

Abstract
We collected and analyzed genomic sequencing data from individuals with clinician-diagnosed early-onset or atypical dementia. Thirty-two patients were previously described, with 68 newly described in this report. Of those 68, 62 patients self-reported white, non-Hispanic ethnicity and 6 reported as African–American, non-Hispanic. Fifty-three percent of patients had a returnable variant. Five patients harbored a pathogenic variant as defined by the American College of Medical Genetics criteria for pathogenicity. A polygenic risk score (PRS) was calculated for Alzheimer's patients in the total cohort and compared to the scores of a late-onset Alzheimer's cohort and a control set. Patients with early-onset Alzheimer's had higher non-APOE PRSs than patients with late-onset Alzheimer's, supporting the conclusion that both rare and common genetic variation associate with early-onset neurodegenerative disease risk.
Keywords: Alzheimer disease, frontotemporal dementia
INTRODUCTION
Dementia affects more than 55 million people worldwide; 9% of these patients are less than 65 years old (Dua et al. 2017). Rare variants in three genes, PSEN1, PSEN2, and APP, are associated with autosomal dominant early-onset Alzheimer's disease (EOAD); however, they only explain ∼10% of genetic cases (Stoychev et al. 2019). The use of genome sequencing allows identification of rare variants not included in some targeted gene panel testing, as well as variation that is the purview of future research such as including noncoding variants and variants in novel risk loci. In our previous study, we reported on the use of genome sequencing in 32 individuals with early-onset and/or atypical dementia and described several pathogenic variants associated with the disease, including combinations of disease-associated risk variants. Our previous work confirmed the value of genetic assessment and identified contributing genetic variation for more than one-half of the cohort (Cochran et al. 2019).
The American College of Medical Genetics (ACMG) criteria was used to assess pathogenicity and pathogenic/likely pathogenic variants were returned. Additional criteria to return variants included (1) any variant with a disease-established odds ratio of >2 described in multiple reports, which we defined as an “established risk variant,” (2) the presence of one or two APOE ε4 alleles in a patient with EOAD or atypical dementia likely due to EOAD, or (3) one strong report with a disease-associated odds ratio >2 with replication included in the study design, which we defined as a “possible risk variant.”
In this report, we also assessed the dementia risk related to common variation for the enrolled patients by calculating polygenic risk scores (PRSs). Our results highlight the complex genetic etiology of early-onset and/or atypical dementia.
RESULTS
Here, we report on 68 additional patients collected as a continuation of the previously published cohort. For each patient with clinician-diagnosed early-onset or atypical dementia, we collected and analyzed genomic sequencing data. We identified returnable, primary findings for 53% of patients (Table 1; Fig. 1A). Including the initial 32 probands described by Cochran et al. in our previous report, a total of 100 patients have been enrolled through the Brain Aging and Memory Clinic at the University of Alabama at Birmingham (Fig. 1B).
Table 1.
Variant table
| Gene | Chromosome | HGVS DNA | HGVS protein | Variant type | Predicted effect | Pathogenicity classification | CADD score | gnomAD allele counts |
|---|---|---|---|---|---|---|---|---|
| APOE | 19 | NM_000041.3:c.388T > C | p.(Cys130Arg) | SNV | Missense | Established risk | 16.65 | 23,875 |
| APOE | 19 | NM_000041.3:c.526C > T | p.(Arg176Cys) | SNV | Missense | Established risk | 26 | 31,094 |
| SORL1 | 11 | NM_003105.6:c.3856C > T | p.(Arg1286Cys) | SNV | Missense | Possible risk | 29.3 | 1 |
| PRNP | 20 | NM_183079.3:c.416T > C | p.(Ile139Thr) | SNV | Missense | Possible risk | 18.98 | 0 |
| SORL1 | 11 | NM_003105.6:c.6194A > T | p.(Asp2065Val) | SNV | Missense | VUS | 28.8 | 347 |
| ABCA7 | 19 | NM_019112.3:c.4416 + 1G > T | NA | SNV | Splice | Established risk | 26.3 | 6 |
| PAH | 12 | NM_000277.3:c.117C > G | p.(Phe39Leu) | SNV | Missense | Pathogenic | 25.6 | 13 |
| PAH | 12 | NM_000277.3:c.194T > C | p.(Ile65Thr) | SNV | Missense | Pathogenic | 27.2 | 45 |
| GRN | 17 | NM_002087.3:c.26C > A | p.(Ala9Asp) | SNV | Missense | Pathogenic | 23.6 | 0 |
| MAPT | 17 | NM_005910.5:c.1216C > T | p.(Arg406Trp) | SNV | Missense | Pathogenic | 23.5 | 4 |
| PSEN1 | 14 | NM_000021.4:c.236C > T | p.(Ala79Val) | SNV | Missense | Pathogenic | 26.8 | 5 |
| PSEN2 | 1 | NM_000447.3:c.712C > T | p.(Leu238Phe) | SNV | Missense | VUS | 25.3 | 3 |
| PSEN2 | 1 | NM_000447.3:c.211C > T | p.(Arg71Trp) | SNV | Missense | Possible risk | 22.4 | 513 |
| ABCA7 | 19 | NM_019112.3:c.4010C > T | p.(Thr1337Ile) | SNV | Missense | Possible risk | 23.9 | 0 |
| TSC2 | 16 | NM_000548.5:c.3847_3856del | p.(Cys1283SerfsTer39) | Deletion | INDEL | VUS | NA | 0 |
| SCN5A | 3 | NM_198056.2:c.673C > T | p.(Arg225Trp) | SNV | Missense | Pathogenic | 23.4 | 12 |
| KCTD17 | 22 | NM_001282684.2:c.299G > C | p.(Gly100Ala) | SNV | Missense | VUS | 34 | 2 |
| SORL1 | 11 | NM_003105.6:c.2711G > A | p.(Arg904Gln) | SNV | Missense | VUS | 31 | 20 |
All variants were observed in the heterozygous state except for APOE (NM_000041.3:c.388T > C, p.(Cys130Arg)), which was observed in both the heterozygous and homozygous state. APOE (NM_000041.3:c.526C > T, p.(Arg176Cys)) is also noted here, because confirmation of its absence along with APOE (NM_000041.3:c.388T > C, p.(Cys130Arg)) indicates the APOE ɛ4 allele. (HGVS) Human Genome Variation Society, (dbSNP) single-nucleotide polymorphism database, (gnomAD) Genome Aggregation Database, (CADD) Combined Annotation-Dependent Depletion, (SNV) single-nucleotide variant, (VUS) variant of uncertain significance.
