


Keywords: α-cell function, β-cell function, impaired fasting glucose, impaired insulin action, insulin secretion
Abstract
Elevated fasting free fatty acids (FFAs) and fasting glucose are additively associated with impaired glucose tolerance (IGT) and decreased β-cell function [quantified as disposition index (DI)]. We sought to examine how changes in fasting FFA and glucose alter islet function. We studied 10 subjects with normal fasting glucose (NFG) and normal glucose tolerance (NGT) on two occasions. On one occasion, Intralipid and glucose were infused overnight to mimic conditions present in IFG/IGT. In addition, we studied seven subjects with IFG/IGT on two occasions. On one occasion, insulin was infused to lower overnight FFA and glucose concentrations to those observed in people with NFG/NGT. The following morning, a labeled mixed meal was used to measure postprandial glucose metabolism and β-cell function. Elevation of overnight fasting FFA and glucose in NFG/NGT did not alter peak or integrated glucose concentrations (2.0 ± 0.1 vs. 2.0 ± 0.1 Mol per 5 h, Saline vs. Intralipid/glucose, P = 0.55). Although overall β-cell function quantified by the Disposition Index was unchanged, the dynamic component of β-cell responsivity (ϕd) was decreased by Intralipid and glucose infusion (9 ± 1 vs. 16 ± 3 10−9, P = 0.02). In people with IFG/IGT, insulin did not alter postprandial glucose concentrations or indices of β-cell function. Endogenous glucose production and glucose disappearance were also unchanged in both groups. We conclude that acute, overnight changes in FFA, and glucose concentrations do not alter islet function or glucose metabolism in prediabetes.
NEW & NOTEWORTHY This experiment studied the effect of changes in overnight concentrations of free fatty acids (FFAs) and glucose on β-cell function and glucose metabolism. In response to elevation of these metabolites, the dynamic component of the β-cell response to glucose was impaired. This suggests that in health overnight hyperglycemia and FFA elevation can deplete preformed insulin granules in the β-cell.
INTRODUCTION
Prediabetes is a metabolic state with a high, but heterogeneous, rate of progression to type 2 diabetes (1). This is a disease which causes morbidity, mortality, and costs in excess of ∼$170 billion/year (2). The heterogeneity with which prediabetes transitions to type 2 diabetes can be explained in part by lifestyle and demographic factors. However, there is also significant variability in islet function among people classified as having prediabetes (3). β-Cell function is optimally quantified by measures that express insulin secretion as a function of the prevailing insulin action. The resulting Disposition Index (DI) is a well-established measure of β-cell function and, unlike other qualitative measures, it can predict progression to type 2 diabetes (4). It quantifies the integrated response to an oral or intravenous glucose challenge (with similar results regardless of the stimulus) (5).
In a subset of prediabetes, subjects with impaired fasting glucose (IFG) and normal glucose tolerance (NGT), i.e. IFG/NGT, have DI values similar to those with normal fasting glucose (NFG)/NGT (3, 6). This may reflect isolated dysfunction of α-cell glucose sensing (7). On the other hand, subjects with impaired glucose tolerance (IGT) exhibit decreased DI with deficits that are more marked in people who have both IFG and IGT rather than IGT alone (3). We (8), and others (9, 10), have noted that IGT and defects in insulin secretion and action are associated with elevated fasting free fatty acid (FFA) concentrations. Increased FFA concentrations reflect increased adipose tissue lipolysis in the fasting state (11) and are affected by fat distribution. Decreased fatty acid oxidation has also been reported in obesity (12). Upper body obesity, but not lower body obesity, is associated with decreased insulin action, increased FFA, and diabetes risk (13).
The observation that combined fasting hyperglycemia and elevated FFA are associated with decreased DI raises the possibility that fasting substrate concentrations could impair subsequent prandial insulin secretion. Indeed, in humans, exercise or changes in glycemic control alter insulin secretion (14, 15). FFAs have dose-dependent effects on insulin action (16). Lowering of FFA concentrations for 48 h with acipimox, a niacin analog, improves insulin secretion and action in nondiabetic subjects (17). Prolonged FFA elevation increases insulin secretion transiently after which it declines (18–20). Others have suggested that the diurnal rise in FFA stimulates β-cell function (21, 22). On the other hand, we demonstrated that high physiologic FFA elevation (∼1,100 μmol/L) acutely impairs both β-cell responsivity (Φ) and insulin action (Si) (23).
It is therefore uncertain if the fasting milieu (specifically fasting glucose and FFA concentrations) prepare (21, 24) or hinder (25) the β-cell response to a subsequent oral challenge. If fasting metabolic abnormalities can acutely alter β-cell function, this could have significant implications for preventing individuals with prediabetes from progressing to T2DM.
To address these questions, we used a nocturnal infusion of Intralipid and dextrose to maintain overnight fasting FFA and glucose in people with normal fasting glucose and normal glucose tolerance (NFG/NGT) at concentrations like those observed in people with IFG/IGT. Conversely, in a group with IFG/IGT, we lowered overnight fasting FFA and glucose concentrations to those seen in people with NFG/NGT. Subsequently, we used a labeled mixed meal test to measure glucose turnover and islet function. We report that acute, overnight changes in FFA, and glucose concentrations do not alter (postprandial) glucose metabolism in people without T2DM. However, the increase in fasting insulin secretion caused by glucose and Intralipid infusion decreased the dynamic component (ϕd) of the overall β-cell response to glucose in the NFG/NGT group despite an unchanged DI.
METHODS
Screening
After approval from the Mayo Clinic Institutional Review Board, we recruited subjects through a combination of intramural and extramural advertising. To be eligible, subjects had to have no history of chronic illness or upper gastrointestinal surgery. In addition, they were not taking medications that could affect glucose metabolism. Potentially eligible subjects who showed interest in participating were then invited to the Clinical Research and Trials Unit (CRTU) for a screening visit. After written, informed consent was obtained, participants underwent a 2-h 75 g oral glucose tolerance test (OGTT) to characterize their glucose tolerance status as previously described (6). All subjects were instructed to follow a weight-maintenance diet containing 55% carbohydrate, 30% fat, and 15% protein for at least 3 days before the study (Supplemental Figs. S1 and S2). Body composition was measured at the time of screening using dual-energy X-ray absorptiometry (Lunar, Madison, WI).
Experimental Design
Participants were admitted to the CRTU at 1700 on the day before the study. After consuming a standard 10 kcal/kg caffeine-free meal (55% carbohydrate, 30% fat, and 15% protein), they fasted overnight (Supplementary Fig. S3). At 2230, a dorsal hand vein was cannulated and placed in a heated Plexiglas box maintained at 55°C to allow sampling of arterialized venous blood. The contralateral forearm vein was cannulated to allow tracer, glucose, and hormone infusions.
To examine the combined effect of hyperglycemia and FFA elevation (FFA/Glucose Day) in subjects with NFG/NGT, at 2300 (−480 min), 50% dextrose [containing [6,6-2H2] glucose to minimize fluctuations in Specific Activity (26)] was infused as needed to maintain glucose at ∼6.7 mmol/L until 0700 (0 min) (27). In addition, an infusion of Intralipid (20%, 0.015 mL/kg/min; Baxter, Healthcare, Deerfield, IL) was started at 2300 and continued until 0700 (0 min) to prevent the fall in FFA concentrations that would normally occur because of the increase in insulin secretion in response to elevated glucose concentrations. On a separate study day (Saline Day), saline, rather than Intralipid and dextrose, was infused from 2300 to 0700 the following morning. The order of the study days (Saline vs. FFA/Glucose) was random and they were performed 14 days apart.
To examine the effect of glucose and FFA lowering (Insulin Study Day) in subjects with IFG/IGT, human insulin (0.1 U/mL Humulin R; Eli Lilly, Indianapolis, IN) was infused, as before (28, 29), in an amount sufficient to maintain glucose at ∼5 mmol/L until 0630 (−30 min). On a separate study day (Saline Day), saline, rather than insulin, was infused from 2300 to 0630 the following morning. The order of the study days (Saline vs. Insulin) was random, and they were performed 14 days apart.
On all the study days, at 0400 (−180 min), a primed, continuous infusion of [6,6-2H2] glucose commenced and was used to trace endogenous glucose production (EGP). Note that on the FFA/glucose day in subjects with NFG/NGT, after 0400, all infused glucose was also enriched with [6,6-2H2] glucose to avoid fluctuations in tracer enrichment. At 0700 (0 min), subjects ingested a mixed meal consisting of 50 g Canadian bacon (<1% carbohydrate and fat) DiLusso Deli Co., CA), 2 Grade A scrambled eggs (no milk added), and Jell-O containing 75 g glucose labeled with [1-13C] glucose (4%) enrichment over a 10-min period (Supplementary Fig. S4). As before, an infusion of [6-3H] glucose was also started to trace meal appearance (30). The experiment ended at 1200 (300 min) when all infusions were stopped, participants consumed a late lunch, and left the CRTU.
