ACP reductase inhibitors. Journal of Computer-Aided Molecular Design 29(1), 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW and Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews 46(1–3), 3–26. [DOI] [PubMed] [Google Scholar]
- Litsa EE, Das P and Kavraki LE (2021) Machine learning models in the prediction of drug metabolism: Challenges and future perspectives. Expert Opinion on Drug Metabolism & Toxicology 17(11), 1245–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Sun X, Jia L, Ma J, Xing H, Wu J, Gao H, Sun Y, Boulnois F and Fan J (2019) Chemi-Net: A molecular graph convolutional network for accurate drug property prediction. International Journal of Molecular Sciences 20(14), 3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Terayama K, Matsumoto S, Isaka Y, Sasakura Y, Iwata H, Araki M and Okuno Y (2021) Structure-based de novo molecular generator combined with artificial intelligence and docking simulations. Journal of Chemical Information and Modeling 61(7), 3304–3313. [DOI] [PubMed] [Google Scholar]
- Mardt A, Pasquali L, Wu H and Noé F (2018) VAMPnets for deep learning of molecular kinetics. Nature Communications 9(1), 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martis E, Radhakrishnan R and Badve R (2011) High-throughput screening: The hits and leads of drug discovery – An overview. Journal of Applied Pharmaceutical Science 1(1), 2–10. [Google Scholar]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS and Olson AJ (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry 30(16), 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muegge I (2006) PMF scoring revisited. Journal of Medicinal Chemistry 49(20), 5895–5902. [DOI] [PubMed] [Google Scholar]
- Mylonas SK, Axenopoulos A and Daras P (2021) DeepSurf: A surface-based deep learning approach for the prediction of ligand binding sites on proteins. Bioinformatics 37, 1681–1690. [DOI] [PubMed] [Google Scholar]
- Mysinger MM, Carchia M, Irwin JJ and Shoichet BK (2012) Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. Journal of Medicinal Chemistry 55(14), 6582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil D, Segler M, Guasch L, Ahmed M, Plumbley D, Sellwood M and Brown N (2018) Exploring deep recurrent models with reinforcement learning for molecule design.
- Nishibata Y and Itai A (1991) Automatic creation of drug candidate structures based on receptor structure. Starting point for artificial lead generation. Tetrahedron 47(43), 8985–8990. [Google Scholar]
- Nunes-Alves A, Kokh DB and Wade RC (2020) Recent progress in molecular simulation methods for drug binding kinetics. Current Opinion in Structural Biology 64, 126–133. [DOI] [PubMed] [Google Scholar]
- Obeid S, Madžarević M, Krkobabić M and Ibrić S (2021) Predicting drug release from diazepam FDM printed tablets using deep learning approach: Influence of process parameters and tablet surface/volume ratio. International Journal of Pharmaceutics 601, 120507. [DOI] [PubMed] [Google Scholar]
- Olivecrona M, Blaschke T, Engkvist O and Chen H (2017) Molecular de-novo design through deep reinforcement learning. Journal of Cheminformatics 9(1), 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panjkovich A and Daura X (2014) PARS: A web server for the prediction of protein allosteric and regulatory sites. Bioinformatics 30(9), 1314–1315. [DOI] [PubMed] [Google Scholar]
- Patel L, Shukla T, Huang X, Ussery DW and Wang S (2020) Machine learning methods in drug discovery. Molecules 25(22), 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul D, Sanap G, Shenoy S, Kalyane D, Kalia K and Tekade RK (2021) Artificial intelligence in drug discovery and development. Drug Discovery Today 26(1), 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlman DA and Murcko MA (1996) CONCERTS: Dynamic connection of fragments as an approach to de novo ligand design. Journal of Medicinal Chemistry 39(8), 1651–1663. [DOI] [PubMed] [Google Scholar]
- Popova M, Isayev O and Tropsha A (2018) Deep reinforcement learning for de novo drug design. Science Advances 4(7), eaap7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroga R and Villarreal MA (2016) Vinardo: A scoring function based on autodock vina improves scoring, docking, and virtual screening. PLoS One 11(5), e0155183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindranath PA and Sanner MF (2016) AutoSite: An automated approach for pseudo-ligands prediction-from ligand-binding sites identification to predicting key ligand atoms. Bioinformatics 32(20), 3142–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindranathan KP, Mandiyan V, Ekkati AR, Bae JH, Schlessinger J and Jorgensen WL (2010) Discovery of novel fibroblast growth factor receptor 1 kinase inhibitors by structure-based virtual screening. Journal of Medicinal Chemistry 53(4), 1662–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren S, He K, Girshick R and Sun J (2015) Faster R-CNN: Towards Real-Time Object Detection with Region Proposal Networks, vol. 28 (ed. Cortes C, Lawrence N, Lee D, Sugiyama M and Garnett R). In 2015 Advances in neural information processing system. pp. 91–99. [Google Scholar]