Figure 1.

Summary of genomic sequencing findings for the (A) 68-proband cohort and (B) all 100 enrolled probands including the 32 described in Cochran et al. Patients carrying one APOE ε4 allele are noted as APOE ε4 Het. and those carrying two alleles are noted as APOE ε4 Hom. Patients carrying a risk allele in addition to one or two APOE ε4 allele are listed as APOE ε4 Het. or Hom. + risk. (VUS) Variant of uncertain significance.
Clinical Presentation and Family History
Of the 68 additional patients described here, 40 patients were male and 28 patients were female. Ethnicity was self-reported of which 62 patients reported white and 6 reported African–American. All patients reported non-Hispanic ethnicity. One patient reported age of onset in their 80s, 4 in their 70s, 23 in their 60s, 29 in their 50s, 10 in their 40s, and one in their 30s (Supplemental Table 1). The nine patients with an age of onset older than 65 had moderate to strong family histories of dementia.
We generated a family history score for each patient that is a derivative of the scoring system first used by Goldman et al. (2005). The modified Goldman score was generated as follows: (1) At least three people in two generations affected with EOAD, frontotemporal dementia (FTD), amyotrophic lateral sclerosis (ALS), corticobasal degeneration (CBD), Parkinson's disease (PD), or progressive supranuclear palsy (PSP) with one person being a first-degree relative of the other two; (1.5) criteria matching (1) but with late-onset Alzheimer's disease (LOAD) instead of EOAD; (2) at least two relatives with dementia, FTD, ALS, CBD, PD, or PSP and criteria for autosomal dominant inheritance were not met; (3) a single affected first- or second-degree family member with early-onset dementia or at least two with FTD, ALS, CBD, PD, mild cognitive impairment (MCI), or PSP; (3.5) a single affected first- or second-degree family member with late-onset dementia, FTD, ALS, CBD, PD, MCI, or PSP; and (4) noncontributory family history or unknown family history. We classified patients with a family history score of 1 or 1.5 as strong family history, 2, 3, or 3.5 as moderate family history, and those with a score of 4 had no contributory or known family history. All family history scores are included in Supplemental Table 1. We note that the researchers do not have the ability to identify patients based on IDs, as the key is kept at the clinical enrollment site.
Genomic Analyses
Variants were evaluated using ACMG criteria (Richards et al. 2015) for pathogenicity and the ACMG evidence codes for all pathogenic variants are detailed in Supplemental ACMG Pathogenicity Evidence Details.
C9orf72 Expansion Testing
We tested for pathogenic G4C2 hexanucleotide expansion at the C9orf72 locus, which is associated with FTD and ALS, either as described in Cochran et al. (2019) (using a separately obtained test from GeneDx) or, for samples that were sequenced with a PCR-free genome, using ExpansionHunter (Dolzhenko et al. 2017). All patients in this cohort expansion of 68 were negative for C9orf72 repeat expansion.
Pathogenic Diagnoses
Two Pathogenic Variants in PAH in a Patient with Phenylketonuria
A patient with phenylketonuria (PKU) with onset in their early 40s had two pathogenic variants in PAH (NM_000277.3:c.117C > G, p.(Phe39Leu)) and (NM_000277.3:c.194T > C, p.(Ile65Thr)). Segregation analysis of the family showed the PAH variants were in trans.
Both variants have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) as pathogenic for PKU, by multiple submitters (VCV000000605.15 and VCV000000636.18). PKU is an autosomal recessive disease that can cause brain dysfunction due to increased phenylalanine concentrations (van Spronsen et al. 2021). Adult-onset PKU is rare and can present with symptoms of dementia and parkinsonism, making it difficult to quickly diagnose. If diagnosed correctly, PKU can be treated with a phenylalanine-restricted diet (Rosini et al. 2014; Tufekcioglu et al. 2016).
A Pathogenic GRN Variant in a Family with FTD
A patient with an onset of MCI in their early 50s had a pathogenic variant in GRN (NM_002087.3:c.26C > A, p.(Ala9Asp)). This variant is absent from gnomAD and has been reported by several laboratories as pathogenic in ClinVar (VCV000016013.12). This variant segregates with two affected family members. We classified this variant as pathogenic as additional studies have shown this variant to segregate with FTD (Mukherjee et al. 2008; Shankaran et al. 2008).
A Rare MAPT Variant in Five Early-Onset Alzheimer's Disease Patients
Two patients with EOAD (one patient also exhibited signs of FTD) with age of onset in their 50s have a pathogenic variant in MAPT (NM_005910.5:c.1216C > T, p.(Arg406Trp)). This rare variant has a minor allele frequency of 0.000016% and was reported by several laboratories as pathogenic in ClinVar (VCV000014247.18). In our previous study (Cochran et al. 2019), we identified three patients with this variant in MAPT. The presence of the same rare variant in a total of five patients enrolled at the same clinic suggests a common ancestry is likely (Fig. 2A). We inferred the relatedness of patients using KING (Manichaikul et al. 2010) and determined a kinship coefficient for each pair of relationships and determined that three probands have a kinship coefficient of 0.06 or greater, reaching the criteria for third-degree relation (Fig. 2B). The other values did not reach the kinship coefficient of 0.06 criteria but were near that threshold and might reflect a more distant relationship.
Figure 2.