Analytic Techniques
All blood was immediately placed on ice after collection, centrifuged at 4°C, separated, and stored at −80°C until assay. Plasma glucose concentrations were measured using a Yellow Springs glucose analyzer. Plasma insulin concentrations were measured using a chemiluminescence assay (Access Assay, Beckman, Chaska, MN). Plasma C-peptide was measured using a 2-site immunoenzymatic sandwich assay (Roche Diagnostics, Indianapolis, IN). Glucagon was measured using a two-site ELISA (Mercodia, Winston Salem, NC) in accordance with the manufacturer’s instructions. Proinsulin concentrations were measured using a sequential 2-site ELISA used clinically (31) (Mayo Medical Labs, Rochester, MN). Assay calibration is performed against the WHO 1st International Standard for human Proinsulin NIBSC code: 09/296. This assay demonstrates no cross-reactivity with insulin or C-peptide. Nonesterified free fatty acids were measured using the WAKO NEFA kit (WAKO Chemicals Inc., Richmond, VA), as previously described (32).
Plasma [6,6-2H2] glucose and [1-13C] glucose enrichments were measured using GC/MS (Thermoquest, San Jose, CA) to simultaneously monitor the C-1 and C-2 and C-3 to C-6 fragments (33). [6-3H] glucose-specific activity was measured by liquid scintillation counting after deproteinization and passage over anion- and cation-exchange columns (30).
Calculations
The systemic rates of meal glucose appearance (Meal Ra), endogenous glucose production (EGP), and glucose disappearance (Rd) were calculated using Steele’s two-compartment model (34). Meal Ra was calculated by multiplying rate of appearance of [1-13C] glucose (obtained from the infusion rate of [6-3H] glucose and the clamped plasma ratio of [6-3H] glucose and [1-13C] glucose) by the meal enrichment. EGP was calculated from the infusion rate of [6,62H2] glucose and the ratio of [6,62H2] glucose to endogenous glucose concentration. Rd was calculated by subtracting the change in glucose mass from the overall rate of glucose appearance (i.e., Meal Ra + EGP).
Net insulin action (Si) was estimated from glucose and insulin concentrations using the oral minimal model (4, 35). β-Cell responsivity indices were estimated using the oral C-peptide minimal model (36), incorporating age-associated changes in C-peptide kinetics (37). The model derives total β-cell responsivity to glucose (Φ) from the static (ϕs) and dynamic (ϕd) contributions, as previously described (4). Disposition indices (DI) were subsequently calculated by multiplying Φ by Si.
Statistical Analysis
Assuming variation (421 ± 62 mmol/L per 6 h—means ±SD, respectively) in integrated post meal glucose concentrations as previously (38), 10 subjects in the NFG/NGT and 7 subjects in the IFG/IGT groups would provide 80% power (at a two-sided 0.05 α level), to detect a biologically-significant (13% in NFG/NGT, 16% in IFG/IGT) change in integrated glucose concentrations. All continuous data are summarized as means ± SE. Area under the curve (AUC) or area above basal (AAB) were calculated using the trapezoidal rule. A paired, two-way Student’s t test (parametric) or a Wilcoxon matched-pairs signed rank test (nonparametric) was used to examine differences within groups (IFG/IGT and NFG/NGT). Between-group differences were assessed using an unpaired, two-way Student’s t test (parametric) or a Mann–Whitney U test (nonparametric). When necessary, multivariate analysis adjusting for the effects of age, sex, and weight was performed. The D’Agostino & Pearson omnibus normality test implemented in Prism 5 (GraphPad Software, San Diego, CA) was utilized to determine if data were normally distributed. A P value <0.05 was considered statistically significant.
RESULTS
Subject Characteristics by Fasting Glucose and by Glucose Tolerance Status
A total of 17 subjects were studied. At the time of screening, 10 subjects were classified as NFG/NGT and the remainder as IFG/IGT (Table 1). The latter group exhibited defects in insulin secretion and action as quantified by the oral minimal model at the time of screening—as expected from prior work (3).
Table 1.
Participant characteristics at the time of screening and baseline β-cell function on the saline study days
| NFG/NGT | IFG/IGT | P Value | |
|---|---|---|---|
| n | 10 | 7 | |
| Age, years | 44 ± 4 | 53 ± 5 | 0.16 |
| Sex (M/F) | 3/7 | 2/5 | |
| Total body mass, kg | 89 ± 6 | 91 ± 8 | 0.82 |
| BMI, kg/m2 | 31 ± 2 | 31 ± 2 | 0.97 |
| LBM, kg | 51 ± 2 | 52 ± 4 | 0.84 |
| Fasting glucose, mmol/L | 5.0 ± 0.1 | 6.3 ± 0.2 | <0.01 |
| Peak glucose, mmol/L | 9.4 ± 0.5 | 12.3 ± 0.9 | <0.01 |
| 120-min glucose, mmol/L | 7.8 ± 0.4 | 11.4 ± 0.9 | <0.01 |
| Si (10−4 dL/kg/min per μU/mL) | 9 ± 3 | 2 ± 1 | 0.09 |
| ϕbasal (10−9 min−1) | 9 ± 1 | 10 ± 1 | 0.67 |
| ϕd (10−9) | 810 ± 73 | 443 ± 129 | 0.04 |
| ϕs (10−9 min−1) | 51 ± 6 | 33 ± 6 | 0.05 |
| Φ (10−9 min−1) | 61 ± 8 | 38 ± 7 | 0.05 |
| DI (10−14 dL/kg/min/pmol) | 868 ± 303 | 117 ± 41 | 0.04 |
Data represent means ± SE. BMI, body mass index; IFG, impaired fasting glucose; IGT, impaired glucose tolerance; LBM, lean body mass; NFG, normal fasting glucose; NGT, normal glucose tolerance. P values represent results of a two-way unpaired t test or a Mann–Whitney test.
Overnight Glucose and Free Fatty Acids
On the saline day of the experiment in people with NFG/NGT, overnight glucose concentrations averaged 4.9 ± 0.5 mmol/L. By design, during FFA and glucose elevation, glucose rose to an average of 6.6 ± 0.1 mmol/L (P < 0.01, Fig. 1A). In people with IFG/IGT, overnight glucose concentrations were lowered from 6.1 ± 0.2 to 5.2 ± 0.1 mmol/L (P < 0.01, Fig. 1B), by insulin infusion.
Figure 1.
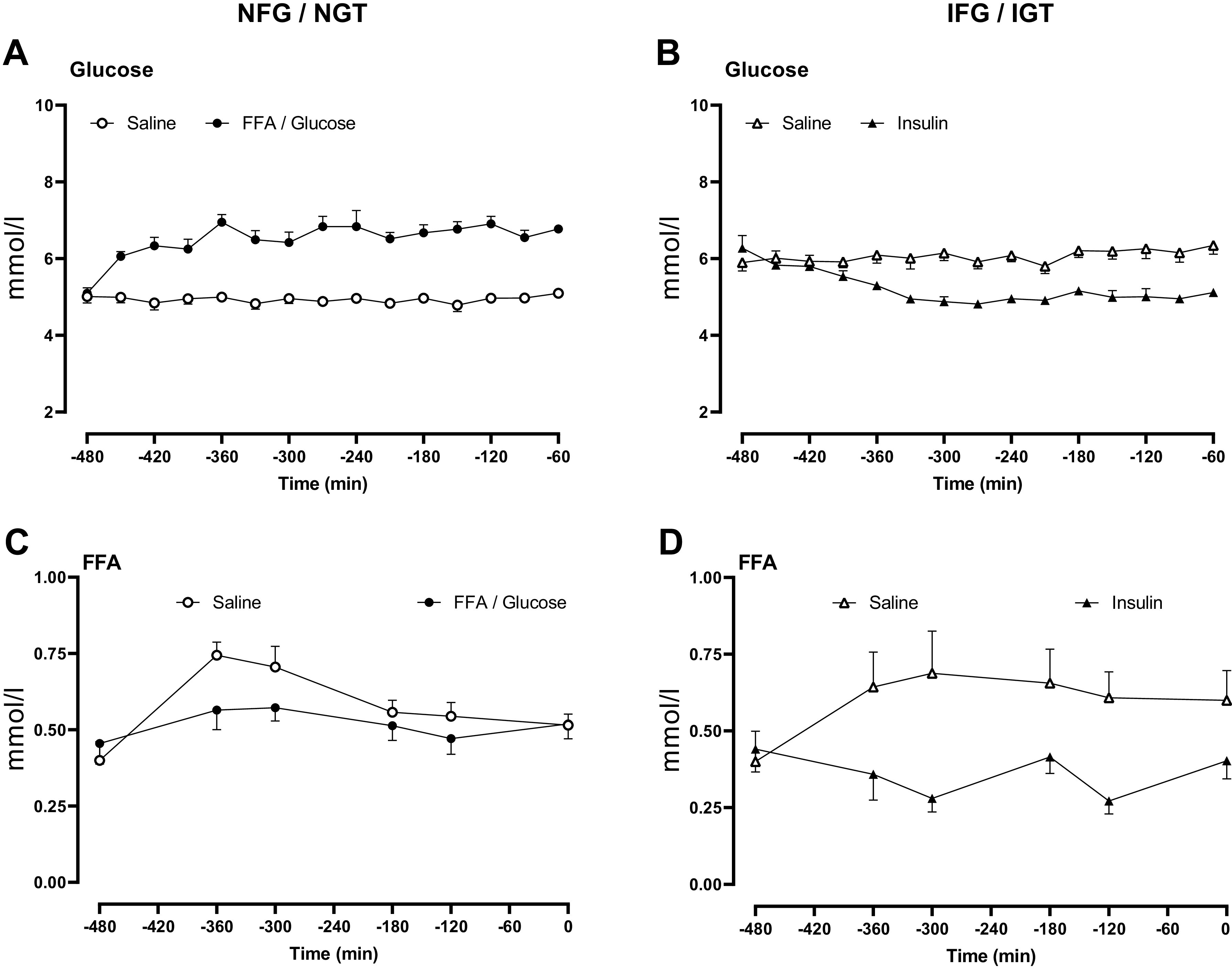
Overnight glucose (A and B) and free fatty acid concentrations (C and D) in people with normal fasting glucose and normal glucose tolerance and in people with impaired fasting glucose and impaired glucose tolerance. Concentrations are shown during saline infusion (○ or in the case of IFG/IGT Δ), Intralipid and glucose infusion (•), and during insulin infusion (▴).