- Rial R, González-Durruthy M, Liu Z and Ruso JM (2022) Conformational binding mechanism of lysozyme induced by interactions with penicillin antibiotic drugs. Journal of Molecular Liquids 358, 119081. [Google Scholar]
- Ronneberger O, Fischer P and Brox T (2015) U-net: Convolutional networks for biomedical image segmentation. In International Conference on Medical Image Computing and Computer-Assisted Intervention. Munich, Germany: Springer, pp. 234–241. [Google Scholar]
- Roy A, Yang J and Zhang Y (2012) COFACTOR: An accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Research 40, W471–W477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Carmona S, Alvarez-Garcia D, Foloppe N, Garmendia-Doval AB, Juhos S, Schmidtke P, Barril X, Hubbard RE and Morley SD (2014) rDock: A fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLOS Computational Biology 10(4), e1003571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salsbury FR (2010) Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Current Opinion in Pharmacology 10(6), 738–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scafuri B, Verdino A, D’arminio N and Marabotti A (2022) Computational methods to assist in the discovery of pharmacological chaperones for rare diseases. Briefings in Bioinformatics, bbac198 (doi: 10.1093/bib/bbac198). [DOI] [PubMed] [Google Scholar]
- Scarselli F, Gori M, Tsoi AC, Hagenbuchner M and Monfardini G (2009) The graph neural network model. IEEE Transactions on Neural Networks 20(1), 61–80. [DOI] [PubMed] [Google Scholar]
- Schmidhuber J (2015) Deep learning in neural networks: An overview. Neural Networks 61, 85–117. [DOI] [PubMed] [Google Scholar]
- Schneider G and Fechner U (2022) Computer-based de novo design of drug-like molecules. Nature Reviews Drug Discovery 4(8), 649–663. [DOI] [PubMed] [Google Scholar]
- Seffernick JT and Lindert S (2020) Hybrid methods for combined experimental and computational determination of protein structure. The Journal of Chemical Physics 153(24), 240901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T, Yang Y, Huang S, Chen L, Kuang Z, Heng Y and Mei H (2019) Molecular image-based convolutional neural network for the prediction of ADMET properties. Chemometrics and Intelligent Laboratory Systems 194, 103853. [Google Scholar]
- Shin M, Jang D, Nam H, Lee KH and Lee D (2018) Predicting the absorption potential of chemical compounds through a deep learning approach. IEEE/ACM Transactions on Computational Biology and Bioinformatics 15(2), 432–440. [DOI] [PubMed] [Google Scholar]
- Shivakumar D, Williams J, Wu Y, Damm W, Shelley J and Sherman W (2010) Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. Journal of Chemical Theory and Computation 6(5), 1509–1519. [DOI] [PubMed] [Google Scholar]
- Singh N, Malik S, Gupta A and Srivastava KR (2021) Revolutionizing enzyme engineering through artificial intelligence and machine learning. Emerging Topics in Life Sciences 5(1), 113–125. [DOI] [PubMed] [Google Scholar]
- Sinko W, Wang Y, Zhu W, Zhang Y, Feixas F, Cox CL, Mitchell DA, Oldfield E and Mccammon JA (2014) Undecaprenyl diphosphate synthase inhibitors: Antibacterial drug leads. Journal of Medicinal Chemistry 57(13), 5693–5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song K, Liu X, Huang W, Lu S, Shen Q, Zhang L and Zhang J (2017) Improved method for the identification and validation of allosteric sites. Journal of Chemical Information and Modeling 57(9), 2358–2363. [DOI] [PubMed] [Google Scholar]
- Srinivasan B, Forouhar F, Shukla A, Sampangi C, Kulkarni S, Abashidze M, Seetharaman J, Lew S, Mao L, Acton TB, Xiao R, Everett JK, Montelione GT, Tong L and Balaram H (2014) Allosteric regulation and substrate activation in cytosolic nucleotidase II from Legionella pneumophila. The FEBS Journal 281(6), 1613–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ståhl N, Falkman G, Karlsson A, Mathiason G and Boström J (2019) Deep reinforcement learning for multiparameter optimization in de novo drug design. Journal of Chemical Information and Modeling 59(7), 3166–3176. [DOI] [PubMed] [Google Scholar]
- Stahl M and Rarey M (2001) Detailed analysis of scoring functions for virtual screening. Journal of Medicinal Chemistry 44(7), 1035–1042. [DOI] [PubMed] [Google Scholar]
- Stepniewska-Dziubinska MM, Zielenkiewicz P and Siedlecki P (2018) Development and evaluation of a deep learning model for protein-ligand binding affinity prediction. Bioinformatics 34(21), 3666–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepniewska-Dziubinska MM, Zielenkiewicz P and Siedlecki P (2020) Improving detection of protein–ligand binding sites with 3D segmentation. Scientific Reports 10(1), 5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SYBYL (2022) Version 6.8. Tripos Inc. Available at http://www.tripos.com/.