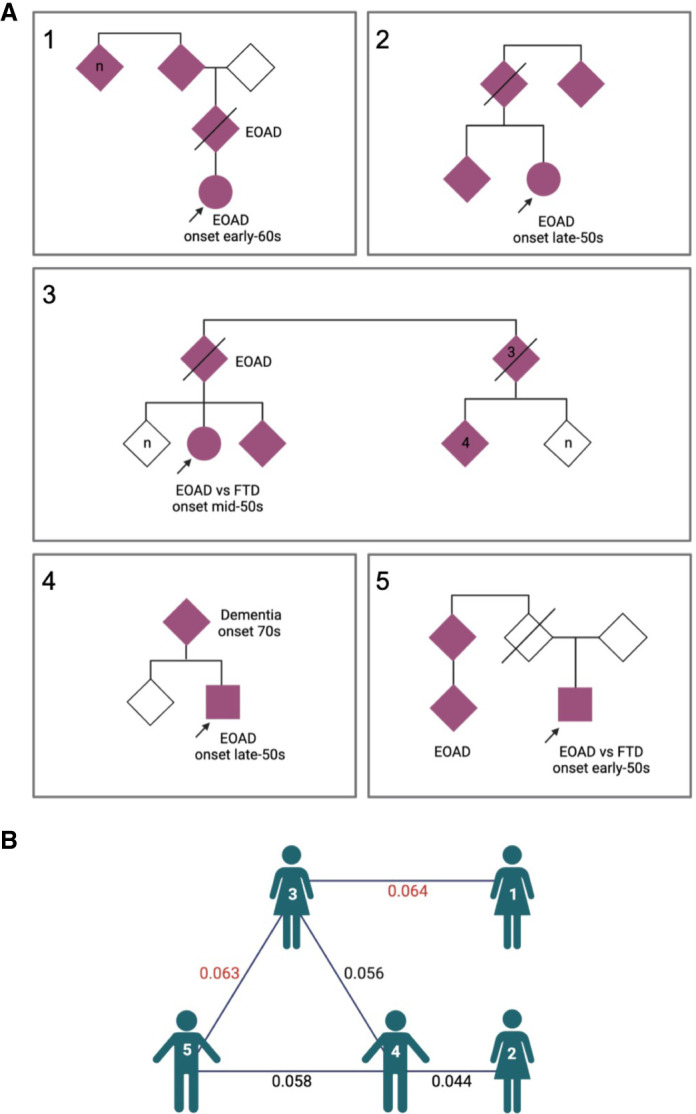
Evaluation of five probands with a MAPT R406W variant. (A) Pedigrees for the five probands with the MAPT variant. Probands are marked with arrow. Affected family members are solid pink. Patients 1–3 were identified in our previous study (Cochran et al. 2019). (B) Diagram showing the KING kinship coefficients between probands. Values in red denote third-degree relatives. Figure was generated at biorender.com. (EOAD) Early-onset Alzheimer's disease, (FTD) frontotemporal dementia.
A PSEN1 Variant in a Patient with Mild Cognitive Impairment
An individual with symptoms of MCI and a strong family history of Alzheimer's disease (AD) had a pathogenic variant in PSEN1 (NM_000021.4:c.236C > T, p.(Ala79Val)). Onset of symptoms began in their 60s and multiple affected family members had onset in 60s. This pathogenic variant has been previously reported in ClinVar (VCV000018157.5) in patients with AD. Variants in PSEN1 are the most common cause of autosomal dominant familial AD (Kelleher and Shen 2017).
Compound Heterozygous Risk Factors
APOE ε4 Risk Factors
The APOE ε4 allele is the most common strong risk factor for AD, the most common form of dementia. Patients carrying one APOE ε4 allele are noted as APOE ε4 heterozygous and those carrying two alleles are noted as APOE ε4 homozygous. We identified 25 (36%) patients with at least one APOE ε4 allele. Several of them, described here, also carried other risk variants.
APOE ε4 with SORL1 Risk Variants
A patient presenting with symptoms of EOAD with onset in their early 50s and a strong family history of AD had one APOE ε4 allele, a “possible risk variant” in SORL1 (NM_003105.6:c.3856C > T, p.(Arg1286Cys)), and a variant in PRNP (NM_183079.3:c.416T > C, p.(lle139Thr)) (returned as a variant of uncertain significance [VUS]). Pathogenic PRNP variants are typically missense variants and are associated with prion diseases (Piazza et al. 2020). To our knowledge, the c.416T > C variant in PRNP has not been previously published or reported in gnomAD (Karczewski et al. 2020). This variant was returned as a VUS because the family history of a prion disease could not be determined. The c.3856C > T SORL1 variant has a minor allele frequency of 0.0007% and is predicted damaging by PolyPhen-2 (Adzhubei et al. 2010) and SIFT (Ng and Henikoff 2003), with a Combined Annotation-Dependent Depletion (CADD) (Kircher et al. 2014) score of 29. Loss-of-function (LOF) SORL1 variant carriers are present at an odds ratio of ∼4 compared to population databases (Raghavan et al. 2018). The odds ratio for LOF variants in this gene could be as high as 12.3 for AD and 27.5 for EOAD. For rare missense variants, the odds ratio could be as high as 3.14 for EOAD (Campion et al. 2019). Other similar variants in SORL1, in aggregate, are enriched in patients with early-onset dementia by at least twofold in comparison to individuals without dementia (Holstege et al. 2017).
Two patients in our study harbored one APOE ε4 allele and the same “possible risk variant” in SORL1 (NM_003105.6:c.6194A > T, p.Asp2065Val). This variant in the SORL1 gene has not been extensively described, to our knowledge. One of these two patients was diagnosed with EOAD, with an onset in their late 40s. The other enrolled patient harboring this variant presented with MCI that was amnestic in character with an age of onset in their 70s and a striking family history of dementia. This patient also had a rare variant in ECE2 (NM_001037324.2:c.1120C > T, p.(Arg374*)) that was not returned but had a CADD score of 39 and had a predicted effect of a stop gained. Variants in ECE2 have been implicated in AD risk, and variants in the peptidase domain were shown to impair the enzymatic activity of ECE2 in Aβ degradation (Liao et al. 2020).
APOE ε4 Heterozygote with ABCA7 Risk Allele
An African–American patient diagnosed with MCI with onset in their late 50s harbored an “established risk” LOF splice variant in ABCA7 (NM_019112.3:c.4416 + 1G > T) and one APOE ε4 risk allele. The ABCA7 splice variant had a CADD score of 26 and is predicted to affect splicing by multiple splicing prediction tools (Jian et al. 2014; Pashaei et al. 2016). The c.4416 + 1G > T variant has been identified in gnomAD six times; however, it has only been identified in African–Americans. Variants in ABCA7 have been implicated as strong genetic predictors of AD in African–Americans (Reitz et al. 2013) and have been associated with the progression and development of AD (Sinha et al. 2019). These variants likely contribute to the individual's strong family history of AD.
Rare Variants of Uncertain Significance
We returned a VUS to seven individuals which are described in detail in Supplemental VUS Details. We highlight one patient below that shows the importance of comprehensive genetic testing through a secondary finding.