In people with NFG/NGT during the saline infusion, FFA concentrations peaked at −360 min (Fig. 1C). Average FFA concentrations did not differ on the FFA/glucose day (0.56 ± 0.03 vs. 0.51 ± 0.03 mmol/L, P = 0.11), despite the presence of hyperglycemia and increased insulin secretion (Fig. 2A). In contrast, in subjects with IFG/IGT, average FFA concentrations were significantly decreased by insulin infusion (0.60 ± 0.10 vs. 0.36 ± 0.04 mmol/l, P = 0.04, Fig. 2D) compared with the saline day.
Figure 2.
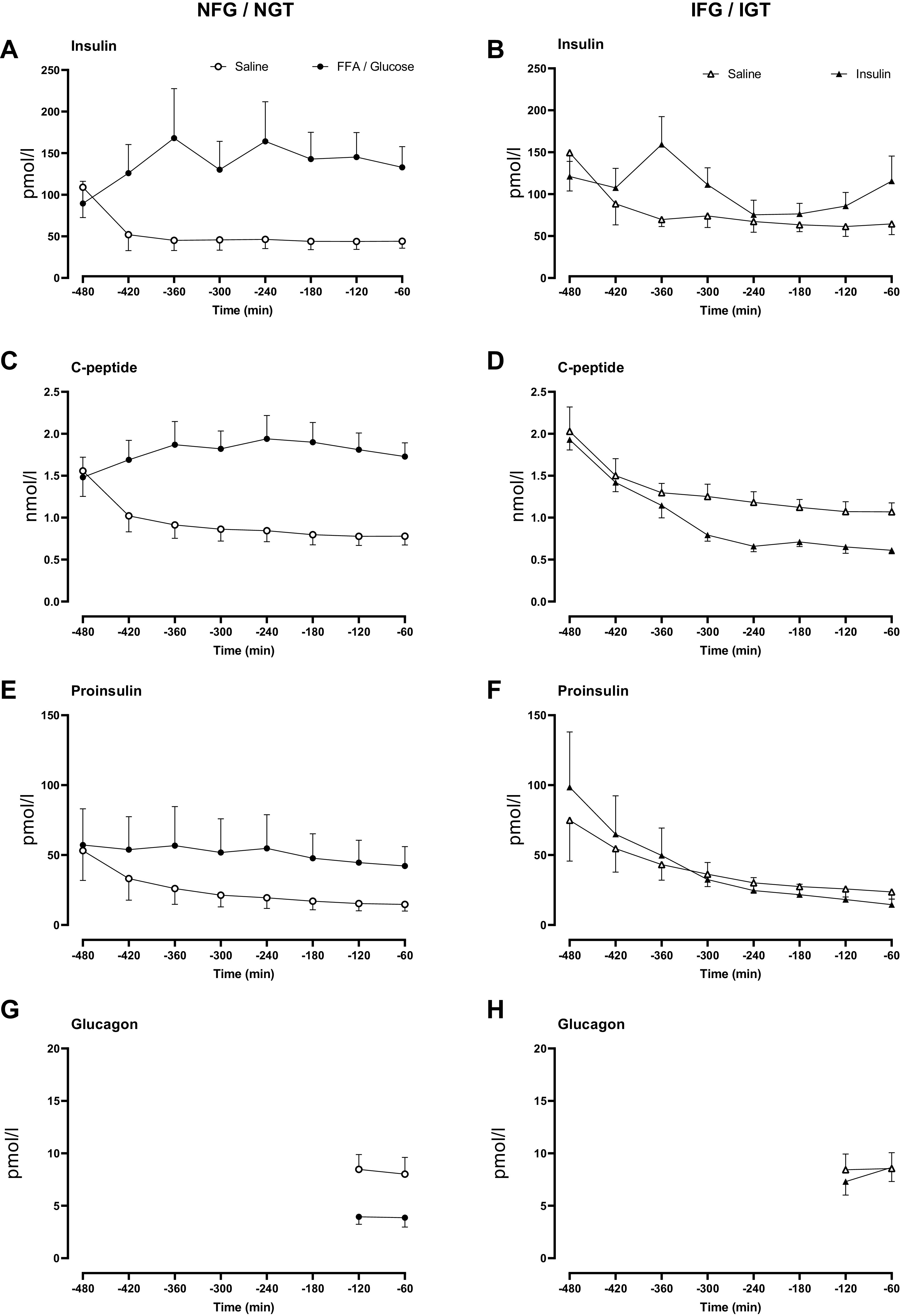
Overnight insulin (A and B), C-peptide (C and D), proinsulin (E and F), and glucagon (G and H) concentrations in people with normal fasting glucose and normal glucose tolerance and in people with impaired fasting glucose and impaired glucose tolerance. Concentrations are shown during saline infusion (○ or in the case of IFG/IGT Δ), Intralipid and glucose infusion (•), and during insulin infusion (▴).
Overnight Insulin, C-Peptide, Proinsulin, and Glucagon Concentrations
Insulin concentrations at the start of the experiment (−480 min) did not differ between study days in the NFG/NGT group (109 ± 36 vs. 90 ± 27 pmol/L, saline vs. FFA/glucose respectively, P = 0.19). As expected, average overnight insulin concentrations were greater during FFA/glucose exposure (46 ± 12 vs. 144 ± 35 pmol/L, P < 0.01, Fig. 2A). In the IFG/IGT group, average insulin concentrations at the start and during the experiment did not differ between study days. However, by design, overnight insulin concentrations were greater at several timepoints during the insulin study day (Fig. 2B).
C-peptide concentrations at the start of the experiment (−480 min) did not differ between study days in the NFG/NGT group but increased in response to FFA/glucose (0.8 ± 0.2 vs. 1.9 ± 0.3 nmol/L, P < 0.01, Fig. 2C). In the IFG/IGT group, C-peptide concentrations at the start of the experiment (−480 min) also did not differ between study days. Insulin infusion decreased endogenous insulin secretion, as indicated by average overnight C-peptide concentrations (1.2 ± 0.1 vs. 0.9 ± 0.1 nmol/L, P = 0.01, Fig. 2D).
Proinsulin concentrations at the start of the experiment (−480 min) did not differ between study days in the NFG/NGT group, but increased in response to FFA/glucose (21 ± 8 vs. 50 ± 20 pmol/L, P = 0.05, Fig. 2E). In subjects with IFG/IGT, differences in proinsulin concentrations were only apparent from −120 min onward, with a slight, but significant, decrease in proinsulin concentrations in the presence of exogenous insulin (22 ± 5 vs. 14 ± 4 pmol/L, P < 0.01, Fig. 2F).
In response to FFA/glucose elevation, fasting glucagon concentrations decreased in the subjects with NFG/NGT (8.7 ± 1.8 vs. 4.7 ± 1.3 pmol/L, P = 0.02). These differences persisted till meal ingestion (Fig. 2G). Fasting glucagon concentrations did not differ between study days in subjects with IFG/IGT (Fig. 2H).