- Tang Y, Guo C, Yang Z, Wang Y, Zhang Y and Wang D (2022) Identification of a tumor immunological phenotype-related gene signature for predicting prognosis, immunotherapy efficacy, and drug candidates in hepatocellular carcinoma. Frontiers in Immunology 13, 862527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H, Jiang X and Tao P (2021) PASSer: Prediction of allosteric sites server. Machine Learning: Science and Technology 2(3), 035015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari V (2022) Pharmacophore screening, denovo designing, retrosynthetic analysis, and combinatorial synthesis of a novel lead VTRA1.1 against RecA protein of Acinetobacter baumannii. Chemical Biology & Drug Design 99(6), 839–856. [DOI] [PubMed] [Google Scholar]
- Trott O and Olson AJ (2010) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry 31(2), 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujikawa H, Sato K, Wei C, Saad G, Sumikoshi K, Nakamura S, Terada T and Shimizu K (2016) Development of a protein–ligand-binding site prediction method based on interaction energy and sequence conservation. Journal of Structural and Functional Genomics 17(2–3), 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbina F, Lentzos F, Invernizzi C and Ekins S (2022) Dual use of artificial-intelligence-powered drug discovery. Nature Machine Intelligence 4(3), 189–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdonk ML, Cole JC, Hartshorn MJ, Murray CW and Taylor RD (2003) Improved protein-ligand docking using GOLD. Proteins 52(4), 609–623. [DOI] [PubMed] [Google Scholar]
- Wang R, Fang X, Lu Y, Yang CY and Wang S (2005) The PDBbind database: Methodologies and updates. Journal of Medicinal Chemistry 48(12), 4111–4119. [DOI] [PubMed] [Google Scholar]
- Wang R, Lai L and Wang S (2002) Further development and validation of empirical scoring functions for structure-based binding affinity prediction. Journal of Computer-Aided Molecular Design 16(1), 11–26. [DOI] [PubMed] [Google Scholar]
- Wang D, Liu W, Shen Z, Jiang L, Wang J, Li S and Li H (2020) Deep learning based drug metabolites prediction. Frontiers in Pharmacology 10, 1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wu Y, Deng Y, Kim B, Pierce L, Krilov G, Lupyan D, Robinson S, Dahlgren MK, Greenwood J, Romero DL, Masse C, Knight JL, Steinbrecher T, Beuming T, Damm W, Harder E, Sherman W, Brewer M, Wester R, Murcko M, Frye L, Farid R, Lin T, Mobley DL, Jorgensen WL, Berne BJ, Friesner RA and Abel R (2015) Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. Journal of the American Chemical Society 137(7), 2695–2703. [DOI] [PubMed] [Google Scholar]
- Weisel M, Proschak E and Schneider G (2007) PocketPicker: Analysis of ligand binding-sites with shape descriptors. Chemistry Central Journal 1, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter R, Montanari F, Steffen A, Briem H, Noé F and Clevert DA (2019) Efficient multi-objective molecular optimization in a continuous latent space. Chemical Science 10(34), 8016–8024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SS, Luo W and Chan KC (2011) EvoMD: An algorithm for evolutionary molecular design. IEEE/ACM Transactions on Computational Biology and Bioinformatics 8(4), 987–1003. [DOI] [PubMed] [Google Scholar]
- Workman P (2003) How much gets there and what does it do?: The need for better pharmacokinetic and pharmacodynamic endpoints in contemporary drug discovery and development. Current Pharmaceutical Design 9(11), 891–902. [DOI] [PubMed] [Google Scholar]