An individual diagnosed with moderate dementia with age of onset in their 60s had a rare deletion in TSC2 (NM_000548.5:c.3846_3855delCTCCAAGGA, p.(Ser1282Arg_delC1283–G1285)) (returned as a VUS). This individual also had a secondary finding: a pathogenic variant in SCN5A (NM_000335.4:c.673C > T, p.(Arg225XTrp)). This specific SCN5A variant has been reported to cause Brugada syndrome (Kapplinger et al. 2010; Beckermann et al. 2014). Brugada syndrome is an inherited cardiac arrhythmia condition, and management includes prevention of cardiac arrest. The individual's sibling was reported to have died from cardiac arrest in their 60s.
Polygenic Risk Score
A PRS represents a statistical estimate of disease risk calculated by combining information about multiple risk variants throughout the genome. We sought to understand the contribution of common variation to AD risk in this cohort. Using the PRS generated by Cruchaga et al. (2018), we computed a PRS for each proband in our study (Supplemental Table 1). We also calculated scores for 880 LOAD cases (818 ADSP, 62 University of Alabama at Birmingham [UAB]), 77 EOAD (UAB early-onset or atypical dementia) cases that are the focus of this study, and 5179 healthy controls (5043 Alzheimer's Disease Sequencing Project [ADSP], 136 other Alabama-based study). We calculated PRS for all individuals using two methods: one that incorporates APOE status and one that omits it. APOE is a strong known risk factor for AD; however, when including it in the PRS there was not a significant difference between dementia cohorts (Supplemental Fig. 1). We did observe differences in the PRS of these cohorts when APOE was excluded (Fig. 3). We observed that the median PRS of the UAB EOAD cohort was higher than the LOAD cohort upon APOE exclusion. These results are consistent with the possibility that APOE status alone could be a stronger association for LOAD; in contrast, although APOE remains a critical contributor to EOAD, additional deleterious genetic variation may be more likely to contribute to the emergence of an EOAD presentation. We also showed that the PRS in all AD cohorts was higher than our control cohort of unaffected individuals (Fig. 3). Because we combined two control cohorts and two LOAD cohorts we also compared the PRS of each cohort separately and did not observe any significant differences between control or LOAD groups (Supplemental Fig. 2). In the 42% of UAB EOAD patients with no returnable findings, we hypothesized that PRS might be higher than those patients with a known pathogenic variant because of the combination of multiple genetic factors contributing to dementia risk. That would indicate that perhaps common variation rather than rare variation was contributing to risk in these cases. However, in the UAB EOAD cohort, we did not see a significant difference in PRS between patients with a returnable pathogenic variant based on genomic sequencing compared to those with no returnable variants (Supplemental Fig. 3).
Figure 3.
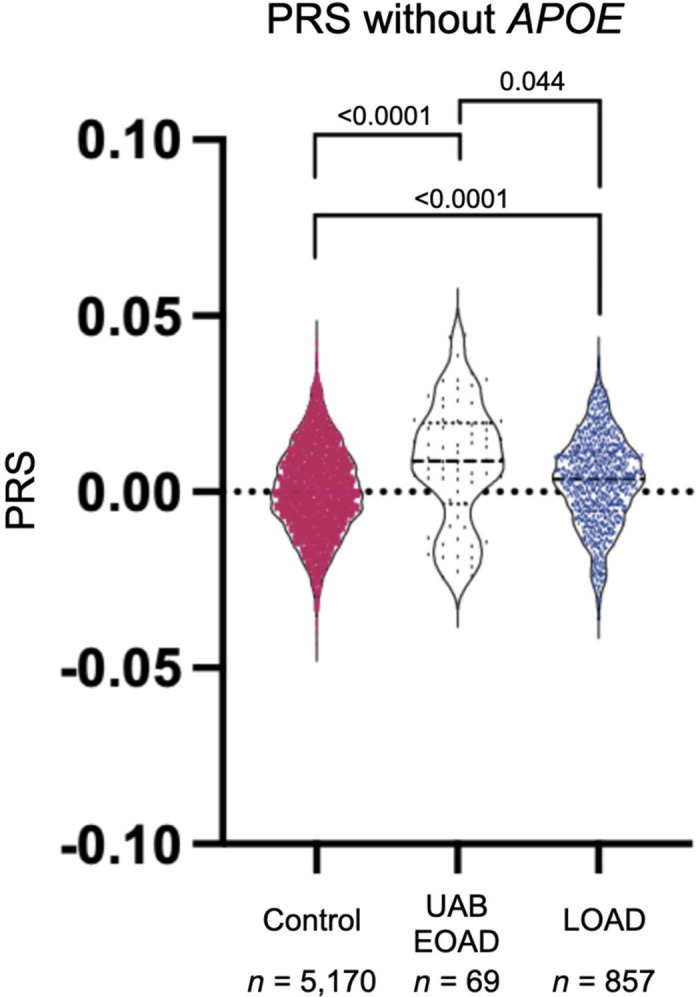
Violin plot comparing PRS of the University of Alabama at Birmingham (UAB) early-onset dementia (UAB EOAD) cohort to patients with a late-onset dementia (LOAD) cohort, and the control (Control) cohort. This PRS excludes APOE status. Only individuals with a self-reported ancestry of self-reported white, non-Hispanic were included in this analysis. ANOVA with Kruskal–Wallis test was used to calculate adjusted P-values.
Individuals with the strongest family history are more likely to have a returnable result (regardless of PRS), but 71% of non-Hispanic white patients with the strongest family history of early-onset or atypical dementia still do not have a known pathogenic mutation. This led us to consider if combinations of common variants can explain risk in these individuals. Indeed, there is a correlation in which a more extensive family history of dementia is associated with significantly higher APOE− PRS (Supplemental Fig. 4A), but there is no effect on APOE+ PRS (Supplemental Fig. 4B).
Individuals that self-identify as black/African–American have a higher prevalence of AD than individuals that self-identify as non-Hispanic white; however, there are disparities in the amount of genetic data available from this population (Clark et al. 2022). Both early-onset and late-onset dementia cohorts had a very small African–American population making it difficult to compare PRS between populations. The generation of PRSs is typically performed using white, non-Hispanic populations. As others have observed (Clark et al. 2022), we saw that existing PRS do not capture variation contributing to AD risk in African–Americans (Supplemental Fig. 5), pointing to the importance of developing ancestry-specific and/or ancestry-adjusted PRS.