Glucose, Insulin, C-Peptide, Proinsulin, and Glucagon Concentrations during the Mixed Meal
By design (Fig. 3A), plasma glucose concentrations before the meal challenge were greater in the NFG/NGT group on the FFA/glucose day (5.2 ± 0.1 vs. 6.4 ± 0.2 mmol/L, P < 0.01). Peak and integrated (We used AUC, see Supplemental Data for rationale) postprandial glucose concentrations did not differ significantly between study days. Similarly, the overnight insulin infusion decreased fasting glucose in IFG/IGT subjects (6.6 ± 0.2 vs. 5.3 ± 0.1 mmol/L, P < 0.01, Fig. 3B). Overnight insulin also did not change peak or integrated (AUC) postprandial glucose concentrations in IFG/IGT subjects.
Figure 3.

Meal glucose (A and B), insulin (C and D), C-peptide (E and F), proinsulin (G and H), and glucagon (I and J) concentrations in people with normal fasting glucose and normal glucose tolerance and in people with impaired fasting glucose and impaired glucose tolerance. Concentrations are shown during the meal after saline infusion (○ or in the case of IFG/IGT Δ), Intralipid and glucose infusion (•), and during insulin infusion (▴).
Fasting insulin concentrations were increased on the FFA/glucose day in the NFG/NGT group (46 ± 9 vs. 118 ± 24 pmol/L, P < 0.01). Peak and integrated (AUC) postprandial insulin concentrations did not differ between the two study days (Fig. 3C). Fasting, peak, and integrated (AUC) postprandial insulin concentrations did not differ between study days in the IFG/IGT group (Fig. 3D).
Fasting C-peptide concentrations were increased on the FFA/glucose day in the NFG/NGT group (0.8 ± 0.1 vs. 1.5 ± 0.2 nmol/L, P < 0.01). Peak and integrated (AUC) postprandial concentrations did not differ between the two study days (Fig. 3E). In IFG/IGT subjects, overnight insulin decreased fasting C-peptide (1.0 ± 0.1 vs. 0.6 ± 0.1 nmol/l, P < 0.01) with no subsequent change in peak and integrated (AUC) postprandial concentrations (Fig. 3F).
Fasting proinsulin concentrations were increased on the FFA/glucose day in the NFG/NGT group (14 ± 4 vs. 41 ± 13 pmol/L, P = 0.01). Peak and integrated (AUC) postprandial concentrations did not differ between the two study days (Fig. 3G). Fasting proinsulin concentrations were decreased by overnight insulin in the IFG/IGT group (22 ± 5 vs. 14 ± 4 pmol/L, P = 0.01), but this had no effect on postprandial peak and integrated (AUC) concentrations (Fig. 3H).
Nadir and integrated (AUC) post meal concentrations of glucagon did not differ between study days in both NFG/NGT and IFG/IGT groups (Fig. 3, I and J, respectively).
Endogenous Glucose Production, Meal Rate of Appearance, and Rates of Glucose Disappearance during a Mixed Meal
Fasting EGP was not significantly decreased by overnight FFA/glucose in the NFG/NGT group (14.4 ± 1.1 vs. 12.8 ± 0.8 μmol/kg/min, P = 0.10). Postprandial suppression of EGP also did not differ between study days (Fig. 4A). Overnight insulin did not alter fasting and nadir EGP in the IFG/IGT group (Fig. 4B).
Figure 4.
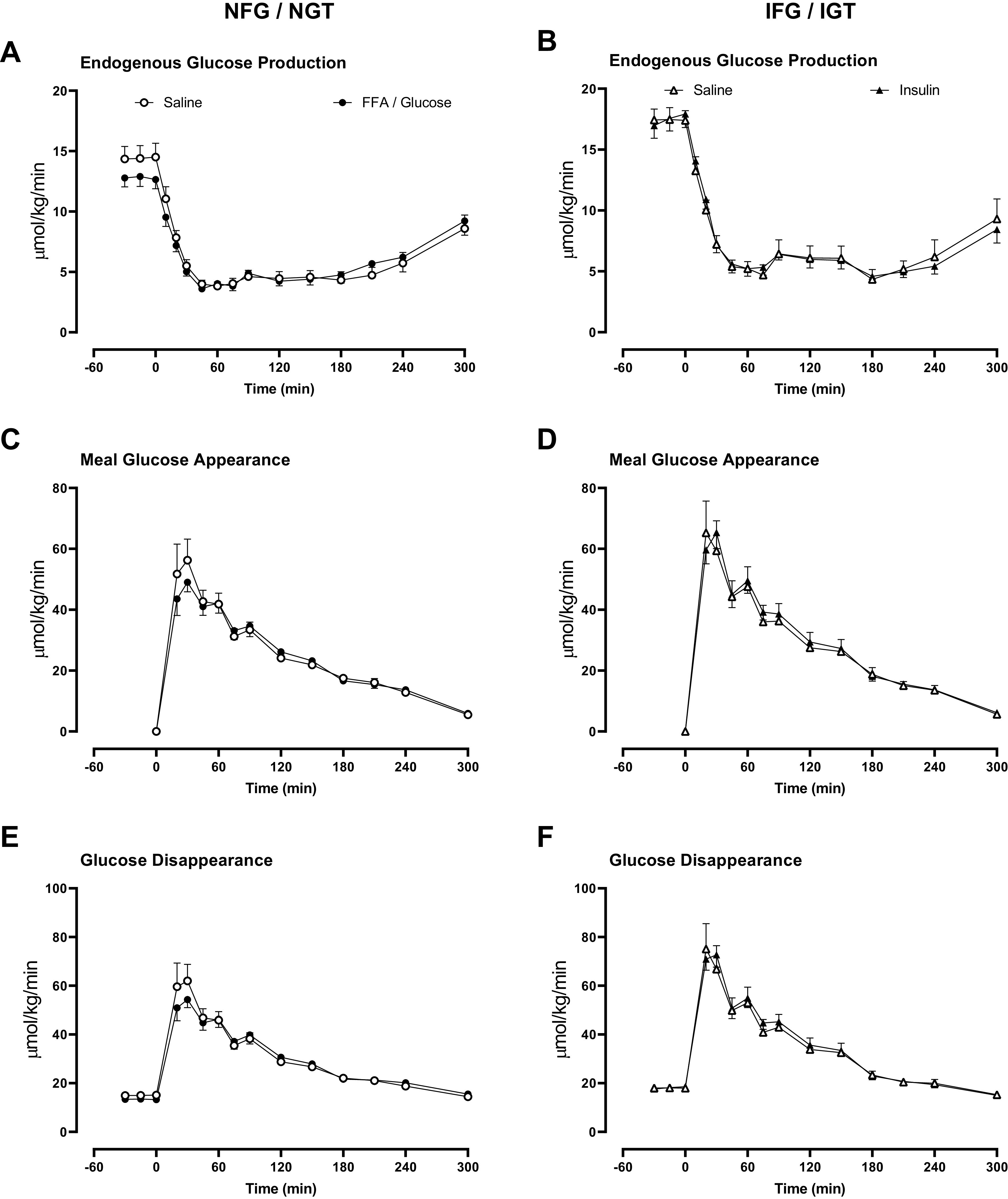
Rates of endogenous glucose production (A and B), meal glucose appearance (C and D), and glucose disappearance (E and F) in people with normal fasting glucose and normal glucose tolerance and in people with impaired fasting glucose and impaired glucose tolerance. Rates are shown during the meal after saline infusion (○ or in the case of IFG/IGT Δ), Intralipid and glucose infusion (•), and during insulin infusion (▴).
Peak and integrated meal appearance did not differ between study days in both the NFG/NGT (Fig. 4C) and the IFG/IGT (Fig. 4D) groups.
Peak glucose disappearance was not significantly changed by overnight FFA/glucose in the NFG/NGT group (69.7 ± 7.7 vs. 56.0 ± 3.6 μmol/kg/min 5 h, P = 0.07). Integrated glucose disappearance did not differ between study days in both the NFG/NGT (Fig. 4E) and the IFG/IGT (Fig. 4F) groups.
Indices of Insulin Secretion and Action
In response to the mixed meal test, insulin action did not differ between the two study days in subjects with NFG/NGT (Fig. 5A). Similarly, overnight infusion of insulin did not alter insulin action in the IFG/IGT group (Fig. 5B).
Figure 5.