- Wu Q, Peng Z, Zhang Y and Yang J (2018) COACH-D: Improved protein-ligand binding sites prediction with refined ligand-binding poses through molecular docking. Nucleic Acids Research 46(W1), W438–W442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Jiang X and Niu Y (2022) Study on the interaction of sweet protein (thaumatin) with key aroma compounds in passion fruit juice using electronic nose, ultraviolet spectrum, thermodynamics, and molecular docking. LWT 162, 113463. [Google Scholar]
- Xie S, Girshick R, Dollár P, Tu Z and He K (2017) Aggregated residual transformations for deep neural networks. In 2017 IEEE Conference on Computer Vision and Pattern Recognition (CVPR). Honolulu, HI, USA: IEEE, pp. 5987–5995. [Google Scholar]
- Yang X, Zhang J, Yoshizoe K, Terayama K and Tsuda K (2017) ChemTS: An efficient python library for de novo molecular generation. Science and Technology of Advanced Materials 18(1), 972–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Zhou Y, Tanaka I and Yao M (2010) Roll: A new algorithm for the detection of protein pockets and cavities with a rolling probe sphere. Bioinformatics 26(1), 46–52. [DOI] [PubMed] [Google Scholar]
- Zhang J, Liu Q Fan XX, Leung EL, Yao XJ and Liu L (2022) Resistance looms for KRAS G12C inhibitors and rational tackling strategies. Pharmacology & Therapeutics 229, 108050. [DOI] [PubMed] [Google Scholar]
- Zhang S, Tong H, Xu J and Maciejewski R (2019) Graph convolutional networks: A comprehensive review. Computational Social Networks 6(1), 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhou X, Lin M and Sun J (2018) Shufflenet: An extremely efficient convolutional neural network for mobile devices. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition. Salt Lake City, UT, USA: IEEE, pp. 6848–6856. [Google Scholar]
- Zhavoronkov A, Ivanenkov YA, Aliper A, Veselov MS, Aladinskiy VA, Aladinskaya AV, Terentiev VA, Polykovskiy DA, Kuznetsov MD, Asadulaev A, Volkov Y, Zholus A, Shayakhmetov RR, Zhebrak A, Minaeva LI, Zagribelnyy BA, Lee LH, Soll R, Madge D, Xing L, Guo T and Aspuru-Guzik A (2019) Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nature Biotechnology 37(9), 1038–1040. [DOI] [PubMed] [Google Scholar]
- Zheng L, Fan J and Mu Y (2019) OnionNet: A multiple-layer intermolecular-contact-based convolutional neural network for protein-ligand binding affinity prediction. ACS Omega 4(14), 15956–15965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Kearnes S, Li L, Zare RN and Riley P (2019) Optimization of molecules via deep reinforcement learning. Scientific Reports 9(1), 10752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Wang J, Wang X, Gao M, Guo B, Gao M, Liu J, Yu Y, Wang L, Kong W, An Y, Liu Z, Sun X, Huang Z, Zhou H, Zhang N, Zheng R and Xie Z (2021) Prediction of drug efficacy from transcriptional profiles with deep learning. Nature Biotechnology 39(11), 1444–1452. [DOI] [PubMed] [Google Scholar]
- Zhu W, Zhang Y, Sinko W, Hensler ME, Olson J, Molohon KJ, Lindert S, Cao R, Li K, Wang K, Wang Y, Liu YL, Sankovsky A, De Oliveira CA, Mitchell DA, Nizet V, Mccammon JA and Oldfield E (2013) Antibacterial drug leads targeting isoprenoid biosynthesis. Proceedings of the National Academy of Sciences of the United States of America 110(1), 123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotnikov ID and Kudryashova EV (2022) Computer simulation of the receptor-ligand interactions of mannose receptor CD206 in comparison with the lectin concanavalin a model. Biochemistry (Moscow) 87(1), 54–69. [DOI] [PMC free article] [PubMed] [Google Scholar]