DISCUSSION
In this study, genome sequencing revealed variants contributing to dementia risk that would be missed by targeted panel testing, affirming the need for comprehensive genetic approaches. For example, MAPT variants are typically associated with FTD; however, there have been multiple reports of MAPT variants in patients with AD (Reed et al. 1997; Rademakers et al. 2003; Cruts et al. 2012). Panels that are specific to AD do not include MAPT variants; thus, this variant would not be detected. Whole-genome sequencing provides a solution to this challenge. Some commercially available panels address this issue by assessing genes associated with multiple neurodegenerative diseases on the same panel. Six percent of our 100-patient cohort had variation in MAPT that contributes to AD risk, however, in this cohort, we hypothesized that the five patients with the MAPT R406W variant likely shared common ancestry limiting the interpretation of the frequency of mutations in this gene leading to AD risk.
To better assess the risk of dementia patients with multiple contributory factors we calculated a PRS for individuals with early-onset or atypical dementia that was either definitively or most likely due to AD and late-onset AD, as well as a control cohort. The PRS we used was used in a previous study (Cruchaga et al. 2018). Similar to Cruchaga et al. we showed an increased PRS in patients with an earlier onset of disease when considering APOE− PRS; however, we did not observe a significant difference for APOE+ PRS. Intriguingly, a recent study (Polsinelli et al. 2023) observed a correlation with earlier age of onset with APOE ε4 status on age of onset in LOAD, but no effect in sporadic EOAD males and a later age of onset in sporadic EOAD females. We also recently observed a shift toward earlier age of onset in dominant AD with APOE ε4 status (Cochran et al. 2023). Taken together, these results point to the importance of further studies to clarify the applicability of LOAD PRS as well as the effect of APOE ε4 in EOAD, and suggests that EOAD-specific PRS could be informative as EOAD cohort sizes grow to a size that permits the generation of a reliable EOAD-specific score. In our cohort, African–American patients with LOAD did not have a significantly different PRS than the controls. African–Americans have twice the risk of developing AD as white individuals (Alzheimer's Association 2020). Future studies generating PRS using genetic data from African–Americans and other non-Europeans is necessary to more accurately determine the contribution of common variation in African–American individuals, but the latest genome-wide association study (GWAS) for African–Americans with AD is still underpowered for this analysis, pointing to the critical nature of further study in this population (Kunkle et al. 2021).
In this study, we showed that multiple genetic factors, both rare and common, may contribute to an individual's dementia risk. Our study has limitations. First, variants were not returned to many patients with no returnable findings. Second, our PRS findings are limited by the application of a LOAD PRS to early-onset AD in this cohort, as well as the small size of our cohort. It is possible that a larger cohort would reveal small effects of PRS in patients with no returnable findings. A PRS for a given ancestral population performs increasingly better with larger numbers, with numbers in the high 1000s–10,000+ likely needed. Data from the ADSP is beginning to address the shortfall of non-European ancestry individuals in AD studies and will permit the development of better non-European population-specific and cross-ancestry PRS scores for AD (Sariya et al. 2021; Lake et al. 2023). Only having one recruitment site limited our cohort size and the expansion to multiple enrollment sites could increase the cohort number and data from non-European populations. Despite the limitation of patient number, this study also has strengths. We identified and returned variants that contribute to the genetic explanation of patients’ symptoms for more than half of the cohort. This study contributes to the body of work showing the value of comprehensive genetic testing in identifying variants contributing to early-onset dementia risk (Cochran et al. 2019; Huq et al. 2022) and highlights that both rare and common genetic variation can associate with early-onset and/or atypical dementia.
METHODS
C9orf72 Expansion Testing
ExpansionHunter (Dolzhenko et al. 2019) was used to detect repeat expansions of C9orf72 for samples with polymerase chain reaction (PCR)-free genomes (21 individuals). Prior to the implementation of PCR-free genomes, C9orf72 expansions were assessed using a separate clinical test (GeneDx).
Polygenic Risk Score
PRS was calculated with and without APOE using PLINK 1.9 score with the no-mean-imputation option. Odds ratios for the single-nucleotide polymorphisms (SNPs) were collected from the IGAP study (Lambert et al. 2013), and a log2 transformation was used on the odds ratios. APOE risk was calculated through the use of artificial SNPs such that each SNP represented the combination of rs429358 and rs7412 alleles. The odds ratios for the PRS scores with APOE used odds ratios from Farrer et al. (1997) for APOE status (ε3/ε4, ε4/ε4, ε2/ε3, ε2/ε4, ε2/ε2) instead of each SNP independently.
The HudsonAlpha CSER study enrolled and sequenced children with an early-onset neurodevelopmental disorder (NDD) with symptoms such as intellectual disability, seizures, developmental delays, etc. Many of these children's parents were also sequenced, and we used these parents as controls. Although these individuals were ascertained as a result of having a child with an NDD, the parents themselves were generally healthy. In that context, it is important to note that the vast majority of the disease-associated variation found in the CSER study was either de novo (i.e., the disease-associated allele in the proband is absent from his/her parents), recessive (i.e., each parent was a heterozygous, unaffected carrier of a disease-associated allele), or X-linked (i.e., the proband is hemizygous for a disease-associated allele inherited from an unaffected heterozygous mother). To the extent that some of these parental individuals harbor symptoms and/or dominant or incompletely penetrant risk factors of NDDs, such conditions are phenotypically and genetically distinct from the neurodegenerative diseases we are studying here. Finally, we note that a small amount of phenotypic data was collected for each of these parents, and none of them were known to have a neurodegenerative disease at enrollment.
Patients with a clinical diagnosis of FTD, CAA, MSA, PCA, CBS, white matter disease, progressive spastic dysarthria, or MS were excluded for PRS calculation.
Genome Sequencing
Genome sequencing was performed at the HudsonAlpha Institute for Biotechnology on the NovaSeq platform using paired-end 150-bp reads. Sequencing libraries were prepared by Covaris shearing, end repair, adapter ligation, and PCR using standard protocols. Library concentrations were normalized using KAPA qPCR prior to sequencing. All sequencing variants returned to patients were validated by Sanger in a CAP/CLIA laboratory.
Data Processing and Quality Control
Quality control included confirmation of each sample's expected biological sex based on counts of Chr X heterozygous variants. All samples were processed through a unified sequence alignment and variant calling pipeline. Variants were called with Strelka v2.9.10. Sequence reads were aligned to GRCh38.p12 (with HudsonAlpha Clinical Sequencing Lab customized ALT mappings) using the Sentieon v201808.07 (Kendig et al. 2019) implementation of BWA-MEM (Li 2013) and command line option -M -K 10000000. BWAKit was used for post-alt processing of the alternate contig alignments. Duplicate reads were marked and base quality scores were recalibrated with Sentieon v201808.07 using dbSNP v146, Mills, and 1000G gold standard indels as training data. Variants were called on the hg38 primary contigs (Chr 1–Chr 22, Chr X, Chr Y, Chr M) using Strelka v2.9.10 (Kim et al. 2018) in germline single-sample analysis mode.