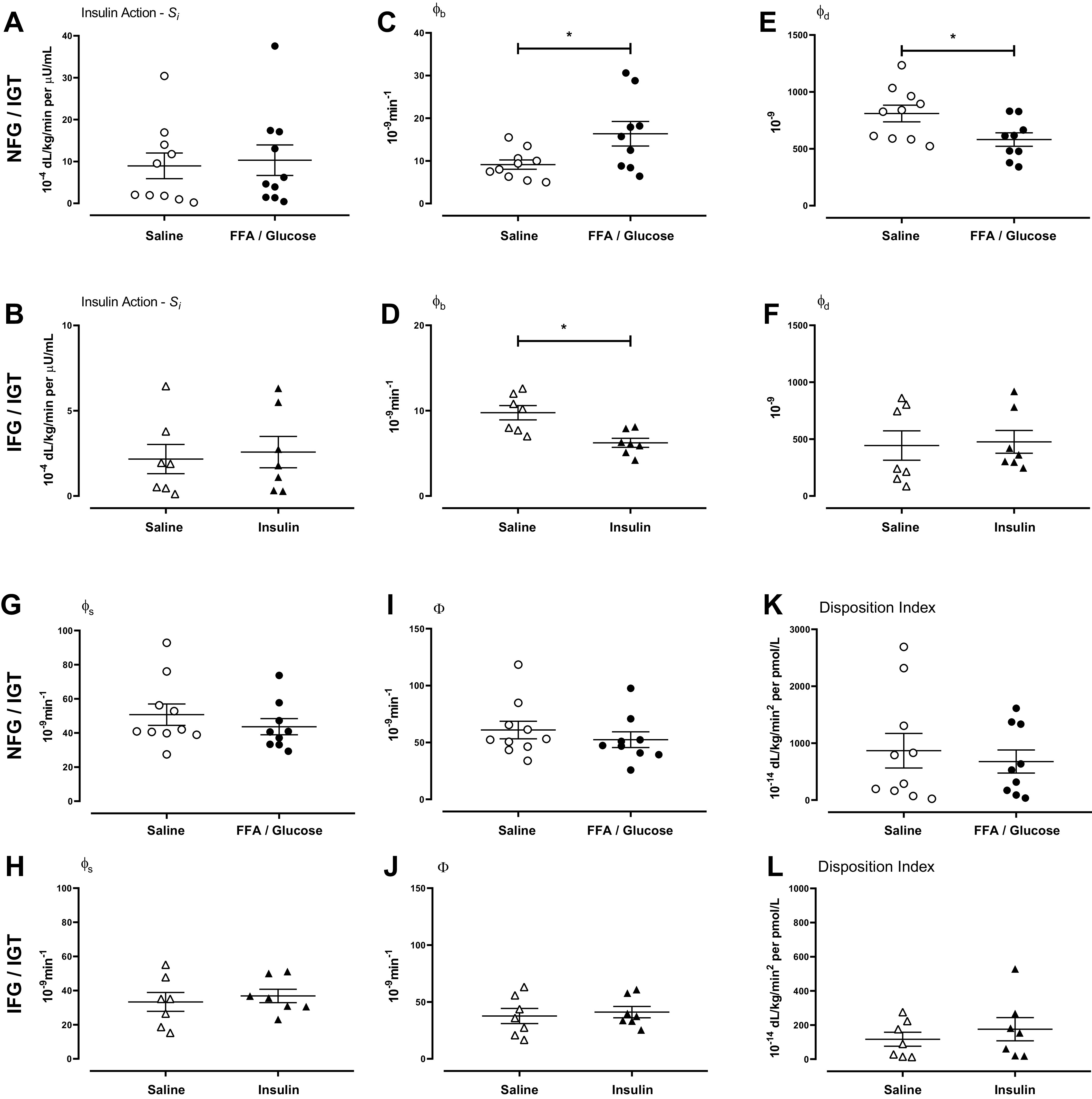
Insulin action (A and B), basal β-cell responsivity (ϕb, C and D), dynamic β-cell responsivity (ϕd, E and F), static β-cell responsivity (ϕs, G and H), total β-cell responsivity (Φ, I and J), and disposition index (K and L) in people with normal fasting glucose and normal glucose tolerance and in people with impaired fasting glucose and impaired glucose tolerance. Indices are shown during the meal challenge after saline infusion (○ or in the case of IFG/IGT Δ), Intralipid and glucose infusion (•), and after insulin infusion (▴).
Glucose and Intralipid infusion increased ϕb compared with the saline infusion (9 ± 1 vs. 16 ± 3 10−9min−1, P < 0.01, Fig. 5C) in people with NFG/NGT. Overnight infusion of insulin decreased ϕb in the IFG/IGT group (10 ± 1 vs. 6 ± 1 10−9min−1, P < 0.01, Fig. 5D).
Glucose and Intralipid infusion decreased ϕd compared with the saline infusion (9 ± 1 vs. 16 ± 3 10−9, P = 0.02, Fig. 5E) in people with NFG/NGT. Overnight infusion of insulin did not alter ϕd in the IFG/IGT group (10 ± 1 vs. 6 ± 1 10−9, P < 0.01, Fig. 5F).
ϕs did not differ between the experimental days in both the NFG/NGT and in the IFG/IGT group (Fig. 5, G and H, respectively). This was also the case for Φ (Fig. 5, I and J, respectively) and for DI (Fig. 5, K and L, respectively).
Ratio of Proinsulin/C-Peptide
Overnight FFA and glucose elevation in NFG/NGT subjects did not alter the proinsulin to C-peptide ratio (Fig. 6A). Similarly, in people with IFG/IGT, overnight insulin infusion did not alter this ratio (Fig. 6B).
Figure 6.
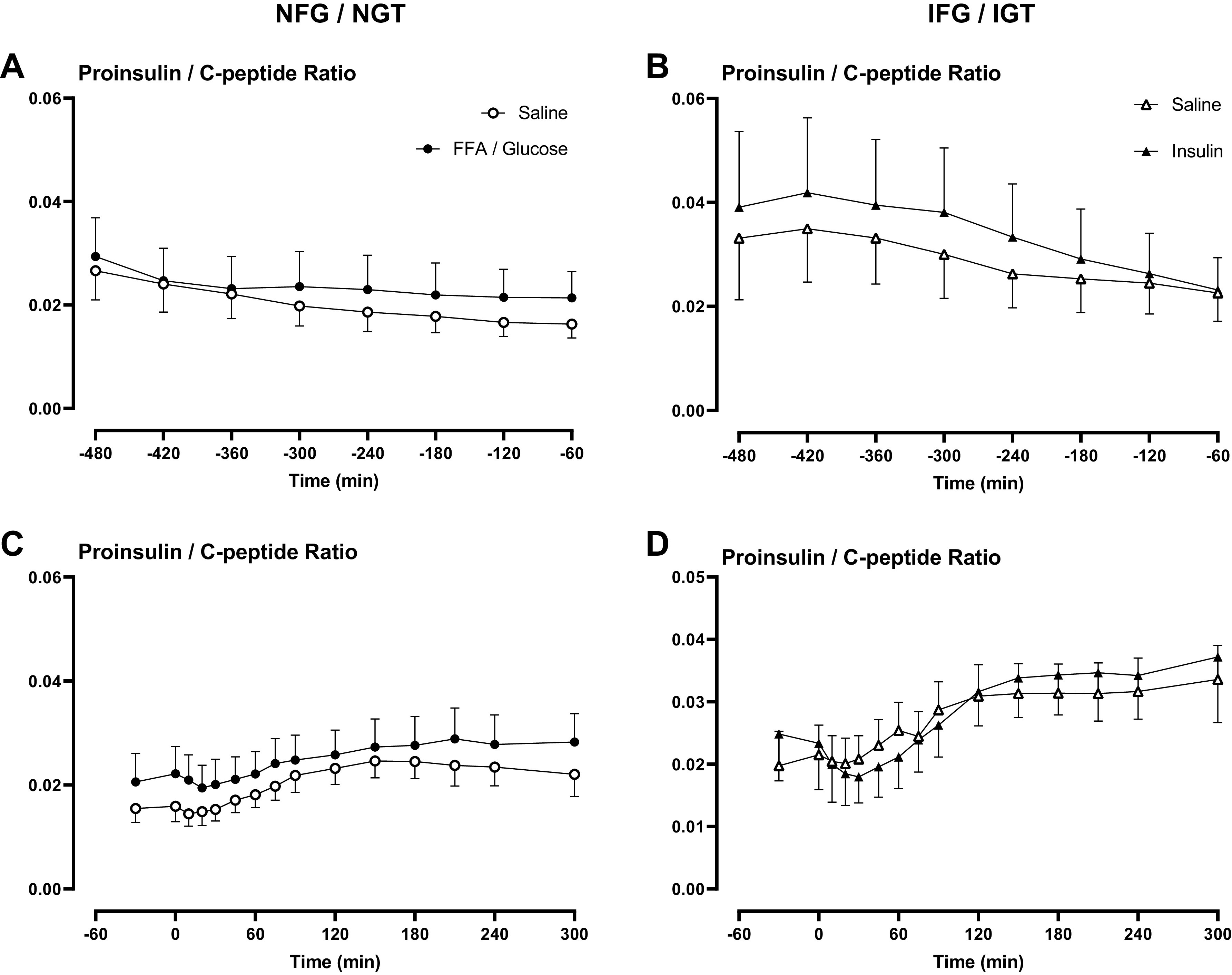
Ratio of proinsulin/C-peptide overnight (A and B) and during the meal challenge (C and D) in people with normal fasting glucose and normal glucose tolerance and in people with impaired fasting glucose and impaired glucose tolerance. Ratios are shown during saline infusion (○ or in the case of IFG/IGT Δ), Intralipid and glucose infusion (•), and during insulin infusion (▴).