Data Analysis
Statistical analyses were conducted using GraphPad Prism 9 (version 9.3.1) and in R (version 3.6.1) using ggplot2 (version 3.3.6) (Wickham 2016).
Vcftools (version 0.1.16) (Danecek et al. 2011) was used for relationship inference analyses using the ‐‐relatedness2 command line option.
GraphPad Prism 9 (version 9.3.1) was used for plotting violin plots, and ANOVA with Kruskal–Wallis tests performed in GraphPad were unpaired, nonparametric, two-tailed with 95% confidence interval.
Genomic Analysis
The HudsonAlpha-developed Codicem application was used to analyze and support the interpretation of the variant data (described elsewhere [Holt et al. 2019]). Simple filtering for population allele frequencies (i.e., gnomAD and TOPMed Bravo [NHLBI 2018]), in silico deleteriousness scores (i.e., CADD, PolyPhen-2, and SIFT), and gene lists relevant to the phenotype of interest would recapitulate our findings using any suitable software package or even by a command line interface. In addition to searching for single-nucleotide variants and small indels, we also searched for large copy-number variations using four callers (DELLY (Rausch et al. 2012), ERDS (Zhu et al. 2012), CNVnator (Abyzov et al. 2011), and BIC-seq2 (Xi et al. 2016)), but did not identify any relevant to patient phenotypes (including the absence of APP duplications).
Return of Results
Results meeting criteria for return were delivered to patients by clinicians in the UAB Brain Aging and Memory Clinic through letters written by a genetic counselor. Letters included information on the variant, associated disease, recurrence risk, and management recommendations. Patients were given the option to have a genetic counselor present for the return of results via phone or videoconference or to follow up with a genetic counselor after delivery of the results. Primary results were provided only to probands. Although a secondary result was identified in only one participant who was a patient, we did also offer nonpatient participants (family members) receipt of actionable secondary findings (ACMG SF v3.0) if such a result had been identified. Family members of patients that received diagnostic results were provided with information to seek out clinical genetic counseling and targeted testing for familial variants if they desired.
Patients were able to opt into receiving secondary findings. To return secondary findings we followed the ACMG classification criteria. Secondary variants had to reach a classification of likely pathogenic or pathogenic and must be in a gene on the ACMG SF v3.0 list.
ADDITIONAL INFORMATION
Data Deposition and Access
Data from the participants enrolled in this study are deposited at NIAGADS under project NG00082—UAB/HudsonAlpha Families with Neurodegenerative Diseases and NG00135—UAB ADRC. We combined two cohorts of LOAD patients. One LOAD cohort was collected at UAB and a Global Diversity (Illumina product #20031816) plus NeuroBooster microarray was run on the samples. The other LOAD samples were from the ADSP (NIAGIDS accession number: NG00067.v9). Control samples were from the HudsonAlpha CSER study (Bowling et al. 2017), dbGap study accession number phs001089.v3.p1 and from ADSP (NIAGIDS accession number NG00067.v9).
Ethics Statement
This study was approved by UAB IRB protocol X161202004, “Evaluation of Genomic Variants in Patients with Neurologic Diseases” and UAB IRB protocol #300000169, “UAB Alzheimer's Disease Research Center/Brain Aging and Memory in the South (BAMS) Study.”
Acknowledgments
We thank the Clinical Services Laboratory at HudsonAlpha for DNA isolations, library generation, quality control, and sequencing and the Codicem software development team at HudsonAlpha for genome analysis software. We thank Dr. Emily Gordon for her assistance with pedigree visualization. We also thank the Greg Cooper Lab, including Drs. Michelle Thompson, Susan Hiatt, and Don Latner, for helpful discussions about variant interpretation. (Also see Supplemental Extended Acknowledgments.)
Author Contributions
J.N.C. and E.D.R. designed the study. J.N.C., E.D.R., and R.M.M. secured funding. J.N.C. and E.D.R. wrote the IRB protocol. V.S. and P.C. coordinated all aspects of patient interaction. J.M.J.L., Z.T.B., and J.W.T. processed sequencing data. C.A.W., J.N.C., M.D.A., and B.A.M. analyzed genomes. C.A.W. coordinated genomic sequencing and led variant review committee meetings. C.A.W. and J.W.T. conducted other analyses. M.C. wrote clinical letters and provided genetic counseling. E.D.R., D.S.G., and M.N.L. recruited participants and returned results. C.A.W. wrote the manuscript, with edits by S.J.C. and J.N.C. All authors approved the final manuscript.
Funding
Funding for genomes sequenced at HudsonAlpha was generously provided by the Daniel Foundation of Alabama and donors to the HudsonAlpha Foundation Memory and Mobility Program. Additional funding was provided by National Institutes of Health (NIH) grant R00AG068271 (J.N.C.), and NIH grant 5P20AG068024 (E.D.R., D.S.G., M.N.L, J.N.C., and J.W.T.).