The subsequent Proinsulin/C-peptide ratios in response to a mixed meal were unaffected by prior lipid and glucose infusion in people with NFG/NGT (Fig. 6C). Likewise, prior insulin infusion did not alter this ratio in response to a meal challenge in people with IFG/IGT (Fig. 6D). Data supplements can be accessed here: https://doi.org/10.6084/m9.figshare.22961753.
DISCUSSION
We tested the primary hypothesis that the fasting overnight milieu can alter the glycemic response to a subsequent meal—in part by changing overnight insulin secretion. Secondary hypotheses were that 1) the effect of manipulation of the fasting milieu might be modified by antecedent β-cell function and 2) that this might directly alter a specific component of the β-cell response to glucose. To do so, we studied people with NFG/NGT and intact β-cell function at baseline and after infusion of glucose and lipid emulsion (to raise fasting glucose and FFA). Conversely, in people with IFG/IGT and impaired β-cell function, we lowered fasting glucose and FFA concentrations by infusing insulin overnight. Our data demonstrate that manipulation of the fasting milieu did not alter glycemic excursion or overall β-cell function, irrespective of baseline status. However, the dynamic component of β-cell responsivity (4) was decreased by an overnight infusion of glucose and Intralipid.
Insulin secretion in response to an oral or intravenous glucose challenge represents insulin release from two distinct populations—preformed and docked insulin granules—referred to as the first phase of insulin secretion (ϕ1) in response to an IVGTT (39). This correlates with the dynamic component of β-cell function (DId = ϕd*Si, where Si represents insulin action) measured by the oral minimal model. The second phase (ϕ2), originally thought to represent insulin synthesized and secreted in response to sustained hyperglycemia, may represent secretion from a preformed reserve pool (40). This correlates with the static component of DI (DIs) (4). Loss of ϕ1 after an IVGTT is an early hallmark of diabetes (41). It appears that acute nocturnal manipulation of FFA and glucose sufficient to double insulin secretion in NFG/NGT subjects depletes the insulin pool mobilized in response to rising glucose concentrations. However, we did not observe the converse in people with IFG/IGT. Whether this is because we were underpowered to detect such an effect or whether this reflects chronic changes that accompany the disordered β-cell function observed in this group remains to be ascertained.
β-cells can synthesize, process, degrade, and secrete >104 peptide molecules per min including insulin, proinsulin, and Islet Amyloid Polypeptide in the basal state (42). This greatly expands during the demands associated with obesity, inflammation, and increased FFA (43). In situations of increased demand, β-cell protein processing increases the accumulation of misfolded proteins (44). Efficient removal of misfolded proteins prevents the endoplasmic reticulum stress (45) that is characteristics of β-cell dysfunction and apoptosis in T2DM (46). Monogenic impairments of the response to protein misfolding cause T2DM (47). Several common genetic variants associated with T2DM are in loci that are part of this response, underlining the protective role of this response for the β-cell (48, 49) especially during high metabolic demand (50).
In vivo, increased proinsulin secretion (as a proportion of insulin secretion) in rodents (51) and humans (52) in response to insulin resistance has been used as a surrogate of increased protein misfolding in β-cells. Proinsulin concentrations are increased in T2DM (53). The proinsulin/insulin ratio has various limitations given differences in half-life of the two peptides, suggesting that a proinsulin/C-peptide ratio may be a better marker of β-cell health. However, prior work has suggested that at least in subjects without diabetes, these ratios do not meaningfully predict defects in β-cell function as quantified by a disposition index (54). In the current experiments, manipulation of fasting FFA and glucose did not alter overnight proinsulin secretion—in absolute terms as well as a function of overnight insulin secretion. More germane to the questions posed before the experiment, the prandial secretion of proinsulin was also unchanged. These data suggest that despite changes in secretory demand—the increase in overnight fasting insulin secretion in people with NFG/NGT is insufficient to stress β-cell insulin synthesis or, possibly, deplete all preformed releasable pools of insulin secretion (40)—beyond that measured as ϕd.
The changes in fasting glucagon concentrations that accompanied the overnight changes are notable. We previously showed that people with IFG have impaired α-cell responsivity to glucose (7). Indeed, in this study, people with IFG/IGT did not alter fasting glucagon concentrations despite lower fasting glucose during overnight insulin infusion. In another set of experiments where FFAs were increased threefold, basal fasting glucagon rose in response (55). However, in this experiment, in people with NFG/NGT, α-cell responsivity to glucose was preserved so that mild hyperglycemia suppressed fasting glucagon concentrations.
There are some caveats to these observations, the main limitation being the acute nature of the intervention used in this situation. This study establishes that short-term manipulation of FFA and glucose does not alter overall glucose metabolism, but it does not exclude the possibility that daily normalization (or elevation) of nocturnal concentrations might affect the β-cell in the longer term. Prior experiments with greater and more prolonged elevations in FFA produce significant alterations in insulin secretion and action (23). However, these elevations were supraphysiologic and do not replicate in vivo conditions.
In the current experiment, we manipulated both FFA and glucose in part because insulin can be used to lower both simultaneously, while avoiding potential off-target effects of pharmacotherapy such as acipimox or niacin that might directly affect β-cell secretion or insulin signaling. A consequence of the experimental design was that glucose infusion in people with NFG/NGT stimulated insulin secretion, which normally would have suppressed FFA. We prevented the decrease in FFA concentrations by infusing a lipid emulsion, creating a state of greater glucose and FFA concentrations than is normal for the degree of hyperinsulinemia. Nevertheless, the intervention altered fasting insulin secretion, allowing us to at least examine the relationship between an acute change in overnight insulin secretion and the subsequent prandial response.
Another consideration is the presence of glycerol in the lipid emulsion we used in the experiment. Previously, in keeping with prior findings (56), we showed that glycerol per se has no direct effect on β-cell function (23). Despite hyperinsulinemia and hyperglycemia, the presence of FFA prevented significant overnight suppression of EGP—again implying a biological effect of FFA manipulation. An alternative explanation is that the provision of glycerol stimulated gluconeogenesis and fasting EGP (57). Nevertheless, postprandial insulin action and suppression of EGP were unchanged despite the prior exposure to Intralipid (and glycerol).
Sustained hyperinsulinemia to a greater degree and duration than utilized here can produce impaired insulin action (58). On the other hand, in people with type 2 diabetes, overnight insulin infusion to produce normoglycemia improved hepatic insulin action (59). In this experiment, we observed no effect of insulin on the suppression of postprandial EGP or insulin action. It is therefore unlikely that the use of insulin to lower glucose and FFA in this experiment interfered with our conclusions.
We conclude that short-term manipulation of the fasting milieu by using insulin to lower FFA and glucose in people with IFG/IGT (and decrease insulin secretion) or conversely raising glucose and supplying FFA overnight to stimulate insulin secretion does not alter the overall subsequent prandial response. However, the change in ϕd suggests that even in people without diabetes, short-term increases in secretory demand might alter some of the preformed pools of insulin within the β-cell.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Figs. S1–S4: https://doi.org/10.6084/m9.figshare.22961753.
GRANTS
The authors acknowledge the support of the Mayo Clinic General Clinical Research Center (DK TR000135). Dr Vella is supported by DK78646, DK116231, and DK126206. Dr Jensen is supported by DK40484 and DK45343. Dr Dalla Man was supported by MIUR (Italian Minister for Education) under the initiative “Departments of Excellence” (Law 232/2016).
DISCLOSURES
Dr Vella is the recipient of an investigator-initiated grant from Novo Nordisk and has consulted for Zeeland Pharmaceuticals, Hanmi Pharmaceuticals, Crinetics and Rezolute. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
M.D.J. and A.V. conceived and designed research; D.S.W., A.M.E., A.A.W., and A.V. performed experiments; D.S.W., M.C.L., Y.S., K.R.B., C.C., C.D.M., and A.V. analyzed data; M.C.L., K.R.B., and A.V. interpreted results of experiments; M.C.L. and A.V. prepared figures; A.V. drafted manuscript; D.S.W., M.C.L., Y.S., A.M.E., A.A.W., K.R.B., C.C., C.D.M., M.D.J., and A.V. edited and revised manuscript; D.S.W., M.C.L., Y.S., A.M.E., A.A.W., K.R.B., C.C., C.D.M., M.D.J., and A.V. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge the excellent editorial assistance of M. M. Davis, Endocrine Research Unit, Mayo Clinic, Rochester, MN.