Competing Interest Statement
The authors have declared no competing interest.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Abyzov A, Urban AE, Snyder M, Gerstein M. 2011. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21: 974–984. 10.1101/gr.114876.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer's Association. 2020. 2020 Alzheimer's disease facts and figures. Alzheimers Dement 10.1002/alz.12068 [DOI] [PubMed] [Google Scholar]
- Beckermann TM, McLeod K, Murday V, Potet F, George AL Jr. 2014. Novel SCN5A mutation in amiodarone-responsive multifocal ventricular ectopy-associated cardiomyopathy. Heart Rhythm 11: 1446–1453. 10.1016/j.hrthm.2014.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowling KM, Thompson ML, Amaral MD, Finnila CR, Hiatt SM, Engel KL, Nicholas Cochran J, Brothers KB, East KM, Gray DE, et al. 2017. Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med 9: 43. 10.1186/s13073-017-0433-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campion D, Charbonnier C, Nicolas G. 2019. SORL1 genetic variants and Alzheimer disease risk: a literature review and meta-analysis of sequencing data. Acta Neuropathol 138: 173–186. 10.1007/s00401-019-01991-4 [DOI] [PubMed] [Google Scholar]
- Clark K, Leung YY, Lee W-P, Voight B, Wang L-S. 2022. Polygenic risk scores in Alzheimer's disease genetics: methodology, applications, inclusion, and diversity. J Alzheimers Dis 89: 1–12. 10.3233/JAD-220025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran JN, McKinley EC, Cochran M, Amaral MD, Moyers BA, Lasseigne BN, Gray DE, Lawlor JMJ, Prokop JW, Geier EG, et al. 2019. Genome sequencing for early-onset or atypical dementia: high diagnostic yield and frequent observation of multiple contributory alleles. Cold Spring Harb Mol Case Stud 5: a003491. 10.1101/mcs.a003491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran JN, Acosta-Uribe J, Esposito BT, Madrigal L, Aguillón D, Giraldo MM, Taylor JW, Bradley J, Fulton-Howard B, Andrews SJ, et al. 2023. Genetic associations with age at dementia onset in the PSEN1 E280A Colombian kindred. Alzheimers Dement 10.1002/alz.13021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruchaga C, Del-Aguila JL, Saef B, Black K, Fernandez MV, Budde J, Ibanez L, Deming Y, Kapoor M, Tosto G, et al. 2018. Polygenic risk score of sporadic late-onset Alzheimer's disease reveals a shared architecture with the familial and early-onset forms. Alzheimers Dement 14: 205–214. 10.1016/j.jalz.2017.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Theuns J, Van Broeckhoven C. 2012. Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat 33: 1340–1344. 10.1002/humu.22117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. 2011. The variant call format and VCFtools. Bioinformatics 27: 2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolzhenko E, van Vugt JJFA, Shaw RJ, Bekritsky MA, van Blitterswijk M, Narzisi G, Ajay SS, Rajan V, Lajoie BR, Johnson NH, et al. 2017. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res 27: 1895–1903. 10.1101/gr.225672.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolzhenko E, Deshpande V, Schlesinger F, Krusche P, Petrovski R, Chen S, Emig-Agius D, Gross A, Narzisi G, Bowman B, et al. 2019. ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 35: 4754–4756. 10.1093/bioinformatics/btz431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dua T, Seeher KM, Sivananthan S, Chowdhary N, Pot AM, Saxena S. 2017. [FTS5–03–01]: World Health Organization's global action plan on the public health response to dementia 2017–2025. Alzheimers Dement 13. 10.1016/j.jalz.2017.07.758 [DOI] [Google Scholar]
- Farrer LA, Adrienne Cupples L, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM. 1997. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis. J Am Med Assoc 278: 1349–1356. 10.1001/jama.1997.03550160069041 [DOI] [PubMed] [Google Scholar]
- Goldman JS, Farmer JM, Wood EM, Johnson JK, Boxer A, Neuhaus J, Lomen-Hoerth C, Wilhelmsen KC, Lee VM-Y, Grossman M, et al. 2005. Comparison of family histories in FTLD subtypes and related tauopathies. Neurology 65: 1817–1819. 10.1212/01.wnl.0000187068.92184.63 [DOI] [PubMed] [Google Scholar]
- Holstege H, van der Lee SJ, Hulsman M, Wong TH, van Rooij JG, Weiss M, Louwersheimer E, Wolters FJ, Amin N, Uitterlinden AG, et al. 2017. Characterization of pathogenic SORL1 genetic variants for association with Alzheimer's disease: a clinical interpretation strategy. Eur J Hum Genet 25: 973–981. 10.1038/ejhg.2017.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt JM, Wilk B, Birch CL, Brown DM, Gajapathy M, Moss AC, Sosonkina N, Wilk MA, Anderson JA, Harris JM, et al. 2019. VarSight: prioritizing clinically reported variants with binary classification algorithms. BMC Bioinformatics 20: 496. 10.1186/s12859-019-3026-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huq AJ, Thompson B, Bennett MF, Bournazos A, Bommireddipalli S, Gorelik A, Schultz J, Sexton A, Purvis R, West K, et al. 2022. Clinical impact of whole-genome sequencing in patients with early-onset dementia. J Neurol Neurosurg Psychiatry 10.1136/jnnp-2021-328146 [DOI] [PubMed] [Google Scholar]
- Jian X, Boerwinkle E, Liu X. 2014. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucl Acids Res 42: 13534–13544. 10.1093/nar/gku1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, Brugada P, Fressart V, Guerchicoff A, Harris-Kerr C, et al. 2010. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 7: 33–46. 10.1016/j.hrthm.2009.09.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. 2020. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581: 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ III, Shen J. 2017. Presenilin-1 mutations and Alzheimer's disease. Proc Natl Acad Sci 114: 629–631. 10.1073/pnas.1619574114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendig KI, Baheti S, Bockol MA, Drucker TM, Hart SN, Heldenbrand JR, Hernaez M, Hudson ME, Kalmbach MT, Klee EW, et al. 2019. Sentieon DNASeq variant calling workflow demonstrates strong computational performance and accuracy. Front Genet 10: 736. 10.3389/fgene.2019.00736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Källberg M, Chen X, Kim Y, Beyter D, Krusche P, et al. 2018. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 15: 591–594. 10.1038/s41592-018-0051-x [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46: 310–315. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkle BW, Schmidt M, Klein H-U, Naj AC, Hamilton-Nelson KL, Larson EB, Evans DA, De Jager PL, Crane PK, Buxbaum JD, et al. 2021. Novel Alzheimer disease risk loci and pathways in African American individuals using the African genome resources panel: a meta-analysis. JAMA Neurol 78: 102–113. 10.1001/jamaneurol.2020.3536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake J, Warly Solsberg C, Kim JJ, Acosta-Uribe J, Makarious MB, Li Z, Levine K, Heutink P, Alvarado CX, Vitale D, et al. 2023. Multi-ancestry meta-analysis and fine-mapping in Alzheimer's disease. Mol Psychiatry 10.1038/s41380-023-02089-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, et al. 2013. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 45: 1452–1458. 10.1038/ng.2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [q-bioGN] 10.48550/arXiv.1303.3997 [DOI] [Google Scholar]
- Liao X, Cai F, Sun Z, Zhang Y, Wang J, Jiao B, Guo J, Li J, Liu X, Guo L, et al. 2020. Identification of Alzheimer's disease-associated rare coding variants in the ECE2 gene. JCI Insight 5: e135119. 10.1172/jci.insight.135119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen W-M. 2010. Robust relationship inference in genome-wide association studies. Bioinformatics 26: 2867–2873. 10.1093/bioinformatics/btq559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee O, Wang J, Gitcho M, Chakraverty S, Taylor-Reinwald L, Shears S, Kauwe JSK, Norton J, Levitch D, Bigio EH, et al. 2008. Molecular characterization of novel progranulin (GRN) mutations in frontotemporal dementia. Hum Mutat 29: 512–521. 10.1002/humu.20681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. 2003. SIFT: predicting amino acid changes that affect protein function. Nucl Acids Res 31: 3812–3814. 10.1093/nar/gkg509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- NHLBI UM. 2018. The NHLBI Trans-Omics for Precision Medicine (TOPMed) Whole Genome Sequencing Program. BRAVO variant browser.