REFERENCES
- 1. Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, Nathan DM; Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 346: 393–403, 2002. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Albright AL, Gregg EW. Preventing type 2 diabetes in communities across the U.S.: the National Diabetes Prevention Program. Am J Prev Med 44: S346–S351, 2013. doi: 10.1016/j.amepre.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bock G, Dalla Man C, Campioni M, Chittilapilly E, Basu R, Toffolo G, Cobelli C, Rizza R. Pathogenesis of pre-diabetes: mechanisms of fasting and postprandial hyperglycemia in people with impaired fasting glucose and/or impaired glucose tolerance. Diabetes 55: 3536–3549, 2006. doi: 10.2337/db06-0319. [DOI] [PubMed] [Google Scholar]
- 4. Cobelli C, Dalla Man C, Toffolo G, Basu R, Vella A, Rizza R. The oral minimal model method. Diabetes 63: 1203–1213, 2014. doi: 10.2337/db13-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bock G, Dalla Man C, Campioni M, Chittilapilly E, Basu R, Toffolo G, Cobelli C, Rizza R. Effects of nonglucose nutrients on insulin secretion and action in people with pre-diabetes. Diabetes 56: 1113–1119, 2007. doi: 10.2337/db06-1272. [DOI] [PubMed] [Google Scholar]
- 6. Sathananthan A, Man CD, Zinsmeister AR, Camilleri M, Rodeheffer RJ, Toffolo G, Cobelli C, Rizza RA, Vella A. A concerted decline in insulin secretion and action occurs across the spectrum of fasting and postchallenge glucose concentrations. Clin Endocrinol (Oxf) 76: 212–219, 2012. doi: 10.1111/j.1365-2265.2011.04159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kohlenberg JD, Laurenti MC, Egan AM, Wismayer DS, Bailey KR, Cobelli C, Man CD, Vella A. Differential contribution of α and β cell dysfunction to impaired fasting glucose and impaired glucose tolerance. Diabetologia 66: 201–212, 2023. doi: 10.1007/s00125-022-05794-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lu J, Varghese RT, Zhou L, Vella A, Jensen MD. Glucose tolerance and free fatty acid metabolism in adults with variations in TCF7L2 rs7903146. Metabolism 68: 55–63, 2017. doi: 10.1016/j.metabol.2016.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Staaf J, Ubhayasekera SJ, Sargsyan E, Chowdhury A, Kristinsson H, Manell H, Bergquist J, Forslund A, Bergsten P. Initial hyperinsulinemia and subsequent β-cell dysfunction is associated with elevated palmitate levels. Pediatr Res 80: 267–274, 2016. doi: 10.1038/pr.2016.80. [DOI] [PubMed] [Google Scholar]
- 10. Rebelos E, Seghieri M, Natali A, Balkau B, Golay A, Piatti PM, Lalic NM, Laakso M, Mari A, Ferrannini E. Influence of endogenous NEFA on β cell function in humans. Diabetologia 58: 2344–2351, 2015. doi: 10.1007/s00125-015-3685-6. [DOI] [PubMed] [Google Scholar]
- 11. Jensen MD, Haymond MW, Gerich JE, Cryer PE, Miles JM. Lipolysis during fasting. Decreased suppression by insulin and increased stimulation by epinephrine. J Clin Invest 79: 207–213, 1987. doi: 10.1172/JCI112785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thyfault JP, Kraus RM, Hickner RC, Howell AW, Wolfe RR, Dohm GL. Impaired plasma fatty acid oxidation in extremely obese women. Am J Physiol Endocrinol Physiol 287: E1076–E1081, 2004. doi: 10.1152/ajpendo.00177.2004. [DOI] [PubMed] [Google Scholar]
- 13. Jensen MD, Haymond MW, Rizza RA, Cryer PE, Miles JM. Influence of body fat distribution on free fatty acid metabolism in obesity. J Clin Invest 83: 1168–1173, 1989. doi: 10.1172/JCI113997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Malin SK, Rynders CA, Weltman JY, Barrett EJ, Weltman A. Exercise intensity modulates glucose-stimulated insulin secretion when adjusted for adipose, liver and skeletal muscle insulin resistance. PLoS One 11: e0154063, 2016. doi: 10.1371/journal.pone.0154063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pennartz C, Schenker N, Menge BA, Schmidt WE, Nauck MA, Meier JJ. Chronic reduction of fasting glycemia with insulin glargine improves first- and second-phase insulin secretion in patients with type 2 diabetes. Diabetes Care 34: 2048–2053, 2011. doi: 10.2337/dc11-0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Belfort R, Mandarino L, Kashyap S, Wirfel K, Pratipanawatr T, Berria R, Defronzo RA, Cusi K. Dose-response effect of elevated plasma free fatty acid on insulin signaling. Diabetes 54: 1640–1648, 2005. doi: 10.2337/diabetes.54.6.1640. [DOI] [PubMed] [Google Scholar]
- 17. Cusi K, Kashyap S, Gastaldelli A, Bajaj M, Cersosimo E. Effects on insulin secretion and insulin action of a 48-h reduction of plasma free fatty acids with acipimox in nondiabetic subjects genetically predisposed to type 2 diabetes. Am J Physiol Endocrinol Physiol 292: E1775–E1781, 2007. doi: 10.1152/ajpendo.00624.2006. [DOI] [PubMed] [Google Scholar]
- 18. Boden G, Chen X. Effects of fatty acids and ketone bodies on basal insulin secretion in type 2 diabetes. Diabetes 48: 577–583, 1999. doi: 10.2337/diabetes.48.3.577. [DOI] [PubMed] [Google Scholar]
- 19. Boden G, Chen X, Rosner J, Barton M. Effects of a 48-h fat infusion on insulin secretion and glucose utilization. Diabetes 44: 1239–1242, 1995. doi: 10.2337/diab.44.10.1239. [DOI] [PubMed] [Google Scholar]
- 20. Carpentier A, Mittelman SD, Lamarche B, Bergman RN, Giacca A, Lewis GF. Acute enhancement of insulin secretion by FFA in humans is lost with prolonged FFA elevation. Am J Physiol Endocrinol Physiol 276: E1055–E1066, 1999. doi: 10.1152/ajpendo.1999.276.6.E1055. [DOI] [PubMed] [Google Scholar]
- 21. Broussard JL, Kolka CM, Castro AVB, Asare Bediako I, Paszkiewicz RL, Szczepaniak EW, Szczepaniak LS, Knutson KL, Kim SP, Bergman RN. Elevated nocturnal NEFA are an early signal for hyperinsulinaemic compensation during diet-induced insulin resistance in dogs. Diabetologia 58: 2663–2670, 2015. doi: 10.1007/s00125-015-3721-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miller MR, Pereira RI, Langefeld CD, Lorenzo C, Rotter JI, Chen YD, Bergman RN, Wagenknecht LE, Norris JM, Fingerlin TE. Levels of free fatty acids (FFA) are associated with insulin resistance but do not explain the relationship between adiposity and insulin resistance in Hispanic Americans: the IRAS Family Study. J Clin Endocrinol Metab 97: 3285–3291, 2012. doi: 10.1210/jc.2012-1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shah M, Varghese RT, Miles JM, Piccinini F, Dalla Man C, Cobelli C, Bailey KR, Rizza RA, Vella A. TCF7L2 genotype and α-cell function in humans without diabetes. Diabetes 65: 371–380, 2016. doi: 10.2337/db15-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dobbins RL, Chester MW, Stevenson BE, Daniels MB, Stein DT, McGarry JD. A fatty acid- dependent step is critically important for both glucose- and non-glucose-stimulated insulin secretion. J Clin Invest 101: 2370–2376, 1998. doi: 10.1172/JCI1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boden G. Obesity, insulin resistance and free fatty acids. Curr Opin Endocrinol Diabetes Obes 18: 139–143, 2011. doi: 10.1097/MED.0b013e3283444b09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vella A, Rizza RA. Application of isotopic techniques using constant specific activity or enrichment to the study of carbohydrate metabolism. Diabetes 58: 2168–2174, 2009. doi: 10.2337/db09-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vella A, Reed AS, Charkoudian N, Shah P, Basu R, Basu A, Joyner MJ, Rizza RA. Glucose-induced suppression of endogenous glucose production: dynamic response to differing glucose profiles. Am J Physiol Endocrinol Physiol 285: E25–E30, 2003. doi: 10.1152/ajpendo.00530.2002. [DOI] [PubMed] [Google Scholar]
- 28. Vella A, Shah P, Basu R, Basu A, Camilleri M, Schwenk WF, Rizza RA. Type I diabetes mellitus does not alter initial splanchnic glucose extraction or hepatic UDP-glucose flux during enteral glucose administration. Diabetologia 44: 729–737, 2001. doi: 10.1007/s001250051682. [DOI] [PubMed] [Google Scholar]
- 29. Vella A, Shah P, Basu R, Basu A, Holst JJ, Rizza RA. Effect of glucagon-like peptide 1(7-36) amide on glucose effectiveness and insulin action in people with type 2 diabetes. Diabetes 49: 611–617, 2000. doi: 10.2337/diabetes.49.4.611. [DOI] [PubMed] [Google Scholar]
- 30. Smushkin G, Sathananthan M, Piccinini F, Dalla Man C, Law JH, Cobelli C, Zinsmeister AR, Rizza RA, Vella A. The effect of a bile acid sequestrant on glucose metabolism in subjects with type 2 diabetes. Diabetes 62: 1094–1101, 2013. doi: 10.2337/db12-0923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Placzkowski KA, Vella A, Thompson GB, Grant CS, Reading CC, Charboneau JW, Andrews JC, Lloyd RV, Service FJ. Secular trends in the presentation and management of functioning insulinoma at the Mayo Clinic, 1987–2007. J Clin Endocrinol Metab 94: 1069–1073, 2009. doi: 10.1210/jc.2008-2031. [DOI] [PubMed] [Google Scholar]
- 32. Song Y, Zhou L, Jensen MD. Errors in measuring plasma free fatty acid concentrations with a popular enzymatic colorimetric kit. Clin Biochem 66: 83–90, 2019. doi: 10.1016/j.clinbiochem.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Beylot M, Previs SF, David F, Brunengraber H. Determination of the 13C-labeling pattern of glucose by gas chromatography-mass spectrometry. Anal Biochem 212: 526–531, 1993. doi: 10.1006/abio.1993.1363. [DOI] [PubMed] [Google Scholar]
- 34. Steele R, Bjerknes C, Rathgeb I, Altszuler N. Glucose uptake and production during the oral glucose tolerance test. Diabetes 17: 415–421, 1968. doi: 10.2337/diab.17.7.415. [DOI] [PubMed] [Google Scholar]
- 35. Dalla Man C, Caumo A, Basu R, Rizza R, Toffolo G, Cobelli C. Measurement of selective effect of insulin on glucose disposal from labeled glucose oral test minimal model. Am J Physiol Endocrinol Physiol 289: E909–E914, 2005. doi: 10.1152/ajpendo.00299.2004. [DOI] [PubMed] [Google Scholar]
- 36. Breda E, Cavaghan MK, Toffolo G, Polonsky KS, Cobelli C. Oral glucose tolerance test minimal model indexes of β-cell function and insulin sensitivity. Diabetes 50: 150–158, 2001. doi: 10.2337/diabetes.50.1.150. [DOI] [PubMed] [Google Scholar]
- 37. Van Cauter E, Mestrez F, Sturis J, Polonsky KS. Estimation of insulin secretion rates from C-peptide levels. Comparison of individual and standard kinetic parameters for C-peptide clearance. Diabetes 41: 368–377, 1992. doi: 10.2337/diab.41.3.368. [DOI] [PubMed] [Google Scholar]
- 38. Sharma A, Laurenti MC, Dalla Man C, Varghese RT, Cobelli C, Rizza RA, Matveyenko A, Vella A. Glucose metabolism during rotational shift-work in healthcare workers. Diabetologia 60: 1483–1490, 2017. doi: 10.1007/s00125-017-4317-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cobelli C, Man CD, Sparacino G, Magni L, De Nicolao G, Kovatchev BP. Diabetes: models, signals, and control. IEEE Rev Biomed Eng 2: 54–96, 2009. doi: 10.1109/RBME.2009.2036073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bratanova-Tochkova TK, Cheng H, Daniel S, Gunawardana S, Liu YJ, Mulvaney-Musa J, Schermerhorn T, Straub SG, Yajima H, Sharp GW. Triggering and augmentation mechanisms, granule pools, and biphasic insulin secretion. Diabetes 51, Suppl 1: S83–S90, 2002. doi: 10.2337/diabetes.51.2007.s83. [DOI] [PubMed] [Google Scholar]
- 41. Gandasi NR, Yin P, Omar-Hmeadi M, Ottosson Laakso E, Vikman P, Barg S. Glucose-dependent granule docking limits insulin secretion and is decreased in human type 2 diabetes. Cell Metab 27: 470–478.e4, 2018. doi: 10.1016/j.cmet.2017.12.017. [DOI] [PubMed] [Google Scholar]
- 42. Costes S, Langen R, Gurlo T, Matveyenko AV, Butler PC. β-Cell failure in type 2 diabetes: a case of asking too much of too few? Diabetes 62: 327–335, 2013. doi: 10.2337/db12-1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rivera JF, Costes S, Gurlo T, Glabe CG, Butler PC. Autophagy defends pancreatic β cells from human islet amyloid polypeptide-induced toxicity. J Clin Invest 124: 3489–3500, 2014. doi: 10.1172/JCI71981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest 110: 1389–1398, 2002. doi: 10.1172/JCI0216886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with β-cell failure and diabetes. Endocr Rev 29: 317–333, 2008. [Erratum in Endocr Rev 29: 631, 2008]. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Haataja L, Gurlo T, Huang CJ, Butler PC. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr Rev 29: 303–316, 2008. doi: 10.1210/er.2007-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fonseca SG, Ishigaki S, Oslowski CM, Lu S, Lipson KL, Ghosh R, Hayashi E, Ishihara H, Oka Y, Permutt MA, Urano F. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J Clin Invest 120: 744–755, 2010. doi: 10.1172/JCI39678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bonnefond A, Froguel P, Vaxillaire M. The emerging genetics of type 2 diabetes. Trends Mol Med 16: 407–416, 2010. doi: 10.1016/j.molmed.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 49. Dombroski BA, Nayak RR, Ewens KG, Ankener W, Cheung VG, Spielman RS. Gene expression and genetic variation in response to endoplasmic reticulum stress in human cells. Am J Hum Genet 86: 719–729, 2010. doi: 10.1016/j.ajhg.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Herbert TP, Laybutt DR. A reevaluation of the role of the unfolded protein response in islet dysfunction: maladaptation or a failure to adapt? Diabetes 65: 1472–1480, 2016. doi: 10.2337/db15-1633. [DOI] [PubMed] [Google Scholar]
- 51. Alarcon C, Boland BB, Uchizono Y, Moore PC, Peterson B, Rajan S, Rhodes OS, Noske AB, Haataja L, Arvan P, Marsh BJ, Austin J, Rhodes CJ. Pancreatic β-cell adaptive plasticity in obesity increases insulin production but adversely affects secretory function. Diabetes 65: 438–450, 2016. doi: 10.2337/db15-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ward WK, LaCava EC, Paquette TL, Beard JC, Wallum BJ, Porte D Jr.. Disproportionate elevation of immunoreactive proinsulin in type 2 (non-insulin-dependent) diabetes mellitus and in experimental insulin resistance. Diabetologia 30: 698–702, 1987. doi: 10.1007/BF00296991. [DOI] [PubMed] [Google Scholar]
- 53. DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 28: 1092–1100, 2005. doi: 10.2337/diacare.28.5.1092. [DOI] [PubMed] [Google Scholar]
- 54. Egan AM, Laurenti MC, Hurtado Andrade MD, Dalla Man C, Cobelli C, Bailey KR, Vella A. Limitations of the fasting proinsulin to insulin ratio as a measure of β-cell health in people with and without impaired glucose tolerance. Eur J Clin Invest 51: e13469, 2021. doi: 10.1111/eci.13469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sharma A, Varghese RT, Shah M, Man CD, Cobelli C, Rizza RA, Bailey KR, Vella A. Impaired insulin action is associated with increased glucagon concentrations in non-diabetic humans. J Clin Endocrinol Metab 103: 314–319, 2018. doi: 10.1210/jc.2017-01197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Noel RJ, Antinozzi PA, McGarry JD, Newgard CB. Engineering of glycerol-stimulated insulin secretion in islet β cells. Differential metabolic fates of glucose and glycerol provide insight into mechanisms of stimulus-secretion coupling. J Biol Chem 272: 18621–18627, 1997. doi: 10.1074/jbc.272.30.18621. [DOI] [PubMed] [Google Scholar]
- 57. Baba H, Zhang XJ, Wolfe RR. Glycerol gluconeogenesis in fasting humans. Nutrition 11: 149–153, 1995. doi: 10.1016/j.metabol.2022.155214. [DOI] [PubMed] [Google Scholar]
- 58. Rizza RA, Mandarino LJ, Genest J, Baker BA, Gerich JE. Production of insulin resistance by hyperinsulinaemia in man. Diabetologia 28: 70–75, 1985. doi: 10.1007/BF00279918. [DOI] [PubMed] [Google Scholar]
- 59. Wise SD, Nielsen MF, Cryer PE, Rizza RA. Overnight normalization of glucose concentrations improves hepatic but not extrahepatic insulin action in subjects with type 2 diabetes mellitus. J Clin Endocrinol Metab 83: 2461–2469, 1998. doi: 10.1210/jcem.83.7.4976. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figs. S1–S4: https://doi.org/10.6084/m9.figshare.22961753.
Data Availability Statement
Data will be made available upon reasonable request.