- Pashaei E, Ozen M, Aydin N. 2016. Splice sites prediction of human genome using AdaBoost. In 2016 IEEE-EMBS International Conference on Biomedical and Health Informatics (BHI), pp. 300–303. IEEE, Piscataway, NJ. [Google Scholar]
- Piazza M, Prior TW, Khalsa PS, Appleby B. 2020. A case report of genetic prion disease with two different PRNP variants. Mol Genet Genomic Med 8: e1134. 10.1002/mgg3.1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polsinelli AJ, Lane KA, Manchella MK, Logan PE, Gao S, Apostolova LG. 2023. APOE ε4 is associated with earlier symptom onset in LOAD but later symptom onset in EOAD. Alzheimers Dement 19: 2212–2217. 10.1002/alz.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R, Dermaut B, Peeters K, Cruts M, Heutink P, Goate A, Van Broeckhoven C. 2003. Tau (MAPT) mutation Arg406Trp presenting clinically with Alzheimer disease does not share a common founder in Western Europe. Hum Mutat 22: 409–411. 10.1002/humu.10269 [DOI] [PubMed] [Google Scholar]
- Raghavan NS, Brickman AM, Andrews H, Manly JJ, Schupf N, Lantigua R, Wolock CJ, Kamalakaran S, Petrovski S, Tosto G, et al. 2018. Whole-exome sequencing in 20,197 persons for rare variants in Alzheimer's disease. Ann Clin Transl Neurol 5: 832–842. 10.1002/acn3.582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. 2012. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28: i333–i339. 10.1093/bioinformatics/bts378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed LA, Grabowski TJ, Schmidt ML, Morris JC, Goate A, Solodkin A, Van Hoesen GW, Schelper RL, Talbot CJ, Wragg MA, et al. 1997. Autosomal dominant dementia with widespread neurofibrillary tangles. Ann Neurol 42: 564–572. 10.1002/ana.410420406 [DOI] [PubMed] [Google Scholar]
- Reitz C, Jun G, Naj A, Rajbhandary R, Vardarajan BN, Wang L-S, Valladares O, Lin C-F, Larson EB, Graff-Radford NR, et al. 2013. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E ɛ4, and the risk of late-onset Alzheimer disease in African Americans. J Am Med Assoc 309: 1483–1492. 10.1001/jama.2013.2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosini F, Rufa A, Monti L, Tirelli L, Federico A. 2014. Adult-onset phenylketonuria revealed by acute reversible dementia, prosopagnosia and parkinsonism. J Neurol 261: 2446–2448. 10.1007/s00415-014-7492-7 [DOI] [PubMed] [Google Scholar]
- Sariya S, Felsky D, Reyes-Dumeyer D, Lali R, Lantigua RA, Vardarajan B, Jiménez-Velázquez IZ, Haines JL, Shellenberg GD, Pericak-Vance MA, et al. 2021. Polygenic risk score for Alzheimer's disease in Caribbean Hispanics. Ann Neurol 90: 366–376. 10.1002/ana.26131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankaran SS, Capell A, Hruscha AT, Fellerer K, Neumann M, Schmid B, Haass C. 2008. Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem 283: 1744–1753. 10.1074/jbc.M705115200 [DOI] [PubMed] [Google Scholar]
- Sinha N, Reagh ZM, Tustison NJ, Berg CN, Shaw A, Myers CE, Hill D, Yassa MA, Gluck MA. 2019. ABCA7 risk variant in healthy older African Americans is associated with a functionally isolated entorhinal cortex mediating deficient generalization of prior discrimination training. Hippocampus 29: 527–538. 10.1002/hipo.23042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoychev KR, Stoimenova-Popova M, Chumpalova P, Ilieva L, Swamad M, Kamburova-Martinova Z. 2019. A clinical case of patient carrying rare pathological PSEN1 gene mutation (L424V) demonstrates the phenotypic heterogenity of early onset familial AD. Front Psychiatry 10: 857. 10.3389/fpsyt.2019.00857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufekcioglu Z, Cakar A, Bilgic B, Hanagasi H, Gurvit H, Emre M. 2016. Adult-onset phenylketonuria with rapidly progressive dementia and parkinsonism. Neurocase 22: 273–275. 10.1080/13554794.2016.1142567 [DOI] [PubMed] [Google Scholar]
- van Spronsen FJ, Blau N, Harding C, Burlina A, Longo N, Bosch AM. 2021. Phenylketonuria. Nat Rev Dis Primers 7: 36. 10.1038/s41572-021-00267-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. 2016. ggplot2: elegant graphics for data analysis. Springer International, New York. [Google Scholar]
- Xi R, Lee S, Xia Y, Kim T-M, Park PJ. 2016. Copy number analysis of whole-genome data using BIC-seq2 and its application to detection of cancer susceptibility variants. Nucl Acids Res 44: 6274–6286. 10.1093/nar/gkw491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Need AC, Han Y, Ge D, Maia JM, Zhu Q, Heinzen EL, Cirulli ET, Pelak K, He M, et al. 2012. Using ERDS to infer copy-number variants in high-coverage genomes. Am J Hum Genet 91: 408–421. 10.1016/j.ajhg.2012.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data from the participants enrolled in this study are deposited at NIAGADS under project NG00082—UAB/HudsonAlpha Families with Neurodegenerative Diseases and NG00135—UAB ADRC. We combined two cohorts of LOAD patients. One LOAD cohort was collected at UAB and a Global Diversity (Illumina product #20031816) plus NeuroBooster microarray was run on the samples. The other LOAD samples were from the ADSP (NIAGIDS accession number: NG00067.v9). Control samples were from the HudsonAlpha CSER study (Bowling et al. 2017), dbGap study accession number phs001089.v3.p1 and from ADSP (NIAGIDS accession number NG00067.v9).